
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in the total synthesis of alkaloids using chiral secondary amine organocatalysts
Yuta
Nakashima
,
Kenta
Rakumitsu
 and
Hayato
Ishikawa
*
and
Hayato
Ishikawa
*
Graduate School of Pharmaceutical Sciences, Chiba University, 1-8-1, Inohana, Chuo-ku, Chiba 260-8675, Japan. E-mail: h_ishikawa@chiba-u.jp
First published on 5th November 2024
Abstract
Since the early 21st century, organocatalytic reactions have undergone significant advancements. Notably, numerous asymmetric reactions utilizing chiral secondary amine catalysts have been developed and applied in the total synthesis of natural products. In this review, we provide an overview of alkaloid syntheses reported since 2017, categorized by scaffold, with a focus on key steps involving asymmetric reactions catalyzed by secondary amine organocatalysts.
 Yuta Nakashima | Yuta Nakashima was born in Kagoshima prefecture, Japan. He received his BSc degree from Kumamoto University in 2020 and studied for two years as a master's student there. He then moved to Chiba University and is currently a graduate student in Professor Hayato Ishikawa's group. He focuses on the total synthesis of natural products using organocatalytic reactions. |
 Kenta Rakumitsu | Kenta Rakumitsu was born in Fukuoka Prefecture, Japan. He received his BSc and PhD degrees in science from Kumamoto University in 2016 and 2021, respectively. In 2021, he moved to Seikei University of Science and Technology as an assistant professor. In 2024, he joined the Faculty of Pharmaceutical Sciences, Chiba University as an assistant professor. He focuses on the total synthesis of natural products and the structural chemistry of organic compounds. |
 Hayato Ishikawa | Hayato Ishikawa received a PhD degree from Chiba University in 2004. After postdoctoral work (2004–2006) in the Scripps Research Institute, he was promoted to the rank of assistant professor at the same institute (2006–2007). In 2007, he moved to Tokyo University of Science as an assistant professor. In 2011, he moved to Kumamoto University and was promoted to associate professor. He was promoted to full professor at Kumamoto University in 2018. He joined the Faculty of Pharmaceutical Sciences, Chiba University in 2021 as a full professor. |
Introduction
Originally, the term ‘alkaloid’ referred to a basic compound isolated from plants. However, it is now used more broadly to describe nitrogen-containing secondary metabolites, which are found not only in plants but also in bacteria and animals. Among the vast number of alkaloids discovered in nature, many have already been utilized as medicines due to their potent biological activities. Due to their complex structures and significant biological activities, many alkaloids have become popular targets for total synthesis, driving advancements in synthetic organic chemistry.Since the early 21st century, organocatalytic reactions have undergone significant advancements. In particular, chiral secondary amine organocatalysts, such as proline (1),1 the diphenylprolinol trimethylsilyl ether catalyst (2, Hayashi–Jørgensen catalyst),2 and the 2,3,5-trisubstituted-4-imidazolidinone catalyst (5-benzyl-2-(tert-butyl)-3-methylimidazolidin-4-one (3), MacMillan catalyst),3 have been employed to catalyze several key enantioselective reactions, including asymmetric aldol, Michael, Mannich, Diels–Alder, and ene reactions, as well as α-oxidation and epoxidation (Fig. 1).4 These reactions consistently proceed with high stereoselectivity (both enantio- and diastereoselectivity) and do not require stringent conditions, such as anhydrous or anoxic environments. Furthermore, organocatalytic reactions are easily adapted to domino reactions, where multiple reactions occur in succession, as well as to one-pot reactions, where several chemical transformations are carried out simultaneously in a single flask.5 This adaptability is due to the catalyst, which is a small molecule with a secondary amine moiety and does not interfere with subsequent reactions. Therefore, they are widely employed in the total synthesis of natural products and pharmaceuticals.6 In 2016, we published a review article on the asymmetric total synthesis of 21 alkaloids utilizing chiral secondary amine organocatalysts.7 This perspective article outlines synthetic strategies and highlights the use of asymmetric organocatalytic reactions as a key step in the synthesis of a total of 24 alkaloids published from 2017 to early 2024.
 | ||
| Fig. 1 Representative chiral secondary amine organocatalysts. | ||
Indole alkaloids
Indole alkaloids represent one of the largest classes of alkaloids and are predominantly biosynthesized from tryptophan.8 Several of these alkaloids, such as vinblastine, vincristine, and reserpine, have important bioactivities and are already employed as therapeutic agents. Additionally, a wide variety of scaffolds are found due to the high reactivity of the indole core. As a result, indole alkaloids continue to capture the attention of synthetic chemists.In 2017, divergent total syntheses of naucleamide-related alkaloids were reported by Jia and coworkers (Scheme 1).9 The key reaction in their synthesis was a contiguous organocatalyzed asymmetric Michael addition/Pictet–Spengler cyclization to construct a tetracyclic ring system. Thus, an α,β,γ,δ-unsaturated aldehyde 9, the precursor for the Michael reaction, was synthesized in four steps from commercially available crotonaldehyde (7). These steps included α-iodination, reduction of the aldehyde followed by silylation of the resulting primary alcohol, and a Heck reaction to couple with acrolein. The Michael reaction, catalyzed by secondary amine 11, between aldehyde 9 and indole-containing malonamate 10 in the presence of catalytic benzoic acid proceeded selectively via 1,4-addition, rather than 1,2- or 1,6-addition, to generate the iminium ion intermediate in situ. Spontaneous cyclization through nucleophilic addition of the amine to the aldehyde produced hemiaminal 12 in the E form and its geometrical isomer (Z form) as a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture in moderate yield. The subsequent Pictet–Spengler reaction under acidic conditions yielded 13, possessing the C3S configuration, in good yield and selectively. At this stage, the enantiomeric excess was found to be 96%. After removal of the silyl group, the generated alcohol spontaneously cyclized to form lactone 5 along with the isomerized methoxycarbonyl group at C16. This lactone 5 is a monoterpenoid indole alkaloid found in plants of the genus Nauclea and named naucleamide C. Reduction of natural product 5 with NaBH4 resulted in a lactone-selective reduction, followed by thermodynamic isomerization at the C16 position to yield the natural product naucleamide A (4) in moderate yield. The total synthesis of naucleamide E (6) was achieved by a biomimetic oxidation using 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) with 4 as the substrate. In addition, Jia and coworkers also synthesized geissoschizine and three related alkaloids from the key intermediate 13. Their synthetic strategy allows access to natural products in a reduced number of steps (6–11 steps from commercially available materials) and enables divergent total syntheses of naucleamide- and geissoschizine-related monoterpenoid indole alkaloids.
:1 mixture in moderate yield. The subsequent Pictet–Spengler reaction under acidic conditions yielded 13, possessing the C3S configuration, in good yield and selectively. At this stage, the enantiomeric excess was found to be 96%. After removal of the silyl group, the generated alcohol spontaneously cyclized to form lactone 5 along with the isomerized methoxycarbonyl group at C16. This lactone 5 is a monoterpenoid indole alkaloid found in plants of the genus Nauclea and named naucleamide C. Reduction of natural product 5 with NaBH4 resulted in a lactone-selective reduction, followed by thermodynamic isomerization at the C16 position to yield the natural product naucleamide A (4) in moderate yield. The total synthesis of naucleamide E (6) was achieved by a biomimetic oxidation using 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) with 4 as the substrate. In addition, Jia and coworkers also synthesized geissoschizine and three related alkaloids from the key intermediate 13. Their synthetic strategy allows access to natural products in a reduced number of steps (6–11 steps from commercially available materials) and enables divergent total syntheses of naucleamide- and geissoschizine-related monoterpenoid indole alkaloids.
 | ||
| Scheme 1 Synthesis of naucleamide-related indole alkaloids by Jia et al. in 2017. | ||
Yang and coworkers achieved the total synthesis of actinophyllic acid (14), a unique monoterpenoid indole alkaloid containing a 1-azabicyclo[4.2.1]nonane ring system (Scheme 2).10 To date, several asymmetric total syntheses and formal total syntheses of 14 have been reported by the groups of Overman,11 Kwon,12 and Qin.13 The key reaction in Yang's synthesis is the modified dual iridium/amine catalytic allylation developed by Carreira's group.14 In this reaction, indole-containing allyl alcohol 15 and aldehyde 16 were treated with an iridium catalyst (iridium and chiral phosphine ligand 17) and an amine catalyst 18 in the presence of (MeO)2PO2H as a promoter, resulting in the coupling product 19 in excellent yield, enantioselectivity (>99% ee), and diastereoselectivity (dr = 12:1). Subsequent diastereoselective addition of the enolate derived from 20 to the aldehyde of 19 followed by TBS protection afforded 21 as a single isomer, with four contiguous stereocenters being controlled. The stereocontrol at C20 was explained using the Felkin–Anh model. For the inversion of the C19 stereocenter, kinetic protonation at low temperature was conducted. Next, the vinyl group of 22 was converted into a methoxycarbonyl group in a three-step transformation involving oxidative cleavage of the olefin moiety, oxidation of the resulting aldehyde to carboxylic acid, and methylation with TMSCHN2 (from compound 22 to 23). The construction of the seven-membered ring was achieved by amide-selective partial reduction with LiBHEt3 and Mannich-like reaction with Tf2O on the generated hemiaminal (from compound 23 to 24). To close the 1-azabicyclo[4.2.1]nonane ring system, the mesyl group was attached as the leaving group in two steps. Following selective deprotection of N-Boc on the pyrrolidine core followed by cyclization under basic condition afforded pentacyclic product 25 in 70% yield for four steps. Finally, actinophyllic acid (14) was accessed after desilylation, oxidation of the resulting alcohol, deprotection of N-Boc, Overman's aldol reaction with formaldehyde for installation of the tetrahydrofuran ring, and hydrolysis.11
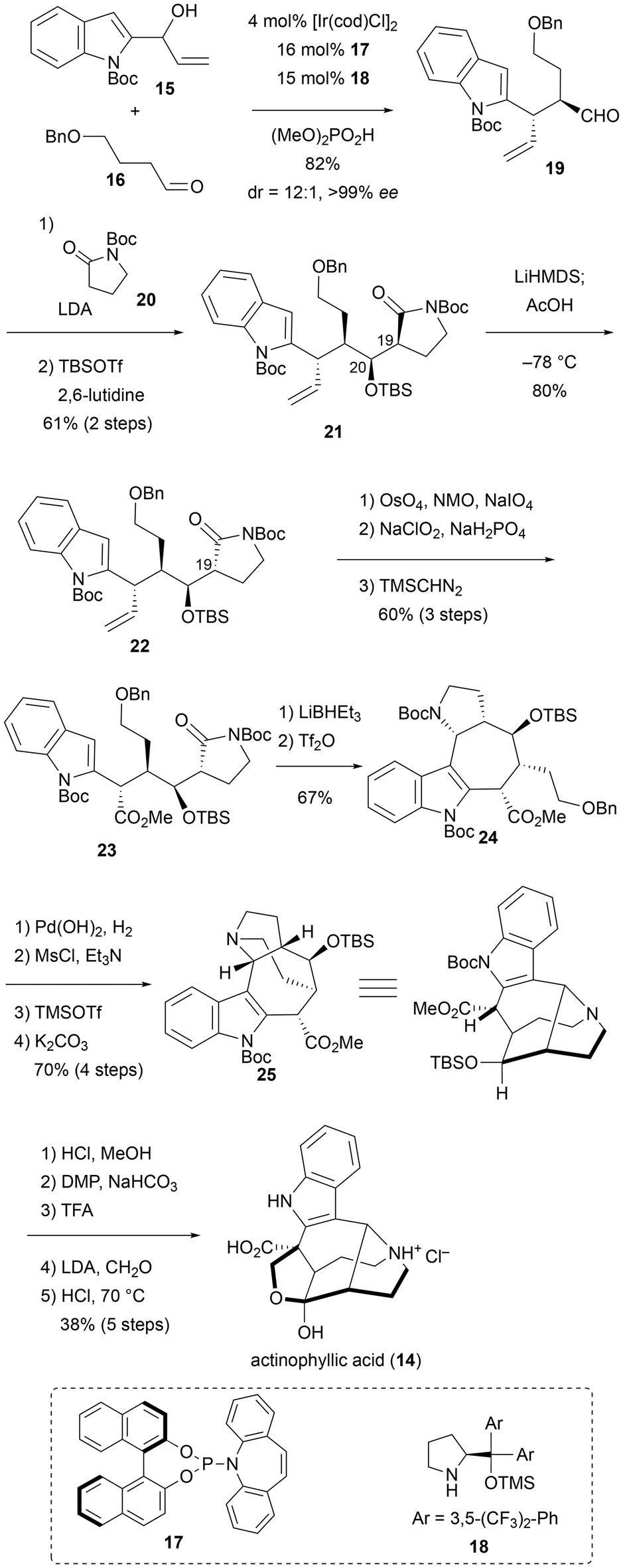 | ||
| Scheme 2 Synthesis of actinophyllic acid by Yang et al. in 2018. | ||
In recent years, concerted asymmetric reactions involving secondary amine catalysts and metal catalysts have made significant progress. In particular, the dual iridium/amine catalytic allylation developed by Carreira et al. has been employed in numerous total syntheses. In the synthesis by Yang et al., this reaction was carried out in the presence of an indole ring and the approach proved to be a powerful tool for the total synthesis of indole alkaloids.
In 2018, Xu and coworkers reported the total synthesis of the Strychnos alkaloid strychnofoline (26), which possesses significant anticancer activity (Scheme 3).15 First, the one-pot acylation/asymmetric Michael reaction/Pictet–Spengler reaction developed by Franzén et al. in 2011 was performed to construct the pentacyclic ring system.16 Thus, 6-methoxytryptamine (27) was treated with diketene to afford β-ketoamide 28. Next, the addition of α,β-unsaturated aldehyde 29 and the Hayashi–Jørgensen catalyst (ent-2) to the same reaction vessel initiated an asymmetric Michael addition, yielding 30, followed by spontaneous cyclization to give intermediate lactol 31. The subsequent addition of AcCl to lactol 31 led to the ring opening of the lactol, hemiaminal formation, and a diastereoselective Pictet–Spengler reaction via intermediate 32, resulting in the formation of 33 in good yield with excellent enantioselectivity (67% yield, >99% ee). Treatment of tBuOCl with indole 33 yielded chloroindolenine intermediate, which subsequently underwent a rearrangement reaction under acidic methanol conditions to produce oxindole 34 as a single isomer. Following selective reduction of the C21 amide using DIBAL, hydrolysis of the cyclic enol ether under acidic conditions, acetylation of the generated primary alcohol, and transformation of the ketone to tosylhydrazone, hydrazone 35 was obtained. After the Shapiro reaction and deacetylation of 35, the resulting primary alcohol was oxidized with Dess–Martin reagent to produce aldehyde 36. Finally, the total synthesis of strychnofoline (26) was achieved through the Pictet–Spengler reaction with N-methyltryptamine, followed by demethylation using BBr3. In this total synthesis, the one-pot cascade reaction, which included the asymmetric Michael addition and Pictet–Spengler reaction to construct the pentacyclic system, enabled the efficient synthesis of strychnofoline (26) from simple 6-methoxytryptamine (27) in just nine steps.
 | ||
| Scheme 3 Synthesis of strychnofoline by Xu et al. in 2018. | ||
In 2019, Ishikawa and coworkers reported the first total synthesis of secologanin (37), the monoterpene moiety in the biosynthesis of monoterpenoid indole alkaloids (Scheme 4).17 In addition, the Ishikawa group achieved the synthesis of strictosidine (38), a common intermediate in the biosynthesis of more than 3000 monoterpenoid indole alkaloids.18a Having established a straightforward synthetic method for secologanin (37) and strictosidine (38), they successfully achieved the total synthesis of 33 indole alkaloids by fully utilizing bioinspired reactions.17,18 The key reactions in the synthesis of 37 were contiguous organocatalytic Michael addition followed by Fukuyama reduction19 and spontaneous cyclization to construct the dihydropyran ring. The precursor of the Michael reaction 41 was synthesized by Knoevenagel condensation of commercially available aldehyde 39 and a malonic acid derivative 40. Subsequent enantioselective Michael reaction using electrophile 41 and aldehyde 42 containing a sulfide moiety as a nucleophile in the presence of catalyst 2 provided Michael adduct 43. An important factor in achieving the unusual anti-selectivity required for the total synthesis was the substitution of an alkynyl group on the electrophile (TS1). Without purification, reduction of adduct 43 under Fukuyama conditions selectively converted the thioester into the aldehyde followed by spontaneous cyclization to provide dihydropyran 44 in good yield with excellent enantio- and diastereoselectivity. When hemiacetal 44 was treated with glycosyl donor 45 in the presence of BF3·Et2O, diastereoselective glycosylation proceeded to afford 46 as a single isomer in 87% yield. A further three-step transformation to prepare the aldehyde and terminal olefin, including deprotection of TMS group, hydroboration-oxidation, and elimination of sulfoxide by heating, provided secologanin tetraacetate (47), which is a key intermediate for the collective synthesis of monoterpenoid indole alkaloids on the decagram scale. Finally, the total synthesis of secologanin (37) was achieved by removing the acetyl group from the sugar moiety. The total synthesis of strictosidine (38) was subsequently achieved as shown in Scheme 4. Thus, the diastereoselective Pictet–Spengler reaction with 47 and a α-cyanotryptamine 48 afforded 49 as almost single isomer (dr = >10:1). The reasons for this intriguing diastereoselectivity have been experimentally and computationally verified; for details, see the original report.18a Next, the reductive decyanation of 49 with sodium cyanoborohydride in the presence of acetic acid provided strictosidine tetraacetate (50) in excellent yield (85% over two steps). Finally, removal of the four acetyl groups yielded strictosidine (38). The total synthesis of 38 was accomplished with a yield of 20% over ten steps and was achieved on a gram scale.
 | ||
| Scheme 4 Syntheses of secologanin and strictosidine by Ishikawa et al. in 2019. | ||
Tu and coworkers reported the total syntheses of naucleofficine I (51) and II (52) (Scheme 5).20 The key reaction was a four-component asymmetric Mannich/acylation/Wittig reaction, catalyzed by the originally developed bifunctional spirocyclic pyrrolidine 54. Thus, 3,4-dihydro-β-carboline 53 was treated with acetaldehyde in the presence of secondary amine catalyst 54, which contains a sulfonamide moiety, to yield 55. Acyl chloride 56 and Wittig reagent 57 were then added to the same reaction vessel, resulting in the desired compound 58 in 42% yield over three steps and 91% ee. Subsequently, the intramolecular hetero-Diels–Alder reaction with ZnBr2 gave pentacyclic product 59 in 51% yield and superb diastereoselectivity (dr >20:1). Then, recrystallization of 59 enhanced the enantio-purity to >99% ee in 85% yield. X-ray crystallographic analysis confirmed the absolute configuration. After a Vilsmeier–Haack reaction, methylation with Me2CuLi afforded the diastereomeric products 61–63. Undesired 63 could be converted into 62 by isomerization of the α position of aldehyde with 1,8-diazabicyclo[5.4.0]-7-undecene (DBU). A further two-step transformation including reduction of the aldehyde to alcohol followed by elimination of the methoxy group with 1,1,1,3,3,3-hexamethyldisilazane (HMDS) via the oxonium cation provided naucleofficine I (51) and II (52) from 61 and 62 in good yield, respectively. A one-pot, four-component asymmetric reaction using a secondary amine catalyst developed by Tu et al. completed the total synthesis of the complex pentacyclic alkaloids 51 and 52 in just seven steps.
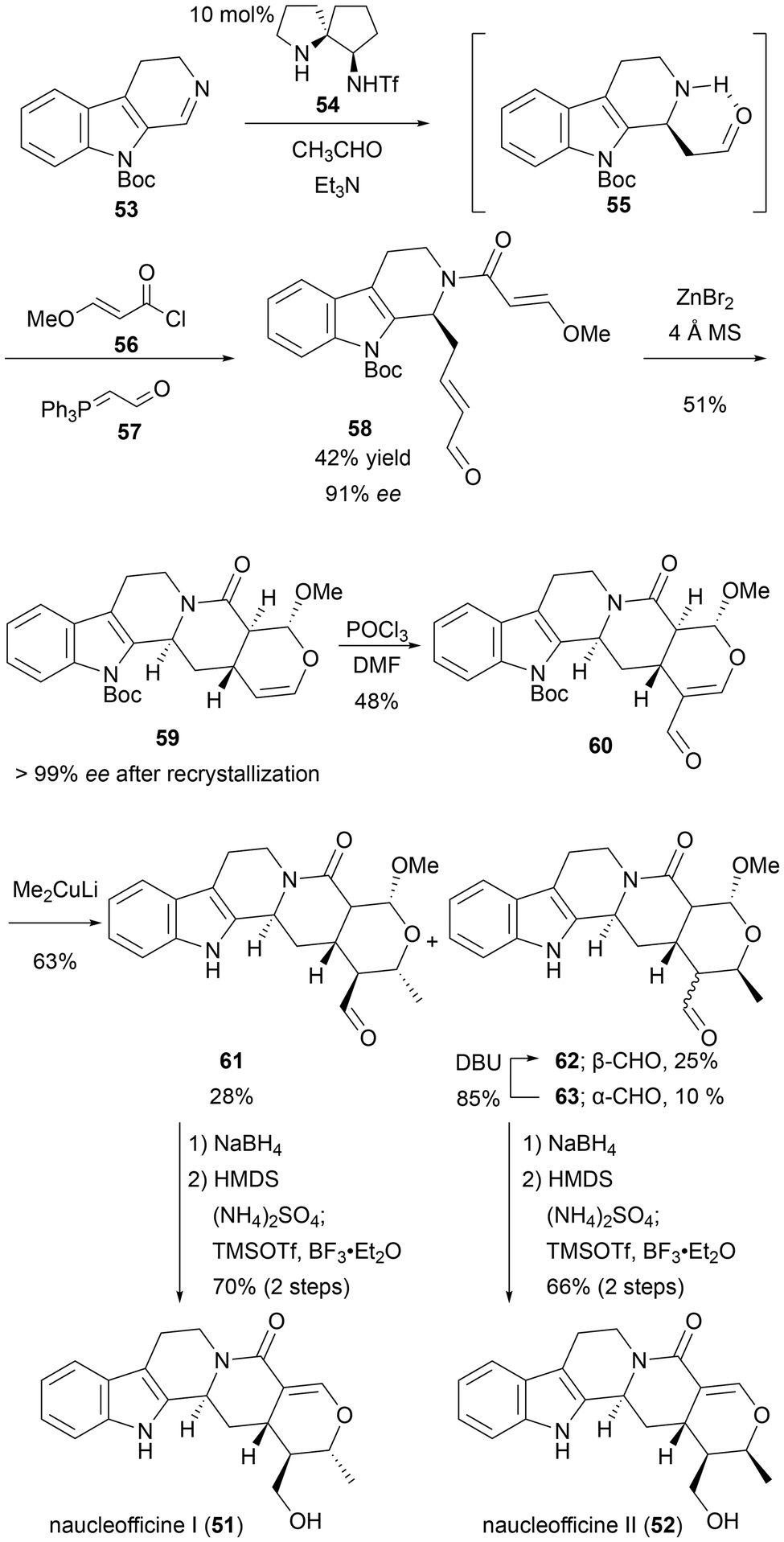 | ||
| Scheme 5 Syntheses of naucleofficine I and II by Tu et al. in 2019. | ||
The Cho group reported the total synthesis of uleine (64) with modified Fischer indolization and asymmetric organocatalytic reaction as the key steps (Scheme 6).21 Their synthesis started from Ma's organocatalytic chiral cyclohexanone synthesis.22 Thus, γ-keto-α,β-unsaturated ester 65 was treated with n-butanal in the presence of catalyst 2 to afford the Michael adduct 66. Subsequently, the adduct 66 underwent intramolecular aldol reaction in the presence of DBU. Without purification, a mesylation of the generated alcohol gave cyclohexanone 67 in good yield and excellent enantioselectivity (49% over three steps and 93% ee). Conversion of the ketone into the cyclic acetal and reduction of the ester provided alcohol 68. The installation of the cyano group onto compound 68 was achieved via the Mitsunobu reaction, utilizing acetone cyanohydrin as the cyanide source to yield compound 69. Following a two-step transformation, which involved the reduction of the cyano group and the protection of the resulting primary amine with CbzCl, compound 70 was obtained. Subsequently, the removal of the acetal protection, followed by a spontaneous intramolecular aza-Michael addition, yielded the 2-azabicyclo[3.3.1]nonane core 71, with an overall yield of 60% over four steps. For regioselective indolization, the ene-hydrazide 72 was prepared by palladium-catalyzed Buchwald–Hartwig coupling reaction via the enol triflate. The Zn-mediated indolization proceeded efficiently to afford the desired indole 73 in 76% yield. Regioselective oxidation of the benzyl position was achieved with PCC. Next, continuous deprotection of N-Cbz and in situ reductive methylation afforded 74 in good yield. The final two-step transformation, including addition of MeLi and dehydration under acidic conditions, gave uleine (64). The Cho group also reported the formal synthesis of tubifolidine, an indole alkaloid containing a pentacyclic core. Cho et al. efficiently synthesized the 2-azabicyclo[3.3.1]nonane core from the chiral cyclohexanone developed by Ma. Modified Fishier indole synthesis thus enables the late-stage introduction of indole using ketones as substrates, where regioselectivity is an issue.
 | ||
| Scheme 6 Synthesis of uleine by Cho et al. in 2020. | ||
More recently, the Ishikawa group reported the first total synthesis of 20-episilicine (75) using an organocatalytic reaction they had previously developed (Scheme 7).23 Their synthesis started from the preparation of 79, a substrate for the key organocatalytic reaction. First, Noyori's protocol24 achieved indole-conjugated ketone selective acetalization using β-esters 76. Reduction of the ester and Wittig reaction of resulting aldehyde afford α,β-unsaturated ester 77 in good yield (67% over three steps). Next, the ester was reduced, and the generated alcohol was protected with an acetyl group. The following protection with PMBCl and NaH provided PMB-protected indole 78 in 77% yield over three steps. The three-step conversion, including the Vilsmeier–Haack reaction to install a formyl group, removal of the acetyl group, and oxidation with MnO2 of the yielded primary alcohol, gave the α,β-unsaturated aldehyde 79 as an E/Z mixture. Treatment of the mixture of 79 and thiomalonamate 80 with secondary amine catalyst 81 in the presence of benzoic acid and three equivalents of MeOH in CH2Cl2 constructed the chiral piperidine ring of intermediate 82. Without purification, crude 82 was treated with trifluoroacetic acid (TFA) to convert the hemiaminal into an enamine. Subsequently, the mixture of stereoisomers of the methoxycarbonyl group was epimerized with DBU to afford 83 as a single isomer. The chemical yield of these three key steps was 72%, and the enantiomeric excess was 93%. After reducing the aldehyde of 83, the construction of the seven-membered ring was accomplished under heating in the presence of MgBr2 to provide 84 in 73% yield over two steps. Reduction of 84, containing a methoxycarbonyl group, with nickel boride selectively reduced the thiocarbonyl group and enamine to give 85 as a single isomer. The conversion of the methoxycarbonyl group into an ethyl group was accomplished by a three-step reaction, including reduction with DIBAL, transformation of the resulting aldehyde into a terminal alkene with Tebbe reagent, and reduction under hydrogenation conditions. Finally, 20-episilicine (75) was obtained by reduction, removing the PMB group, and conversion of the acetal into a carbonyl group. In addition, the Ishikawa group achieved the total synthesis of silicine from common intermediate 84. Ishikawa et al. achieved an efficient total synthesis of indole alkaloids, indicating the applicability of the originally developed organocatalytic piperidine ring synthesis.
 | ||
| Scheme 7 Synthesis of 20-episilicine by Ishikawa et al. in 2023. | ||
Cinchona alkaloids
Cinchona alkaloids, which belong to the monoterpenoid indole alkaloids in biosynthesis, are historically significant molecules.25 For instance, quinine (86) was one of the most effective medicines for treating malaria. Additionally, Cinchona alkaloids such as quinine (86), quinidine, and their derivatives function as organocatalysts, promoting several important reactions.26In 2019, Córdova et al. reported the enantioselective synthesis of quinine (86) utilizing the diphenylprolinol silyl ether catalyst 2 (Scheme 8).27 The key step in this synthesis is an enantioselective Michael addition followed by imine formation, reductive amination, and lactamization in a cascade reaction (Scheme 8a). Thus, when α,β-unsaturated aldehyde 87 and dimethyl malonate are used as substrates, and 10 mol% of secondary amine catalyst 2 is employed in ethanol, the Michael addition proceeds stereoselectively to form the Michael adduct 88. After the reaction is complete, the solvent is removed under reduced pressure, and the resulting crude product is stirred with magnesium sulfate and benzylamine in dichloromethane to form an imine, which is subsequently converted into the corresponding benzyl amine by reduction with sodium triacetoxyborohydride. The reaction mixture is then stirred at room temperature for an additional 17 hours to allow lactamization, yielding compound 89 in high yield (85% over two steps) and excellent enantioselectivity (97% ee). The ester of compound 89 is formed in a 6:1 ratio of the anti and syn forms. However, since the syn form was required for the synthesis of quinine (86), epimerization was carried out using the following three-step chemical transformations. First, the ester and amide groups of 89 were simultaneously reduced with LiAlH4 to give the corresponding amino alcohol. This was then oxidized to the aldehyde via a Swern oxidation reaction, followed by mild epimerization with pyrrolidine, producing a diastereomeric mixture of 90 and 91 with an anti/syn ratio of 3:1. The mixture was purified using silica gel column chromatography, isolating the pure syn form 91 (dr >21:1). The conversion of aldehyde 91 into quinine (86) was carried out by following the route developed by Kobayashi et al.28 (Scheme 8b). First, 91 was reduced to the corresponding alcohol and protected with a silyl group. The nitrogen protecting group was then switched from a benzyl group to a Teoc group, followed by Swern oxidation to form aldehyde 92. Aldehyde 92 was converted into the coupling product 94 through a Horner–Wadsworth–Emmons (HWE) reaction with phosphate 93. The conversion of 94 into quinine (86) was achieved in six steps, including Sharpless asymmetric dihydroxylation and quinuclidine formation via an SN2 reaction, with a 45% yield, by following the method of Jacobsen et al.29 Additionally, the Córdova group synthesized quinidine from synthetic intermediate 91. This synthesis serves as a good example of the application of one-pot cascade reactions, where organocatalysis plays a key role in total synthesis.
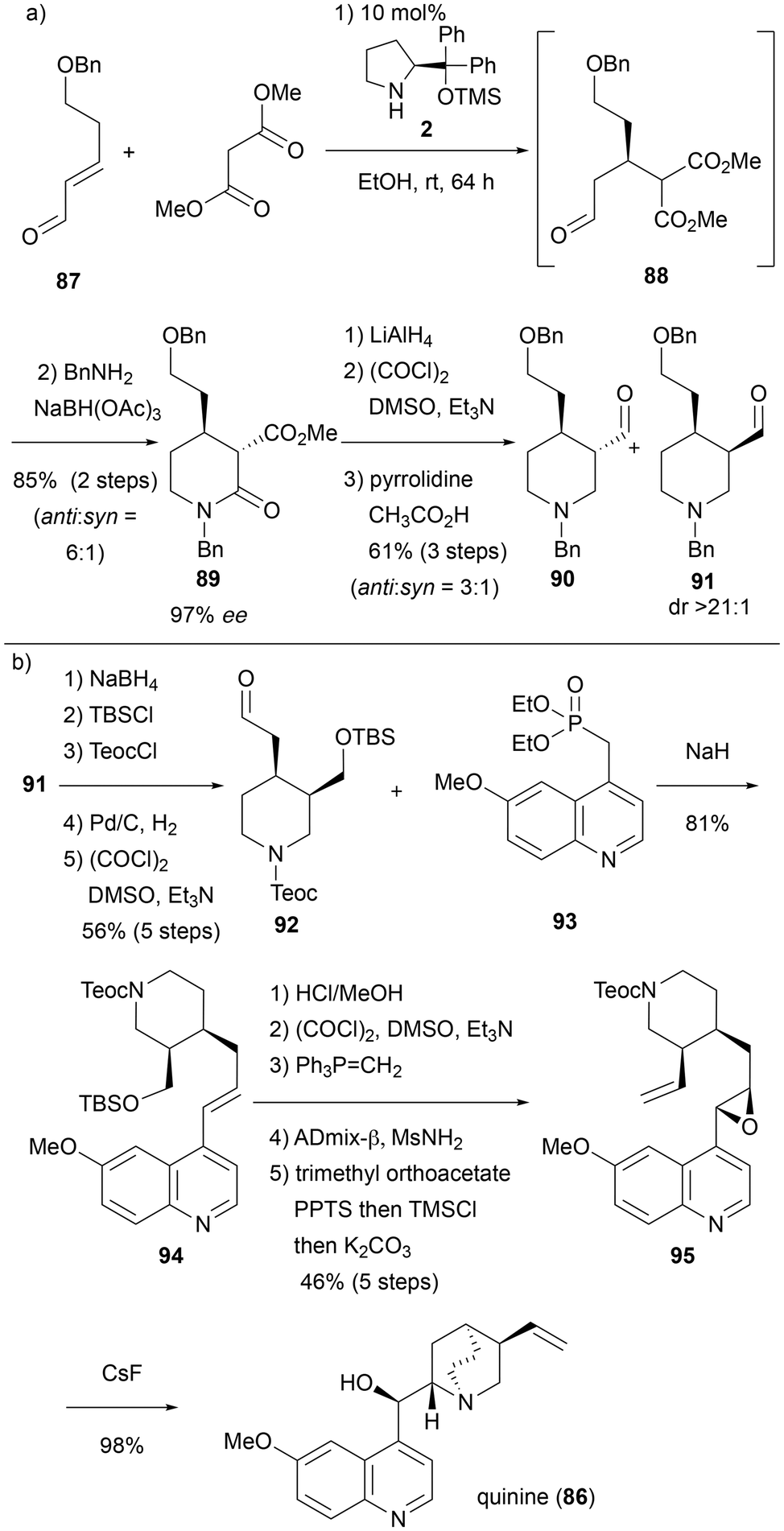 | ||
| Scheme 8 Synthesis of quinine by Córdova et al. in 2019. | ||
The supply of cinchona alkaloids still relies on isolation from plants, making it challenging to obtain the enantiomers of quinine (86) and quinidine. However, this does not pose a major obstacle to developing asymmetric reactions, as cinchonidine and cinchonine, also isolated from the same plant, can serve as pseudoenantiomers of quinine (86) and quinidine. Nonetheless, quinine (86) and quinidine are diastereomers, and their chemical reactivity and ability to induce chirality are not identical. Therefore, efficient preparation of the enantiomers of quinine (86) and quinidine through total synthesis is highly significant. In 2019, Ishikawa and coworkers achieved the asymmetric total synthesis of ent-86 using less than 1 mol% of catalysts in key asymmetric organocatalytic reactions, aiming to provide a quantitative supply of unnatural quinine (ent-86) through chemical synthesis (Scheme 9).30 The key reaction in Ishikawa's synthesis was an enantioselective Michael addition/hemiamination/Strecker reaction, catalyzed by Hayashi–Jørgensen catalyst using thiomalonamate as the nucleophile. Thus, when α,β-unsaturated aldehyde 96 and thiomalonamate 97 react with secondary amine catalyst 81 in the presence of benzoic acid and methanol, the asymmetric Michael addition was followed by a hemiamination reaction to produce thiolactam 98. Stirring the crude materials with trimethylsilyl cyanide under solvent-free conditions results in the rapid silylation of the hemiacetal. Upon completion of this reaction, the addition of boron trifluoride diethyl ether complex at low temperature promoted the Strecker reaction, yielding the desired cyanothiolactam 99 in two steps in 90% yield. To obtain key intermediate 102 from 99, a stepwise and functional group-selective reduction of the ester, nitrile, and thiolactam was essential. This challenge was addressed by converting the cyano group into an imidate group. Thus, when 99 was stirred in methanol under basic conditions, imidate 100 was obtained as a diastereomeric mixture (α/β = 3:1). The enantioselective excess of 100 was 94%, confirming that the initial catalytic asymmetric reaction proceeded with high enantioselectivity. Imidate 100 was then converted into piperidine 101 through thiolactam-selective reduction using nickel boride. When the obtained amine intermediate was treated with DIBAL at low temperature, only the ester was reduced to an aldehyde. This imidate moiety was then rapidly hydrolyzed to an ester under acidic conditions in aqueous THF, resulting in piperidine-5-carbaldehyde 101. The aldehyde in 101 was converted into a terminal double bond using Tebbe reagent. Subsequent reduction of the ester with DIBAL yielded the trisubstituted piperidine 102 in good yield (5 steps, 45%). During purification, the aldehyde moiety of 102 was isomerized to the equatorial position, adopting a thermodynamically stable chair configuration due to the epimerization process; as a result, it was obtained as a single isomer required for the synthesis of ent-86. In the subsequent reaction to introduce the quinoline ring, a 1,2-dihydroquinoline derivative 103, which lacks quinoline resonance to enhance reactivity, was lithiated and reacted with 102. As a result, the desired coupling product 104 was obtained in 72% yield. Although the hydroxyl group of 104 was generated as a 1:1 ratio of α- and β-isomers, the synthesis proceeded with this diastereomeric mixture. The secondary hydroxyl group of 104 was first acetylated, and the silyl group was removed using hydrochloric acid/methanol to yield 105. Compound 105 was then mesylated and heated at 120 °C in toluene, facilitating an intramolecular SN2 reaction. The 1,3-dimethoxybenzyl (DMB) group of the ammonium salt produced during cyclization was captured by added anisole, resulting in a quinuclidine intermediate. This process combined cyclization and deprotection in a single step. Finally, the removal of the acetyl group and the simultaneous desulfonation/oxidation of dihydroquinoline were carried out, leading to the total synthesis of unnatural quinine (ent-86) and 9-epi-quinine (106) in three steps with a 77% yield. 9-epi-Quinine (106) is also an important molecule as an organocatalyst.25 The synthesis of ent-86 and 106 could be interconverted with high yield via the Mitsunobu reaction. The synthesis of (+)-quinine (ent-86) is highly efficient, involving a total of 15 steps, including Mitsunobu reactions, with an overall yield of 16%. Furthermore, a synthesis that uses only 0.5 mol% of catalyst to construct chiral carbon centers is considered excellent for asymmetric total synthesis.
 | ||
| Scheme 9 Synthesis of unnatural enantiomers of quinine by Ishikawa et al. in 2019. | ||
In 2022, Hayashi and Terunuma reported an extremely efficient total synthesis of quinine (86) using their originally developed asymmetric organocatalytic reaction and one-pot protocols (Scheme 10).31 The key step for introducing the chiral carbon centers was the asymmetric Michael reaction, catalyzed by 2, using sulfide-containing aldehyde 107 and nitroalkene 108 as substrates. The resulting intermediate 109 was then treated with an appropriate reactive imine 110 which was prepared in situ in the presence of DBU. This allows the successive aza-Henry reaction and hemiaminalization to proceed. The cyclized product 111 was kept under basic conditions, leading to the elimination of HNO2 and affording the tetrahydropyridine derivative 112 in 66% yield and 98% ee. In this first one-pot reaction, five transformations occurred, including the preparation of compound 110, with the reaction conducted on a decagram scale. The second one-pot reaction was an ingenious five-step process that began with the reductive removal of the hydroxyl group at the C2 position. Cyclized compound 112 was treated with Et3SiH in the presence of TFA to remove both the Boc and hydroxyl groups, and the enamine double bond was further reduced using NaBH(OAc)3. At this stage, the ethoxycarbonyl group adopted a thermodynamically stable S-form. The excess reducing agent was then decomposed with acetaldehyde, and volatile reagents were removed under reduced pressure. Finally, the Boc group was reintroduced, affording compound 113 in 78% yield with full control over the three stereocenters. The third one-pot reaction involved the construction of a terminal double bond and the conversion of an ester to an aldehyde. The allyl thioether moiety of compound 113 was converted into an alkene using the elimination reaction conditions developed by Matsuo et al.32 Additionally, DIBAL was directly introduced into the reaction vessel, converting the ester into an aldehyde. This one-pot reaction proceeded in 77% yield. The fourth one-pot reaction involved the introduction of the quinoline ring. Hayashi et al. successfully incorporated 2,4-dibromoquinoline 116 into the aldehyde of compound 115 using an organolanthanide species developed by Knochel et al.33 The diastereoselectivity at the C9 position was nearly 1:1, but the undesired 9-epi-117 was converted into the desired 117 through oxidation and stereoselective reduction. The final one-pot reaction for quinine synthesis involves five steps, including the construction of the quinuclidine ring. First, the bromine on the quinoline ring of 117 was removed under zinc/acetic acid conditions. Next, AcCl was added in methanol to generate hydrochloric acid, which removed the Boc and TBDPS groups. MsCl was used for selective mesylation of the primary hydroxyl group, followed by treatment with ammonia to activate the nitrogen on the piperidine ring, which was temporarily protected as a hydrochloride salt. Finally, the quinuclidine ring was constructed via an SN2 reaction, completing the total synthesis of quinine (86). The quinine synthesis reported by Hayashi and Terunuma was achieved through a 17-step chemical transformation. Compared to previous total syntheses, this 17-step process is relatively short and efficient. Additionally, they refined the synthesis to be completed using only five reaction vessels. The overall yield was 14%, demonstrating the practicality of the approach. Notably, the first asymmetric organocatalytic reaction and the aza-Henry reaction were crucial and unique steps that significantly contributed to the success of this total synthesis.
 | ||
| Scheme 10 Synthesis of quinine by Hayashi and Terunuma in 2022. | ||
Stemona alkaloids
Stemona alkaloids were primarily isolated from stemonaceous plants, which were used in folk medicine in East Asian countries.34 In addition, those alkaloids have attractive skeletons characterized by a 5/7/5 tricyclic core. Therefore, they have historically been the target of total synthesis studies.In 2020, Prasad et al. reported the formal total synthesis of stemoamide (118) using sequential organocatalytic α-oxyamination and originally developed the conjugate addition of aza-allylanion (Scheme 11).35 Their synthesis commenced from MacMillan sequencing to afford chiral γ-hydroxy α,β-unsaturated ester 121.36 Thus, treatment of the known aldehyde 119 with nitrosobenzene in the presence of D-proline furnished α-oxyamino aldehyde 120, which upon in situ HWE olefination followed by reduction of the N–O bond with Cu(OAc)2 gave 121 in 36% yield over three steps and 98% ee. After protection of the secondary alcohol with the TBS group, addition of the lithium anion of 2-aza-pentadiene 122 to the unsaturated ester diastereoselectively provided 123 in 47% yield. Deprotection of the TBS group proceeded with successive lactonization, and subsequent generation of free amine with hydroxylamine and treatment with acryloyl chloride gave unsaturated amide 124 in 35% yield over three steps. The three-step conversion, including ring-closing metathesis (RCM), simultaneous reduction of resulting olefine and benzyl group, and mesylation, afforded lactam 125 (54% over three steps), which is an intermediate in Narasaka's total synthesis of stemoamide (118).37 Therefore, the formal total synthesis was accomplished.
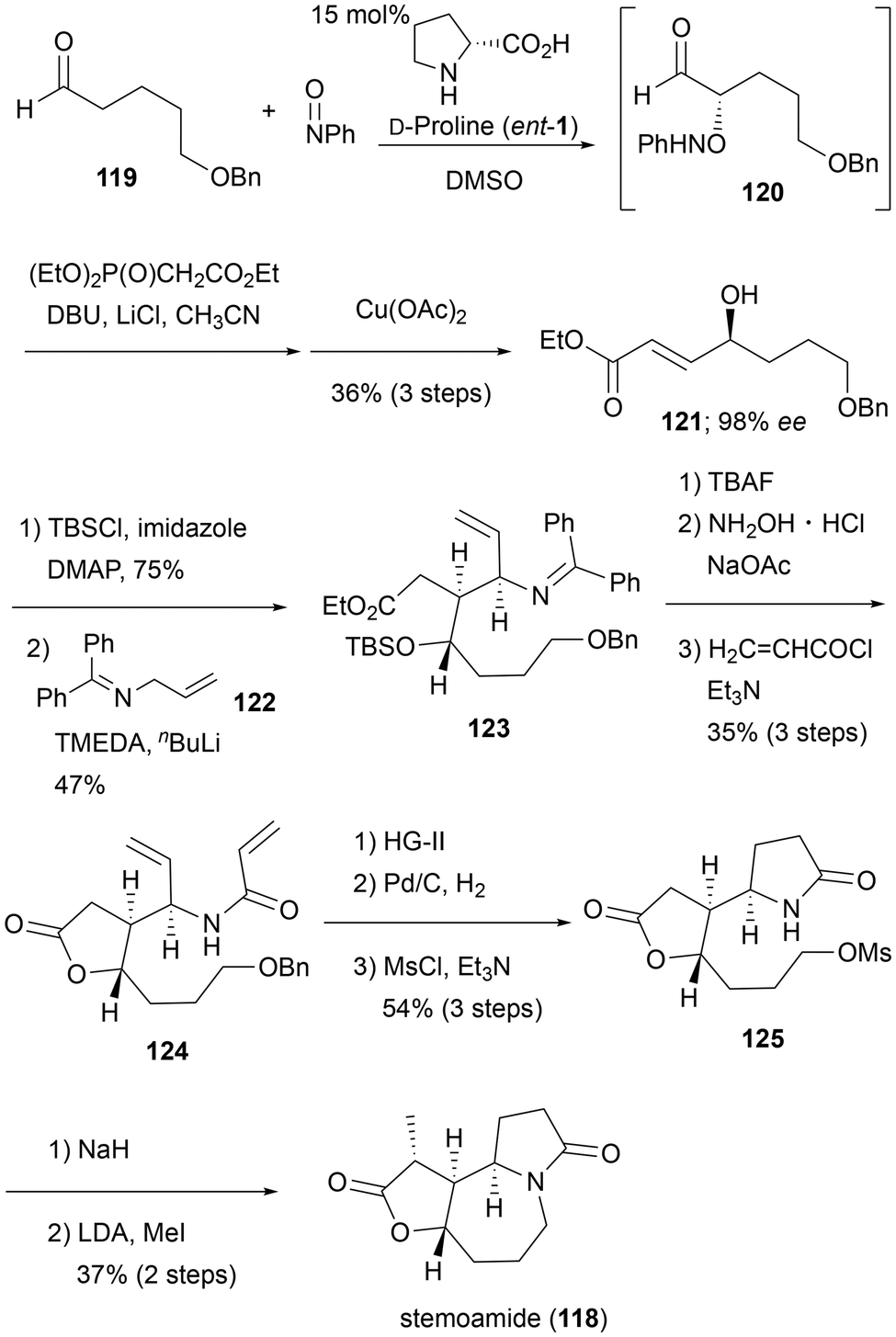 | ||
| Scheme 11 Synthesis of stemoamide by Prasad et al. in 2020. | ||
Elegant collective total synthesis of Stemona alkaloids using tailored dyotropic rearrangement of β-lactone was reported by Tang in 2021.38Scheme 12 shows the total synthesis of bisdehydroneostemoninine (126). First, MacMillan's organo-SOMO catalyzed α-allylation39 using ceric ammonium nitrate (CAN) as a single-electron oxidant and subsequent reduction converted aldehyde 127 into 129 in high yield and excellent enantioselectivity (72%, 93% ee). Reduction of amide 129 with alane provided 130. Due to the instability of the produced aldehyde, the oxidation and subsequent conversion of 130 were extremely difficult. After extensive trials, the synthesis of 133 was achieved by modified Ley–Griffith oxidation followed by rhodium-catalyzed reductive aldol reaction with α,β-unsaturated ester 132 in the presence of catalyst 131 in 30% yield for two steps. β-Lactone 134, obtained by hydrolysis of 133 followed by lactonization reactions, underwent dyotropic rearrangement using silica gel in the absence of acid (TS2). In this reaction, the electron-rich pyrrole ring rearranged and generated 135 in a nearly quantitative yield. Methylenation of 135 with Petasis reagent gave enol ether 136. Oxaspirolactone was constructed by photoredox-catalyzed formal [3 + 2] cycloaddition with α-bromocarboxylic acid 137, which afforded two pairs of diastereomer 138a/b and 139a/b in 63% and 22% yields, respectively. The undesired isomer 138a/b could be converted with TFA into the desired 139a/bvia ring opening/closing equilibrium. Finally, the total synthesis of bisdehydroneoustemoninine (126) was accomplished by β-elimination with DBU followed by ruthenium-catalyzed double-bond isomerization (from compound 139 to 126via140). In addition, Tang reported not only the total syntheses of Stemona alkaloids including stemoamide, tuberostemospiroline, saxorumamide, isosaxorumamide, and stemonine, but also the application of photoredox-catalyzed formal [3 + 2] cycloaddition with eight substrates. Their synthesis of 126 was accomplished in 12 steps from 127 and the overall yield was 5.9%. The reader, interested in the dyotropic rearrangement of β-lactone, is referred to the original report by Tang et al.38
 | ||
| Scheme 12 Synthesis of bisdehydroneostemoninine by Tang et al. in 2021. | ||
Yang and coworkers reported an efficient total synthesis of bisdehydrotuberostemonine D (141) (Scheme 13).40 These two alkaloids in a diastereomeric relationship were synthesized via Carreira's Ir/amine dual catalytic stereodivergent reaction, respectively. Thus, 143, converted by N-alkylation and addition of vinylmagnesium bromide from 142, and butanal were treated with catalytic Ir/ent-17 and 18 in the presence of malonic acid to obtain 144 with two newly generated stereocenters in good yield and superb enantio- and diastereoselectively (70%, dr >50:1, 99% ee). The RCM reaction of 144 using Grubbs’ second-generation catalyst proceeded with excellent yield to form a seven-membered ring, resulting in the formation of 145. Subsequently, Krische's allylation41 using Ir-catalyst 147 with acrylic acid derivative 146 and spontaneous lactonization gave γ-butyrolactone 148 as a single isomer in 80% yield. After ruthenium-catalyzed isomerization of exo-methylene, site-selective iodination followed by intramolecular Heck reaction afforded tetracyclic 150via compound 149. The aldehyde 151, the precursor for the second Krische allylation, was provided by a three-step transformation including the hydrogenation of two double bonds, reduction of the methoxycarbonyl group, and oxidation of the generating primary alcohol. Finally, the second Krische allylation followed by hydrogenation under Takaya's protocol42 gave bisdehydrotuberostemonine D (141) in 32% yield over two steps. Their stereodivergent syntheses using one Carreira Ir/amine dual catalytic reaction and two Krische allylations allowed them to efficiently provide pyrrole Stemona alkaloids.
 | ||
| Scheme 13 Synthesis of bisdehydrotuberostemonine D by Yang et al. in 2021. | ||
More recently, Cernak and coworkers reported a short and straightforward total synthesis of stemoamide (118), utilizing a computer-aided key step and enhancements by chemists (Scheme 14).43 Their synthesis started from proline-catalyzed self-Mannich reaction developed independently by Hayashi and Barbas in 2003.44 Thus, two equivalents of aldehyde 152 and one equivalent of p-toluidine were treated with 20 mol% of L-proline (1), affording 153 through the addition of the enamine, formed from L-proline (1) to the imine formed from p-toluidine (TS3). Subsequently, the direct addition of allyl bromide, zinc, and bismuth chloride to the reaction vessel produced lactone 154. Without purification, the crude mixture of 154 was treated with TFA for lactamization, yielding 155. This two-step sequence rapidly formed three contiguous stereocenters and two rings in 33% yield with excellent enantioselectivity (99% ee). Next, the alkene was converted into an alcohol via hydroboration using [Ir(cod)Cl]2, followed by bromination and in situ removal of the PMP group to yield 156 in 40% over two steps. The two known reactions, the construction of the azepane ring and the diastereoselective introduction of a methyl group, led to the total synthesis of stemoamide (118). In addition, they accomplished a three-step total synthesis of 118 using an alternative protocol, which included allylation, Michael addition, and the Schmidt–Aubé rearrangement, following a computer-suggested synthetic route. Their synthesis was the shortest of the 32 total syntheses of natural product 118 reported to date, an impressive achievement made possible with the assistance of computational science.
 | ||
| Scheme 14 Synthesis of stemoamide by Cernak et al. in 2023. | ||
Daphniphyllum alkaloids
Daphniphyllum alkaloids, isolated from the plant genus Daphniphyllum, are part of a group of over 320 natural triterpenoids.45 They share a distinctive polycyclic ring-fused framework with multiple contiguous stereogenic centers and exhibit potential bioactivity against tumors and the human immunodeficiency virus. Their fascinating structures and promising bioactivities have made these molecules highly attractive to synthetic chemists and biologists alike. In this perspective article, two total syntheses of Daphniphyllum alkaloids are presented, each incorporating asymmetric reactions using secondary amine catalysts in the initial stages.Himalensine A (157) is an alkaloid with a [6-6-5-7-5] pentacyclic core, belonging to the calyciphylline A sub-family. In 2023, Dixon et al. reported the synthesis of this complex alkaloid using a practical and efficient dual amine/palladium catalyst system to construct the morphan skeleton (Scheme 15).46 The synthesis of the substrate for the key desymmetrization reaction began with a monoprotected diketone 158. Thus, arylamine 159 was condensed with ketone 158 in a reductive alkylation reaction, and the cyclic acetal moiety was reverted to a ketone. The secondary amine was then protected with a tosyl group, yielding substrate 160. A considerable amount of examination was needed to develop the novel asymmetric carbon–carbon bond-formation reaction the authors were aiming for. Ultimately, they identified optimal conditions: 20 mol% of cis-4-hydroxyproline 161 as a secondary amine catalyst, 5 mol% of Pd(OAc)2 and 15 mol% tris [p-(trifluoromethyl)phenyl] phosphine as metal catalysts, with dipotassium hydrogen phosphate as a base, and the reaction conducted in methanol at 85 °C. As a result, an optically active morphan compound 162, with the α-position of the ketone coupled to the vinyl bromide, was obtained in excellent yields and enantioselectivity on a gram scale (92%, 94% ee). The reaction mechanism was analyzed using computational chemistry, and an impressive transition state (TS4) was proposed, in which the carboxylic acid of the secondary amine catalyst forms a bridge with Pd. To construct the methyl group at C18 of the natural product, a diastereoselective reduction was performed using Crabtree's catalyst in a hydrogenation reaction. It was subsequently converted into compound 163 using the catalytic enone synthesis protocol developed by Stahl et al.47 Enone 163 underwent 1,4-addition with methylcopper reagent, followed by silyl enol formation with TMSCl, bromination and elimination at the α-position, and removal of the tosyl group with sodium naphthalenide with the enone moiety protected as an enolate. This sequence of reactions yielded cyclic amine 164 in high yield (87%, 4 steps). The construction of the C-ring was achieved through the condensation of malonic acid derivative and intramolecular Michael reaction. Thus, dehydration condensation of allylated malonate 165 with 164, followed by treatment with a base, leads to a diastereoselective intramolecular Michael reaction. This process results in compound 166, where the C-ring is constructed along with the formation of two contiguous quaternary chiral carbon centers. Next, they developed and applied the O-allylation/Claisen rearrangement/RCM sequence for constructing the D-ring. First, ketone 166 was treated with potassium hexamethyldisilazide (KHMDS) at low temperature, followed by the addition of 167, resulting in the O-allylated product 168. When 168 was heated to 200 °C in mesitylene, a diastereoselective Claisen rearrangement reaction proceeded, yielding 169 in good yield. Following the ring-closing olefin metathesis reaction for D-ring construction, decarboxylation and removal of the silyl group were carried out, resulting in the preparation of the pentacyclic compound 170. Finally, after oxidation of the secondary alcohol 170, subsequent isomerization of the double bond, and amide-selective reduction with Vaska's complex,48 the total synthesis of himalensine A (157) was achieved. This synthesis involves 19 steps, which is highly efficient given the structural complexity of 157. The success of this impressive total synthesis is largely attributed to the discovery of a novel concerted reaction between secondary amines and transition-metal catalysts.
 | ||
| Scheme 15 Synthesis of himalensine A by Dixon et al. in 2023. | ||
Most recently, Yang and coworkers reported the total synthesis of daphenylline (171), which features a benzene ring—an uncommon characteristic for a Daphniphyllum alkaloid (Scheme 16).49 The key reaction for introducing the asymmetric carbon center is the Ir/amine dual-catalyzed allylation methodology developed by Carreira's group, which controls the stereochemistry of the methyl group at C18 on the D-ring. Thus, subjecting phenyl vinyl carbinol 172 and propanal to conditions using Ir/ent-17 and secondary amine ent-18 as catalysts, with dichloroacetic acid as the promoter, yielded aldehyde 173 in 64% yield and >99% ee. The optically active 173 was converted into the Boc-protected amine via reductive amination with BocNH2, followed by transformation of the terminal double bond into a carboxylic acid using the radical hydrocarboxylation reaction developed by Wickens et al.50 A subsequent intramolecular Friedel–Crafts acylation was achieved by heating carboxylic acid 175 in the presence of polyphosphoric acid (PPA), resulting in the formation of a cyclized ketone with good conversion. The construction of the morphan skeleton was achieved by applying the copper-catalyzed α-amination reaction developed by MacMillan et al. to an intramolecular reaction.51 The ketone was then converted into allylic bridgehead amine 176via a Wittig reaction. The secondary amine of compound 176 was condensed with an alkynyl carboxylic acid 177 to afford precursor 178 for reductive 1,6-enyne cyclization. Krische's Rh-catalyzed reductive 1,6-enyne cyclization was used to construct the C-ring.52 The cyclization reaction of 178 proceeded stereoselectively, yielding tetracyclic compound 179 in high yield. The stereoselective reduction of the double bond and simultaneous removal of the silyl group are carried out. The resulting primary alcohol was then oxidized to a carboxylic acid, which was subsequently converted into an acid chloride. An intramolecular Friedel–Crafts reaction was performed to construct the D-ring, and finally, the ketone was converted into an alkene, completing the synthesis of 180. Again, compound 180 underwent a radical hydrocarboxylation reaction to introduce the carboxylic acid moiety and achieve a one-carbon elongation and yielding compound 181. Furthermore, a third intramolecular Friedel–Crafts reaction was employed to construct the E-ring. After decarbonylation, the total synthesis of daphenylline (171) was completed. The total synthesis was completed in just 14 steps, starting with Carreira's Ir/amine dual-catalyzed allylation. The impressive efficiency is due to the use of multiple catalytic reactions and the central benzene ring in the Friedel–Crafts reactions to form three key C–C bonds.
 | ||
| Scheme 16 Synthesis of daphenylline by Yang et al. in 2024. | ||
Pyrrolizidine and quinolizidine alkaloids
Multiply substituted chiral pyrrolizidine and quinolizidine ring systems are found in a wide variety of natural products, and several of these alkaloids have important biological activities such as sedative, anticonvulsant, anti-inflammatory, and antivirus actions.53 These alkaloids are mainly biosynthesized from L-lysine or L-ornithine. In addition, these alkaloids have a wide range of structural diversity and substituent variation. Therefore, numerous total syntheses of these alkaloids or the construction of their scaffolds have been reported.In 2020, Appayee et al. reported the total syntheses of all four stereoisomers of 1-hydroxymethylpyrrolizine alkaloids, namely, (+)- and (−)-isoretronecanol (182 and ent-182), (+)-laburnine (183), and (−)-trachelanthamidine (ent-183) using organocatalytic self-Mannich reactions (Scheme 17).54 When 4-oxobutanoate 152 and p-toluidine were treated with 10 mol% catalyst 2 in the presence of benzoic acid, the anti-selective self-Mannich reaction proceeded to provide aldehyde 184 (Scheme 17a). After completion of the Mannich reaction, aldehyde 184 was reduced with NaBH4, followed by lactonization under acidic conditions to provide lactam 185 in excellent yield with high enantio- and diastereoselectivity (80% yield, dr = 4:1, 98% ee). After removal of the PMP group, a selective lactam reduction using Meerwein reagent (Me3OBF4) and NaBH4, and a cyclization, primary alcohol was protected with the benzoyl group to provide bicyclic lactam 186. Finally, the reduction of the lactam followed by removal of the benzoyl group gave (+)-isoretronecanol (182). Next, the total synthesis of (−)-isoretronecanol (ent-182) was also accomplished. Thus, in the asymmetric Mannich reaction, the opposite enantiomer ent-2 was used as an organocatalyst to synthesize (−)-isoretronecanol (ent-182). In addition, total syntheses of (+)-laburnine (183) and (−)-trachelanthamidine (ent-183) using an organocatalytic syn-selective self-Mannich reaction were accomplished (Scheme 17b). Thus, when L-proline (1) was employed as a catalyst in the asymmetric Mannich reaction, the selectivity was reversed from that of the diphenylprolinol silyl ether catalyst 2, and syn-self-Mannich reaction proceeded, followed by reduction using NaBH4 and lactone formation to afford lactam 187 in excellent yield with high enantio- and diastereoselectivity (80% yield, dr = 7:1, 93% ee). Subsequent removal of the PMP group, lactam selective reduction, formation of the bicyclic lactam, and protection of primary alcohol proceeded. Reduction of the lactam and removal of the benzoyl group then afforded (+)-laburnine (184). The total synthesis of (–)-trachelanthamidine (ent-183) was also achieved by using D-proline in the syn-selective self-Mannich reaction. By using a different catalyst, namely, diphenylprolinol silyl ether catalyst 2 and proline in a self-Mannich reaction, the total synthesis of four 1-hydroxymethylpyrrolizine alkaloids was achieved in eight steps and 20–21% overall yield. This difference in stereoselectivity arises because in proline catalysts, the carboxylic acid moieties form hydrogen bonds, whereas in diphenylprolinol silyl ether catalysts, selectivity is influenced by the steric bulk of the substituents. Using different catalysts allows for the collective total synthesis of diastereomers.
 | ||
| Scheme 17 Synthesis of isoretronecanol, laburnine, and trachelanthamidine by Appayee et al. in 2020. | ||
In 2021, Rawal et al. reported excellent total synthesis of the steroidal alkaloid heilonine (189) (Scheme 18).55 One of the key steps was an organocatalytic Diels–Alder reaction using siloxydiene 190 and commercially available ethyl trans-4-oxo-2-butenoate (191) developed by the Hayashi group.56 Thus, when siloxydiene 190 was treated with aldehyde 191 and TFA in the presence of catalyst 192, regio- and stereoselective asymmetric Diels–Alder reaction proceeded to provide cycloadduct 193 in 67% yield and 90% ee. Subsequent Gilbert–Seferth reaction using Ohira–Besmann reagent, reduction of ethyl ester, removal of the TBS group, and protection of the primary alcohol afforded cyclohexanone 194 as a mixture of diastereomers at the α-methyl group. After conversion into its thermodynamic silyl enol ether using TBSI and HMDS, the Mukaiyama–Michael reaction with methyl vinyl ketone (MVK) and subsequent aldol/dehydration sequence afforded bicyclic ketone 195.57 In this three-step Robinson annulation protocol, the Mukaiyama–Michael reaction afforded an inseparable mixture of diastereomer (3:1); however, only the major (desired) isomer cyclized to provide 195. Subsequent protection of the ketone moiety with neopentyl glycol, removal of the silyl group, and sulfonylation of the primary alcohol provided the tosylated compound. The coupling reaction with lithium acetylide 196 proceeded smoothly, and subsequent removal of the THP group and Appel reaction transformed diyne 197 in high yield (46%, 6 steps). The coupling reaction between propargyl bromide 197 and alkyne-containing lactam 198 (synthesized by seven-step protocol from commercially available materials) was carried out using NaH to provide triyne 199 in 93% yield. To construct a benzene ring, the authors performed [2 + 2 + 2] cycloaddition using a transition-metal catalyst. Thus, when triyne 199 was treated with 10 mol% RhCl(PPh3)3 in EtOH at 80 °C, alkyne trimerization proceeded smoothly to afford the six-membered ring product 200 in 89% yield. Further diastereoselective introduction of a methyl group at the α-position was carried out using MeI and LiTMP as a base, followed by removal of the acetal moiety using p-TsOH and enolization using TMSCl, NaI and Ac2O to afford dienol acetate. Subsequent oxidation of the γ-position was performed with mCPBA, followed by isomerization to the γ-diketone using HBr as a catalyst and reduction of two ketones with LiAlH4 to accomplish the total synthesis of heilonine (189) in a total of 21 steps from ethyl trans-4-oxo-2-butenoate (191). This elegant approach to the coupling of the two fragments and subsequent construction of the benzene ring in the late stage enabled the first total synthesis of heilonine (189).
 | ||
| Scheme 18 Synthesis of heilonine by Rawal et al. in 2021. | ||
The total synthesis of senepodine F (201) using an organocatalytic asymmetric Diels–Alder reaction was reported by Ishikawa and coworkers in 2023 (Scheme 19).58 Their key organocatalytic reaction was originally developed as a Diels–Alder reaction using a 5-nitro-2,3-dihydro-4-pyridone derivative 202 as a dienophile. Thus, pyridone derivative 202 was treated with α,β,γ,δ-unsaturated aldehyde 203 and benzoic acid in the presence of 5 mol% of catalyst 204, to afford the optically active endo-cycloadduct 205 in excellent yield with high regio-, enantio-, and diastereoselectivity (88% yield, dr 4.4:1, 96% ee). In this reaction, the trienamine intermediate generated from aldehyde 203 and catalyst 204 induced high stereoselectivity (TS5). Further, a denitration reaction under reductive conditions (nBu3SnH, cat. AIBN) followed by isomerization to the thermodynamically stable trans-fused octahydroquinoline under basic conditions and a diastereoselective hydrogen atom transfer (HAT) reaction afforded 7R-decahydroquinoline 206 in high yield and diastereoselectivity (70% in 3 steps, dr at C7 = 5:1). Following reduction of the ketone using NaBH4, thiocarbonylation of a secondary alcohol, deoxygenation using nBu3SnH, and removal of the acetal moiety under acidic hydrolysis conditions provided aldehyde 207. After a coupling reaction with piperidine fragment 208 (synthesized in eight steps from commercially available materials) was achieved using nBuLi as a base, Dess–Martin oxidation of the secondary alcohol was performed to provide the coupling product 209. Subsequent diastereoselective reduction of the ketone moiety under Noyori reduction conditions59 using catalyst 210, reduction of the alkyne and removal of the Cbz group under catalytic hydrogenation conditions, protection of the amine afforded a precursor of quinolizidine cyclization. Mesylation of the secondary hydroxyl group followed by deprotection of the Fmoc group and SN2 cyclization using DBU were carried out to successfully provide 211 in 40% over five steps as a single isomer. Finally, the deprotection of the Boc group and acetylation sequence afforded senepodine F (201). The asymmetric Diels–Alder reaction developed by them is a powerful method to provide the optically active decahydroquinoline scaffold and will be used in the synthesis of other alkaloids with this scaffold in near future.
 | ||
| Scheme 19 Synthesis of senepodine F by Ishikawa et al. in 2023. | ||
Other alkaloids
In 2017, Chen and coworkers achieved the total synthesis of communesin F (212), a member of the communesin family of polycyclic bis-aminal alkaloids (Scheme 20).60 In the synthesis of communesins, the construction of a contiguous quaternary chiral center within the ring is crucial. Chen et al. elegantly constructed this challenging contiguous stereocenter using a secondary amine organocatalyst. The electrophile of the key organocatalytic reaction was an oxindole derivative 213, which has a methoxy group—a leaving group—at the β-position of the aldehyde. The nucleophile was oxindole 214, with an azide side chain. When the substrates and catalyst 11 were mixed in ethanol and stirred at low temperature, 213 and 11 condensed to form activated α,β-unsaturated iminium ion intermediates. Subsequently, another oxindole 214 was added, leading to the construction of a contiguous quaternary chiral center. The aldehyde of the generated intermediate 215 was reduced and isolated as alcohol 216. This challenging coupling reaction proceeded with high enantioselectivity in moderate yield (86% ee, 55%). The azide group of 216 was reduced, and a p-nitrobenzyl group was introduced into the resulting amine. Subsequent treatment with the Ti(OiPr)4 as Lewis acid induced skeletal rearrangement to form a pentacyclic lactam 218via intermediate 217. In this rearrangement, part of the originally present MOM group was converted into an isopropoxymethyl group. The primary hydroxyl group in compound 218 was converted into a primary amine in three steps. This was followed by the introduction of diisopropyl oxalamide, a directing group for C–H functionalization as reported by Zhao et al.61 The second key reaction in this total synthesis is the introduction of the acrylic acid unit via C–H activation at C11a. Thus, methyl acrylate was introduced at the C11a position of 219, with direct activation using palladium acetate and silver carbonate. The etheric protecting group was then removed using hydrochloric acid, and the resulting amino group was protected with a Boc group. The imidate moiety linked to aniline was stereoselectively reduced to form 220. The acrylic acid moiety of 220 was cleaved by dihydroxylation/oxidation process to yield an aldehyde. The diisopropyl oxalamide group, used as a directing group, was then removed under hydrolysis conditions. Subsequently, the alkynyl group was introduced into the aldehyde to produce alcohol 221. The Au-catalyzed Meyer–Schuster rearrangement62 using alkynes was performed to construct additional ring structures. The resulting α,β-unsaturated ketone was then converted into 2-methylpropene moiety through methylation and dehydration. Subsequently, selective removal of the Boc group, followed by intramolecular dehydration and cyclization, led from the lactam ring to the cyclic imidate 222. The total synthesis of communesin F (212) was achieved in three steps, including the reduction and acetylation of the imidate. Alkaloids from the communesin family consistently feature a contiguous quaternary chiral center. The organocatalytic protocols developed by Chen et al. will be highly useful for synthesizing these alkaloids. | ||
| Scheme 20 Synthesis of communesin F by Chen et al. in 2017. | ||
In 2019, Hu's group achieved the total syntheses of two marine benzoxazole alkaloids, pseudopteroxazole (223) and ileabethoxazole (224), which were isolated from the Caribbean sea whip Pseudopterogorgia elisabethae (Scheme 21).63 The key reaction for introducing the chiral centers is Carreira's asymmetric dual catalytic allylation, which utilizes allyl alcohol 225. The combination of amine catalyst 18 and [Ir/ent-17] was employed to introduce the two desired stereocenters in 223. In this reaction, the addition of various acids was explored to improve diastereoselectivity and yields. Ultimately, excellent selectivity and chemical yields were achieved with the addition of difluoroacetic acid (dr = 10:1, 83%). The enantioselectivity was excellent (94% ee). Subsequently, the aldehyde 226 was converted into 228, featuring a diene structure, through Wittig–Vedejs E-selective olefination with 227.64 The construction of the C-ring proceeded diastereoselectively by treating 228 with methanesulfonic acid. Hu et al. proposed an interesting reaction mechanism, involving a 1,2-shift pathway. The terminal double bond was elongated via a cross-metathesis reaction with methyl acrylate, followed by 1,4-reduction and hydrolysis to yield carboxylic acid 229. B-Ring construction was achieved through intramolecular Friedel–Crafts acylation, and the resulting ketone was converted into 230via methylation followed by reduction. Finally, the D-ring was constructed using the oxazole motif synthesis reported by Kerr et al.,65 completing the total synthesis of pseudopteroxazole (223). Using the same strategy, they also achieved the total synthesis of ileabethoxazole (224), which features a five-membered C-ring. The total synthesis is a concise and practical process that effectively utilizes the aldehydes and terminal double bonds generated through asymmetric dual catalytic allylation, a concerted reaction involving both organocatalysts and metal catalysts.
 | ||
| Scheme 21 Synthesis of pseudopteroxazole by Hu et al. in 2019. | ||
Morphine (231) is a crucial drug used to alleviate all types of pain. Additionally, codeine (232), a derivative of morphine, is commonly used as a cough suppressant. These alkaloids have a pentacyclic structure with five contiguous chiral centers, one of which is a quaternary carbon center. In 2019, Tu, Zhang, and coworkers synthesized this important drug using intramolecular asymmetric organocatalytic Michael reaction (Scheme 22).66 The secondary amine catalyst used in the organocatalytic reactions was originally developed by Tu et al.66 It efficiently induces chirality through its rigid spirocycles and the siloxy groups extending from them. The key organocatalytic reaction precursor, aldehyde 233, was prepared in four steps starting from commercially available materials. When catalyst 234 and bulky 2,4,6-triisopropylbenzoic acid are added to compound 233 in CH2Cl2 at low temperature, the enantioselective intramolecular Michael reaction proceeded, leading to the formation of an all-carbon quaternary chiral center. The crude mixture of 235 was directly treated with p-toluenesulfonic acid (PTSA) under heating conditions, resulting in an aldol condensation reaction that provided the tricyclic product 236. The enantioselectivity of 236 was 94% ee, which was further improved by recrystallization. The α-position of the ketone moiety of compound 236 was subsequently allylated to give 237 as a diastereomeric mixture. Next, selective ozonolysis of the electron-rich double bond of 237 was performed, followed by intramolecular Friedel–Crafts-type cyclization using a catalytic amount of PPA, yielding phenanthrofuran 238 as a single diastereomer. Then, site- and diastereoselective epoxidation of the α,β-unsaturated ketone, followed by the Wharton transposition sequence was carried out to afford allyl alcohol. Subsequent debenzylation yielded diol 239. Selective sulfonamidation of the primary hydroxyl group under Mitsunobu conditions, followed by a sequential one-pot oxidation–reduction process, generated Guillou's intermediate 240.67 The total synthesis of codeine (232) was achieved through one-electron reduction of compound 240, followed by a hydroamination reaction. Further demethylation then led to the total synthesis of morphine (231). The intramolecular Michael reaction catalyzed by spiro-secondary amines, developed by Tu, Zhang, and coworkers is highly useful for synthesizing natural products with a cis-hydroxybenzofuran skeleton. In fact, they also reported the total syntheses of galanthamine and lycoramine, which have similar structural motifs, shortly after their synthesis of morphine.68
 | ||
| Scheme 22 Synthesis of morphine and codeine by Tu et al. in 2019. | ||
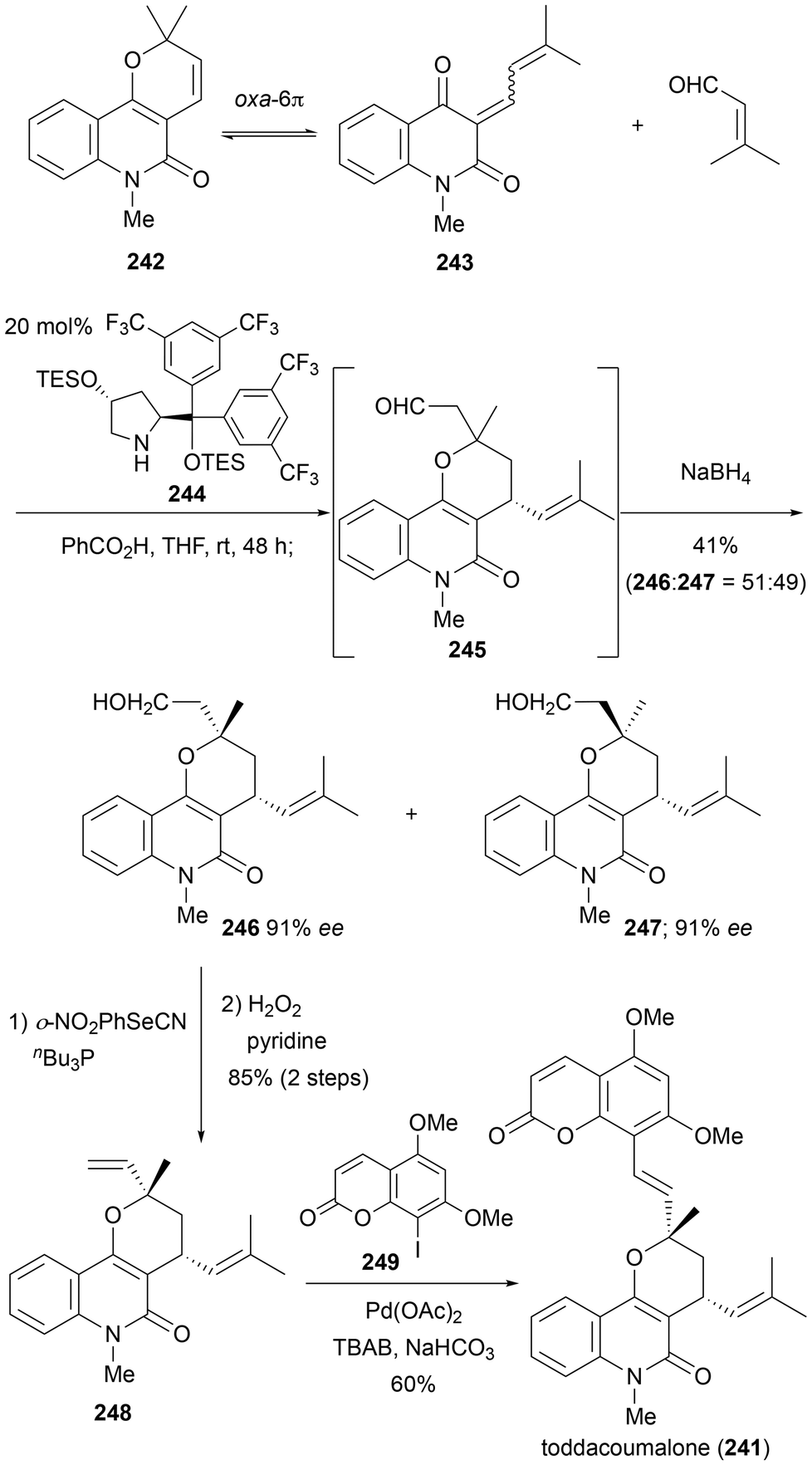
Toddacoumalone (241) is a known phosphodiesterase-4 (PDE4) inhibitor and features a dimeric structure composed of coumarin and quinolone.69 Since natural product 241 was isolated as a racemic mixture, structure–activity relationship studies using the optically active forms and its diastereomers are desired. In 2020, Xiong et al. developed an intriguing organocatalytic asymmetric inverse electron-demand Diels–Alder reaction, which was employed to achieve the first asymmetric total synthesis of toddacoumalone (241) (Scheme 23).70 They found that the non-isolable diene 243 exists as a stable and isolable ring-closing pyranoquinolinone 242, which could be in equilibrium via an oxa-6π electrocyclic pathway. They then investigated the asymmetric Diels–Alder reaction using this diene and 3-methyl-2-butenal as the dienophile. After careful catalyst screening, catalyst 244, derived from trans-4-hydroxyproline, was identified. The Diels–Alder reaction with the highly reactive quinolinone-derived diene produced the cyclized product 245. Subsequent reduction of the aldehyde moiety yielded the alcohol 246 and 247 as diastereomers. The chemical yield was 41%, with a diastereomeric ratio of 1:1 at the quaternary carbon center; each diastereomer having an enantiomeric excess of 91% ee. The resulting primary alcohol was converted into a terminal double bond using the Grieco–Nishizawa protocol, yielding compound 248. This was followed by the condensation of the known coumarin derivative 249via the Heck reaction, leading to the total synthesis of 241. Xiong et al. subsequently prepared enantiomers, diastereomers, and their derivatives for structure–activity relationship studies. This is a notable achievement as it involves an organocatalytic reaction utilizing active intermediates that are not visible as isolated compounds (such intermediates might be inferred from biosynthesis).
 | ||
| Scheme 23 Synthesis of toddacoumalone by Xiong et al. in 2020. | ||
The total synthesis of δ-lycorane (250) was reported by Koley and coworkers in 2023 (Scheme 24).71 One of the key reactions was a diastereoselective organocatalytic Mannich reaction using optically active δ-methoxylactam 252 (prepared by a four-step transformation from commercially available materials) and phenylacetaldehyde 251 to construct 5-substituted-2-pyrrolidone derivative 254. Thus, treatment of the lactam 252 with the aldehyde 251 in the presence of 10 mol% organocatalyst 253 and 5 mol% Ni(OTf)2, proceeded a diastereoselective Mannich reaction, followed by reduction using NaBH4 to provide 5-substituted-2-pyrrolidone derivative 255 with good yield and diastereoselectivity (67% yield, dr = 40:4:1:1). The diastereoselectivity in this reaction was proposed by Koley to stem from the most sterically vacant Re face of enamines generated from organocatalyst and aldehyde reacting with the Re face of N-acyliminium (TS6). The lactam 255 was treated with TFA/triisopropylsilane followed by hydrogenation using Pd(OH)2, and PMB protection of alcohol and amide. Subsequent α-selenylation and oxidation were performed to afford enamide 256. The diastereoselective 1,4-addition of enamide 256 was carried out using an allyl magnesium reagent, followed by treatment of the crude reaction mixture with In(OTf)3 and triisopropylsilane to decompose the inseparable byproduct and afford the adduct. After the PMB group on alcohol was removed, Dess–Martin oxidation and Wittig reaction provided dialkene 257. Subsequent RCM with Hoveyda–Grubbs II catalyst, reduction of amide, reduction of the alkene, removal of the PMB group under catalytic hydrogenation conditions, and formation of the carbamate afforded a precursor of cyclization 258. Finally, the total synthesis of δ-lycorane (250) was achieved by a two-step transformation. Thus, the Bischler–Napieralski reaction of 258 was performed in the presence of POCl3 followed by amide reduction to provide δ-lycorane (250). The two vicinal stereocenters of 250 were efficiently constructed by Mannich reaction using organocatalysts 253 and Ni(OTf)2, and this method could also be used for the total synthesis of other Amaryllidaceae alkaloids.
 | ||
| Scheme 24 Synthesis of δ-lycoran by Koley et al. in 2023. | ||
Conclusion
In the alkaloid syntheses listed in this article, many examples of domino and one-pot reactions were used to significantly increase the efficiency of the total synthesis. Since many alkaloids contain polycyclic structures, these sequential reactions are particularly effective for constructing multifunctionalized ring systems. This effectiveness may be due to the fact that the secondary amine catalyst does not inhibit subsequent domino or one-pot reactions. Furthermore, of the 24 cases discussed in this article, 7 involve concerted reactions between organocatalysts and metal reagents (mostly catalysts). Notably, the use of dual iridium/amine catalytic allylation, developed by Carreira et al., is used in four of these cases. The terminal double bonds produced in this reaction can be utilized in hydroboration, hydrocarbonylation, olefin metathesis, and other transformations, thereby introducing diversity into synthetic strategies. In all syntheses, high enantioselectivity was achieved through organocatalysis, highlighting the reliability of organocatalysis, which has been well-established science around 2000. The full potential of chiral secondary-amine-catalyzed asymmetric reactions for total synthesis has not been realized yet. Currently, advanced research is underway to apply the active species generated by secondary amine organocatalysts to photoreactions and radical reactions, with the expectation that these methods will soon be applied to alkaloid synthesis. Consequently, elegant and attractive syntheses of alkaloids using secondary amine catalysts are expected to continue appearing.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the JSPS KAKENHI Grant No. JP24K02141 (Scientific Research (B)), Grant-in-Aid for Transformative Research Areas (A) “Latent Chemical Space” [JP24H01749].References
- (a) B. List, R. A. Lerner and C. F. Barbas III, J. Am. Chem. Soc., 2000, 122, 2395–2396 CrossRef CAS; (b) W. Notz and B. List, J. Am. Chem. Soc., 2000, 122, 7386–7387 CrossRef CAS.
- (a) M. Marigo, T. C. Wabnitz, D. Fielenbach and K. A. Jørgensen, Angew. Chem., Int. Ed., 2005, 44, 794–797 CrossRef CAS PubMed; (b) Y. Hayashi, H. Gotoh, T. Hayashi and M. Shoji, Angew. Chem., Int. Ed., 2005, 44, 4212–4215 CrossRef CAS PubMed; (c) J. Franzén, M. Marigo, D. Fielenbach, T. C. Wabnitz, A. Kjærsgaard and K. A. Jørgensen, J. Am. Chem. Soc., 2005, 127, 18296–18304 CrossRef PubMed.
- K. A. Ahrendt, C. J. Borths and D. W. C. MacMillan, J. Am. Chem. Soc., 2000, 122, 4243–4244 CrossRef CAS.
- (a) P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2004, 43, 5138–5175 CrossRef CAS PubMed; (b) D. W. C. MacMillan, Nature, 2008, 455, 304–308 CrossRef CAS PubMed; (c) B. Han, X.-H. He, Y.-Q. Liu, G. He, C. Peng and J.-L. Li, Chem. Soc. Rev., 2021, 50, 1522–1586 RSC.
- Y. Hayashi, Chem. Sci., 2016, 7, 866–880 RSC.
- D. L. Hughes, Org. Process Res. Dev., 2018, 22, 574–584 CrossRef CAS.
- H. Ishikawa and S. Shiomi, Org. Biomol. Chem., 2016, 14, 409–424 RSC.
- F. Omar, A. M. Tareq, A. M. Alqahtani, K. Dhama, M. A. Sayeed, T. B. Emran and J. Simal-Gandara, Molecules, 2021, 26, 2297 CrossRef CAS PubMed.
- L. Li, P. Aibibula, Q. Jia and Y. Jia, Org. Lett., 2017, 19, 2642–2645 CrossRef CAS PubMed.
- X. Liang, T.-Y. Zhang, C.-Y. Meng, X.-D. Li, K. Wei and Y.-R. Yang, Org. Lett., 2018, 20, 4575–4578 CrossRef CAS PubMed.
- (a) C. L. Martin, L. E. Overman and J. M. Rohde, J. Am. Chem. Soc., 2008, 130, 7568–7569 CrossRef CAS PubMed; (b) C. L. Martin, L. E. Overman and J. M. Rohde, J. Am. Chem. Soc., 2010, 132, 4894–4906 CrossRef CAS PubMed.
- L. Cai, K. Zhang and O. Kwon, J. Am. Chem. Soc., 2016, 138, 3298–3301 CrossRef CAS PubMed.
- F. Xue, H. Lu, L. He, W. Li, D. Zhang, X.-Y. Liu and Y. Qin, J. Org. Chem., 2018, 83, 754–764 CrossRef CAS PubMed.
- S. Krautwald, D. Sarlah, M. A. Schafroth and E. M. Carreira, Science, 2013, 340, 1065–1068 CrossRef CAS PubMed.
- P. Guo, J. Jian, Y. Chen and J. Xu, Chem. Commun., 2018, 54, 1125–1128 RSC.
- W. Zhang, J. Bah, A. Wohlfarth and J. Franzén, Chem. – Eur. J., 2011, 17, 13814–13824 CrossRef CAS PubMed.
- K. Rakumitsu, J. Sakamoto and H. Ishikawa, Chem. – Eur. J., 2019, 25, 8996–9000 CrossRef CAS PubMed.
- (a) J. Sakamoto, Y. Umeda, K. Rakumitsu, M. Sumimoto and H. Ishikawa, Angew. Chem., Int. Ed., 2020, 59, 13414–13422 CrossRef CAS PubMed; (b) J. Sakamoto and H. Ishikawa, Chem. – Eur. J., 2022, 28, e202104052 CrossRef CAS PubMed; (c) N. Nakashima, J. Sakamoto, K. Rakumitsu, M. Kitajima, L. D. Juliawaty and H. Ishikawa, Chem. Pharm. Bull., 2022, 70, 187–191 CrossRef CAS PubMed; (d) J. Sakamoto, M. Kitajima and H. Ishikawa, Chem. Pharm. Bull., 2022, 70, 662–668 CrossRef CAS PubMed; (e) J. Sakamoto, M. Kitajima and H. Ishikawa, Chem. – Eur. J., 2023, 29, e202300179 CrossRef CAS PubMed; (f) A. Yoshidome, J. Sakamoto, M. Kohara, S. Shiomi, M. Hokaguchi, Y. Hitora, M. Kitajima, S. Tsukamoto and H. Ishikawa, Org. Lett., 2023, 25, 347–352 CrossRef CAS PubMed; (g) J. Sakamoto and H. Ishikawa, Synlett, 2023, 34, 2293–2303 CrossRef CAS; (h) S. Imaoka, Y. Nakashima, M. Kitajima and H. Ishikawa, Chem. Pharm. Bull., 2024, 72, 68–74 CrossRef CAS PubMed; (i) J. Sakamoto, D. Hiruma, M. Kitajima and H. Ishikawa, Synlett, 2024, 35, 576–581 CrossRef CAS.
- (a) T. Fukuyama, S.-C. Lin and L. Li, J. Am. Chem. Soc., 1990, 112, 7050–7051 CrossRef CAS; (b) H. Tokuyama, S. Yokoshima, T. Yamashita, S.-C. Lin, L. Li and T. Fukuyama, Synthesis, 2002, 1121–1123 CrossRef CAS.
- Y.-H. Yuan, X. Han, F.-P. Zhu, J.-M. Tian, F.-M. Zhang, X.-M. Zhang, Y.-Q. Tu, S.-H. Wang and X. Guo, Nat. Commun., 2019, 10, 3394 CrossRef PubMed.
- D.-H. Kim, J.-H. Kim, T.-H. Jeon and C.-G. Cho, Org. Lett., 2020, 22, 3464–3468 CrossRef CAS PubMed.
- J. Wang, A. Ma and D. Ma, Org. Lett., 2008, 10, 5425–5428 CrossRef CAS PubMed.
- (a) S. Shiomi, E. Sugahara and H. Ishikawa, Chem. – Eur. J., 2015, 21, 14758–14763 CrossRef CAS PubMed; (b) Y. Ataka, M. Kitajima and H. Ishikawa, Org. Lett., 2023, 25, 7601–7605 CrossRef CAS PubMed.
- T. Tsunoda, M. Suzuki and R. Noyori, Tetrahedron Lett., 1980, 21, 1357–1358 CrossRef CAS.
- P. J. Boratyński, M. Zielińska-Błajet and J. Skarżewski, The Alkaloids: Chemistry and Biology: Chapter Two-Chinchona Alkaloids-Derivatives and Applications, Elsevier. B. V., 2019, vol. 82, pp. 29–145 Search PubMed.
- E. S. Choong, Cinchona Alkaloids in Synthesis & Catalysis: Ligands, Immobilization and Organocatalysis, Wiley-VCH, 2009 Search PubMed.
- Y. Jiang, L. Deiana, K. Zhang, S. Lin and A. Córdova, Eur. J. Org. Chem., 2019, 6016–6023 CrossRef CAS.
- (a) J. Igarashi, M. Katsukawa, Y.-G. Wang, H. P. Acharya and Y. Kobayashi, Tetrahedron Lett., 2004, 45, 3783–3786 CrossRef CAS; (b) J. Igarashi and Y. Kobayashi, Tetrahedron Lett., 2005, 46, 6381–6384 CrossRef CAS.
- I. T. Raheem, S. N. Goodman and E. N. Jacobsen, J. Am. Chem. Soc., 2004, 126, 706–707 CrossRef CAS PubMed.
- S. Shiomi, R. Misaka, M. Kaneko and H. Ishikawa, Chem. Sci., 2019, 10, 9433–9437 RSC.
- T. Terunuma and Y. Hayashi, Nat. Commun., 2022, 13, 7503 CrossRef CAS PubMed.
- J. Matsuo, T. Kozai and H. Ishibashi, Org. Lett., 2006, 8, 6095–6098 CrossRef CAS PubMed.
- A. Krasovskiy, F. Kopp and P. Knochel, Angew. Chem., Int. Ed., 2006, 45, 497–500 CrossRef CAS PubMed.
- Reviews of Stemona alkaloids: (a) R. A. Pilli and M. C. F. de Oliveira, Nat. Prod. Rep., 2000, 17, 117–127 RSC; (b) R. A. Pilli, G. B. Rosso and M. C. F. de Oliveira, Nat. Prod. Rep., 2010, 27, 1908–1937 RSC; (c) H. Greger, Phytochem. Rev., 2019, 18, 463–493 CrossRef CAS.
- (a) M. B. Uphade and K. R. Prasad, Tetrahedron, 2020, 76, 131623 CrossRef CAS; (b) K. R. Prasad and M. B. Uphade, J. Org. Chem., 2019, 84, 9648–9660 CrossRef CAS PubMed.
- (a) S. P. Brown, M. P. Brochu, C. J. Sinz and D. W. C. MacMillan, J. Am. Chem. Soc., 2003, 125, 10808–10809 CrossRef CAS PubMed; (b) I. K. Mangion and D. W. C. MacMillan, J. Am. Chem. Soc., 2005, 127, 3696–3697 CrossRef CAS PubMed.
- Y. Kohno and K. Narasaka, Bull. Chem. Soc. Jpn., 1996, 69, 2063–2070 CrossRef CAS.
- Z. Guo, R. Bao, Y. Li, Y. Li, J. Zhang and Y. Tang, Angew. Chem., Int. Ed., 2021, 60, 14545–14553 CrossRef CAS PubMed.
- (a) T. D. Beeson, A. Mastracchio, J.-B. Hong, K. Ashton and D. W. C. MacMillan, Science, 2007, 316, 582–585 CrossRef CAS PubMed; (b) J. C. Conrad, J. Kong, B. N. Laforteza and D. W. C. MacMillan, J. Am. Chem. Soc., 2009, 131, 11640–11641 CrossRef CAS PubMed.
- Y. Deng, X. Liang, K. Wei and Y.-R. Yang, J. Am. Chem. Soc., 2021, 143, 20622–20627 CrossRef CAS PubMed.
- (a) T. P. Montgomery, A. Hassan, B. Y. Park and M. J. Krische, J. Am. Chem. Soc., 2012, 134, 11100–11103 CrossRef CAS PubMed; (b) S. W. Kim, W. Zhang and M. J. Krische, Acc. Chem. Res., 2017, 50, 2371–2380 CrossRef CAS PubMed.
- T. Ohta, T. Miyake, N. Seido, H. Kumobayashi and H. Takaya, J. Org. Chem., 1995, 60, 357–363 CrossRef CAS.
- Y. Lin, R. Zhang, D. Wang and T. Cernak, Science, 2023, 379, 453–457 CrossRef CAS PubMed.
- (a) Y. Hayashi, W. Tsuboi, I. Ashimine, T. Urushima, M. Shoji and K. Sakai, Angew. Chem., Int. Ed., 2003, 42, 3677–3680 CrossRef CAS PubMed; (b) W. Notz, F. Tanaka, S. Watanabe, N. S. Chowdari, J. M. Turner, R. Thayumanavan and C. F. Barbas III, J. Org. Chem., 2003, 68, 9624–9634 CrossRef CAS PubMed.
- J. Zhong, H. Wang, Q. Zhang and S. Gao, The Alkaloids: Chapter Two-the Chemistry of Daphniphyllum alkaloids, Elsevier. B. V., 2019, vol. 85, pp. 113–176 Search PubMed.
- R. Kučera, S. R. Ellis, K. Yamazaki, J. H. Cooke, N. Chekshin, K. E. Christensen, T. A. Hamlin and D. J. Dixon, J. Am. Chem. Soc., 2023, 145, 5422–5430 CrossRef PubMed.
- T. Daio and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 14566–14569 CrossRef PubMed.
- Y. Motoyama, M. Aoki, N. Takaoka, R. Aoto and H. Nagashima, Chem. Commun., 2009, 1574–1576 RSC.
- B.-L. Wu, J.-N. Yao, X.-X. Long, Z.-Q. Tan, X. Liang, L. Feng, K. Wei and Y.-R. Yang, J. Am. Chem. Soc., 2024, 146, 1262–1268 CrossRef CAS PubMed.
- S. N. Alektiar, J. Han, Y. Dang, C. Z. Rubel and Z. K. Wickens, J. Am. Chem. Soc., 2023, 145, 10991–10997 CrossRef CAS PubMed.
- R. W. Evans, J. R. Zbieg, S. Zhu, W. Li and D. W. C. MacMillan, J. Am. Chem. Soc., 2013, 135, 16074–16077 CrossRef CAS PubMed.
- H.-Y. Jang, F. W. Hughes, H. Gong, J. Zhang, J. S. Brodbelt and M. J. Krische, J. Am. Chem. Soc., 2005, 127, 6174–6175 CrossRef CAS PubMed.
- J. Zhang, Y.-Q. Liu and J. Fang, The Alkaloids: Chemistry and Biology: Chapter one-The biological activities of quinolizidine alkaloids, Elsevier. B. V., 2023, vol. 89, pp. 1–37 Search PubMed.
- A. M. Sarkale and C. Appayee, Org. Lett., 2020, 22, 4355–4359 CrossRef CAS PubMed.
- K. J. Cassaidy and V. H. Rawal, J. Am. Chem. Soc., 2021, 143, 16394–16400 CrossRef CAS PubMed.
- H. Gotoh and Y. Hayashi, Org. Lett., 2007, 9, 2859–2862 CrossRef CAS PubMed.
- P. Duhamel, G. Dujardin, L. Hennequin and J.-M. Poirier, J. Chem. Soc., Perkin Trans. 1, 1992, 387–396 RSC.
- (a) T. Inoshita, K. Goshi, Y. Morinaga, Y. Umeda and H. Ishikawa, Org. Lett., 2019, 21, 2903–2907 CrossRef CAS PubMed; (b) Y. Nakashima, T. Inoshita, M. Kitajima and H. Ishikawa, Org. Lett., 2023, 25, 1151–1155 CrossRef CAS PubMed.
- (a) K. Matsumura, S. Hashiguchi, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1997, 119, 8738–8739 CrossRef CAS; (b) M. Barbazanges, C. Meyer and J. Cossy, Org. Lett., 2008, 10, 4489–4492 CrossRef CAS PubMed.
- J. Park, A. Jean and D. Y.-K. Chen, Angew. Chem., Int. Ed., 2017, 56, 14237–14240 CrossRef CAS PubMed.
- Q. Wang, J. Han, C. Wang, J. Zhang, Z. Huang, D. Shi and Y. Zhao, Chem. Sci., 2014, 5, 4962–4967 RSC.
- R. S. Ramón, N. Marion and S. P. Nolan, Tetrahedron, 2009, 65, 1767–1773 CrossRef.
- X. Zhang, X. Fang, M. Xu, Y. Lei, Z. Wu and X. Hu, Angew. Chem., Int. Ed., 2019, 58, 7845–7849 CrossRef CAS PubMed.
- (a) E. Vedejs and H. Fang, J. Org. Chem., 1984, 49, 210–212 CrossRef CAS; (b) Y. Ribeill and H.-J. Cristau, Synthesis, 1988, 911–912 Search PubMed.
- M. W. B. McCulloch, F. Berrue, B. Haltli and R. G. Kerr, J. Nat. Prod., 2011, 74, 2250–2256 CrossRef CAS PubMed.
- Q. Zhang, F.-M. Zhang, C.-S. Zhang, S.-Z. Liu, J.-M. Tian, S.-H. Wang, X.-M. Zhang and Y.-Q. Tu, Nat. Commun., 2019, 10, 2507 CrossRef PubMed.
- M. Varin, E. Barré, B. Iorga and C. Guillou, Chem. – Eur. J., 2008, 14, 6606–6608 CrossRef CAS PubMed.
- Q. Zhang, F.-M. Zhang, C.-S. Zhang, S.-Z. Liu, J.-M. Tian, S.-H. Wang, X.-M. Zhang and Y.-Q. Tu, J. Org. Chem., 2019, 84, 12664–12671 CrossRef CAS PubMed.
- T.-T. Lin, Y.-Y. Huang, G.-H. Tang, Z.-B. Cheng, X. Liu, H.-B. Luo and S. Yin, J. Nat. Prod., 2014, 77, 955–962 CrossRef CAS PubMed.
- K.-Q. Hou, X.-P. Chen, Y. Huang, A. S. C. Chan, H.-B. Luo and X.-F. Xiong, Org. Lett., 2020, 22, 584–588 CrossRef CAS PubMed.
- J. Shukla, M. K. Gangwar and D. Koley, New J. Chem., 2023, 47, 18900–18904 RSC.
| This journal is © The Royal Society of Chemistry 2024 |