DOI:
10.1039/D4OB01326C
(Review Article)
Org. Biomol. Chem., 2024,
22, 9283-9318
Synthesis of metal-binding amino acids
Received
9th August 2024
, Accepted 28th September 2024
First published on 30th September 2024
Abstract
The ability for amino acid residues to bind metals underpins the functions of metalloproteins to conduct a plethora of critical processes in living organisms as well as unnatural applications in the fields of catalysis, sensing and medicinal chemistry. The capability to access metal-binding peptides heavily relies on the ability to generate appropriate building blocks. This review outlines recently developed strategies for the synthesis of metal binding non-proteinogenic amino acids. The chemistries to access, as well as to incorporate these amino acids into peptides is presented herein.
 Katherine E. V. Deck (Right) and William D. G. Brittain (Left) | Katherine obtained a MSci degree in Natural Sciences from Durham University in 2023, specialising in chemistry and biology with a final year project in the O'Donoghue group focusing on stable radical bioconjugation. As an undergraduate she carried out two summer research projects under the supervision of Dr William Brittain, for which she was awarded the prize of Student Employee of the Year 2023. She has since joined the Brittain Group as a PhD student as part of MoSMed CDT in collaboration with Dr Patricia Muller (Department of Biosciences) where she is working on designing novel anticancer peptides. Will completed his PhD at the University of Birmingham working with Prof. John Fossey and Prof. Ben Buckley in 2017. He then moved to Durham University to undertake postdoctoral studies with Prof. Steven Cobb. In 2021 he began a Leverhulme Early Career Fellowship studying metal binding peptides before being appointed as an Assistant Professor of Organic Chemistry in 2024. Later in 2024 he was promoted to Associate Professor at Durham University. His group specialises in organofluorine chemistry and peptide synthesis. |
1. Introduction
There are many examples of naturally occurring peptides and proteins which are capable of binding metal ions. In fact, metalloproteins carry out many important roles within living organisms from oxygen transport to tumor suppression, which depend on the proteins haemoglobin and p53 respectively Nature utilizes its collection of proteinogenic amino acids to facilitate metal binding with residues such as histidine, methionine and cysteine commonly employed. These naturally occurring residues have been exploited in de-novo protein design and synthesis to allow access to a range of metal binding peptides.1,2a,b This has been a highly fruitful area with systems such as peptide coiled-coils showing high affinities for metal binding in combination with the ability to accommodate metals that are not commonly found in biological environments (e.g. lanthanides/actinides).3 De-novo peptide design allows for applications that natural proteins are not suitable for such as catalysis, metallodrugs or contrast agents.4 Whilst these proteinogenic residues can bind metals they are limited in their scope and flexibility. In the area of synthetic chemistry many motifs have been developed which are capable of binding metal ions with high affinity. Many of these motifs have then been applied successfully in areas such as transition metal catalysis, chemical sensing and medicinal chemistry. In particular these advances have allowed for insights to be gained into structural biology and biosynthetic pathway elucidation revealing the critical roles that metalloproteins play with organisms. The motifs and ligands do not have to be confined to the naturally occurring functional groups found within genetically encoded amino acids which means they can have increased function and flexibility that proteinogenic amino acids are incapable of. Therefore, a growing area of interest is the incorporation of novel metal-binding residues and motifs into peptides and peptidomimetics.
Non-canonical amino acid building blocks comprise a vital part of the repertoire of methodology for engineering synthetic metallopeptides and peptidomimetics. The demand for readily accessible building blocks with simple installation has grown over the past decade as the interest in metal-binding peptide materials for wide-ranging applications continues to increase. This review will discuss the development of methodology for the synthesis of metal-binding amino acids, and examples of how these building blocks have been utilized both in peptide synthesis and in wider applications. Amino acid examples will be categorized either by the identity of the metal binding atoms or metal-binding functional group.
2. Phosphorus-containing amino acids
2.1. β-Phosphino amino acids
Owing to their electronic and steric tunability, phosphine ligands are highly versatile within transition metal catalysis and applicable to a wide range of organic transformations. These range from C–C bond forming reactions, e.g. Suzuki and Heck couplings, to Rh(I)-catalysed olefin hydrogenation.5a,b Functionalisation of amino acids with phosphine moieties was pioneered by Gilbertson and co-workers in 1994, who first reported a synthetic route to diphenylphosphinoserine (Pps) sulfide 5 using Evan's chiral oxazolidinone chemistry (Scheme 1).6 Treatment of acrylic acid 1 with diphenylphosphine under basic conditions afforded carboxylic acid 2 in 96% yield, from which oxazolidinone 4 was formed through coupling to 3. The diastereomeric azide 5, formed by reaction of triszyl azide with the enolate of 4, was separated chromatographically and treated with lithium hydroxide to cleave the chiral auxiliary. Reduction to the amine with tin(II) chloride followed by installation of the Fmoc protecting group gave the desired amino acid 7 suitable for direct incorporation into a peptide by solid phase peptide synthesis (SPPS).
 |
| | Scheme 1 First reported synthesis of an amino acid functionalised with a phosphine moiety – Gilbertson and co-workers.6 | |
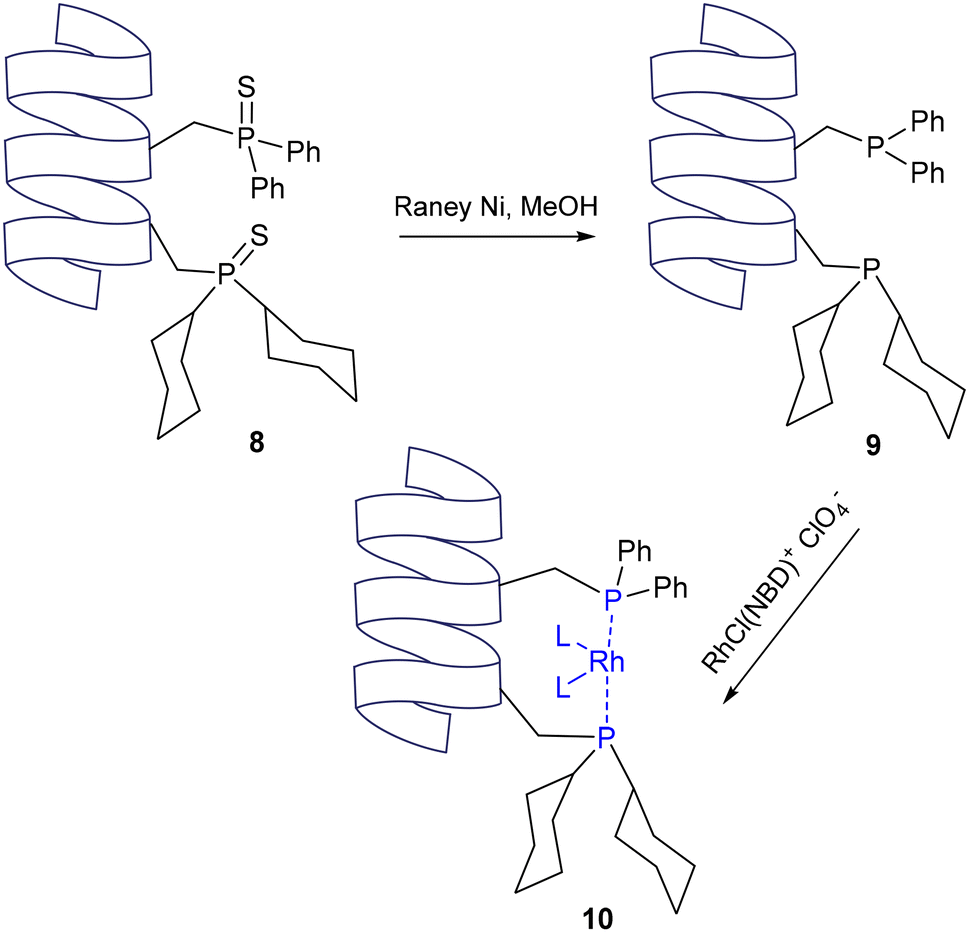
The same was applied soon after to synthesize (dicyclohexylphosphino)serine (Cps) and its sulfide precursor, which was incorporated into a 12mer peptide sequence along with the previously reported Pps sulfide.7 This peptide (8) was then treated with RANEY® nickel and methanol to reduce the sulfide-bearing P(V) centre to the corresponding P(III) phosphine, enabling subsequent metallation by addition of a Rh(I) salt. By placing these residues at the i and i + 4 positions of the α-helical peptide, the phosphine moieties were brought into the correct orientation to allow for metal coordination via the phosphorus atoms (Scheme 2).
 |
| | Scheme 2 Formation of a Rh(I) complex within an alpha helical peptide containing phosphine-functionalised residues at the I and I + 4 positions – Gilbertson and co-workers.7 | |
To structurally diversify the peptides obtained from incorporating these amino acids, Gilbertson and co-workers sought to devise a synthetic route to Fmoc-protected phosphanyl-substituted proline 16, with the aim of inducing a β-turn structure upon its incorporation into a peptide tetramer.8 The original strategy started with 4-hydroxyproline 11, from which the N-protected compound 12 was formed via mesylation with methanesulfonyl chloride (Scheme 3A). Additionally, protection of the carboxylic acid moiety was required to avoid reaction with the phosphide nucleophile – initial attempts using a variety of ester protecting groups were unsuccessful, therefore it was decided to convert the acid substrate into the corresponding TBDMS-protected alcohol 13 to enable selective attack of the phosphide at the 4-hydroxy position. Subsequent removal of the N- and O-protecting groups gave 14 which was readily converted to either the Fmoc- or Boc-protected amino alcohol. Although ultimately successful in generating both protected proline derivatives, this approach necessitated a challenging selective oxidation sequence to reform the carboxylic acid while leaving the phosphine sulphide group intact. A more streamlined approach was later developed by the same group that utilised an oxazoline acid protecting group to allow for nucleophilic substitution of the mesyl-alcohol 20 with sodium diphenylphosphide, followed by acidic hydrolysis to reform the carboxylic acid 22 as desired (Scheme 3B).9
 |
| | Scheme 3 Synthesis of phosphino-proline derivatives via (A) nucleophilic displacement and (B) chiral oxazolidone – Gilbertson and co-workers.8,9 | |
Following on from these examples of peptidic phosphine ligands with rhodium-coordinating capabilities, a 63-member library of diphosphine peptides was produced using combinations of Pps and Cps residues in varying orientations, with α-aminoisobutyric acid (Aib) residues included to enforce a helical secondary structure. Each of these peptides were complexed to rhodium and screened for their catalytic activity in the asymmetric hydrogenation of methyl 2-acetamidoacrylate.10 Although some degree of selectivity was observed, and later improved upon in a subsequent 2nd generation library,11 the consistently low to moderate enantiomeric excesses (up to 36% ee) led the authors to change their focus towards peptides featuring a β-turn secondary structural element.12 This proved fruitful, with peptides containing the well-known β-turn forming motif –Pro-D-Yyy– (where Yyy is a D-amino acid)13a,b flanked by diphenylphosphinoserine 7 showing up to 95% ee in the Pd-catalysed addition of dimethylmalonate 24 to 3-acetoxycyclopentene 23 (Scheme 4).14
 |
| | Scheme 4 Palladium-catalysed allylic alkylation of dimethylmalonate with cyclic substrate 3-acetoxycyclopentene using the phosphine-containing β-turn peptide Boc–D-Phg–Xps–Pro–D-Val–Pps–D-Leu–OMe – Gilbertson and co-workers.14 | |
An alternative strategy towards β-phosphino amino acids was reported in 1998 by Burgess and co-workers, enabling access to the D-enantiomer of Pps(sulfide) starting from L-serine (Scheme 5).15 Using a previously reported procedure to form Boc-protected oxazolidine 27,16 tosylation to give 28 enabled installation of the diphenylphosphine group through nucleophilic substitution with the corresponding phosphide. Subsequent sulphuration of the phosphorus centre gave 29, of which the oxazolidine ring could be cleaved by treatment with aqueous acid. Despite the need to reintroduce the Boc protecting group, 30 was obtained in high yield, from which 31 was formed in 99% ee by oxidation of the alcohol using pyridinium dichromate. A similar approach was later attempted by Stelzer and co-workers with the intention of developing a strategy applicable to a larger scale (Scheme 6).17 Use of a Ph2PH/K2CO3 heterogeneous system under solid–liquid phase transfer conditions enabled in situ generation of the anionic phosphide nucleophile, which reacted readily with L-iodoalanine 34, formed through treatment of O-tosylated N-Boc-L-serine 33 with sodium iodide. Although the corresponding phosphines were obtained in high yield, racemisation at the α-carbon was a major issue.
 |
| | Scheme 5 Synthesis of the D-enantiomer of the phosphine-containing amino acid Pps – Burgess and co-workers.15 | |
 |
| | Scheme 6 Synthesis of Pps using a heterogenous catalysis approach – Stelzer and co-workers.17 | |
Based on precedents set for the use of iodozinc reagents in the synthesis both of phosphines and unnatural amino acids,18a,b Gilbertson and co-workers demonstrated that a range of phosphinoserine derivatives could be efficiently synthesized by coupling nucleophilic zinc/copper reagent 38 to an electrophilic chlorophosphine (Scheme 7).19 Using Knochel's procedure for the preparation of functionalised copper reagents,20 iodozinc 37 is readily accessible from treatment of commercially available N-Boc iodoalanine 34 with activated zinc, which can subsequently be converted into 38 following stoichiometric addition of the copper salt. Treatment of this reactive intermediate with various aryl and alkyl chlorophosphine reagents afforded a new library of phosphine-containing amino acids 39a-I in yields ranging between 40% and 75%. This method would later be implemented in 2015 by Roy and co-workers in the design of diiron analogues of [FeFe]-hydrogenases, who demonstrated the incorporation of the diphenyl-, diethyl- or previously unreported diisopropylphosphine-functionalised amino acid 40 into a model hydrophobic tripeptide sequence (41) using solution-phase peptide synthesis. Metalation with (μ-pdt)Fe2(CO)6 afforded the desired phosphine-substituted diiron complex 42, which was subsequently found to exhibit electrocatalytic activity in the presence of acetic acid in mixed aqueous–organic solutions (Scheme 8).21
 |
| | Scheme 7 Synthesis of aryl and alkyl phosphinoserine derivatives via zinc/copper mediated P–C coupling – Gilbertson and co-workers.19 | |
 |
| | Scheme 8 Incorporation of a diiron complex into a tripeptide via a phosphine moiety for the formation of a biomimetic model of a [FeFe]–hydrogenase – Roy and co-workers.21 | |
2.2. β-Phosphino derivatives of aromatic amino acids
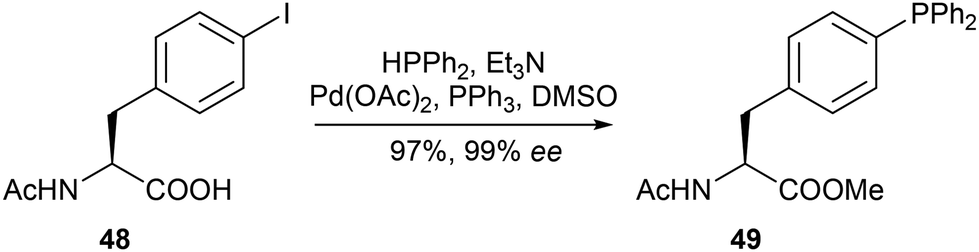
Following their successful synthesis of serine derivatives, Gilbertson and coworkers sought to expand the selection of phosphine-bearing building blocks to include aromatic amino acids. Taking inspiration from the well-established Stille coupling of aryl halides with diphenylphosphines,22 combined with the existing precedent of palladium-catalysed couplings with aryl triflates,23 a method was developed for the catalytic conversion of the phenolic hydroxyl group of tyrosine 43 to the arylphosphine ligand 45via the corresponding aryl triflate 44 (Scheme 9).24 Additionally, this method was applied directly to a pentapeptide (46) bearing a tyrosine residue, demonstrating a convenient alternative to the individual preparation and installation of phosphine amino acids into peptides. Kraatz reported a similar method utilising palladium catalysis to couple iodophenylalanine 48 to diphenylphosphine, giving rise to N-acetyl protected diphenylphosphanyl derivatives of D- and L-phenylalanine (49) without racemisation (Scheme 10).25
 |
| | Scheme 9 Transformation of (A) N-protected tyrosine esters and (B) a tyrosine residue in the peptide Boc–Ala–Tyr–Ala–Aib–Ala–OBn, into aryl phosphine ligands via the corresponding triflate – Gilbertson and co-workers.24 | |
 |
| | Scheme 10 Palladium-catalysed synthesis of phosphine-containing aromatic amino acids – Kraatz and co-workers.25 | |
Contrastingly, phosphinophenylglycine and -alanine derivatives 51a–b were synthesized by Stelzer and co-workers via nucleophilic aromatic substitution starting from the sodium salts of 4-fluoro-α-phenylglycine (n = 0) and -alanine (n = 1).26 Treatment of salts 50a–b with potassium diphenylphosphide leads to attack of the phosphide anion at the 4-position with loss of fluoride to give para-substituted phosphinophenylamino acids 51a–b respectively (Scheme 11). This methodology was later extended to include the analogous ortho-substituted derivatives, synthesized from the corresponding 2-fluoro-α-phenylglycine and alanine salts.27
 |
| | Scheme 11 Synthesis of diphenylphosphine derivatives of phenylglycine and -alanine derivatives via SnAr chemistry – Stelzer and coworkers.26 | |
2.3. Other phosphorus-containing amino acids
While the phosphine moiety makes up a considerable portion of phosphorus-based ligand chemical space, several other ligand classes have appeared in the literature for incorporation into peptides for various metal-binding applications. The following section includes discussion of amino acids functionalised with phosphole, phosphinite and phosphate moieties – for a recent in depth review on the synthesis of other phosphorus-containing amino acids see the provided ref. 28.
2.4. Phosphole amino acids
Like the phosphine group, phospholes comprise an important class of phosphorus(III) donor ligand broadly applicable to a variety of metal-catalysed transformations.29 The use of these P-heterocyclic ligands has been widely reported both in standard and asymmetric reactions, from their initial emergence in rhodium-catalysed hydrogenation30 and hydroformylation processes,31 to more recent examples such as catalytically active phosphole-containing porphyrin-derived platforms32 and phosphole-embedded helicenes for use in enantioselective gold catalysis.33 Aside from catalysis, phosphole metal complexes have been used in biomedical research, owing to their well-documented activity as disulphide reductase inhibitors.34 Bioconjugation is a well-established strategy for the development of metallopharmaceuticals with suitable pharmacological characteristics. A notable example of this is the anti-arthritic drug Auranofin (52), approved by the FDA in 1985, which is comprised of a thiosugar and phosphine ligand coordinated to a Au(I) centre (Fig. 1).35 More recently, modification of a phosphole Au(I) complex with a thiosugar (53) was demonstrated to increase bioavailability whilst retaining antiproliferative potency.36
 |
| | Fig. 1 Auranofin 52 and phosphole analogue 53 – Bagrel and co-workers.36 | |
The first reported phosphole-containing amino acid 58 was synthesized in 2007 by Le Floch and co-workers, starting from N-Boc serine 54 (Scheme 12).37 The carboxylic acid was converted to the corresponding methyl ester using methyl iodide, followed by treatment with triphenylphosphine and iodine in the presence of imidazole to form the iodoalanine methyl ester (34). The phosphole species (56) was then generated by reacting 34 with the lithium salt of 3,4-dimethylphospholide (55). As with the phosphine-containing amino acids described in section, the phosphorus(III) centre in 56 is highly prone to oxidation, therefore protection of the lone pair was carried out with elemental sulphur to form the sulfide derivative 57. This sulphide-protected phosphole moiety was found to be highly stable throughout manipulation of the amino nitrogen protecting groups, ultimately enabling formation of the desired Fmoc-protected amino acid 58 for potential applications in peptide synthesis.
 |
| | Scheme 12 Synthesis of a phosphole-containing amino acid 58 – Le Floch and co-workers.37 | |
In 2010, the same group reported two new synthetic strategies towards the incorporation of the phosphole moiety into aromatic amino acid sidechains.38 Functionalisation of the tyrosine hydroxyl group was envisaged via a nucleophilic substitution approach – a method frequently seen for the installation of an alkoxy group to a phosphorus centre,39a,b although not previously applied to the phosphole functionality specifically. The key component of the synthesis, chlorophosphole 61, was accessed via metallacycle transfer reaction of the corresponding organozirconium 60, prepared from Cp2ZrCl2 and n-butyllithium as reported by Negishi in 1986.40 Reaction of chlorophosphole 61 with readily available Cbz-protected tyrosine methyl ester at −78 °C in the presence of base led to formation of the desired amino acid 62, albeit in a relatively low yield (Scheme 13A). The second strategy utilised a Pd-catalysed Stille cross-coupling between the N-protected amino acid methyl ester 63 and substituted stannylphosphole reagent 64. While no reaction was observed for attempted couplings with the N-Cbz tyrosine triflate methyl ester, even in the presence of catalytic amounts of Cu(I) halide salts, full conversion was seen when N-Boc 4-iodophenylalanine methyl ester 63 was instead used (Scheme 13B). The phenylalanine phospholyl-derivative 65 was ultimately obtained following protection of the phosphorus lone pair with a sulphide group.
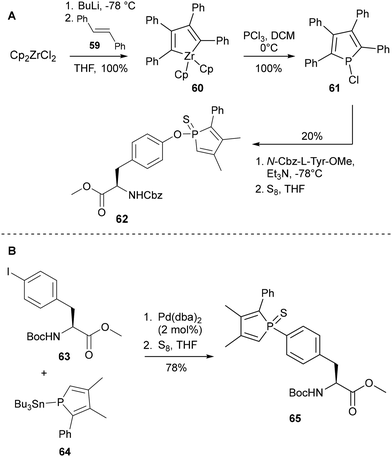 |
| | Scheme 13 Functionalisation of aromatic amino acids with a phosphole moiety via (A) nucleophilic substitution and (B) Stille cross-coupling – Le Floch and co-workers.38 | |
Although the sulphide group confers adequate stability to the otherwise free P(III) centre of the amino acid, the occurrence of racemisation during manipulation of this protecting group cannot be fully ruled out. Convincing data surrounding the enantiomeric purity of these amino acids has remained sparse, and the reported deprotection procedure via reductive desulphurisation with RANEY® nickel has been previously reported to cause racemization at the α-carbon of phosphine-based amino acids, ultimately leading to the generation of diastereomeric peptides.21 More recently, Cavelier and co-workers detailed a modified approach to the preparation of phospholyl amino acids and peptides, in which the limitations of previous methodology were addressed.41 Specifically, substitution of SPPS-compatible N-protected iodo amino esters 67 with deprotonated phospholide 66 followed by addition of borane dimethyl sulphide led to the formation of N-protected phospholyl(borane) amino esters 68 in up to 99% ee, as determined by chiral HPLC (Scheme 14). The applicability to peptide synthesis was effectively demonstrated through the formation of dibenzophospholyl(borane) dipeptide derivatives through N- and C-amide coupling with an alanine residue. The use of borane as a complexing agent enabled the racemization-free transformation of the phospholyl amino acid derivatives into the corresponding free P(III) amino ester, oxide, sulfide, and notably, complexation to an Au(I) centre. Although this work is focused primarily on the photophysical properties of phosphole amino acids, the demonstration of their Au(I)-coordinating ability combined with the known propensity of phospholes to bind transition metals opens the door for future investigation into phosphole-containing peptides both for their fluorophoric and metal-binding characteristics.
 |
| | Scheme 14 Synthesis of N-protected phospholyl(borane) amino esters – Cavelier and co-workers.41 | |
2.5. Phosphinite amino acids
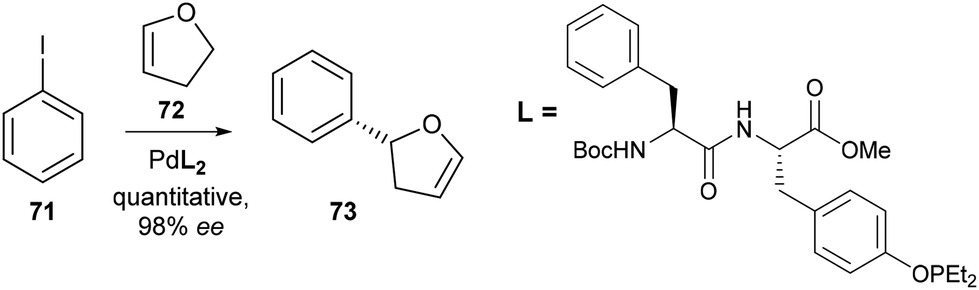
The phosphinite group can be represented as PR2OR, where the P(III) centre is bonded to one oxygen atom, and two alkyl or aryl groups. In the context of transition metal catalysis, this functionality has proven particularly prevalent in the design of iridium catalysts,42 with examples including iridium bis(phosphinite) pincer complexes for alkane transfer dehydrogenation,43 and pyridine–phosphinite ligands for the asymmetric hydrogenation of olefins.44 Whilst amino acids have been utilised as important chiral starting materials for the synthesis of phosphinite ligands, for example, the use of prolinol-based amidophosphine–phosphinite ligands in Rh-catalysed asymmetric hydrogenation reactions,45a,b this class of ligand has scarcely been explored in the context of metal-binding peptides. In 2003, Kraatz and co-workers detailed the first preparation of phosphinite-containing amino acids, based on the modification of sidechain hydroxyl groups. Treatment of Boc-protected serine methyl ester 32 with chlorodiphenylphosphine under basic conditions afforded the corresponding phosphinite 69 in 60% yield (Scheme 15) which coordinated in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio with Pt(II) to form complex 70.46 Additional phosphinite analogues based on the hydroxyl-containing amino acids threonine and tyrosine were also prepared using this method, each of which were demonstrated to coordinate to Pt(II) and Pd(II) ions with square planar geometry. Later work by the same group found Pd complexes with peptide ligands comprising of similar phosphinite amino acids were effective in catalysing asymmetric Heck reactions between iodobenzene 71 and dihydrofuran 73 with up to 98% ee reported (Scheme 16).47
:1 ratio with Pt(II) to form complex 70.46 Additional phosphinite analogues based on the hydroxyl-containing amino acids threonine and tyrosine were also prepared using this method, each of which were demonstrated to coordinate to Pt(II) and Pd(II) ions with square planar geometry. Later work by the same group found Pd complexes with peptide ligands comprising of similar phosphinite amino acids were effective in catalysing asymmetric Heck reactions between iodobenzene 71 and dihydrofuran 73 with up to 98% ee reported (Scheme 16).47
 |
| | Scheme 15 Synthesis of a serine-derived phosphinite ligand and its coordination to Pt(II) – Kraatz and co-workers.46 | |
 |
| | Scheme 16 Asymmetric Heck reaction catalysed by a Pd(II) complex containing the dipeptide phosphinite ligand Boc–Phe–Tyr(OPEt2)OMe – Kraatz and co-workers.47 | |
2.6. Phosphorylated amino acids
With critical roles across a wide array of cell-signalling processes, phosphorylation has been indicated to be the most commonly occurring post-translational modification (PTM) in the eukaryotic proteome.48 In metalloproteins, phosphorylation of particular amino acid residues often modulates both the metal binding affinities and the overall properties of the protein as a whole,49a,b both from the local effect of altered residue charges alteration and metal coordination geometry, and global conformational changes to the protein that may distort metal-binding sites or expose sites previously occluded.50 A prominent example of such a protein is α-synuclein, a metal-binding protein found primarily in the brain and an important hallmark of Parkinson's disease pathology. Several studies have probed the impact of Ser and Tyr hydroxyl group phosphorylation in the C-terminal region of α-synuclein, reporting profound effects on the peptide's metal binding affinity and specificity,51a,b as well as the conformation and location of the peptide's metal-binding sites.52 Given the inaccessibility of commercially available phosphorylated amino acids, reliable methodology for their preparation is of significant demand for the study of metalloprotein phosphorylation events.
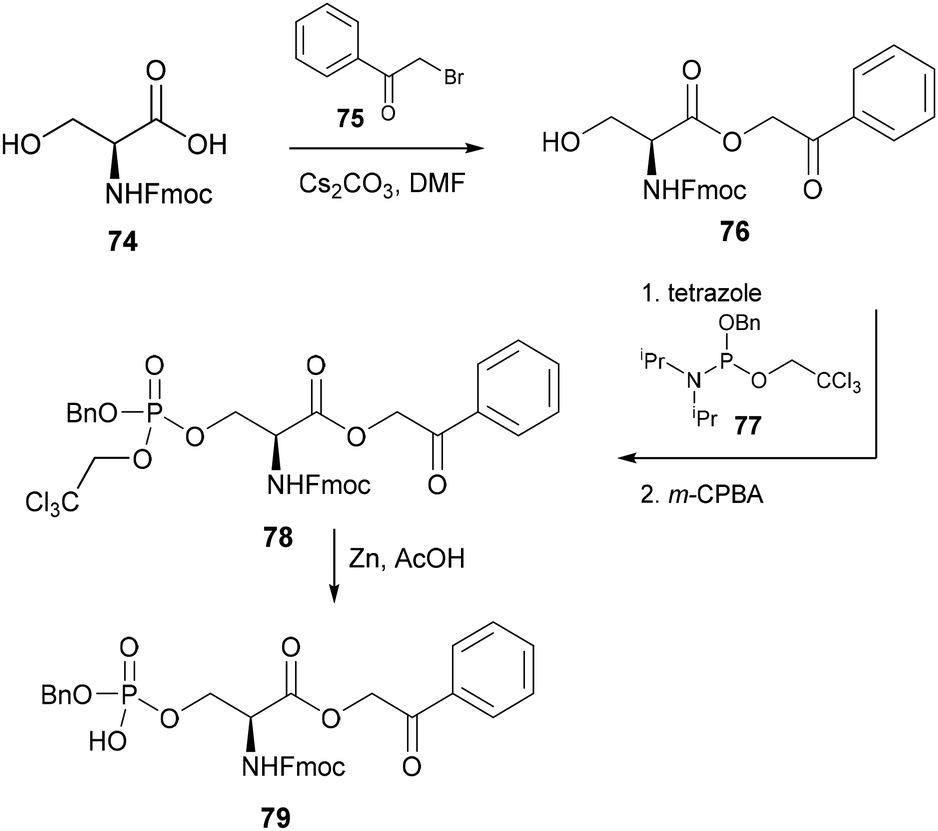
First reported by Wakamiya and co-workers in 1994, the original synthesis of phosphorylated serine 79 involved the reaction of phenacyl-protected serine derivative 76 with dialkyl N,N-phosphoramidite 77 in the presence of tetrazole to form the phosphite intermediate 78, which was ultimately converted to the desired phosphoamino acid 79 following oxidation with m-CPBA and removal of the phenacyl and trichloroethyl groups (Scheme 17).53
 |
| | Scheme 17 Synthesis of phosphorylated serine 79 using phosphoramidite chemistry – Wakamiya and co-workers.53 | |
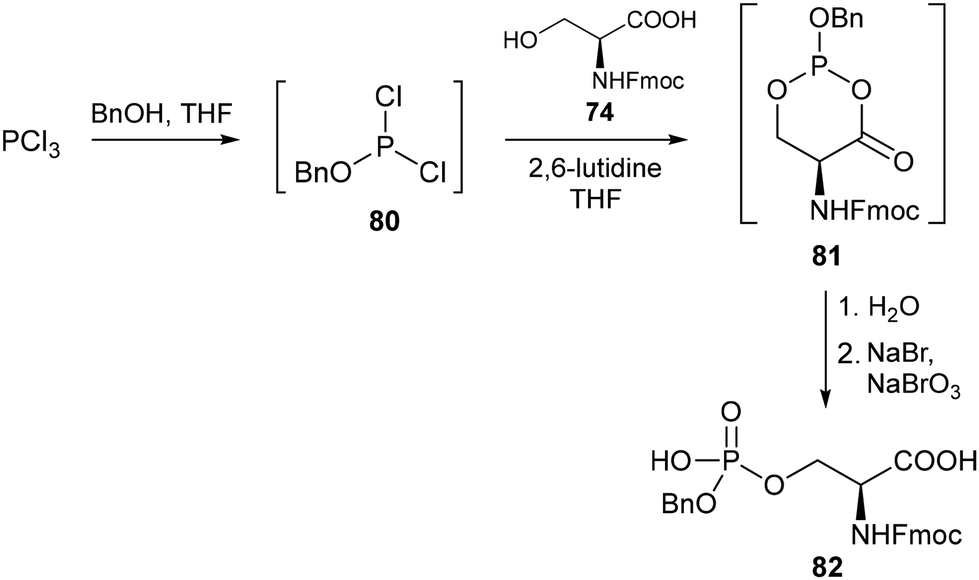
As a more efficient alternative to the established phosphoramidite-based approach, Bakale and co-workers reported a one-pot synthesis of protected phosphoserine derivatives involving convenient in situ generation of the phosphorylating agent whilst removing the need for tetrazole, a highly explosive heterocycle used for the promotion of phosphitylation.54 The reaction of phosphorus trichloride with benzyl alcohol gave rise to 80, which was sufficiently reactive towards the serine hydroxyl group without the use of a tetrazole activator, after which the resulting cyclic mixed phosphite intermediate 81 was subsequently oxidised into the target phosphorylated amino acid 82 (Scheme 18). Further development of this method gave rise to a procedure enabling the synthesis of Fmoc-O-benzyl-L-phosphoserine on a kilogram scale.55
 |
| | Scheme 18 One-pot process for the synthesis of phosphoserine derivatives – Bakale and co-workers.54 | |
3. Metal coordination via nitrogen
In the context of metal coordination, N-donor ligands represent a vast range of compounds with diverse properties. The pyridine ring is an example of a highly pervasive structural motif across transition metal catalysis, owing both to the availability of its nitrogen lone pair for metal coordination and to the overall stability of its planar ring structure. In ligand design, pyridine is commonly found within an extended conjugated system, e.g. bipyridine or phenanthroline, or as part of a mixed heteroatomic ligand such as pyridine-oxazolines (PyOx). Due to the growing prominence of artificial metalloenzymes and other peptide-based catalysts, reliable methodology for incorporating metal-binding functionalities into peptides is of great interest. Accordingly, numerous examples of unnatural amino acids with such functionalities have emerged in the literature for various applications such as the design of metal ion sensors and the study of electron transfer in peptides.56a,b Given the wide range of N-donor ligand architectures that have been incorporated into the design of noncanonical amino acids, this section has been organised into further subcategories based upon structural features, namely denticity and the aromatic or aliphatic nature of the ligand (Scheme 19).
 |
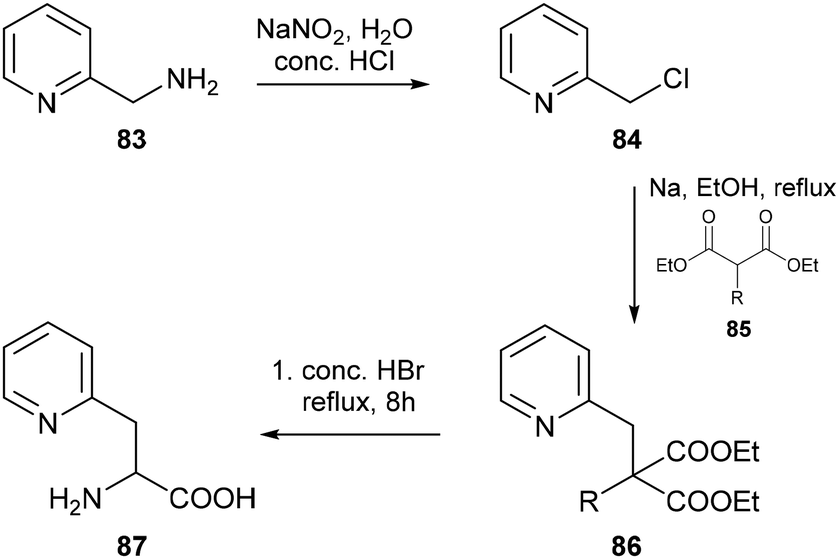
| | Scheme 19 Synthesis of 2-pyridylalanine 87via the malonic ester method, where R = pthalamido, benzamido or acetamido. | |
3.1. Pyridylalanine derivatives
Early syntheses of pyridylalanine derivatives typically relied upon one of two approaches: synthesis via a malonic ester (Scheme 19) or via an azlactone intermediate (Scheme 20). The former method involves alkylation of an amido-functionalised malonic ester (85) at the α-carbon position using picolyl chloride 84, readily accessible from the corresponding amine 83, followed by acidic hydrolysis under reflux to access the desired amino acid 87 from the condensation product 86, as shown in Scheme 19. The earliest synthesis of a pyridyl-functionalised alanine derivative was reported by Overhoff and coworkers in 1936, who first isolated 2-pyridylalanine as a racemate.57 Subsequent efforts towards increasing both the yield and scale of the synthesis included replacement of the original pthalimidomalonic ester with benzamido- or acetamido-malonic esters, use of ethanol instead of xylene as a solvent for the condensation reaction, and refluxing with hydrobromic versus hydrochloric acid as originally reported by Overhoff.58a,b The applicability of the 2-pyridylalanine derivative prepared via this method for peptide synthesis was later demonstrated by Watanabe and coworkers in 1968, who successfully isolated the corresponding dipeptide benzyl ester in 75% yield upon carbobenzoxylation of 2-pyridylalanine and subsequent amide coupling to glycine benzyl ester using dicyclohexylcarbodiimide.59
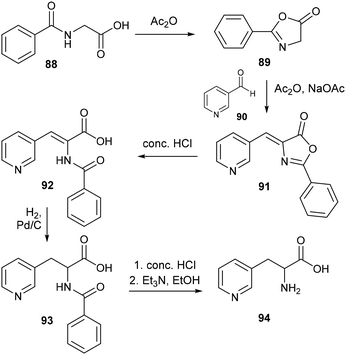 |
| | Scheme 20 Synthesis of 3-pyridylalanines via the azlactone method. | |
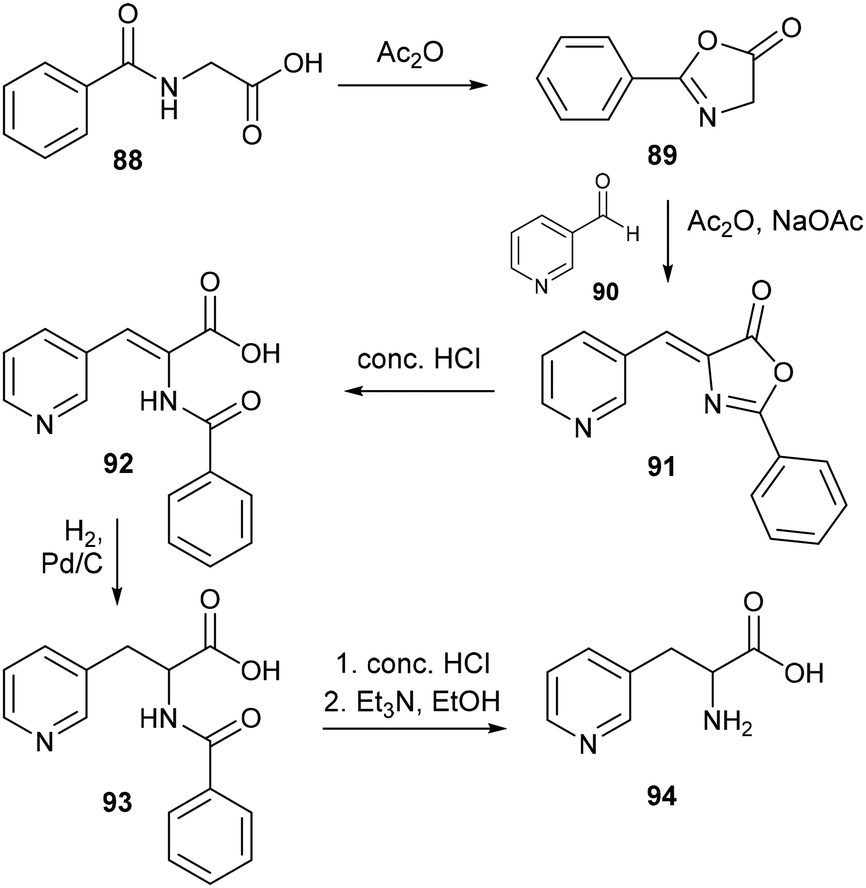
Comparatively, methodology for synthesizing pyridylalanines via an azlactone intermediate has experienced greater development. Based upon the well-known Erlenmeyer synthesis of aromatic amino acids,60 the azlactone method involves firstly the formation of 2-phenyl-oxazolone 89 from N-benzoylglycine 88, which cyclises in the presence of acetic anhydride and a fused sodium acetate catalyst – this reactive intermediate then undergoes condensation with an aromatic aldehyde 90 to form the azlactone species 91 as shown in Scheme 20. For both methods, acidic hydrolysis is used to access the desired amino acid from the condensation product, while the azlactone approach additionally requires catalytic hydrogenation to reduce the enamide bond still present in the acrylic acid 92 prior to hydrolysis. The azlactone method was first applied to the synthesis of 3-pyridylalanine by Wibaut and coworkers in 1955,61 who obtained the racemic amino acid in up to 68% yield, albeit on a relatively small, sub-gram scale. This method was soon extended to access the isomeric 2- and 4-pyridylalanines,62 while modification to the original reaction conditions in subsequent reports led to improved outcomes,63 such as the introduction of anhydrous potassium carbonate as a catalyst for azlactone formation, which ultimately enabled the gram scale synthesis of DL-3-pyridylalanine in 95% yield.64
In 1984, Cativiela and coworkers incorporated chiral phosphine-containing rhodium complexes in the synthesis of 3-pyridylalanine among various hetaryl-functionalised amino acids.65 In this work, prochiral acetamido-hetarylacrylic acids were formed upon hydrolysis of the azlactone condensation product of the corresponding hetaryl-aldehyde with N-acetyl- or benzoyl-glycine – these prochiral precursors were then subjected to Rh-catalysed hydrogenation to generate 3-pyridylalanine with moderate enantioselectivity. A similar approach was later taken by Döbler and coworkers in 1996, who used a rhodium catalyst in the presence of HBF4 to form enantiomerically pure 3- and 4-pyridylalanines in up to 90% ee.66 A variation of this method was reported by Bozell and coworkers, who synthesized enantiopure pyridylalanines via a Heck-like Pd-catalysed coupling of 4-bromopyridine 95 with olefin 96 (Scheme 21), followed by Rh-catalysed asymmetric hydrogenation and acidic hydrolysis.67 Although initial attempts using conditions originally reported by Heck68 led to the undesired bipyridyl homocoupling product, implementation of more dilute reaction conditions gave the prochiral enamide 97 which was subsequently converted to the L- or D-enantiomers of 4-pyridylalanine (99) in up to 96% ee. While no analogous asymmetric syntheses have emerged from the malonic ester methods, enantiopure pyridylalanines have been obtained in conjunction with either azlactone or malonic ester approaches through the use of enzymatic resolution via subtilisin69 or chymotrypsin.70a–c
 |
| | Scheme 21 Asymmetric synthesis of L-4-pyridylalanine – Bozell and co-workers.67 | |
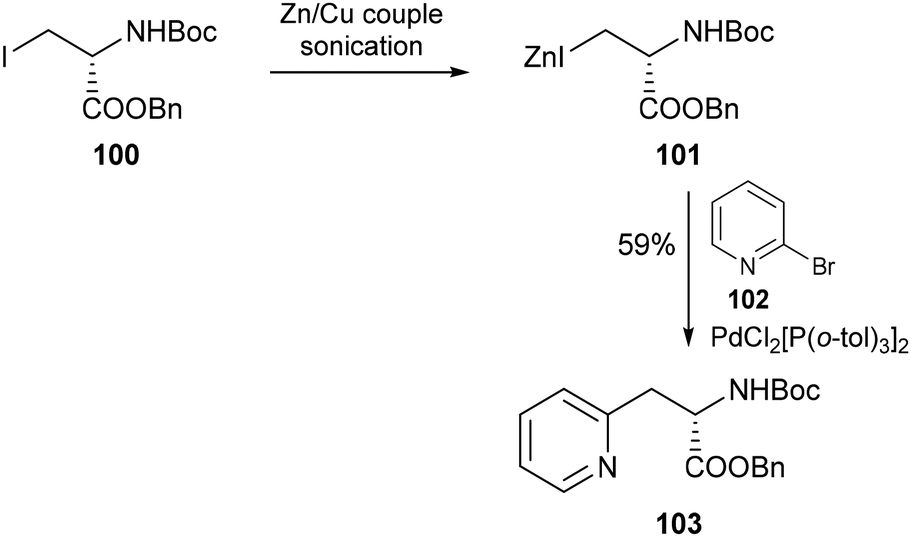
A growing number of methods have since emerged for the incorporation of pyridyl moieties into amino acid scaffolds. One widely utilised strategy is the used of the Negishi cross-coupling, a C–C bond forming reaction pervasive across synthetic chemistry. The application of this reaction to the area of amino acid synthesis was pioneered by Jackson and co-workers in 1989.71 In their seminal report, an organozinc reagent derived from Boc-protected iodoalanine was demonstrated to effectively undergo palladium-catalysed coupling to a variety of aryl iodides to generate enantiomerically pure β-aryl amino acids. The scope of the transformation was additionally extended to include heteroaryl halides such as 2-bromopyridine (102) to give the corresponding β-(2-pyridyl)alanine 103 in 59% yield, alongside numerous further examples containing substituted pyridines which were used in the construction of catalytic tripeptides (Scheme 22).72 Since this first report, Negishi cross-coupling has been widely applied to the synthesis of pyridyl-containing amino acids, with examples including the antitumour antibiotic azatyrosine,73a,b the chloroazatyrosyl napthoamide fragment of the kedarcidin chromophore,74 and tridentate 2,6-bis(ethoxycarbonyl)pyridin-4-yl alanine ligands.75 An in-depth discussion of the use of Negishi cross-couplings for the synthesis of unnatural amino acids can be found in a previous review by Brittain and Cobb.76
 |
| | Scheme 22 Synthesis of β-(2-pyridyl)alanine via palladium-catalysed cross coupling of the organozinc reagent 100 with 2-bromopyridine 101 – Jackson and co-workers.72 | |
Other synthetic routes that have been employed for the synthesis of pyridylalanine derivatives include the alkylation of chiral glycine equivalents with pyridyl halides77 or carbinols78 followed by acidic hydrolysis, photoredox-catalysed radical conjugate addition of halopyridines to dehydroalanine,79 and a two-step strategy comprised of a lanthanide-catalysed hetero Diels–Alder and subsequent Knoevenagel-Stobbe reaction.80 Additionally, several reports have detailed the use of a chemoenzymatic approach to access unnatural amino acids with high enantioselectivity. In particular, the enzyme phenylalanine ammonia lyase (PAL) has emerged as an effective biocatalyst for the synthesis of L-pyridylalanine analogues. In 2016, Turner and co-workers reported a one-pot telescopic synthesis comprising of two steps: firstly a Knoevenagel-Doebner condensation between a substituted pyridyl–aldehyde and malonic acid in the presence of piperidine to form the corresponding pyridylacrylic acid, followed by biocatalytic hydroamination using bacterial PAL from Anabaena variabilis.81 Notably, this biocatalyst offered conversions of up to 95%, and excellent enantioselectivity of >99% ee. The use of other PAL enzymes as well as a tyrosine phenol lyase have also been documented for the biosynthetic synthesis of pyridylalanine derivatives.82a–c
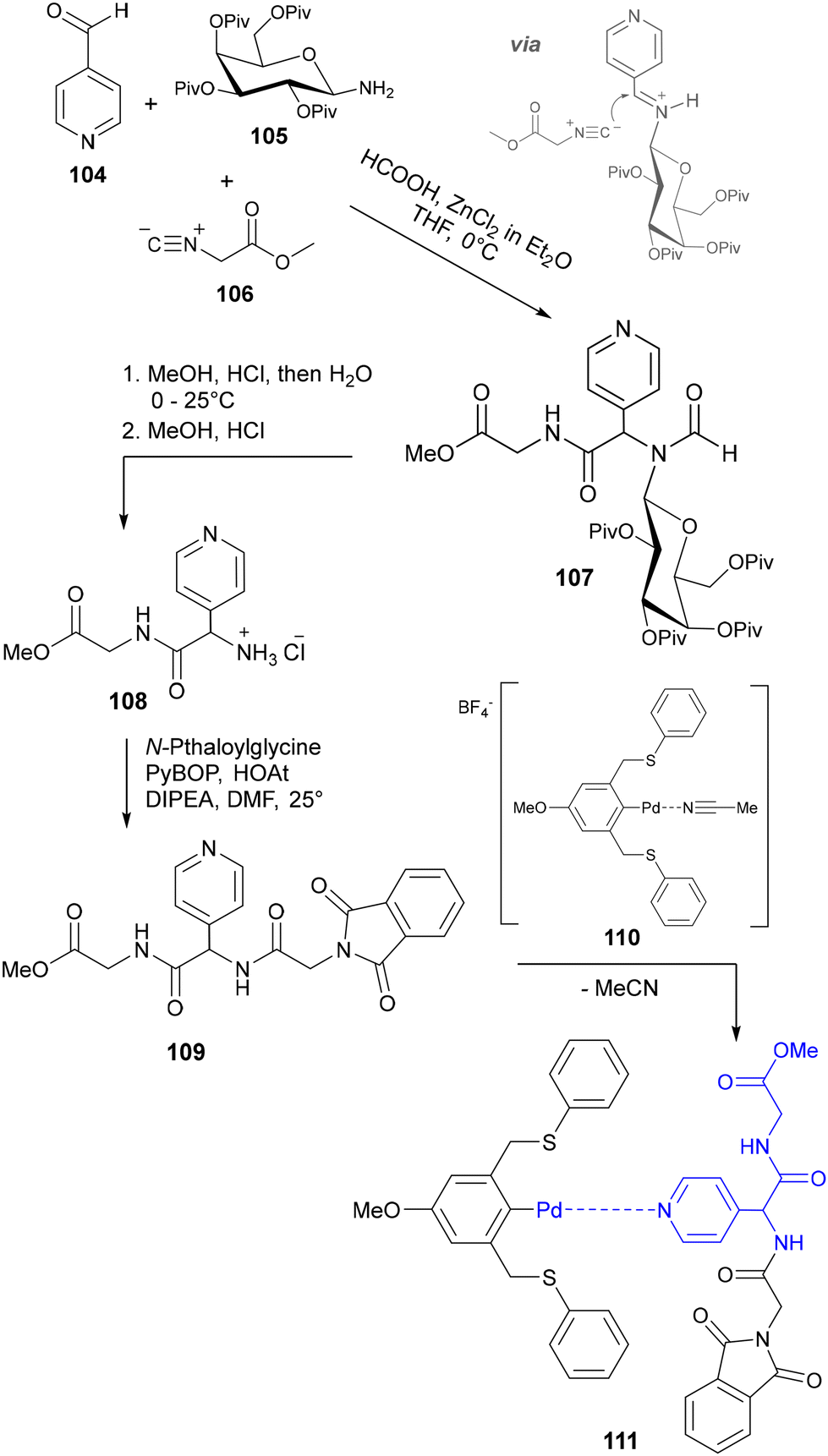
In contrast with the aforementioned pyridylalanines, very few examples have been reported of analogous glycine derivatives, i.e. where the pyridyl moiety is attached to the amino acid via the α-carbon. Although several syntheses of N-terminally substituted glycine derivatives have been reported,83a–c strategies towards a pyridylglycine derivative suitable for peptide synthesis have remained sparse in the literature. In 2006, Gerhardt and Weck detailed the design of a series of self-assembling biological-hybrid scaffolds comprised of a p-methoxy SCS–Pd complex coordinated to a tripeptide bearing a central pyridylalanyl or pyridylglycyl residue.84 While synthesis of the pyridylalanine-containing tripeptide posed no issues, installation of the pyridylglycine moiety proved problematic due to the susceptibility of the pyridylglycyl carboxylic acid to spontaneous decarboxylation. To circumvent this issue, they proposed synthesizing a stable dipeptide unit containing a protected 3- or 4-pyridylglycine moiety, which could then undergo further amide coupling to facilitate peptide extension. Their approach involved a method based on a modified Strecker, Ugi-type four-component synthesis developed by Kunz and co-workers,85a–c where the pyridine carboxaldehyde precursor 104 is reacted with protected amino sugar auxiliary 105 to form an imine intermediate (Scheme 23) which subsequently undergoes attack by isonitrile 106 and methanoic acid. Following acidic hydrolysis of the resulting dipeptide 107 to liberate the N-terminal amine, further amide coupling was successfully achieved to give the desired N-pthaloyl-, C-methyl ester protected tripeptide 109. The pyridyl-containing peptides were subsequently coordinated to a complementary recognition unit comprised of a SCS–Pd pincer complex (110). While previous examples of pincer amino acids featured an N-terminal metal-binding moiety,86 limiting their versatility as functional synthons for the construction of supramolecular peptide-based architectures, the design of 108 conveniently enables further coupling to either terminus of a growing peptide.
 |
| | Scheme 23 Synthesis and incorporation of 4-pyridylglycine residue into a peptide for binding to a palladium pincer complex – Gerhardt and co-workers.84 | |
In 2016, Marsden and co-workers reported a synthetic route to α-pyridyl, α-substituted amino acids of type 114 (Scheme 24).87 Based upon recent reports from Londregan and co-workers involving the PyBroP-activated reaction of pyridine N-oxides with a range of nucleophiles,88a–c the authors reasoned that azlactones of comparable pKa89 such as 112 may serve as suitable nucleophiles for reaction with activated electrophile pyridine N-oxide 113, giving access to amino acids 114via a direct pyridylation approach. With the aim of using greener reaction conditions, ethyl acetate was found to enable the reaction with efficiency comparable to that of THF, when used in conjunction with TsCl as an activating agent.
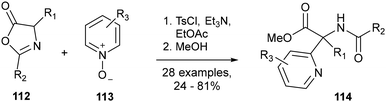 |
| | Scheme 24 Synthesis of α-pyridyl, α-substituted amino acids – Marsden and co-workers.87 | |
3.2. Bipyridyl- and extended pyridyl-amino acids
The first example of a bipyridyl-functionalised amino acid was reported in 1992 by Imperiali and coworkers, who synthesized Fmoc-protected bipyridin-6-yl 119 starting from the commercially available benzophenone imine glycine derivative 115 (Scheme 25A).90 Using a method previously reported by O'Donnell for the synthesis of α-amino acids,91 asymmetric alkylation of 115 with 6-(bromomethyl)-2,2′-bipyridine 116 was achieved using a N-benzylcinchonidinium chloride phase transfer catalyst, giving the S enantiomer of 117 with a moderate ee of 53%. Following crystallisation to resolve the racemate, enantiomerically pure 117 was hydrolysed under acidic conditions to afford the free amino acid 118. Fmoc azide was used for installation of the amine protecting group, enabling direct use of 119 as a building block for the solid-phase synthesis of metallopeptides. By incorporating either one or two (Scheme 25B) bipyridin-6-yl moieties into the peptide, alongside a β-turn-favouring D-proline residue, the relative contributions of both inter- and intramolecular coordination towards metal binding could be investigated. Standard spectroscopy methods were used to evaluate the interactions between these peptides and a series of divalent metal cations: UV-Vis titrations were used to derive a series of metal–peptide dissociation constants, while spectra obtained by circular dichroism indicated the formation of a chiral complex upon addition of Cu2+ to peptide 120.92
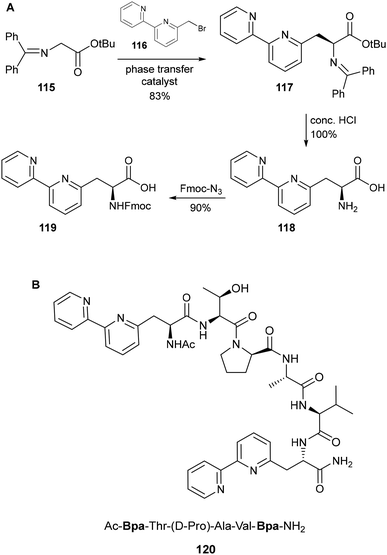 |
| | Scheme 25 (A) Synthesis of the 6-bipyridyl-containing amino acid 6Bpa and (B) structure of a peptide incorporating the 6Bpa residue – Imperiali and co-workers.90 | |
Given the sizable impact of varying bipyridine ring substitution on the metal binding properties of a bipyridyl ligand, Imperiali and coworkers sought to develop a means of accessing the bipyrid-4-yl and 5-yl isomers of 6Bpa. Racemic 5Bpa was obtained using the previously reported asymmetric alkylation approach (Scheme 26), using 5-(bromomethyl)-2,2′-bipyridine with the same commercially available glycine derivative and phase transfer catalyst.93 Synthesis of the bipyrid-4-yl derivative 127 was carried out via a route based on the Erlenmeyer synthesis of aromatic amino acids reported in 1893.60 Treatment of the unsymmetrical bipyridine 121 with selenium dioxide enabled transformation of the 4-methyl group to the corresponding formyl group of aldehyde 122. Reaction of 122 with N-benzoyl glycine (88) generates a benzamide intermediate, which undergoes cyclisation in the presence of acetic anhydride to give oxazolone 123. Simultaneous reduction and hydrolytic cleavage of the oxazolone moiety using hydroiodic acid with red phosphorus afforded the desired racemic amino acid, which was converted to the corresponding methyl ester 124 by treatment with methanol under acidic conditions. Unlike with bipyridin-6-yl 119, enantiomerically pure material could not be obtained by crystallisation for the 4Bpa and 5Bpa derivatives. Instead, resolution was achieved enzymatically using a previously reported method for the enantioselective hydrolysis of racemic amino acid esters,94 in which a mixture of enzymes is used with the alkaline serine protease Subtilisin Carlsberg as the major component.
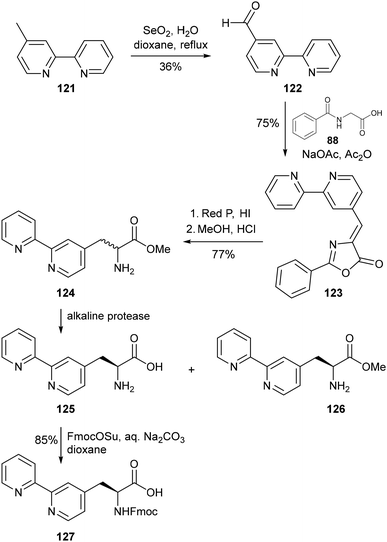 |
| | Scheme 26 Synthesis and enzymatic resolution of the 5-bipyridyl-functionalised amino acid 5Bpa – Imperiali and co-workers.93 | |
The Imperiali group subsequently used amino acids 119 and 127 to enable the construction of mutant horse heart cytochrome c (hh cyt c) proteins containing a redox-active Ru2+ centre.95 The native heme-containing fragment (residues 1–65) was obtained from the wildtype protein by cyanogen bromide cleavage at Met65, resulting in the formation of a C-terminal homoserine lactone functionality, while the second fragment, containing either 6Bpa (119) or 4Bpa (127) at the 72 position, was assembled on solid phase. Incubation of the two purified fragments under neutral reducing conditions enabled amide bond formation between the C-terminal lactone and N-terminal amine. Treatment of the resulting proteins with excess Ru(bpy)2CO3 gave rise to metal complexes of the 4Bpa- and 6Bpa-modified proteins, which demonstrated significantly increased rates of electron transfer via direct photoinduced (DP) and flash quench (FQ) techniques when compared with an analogous His72 cyt c mutant. Use of the 5-pyridyl amino acid was later reported by Yoshikawa in 1996 for the design of a Ni2+-binding 3α-helix bundle: placement of 5Bpa in the middle of a 13mer peptide with flanking hydrophobic cyclohexylalanine (Cha) and leucine residues promoted the formation of a tight hydrophobic core, which, upon assembly of the 3α-helical structure, would in turn surround the hydrophilic trisbipyridine nest, in which the Ni2+ ion is accommodated.96
Following Imperiali's original syntheses of bipyridinyl α-amino acids, Bowler and coworkers desired a simplified method for making an amino acid attached to the bipyridine ring via the 4 position that avoided the use of selenium dioxide, as well as the laborious need to construct the bipyridine ring system.97 The first step involved formation of bipyridine 129via asymmetric bromination of commercially available 4,4′-dimethyl-2,2′-bipyridine 128 using N-bromosuccinimide (Scheme 27). The brominated bipyridine was then converted into the desired amino acid 132 in an analogous fashion to the original method:90 treatment with the glycine benzophenone imine derivative 115 and phase transfer catalyst, subsequent acidic hydrolysis and esterification of 130, with enzyme-mediated resolution bringing the final enantiomeric excess up from 65% to 95%. Boc-protected 132 facilitated the formation of an α-helix secondary structure upon SPPS installation into a predominantly alanine-containing peptide.
 |
| | Scheme 27 Synthesis of an N-Boc protected 4-methyl-2,2′-bipyridyl amino acid – Bowler and co-workers.97 | |
Seeking to expand the repertoire of amino acids with sidechain N-heterocyclic scaffolds Imperiali and coworkers reported the synthesis of α-amino acids containing phenanthroline and 2,9-dimethyl-1,10-phenanthroline (neocuproine) functionalities for incorporation into small peptides.98 Applying methodology previously reported by Sörensen,99 chloromethyl derivatives 133a–b were converted into 135a–b by reaction with excess diethyl acetamidomalonate (134) in ethanol under reflux (Scheme 28). Subsequent refluxing with concentrated hydrochloric acid and esterification with acidic methanol afforded the phenanthrolyl- and neocuprionyl-amino acid methyl esters 136a and 136b respectively. Stereoselective hydrolysis of the racemic methyl esters was achieved using alkaline protease giving the amino acids 137a and 137b with >98% ee.
 |
| | Scheme 28 Synthesis of a phenanthroline-containing amino acid – Imperiali and co-workers.98 | |
All amino acids discussed thus far have fallen into the category of α-amino acids, meaning that the amino group and side chain R group (in this case, bipyridyl) are attached to the α-carbon i.e. the central backbone carbon atom adjacent to the carbonyl carbon of the carboxylic acid group. While the standard proteinogenic amino acids fit into this category, hundreds of other naturally-occurring amino acids exist in which the location of the core amino functional group relative to the alpha carbon is varied. This structural variation is classified using the Greek letters α, β, γ, δ … ω to denote the position of the amine terminus along a carbon chain of increasing length, for example β-phenylalanine 139 and γ-aminobutyric acid 141 – also known as GABA, the most common central nervous system inhibitory neurotransmitter (Fig. 2). The carbon at the final position is sometimes designated lower-case omega (ω) irrespective of chain length. In the context of metal-binding residues, the bipyridine containing 140 (Fig. 2) is therefore classified as an ω-amino acid, in contrast with the α-amino acid 138 containing a sidechain bipyridine moiety. Synthesis of SPPS-compatible amino acids resembling 140 therefore allows for the incorporation of a N,N bidentate unit into a peptide backbone, in turn unlocking metal-binding architectures with unique three-dimensional characteristics.
 |
| | Fig. 2 Classifications of amino acids: bipyridine-containing α- and ω-amino acids Y and X, naturally occurring β- and γ-amino acids A and B. | |
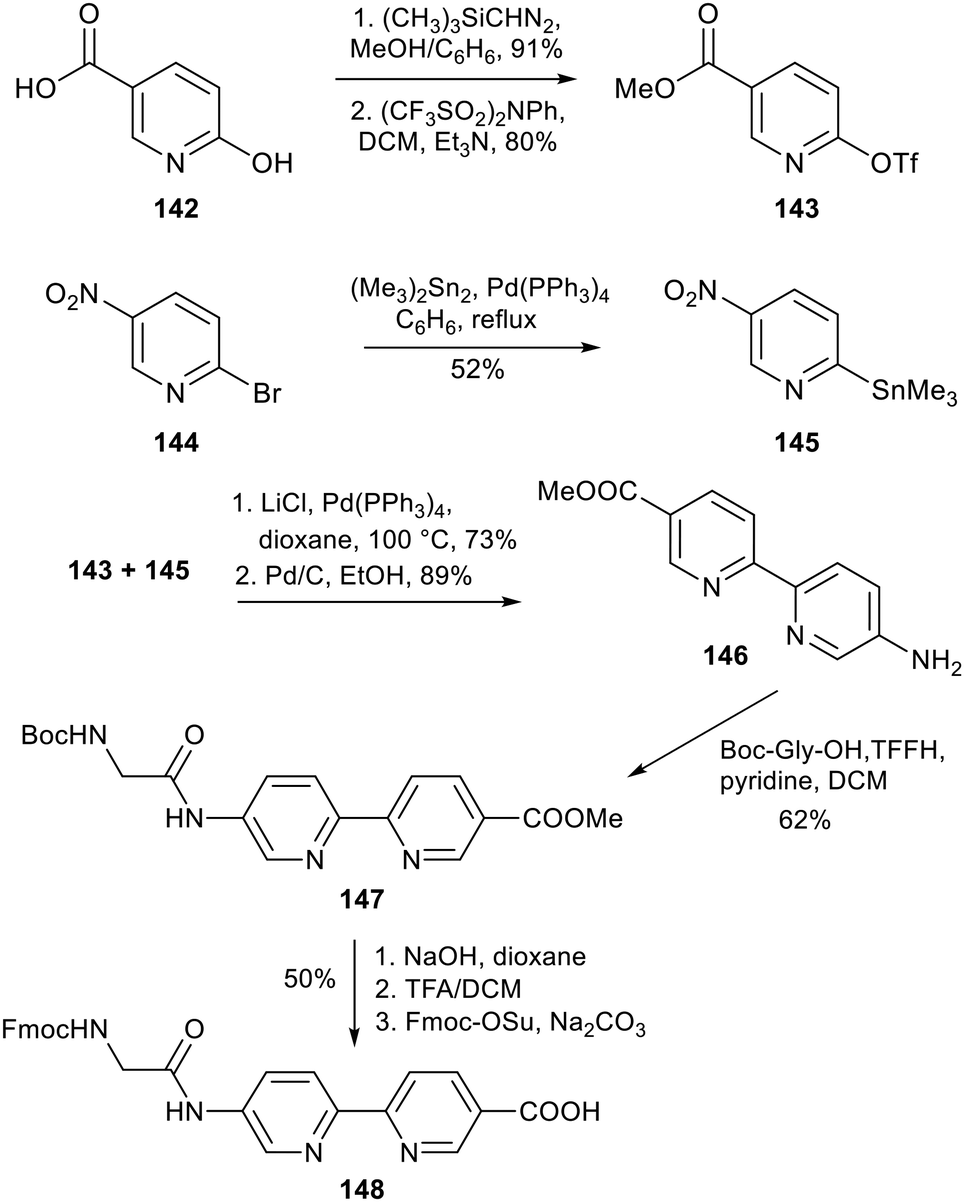
Synthesis of this type of bipyridyl amino acid was first reported by Imperiali and coworkers in 1996, who derivatised 146 into the building block Fmoc-XBp (148) for use in the design of fluorescent peptide-based metal ion sensors alongside a cyanoanthracene fluorophore Fmoc–Flu.100 The synthesis of 148 initially comprised three key stages: preparation and palladium-catalysed Stille coupling of the triflate and stannane pyridine derivatives 143 and 145, and reduction of the nitro group via catalytic hydrogenation to give 146 – the methyl ester of amino acid 148. The researchers had intended to protect and introduce the ω-bipyridine amino acid directly into a peptide, however, attempts to install an Fmoc group were unsuccessful even under forcing conditions. Due to the poor reactivity of its anilinic amine, 146 was not considered suitable for solid phase peptide synthesis, hence necessitating further modification: 146 was thus transformed into the pseudo-dipeptide 147 by coupling of Boc–glycine via in situ formation of a reactive acyl fluoride intermediate. The desired amino acid 148 was obtained following Boc removal, ester hydrolysis and installation of the Fmoc group (Scheme 29).
 |
| | Scheme 29 Synthesis of the ω-amino acid XBp containing a bipyridyl group – Imperiali and co-workers.100 | |
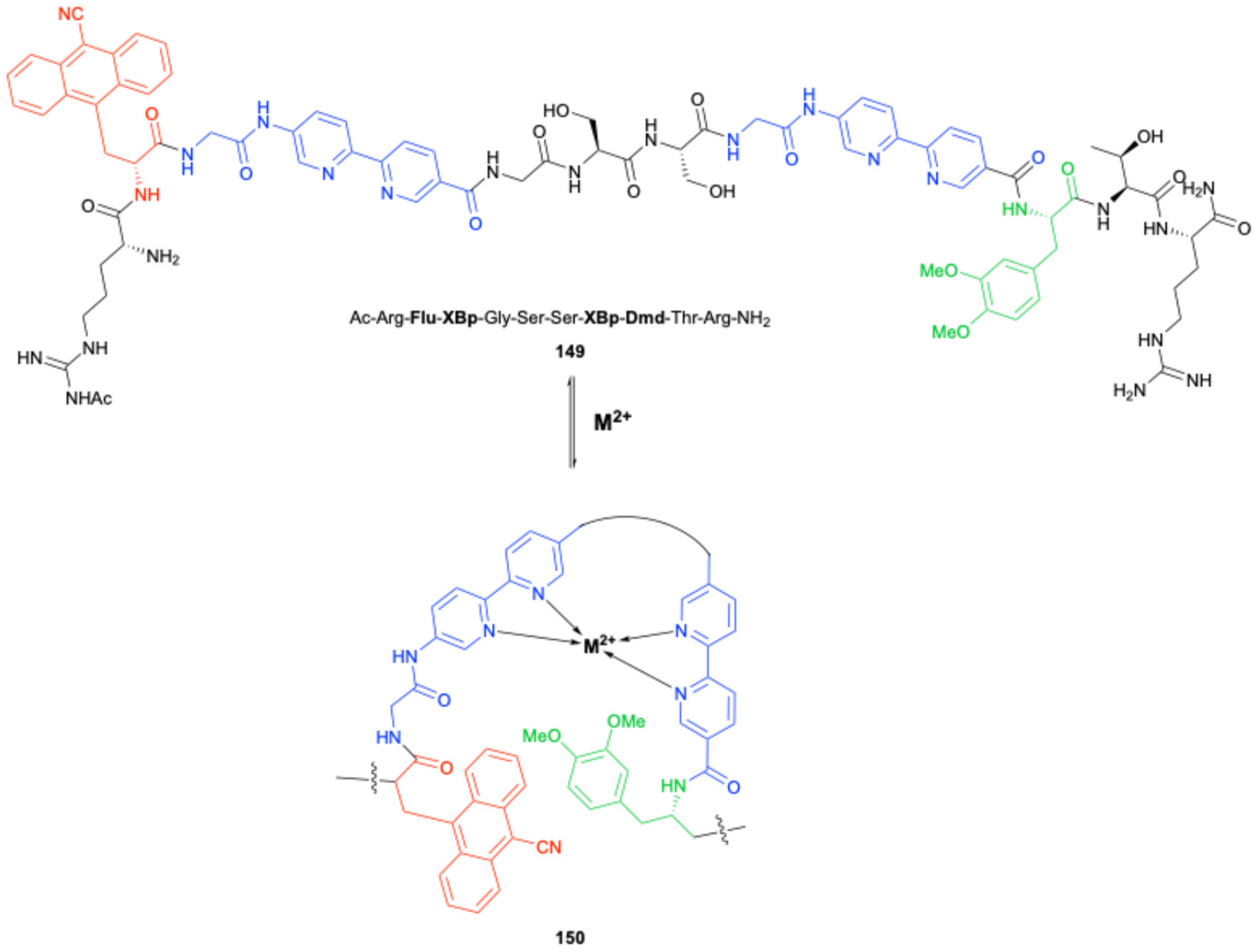
This building block (148) was in turn compatible with SPPS methodology, as shown by its successful incorporation into the peptide 149, alongside the cyanoanthracene fluorophore Fmoc–Flu Q and fluorescence quencher L-3,4-dimethoxy-DOPA (Dmd) shown in Scheme 30. The absence of metal ions, FXR03 is found in a random coil conformation and exhibits strong fluorescence on account of the Flu sidechain – upon addition of aqueous ZnCl2, binding of Zn2+ to the bipyridine units induces a conformational change that brings the fluorescent Flu sidechain in close proximity to Dmd. This in turn triggers a metal-dependent photoinduced electron transfer (PET) event from the excited state Dmd to Flu, monitorable by fluorescence emission spectroscopy.
 |
| | Scheme 30 Metal ion-induced conformational change of a metal-dependent peptide PET sensor; unnatural amino acids with fluorescent, fluorescence-quenching, and metal-binding properties are shown in red, green and blue respectively – Imperiali and co-workers. | |
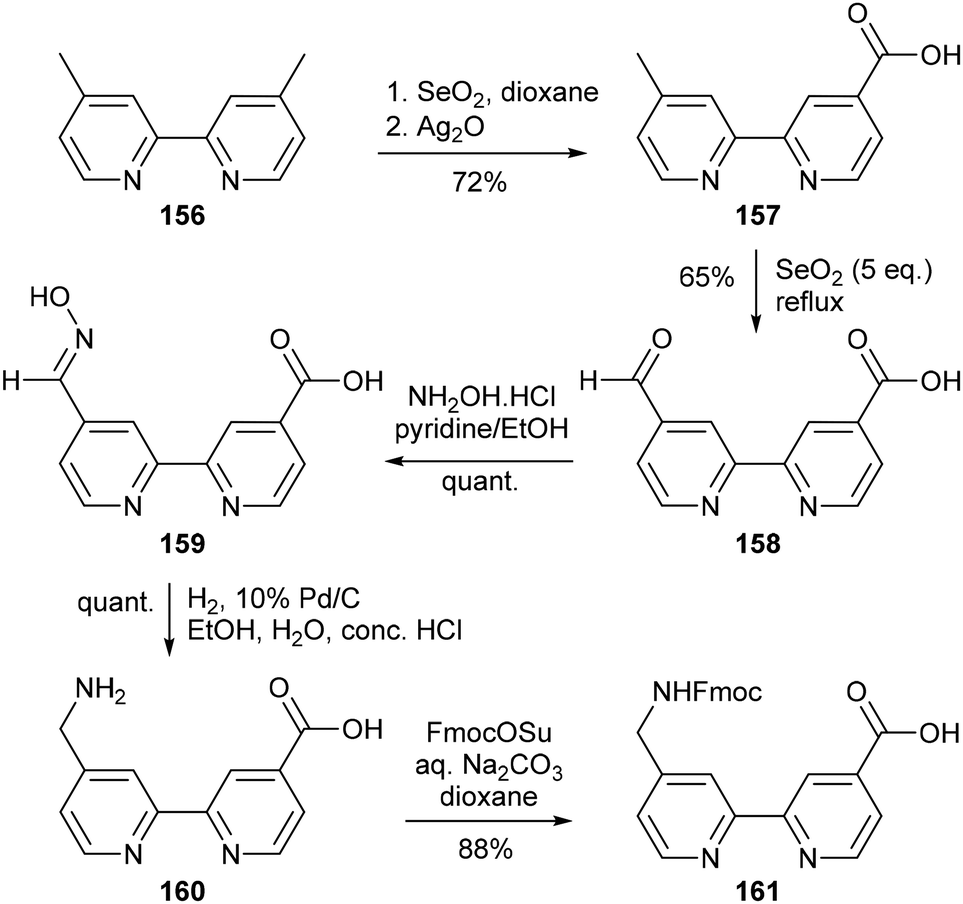
An alternative route to this bipyridyl amino acid was described by Patri and coworkers the following year, starting from the symmetrical ester diethyl 2,2′-bipyridine—5,5′-dicarboxylate 151, which was converted into 152 by treatment with 1.5 equiv. hydrazine hydrate (Scheme 31).101 Due to the inherent low solubility of carbohydrazides, it was possible to selectively form the asymmetric monocarbohydrazide (152) in 85% yield by adjusting the reaction temperature and the polarity of the ethanol/toluene solvent system. Addition of sodium nitrite to a solution of 152 in concentrated hydrochloric acid converted 152 into the corresponding carbazide 153, which formed urethane 154 upon Curtius rearrangement. From here, the desired amino acid 155 was finally obtained as the hydrochloride salt by saponification of 154. A similar Fmoc-protected derivative was reported by Erickson and co-workers in 2000, where the amino acid Fmoc–Abc (161) was obtained from 4,4′-dimethyl-2,2′-bipyridine 156 using a selective dual oxidation strategy (Scheme 32).102 This was achieved by firstly using selenium dioxide to selectively convert the 4′-methyl group into an aldehyde, followed by oxidation with Ag2O to access the mono-acid 157 from the monoaldehyde.103 Subsequent use of selenium dioxide in a fivefold excess enabled oxidation of the remaining methyl group to give 158, from which the required nitrogen functionality was installed by formation of the oxime 159 followed by catalytic hydrogenolysis to afford the target amino acid 160.
 |
| | Scheme 31 Synthesis of XBp from a symmetrical bipyridine diester – Patri and co-workers.101 | |
 |
| | Scheme 32 Synthesis of the bipyridyl amino acid Fmoc–Abc – Erickson and co-workers.102 | |
In 2004, Ishida and coworkers reported the synthesis of a bipyridine amino acid building block for the incorporation into ruthenium-binding peptides.104 As encountered previously by Imperiali,100 the poor reactivity of the aromatic amine rendered the amino acid unusable for incorporation with Fmoc peptide chemistry. Accordingly, SPPS installation of bipyridine into a peptide was facilitated using an approach involving condensation of the unreactive amine with an activated intermediate derived from a canonical amino acid, in this case alanine. The route described by Ishida accesses the desired Fmoc-protected 164 through activation of Fmoc-protected alanine (162) with thionyl chloride, in turn making the corresponding acyl chloride 163 sufficiently reactive to undergo an amide coupling with the aromatic amine of H–5Bpy–OH (140) under basic conditions (Scheme 33). The resulting dipeptide unit (164) was introduced into a peptide by standard Fmoc SPPS with HBTU and HOBt as coupling reagents to form the peptide Ac–(Ala)3–5Bpy–(Ala)3–OH (165). By using the unprotected bipyridyl amino acid and Fmoc-protected alanine, the overall synthesis is shortened as the need for Boc removal, Fmoc installation and ester hydrolysis is circumvented. This approach was later implemented in the design of an artificial metalloprotein containing a previously unreported ruthenium(II) tris(bipyridyl) complex at the core.105
 |
| | Scheme 33 Preparation of a bipyridyl dipeptide building block for Fmoc peptide synthesis – Ishida and co-workers.104 | |
In 2012, an analogous synthesis was reported by the same group, using instead the acyl chloride of Fmoc-protected leucine to couple to the bipyridine amino group (Scheme 34). This ultimately allowed for the construction of a peptide of desired sequence Ac–Ala–Leu–5Bpy–Val–Phe–Gly–NH2 (172) which was subsequently complexed to Ru2+ by refluxing the peptide in ethanol with equimolar Ru(bpy)2Cl2.106 Shortly afterwards, the same authors reported the design of Fmoc–O1PenBpy–OH (Scheme 35), another bipyidine-containing building block compatible with SPPS methodology.107 The bipyridyl unit 168 was synthesized starting from the commercially available 5,5′-dimethyl-2,2′-bipyridine 174 over six steps, and coupled to Fmoc-protected 5-amino-3-oxapentanoic acid (O1Pen 175) to give the desired product 176 in moderate yield on a multigram scale.
 |
| | Scheme 34 Incorporation of a bipyridyl dipeptide unit into a peptide for preparation of a ruthenium complex – Ishida and co-workers.106 | |
 |
| | Scheme 35 Synthesis of Fmoc–O1PenBpy–OH – Ishida and co-workers.108 | |
Terpyridines represent another class of nitrogen-containing extended heterocycle that are particularly prominent across the area of transition metal catalysis. Over the past decade, the biocompatibility of the terpyridine (terpy) functionality has been exploited for applications in biocatalysis, with examples including nucleic acid-based catalysts containing Cu(II)–terpy and Fe(III)–terpy functionalities,109a,b a terpy–Mn(II) cofactor nitrobindin conjugate with broad oxygenation capabilities,110 and a terpy–Cu(II) G-quadruplex DNA metalloenzyme for application in enantioselective Diels–Alder reactions.111 While terpy–metal complexes have been conjugated to genetically encoded proteins for use as catalysts,112 a report has yet to disclosed of direct incorporation of a terpy amino acid into such a scaffold via orthogonalized protein translation, a highly efficient in vivo approach for incorporating unnatural amino acids into a protein. This approach has so far only been applied to a small number of chelating amino acids,113a–c since the engineering of aminoacyl–tRNA synthetases for this process requires a reliable supply of the unnatural amino acid of interest, which may only be available via complex synthetic routes inaccessible to chemical biologists.
In 2019, Budisa and co-workers reported the first synthetic route to a terpyridyl-containing amino acid 185 and its binding properties to a range of bivalent metal ions (Scheme 36).114 Firstly, the terpyridyl carboxylic acid 180 was synthesized from the corresponding furan-2-yl derivative 179 using a previously reported procedure by Kühn and co-workers,115 wherein a bis-pyridyl intermediate formed through an aldol condensation of 2-furaldehyde (177) with the first equivalent of 2-acetylpyridine (178) followed by Michael addition of the second. This approach was later used by Garoufis and co-workers to obtain the multi-aromatic terpyridine-containing amino acid 187 shown in Scheme 37.
 |
| | Scheme 36 Synthesis of a terpyridine-functionalised amino acid – Budisa and co-workers.114 | |
 |
| | Scheme 37 Synthesis of a phenylterpyridyl amino acid – Garoufis and co-workers. | |
3.3. Other pyridyl amino acids
Tripodal ligands are a distinct class of polydentate ligand typically associated with particularly high metal binding affinities. Among a series of other N-donor tripodal ligands, Canary and Chiu first reported the synthesis of bispyridyl glycine residue 188 (Fig. 3) in 2003 via the reaction of glycine with picolyl chloride under basic conditions.116 In 2019, this carboxylic acid was complexed to Pd(II) and conjugated to a transferrin receptor binding peptide sequence via the N terminus, giving rise to the first example of a Pd(II) metallopeptide with anticancer activity.117
 |
| | Fig. 3 Bispyridylglycine A and a Pd complex of its conjugate to a peptide. | |
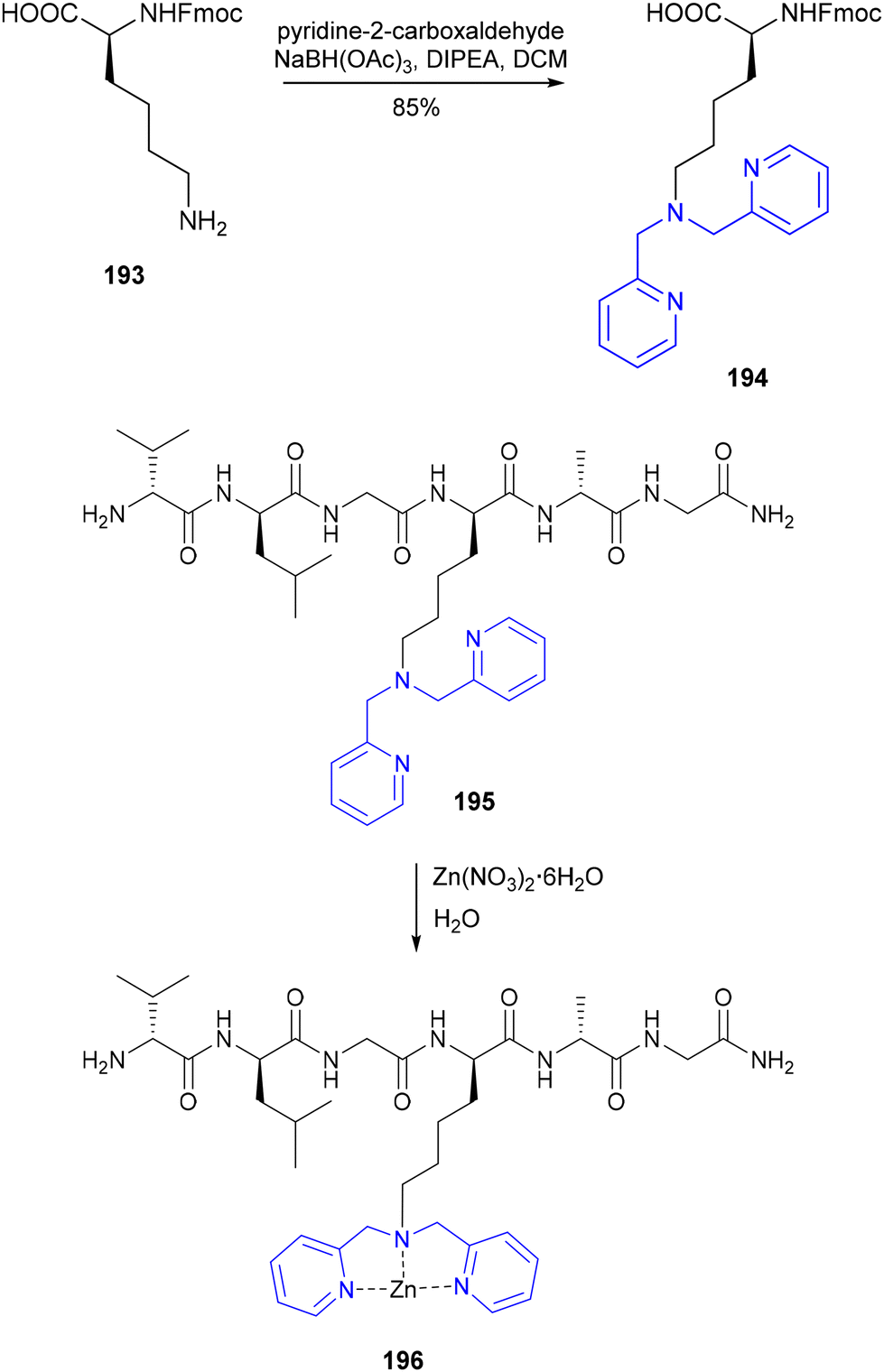
While several other examples of metal-binding amino acid-based polypyridyl ligands and related N-terminally modified metallopeptides exist in the literature,118a–c to the best of our knowledge only two examples of an amino acid building block with functional N- and C-termini have been reported. In 2007, Alsfasser and co-workers detailed the design of Boc–Asp(Bpa)OBz 192 by functionalisation of C- and N-protected aspartic acid with 2,2′-dipicolylamine 191 (Scheme 38).119 As coordination of the resulting chelating amino acid 192 to Zn2+ and Cd2+ was shown to sufficiently reduce the rotational barrier of the N-Boc amide to permit isomerisation about the C–N bond, amino acid 192 has been proposed for potential applications in the design of metal ion responsive peptide-based conformational switches. Around the same time, König and co-workers detailed the synthesis of several metal-chelating amino acids and peptides,120 including the lysine derivative 194, where a dipicolylamine moiety was installed at the ε-amine by reductive amination using NaBH(OAc)3 (Scheme 39).121194 was incorporated into a peptide using standard SPPS methodology, which formed the Zn2+ complex 196 upon stirring with an aqueous solution of a zinc nitrate salt.
 |
| | Scheme 38 Synthesis of an amino acid containing a tripodal dipicolylamine ligand functionality – Alsfasser and co-workers. | |
 |
| | Scheme 39 Synthesis of lysine derivative 194 containing a metal-binding dipicolylamine moiety (highlighted in blue) and its use in the preparation of the Zn2+-binding peptide Val–Leu–Gly-194–Ala–Gly–NH2 – König and co-workers.120 | |
Another example of a structurally unique unnatural amino acid is Boc–Daf–OMe 201, the first reported α-disubstituted glycine derivative containing a rigid bipyridine moiety, synthesized using a transamination–carboxymethoxylation strategy (Scheme 40).122 Preparation of 201 began with the conversion of the fluorenone 197, prepared by the oxidation of phenanthroline,123 into the benzyl-imine 198. Deprotonation of 198 using NaHMDS followed by addition of excess methyl chloroformate (199) effectively introduced a carbomethoxy group at the 9-fluorenyl position, eventually forming the C-protected amino acid 200 after acidic hydrolysis. Further installation of a Boc group allowed for coupling of an alanine residue to either terminus of the amino acid,124 demonstrating the possibility for the use of 201 in the construction of metal-binding peptide architectures.
 |
| | Scheme 40 Synthesis of an α-disubstituted amino acid functionalised with a rigid 2,2′-bipyridine ligand – Toniolo and co-workers.124 | |
4. Metal coordination via carbon
4.1. N-Heterocyclic carbene derived amino acids
N-Heterocyclic carbenes, commonly referred to as NHCs, represent an increasingly prominent class of neutral donor ligand. Typically formed upon C2 deprotonation of an N-alkylated imidazolium salt,125a,b the imidazole-2-ylidene moiety features extensively across the areas of organic synthesis, catalysis, and organometallic chemistry.126 Considering the nature of its sidechain, histidine has emerged as a convenient precursor for the design of imidazolium-based NHC metal complexes within the area of bioorganometallic chemistry. Given their established affinity towards various metals, several groups have investigated modes of incorporating NHC motifs into peptides as one way of creating artificial metallopeptides. This concept was first investigated in 2005 by Erker and co-workers, who first reported the synthesis of histidine-derived NHC ligands and their complexes to palladium and rhodium.127 Starting with N-and C-protected L-histidine, di-alkylation of the imidazole nitrogen atoms was initially carried out by treatment with excess Meerwein's reagent (Et3O+BF4−), however this method led to both unwanted O-alkylation, as well as racemisation of the desired product. Upon modification of the reaction conditions to instead use n-propyl bromide or isopropyl iodide with sodium bicarbonate3, alkylation proceeded cleanly to give di(n-propyl)- and diisopropyl-histidinium salts 204 and 205 respectively, both of which were confirmed to be optically active (Scheme 41). To demonstrate their suitability as metal ligands, 204 and 205 were converted into their corresponding Ag(I) complexes with Ag2O, followed by addition of either Pd(CH3CN)2Cl2 or [RhCl(cod)]2 to affect the transmetallation process.
 |
| | Scheme 41 First reported synthesis of metal-binding imidazolium amino acids from histidine – Erker and co-workers.127 | |
Shortly afterwards, Guillen and co-workers reported a method enabling the introduction of two different alkyl groups at the imidazole nitrogen atoms, also referred to as the wingtip substituents.128 Using an approach based on the methodology of Cohen and coworkers,129 histidine methyl ester was converted into cyclic urea intermediate 208, wherein both the amino and N2 ring nitrogen atoms are protected, allowing for selective methylation solely at the N1 position (Scheme 42). Subsequent base-mediated ring opening with tert-butanol and treatment with n-bromobutane gave the desired chiral imidazolium salt 211. The scope for its application in peptide chemistry was demonstrated by HATU-mediated amide coupling to alanine: acidic hydrolysis of 211 to free amine 212 allowed for coupling to N-Boc–alanine, while coupling to the C-terminus was achieved through saponification of 211 into 214 followed by reaction with the tert-butyl ester of alanine, ultimately forming dipeptides 213 and 215 without racemization.
 |
| | Scheme 42 Synthesis and amide coupling of asymmetrically alkylated histidine-derived amino acids – Guillen and co-workers.128 | |
Albrecht and co-workers have used similar approaches to synthesize a series of histidine-derived metal-binding ligands with the aim of creating bio-inspired NHC-containing catalysts. Their first report in 2011 detailed a route to symmetrically and non-symmetrically alkylated histidinium salts: N,N′-dimethylated 217 was formed by refluxing protected L-histidine 216 with excess methyl iodide, while 219 was accessed through selective Nε alkylation with isopropyl iodide, followed by methylation of the remaining imidazole nitrogen (Scheme 43).130 These racemic histidine derivatives were subsequently transmetallated to give ruthenium complexes which were shown to exhibit moderate catalytic activity in the transfer hydrogenation of benzophenone.
 |
| | Scheme 43 Synthesis and ruthenium complexation of racemic histidine derivatives – Albrecht and co-workers.130 | |
The following year, the same group reported an enantioselective synthesis of the N,N′-di-methylated amino acid 222, using the previously reported methodology of Guillen and co-workers (Scheme 42).128 This amino acid was then used to form complexes with both iridium and rhodium, the latter of which consistently displayed high conversion in the catalytic hydrosilylation of ketones.131 Albrecht and co-workers further showed that incorporation of amino acid 222 into the tetrapeptide Boc–Met–Ala–Ala-X–OMe (223) followed by metalation with [RhCl(cod)]2 gives rise to a macrocyclic chelate (224) that can serve as an efficient hydrosilylation catalyst precursor (Scheme 44).132 Comparison with a tri-alanyl histidinium peptide indicated that chelation of a rhodium centre by histidinium and methionine residues via a C,S-bidentate binding mode enhances catalytic turnover.133 In 2021, they reported an Fmoc-protected analogue for use as a building block for solid phase peptide synthesis. Metallation of the resulting peptide scaffold with iridium generated a protein-like assembly furnished with a metal–carbene active site, which displayed pseudo-reductase activity in the hydrogenation of acetophenone.134
 |
| | Scheme 44 Enantioselective synthesis of histidine derivative X and its incorporation into a chelating peptide for Rh catalysis – Albrecht and co-workers.132 | |
The amino acids and related catalytic metal complexes discussed so far have only included histidine-derived NHC ligands with simple alkyl wingtip substituents. Given the well-established influence of a NHC ligand's wingtip substituents on the catalytic activity of its corresponding metal complex,135a–c it has been reasoned that expanding upon the structural diversity of existing NHC-containing amino acids would broaden their applicability within transition metal-based catalysis. Seeking to expand the diversity of NHC-containing amino acids, Elsevier and co-workers reported new methodology leading to novel benzyl- and aryl-substituted histidinium salts in 2015, which were subsequently investigated as ligands for Pd(II)-catalysed alkyne transfer semihydrogenation.136 Starting from Boc-protected histidine methyl ester 202, the symmetrically substituted N,N-dibenzyl histidinium bromide 225a was obtained quantitatively through refluxing with benzyl bromide in the presence of sodium bicarbonate. This direct benzylation approach was also used with picolyl bromide; despite the formation of several byproducts, the bis-pyridyl histidinium 225b was isolated in relatively high yield (Scheme 45). In order to access non-symmetrically substituted derivatives, an adapted version of the Chan–Lam–Evans reaction was used.137 While attempting N-arylation of Cbz-protected histidine methyl ester under original Chan–Lam–Evans conditions was low yielding, Elsevier and co-workers found that the methoxyphenyl and mesityl N-arylated histidine derivatives 226 could be accessed in reasonable yield by adapting the improved protocol of Campagne and co-workers,138 specifically by increasing the reaction times and carrying the reactions out at room temperature. These monosubstituted products were then subject to alkylation with a variety of electrophilic halides, ultimately yielding histidinium salts 227a–c in yields between 51% and >99% (Scheme 49).
 |
| | Scheme 45 Synthesis of aryl-substituted histidinium amino acids via – Elsevier 2015.136 | |
Over the past decade, methodology for the synthesis of NHC-functionalised amino acids has diversified beyond the general approach of alkylated histidine derivatives. In 2014, Gilbertson and co-workers reported the design of 231, a building block containing both imidazolium and proline moieties, for use in the construction of rhodium complexes of NHC-containing peptides.139 Starting with C- and N-protected hydroxyproline 228, conversion to the corresponding electron-withdrawing triflate 229 created a substrate suitable for the direct quaternisation of N-mesityl-substituted imidazole (230), ultimately generating the desired amino acid 231 (Scheme 46). The applicability of 231 in peptide synthesis was demonstrated using a standard EDC/HOBt amide coupling protocol at both the C-terminus and proline amine, following removal of the Cbz protecting group, with both the dipeptide 232 and tripeptide 233 effectively complexed to rhodium following transmetallation of the corresponding Ag(I) complexes.
 |
| | Scheme 46 Synthesis of Rh-binding proline-derived amino acids and peptides – Gilbertson and co-workers.139 | |
In 2020, Metzler-Nolte and co-workers expanded upon the toolbox for peptidic metal–NHC complexes (Fig. 4) including rhodium and iridium complexes using derivatives of aspartic acid (n = 1) and glutamic acid (n = 2),140 as well as thiazolylalanine, with which ruthenium complex 236 was prepared.141 These imidazolium-functionalised amino acids were incorporated into peptides via SPPS and successfully transformed into metal–NHC complexes using a standard transmetallation protocol, giving metallopeptide complexes of the type shown in Fig. 4.
 |
| | Fig. 4 Metal complexes of peptides with NHC-containing amino acids derived from aspartic (n = 1) or glutamic acid (n = 2) and thiazolylalanine 236. | |
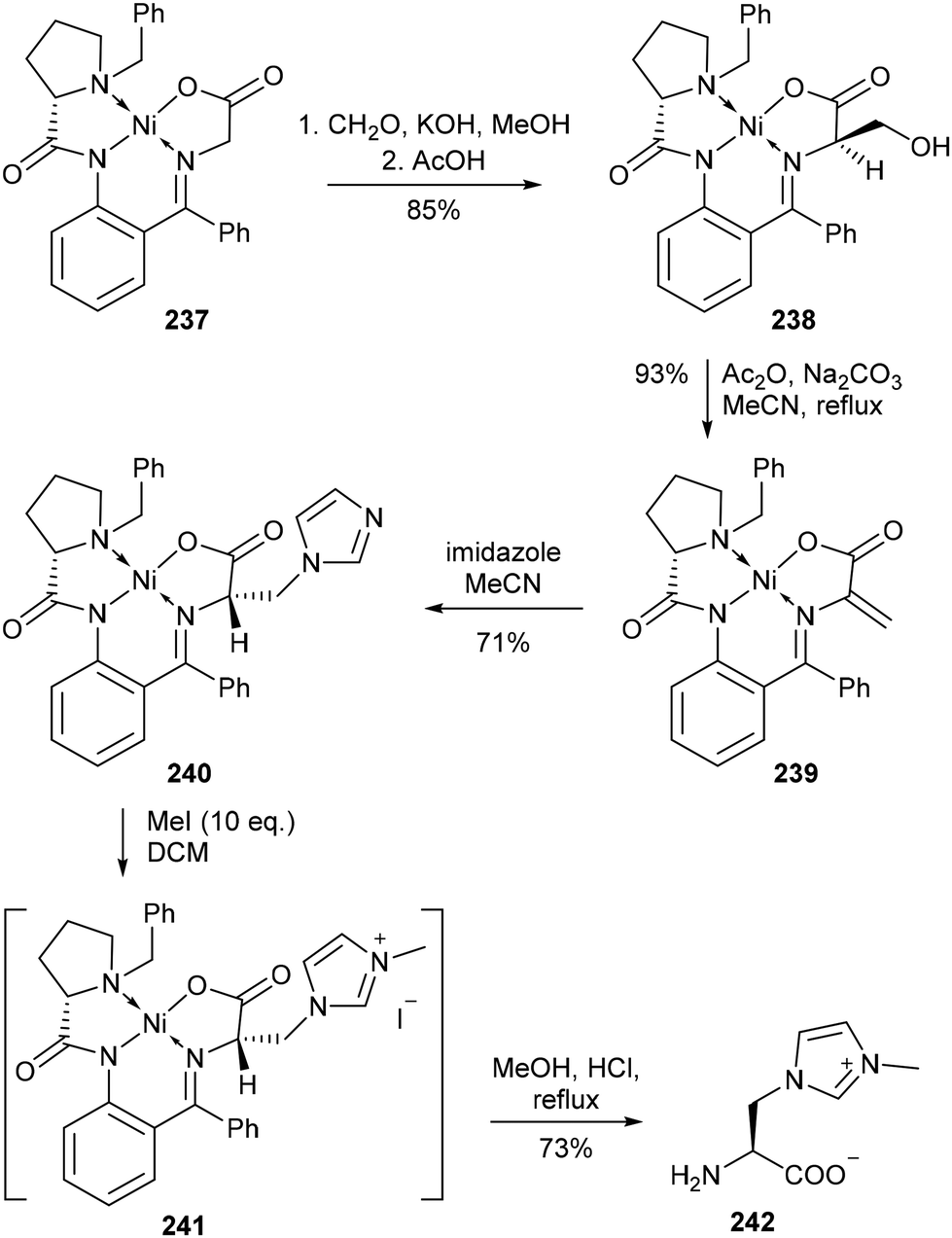
In 2008, Belokon and co-workers reported the synthesis of 242, a novel imidazolyl amino acid with unique connectivity, wherein the amino acid functionality is attached via an imidazole nitrogen (Scheme 47).142 This amino acid was accessed using an approach based on their seminal 1986 report in which the use of Ni(II) complexes of glycine and [N-(benzylprolyl)amino]benzophenone Schiff bases for enantioselective amino acid synthesis was first detailed.143 Following the formation of 238 through base-catalysed condensation of Ni(II)–glycine complex 237 with formaldehyde, dehydration via acetic anhydride with catalytic sodium carbonate gave dehydroalanine complex 239, furnished with a carbon–carbon double bond suitable for a Michael addition reaction with imidazole as a nucleophilic donor component.144 From here, the resulting addition product 240 was treated with methyl iodide to give the corresponding ionic imidazolium complex 241, from which the desired amino acid 242 was obtained by refluxing a methanolic solution of the Ni(II) complex with hydrochloric acid. Reaction of 242 with Boc2O gave the N-Boc-protected amino acid, which was successfully converted into the corresponding silver complex using a routine procedure.145 Shortly after, the same group expanded upon this method both by introducing a variety of substituted imidazole components at the Michael addition step and by varying the alkyl halide in the subsequent alkylation step, however, the coordination of these amino acids to metals was not investigated.146
 |
| | Scheme 47 Synthesis of an imidazolyl amino acid using a chiral masked glycine Ni(II) complex – Belokon and co-workers.142 | |
Since the pioneering work of Belokon and co-workers, the use of Ni(II) complexes of amino acid Schiff bases has emerged as a prominent strategy for the asymmetric synthesis of structurally diverse unnatural α-amino acids, including but not limited to sterically strained,147a,b cyclic,148a,b fluorinated,149a,b fullerene-derivatised,150 spin-labelled,151 and isotopically labelled examples.152a,b Moreover, this methodology has seen significant expansion in scope over the past decade. While the original reports detailed transformation of the glycine moiety by aldol and Michael addition processes,143,144 numerous other examples of reaction types have since been demonstrated, including alkyl halide alkylations, dialkylations, Mannich reactions,153a–c and more recently, Heck,154 Suzuki,155 reductive olefin coupling,156 allylic alkylation,157 cyclopropanation,158 electrochemical thioalkylation159 and radical perfluoroalkylation reactions.160 In-depth discussions of this methodology can be found in the following reviews.161a,b
The use of nickel templating has also widely been applied to the synthesis of amino acids containing metal-binding groups. In 2009, Burck and co-workers utilised the prototypical Belekon template 243a,143 the Ni(II) Schiff base complex of (S)-2-[N-(benzylpropyl)-amino]benzophenone, to create a chiral environment allowing for enantioselective alkylation with a phosphine-containing reagent with varying lengths of alkyl linker (Scheme 48).162 Installation of an Fmoc group into the sulphide-protected derivatives 244 enabled the authors to incorporate these amino acids into cyclic peptides based on the natural product Gramicidin S. Subsequently, Rh(I) and Pd(II) complexes based upon the bisphosphine-functionalised decapeptide were demonstrated to be catalytically active in asymmetric hydrogenation and allylic substitution reactions, showing moderate ee of up to 52% in the hydrogenation of dehydroalanine.163 More recently, Belokon and co-workers reported an efficient synthesis of 1,4-substituted 1,2,3-triazole containing amino acid 245 by combining their Ni(II) template approach with the well-established copper(I)-catalysed azide alkyne cycloaddition (CuAAC) click reaction.164 Following derivatisation with an alkyne side chain, the prototypical Ni(II)–glycine complex 243a was mixed with an alkyl azide and catalytic copper iodide under basic conditions to give the corresponding triazole, which was then subjected to acidic hydrolysis to generate the desired amino acids in high yield (93–96%) and ee (>99%).165 Although their metal-coordinating behaviour was not explored by the authors, the chelating ability of triazole-functionalised amino acids and peptides is well-documented in the literature,166a,b particularly in the context of biomolecule radiolabelling for theranostics applications.167a,b In 2019, Jamieson and coworkers used a chiral Ni(II) Schiff base complex containing a fluorinated analogue of the original BPB chiral auxiliary (243b) in an alkylation reaction with 6-bromo-N-(tert-butoxy)hexanamide.168 Subsequent decomplexation and installation of an Fmoc group afforded Fmoc–Asu(NHOtBu)–OH (246) which was used to construct peptides with zinc-binding hydroxamic acid sidechains for evaluation as HDAC inhibitors.
 |
| | Scheme 48 Application of the Belekon template for the synthesis of amino acids containing metal-binding groups. | |
Aside from catalysis, N,N-functionalised NHC amino acids have been shown to hold potential in biomedical applications, particularly in the development of biocompatible gold nanoparticles (AuNPs). In 2017, Reithofer and co-workers first demonstrated the applicability of NHC ligands derived from N-Boc histidine methyl ester in the stabilisation of AuNPs with optical activity.169 Given the importance of aqueous stability and solubility in the design of biocompatible nanoparticles, the group sought to modify their histidine-derived AuNP. By instead forming the Au(I) complex with N-acetyl protected L-histidine ethyl ester as a precursor, followed by N-alkylation and saponification of the ester, the authors were able to create a water-soluble and pH responsive variant of the NHC-stabilised AuNP.170 These complexes were subsequently evaluated for their pharmacological properties, including toxicity and relative stability in human serum, using inductively coupled plasma mass spectrometry (ICP-MS) methodology.171 In 2020, Kühn and co-workers reported the synthesis of a series of bis-NHC complexes derived from histidine through a benzylation and metal complexation sequence (Scheme 49).172 Subsequent biological evaluation indicated a link between the nature of the wingtip substituent and the antiproliferative activity of the complex, wherein a greater lipophilicity led to greater potency against a selection of cancer cell lines. Notably, multiple complexes including 249 exhibited antiproliferative activity towards MCF7 (breast) cells significantly greater than the widely used metallodrug cisplatin (IC50 = 21 ± 6.3 μM) and comparable to that of auranofin (IC50 = 0.28 ± 6.3 μM).
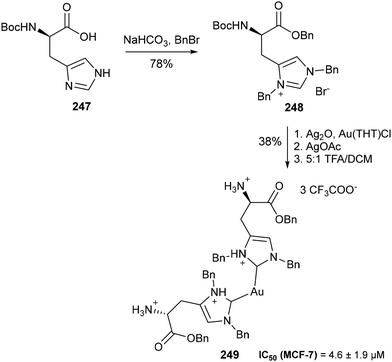 |
| | Scheme 49 Synthesis of a histidine-derived Au(I)-bis NHC complex with potent antiproliferative activity against the MCF7 cell line – Kühn and co-workers.172 | |
4.2. Metallocene amino acids
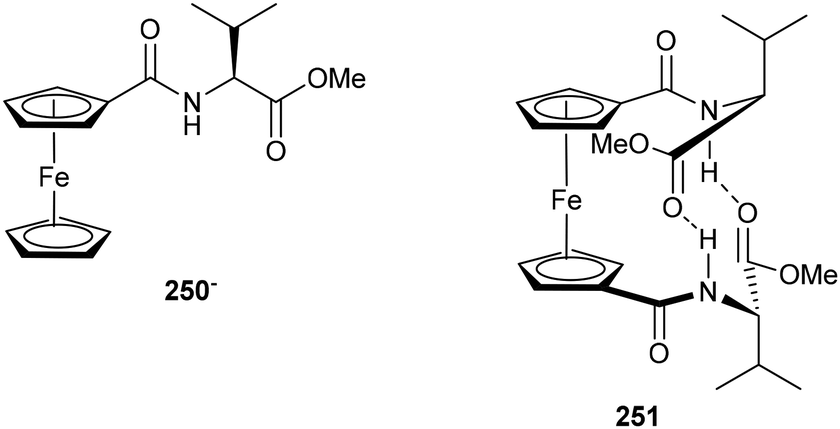
The incorporation of cyclopentadienyl (Cp) rings into peptides has emerged as a promising strategy in the design of β-turn mimetics for the development of functional peptide β-sheet systems. This concept was first recognized in 1996 by Herrick and co-workers, who prepared mono- and bis-amino acid ferrocenyl complexes by reacting the methyl ester hydrochloride of the amino acid with 1,1′-ferrocene mono- or di-carbonyl chloride respectively.173 Subsequent spectroscopic analysis showed lowering of N–H amide and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O ester IR stretches for the bis-compared with the corresponding mono-complexes, as well as deshielding of the 13C NMR resonance observed for the ester carbonyl carbon moving from the mono- to bis-complex, with the carbon of the corresponding amide group remaining virtually unaffected, pointing towards the formation of hydrogen bonding interactions between the NH proton and ester carbonyl, as depicted in Fig. 5.
O ester IR stretches for the bis-compared with the corresponding mono-complexes, as well as deshielding of the 13C NMR resonance observed for the ester carbonyl carbon moving from the mono- to bis-complex, with the carbon of the corresponding amide group remaining virtually unaffected, pointing towards the formation of hydrogen bonding interactions between the NH proton and ester carbonyl, as depicted in Fig. 5.
 |
| | Fig. 5 Mono- (250) and bis- (251) valine ferrocenyl complexes – Herrick and co-workers.173 | |
Modification of the amino acid N-terminus with a metallocene fragment has been widely reported since the first example by Schlögl and co-workers in 1957 for use in peptide labelling and construction of beta turn mimics.174 Fca (254) initially reported in 1998 by Butler and co-workers, represents the first example of a metallocene-containing unit containing both an available N- and C-terminus for peptide bond formation. Among a series of heterosubstituted aminoferrocenes, Fca was synthesized starting from 1,1′-dibromoferrocene 252 (Scheme 50A).175 The corresponding bromo-1′-aminoferrocene 253 was obtained first by monolithiation with nBuLi, followed by reaction with 0.4 equivalents of O-benzylhydroxylamine. Following further addition of excess nBuLi, the reaction was quenched using solid carbon dioxide, which was accompanied by the immediate precipitation of the target amino acid 254. In 2002, Kovac and co-workers proposed an alternative synthesis of Fca starting from the ferrocenyl acid–ester 255 (Scheme 50B). Installation of the azide group to give 256 enabled the formation of carbamate 257via a Curtius rearrangement upon heating with benzyl alcohol, which was followed by alkaline hydrolysis of the methyl ester group to give the acid–carbamate 258. Finally, hydrogenolysis was performed using palladium on carbon to transform the carbamate moiety into an amine group to give the desired amino acid 254,176 which was later used to construct medicinally-relevant β-lactam-containing ferrocenyl peptides exhibiting stabilisation via a novel cooperative three-hydrogen-bond network.177 Metzler-Nolte and coworkers reported the first example of incorporating Boc-protected Fca 259 into an oligopeptide, demonstrating its compatibility with typical peptide coupling conditions via both the N- and C-termini (Scheme 51).178
 |
| | Scheme 50 Synthesis of 1′-aminoferrocene-1-carboxylic acid by (A) lithiation and carboxylation, and (B) Curtius rearrangement and hydrogenolysis.175,176 | |
 |
| | Scheme 51 Incorporation of Fca into a peptide via both C- and N-termini – Metzler-Nolte and co-workers.178 | |
Since the synthesis of Fca (254) a range of structurally diverse amino acids containing the ferrocene moiety have emerged in the literature. The ferrocenophane amino acid 262 (Scheme 52) is an example where the amino and carboxyl groups are homoannularly substituted,179 meaning these two functionalities are connected to the same Cp ring, in contrast to Fca described previously. Further examples of related amino acids include the carbocyclic 265 and alkyl-linked 266 derived from glutamic acid (Fig. 6) – both of which were prepared in N-protected forms and successfully incorporated into peptides via solid phase and solution phase methods respectively.180a,b
 |
| | Scheme 52 Incorporation of a ferrocenophane amino acid into a peptide via both C- and N-termini – Erker and co-workers.179 | |
 |
| | Fig. 6 Examples of structurally diverse ferrocenyl amino acids used for peptide synthesis.180a,b | |
5. Complex heterocyclic
Aside from the pyridyl- and extended pyridyl-systems described in section 3, numerous additional examples of heterocyclic ligands exist in the literature encompassing a mixture of coordinating heteroatoms and denticities. One notable example is 8-hydroxyquinoline, a medicinally-relevant scaffold comprised of a pyridine ring fused to benzene, with a single hydroxyl substituent at a particular location around the ring.181 In the context of peptide chemistry, the first example of an amino acid synthesized with this functionality was reported by Imperiali and co-workers in 1998.182 The 2-substituted amino acid 271 was synthesized with high diastereoselectivity starting from 8-hydroxy-2-methylquinoline 267, using asymmetric alkylation methodology developed by Myers and co-workers involving pseudoephedrine glycinamide as a chiral auxiliary (Scheme 53).183 To begin with, 267 was converted into the corresponding bromide 268 using radical bromination via the protected benzenesulfonate ester. The chiral auxiliary (269), prepared by the selective N-acylation of pseudoephedrine with the mixed anhydride of N-Boc–glycine, was then incorporated to introduce the desired L-configuration to the amino acid, which was subject to alkaline hydrolysis to ultimately give the Fmoc-protected derivative 271 for incorporation into a peptide. Additionally, a 5-substituted analogue was prepared via the same method shown in Scheme 53 using the 5-methyl starting material derivative – these two hydroxyquinoline-containing amino acids were subsequently used for the synthesis of short peptides that displayed significant selectivity for Zn(II) over a selection of other divalent metals, demonstrating potential for application in the design of a Zn(II) chemosensor.
 |
| | Scheme 53 Asymmetric synthesis of an amino acid containing the 8-hydroxyquinoline scaffold – Imperiali and co-workers.182 | |
While 271 was found to show good selectivity for Zn(II), a residue with a longer excitation wavelength was desired to facilitate the construction of a sensitive fluorescent sensor capable of monitoring Zn(II) concentrations in real time. Imperiali and co-workers addressed this by synthesizing the phenyl-substituted derivative 276via a Suzuki-type coupling of benzeneboronic acid to the brominated hydroxymethylquinoline 272, followed by asymmetric alkylation with N-(diphenylmethylene)glycine tert-butyl ester in the presence of a phase transfer catalyst (Scheme 54).184 As expected, extension of the conjugated system in the metal-binding residue induced a red shift of fluorescence emission absorption bands to 269 and 391 nm compared with 251 and 357 nm for an analogous peptide containing amino acid 271.
 |
| | Scheme 54 Synthesis of a hydroxyquinoline-containing amino acid with a phenyl substituent – Imperiali and co-workers.184 | |
Aside from the above examples, a racemic 3-substituted hydroxyquinoline alanine derivative (8HQ–3Ala) has been synthesized by Schultz and co-workers by radical bromination followed by formation and hydrolysis of a malonate (Scheme 55).185 The amino acid (279) was then genetically encoded into E. coli and used for the incorporation of a Zn2+ binding site into O-acetylserine sulphydrylase.
 |
| | Scheme 55 Racemic synthesis of a 3-substituted amino acid containing the 8-hydroxyquinoline scaffold – Schultz and co-workers.185 | |
In 2022, Banwell and co-workers reported a stereodivergent route for accessing the enantiopure forms of the 3-substituted hydroxyquinoline amino acid.186 Their approach centred around the rhodium-catalysed asymmetric hydrogenation of a dehydroamino acid precursor, a widely-precedented strategy in the literature used for obtaining enantiomerically enriched amino acids.187 Using methodology reported by Meth-Cohn for the synthesis of quinolines,188 acetanilide 280 was treated with DMF and phosphorus oxytrichloride to obtain the O-protected hydroxyquinoline scaffold 281 containing an aldehyde moiety. From here the required dehydroamino acid (283) was prepared via the azlactone method, where N-acetyl glycine cyclises and condenses with aldehyde 281 to give the oxazolone 282, which is converted into 283 through ring opening with sodium acetate in methanol (Scheme 56). A rhodium(I) complex combined with a DuPHOS-type ligand was used to catalyse the hydrogenation of 283 into 284 with 98% ee, which was then subject to Pd-catalysed reductive dichlorination to give the N- and C-protected amino acid 285. Although a successful deprotection procedure, treatment of 285 with aqueous HBr led to trace impurities arising from unwanted electrophilic bromination of the phenolic ring, therefore purification was carried out by preparative reversed-phase HPLC to obtain 286 as a bis-TFA salt in 44% yield.
 |
| | Scheme 56 Asymmetric synthesis of 8HQ–3Ala via Rh-catalysed hydrogenation of a dehydroamino acid precursor – Banwell and co-workers.186 | |
Benzoxazole – a heterocyclic aromatic system containing a five-membered oxazole fused to a phenyl ring – represents another important scaffold on account of its diverse bioactivity and propensity towards complexation with a wide range of metals.189 Numerous examples of amino acids containing this moiety have been reported in the literature, following the first synthesis of a benzoxazol-5-yl alanine derivative reported by Guzow and co-workers involving the catalytic reduction of 3-nitro-L-tyrosine 287, formation of a Schiff base (288) and cyclisation into benzoxazole 289 in the presence of lead tetraacetate as an oxidising agent (Scheme 57).190
 |
| | Scheme 57 Synthesis of a benzoxazol-5-yl alanine derivative – Guzow and co-workers.190 | |
This general approach based on the oxidative cyclisation of a Schiff base formed from the reaction of a tyrosine derivative with various aldehydes has been utilised in the design of fluorescent metal sensors. Examples of metal-binding moieties that have been incorporated into benzoxazole amino acids include quinoxaline,191 thiophene,192 and imidazole,193 as highlighted in Fig. 7.
 |
| | Fig. 7 Examples of amino acids containing benzoxazoles functionalised with various metal-binding groups (shown in blue). | |
In 2011, Costa and co-workers reported the design of a series of peptidic metal-ion sensors featuring a benzoxazole chromophore with high sensitivity towards Hg2+ ions.194 The peptide 293 (Fig. 8), synthesized by carbodiimide-mediated coupling of thiophene benzoxazole amino acid 291 to a protected Cys–Ala dipeptide, was able to detect both Hg2+ and Ag+ ions in water when inserted into silica core/PEG shell nanoparticles, demonstrating the potential of such nanoarchitectures as sensitive metal ion sensors in a biological setting.
 |
| | Fig. 8 A benzoxazole-containing fluorescent peptide for the sensing of metal ions – Costa and co-workers.194 | |
An assortment of other unnatural amino acids containing diverse metal-binding heterocycles has been generated from protected 4-formyl phenylalanine 294 as a starting material. Selected examples, summarised in Scheme 58, include the pyridyl-containing trriarylimidazole amino acid 296,195 benzimidazole derivative 298,196 and hydrazone-containing one 300.197
 |
| | Scheme 58 Synthesis of various amino acids with metal sensing properties starting from an aldehyde-functionalised phenylalanine derivative – Costa and co-workers.195–197 | |
6. Macrocyclic
Cyclen is a nitrogen-containing macrocycle known for its strong, multi-dentate, coordinating abilities towards a broad range of metal ions, including both transition metals and lanthanides. In 2003, Suh and co-workers reported the Cu2+- and Co2+-binding cyclen-functionalised lysine derivative 302, prepared by the attachment of a Boc-protected cyclen moiety to the ε-amino group of N-acetylated lysine (Fig. 9).198 In a similar fashion, König and co-workers synthesized a glutamic acid derivative functionalised with a cyclen group via the sidechain carboxylic acid group – the resulting amino acid was further modified with an Fmoc group to facilitate subsequent peptide coupling.199
 |
| | Fig. 9 Cyclen-functionalised derivatives of lysine (302 – Suh and co-workers)198 and glutamic acid (301 – König and co-workers).199 | |
The homoserine analog 306, reported by Yu and co-workers, was prepared by first converting the hydroxyl group of N- and C-protected homoserine into the corresponding bromide 305via the activated tosylated, followed by alkylation with cyclen under reflux and N-Boc protection of the cyclen amines (Scheme 59). Alkaline hydrolysis afforded the desired amino acid 306, which was successfully coupled to L-alanine methyl ester using DCC and HOBt to give the dipeptide in 75% yield.200 In 2006, König and co-workers reported the first application of a Negishi coupling using p-iodoaniline derivatives (Scheme 60).201 Among the several examples of phenylalanine derivatives was the synthesis of cyclen-functionalised 310, where the aryl iodide partner 309 was produced by reacting 1-bromomethyl-4-iodobenzene with cyclen under basic conditions.
 |
| | Scheme 59 Functionalisation of the L-homoserine sidechain with cyclen – Yu and co-workers.200 | |
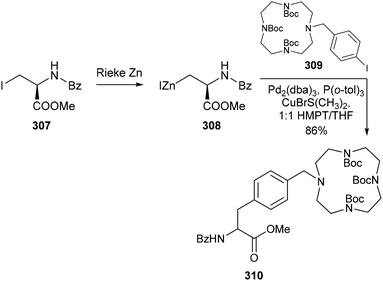 |
| | Scheme 60 Synthesis of a phenylalanine derivative containing a cyclen moiety in the sidechain by Negishi coupling – König and co-workers.201 | |
In 2007, König and co-workers reported the synthesis and incorporation of the bis-cyclen amino acid 312 into a polyalanine peptide (Scheme 61).120 The triazine-bis-cyclen structure 311 was prepared from Boc-protected cyclen and trichlorotriazine as previously reported,202 to which a lysine unit was appended via a nucleophilic aromatic substitution reaction to form the target Fmoc-protected amino acid 312. By using the coupling agents HOAt and HATU with collidine as a base, 312 was successfully coupled to alanine residues at both termini, giving rise to peptide 313 capable of binding two Zn2+ ions via the cyclen moieties.
 |
| | Scheme 61 Preparation of a peptide containing a bis-Zn2+-bis-cyclen functionality – König and co-workers.120 | |
A closely related aza-crown ether, triazacyclononane (tacn), has also been used in the synthesis of metal-binding peptides. The first example of a tacn-functionalised amino acid was reported by Scrimin and co-workers in 1999, wherein the N-Cbz protected amino acid 316 was obtained by the reaction of the doubly protected triazacyclononane 314 with the β-lactone form of L-serine (315) in 80% yield (Scheme 62).203 When incorporated twice into a helix-forming heptapeptide, this residue enabled the formation of a dinuclear zinc complex that showed catalytic activity both in the intramolecular transesterification of a RNA model substrate and in the hydrolysis of plasmid DNA,204a,b with cooperativity observed between the two metal ions. The same group has also reported the design of an artificial metallonuclease with a well-defined helix-loop-helix tertiary structure, where the extent of helical content was dependent on the coordination of four units of the amino acid 316 to zinc ions.205
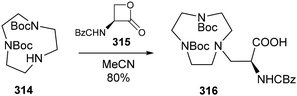 |
| | Scheme 62 Preparation of a triazacyclononane amino acid – Scrimin and co-workers.203 | |
The crown ether is a unique heterocyclic scaffold typically characterised by repeating ether linkages, although substitution of one or more oxygen atoms with other heteroatoms, typically nitrogen and/or sulphur, are well documented. Given their flexible macrocyclic nature, crown ethers are able to interact with a wide range of metal ions with high affinity on account of the chelate effect. Additionally, the tunability of the crown ether cavity size allows for highly specific binding of variously-sized metal cations, e.g. 18-crown-6 has a particularly high affinity for K+, while 15-crown-5 accommodates the smaller Na+ ion. These properties make the crown ether particularly suitable for the design of artificial receptors and chelation-based chemosensors.206 In the area of peptide chemistry, the incorporation of crown ethers has been explored in the design of artificial membrane ion channels,207a,b as well as a means of constructing antimicrobial peptides with cell permeabilising properties.208
In addition to the examples shown in Scheme 58, the aldehyde-functionalised phenylalanine 294 has also been used as a precursor for the preparation of amino acids containing a crown ether moiety. In 2016, Costa and co-workers reported the synthesis of a phenylalanine derivative furnished with a 15-crown-5 macrocycle by the reaction of the aldehyde 294 and the 4-amino-5-nitro aryl crown ether species 317 in the presence of excess sodium thiosulphate (Scheme 63A).209 To further tune the electronic properties of the amino acid, the authors designed further analogues incorporating a thiophene spacer unit. This was achieved first via the Suzuki coupling of para-brominated phenylalanine 319 and the boronic acid of 5-formylthiophene (320) to form aldehyde 321, before the addition of the crown ether reagent 317 as seen in Scheme 63B.
 |
| | Scheme 63 Synthesis of crown ether phenylalanine derivatives starting from (A) 4-formyl phenylalanine, and (B) a thiophene-containing phenylalanine derivative formed via Suzuki cross-coupling – Costa and co-workers.209 | |
In 2023, the same group reported a new series of structurally diverse amino acids for use as fluorometric sensors of metal ions, applying similar methodology as previously described. Comprising a phenylalanine core, each of these analogues was functionalised with a benzoxazole reporting unit, a phenyl or heterocyclic π spacer, and a crown ether moiety of varying size.210 To synthesize 327, a representative example, the benzocrown bromide 323 was first coupled to 5-formyl-2-furanylboronic acid 320 in the presence of catalytic palladium to form the to form the corresponding aldehyde 321, which subsequently underwent oxidative cyclisation with phenylalanine derivative 326 to form the required benzoxazole moiety (Scheme 64).
 |
| | Scheme 64 Synthesis of a benzocrown ether amino acid featuring a furan spacer unit – Costa and co-workers.210 | |
With the aim of expanding the scope of these amino acids beyond the archetypal crown ether, Budisa and co-workers have detailed two synthetic routes enabling access to amino acids with sulphur- and nitrogen-containing crown macrocycles (Scheme 65).211
 |
| | Scheme 65 (A) Synthesis of thia-crown amino acids, and (B) Synthesis of aza-crown amino acids - Budisa and co-workers.211 | |
The first of these methods is based upon the installation of a thiol nucleophile (329) to form a thiacrown from the tosylated precursor 328, followed by conversion of the aldehyde into an amino acid moiety by introduction and alkaline hydrolysis of a diethylamidomalonate group. The second approach, using the linear nitrogen-containing crown ether precursor 335, involved cyclisation of a DOPA analog 334 by nucleophilic displacement of the tosylated groups in 335. Although their metal-binding properties were not investigated, the authors proposed that broadening the scope of available crown ether-based residues should give rise to amino acids with altered ion selectivity, allowing for the construction of more complex metal-sensing peptide architectures.
A prominent agent in the area of theranostics, DOTA is a macrocyclic ligand comprising up to four nitrogen atoms and four carboxylic acid groups capable of chelating a range of metals, particularly lanthanides. As a chelator, it is widely used in the form of a bioconjugate, typically either to enhance the effects of yttrium and lutetium radiotherapy in cancer patients, or to act as a Gd(III) MRI contrast agent.212 Incorporation of DOTA into a peptide sequence is typically achieved through direct modification of the peptide either via the N-terminus, or via a lysine or tyrosine sidechain.213 To increase the versatility of the available methodology, Sherry and co-workers reported the design of DOTA-containing amino acids compatible with standard SPPS conditions, such that the chelating moiety could be incorporated anywhere along a peptide sequence.214 Specifically, this involved the incorporation of a DOTA derivative triply protected with acid-labile protecting groups into the amino acid sidechain, and installation of a Fmoc group onto the α-amino terminus. The phenylalanine derivative 342 was prepared starting from para-nitro phenylalanine (337) which was furnished with Cbz and Bn protecting groups before reduction of the nitro group to give amine 338 (Scheme 66). From here the bromoacetylated intermediate 340 was formed, which was coupled to the free amine of the DO3A tris-tBu ester to give 341. Compound 341 was then transformed into the desired Fmoc-protected amino acid 342 following removal of the Cbz and Bn groups via catalytic hydrogenation. An analogous strategy was utilised to obtain a DOTA–lysine derivative, where the DOTA moiety was introduced through bromoacetylation of the ε-amino group. The utility of 342 as a building block for SPPS was demonstrated through the preparation of three model pentapeptides using HBTU activation chemistry, where deprotection of the DOTA esters was achieved using trifluoroacetic acid, in tandem with the resin cleavage step. Complexation with Gd3+ was carried out after purification of the crude DOTA-containing peptides, and the association constants of the resulting Gd3+–peptide complexes confirmed via fluorescence polarisation, using a competitive binding experiment with an equivalent fluorescin-labelled peptide.
 |
| | Scheme 66 Synthesis of a DOTA phenylalanine derivative for Fmoc peptide synthesis – Sherry and co-workers.214 | |
In 2012, a DOTA–alanine derivative 347 (DOTAla) was reported by Caravan and co-workers for the development of high relaxivity gadolinium-based MRI contrast agents.215 Applying similar methodology to that of Sherry and co-workers, initial attempts to introduce the protected serine derivative 343 onto the t-butyl DO3A ester were unsuccessful. Therefore, a modified approach was taken wherein 343 was used to monoalkylate cyclen to give compound 344, which was converted into the protected DOTA–alanine 346 upon further N-alkylation of the remaining cyclen amine groups using excess t-butyl bromoacetate (Scheme 67A). Following isolation of 346via preparative HPLC, the target Fmoc-protected amino acid was obtained using standard procedures of hydrogenation and Fmoc installation, giving 347 in an overall yield of 15% over five synthetic steps. This amino acid (347) was subsequently used in the construction of a diverse peptide library, including sequences with up to three DOTAla units, in each case flanked by two cysteine residues (Scheme 67B). The cysteine residues were either left with Acm protecting groups intact, or deprotected to induce intramolecular cyclisation, demonstrating scope for the modification of peptide secondary structure. The corresponding Gd chelates were found to possess high thermodynamic stability and kinetic inertness, demonstrating the potential of these peptide frameworks in the design of new contrast agents.
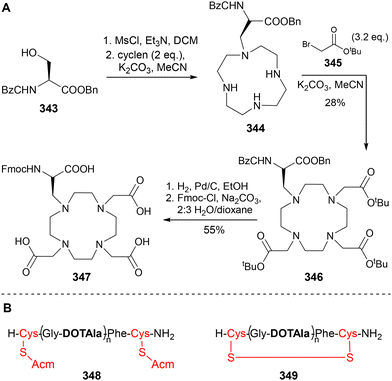 |
| | Scheme 67 (A) Synthesis of DOTAla X and (B) peptide sequences assembled by Fmoc SPPS incorporating up to 3 repeating DOTAla residues – Caravan and co-workers.215 | |
A DOTA–lysine was reported by Linscheid and co-workers in 2014 for the construction of metalated peptides in the design of reporter probes for an ICP-MS-based DNA quantification assay.216 Unlike the synthesis described by Sherry, 351 was synthesized by treating Fmoc–lysine with the N-hydroxysuccinimide ester of DOTA (350) in a TEAB buffer system, followed by complexation to either Tb3+ or Lu3+ (Scheme 68). By preparing the amino acid as a metal complex prior to use in SPPS, this approach allowed for the site-specific introduction of different lanthanides within one peptide sequence.
 |
| | Scheme 68 Synthesis of a lysine–DOTA derivative complexed to a lanthanide metal ion – Linscheid and co-workers.216 | |
7. Linear polydentate
The propensity of metal ions to stabilise the structural elements of synthetic peptides was first explored by the pioneering work of Ruan and co-workers, who observed that the helical content of peptides containing unnatural chelating residues could be drastically increased upon the addition of a divalent metal ion.217 Bearing close resemblance to the classic chelator EDTA, aminodiacetic acid (Ada) 352 was chosen as a starting point for the design of the chelating amino acids 353 containing a (CH2)nN–(CH2COOH)2– sidechain (Fig. 10).
 |
| | Fig. 10 Amino acids containing a chelating sidechain based on aminodiacetic acid 349.217 | |
This design concept was built upon further by Schneider and co-workers in 2006, who reported a library of penta- and hexadentate chelating amino acids with sidechains based upon the aminodiacetic acid motif (Fig. 11).218
 |
| | Fig. 11 Amino acids containing polydentate chelating sidechains – Schneider and co-workers.218 | |
8. Conclusions
There are a wide variety of synthetic strategies to access metal-binding amino acids. This review has demonstrated that synthetic organic chemists have been able to access motifs which are not available to nature and successfully incorporate this into building blocks for peptide chemistry. The flexibility of synthetic approaches means that libraries of building blocks can be readily accessed which would not be practical through biosynthetic approaches, perchance. The chemistry outlined here demonstrates a straightforward way to diversify the functional groups and connectivity within amino acid architectures allowing for researchers to fully explore chemical space. This has been clearly demonstrated in the range of applications that these synthetic amino acids have been so far used in.
There are clear pathways to future impact using these strategies with innovative, sustainable approaches to catalysis being one. The chemistry detailed here is complementary to the work conducted by others, in fields such as directed evolution to allow metalloenzymes to conduct unnatural reactions under green reaction conditions. The use of synthetic amino acids should be further exploited to achieve similar goals. In addition, the strategies outlined here are synergistic with biological approaches for genetic incorporation of unnatural residues into proteins (e.g. stop codon suppression) where reliable synthetic routes to unnatural amino acids are a prerequisite for these processes. Therefore, to expand the toolbox of unnatural metal binding residues further exploration into synthetic methodology should be conducted.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
K. E. V. D. and W. D. G. B. would like to thank Pleco Therapeutics (Netherlands) and the EPSRC for a PhD studentship through the MoSMed CDT programme (EP/S022791/1). Durham University is also thanked for their support.
References
- M. L. Kennedy and B. R. Gibney, Curr. Opin. Struct. Biol., 2001, 11, 485–490 CrossRef CAS PubMed.
-
(a) M. J. Chalkley, S. I. Mann and W. F. DeGrado, Nat. Rev. Chem., 2022, 6, 31–50 CrossRef CAS PubMed;
(b) A. Torrado, G. K. Walkup and B. Imperiali, J. Am. Chem. Soc., 1998, 120, 609–610 CrossRef CAS.
- M. R. Berwick, D. J. Lewis, A. W. Jones, R. A. Parslow, T. R. Dafforn, H. J. Cooper, J. Wilkie, Z. Pikramenou, M. M. Britton and A. F. A. Peacock, J. Am. Chem. Soc., 2014, 136, 1166–1169 CrossRef CAS PubMed.
- M. L. Zastrow, A. F. A. Peacock, J. A. Stuckey and V. L. Pecoraro, Nat. Chem., 2012, 4, 118–123 CrossRef CAS PubMed.
-
(a) A. L. Clevenger, R. M. Stolley, J. Aderibigbe and J. Louie, Chem. Rev., 2020, 120, 6124–6196 CrossRef CAS PubMed;
(b) Y.-M. Li, F.-Y. Kwong, W.-Y. Yu and A. S. C. Chan, Coord. Chem. Rev., 2007, 251, 2119–2144 CrossRef CAS.
- S. R. Gilbertson, G. Chen and M. McLoughlin, J. Am. Chem. Soc., 1994, 116, 4481–4482 CrossRef CAS.
- S. R. Gilbertson and X. Wang, J. Org. Chem., 1996, 61, 434–435 CrossRef CAS PubMed.
- S. R. Gilbertson and R. V. Pawlick, Angew. Chem., Int. Ed. Engl., 1996, 35, 902–904 CrossRef CAS.
- S. R. Gilbertson and D. Xie, Angew. Chem., Int. Ed., 1999, 38, 2750–2752 CrossRef CAS PubMed.
- S. R. Gilbertson and X. Wang, Tetrahedron Lett., 1996, 37, 6475–6478 CrossRef CAS.
- S. R. Gilbertson and X. Wang, Tetrahedron, 1999, 55, 11609–11618 CrossRef CAS.
- S. R. Gilbertson, S. E. Collibee and A. Agarkov, J. Am. Chem. Soc., 2000, 122, 6522–6523 CrossRef CAS.
-
(a) M. D. Struthers, R. P. Cheng and B. Imperiali, Science, 1996, 271, 342–345 CrossRef CAS PubMed;
(b) S. R. Trevino, S. Schaefer, J. M. Scholtz and C. N. Pace, J. Mol. Biol., 2007, 373, 211–218 CrossRef CAS PubMed.
- S. J. Greenfield, A. Agarkov and S. R. Gilbertson, Org. Lett., 2003, 5, 3069–3072 CrossRef CAS PubMed.
- A. M. Porte, W. A. van der Donk and K. Burgess, J. Org. Chem., 1998, 63, 5262–5264 CrossRef CAS.
- A. McKillop, R. J. K. Taylor, R. J. Watson and N. Lewis, Synthesis, 1994, 31–33 CrossRef CAS.
- D. J. Brauer, K. W. Kottsieper, S. Schenk and O. Stelzer, Z. Anorg. Allg. Chem., 2001, 627, 1151–1156 CrossRef CAS.
-
(a) F. Langer, K. Püntener, R. Stürmer and P. Knochel, Tetrahedron: Asymmetry, 1997, 8, 715–738 CrossRef CAS;
(b) H. J. C. Deboves, U. Grabowska, A. Rizzo and R. F. W. Jackson, J. Chem. Soc., Perkin Trans. 1, 2000, 4284–4292 RSC.
- S. J. Greenfield and S. R. Gilbertson, Synthesis, 2001, 2337–2340 CrossRef CAS.
- P. Knochel, M. C. P. Yeh, S. C. Berk and J. Talbert, J. Org. Chem., 1988, 53, 2390–2392 CrossRef CAS.
- S. Roy, T.-A. D. Nguyen, L. Gan and A. K. Jones, Dalton Trans., 2015, 44, 14865–14876 RSC.
- S. E. Tunney and J. K. Stille, J. Org. Chem., 1987, 52, 748–753 CrossRef CAS.
- W. J. Scott, G. T. Crisp and J. K. Stille, J. Am. Chem. Soc., 1984, 106, 4630–4632 CrossRef CAS.
- S. R. Gilbertson and G. W. Starkey, J. Org. Chem., 1996, 61, 2922–2923 CrossRef CAS PubMed.
- H.-B. Kraatz and A. Pletsch, Tetrahedron: Asymmetry, 2000, 11, 1617–1621 CrossRef CAS.
- M. Tepper, O. Stelzer, T. Häusler and W. S. Sheldrick, Tetrahedron Lett., 1997, 38, 2257–2258 CrossRef CAS.
- D. J. Brauer, S. Schenk, S. Roßenbach, M. Tepper, O. Stelzer, T. Häusler and W. S. Sheldrick, J. Organomet. Chem., 2000, 598, 116–126 CrossRef CAS.
- M. Arribat, F. Cavelier and E. Rémond, RSC Adv., 2020, 10, 6678–6724 RSC.
- K. Fourmy, D. H. Nguyen, O. Dechy-Cabaret and M. Gouygou, Catal. Sci. Technol., 2015, 5, 4289–4323 RSC.
- D. G. Holah, A. N. Hughes and B. C. Hui, Can. J. Chem., 1972, 50, 3714–3718 CrossRef CAS.
- D. Neibecker and R. Réau, J. Mol. Catal., 1989, 53, 219–227 CrossRef CAS.
- Y. Matano, T. Miyajima, N. Ochi, T. Nakabuchi, M. Shiro, Y. Nakao, S. Sakaki and H. Imahori, J. Am. Chem. Soc., 2008, 130, 990–1002 CrossRef CAS PubMed.
- K. Yavari, P. Aillard, Y. Zhang, F. Nuter, P. Retailleau, A. Voituriez and A. Marinetti, Angew. Chem., Int. Ed., 2014, 53, 861–865 CrossRef CAS PubMed.
- S. Urig, K. Fritz-Wolf, R. Réau, C. Herold-Mende, K. Tóth, E. Davioud-Charvet and K. Becker, Angew. Chem., Int. Ed., 2006, 45, 1881–1886 CrossRef CAS PubMed.
- S. Gromer, L. D. Arscott, C. H. Williams Jr., R. H. Schirmer and K. Becker, J. Biol. Chem., 1998, 273, 20096–20101 CrossRef CAS PubMed.
- E. Viry, E. Battaglia, V. Deborde, T. Müller, R. Réau, E. Davioud-Charvet and D. Bagrel, ChemMedChem, 2008, 3, 1667–1670 CrossRef CAS PubMed.
- S. van Zutphen, V. J. Margarit, G. Mora and P. Le Floch, Tetrahedron Lett., 2007, 48, 2857–2859 CrossRef CAS.
- F. Bisaro and P. Le Floch, Synlett, 2010, 3081–3085 CrossRef CAS.
-
(a) X. Caldentey and M. A. Pericàs, J. Org. Chem., 2010, 75, 2628–2644 CrossRef CAS PubMed;
(b) M. Godoi, E. E. Alberto, M. W. Paixão, L. A. Soares, P. H. Schneider and A. L. Braga, Tetrahedron, 2010, 66, 1341–1345 CrossRef CAS.
- E.-I. Negishi, F. E. Cederbaum and T. Takahashi, Tetrahedron Lett., 1986, 27, 2829–2832 CrossRef CAS.
- M. Arribat, E. Rémond, S. Clément, A. V. D. Lee and F. Cavelier, J. Am. Chem. Soc., 2018, 140, 1028–1034 CrossRef CAS PubMed.
- J. Margalef, M. Biosca, P. de la Cruz Sánchez, J. Faiges, O. Pàmies and M. Diéguez, Coord. Chem. Rev., 2021, 446, 214120 CrossRef CAS.
- I. Göttker-Schnetmann, P. White and M. Brookhart, J. Am. Chem. Soc., 2004, 126, 1804–1811 CrossRef PubMed.
- S. Kaiser, S. P. Smidt and A. Pfaltz, Angew. Chem., Int. Ed., 2006, 45, 5194–5197 CrossRef PubMed.
-
(a) F. Agbossou, J.-F. Carpentier, C. Hatat, N. Kokel, A. Mortreux, P. Betz, R. Goddard and C. Krueger, Organometallics, 1995, 14, 2480–2489 CrossRef CAS;
(b) A. Roucoux, L. Thieffry, J.-F. Carpentier, M. Devocelle, C. Méliet, F. Agbossou, A. Mortreux and A. J. Welch, Organometallics, 1996, 15, 2440–2449 CrossRef CAS.
- P. W. Galka and H.-B. Kraatz, J. Organomet. Chem., 2003, 674, 24–31 CrossRef CAS.
- D. Jayasinghe and H.-B. Kraatz, Inorg. Chim. Acta, 2006, 359, 3054–3065 CrossRef CAS.
- P. Vlastaridis, P. Kyriakidou, A. Chaliotis, Y. Van de Peer, S. G. Oliver and G. D. Amoutzias, GigaScience, 2017, 6, 1–11 CrossRef PubMed.
-
(a) L. Sauser and M. S. Shoshan, Chimia, 2021, 75, 530–534 CrossRef CAS PubMed;
(b) G. S. Baldwin, M. F. Bailey, B. P. Shehan, I. Sims and R. S. Norton, Biochem. J., 2008, 416, 77–84 CrossRef CAS PubMed.
- C. L. Mathis and A. M. Barrios, J. Inorg. Biochem., 2021, 225, 111606 CrossRef CAS PubMed.
-
(a) L. L. Liu and K. J. Franz, J. Am. Chem. Soc., 2005, 127, 9662–9663 CrossRef CAS PubMed;
(b) L. L. Liu and K. J. Franz, J. Biol. Inorg. Chem., 2007, 12, 234–247 CrossRef CAS PubMed.
- Y. Lu, M. Prudent, B. Fauvet, H. A. Lashuel and H. H. Girault, ACS Chem. Neurosci., 2011, 2, 667–675 CrossRef CAS PubMed.
- T. Wakamiya, K. Saruta, J.-I. Yasuoka and S. Kusumoto, Chem. Lett., 1994, 23, 1099–1102 CrossRef.
- D. E. Petrillo, D. R. Mowrey, S. P. Allwein and R. P. Bakale, Org. Lett., 2012, 14, 1206–1209 CrossRef CAS PubMed.
- D. R. Mowrey, D. E. Petrillo, S. P. Allwein, S. Graf and R. P. Bakale, Org. Process Res. Dev., 2012, 16, 1861–1865 CrossRef CAS.
-
(a) A. Breivogel, C. Kreitner and K. Heinze, Eur. J. Inorg. Chem., 2014, 2014, 5468–5490 CrossRef CAS;
(b) A. Torrado and B. Imperiali, J. Org. Chem., 1996, 61, 8940–8948 CrossRef CAS PubMed.
- J. Overhoff, J. Boeke and A. Gorter, Recl. Trav. Chim. Pays-Bas, 1936, 55, 293–296 CrossRef CAS.
-
(a) C. Niemann, R. N. Lewis and J. T. Hays, J. Am. Chem. Soc., 1942, 64, 1678–1682 CrossRef CAS;
(b) R. L. Bixler and C. Niemann, J. Org. Chem., 1958, 23, 575–584 CrossRef CAS.
- H. Watanabe, S. Kuwata, K. Naoe and Y. Nishida, Bull. Chem. Soc. Jpn., 1968, 41, 1634–1638 CrossRef CAS PubMed.
- E. E. Jun, Justus Liebigs Ann. Chem., 1893, 275, 1–8 CrossRef.
- J. P. Wibaut, H. P. Wallingford, H. J. Rang and D. K. Kettenes, Recl. Trav. Chim. Pays-Bas, 1955, 74, 1049–1055 CrossRef CAS.
- G. Slater and A. W. Somerville, Tetrahedron, 1967, 23, 2823–2828 CrossRef CAS PubMed.
- R. K. Griffith and H. J. Harwood, J. Org. Chem., 1964, 29, 2658–2662 CrossRef CAS.
- M. Ali, N. H. Khan and A. A. Siddiqui, Synth. Commun., 1976, 6, 227–235 CrossRef CAS.
- C. Cativiela, J. A. Mayoral, E. Melendez, L. A. Oro, M. T. Pinillos and R. Uson, J. Org. Chem., 1984, 49, 2502–2504 CrossRef CAS.
- C. Döbler, H.-J. Kreuzfeld, M. Michalik and H. W. Krause, Tetrahedron: Asymmetry, 1996, 7, 117–125 CrossRef.
- J. J. Bozell, C. E. Vogt and J. Gozum, J. Org. Chem., 1991, 56, 2584–2587 CrossRef CAS.
- R. F. Heck, Acc. Chem. Res., 1979, 12, 146–151 CrossRef CAS.
- K. Folkers, T. Kubiak and J. Stepinski, Int. J. Pept. Protein Res., 1984, 24, 197–200 CrossRef CAS PubMed.
-
(a) I. Voskuyl-Holtkamp and C. Schattenkerk, Int. J. Pept. Protein Res., 1979, 13, 185–194 CrossRef CAS PubMed;
(b) J. E. Rivier, J. Porter, C. L. Rivier, M. Perrin, A. Corrigan, W. A. Hook, R. P. Siraganian and W. W. Vale, J. Med. Chem., 1986, 29, 1846–1851 CrossRef CAS PubMed;
(c) A. Moussa, P. Meffre, J. Martinez and V. Rolland, Amino Acids, 2012, 42, 1339–1348 CrossRef CAS PubMed.
- R. F. W. Jackson, M. J. Wythes and A. Wood, Tetrahedron Lett., 1989, 30, 5941–5944 CrossRef CAS.
- S. Tabanella, I. Valancogne and R. F. W. Jackson, Org. Biomol. Chem., 2003, 1, 4254–4261 RSC.
-
(a) B. Ye and T. R. Burke Jr., J. Org. Chem., 1995, 60, 2640–2641 CrossRef CAS;
(b) A. W. Seton, M. F. G. Stevens and A. D. Westwell, J. Chem. Res., 2001, 9, 546–548 CrossRef.
- S. Kawata, S. Ashizawa and M. Hirama, J. Am. Chem. Soc., 1997, 119, 12012–12013 CrossRef CAS.
- B. Schmidt and D. K. Ehlert, Tetrahedron Lett., 1998, 39, 3999–4002 CrossRef CAS.
- W. D. G. Brittain and S. L. Cobb, Org. Biomol. Chem., 2018, 16, 10–20 RSC.
- P. D. Croce, C. La Rosa and E. Pizzatti, Tetrahedron: Asymmetry, 2000, 11, 2635–2642 CrossRef CAS.
- A. F. M. Noisier, C. S. Harris and M. A. Brimble, Chem. Commun., 2013, 49, 7744–7746 RSC.
- R. A. Aycock, D. B. Vogt and N. T. Jui, Chem. Sci., 2017, 8, 7998–8003 RSC.
- A. H. Harkiss, J. D. Bell, A. Knuhtsen, A. G. Jamieson and A. Sutherland, J. Org. Chem., 2019, 84, 2879–2890 CrossRef CAS PubMed.
- S. T. Ahmed, F. Parmeggiani, N. J. Weise, S. L. Flitsch and N. J. Turner, Org. Lett., 2016, 18, 5468–5471 CrossRef CAS PubMed.
-
(a) S. L. Lovelock, R. C. Lloyd and N. J. Turner, Angew. Chem., Int. Ed., 2014, 53, 4652–4656 CrossRef CAS PubMed;
(b) A. Gloge, B. Langer, L. Poppe and J. Rétey, Arch. Biochem. Biophys., 1998, 359, 1–7 CrossRef CAS PubMed;
(c) E. B. Watkins and R. S. Phillips, Bioorg. Med. Chem. Lett., 2001, 11, 2099–2100 CrossRef CAS PubMed.
-
(a) M. Ohta and M. Masaki, Bull. Chem. Soc. Jpn., 1960, 33, 1150–1150 CrossRef CAS;
(b) M. Augustin and H. Dehne, J. Prakt. Chem., 1961, 13, 118–120 CrossRef CAS;
(c) T. Mega, Y. Hamazume and T. Ikenaka, Bull. Chem. Soc. Jpn., 1988, 61, 4315–4321 CrossRef CAS.
- W. W. Gerhardt and M. Weck, J. Org. Chem., 2006, 71, 6333–6341 CrossRef CAS PubMed.
-
(a) H. Kunz and W. Sager, Angew. Chem., Int. Ed. Engl., 1987, 26, 557–559 CrossRef;
(b) H. Kunz and W. Pfrengle, J. Am. Chem. Soc., 1988, 110, 651–652 CrossRef CAS;
(c) H. Kunz, W. Pfrengle, K. Rück and W. Sager, Synthesis, 1991, 1039–1042 CrossRef.
- G. Guillena, G. Rodríguez, M. Albrecht and G. van Koten, Chem. – Eur. J., 2002, 8, 5368–5376 CrossRef CAS PubMed.
- T. C. Johnson and S. P. Marsden, Org. Lett., 2016, 18, 5364–5367 CrossRef CAS PubMed.
-
(a) A. T. Londregan, S. Jennings and L. Wei, Org. Lett., 2010, 12, 5254–5257 CrossRef CAS PubMed;
(b) A. T. Londregan, S. Jennings and L. Wei, Org. Lett., 2011, 13, 1840–1843 CrossRef CAS PubMed;
(c) A. T. Londregan, K. Burford, E. L. Conn and K. D. Hesp, Org. Lett., 2014, 16, 3336–3339 CrossRef CAS PubMed.
- J. De Jersey and B. Zerner, Biochemistry, 1969, 8, 1967–1974 CrossRef CAS PubMed.
- B. Imperiali and S. L. Fisher, J. Org. Chem., 1992, 57, 757–759 CrossRef CAS.
- M. J. O'Donnell, W. D. Bennett and S. Wu, J. Am. Chem. Soc., 1989, 111, 2353–2355 CrossRef.
- B. Imperiali and S. L. Fisher, J. Am. Chem. Soc., 1991, 113, 8527–8528 CrossRef CAS.
- B. Imperiali, T. J. Prins and S. L. Fisher, J. Org. Chem., 1993, 58, 1613–1616 CrossRef CAS.
- S.-T. Chen, K.-T. Wang and C.-H. Wong, J. Chem. Soc., Chem. Commun., 1986, 1514–1516 RSC.
- D. S. Wuttke, H. B. Gray, S. L. Fisher and B. Imperiali, J. Am. Chem. Soc., 1993, 115, 8455–8456 CrossRef CAS.
- N. Nishino, T. Kato, T. Murata, H. Nakayama, T. Arai, T. Fujimoto, H. Yamamoto and S. Yoshikawa, Chem. Lett., 1996, 25, 49–50 CrossRef.
- K. J. Kise and B. E. Bowler, Tetrahedron: Asymmetry, 1998, 9, 3319–3324 CrossRef CAS.
- M. D. Struthers, R. P. Cheng and B. Imperiali, Science, 1996, 271, 342–345 CrossRef CAS PubMed.
- S. P. L. Sörensen, Biol. Chem., 1905, 44, 448–460 CrossRef.
- A. Torrado and B. Imperiali, J. Org. Chem., 1996, 61, 8940–8948 CrossRef CAS PubMed.
- G. R. Newkome, J. Gross and A. K. Patri, J. Org. Chem., 1997, 62, 3013–3014 CrossRef CAS PubMed.
- B. M. Bishop, D. G. McCafferty and B. W. Erickson, Tetrahedron, 2000, 56, 4629–4638 CrossRef CAS.
- D. G. McCafferty, B. M. Bishop, C. G. Wall, S. G. Hughes, S. L. Mecklenberg, T. J. Meyer and B. W. Erickson, Tetrahedron, 1995, 51, 1093–1106 CrossRef CAS.
- H. Ishida, M. Kyakuno and S. Oishi, Biopolymers, 2004, 76, 69–82 CrossRef CAS PubMed.
- H. Ishida, Y. Maruyama, M. Kyakuno, Y. Kodera, T. Maeda and S. Oishi, ChemBioChem, 2006, 7, 1567–1570 CrossRef CAS PubMed.
- Y. Shiina, S. Oishi and H. Ishida, Tetrahedron Lett., 2012, 53, 1249–1252 CrossRef CAS.
- G. Rama, A. Ardá, J.-D. Maréchal, I. Gamba, H. Ishida, J. Jiménez-Barbero, M. E. Vázquez and M. Vázquez López, Chem. – Eur. J., 2012, 18, 7030–7035 CrossRef CAS PubMed.
- G. Rama, A. Ardá, J. D. Maréchal, I. Gamba, H. Ishida, J. Jiménez-Barbero, M. E. Vázquez and M. Vázquez López, Chemistry, 2012, 18, 7030–7035 CrossRef CAS PubMed.
-
(a) Y. Biniuri, B. Albada, M. Wolff, E. Golub, D. Gelman and I. Willner, ACS Catal., 2018, 8, 1802–1809 CrossRef CAS;
(b) G.-F. Luo, Y. Biniuri, M. Vázquez-González, V. Wulf, M. Fadeev, R. Lavi and I. Willner, Adv. Funct. Mater., 2019, 29, 1901484 CrossRef.
- C. Zhang, P. Srivastava, K. Ellis-Guardiola and J. C. Lewis, Tetrahedron, 2014, 70, 4245–4249 CrossRef CAS PubMed.
- Y. Li, M. Cheng, J. Hao, C. Wang, G. Jia and C. Li, Chem. Sci., 2015, 6, 5578–5585 RSC.
- X. Liu, F. Kang, C. Hu, L. Wang, Z. Xu, D. Zheng, W. Gong, Y. Lu, Y. Ma and J. Wang, Nat. Chem., 2018, 10, 1201–1206 CrossRef CAS PubMed.
-
(a) I. Drienovská, R. A. Scheele, C. Gutiérrez de Souza and G. Roelfes, ChemBioChem, 2020, 21, 3077–3081 CrossRef PubMed;
(b) X. Luo, T. A. Wang, Y. Zhang, F. Wang and P. G. Schultz, Cell Chem. Biol., 2016, 23, 1098–1102 CrossRef CAS PubMed;
(c) P. J. Almhjell and J. H. Mills, Curr. Opin. Struct. Biol., 2018, 51, 170–176 CrossRef CAS PubMed.
- T. Schneider, I. Gavrilova and N. Budisa, Tetrahedron Lett., 2019, 60, 906–910 CrossRef CAS.
- E. M. Hahn, N. Estrada-Ortiz, J. Han, V. F. C. Ferreira, T. G. Kapp, J. D. G. Correia, A. Casini and F. E. Kühn, Eur. J. Inorg. Chem., 2017, 2017, 1667–1672 CrossRef CAS.
- Y.-H. Chiu and J. W. Canary, Inorg. Chem., 2003, 42, 5107–5116 CrossRef CAS PubMed.
- C. M. Teles, L. C. Lammoglia, M. A. Juliano, A. L. T. G. Ruiz, T. Z. Candido, J. E. de Carvalho, C. S. P. Lima and C. Abbehausen, J. Inorg. Biochem., 2019, 199, 110754 CrossRef CAS PubMed.
-
(a) Đ. Škalamera, E. Sanders, R. Vianello, A. Maršavelski, A. Pevec, I. Turel and S. I. Kirin, Dalton Trans., 2016, 45, 2845–2858 RSC;
(b) N. Niklas, F. Hampel and R. Alsfasser, Chem. Commun., 2003, 1586–1587, 10.1039/b303172a;
(c) C. Gawlig, J. Jung, D. Mollenhauer and S. Schindler, Z. Anorg. Allg. Chem., 2021, 647, 951–959 CrossRef CAS.
- N. Niklas, A. Zahl and R. Alsfasser, Dalton Trans., 2007, 154–162 RSC.
- G. Dirscherl, R. Knape, P. R. Hanson and B. König, Tetrahedron, 2007, 63, 4918–4928 CrossRef CAS.
- A. F. Abdel-Magid, K. G. Carson, B. D. Harris, C. A. Maryanoff and R. D. Shah, J. Org. Chem., 1996, 61, 3849–3862 CrossRef CAS PubMed.
- J.-P. Mazaleyrat, M. Wakselman, F. Formaggio, M. Crisma and C. Toniolo, Tetrahedron Lett., 1999, 40, 6245–6248 CrossRef CAS.
- L. J. Henderson Jr., F. R. Fronczek and W. R. Cherry, J. Am. Chem. Soc., 1984, 106, 5876–5879 CrossRef.
- J.-P. Mazaleyrat, K. Wright, M. Wakselman, F. Formaggio, M. Crisma and C. Toniolo, Eur. J. Org. Chem., 2001, 1821–1829 CrossRef CAS.
-
(a) A. J. Arduengo III, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef;
(b) V. Nesterov, D. Reiter, P. Bag, P. Frisch, R. Holzner, A. Porzelt and S. Inoue, Chem. Rev., 2018, 118, 9678–9842 CrossRef CAS PubMed.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed.
- F. Hannig, G. Kehr, R. Fröhlich and G. Erker, J. Organomet. Chem., 2005, 690, 5959–5972 CrossRef CAS.
- F. Guillen, D. Brégeon and J.-C. Plaquevent, Tetrahedron Lett., 2006, 47, 1245–1248 CrossRef CAS.
- R. Jain and L. A. Cohen, Tetrahedron, 1996, 52, 5363–5370 CrossRef CAS.
- A. Monney, G. Venkatachalam and M. Albrecht, Dalton Trans., 2011, 40, 2716–2719 RSC.
- A. Monney, E. Alberico, Y. Ortin, H. Müller-Bunz, S. Gladiali and M. Albrecht, Dalton Trans., 2012, 41, 8813–8821 RSC.
- A. Monney and M. Albrecht, Chem. Commun., 2012, 48, 10960–10962 RSC.
- A. Monney, F. Nastri and M. Albrecht, Dalton Trans., 2013, 42, 5655–5660 RSC.
- K. Lenzen, M. Planchestainer, I. Feller, D. R. Padrosa, F. Paradisi and M. Albrecht, Chem. Commun., 2021, 57, 9068–9071 RSC.
-
(a) Q. Zhao, G. Meng, G. Li, C. Flach, R. Mendelsohn, R. Lalancette, R. Szostak and M. Szostak, Chem. Sci., 2021, 12, 10583–10589 RSC;
(b) G. Li, P. Lei, M. Szostak, E. Casals-Cruañas, A. Poater, L. Cavallo and S. P. Nolan, ChemCatChem, 2018, 10, 3096–3106 CrossRef CAS;
(c) D. Canseco-Gonzalez, A. Petronilho, H. Mueller-Bunz, K. Ohmatsu, T. Ooi and M. Albrecht, J. Am. Chem. Soc., 2013, 135, 13193–13203 CrossRef CAS PubMed.
- R. M. Drost, D. L. J. Broere, J. Hoogenboom, S. N. de Baan, M. Lutz, B. de Bruin and C. J. Elsevier, Eur. J. Inorg. Chem., 2015, 2015, 982–996 CrossRef CAS.
- D. M. T. Chan, K. L. Monaco, R.-P. Wang and M. P. Winters, Tetrahedron Lett., 1998, 39, 2933–2936 CrossRef CAS.
- C. DalZotto, J. Michaux, E. Martinand-Lurin and J.-M. Campagne, Eur. J. Org. Chem., 2010, 3811–3814 CrossRef CAS.
- Y. Zhao and S. R. Gilbertson, Org. Lett., 2014, 16, 1033–1035 CrossRef CAS PubMed.
- I. M. Daubit, J. Wolf and N. Metzler-Nolte, J. Organomet. Chem., 2020, 909, 121096 CrossRef CAS.
- J. Lemke and N. Metzler-Nolte, J. Organomet. Chem., 2011, 696, 1018–1022 CrossRef CAS.
- Y. N. Belokon, A. V. Grachev, V. I. Maleev, V. N. Khrustalev, A. S. Peregudov and M. North, Tetrahedron: Asymmetry, 2008, 19, 756–760 CrossRef CAS.
- Y. N. Belokon, A. G. Bulychev, S. V. Vitt, Y. T. Struchkov, A. S. Batsanov, T. V. Timofeeva, V. A. Tsyryapkin, M. G. Ryzhov and L. A. Lysova, J. Am. Chem. Soc., 1985, 107, 4252–4259 CrossRef CAS.
- Y. N. Belokon, A. S. Sagyan, S. M. Djamgaryan, V. I. Bakhmutov and V. M. Belikov, Tetrahedron, 1988, 44, 5507–5514 CrossRef CAS.
- H. M. J. Wang and I. J. B. Lin, Organometallics, 1998, 17, 972–975 CrossRef CAS.
- V. I. Maleev, A. V. Grachev, V. N. Khrustalev and F. M. Dolgushin, Russ. Chem. Bull., 2010, 59, 1273–1283 CrossRef CAS.
-
(a) V. A. Soloshonok, X. Tang and V. J. Hruby, Tetrahedron, 2001, 57, 6375–6382 CrossRef CAS;
(b) J. Wang, D. Lin, S. Zhou, X. Ding, V. A. Soloshonok and H. Liu, J. Org. Chem., 2011, 76, 684–687 CrossRef CAS PubMed.
-
(a) A. Debache, S. Collet, P. Bauchat, D. Danion, L. Euzenat, A. Hercouet and B. Carboni, Tetrahedron: Asymmetry, 2001, 12, 761–764 CrossRef CAS;
(b) T. Yamada, K. Sakaguchi, T. Shinada, Y. Ohfune and V. A. Soloshonok, Tetrahedron: Asymmetry, 2008, 19, 2789–2795 CrossRef CAS.
-
(a) T. Hohmann, M. Dyrks, S. Chowdhary, M. Weber, D. Nguyen, J. Moschner and B. Koksch, J. Org. Chem., 2022, 87, 10592–10604 CrossRef CAS PubMed;
(b) J. L. Aceña, A. E. Sorochinsky, H. Moriwaki, T. Sato and V. A. Soloshonok, J. Fluor. Chem., 2013, 155, 21–38 CrossRef.
- O. A. Levitskiy, Y. K. Grishin, O. O. Semivrazhskaya, A. A. Ambartsumyan, K. A. Kochetkov and T. V. Magdesieva, Angew. Chem., Int. Ed., 2017, 56, 2704–2708 CrossRef CAS PubMed.
- W. Vuong, F. Mosquera-Guagua, R. Sanichar, T. R. McDonald, O. P. Ernst, L. Wang and J. C. Vederas, Org. Lett., 2019, 21, 10149–10153 CrossRef CAS PubMed.
-
(a) A. Popkov and B. De Spiegeleer, Dalton Trans., 2012, 41, 1430–1440 RSC;
(b) R. Takeda, H. Abe, N. Shibata, H. Moriwaki, K. Izawa and V. A. Soloshonok, Org. Biomol. Chem., 2017, 15, 6978–6983 RSC.
-
(a) A. E. Sorochinsky, J. L. Aceña, H. Moriwaki, T. Sato and V. A. Soloshonok, Amino Acids, 2013, 45, 691–718 CrossRef CAS PubMed;
(b) A. E. Sorochinsky, J. L. Aceña, H. Moriwaki, T. Sato and V. Soloshonok, Amino Acids, 2013, 45, 1017–1033 CrossRef CAS PubMed;
(c) J. L. Aceña, A. E. Sorochinsky and V. Soloshonok, Amino Acids, 2014, 46, 2047–2073 CrossRef PubMed.
- S. Parpart, Z. Z. Mardiyan, P. Ehlers, A. Petrosyan, A. F. Mkrtchyan, A. S. Saghyan and P. Langer, Synlett, 2018, 793–798 CAS.
- A. S. Saghyan, A. F. Mkrtchyan, Z. Z. Mardiyan, L. A. Hayriyan, Y. N. Belokon and P. Langer, ChemistrySelect, 2019, 4, 4686–4688 CrossRef CAS.
- V. A. Larionov, N. V. Stoletova, V. I. Kovalev, A. F. Smol'yakov, T. Y. F. Savel'yeva and V. I. Maleev, Org. Chem. Front., 2019, 6, 1094–1099 RSC.
- C. Yu, Y. Yu, L. Sun, X. Li, Z. Liu, M. Ke and F. Chen, Org. Biomol. Chem., 2022, 20, 4894–4899 RSC.
- O. A. Levitskiy, O. I. Aglamazova, Y. K. Grishin, S. E. Nefedov and T. V. Magdesieva, Electrochim. Acta, 2022, 409, 139980 CrossRef CAS.
- O. Levitskiy, Y. Grishin, K. Paseshnichenko, K. Kochetkov and T. Magdesieva, Tetrahedron Lett., 2018, 59, 2831–2834 CrossRef CAS.
- N. V. Stoletova, A. D. Moshchenkov, A. F. Smol'yakov, Z. T. Gugkaeva, V. I. Maleev, D. Katayev and V. A. Larionov, Helv. Chim. Acta, 2021, 104, e2000193 CrossRef CAS.
-
(a) Y. Zou, J. Han, A. S. Saghyan, A. F. Mkrtchyan, H. Konno, H. Moriwaki, K. Izawa and V. A. Soloshonok, Molecules, 2020, 25, 2739 CrossRef CAS PubMed;
(b) Y. Wang, X. Song, J. Wang, H. Moriwaki, V. A. Soloshonok and H. Liu, Amino Acids, 2017, 49, 1487–1520 CrossRef CAS PubMed.
- S. Burck, S. G. A. van Assema, B. Lastdrager, J. C. Slootweg, A. W. Ehlers, J. M. Otero, B. Dacunha-Marinho, A. L. Llamas-Saiz, M. Overhand, M. J. van Raaij and K. Lammertsma, Chem. – Eur. J., 2009, 15, 8134–8145 CrossRef CAS PubMed.
- G. Guisado-Barrios, B. K. Muñoz, P. C. J. Kamer, B. Lastdrager, G. van der Marel, M. Overhand, M. Vega-Vázquez and M. Martin-Pastor, Dalton Trans., 2013, 42, 1973–1978 RSC.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- V. A. Larionov, H. V. Adonts, Z. T. Gugkaeva, A. F. Smol'yakov, A. S. Saghyan, M. S. Miftakhov, S. A. Kuznetsova, V. I. Maleev and Y. N. Belokon, ChemistrySelect, 2018, 3, 3107 CrossRef CAS.
-
(a) S. Mann, A. Kaur, A. Kaur, N. Priyadarshi, B. Goyal, N. K. Singhal and D. Goyal, ACS Chem. Neurosci., 2023, 14, 1631–1645 CrossRef CAS PubMed;
(b) N. Agouram, E. M. El Hadrami and A. Bentama, Molecules, 2021, 26, 2937 CrossRef CAS PubMed.
-
(a) T. L. Mindt, H. Struthers, L. Brans, T. Anguelov, C. Schweinsberg, V. Maes, D. Tourwé and R. Schibli, J. Am. Chem. Soc., 2006, 128, 15096–15097 CrossRef CAS PubMed;
(b) H. Struthers, B. Spingler, T. L. Mindt and R. Schibli, Chemistry, 2008, 14, 6173–6183 CrossRef CAS PubMed.
- A. Mahindra, C. J. Millard, I. Black, L. J. Archibald, J. W. R. Schwabe and A. G. Jamieson, Org. Lett., 2019, 21, 3178–3182 CrossRef CAS PubMed.
- A. J. Young, C. J. Serpell, J. M. Chin and M. R. Reithofer, Chem. Commun., 2017, 53, 12426–12429 RSC.
- A. J. Young, C. Eisen, G. M. D. M. Rubio, J. M. Chin and M. R. Reithofer, J. Inorg. Biochem., 2019, 199, 110707 CrossRef CAS PubMed.
- O. V. Kuznetsova, G. Rubio, B. K. Keppler, J. M. Chin, M. R. Reithofer and A. R. Timerbaev, Anal. Biochem., 2020, 611, 114003 CrossRef CAS PubMed.
- C. H. G. Jakob, B. Dominelli, E. M. Hahn, T. O. Berghausen, T. Pinheiro, F. Marques, R. M. Reich, J. D. G. Correia and F. E. Kühn, Chem. – Asian J., 2020, 15, 2754–2762 CrossRef CAS PubMed.
- R. S. Herrick, R. M. Jarret, T. P. Curran, D. R. Dragoli, M. B. Flaherty, S. E. Lindyberg, R. A. Slate and L. C. Thornton, Tetrahedron Lett., 1996, 37, 5289–5292 CrossRef CAS.
- K. Schlögl, Monatsh. Chem. Verw. Teile Anderer Wiss., 1957, 88, 601–621 Search PubMed.
- I. R. Butler and S. C. Quayle, J. Organomet. Chem., 1998, 552, 63–68 CrossRef CAS.
- L. Barisic, W. Rapic and V. Kovač, Croat. Chem. Acta, 2002, 75, 199–210 CAS.
- V. Kovač, K. Radolović, I. Habuš, D. Siebler, K. Heinze and V. Rapić, Eur. J. Inorg. Chem., 2009, 2009, 389–399 CrossRef.
- L. Barisić, M. Dropucić, V. Rapić, H. Pritzkow, S. I. Kirin and N. Metzler-Nolte, Chem. Commun., 2004, 2004–2005 RSC.
- L. Tebben, K. Bussmann, M. Hegemann, G. Kehr, R. Fröhlich and G. Erker, Organometallics, 2008, 27, 4269–4272 CrossRef CAS.
-
(a) A. Hunold, I. Neundorf, P. James, J. Neudörfl and H.-G. Schmalz, Eur. J. Org. Chem., 2009, 4429–4440 CrossRef CAS;
(b) A. T. Philip, S. Chacko and R. Ramapanicker, J. Pept. Sci., 2015, 21, 887–892 CrossRef CAS PubMed.
- Y. N. Song, H. Xu, W. Chen, P. Zhan and X. Liu, MedChemComm, 2015, 6, 61–74 RSC.
- G. K. Walkup and B. Imperiali, J. Org. Chem., 1998, 63, 6727–6731 CrossRef CAS.
- A. G. Myers, J. L. Gleason, T. Yoon and D. W. Kung, J. Am. Chem. Soc., 1997, 119, 656–673 CrossRef CAS.
- N. Jotterand, D. A. Pearce and B. Imperiali, J. Org. Chem., 2001, 66, 3224–3228 CrossRef CAS PubMed.
- H. S. Lee, G. Spraggon, P. G. Schultz and F. Wang, J. Am. Chem. Soc., 2009, 131, 2481–2483 CrossRef CAS PubMed.
- M. G. Banwell, B. D. Schwartz, A. C. Bissember, T. Herlt, A. C. Willis, M. G. Gardiner, J. Illesinghe and A. J. Robinson, Asian J. Org. Chem., 2022, 11, e202100455 CrossRef CAS.
- M. J. Burk, J. E. Feaster, W. A. Nugent and R. L. Harlow, J. Am. Chem. Soc., 1993, 115, 10125–10138 CrossRef CAS.
- O. Meth-Cohn, B. Narine and B. Tarnowski, J. Chem. Soc., Perkin Trans. 1, 1981, 1520–1530 RSC.
- A. Abdullahi and K. Y. Yeong, Med. Chem. Res., 2024, 33, 406–438 CrossRef CAS.
- K. Guzow, M. Szabelski, J. Malicka and W. Wiczk, Helv. Chim. Acta, 2001, 84, 1086–1092 CrossRef CAS.
- M. Milewska, A. Skwierawska, K. Guzow, D. Szmigiel and W. Wiczk, Inorg. Chem. Commun., 2005, 8, 947–950 CrossRef CAS.
- S. P. G. Costa, E. Oliveira, C. Lodeiro and M. M. Raposo, Sensors, 2007, 7, 2096–2114 CrossRef CAS PubMed.
- C. I. C. Esteves, R. C. M. Ferreira, M. M. M. Raposo and S. P. G. Costa, Dyes Pigm., 2018, 151, 211–218 CrossRef CAS.
- E. Oliveira, D. Genovese, R. Juris, N. Zaccheroni, J. L. Capelo, M. M. M. Raposo, S. P. G. Costa, L. Prodi and C. Lodeiro, Inorg. Chem., 2011, 50, 8834–8849 CrossRef CAS PubMed.
- C. I. C. Esteves, M. M. M. Raposo and S. P. G. Costa, Dyes Pigm., 2016, 134, 258–268 CrossRef CAS.
- R. M. F. Batista, R. C. M. Ferreira, M. M. M. Raposo and S. P. G. Costa, Tetrahedron, 2012, 68, 7322–7330 CrossRef CAS.
- C. I. C. Esteves, M. M. M. Raposo and S. P. G. Costa, Molecules, 2023, 28, 7256 CrossRef CAS PubMed.
- C. E. Yoo, P. S. Chae, J. E. Kim, E. J. Jeong and J. Suh, J. Am. Chem. Soc., 2003, 125, 14580–14589 CrossRef CAS PubMed.
- S. Miltschitzky and B. König, Synth. Commun., 2004, 34, 2077–2084 CrossRef CAS.
- S.-Y. Chen, Y. Huang, G.-L. Zhang, H. Cheng, C.-Q. Xia, L.-J. Ma, H. Yu and X.-Q. Yu, Synthesis, 2005, 888–892 CAS.
- M. Kruppa, G. Imperato and B. König, Tetrahedron, 2006, 62, 1360–1364 CrossRef CAS.
- D. S. Turygin, M. Subat, O. A. Raitman, V. V. Arslanov, B. König and M. A. Kalinina, Angew. Chem., Int. Ed., 2006, 45, 5340–5344 CrossRef CAS PubMed.
- P. Rossi, F. Felluga and P. Scrimin, Tetrahedron Lett., 1998, 39, 7159–7162 CrossRef CAS.
-
(a) P. Rossi, F. Felluga, P. Tecilla, F. Formaggio, M. Crisma, C. Toniolo and P. Scrimin, J. Am. Chem. Soc., 1999, 121, 6948–6949 CrossRef CAS;
(b) C. Sissi, P. Rossi, F. Felluga, F. Formaggio, M. Palumbo, P. Tecilla, C. Toniolo and P. Scrimin, J. Am. Chem. Soc., 2001, 123, 3169–3170 CrossRef CAS PubMed.
- P. Rossi, P. Tecilla, L. Baltzer and P. Scrimin, Chem. – Eur. J., 2004, 10, 4163–4170 CrossRef CAS PubMed.
- J. Li, D. Yim, W.-D. Jang and J. Yoon, Chem. Soc. Rev., 2017, 46, 2437–2458 RSC.
-
(a) N. Voyer, J. Am. Chem. Soc., 1991, 113, 1818–1821 CrossRef CAS;
(b) E. Biron, F. Otis, J.-C. Meillon, M. Robitaille, J. Lamothe, P. Van Hove, M.-E. Cormier and N. Voyer, Bioorg. Med. Chem., 2004, 12, 1279–1290 CrossRef CAS PubMed.
- P.-A. Paquet-Côté, J.-P. Paradis, M. Auger and N. Voyer, Biochim. Biophys. Acta, Biomembr., 2020, 1862, 183261 CrossRef PubMed.
- C. I. C. Esteves, R. M. F. Batista, M. M. M. Raposo and S. P. G. Costa, Dyes Pigm., 2016, 135, 134–142 CrossRef CAS.
- P. M. R. Batista, C. D. F. Martins, M. M. M. Raposo and S. P. G. Costa, Molecules, 2023, 28, 3326 CrossRef CAS PubMed.
- T. Schneider, N. Brüssow, A. Yuvanc and N. Budisa, ChemistrySelect, 2020, 5, 2854–2857 CrossRef CAS.
- L. M. De León-Rodríguez and Z. Kovacs, Bioconjugate Chem., 2008, 19, 391–402 CrossRef PubMed.
- L. M. De León-Rodríguez and Z. Kovacs, Bioconjugate Chem., 2008, 19, 391–402 CrossRef PubMed.
- L. M. De León-Rodriguez, Z. Kovacs, G. R. Dieckmann and A. D. Sherry, Chemistry, 2004, 10, 1149–1155 CrossRef PubMed.
- E. Boros, M. Polasek, Z. Zhang and P. Caravan, J. Am. Chem. Soc., 2012, 134, 19858–19868 CrossRef CAS PubMed.
- K. Brückner, R. Zitterbart, O. Seitz, S. Beck and M. W. Linscheid, Bioconjugate Chem., 2014, 25, 1069–1077 CrossRef PubMed.
- F. Ruan, Y. Chen and P. B. Hopkins, J. Am. Chem. Soc., 1990, 112, 9403–9404 CrossRef CAS.
- C. M. Micklitsch, Q. Yu and J. P. Schneider, Tetrahedron Lett., 2006, 47, 6277–6280 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  * and
William D. G.
Brittain
* and
William D. G.
Brittain