
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn vitro anti-trypanosomal activity of 3-(aryl)-6-piperazin1,2,4-triazolo[3,4-a]phthalazines-loaded ultrathin polymeric particles: effect of polymer type and particle size†
Karina
González
ab,
Ender
Medina
b,
Elena
Aguilera
c,
Gema
González‡
d,
Marcos A.
Sabino
 *b and
Angel H.
Romero
*c
*b and
Angel H.
Romero
*c
aUnidad de Química Medicinal, Facultad de Farmacia, Universidad Central de Venezuela, Los Chaguaramos, 1041-A Caracas, Venezuela
bGrupo de Investigación B5IDA, Departamento de Química, Universidad Simón Bolívar, Sartenejas, 1080 Caracas, Venezuela. E-mail: msabinog@gmail.com
cGrupo de Química Orgánica Medicinal, Instituto de Química Biológica, Facultad de Ciencias, Universidad de la República, Iguá 4225, 11400 Montevideo, Uruguay. E-mail: angel.ucv.usb@gmail.com; aromero@fcien.edu.uy
dCentro de Ingeniería de Materiales y Nanotecnología, Laboratorio de Materiales, Instituto Venezolano de Investigaciones Científicas (IVIC), Carretera Panamericana Km 11, 20632 Caracas, Venezuela
First published on 21st February 2024
Abstract
3-(Aryl)-6-piperazin-1,2,4-triazolo[3,4-a]phthalazines have shown great potential as leishmanicidal agents. Herein, we prepared a series of PLGA-, PLA- and PCL-based-microparticles/nanoparticles of different particle sizes and loaded them with active 3-(aryl)-6-piperazin-1,2,4-triazolo[3,4-a]phthalazines TF1 and TF2. The synthesized microparticles/nanoparticles seek to improve the leishmanicidal activity of 3-(aryl)-6-piperazin-1,2,4-triazolo[3,4-a]phthalazines and extend its effect to the T. cruzi parasite. The encapsulates were prepared using a microemulsification method, achieving an encapsulation percentage between 89% and 99% for PLGA-, PLA- and PCL-microparticles/nanoparticles. The encapsulation of triazolo-phthalazines was confirmed through UV-Vis or EDX analyses. From SEM analysis, two nanoparticle or microparticle/nanoparticle system-loaded TF1 or TF2 with mean sizes of 250, 400, 600–900 or 900–2000 nm were obtained for each of PLGA, PLA and PCL polymeric matrices. TEM analysis revealed that all the prepared microparticles/nanoparticles consisted of particles and not spheres. The microencapsulates/nanoencapsulates showed an acceptable drug release under physiological conditions, achieving a continuous release for up to 96 hours for most of the studied cases. From biological evaluation, encapsulation with PLGA and PLA showed a positive effect against the in vitro model of both parasites showing a decrease in their EC50 values compared with free compounds. Conversely, no improvement in trypanosomaticidal activity was found with PCL encapsulation. Importantly, it was found that that either the small particle size of the capsulate system or facile drug release favored anti-trypanosomatid activity. The three polymeric matrices showed a discrete but slight increase in toxicity toward J774.1 macrophages compared to free compounds. This may be associated with the facile penetration of the polymeric matrix across the macrophage membrane, favoring against intracellular forms of parasites. This study shows that either the particle size or the type of polymer represent key issues for improving the trypanosomaticidal activity of polymeric nanoformulations.
1. Introduction
Chagas disease (CD) and visceral leishmaniasis (VL), which are caused by parasitic trypanosomatids of T. cruzi and L. infantum parasites, respectively, constitute a group of neglected tropical diseases (NTDs). These diseases are widespread in tropical and subtropical areas owing to poor hygiene and insufficient health infrastructures.1 Leishmaniasis affects about 12 million people worldwide with 350 million people under the risk of infection in 98 developing countries annually,2 whereas Chagas disease affects around 8 million people in the Americas—from Mexico and Central America to Argentina and Patagonia.3 In particular, visceral leishmaniasis is one of the clinical manifestations of leishmaniasis, and it may be fatal for 90% of untreated cases.2To date, no registered vaccines are available for both the NTDs. Alternatively, there are a limited number of therapeutic options, and only two approved drugs are currently employed for the treatment of each of these diseases: (i) pentavalent antimonials (e.g., Glucantime and Pentostam) against Leishmaniasis and (ii) benznidazole and nifurtimox against Chagas disease (Chart 1). A second line of leishmanicidal agents, including pentamidine, amphotericin B and miltefosine (Chart 1), as well as anti-Chagas drugs such as azole antifungals (e.g., posaconazole, ketoconazole, ravuconazole), are frequently used when the first line fails.4 However, these drugs present several disadvantages such as toxicity (affecting the heart, liver and kidneys), high cost, low therapeutic efficacy, prolonged treatment times (30–60 days) and parasite resistance. Thus, limited therapeutic options, suboptimal diagnostics, poor community awareness, drug resistance, toxicity, asymptomatic carriers, and, most importantly, absence of vaccines encourage more investigation into a more effective treatment of leishmaniasis and Chagas disease. Moreover, the challenge is even more significant by the rigorous pharmacological requirements, which seek to minimize side-effect risks.
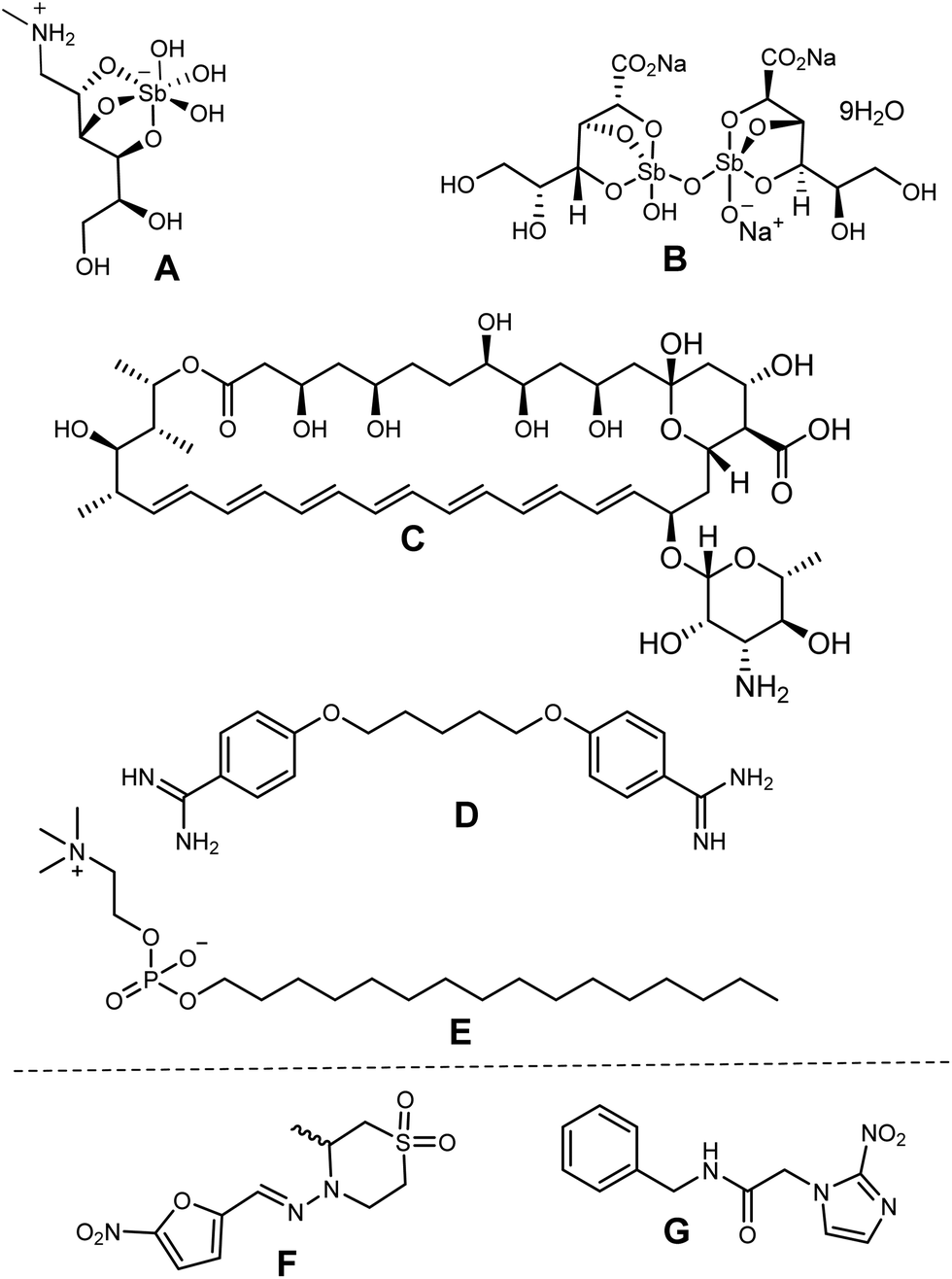 | ||
| Chart 1 Structures of the main approved drugs: (A) Glucantime, (B) Pentostam, (C) amphotericin B, (D) pentamidine and (E) miltefosine against Leishmaniasis; and (F) nifurtimox and (G) benznidazole as anti-Chagas drugs. | ||
In this sense, the development of drug delivery systems (DDSs) is an attractive alternative to design leishmanicidal or antichagasic formulation drugs.5,6 The DDSs favor a controlled drug release post-administration, improving the bioavailability and pharmacokinetic parameters of the encapsulated drug, as well as reducing toxicity side effects.5 These nanoformulations have overcome biological barriers, such as gastrointestinal pH or blood-brain barrier, without any loss of drug efficacy. Concerning the NTDs, as both diseases are characterized by infection of the host cell from the intracellular amastigote, the use of a nanocarrier system containing an anti-trypanosomatid drug represents a promising approach to penetrate the macrophages’ cells via endocytosis7a and reach the infectious parasite, solving one of the typical drawbacks of anti-trypanosomatid drugs related to the penetration of the host cell.7b The latter favors the specific antiparasitic activity and enhances the efficient delivery. The application of nanoformulations has been more extended against leishmaniasis, and the development of their DDSs started in 1992.8 Since then, as the most fundamental component in the construction of nanostructures, many NPs have provided high stability and loading, ease of incorporation of both hydrophilic and hydrophobic substances, feasibility of variable routes of administration, and relevant biological improvements at preclinical levels for leishmanicidal drugs.9,10 Interestingly, encapsulation facilitates the improved and sustained release of cargo at the specific organ or tissues where it is destined, which is essential against diseases such as Chagas disease and Visceral leishmaniasis in organs or issues. Encapsulation of drugs into nanocarriers also provides enhanced protection against intracellular and extracellular degradation compared to non-encapsulated drugs.11,12 It is important to mention that formulation-based targeted drug delivery has helped in overcoming drug resistance for in vitro and in vivo models of leishmaniasis as consequence of the higher drug accumulation inside the macrophage through mucosal penetration.12b Another beneficial aspect of nanoscale delivery is the pH adaptability of the drug moieties in microenvironments.13 This is important since the parasite resides in the acidic phagolysosome compartment within macrophages, where the pH is low. A drug moiety capable of reaching a target should resist the variable pH seen across different tissues or subcellular compartments. Therefore, pH-dependent drug release is very important for the site-specific accumulation and action of drugs. NPs consisting of a drug-loaded polylactic-co-glycolic acid (PLGA) core and wrapped with acid-triggered membrane peptide have shown facilitated internalization into cells within the acidic microenvironment where intracellular conditions degrade the NPs, thereby releasing the chemotherapeutic cargo.14–21
Within the DDSs, those based on polymeric micro- and nanoparticles are ideal for the design of DDSs because it is possible to reach the nano-dimension, which allows for access to small regions of the parasite membrane.9,10 Among the polymeric matrices, biodegradable and biocompatible PLGA, poly-(D,L-lactic acid) (PLA) and poly-caprolactone (PCL) nanoparticles (NPs) are commonly used for the preparation of NPs-loaded drugs. They are approved by the U.S. Food and Drug Administration (FDA) and European Medicines Agency (EMA) as drug carriers for human applications due to their high biocompatibility.22 These polymers present several advantages, including reduction of side effects, improvement of oral bioavailability, sustained and controlled drug release, tissue targeting, improvements in drug transportation, adjuvant co-encapsulation and protection from enzymatic degradation. In particular, the PLGA-based nanoparticle systems, beyond its function as a drug carrier, have demonstrated the ability to induce humoral and cell-mediated specific immune responses via activation of dendritic cells in animals models.14–21 Thus, they are a convenient polymeric matrix for the design of antitrypanosomal agents because it is well documented that these parasites are sensitive to the immunological host's effectors.
As described above, the use of DDS offers a great advantage to improve the biological activity of reference drugs and it could be extended to active leishmanicidal compounds. Recently, we reported that 3-aryl-6-piperazin-1,2,4-triazolo[3,4-a]phthalazines showed good potential as leishmanicidal agents, displaying an IC50 of about 9 μM against intracellular amastigote of L. braziliensis and selectivities indexes between 10 and 15.23 The compounds showed a submicromolar IC50 against promastigote and axenic amastigote, emerging as a potential and safer leishmanicidal agent for further investigation. This chemical system was derived from a optimization of phthalazine derivatives.25–28 These 3-(3-nitrophenyl)-6-piperazin-1,2,4-triazolo[3,4-a]phthalazines have also shown potent cytotoxic activity against cancer strains with a modulation of potassium channels.24 To enhance the potential of 3-(3-nitrophenyl)-6-piperazin-1,2,4-triazolo[3,4-a]phthalazine, we evaluated the effect of a controlled release through the preparation of micro/nanoencapsulates loaded with two active leishmanicidal triazolo-phthalazines, TF1 and TF2, using three types of polymeric matrices, PLGA, PLA and PCL (Chart 2). Then, we evaluated the effect of the entrapment with these three polymeric matrices on the antitrypanosomal activity of the most active 3-(3-nitrophenyl)-6-piperazin-1,2,4-triazolo[3,4-a]phthalazine, TF1, providing a convenient formulation for an enhancement in the in vitro and in vivo activities of these organic compounds against both NDTs. Also, a discretely active triazolo-phthalazine such as 3-(4-fluorophenyl)-6-piperazin-1,2,4-triazolo[3,4-a]phthalazine, TF2, was studied to evaluate the power of the entrapment.
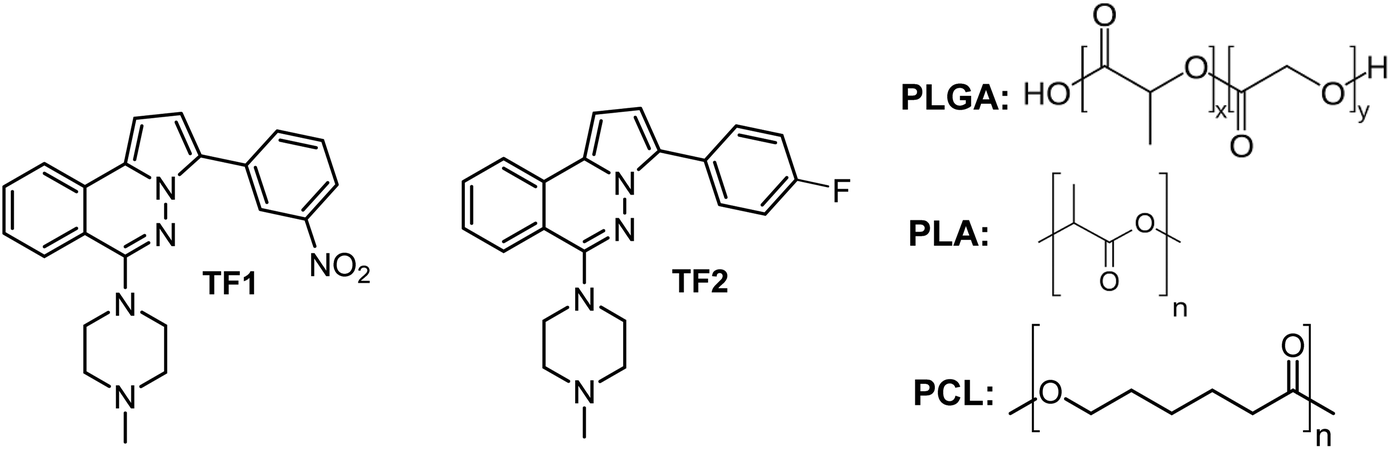 | ||
| Chart 2 Structure of the 3-(3-aryl)-6-piperazin-1,2,4-triazolo[3,4-a]phthalazines, TF1 and TF2, and polymeric matrices of the PLGA copolymer and homopolymers of PLA and PCL. | ||
2. Results and discussion
2.1. Synthesis and characterization of loaded nanoparticles or microparticles/nanoparticles
The use of polymeric matrices for microencapsulation/nanoencapsulation has been shown to improve the effectiveness of diverse therapeutic agents against various type of diseases including leishmaniasis.10 That effectiveness is associated either with the improvement of the therapeutic effect and decrease of toxicity or with the improvement of pharmacokinetic parameters, which could be favored by controlled drug delivery.5 In the current study, we used PLA, PCL and PLGA polymeric matrices to construct the microparticle/nanoparticle encapsulates of two 3-(3-aryl)-6-piperazin-1,2,4-triazolo[3,4-a]phthalazines, seeking to improve its known leishmanicidal activity. The new encapsulate chemical systems were extended to assess their effectiveness against the T. cruzi parasite. The use of the PLGA copolymer has been widely applied for the encapsulation of leishmanicidal drugs,10 whereas the use of PLA and PCL homopolymers is much less common. Thus, their encapsulation will be very interesting to evaluate the effect of the nature of the polymeric matrix. PLA is a more hydrophilic polymer owing to the presence of the hydroxylic moiety, whereas PCL may be defined as a lipophilic polymer by its long aliphatic hydrocarbon chain.Beyond the effect of the polymer, we also analyzed the effect of the particle sizes, for which micronanoparticles/nanoparticles with different mean particles sizes were prepared. In general, the method of preparation involved microemulsification by dripping of the polymer/drug dissolved in a common solvent. This allows for a high degree of encapsulation, the formation of nanodroplets that will later form nanoparticles, and a good dispersion of the drug within the polymeric matrix. This method also plays an important role in terms of the particle size that will be obtained. Many studies have showed that the size, shape and surface coating of NPs are able to modulate the therapeutic and toxicity profile of the active compounds. For example, spherical NPs can generally be comparatively more toxic than larger particles because the former are able to move more freely than bulkier molecules. In addition, the size of the NPs has diverse benefits, such as improvement of cellular uptake, favorable interactions with the protein receptor associated with the immune system, and its favorable clearance from the body.5,29 The latter is of great relevance against diseases such as VL because the parasite primarily infects the macrophages of the liver and spleen. Thus, the partial activation of the host cell from a controlled drug delivery method is an attractive strategy for the design of antileishmanial agents, which could be extended to Chagas disease.5
Based on the aspects mentioned above, either two PLA-NPs or two PCL-NPs were obtained for both TF1 and TF2 to give eight formulations: PLA-TF1-600/1200, PLA-TF1-1200/2000, PCL-TF1-600/2000, PCL-TF1-1200/2000, PLA-TF2-300/600, PLA-TF2-300/3000, PCL-TF2-600–2000 and PCL-TF2-600/3000. For PLGA, only a PLGA-NP system was obtained for each compound, TF1 and TF2. The microencapsulates/nanoencapsulates were prepared from the O/W emulsion-solvent evaporation method with a few modifications, and correctly characterized using SEM and TEM for visualizing the shape, morphology, and size of nanoparticles. Furthermore, UV-Vis or EDX analysis was used to verify the encapsulation of the organic TF1 and TF2 compounds. From SEM analysis, it should be noted that most of the microformulations/nanoformulations consisted of spherical nanoparticles (Fig. 1A–E, 2A–E and 3A–B), which were homogeneous and uniformly distributed. A few agglomerates of particles can be observed, which may be due to the washing and freeze-drying processes. For the PLA-TF1-NPs, PLA-TF1-600/2000 consists of a microformulation/nanoformulation containing compound TF1 loaded into microformulation/nanoparticles with sizes between 600 and 2000 nanometers (Fig. 1B). PLA-TF1-1200/2000 is constituted by microparticles with sizes between 1200 and 2000 nanometers (Fig. 1C). Meanwhile, the PLA-TF2-NPs consisted of a nanoformulation, PLA-TF2-300/600, containing nanoparticles with sizes between 300 and 600 nanometers (Fig. 1D). The other PLA-TF2-300/3000 consisted of a polymeric system containing microparticles/nanoparticles with well-dispersed sizes between 300 and 3000 nanometers (Fig. 1E). The unloaded PLA-NPs exhibited particle sizes between 300 and 6500 nanometers (Fig. 1A).
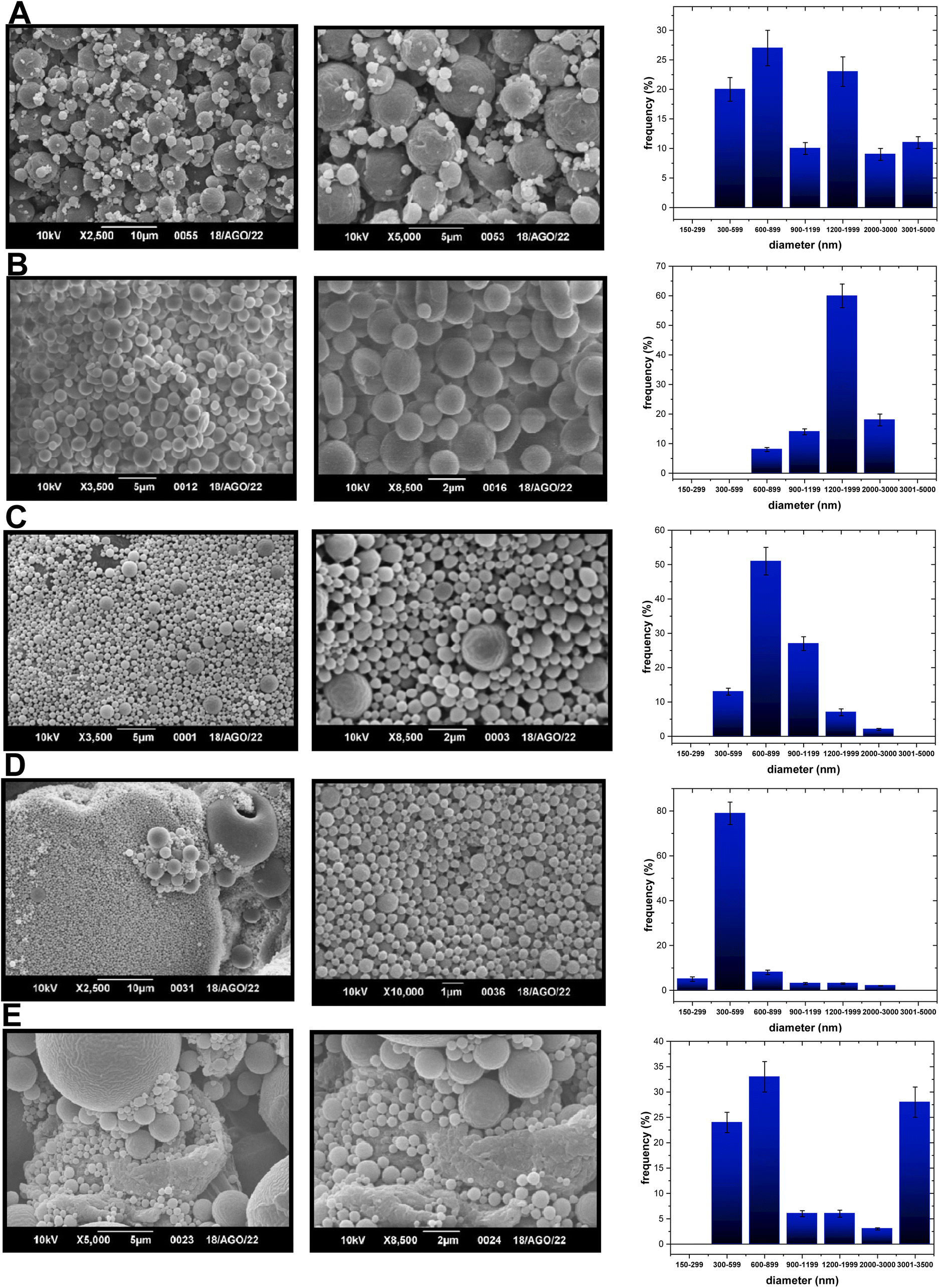 | ||
| Fig. 1 SEM images of (A) PLA-NPs-300/2000, (B) PLA-TP1-NPs-1200/200, and (C) PLA-TP1-NPs-600/1200 at 2, 5 or 10 microns; (D) PLA-TP2-NPs-300/900 and (E) PLA-TP2-NPs-300/3200 at 1, 2, 5 or 10 microns. | ||
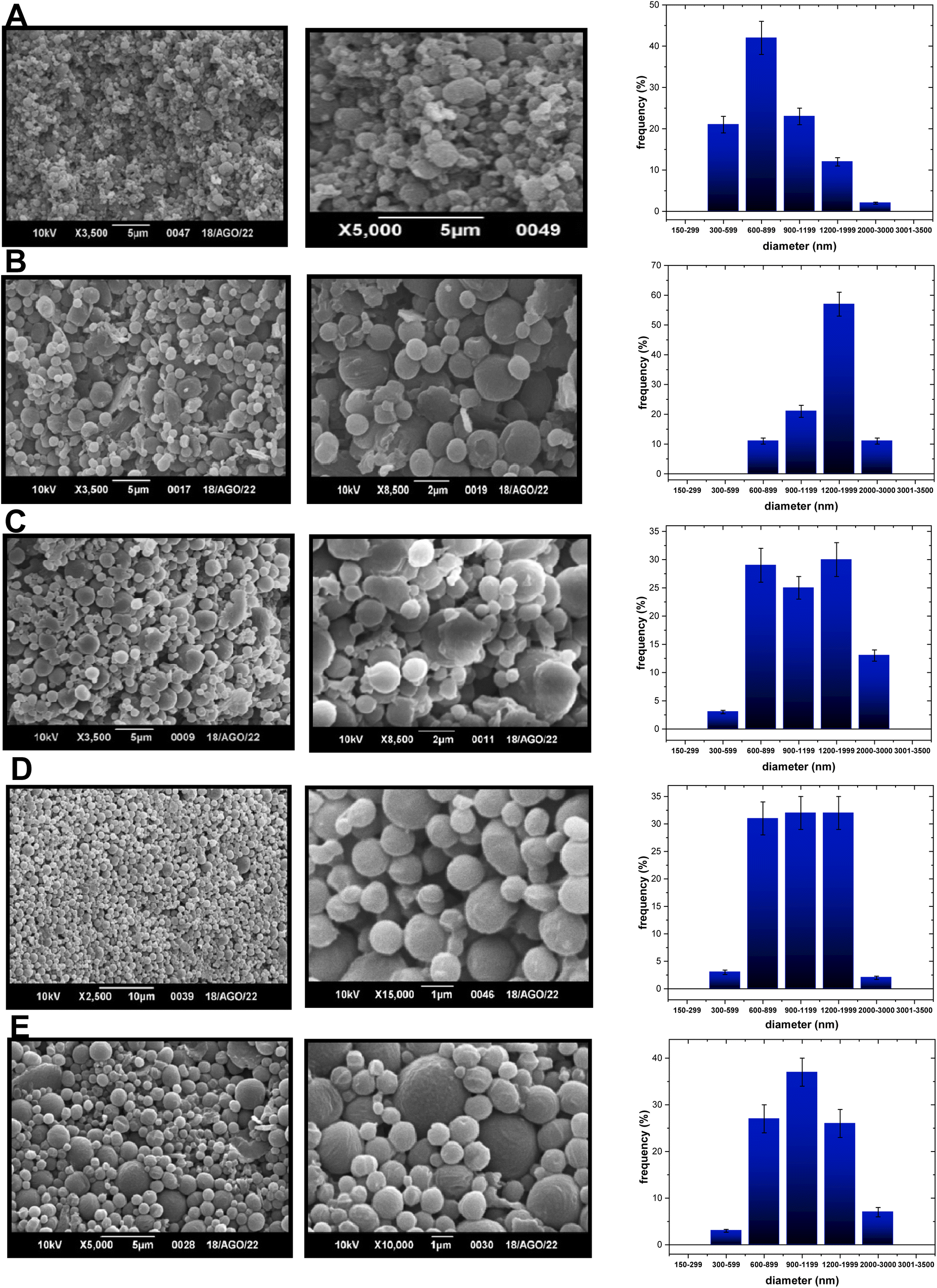 | ||
| Fig. 2 SEM images of (A) PCL-NPs-300/1200, (B) PCL-TP1-NPs-900/2000 and (C) PCL-TP1-NPs-600/2000 at 2 or 5 microns, (D) PCL-TP2-NPs-600/2000, and (E) PCL-TP2-NPs-600/2000 at 1, 5 or 10 microns. | ||
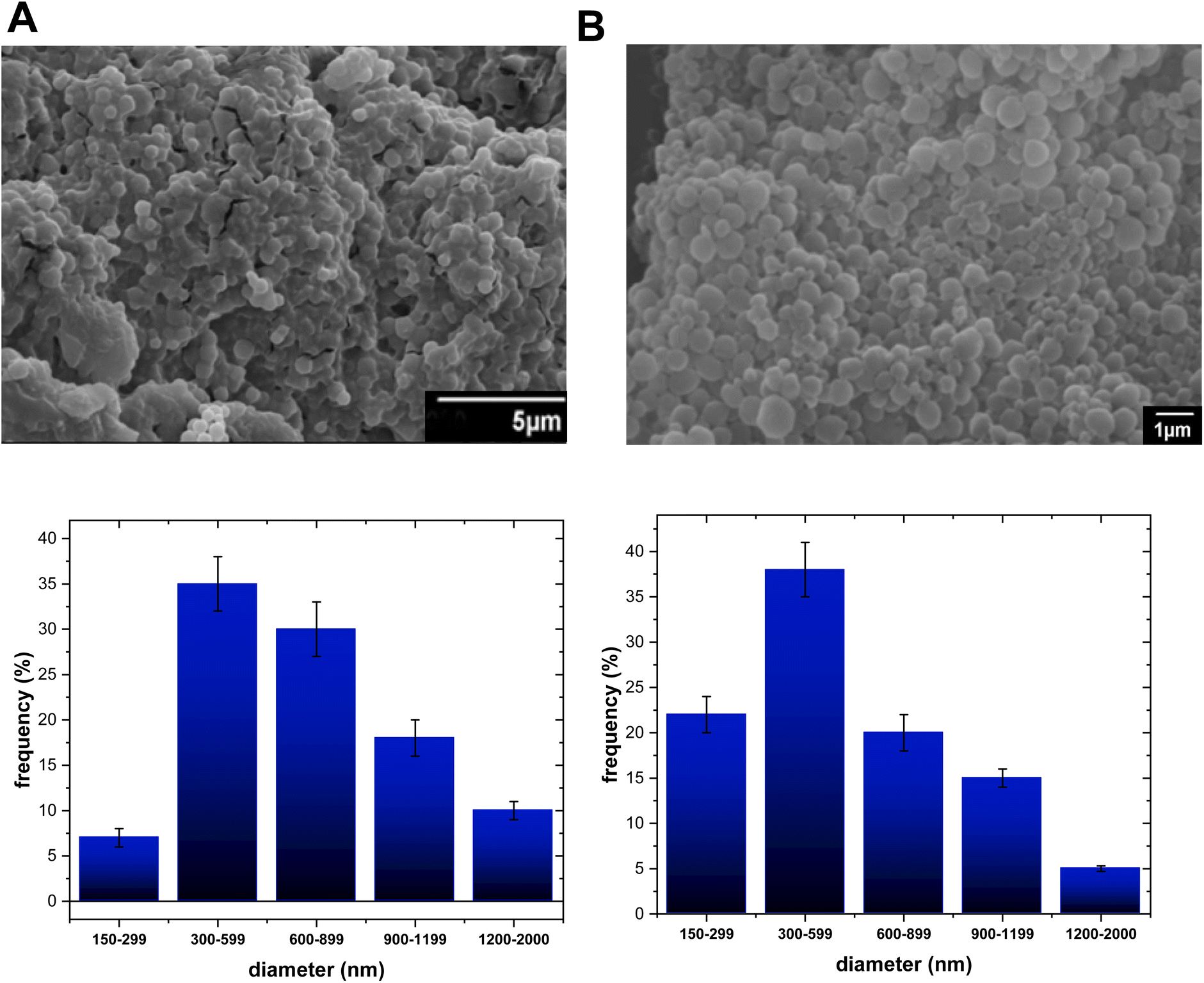 | ||
| Fig. 3 SEM i of (A) PLGA-TP1-NPs-432, and (B) PLGA-TP2-NPs-298 at 1 or 5 microns. | ||
For the PCL nanoformulations, PCL-TF1-600/2000 consists of a nanoformulation containing compound TF1 loaded into nanoparticles that were 600–2000 nanometers in size (Fig. 2B), whereas PCL-TF1-1200/2000 is constituted by microparticles with sizes between 1200 and 2000 nanometers (Fig. 2C). Meanwhile the PCL-TF2-NPs consisted of a nanoformulation (PCL-TF2-600/2000) containing nanoparticles with sizes between 600 and 2000 nanometers (Fig. 2D), whereas the other PCL-TF2-300–3000 consisted of a system containing micro and nanoparticles with well-dispersed sizes between 600 and 3000 nanometers (Fig. 2E). The unloaded PLA-NPs exhibited particle sizes between 600 and 2000 nanometers (Fig. 2A).
Finally, the PLGA NPs consisted of a well-defined mean particle size of about 432 and 298 nm for the TF1 and TF2 compounds, respectively. It can be inferred that the shortest PLGA (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da) and PLA (56000 Da) seem to be more susceptible to generating smaller nanoparticles than the heaviest PCL polymer (91000 Da), which preferred to generate the largest nanoparticles. TEM analysis indicated that at least for the PLGA-(TF1/TF2)-NPs, their structures are composed of nanoparticles (Fig. S1 and S2†).
000 Da) and PLA (56000 Da) seem to be more susceptible to generating smaller nanoparticles than the heaviest PCL polymer (91000 Da), which preferred to generate the largest nanoparticles. TEM analysis indicated that at least for the PLGA-(TF1/TF2)-NPs, their structures are composed of nanoparticles (Fig. S1 and S2†).
For the entrapment efficiency of the encapsulate drug, the identification of the characteristic maximum absorption wavelength of the 3-(3-aryl)-6-piperazin-1,2,4-triazolo[3,4-a] from UV-Vis spectroscopy allowed us to verified its encapsulation and quantification. From UV-Vis spectroscopy, we found that the encapsulation efficiency of the PLGA-PEG NPs was about 99.9% and 99% for TF1 and TF2, respectively (entries 2 and 8, Table 1). The entrapment efficiency of the PLA and PCL microencapsulates/nanoencapsulates ranged from 90% to 95% (entries 3–6 and 9–12, Table 1). The use of PVA as a stabilizing factor leads to reduced drug leakage from NPs and improves the drug loading efficiency. In addition, the encapsulation of TF1 and TF2 into PLA and PCL was confirmed from EDX analysis, which verified the presence of the characteristic elements of the drugs TF1 (nitrogen) and TF2 (nitrogen and fluorine) in the particles. An appreciable nitrogen percentage, combined with changes in the elemental composition of carbon and oxygen elements when compared with the unloaded PLA or PCL, confirmed the encapsulation of the 1,2,4-triazolo-phthalazine TF1 in the PLA- and PCL-NPs. In particular, the detection of fluorine for the encapsulate containing TF2 confirmed the encapsulation of the fluorinated compound, TF2.
| Entries | P-TF-NPs | EE (%) | EDX (%) |
|---|---|---|---|
| EE: entrapment efficiency; EDX: energy-dispersive X-ray spectroscopy. | |||
| 1 | TF1 | — | C, 61.42; H, 4.81; N, 25.06 |
| 2 | PLGA-TF1-432 | 99.9 ± 0.1 | — |
| 3 | PLA-TF1-300/3000 | C: 66.52, O: 27.70, N: 5.78 | |
| 4 | PLA-TF1-300/600 | C: 64.41, O: 30.49, N: 5.10 | |
| 5 | PCL-TF1-600/3000 | C: 60.36, O: 32.64, N: 7.00 | |
| 6 | PCL-TF1-600/2000 | C: 71.15, O: 22.35, N: 6.15 | |
| 7 | TF2 | — | C, 66.23; H, 5.24; N, 23.00 |
| 8 | PLGA-TF2-298 | 99.0 ± 0.1 | — |
| 9 | PLA-TF2-1200/2000 | C: 52.42, O: 42.50, N: 2.42, F: 2.66 | |
| 10 | PLA-TF2-600/1200 | C: 58.47, O: 30.87, N: 8.36, F: 2.30 | |
| 11 | PCL-TF2-1200/2000 | C: 68.00, O: 27.29, N: 2.38, F: 2.33 | |
| 12 | PCL-TF2-600/2000 | C: 70.02, O: 25.36, N: 2.26, F: 2.36 | |
| 13 | PLA | — | C: 68.86, O: 31.14 |
| 14 | PCL | — | C: 72.54, O: 27.46 |
| 15 | PLGA | — | — |
For the controlled release of TF1 and TF2 from the microencapsulated/nanoencapsulated structures, the dialysis method was used at pH 7.4, and at 25 °C and 37 °C. At physiological conditions, the drug release pattern presented a burst release in the first 24 h, followed by a controlled release of the drug for a period of 14 days, although the release rate was shown to be dependent on the particle size and polymer nature. Beginning the analysis with the PLA- and PCL-microencapsulated/nanoencapsulated TF1, it should be noted that PLA-NPs showed a faster release than PCL-NPs with a 50% and 65% release of 1,2,4-triazolophthalazines at 7 days (Fig. 4A), respectively. Interestingly, a more complete release was observed for NPs having larger particle sizes either for PLA- or for PCL-TF1-NPs, although their differences with the PLA- and PCL-NPs having smaller NPs are minimal. Surprisingly, PLA- and PCL-TF2-NPs showed a contrary tendency as the structures with smaller NPs showed a faster and more continuous release than the PLA- and PCL-structures having larger NPs. In general, a first release between 24 and 48 h was observed, followed with a large release (50% or 65%) after 7 days. This could be important for promoting a fast biological therapeutic response, and it seems to barely favor the PLA- and PCL-structures having smaller NPs. The first release could be attributed to a combined effect of pore diffusion, swelling, surface erosion, in situ cross-linking, degradation processes, and the possible presence of the drugs at the surface level of the nanoparticles, which would allow an early release of a certain amount of phthalazines to occur. The initial fast rate was ascribed to the diffusion of drug entrapped close to the surface of NPs, while the subsequent release was because of the slow diffusion of the entrapped drug from the interior core of the polymeric matrix. Regarding the PLGA-NPs, their NPs showed a rapid release (by about 25%) in the first 48 h, followed by a release of about 55% after the first seven days. The rapid release under physiological conditions could be favored by the particle size, where smaller NPs tend to favor the release.
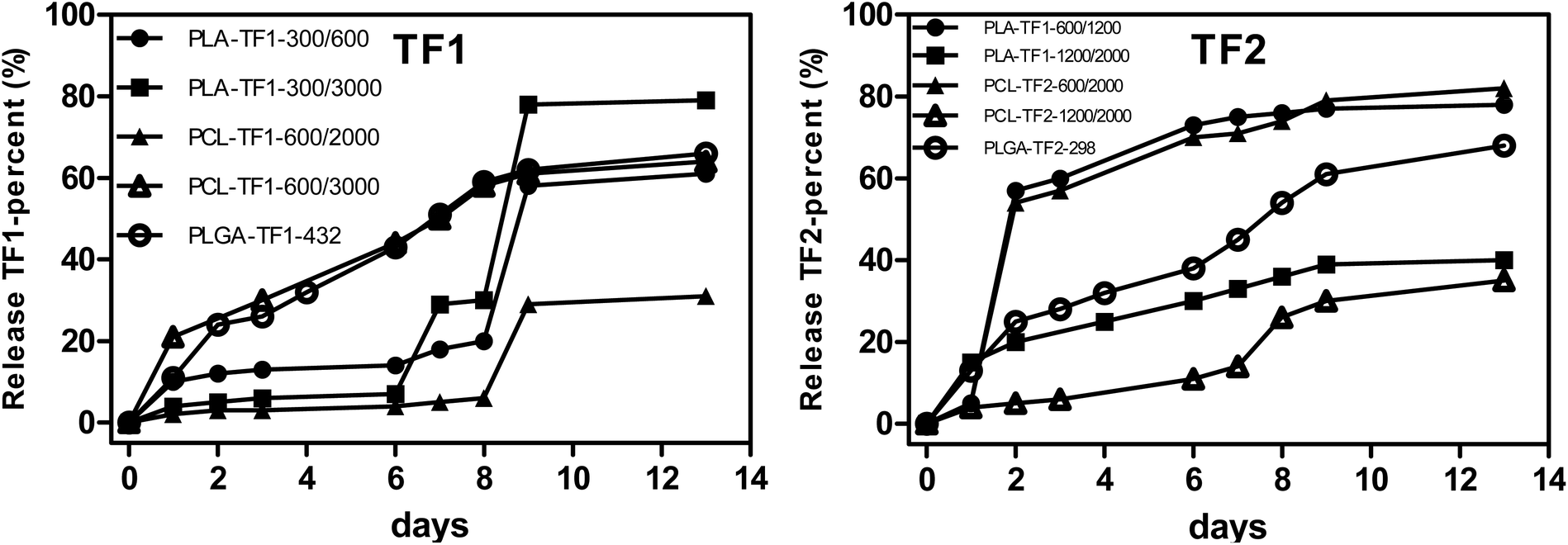 | ||
| Fig. 4 Controlled-release study of PLA-, PCL- and PLGA-microencapsulates/nanoencapsulates loaded with TF1 and TF2 at physiological conditions from 0 to 192 hours. Each point consists of a mean derived from three independent experiments. SD of each point for either TF1 or TF2 was below 5%. | ||
2.2. Biological activity
The 3-aryl-6-piperazin-1,2,4-triazolo[3,4-a]phthalazines, TF1 and TF2, showed good antileishmanial activity against the promastigote and intracellular amastigote of L. braziliensis, a Leishmania specie responsible for Cutaneous Leishmaniasis. Herein, we investigated the potential of these compounds against other trypanosomatids such as L. infantum, responsible for visceral leishmaniasis, and T. cruzi, which is responsible for Chagas disease. Importantly, we also explored the potential of the encapsulation with a focus on: (i) the type of polymer (PLGA, PLA and PCL), and (ii) the size of their nano and microparticles. We first evaluated the therapeutic effect of the compounds TF1 and TF2, alone and with encapsulates, against the promastigote of L. infantum and epimastigote of T. cruzi. The biological effects of the free and encapsulated compounds were collected after 5 days, which is a key point because the controlled release of most of the encapsulated compounds needs at least five days. The five microformulations/nanoformulations (two of PLA and PCL, and one of the PLGA polymer) of each of the compounds TF1 and TF2 in combination with their free forms were evaluated in vitro against both parasites, which sought to study the effect of the polymer and nanoparticle size. The empty PLA-, PCL- and PLGA-microparticles/NPs were innocuous against the parasites. In general, it should be noted that the biological activity showed a dependence on the nature of the polymer and nanoparticle size against both promastigote L. infantum and epimastigote T. cruzi parasites. In addition, the compounds were significantly more active against Leishmania than T. cruzi parasite, by which we focused our analysis on the effect against the L. infantum parasite.As a function of the nanoparticle size, it should be noted that the encapsulation, in general terms, improved the anti-trypanosomatid activity of the compounds TF1 and TF2, with EC50 values lower than those found for the free compounds. The free compounds TF1 and TF2 showed EC50 values of 12.12 and 17.22 μM against L. infantum, and of 11.03 and 23.61 μM against T. cruzi, respectively (entries 1 and 7, Table 2). For compound TF1, PLGA-NPs decreased its EC50 at 10.11 μM (entry 2, Table 2), whereas PLA-NPs displayed only a discrete decrease for the smaller NPs-system (PLA-TF1-300/600), giving an EC50 value of 11.44 μM because the larger NPs-system (PLA-TF1-300/3000) displayed an EC50 value of 12.99 μM (entries 3 and 4, Table 2). Meanwhile, no improvement in the leishmanicidal activity of TF1 was found for the encapsulation with PCL, with an EC50 value of 21.22 and higher than 25 μM for the NPs of PCL-TF1-900/2000 and PCL-TF1-600/2000, respectively (entries 5 and 6, Table 2). For the compound TF2, the EC50 of PLGA-microparticles/nanoparticles decreased to 14.84 μM (entry 8, Table 2), which is lower than the EC50 value of 17.22 μM for the free compound. Meanwhile, the PLA- and PCL-microparticles/nanoparticles, similar to those described above for the TF1 compound, promoted only a decrease in the leishmanicidal activity for the smaller particle size, PLA-TF2-600/1200 (EC50 = 15.98 μM) (entry 10, Table 2). Meanwhile, an increase in the EC50 value (22.32 μM) (entry 9, Table 2) was found for the larger PLA-TF2-NP (PLA-TF2-1200/2000), and no activity was observed for the PCL-TF2-microparticles/nanoparticles at 25 μM (entries 11 and 12, Table 2). A similar tendency in terms of the type of polymer and particle size was found for the microformulations/nanoformulations against T. cruzi, although it is important to mention that T. cruzi was less susceptible than the L. infantum parasite when using either the micronanoparticle/nanoparticle systems or the free compound. Only PLGA-TF1-432, PLA-TF1-300/3000 and PLA-TF1-300/600 displayed the most promising biological response against the T. cruzi epimastigote, giving EC50 values of 10.45, 11.76 and 10.99 μM, which is relatively comparable with the performance of free TF1 (EC50 = 11.03 μM). Taken together, the results confirm the potential of PLGA to construct PLGA-loaded leishmanicidal drugs, which is already widely applied,9 and give some first evidence on the potential of PLA as a polymeric matrix to construct micronanoparticle/nanoparticle systems based on leishmanicidal or Chagas's drugs. Secondly, we showed that the size of the particle in the polymeric microformulations/nanoformulations, as described previously for other types of NP-structures (such as metallic NPs),5 favor the trypanosomaticidal activity. We think that the remarkable activity of the smaller NPs could be attributed to the rapid release of their microformulations/nanoformulations, and possibly to its easy penetration or transportation within the parasite, where the smaller NP size could favor better cell penetration than the microparticle/nanoparticle systems having larger particles. On the other hand, from the nature of the polymer, it seems that hydrophilic polymers such as PLGA and PLA are more convenient than the lipophilic PCL polymer for the construction of microparticle-/nanoparticle-based compounds.
| Entries | Microformulation/nanoformulation | EC50 (μM) | CC50 (μM) | ||
|---|---|---|---|---|---|
| L. infantum (P)a | L. infantum (A)b | T. cruzi (E)c | J774.1d | ||
| a EC50 values against the promastigote of L. infantum of the microencapsulates/nanoencapsulates and free compounds. b EC50 values against the amastigote of L. infantum of the microencapsulates/nanoencapsulates and free compounds. c EC50 values against the epimastigote of T. cruzi of the microencapsulates/nanoencapsulates and free compounds. d CC50 values on the macrophages J774.1. e Drug reference for Leishmaniasis. f Drug reference for Chagas disease. Note: all biological assays were performed in triplicates, and the EC50 and CC50 were reported with a SD below 6% for most of the cases. | |||||
| 1 | TF1 | 12.12 | 18.48 | 11.03 | 123.93 |
| 2 | PLGA-TF1-432 | 10.11 | 16.56 | 10.45 | 93.67 |
| 3 | PLA-TF1-300/3000 | 12.99 | — | 11.76 | — |
| 4 | PLA-TF1-300/600 | 11.44 | >25.00 | 10.99 | 106.59 |
| 5 | PCL-TF1-900/2000 | >25.00 | — | 24.34 | — |
| 6 | PCL-TF1-600/2000 | 21.22 | — | 19.89 | — |
| 7 | TF2 | 17.22 | 23.43 | 23.61 | 103.34 |
| 8 | PLGA-TF2-298 | 14.84 | >25.00 | 21.78 | 96.22 |
| 9 | PLA-TF2-1200/2000 | 22.32 | — | ≥25.00 | — |
| 10 | PLA-TF2-600/1200 | 15.98 | >25.00 | 22.49 | 91.44 |
| 11 | PCL-TF2-1200/2000 | >25.00 | — | >25.00 | — |
| 12 | PCL-TF2-600/2000 | >25.00 | — | >25.00 | — |
| 13 | MLT | 10.01 | 12.11 | — | — |
| 14 | Nfx | — | — | 6.4 | — |
The cytotoxicity of the most active microencapsulates/nanoencapsulates PLGA-TF1-432, PLGA-TF2-298, PLA-TF1-300/600, PLA-TF2-600/1200 and their free compounds TF1 and TF2 was evaluated. In general, the cytotoxic concentration (CC50) values for the compounds TF1 and TF2 was decreased with the use of the PLGA- and PLA-NPs. For the compound TF1, lower CC50 values were found for their microencapsulates/nanoencapsulates PLGA-TF1-432 (CC50 = 93.67 μM), PLA-TF1-300/3000 (CC50 = 106.59 μM) than the toxicity of the free TF1 compound (CC50 = 123.93 μM). For the compound TF2, their microencapsulates/nanoencapsulates PLGA-TF2-298 (CC50 = 96.22 μM) and PLA-TF2-600/1200 (CC50 = 91.44 μM) displayed lower CC50 values than the corresponding free compound (CC50 = 103.34 μM). Thus, the encapsulated PLGA- and PLA-NPs were more cytotoxic than free compounds, which suggests that the cell penetration across the macrophage membrane cell could favor the accumulation into the macrophage cytosol, increasing its effective concentration within the macrophage. The latter effect increases the toxicity of the compounds, and can occur through cell penetration or phagocytosis. However, that facile access within the macrophage is convenient because the infective form of the parasite, intracellular amastigote, lives within the macrophage, facilitating a path for the direct interaction between the drug and compound.
Finally, as the microparticle/nanoparticle construction showed better activity against Leishmania parasites, the most active nanoformulations PLGA-TF1-432, PLGA-TF2-298, PLA-TF1-300/600, PLA-TF2-600/1200 and their free compounds TF1 and TF2 were assayed against the axenic amastigote of L. infantum. In general, only a small improvement was found for the PLGA-TF1-432 (IC50 = 16.56 μM), which was barely lower than that found for the free TF1 compound (IC50 = 18.48 μM). Meanwhile, no improvements in the leishmanicidal activities were found for the remaining NPs-TF1/TF2. We think that that the weak therapeutic effect could be associated with the short time exposure (48 h). At that time, the amount of active compound on the culture is limited, as seen from Fig. 4, where only 20% is expected in solution after 48 h for most of the encapsulated formulations. It represents one of the limitations of the nanoencapsulation for in vitro assays. However, the biological effect against the promastigote strain (collected after 5 days) demonstrates the potential of PLGA and PLA as polymeric matrices, as well as the importance of the size control in the micro-/nano-construction.
3. Conclusions
In summary, TF1 and TF2 were successfully encapsulated into PLGA-, PLA- and PCL-micropartice/-nanoparticle systems, and their in vitro anti-trypanosomatid response against L. infantum and T. cruzi and in vitro toxicity were assayed. From the present investigation, we showed using the triazolo-phthalazine as a drug model that the PLGA- and PLA-NPs can be conveniently used for the construction of a microformulation/nanoformulation with potential leishmanicidal activity, and are able to improve the therapeutic effect. Importantly, we found that the particle sizes also play an important role, with the encapsulated particles of smaller size performing better in improving the therapeutic effect. Our investigation offers a starting point to focus new research on the particle size and type of polymer.4. Materials and methods
4.1. Chemical section
000), PLA (Mn = 56000) and PCL (Mn = 91000) were purchased from Sigma-Aldrich, Nature Works and Union Carbide, respectively. 3-(3-Nitrophenyl)-6-piperazin-1,2,4-triazolo[3,4-a]phthalazine and 3-(4-fluorophenyl)-6-piperazin-1,2,4-triazolo[3,4-a]phthalazine were prepared previously by our group.23,24 The solvents, chloroform HPLC grade (99.9%) and ethanol absolute (99.5%), were purchased from Sigma-Aldrich and Merck, respectively. All other chemicals used in this study were of analytical grade from JT Baker and Merck, and used without further purification. The materials were supplied by Laboratorio de Química Orgánica (Grupo B5IDA, Universidad Simón Bolívar), Laboratorio de Fisicoquímica de Ingeniería de Materiales y Centro de Nanotecnología (Instituto Venezolano de Investigaciones Científicas), Laboratorio de Química General (Facultad de Farmacia, Universidad Central de Venezuela) and Laboratorio de Química Orgánica Medicinal (Facultad de Ciencias, Universidad de la República).
000g, 4 °C, and 15 min), and washed three times with deionized water to give white to yellowish fine solids. The PLA-, PCL and PLGA-microparticles/-nanoparticles were prepared using 1% chloroform and 0.02% PVA. After the particles were precipitated, they were properly washed by dialysis and then lyophilized.
The amount of entrapped 1,2,4-triazolo[3,4-a]phthalazine in PLGA microparticles/nanoparticles was determined from UV-Vis spectroscopy and energy dispersion analysis. Briefly, the nano-suspension was centrifuged at 12000g for 15 min after PLGA-1,2,4-triazolo[3,4-a]phthalazine microparticle/nanoparticle formation. The clear supernatant was then collected, diluted and detected. Then, the entrapment efficiency (EE)% of the compounds was determined indirectly from the quantification of the derivatives present in the washing waters, and obtained after executing the encapsulation process on the micro–nanoparticles. Chloroform and a saturated solution of NaCl were used as organic solvent. The organic phase was dried with a rotary evaporator, and the obtained compound was brought to a volume of 2.0 mL using DMSO to then evaluate the absorption of the UV-Vis radiation. The corresponding calibration curves were obtained. For the measurements, the absorbance at the wavelength of the maximum absorption of each derivative was used (in the range between 347–360 nm). Subsequently, the encapsulation efficiency was determined as follows:31
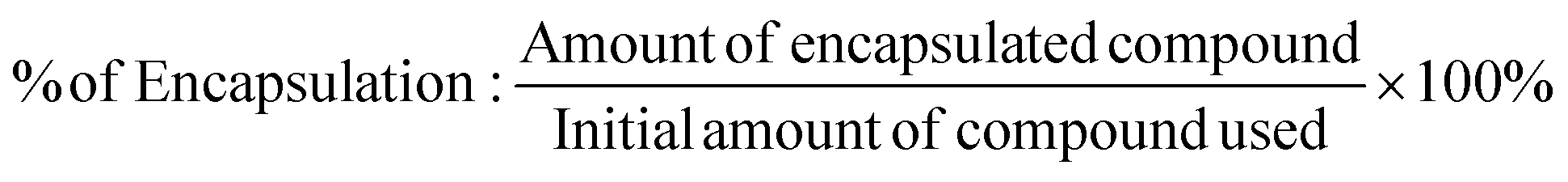 000g for 15 min at 4 °C. The supernatant was then collected, frozen, and immediately replaced with an equal volume of fresh release medium. To determine the TF1 and TF2 concentration, collected supernatants were diluted in the mobile phase and quantified using Thermo Scientific Varioskans Flash Multimode instrument. All assays were performed in triplicate. Alternatively, the resulting pH in solution was measured from NP exposed at 37 °C for different times (6, 12, 24, 48, 72, 96, 120 and 144 h).32
000g for 15 min at 4 °C. The supernatant was then collected, frozen, and immediately replaced with an equal volume of fresh release medium. To determine the TF1 and TF2 concentration, collected supernatants were diluted in the mobile phase and quantified using Thermo Scientific Varioskans Flash Multimode instrument. All assays were performed in triplicate. Alternatively, the resulting pH in solution was measured from NP exposed at 37 °C for different times (6, 12, 24, 48, 72, 96, 120 and 144 h).32
4.2. Biological section
:80 ratio, followed by combined ultrasound and stirring for 40 minutes in order to obtain an emulsion sample. Cell viability was assessed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assays with a few modifications.33 All controls and the tested wells contain no more than 1% DMSO. The screening was performed in 96-well microliter plates maintained at 25 °C. Briefly, 2 × 106 parasites per mL were exposed to increasing concentrations from 5 to 200 mg L−1 of each compound for 72 h at 25 °C. Controls contain 1% DMSO and medium. After incubation, cells were treated with 100 μL of 0.4 mg mL−1 MTT for 4 h at 37 °C. Subsequently, the medium was removed, and 100 μL DMSO was added to the resulting mixture to dissolve the formazan salt. The solubilized formazan product was quantified through absorbance measurements at 570 nm using a Thermo Scientific Varioskan Flash Multimode instrument at 72 h. All experiments were performed in triplicate. Untreated control parasites were used to calculate the relative proliferation. Miltefosine was used as the reference drug. The percentage of parasite inhibition with regard to the controls was calculated as follows: 100 − [(parasite counts in treated cells/parasite counts in untreated cells) − 100]. The EC50 was determined by regression analysis using GraphPad (Prism 5.0), and the means and standard errors from three different experiments were utilized to estimate the final EC50.
Dose–response curves were recorded, and the IC50 values were determined using the GraphPad Prism version 6.00 for Windows (GraphPad Software, La Jolla California, USA).
:80 ratio as solvents. Macrophages were cultured at a density of 50 × 103 cells per well in 96-well plates, and the micro–nanoparticles of the corresponding derivatives were added in a concentration range from 25 to 400 mg L−1. After complete cell adhesion after 24 hours, the cells were treated for 48 hours using concentrations of the micro–nanoparticles in a range of 25 to 400 mg L−1. Then, the cells were washed with PBS and resuspended in MTT solution (0.5 mg mL−1) and incubated at 37 °C for 4 h in the dark. Violet crystals of formazan were formed as a result of the redox reaction between MTT and the mitochondrial enzyme succinate dehydrogenase of macrophages. The crystals formed were dissolved in DMSO. Cell viability was determined by quantifying the intensity of the formazan crystals using the Thermo Scientific Varioskans Flash Multimode spectrophotometer. The results were expressed in terms of the mean cytotoxic concentration (CC50), which represents the concentration required to cause cytotoxicity in 50% of the population. The CC50 was determined by linear regression analysis, taking into account a coefficient of determination greater than 0.9. The tests were carried out in triplicate.
Author contributions
K. N. Gonzalez and E. M. prepared the NP structures and its characterization. A. H. Romero participated in the in vitro evaluation against the parasites and cytotoxicity, analysis of the biological data, and preparation of the manuscript. E. P. Aguilera generally contributed to the evaluation of the biological data. G. Gonzalez and M. Sabino supervised the preparation and characterization of NPs, and provided funding resources.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors gratefully acknowledge support from the Organic Chemistry Laboratory of Grupo B5IDA (USB), the Physicochemistry Laboratory of the Materials Engineering and Nanotechnology Center (IVIC) and the General Chemistry laboratory of the Faculty of Pharmacy (UCV). The investigation was supported in part by the Project CDCH-UCV (grant number ID: PG-09-8819/2013) (Universidad Central de Venezuela, Caracas, Venezuela), and the project ANII (grant number ID: FCE_3_2022_1_172684). A. H. R. and E. A. are grateful to UDELAR (PEDECIBA-QUIMICA) for additional funding. The authors are also grateful to Lic. Gleen Rodriguez from the Surface Lab (Lab E) of the Simon Bolivar University (USB), for the preparation and observation of the samples by SEM. The authors gratefully acknowledge Nicole Lecoq for the collaboration in the TEM analysis.References
- M. De Rycker, S. Wyllie, D. Horn, K. Read and I. Gilbert, Nat. Rev. Microbiol., 2023, 21, 35 CrossRef CAS.
- World Health Organization, Leishmaniasis, https://www.who.int/news-room/fact-sheets/detail/leishmaniasis (accessed November 2023).
- (a) World Health Organization, Chagas Disease, Chagas disease (also known as American trypanosomiasis) (who.int) (accessed November 2023); ; (b) F. F. Tuon, V. A. Neto and V. Sabbaga-Amato, FEMS Immunol. Med. Microbiol., 2008, 54, 158–166 CrossRef CAS PubMed; (c) A. Mannaert, T. Downing, H. Imamura and J. C. Dujardin, Trends Parasitol., 2012, 28, 370 CrossRef CAS PubMed.
- S. Silva-Santos, R. V. de Araújo, J. Giarolla, O. ElSeoud and E. I. Ferreira, Int. J. Antimicrob. Agents, 2020, 55, 105906 CrossRef PubMed.
- D. B. Scariot, A. Staneviciute, J. Zhu, X. Li, E. A. Scott and D. M. Engman, Front. Cell. Infect. Microbiol., 2022, 12, 1000972 CrossRef CAS PubMed.
- C. Quijia Quezada, C. S. Azevedo, S. Charneau, J. M. Santana, M. Chorilli, M. B. Carneiro and I. Marques Dourado, Int. J. Nanomed., 2019, 14, 6407 CrossRef PubMed.
- (a) D. Manzanares and V. Ceña, Pharmaceutics, 2020, 12, 371 CrossRef CAS PubMed; (b) P. Prasanna, P. Kumar, S. Kumar, V. Kumar-Rajana, V. Kant, S. R. Prasad, U. Mohan, V. Ravichandiran and D. Mandal, Biomed. Pharmacother., 2021, 141, 111920 CrossRef CAS PubMed.
- M. Akbari, A. Oryan and G. Hatam, Acta Trop., 2017, 172, 86 CrossRef CAS.
- D. Maza-Vega, M. Di Meglio, S. del Valle Alonso, F. Alvira and J. Montanari, OpenNano, 2023, 12, 100158 CrossRef.
- O. P. Singh, M. R. Gedda, S. L. Mudavath, O. N. Srivastava and S. Sundar, Nanomedicine, 2019, 14, 1911 CrossRef CAS PubMed.
- A. Haleem, M. Javaid, R. Pratap-Singh, S. Rab and R. Suman, Global Heal. J., 2023, 7, 70 CrossRef.
- (a) A. K. Saim, F. N. Kumah and M. N. Oppong, Environ. Eng., 2021, 6, 1–11 CAS; (b) A. Valiallahi, Z. Vazifeh, Z. Rezanejad-Gatabi, M. Davoudi and I. Rezanezhad-Gatabi, Curr. Med. Chem., 2023 DOI:10.2174/0929867331666230823094737.
- M. Borilli-Pereira, B. Gomes-Sydor, K. G. Memare, T. Gomes-Verzignassi, S. M. Alessi-Aristides, E. Monguilhott-Dalmarco, J. J. Vieira-Teixeira, M. Valdrinez Campana Lonardoni and I. Galhardo-Demarchi, Nanomedicine, 2021, 16, 1505 CrossRef.
- R. Nicolete, D. F. dos Santos and L. H. Faccioli, Nanomedicine, 2013, 9, 985 CrossRef PubMed.
- J. Koerner, D. Horvath and M. Groettrup, Front. Immunol., 2019, 10, 707 CrossRef CAS PubMed.
- D. Horvath and M. Basler, Pharmaceutics, 2023, 15, 615 CrossRef CAS PubMed.
- Y. Xiao, W. Yao, M. Lin, W. Huang, B. Li, B. Peng, Q. Ma, X. Zhou and M. Liang, Drug Delivery, 2022, 29, 1712 CrossRef CAS PubMed.
- W. Ma, M. Chen, S. Kaushal, M. McElroy, Y. Zhang, C. Ozkan, M. Bouvet, C. Kruse, D. Grotjahn, T. Ichim and B. Minev, Int. J. Nanomed., 2012, 7, 1475–1487 CrossRef CAS PubMed.
- J. J. Moon, H. Suh, M. E. Polhemus, C. F. Ockenhouse, A. Yadava and D. J. Irvine, PLoS One, 2012, 7, e31472 CrossRef CAS PubMed.
- P. Hartmeier, J. Kosanovich, K. Velankar, J. Armen-Luke, M. Lipp, E. Gawalt, N. Giannoukakis, K. Empey and W. Meng, Mol. Pharmaceutics, 2022, 19, 2638 CrossRef CAS PubMed.
- J. Koerner, D. Horvath and V. L. Herrmann, Nat. Commun., 2021, 12, 2935 CrossRef CAS PubMed.
- D. Bobo, K. J. Robinson, J. Islam, K. J. Thurecht and S. R. Corrie, Pharm. Res., 2016, 33, 2373 CrossRef CAS PubMed.
- A. H. Romero, N. Rodríguez and O. G. Ramírez, New J. Chem., 2020, 44, 13807 RSC.
- A. H. Romero, F. Sojo, F. Arvelo, C. Calderón, A. Morales and S. E. López, Bioorg. Chem., 2020, 101, 104031 CrossRef CAS PubMed.
- A. H. Romero, R. Medina, A. M. Alcala, Y. García-Marchan, J. Nuñez-Duran, J. Leañez, A. Mihoba, C. Ciangherotti, X. Serrano-Martín and S. E. Lopez, Eur. J. Med. Chem., 2017, 127, 606 CrossRef CAS PubMed.
- A. H. Romero, J. Rodriguez, Y. García-Marchan, J. Leañez, X. Serrano-Martín and S. E. Lopez, Bioorg. Chem., 2017, 72, 51 CrossRef CAS PubMed.
- A. H. Romero and S. E. Lopez, J. Mol. Graphics Modell., 2017, 76, 313 CrossRef CAS PubMed.
- A. H. Romero, N. Rodriguez, S. E. Lopez and H. Oviedo, Arch. Pharm., 2019, 352, 1800299 CrossRef PubMed.
- T. Borel and C. M. Sabliov, Annu. Rev. Food Sci. Technol., 2014, 5, 197 CrossRef CAS PubMed.
- (a) S. Chen, J. Zhou, B. Fang, Y. Ying, D. G. Yu and H. He, Macromol. Mater. Eng., 2023, 2300361, DOI:10.1002/mame.202300361; (b) L. Sun, J. Zhou, Y. Chen, D. G. Yu and P. Liu, Front. Bioeng. Biotechnol., 2023, 11, 1308004 CrossRef PubMed; (c) L. Xu, H. He, Y. Du, S. Zhang, D. G. Yu and P. Liu, Pharmaceutics, 2023, 15, 2314 CrossRef CAS PubMed; (d) J. Zhou, L. Wang, W. Gong, B. Wang, D. G. Yu and Y. Zhu, Biomedicines, 2023, 11, 2146 CrossRef CAS PubMed.
- H. C. Quadros, L. M. Ferreira-Santos, C. S. Meira, M. I. Khouri, B. Mattei, M. B. Pereira-Soares, W. Castro-Borges, L. Paiva-Farias and F. Rocha-Formiga, Int. J. Pharm., 2020, 576, 118997 CrossRef CAS PubMed.
- R. A. Rezende, M. A. Sabino, J. Dernowsek, F. Albuquerque Vilalba, V. Mironov and J. V. Lopes-Silva, Int. J. Adv. Med. Biotechnol., 2018, 1, 41 CrossRef.
- A. Dutta, S. Bandyopadhyay, C. Mandal and M. Chatterjee, Parasitol. Int., 2005, 54, 119 CrossRef CAS PubMed.
- Bangs Laboratorie, I., Turbidimetric Assays, doi:IN 46038.
- A. H. Romero, E. Aguilera, L. Gotopo, G. Cabrera, B. Dávila and H. Cerecetto, RSC Med. Chem., 2023, 14, 1992 RSC.
- T. Mosmann, Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays, J. Immunol. Methods, 1983, 65, 55 CrossRef CAS PubMed.
- Graph Pad Prism Software Inc. 4.02 for windows. May 17th 1992–2004.
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental details and further theoretical information. See DOI: https://doi.org/10.1039/d3pm00002h |
| ‡ Current address: Escuela de Fisica y Nanotecnologia, Universidad Yachay, Ecuador. |
| This journal is © The Royal Society of Chemistry 2024 |