
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interrogating the potential of helical aromatic foldamers for protein recognition†
Sunbum
Kwon‡
 ab,
Vasily
Morozov‡
a,
Lingfei
Wang‡
a,
Pradeep K.
Mandal
a,
Stéphane
Chaignepain
c,
Céline
Douat
a and
Ivan
Huc
*a
ab,
Vasily
Morozov‡
a,
Lingfei
Wang‡
a,
Pradeep K.
Mandal
a,
Stéphane
Chaignepain
c,
Céline
Douat
a and
Ivan
Huc
*a
aDepartment of Pharmacy, Ludwig-Maximilians-Universität, Butenandtstraße 5–13, D-81377 München, Germany. E-mail: ivan.huc@cup.lmu.de
bDepartment of Chemistry, Chung-Ang University, 84 Heukseok-ro, Dongjak-gu, Seoul 06974, Republic of Korea
cCBMN (UMR5248), Univ. Bordeaux—CNRS—IPB, 2 rue Robert Escarpit, 33600 Pessac, France
First published on 30th October 2024
Abstract
A biotinylated helical aromatic oligoamide foldamer equivalent in size to a 24mer peptide was designed without any prejudice other than to display various polar and hydrophobic side chains at its surface. It was synthesized on solid phase, its P- and M-helical conformers were separated by HPLC on a chiral stationary phase, and the solid state structure of a non-biotinylated analogue was elucidated by X-ray crystallography. Pull-down experiments from a yeast cell lysate using the foldamer as a bait followed by proteomic analysis revealed potential protein binding partners. Three of these proteins were recombinantly expressed. Biolayer interferometry showed submicromolar binding demonstrating the potential of a given foldamer to have affinity for certain proteins in the absence of design considerations. Yet, binding selectivity was low in all three cases since both P- and M-conformers bound to the proteins with similar affinities.
With their main chain amide functions, their variety of hydrophobic, polar neutral, cationic and anionic side chains arranged at defined positions in space, and their complex shapes, proteins are ideally suited to selectively interact with one another. An illustration of this fact resides in the very propensity of the α-peptide backbone to adopt folded conformations – folding can be viewed as a kind of self-recognition. It follows that peptides, particularly peptide macrocycles,1 and proteins themselves are prime candidates for the selective recognition of protein surfaces. Purposely developed for that task are naturally occurring antibodies as well as recombinant proteins amenable to in vitro display selection, e.g. phage-, ribosome-, or mRNA-display, when their structure is stable enough to withstand the randomization of multiple surface residues.2 To bind a protein surface, being protein-like is not a requirement, as nucleic acid aptamers illustrate,3 but it is certainly an advantage.4 There is thus currently great interest in the design of proteomimetics, synthetic molecules that would reproduce some features of proteins, in particular in the context of molecular recognition.5
We and others have been developing aromatic amide foldamers that adopt stable, predictable, helically folded conformations.6 Recent designs show that such synthetic objects may reach the size of small proteins.7 Although their aromatic backbone and folding propensity are remote from those of proteins, their surface can be decorated with proteinogenic side chains at precise locations.8 Their ability to recognize protein surfaces is currently being explored,9 with emphasis on targeting amyloid proteins10 as well as DNA-binding proteins.11 It is hoped that such large foldamers may become efficient tools to bind sizeable protein surface areas and serve as competitive inhibitors of protein–protein and protein–nucleic acid interactions for pharmacological or even therapeutic applications, complementing antibodies and aptamers in such tasks. Indeed, abiotic foldamers may even overcome some of the disadvantages of proteins that can hamper their practical use. For example, proteins may be susceptible to proteolytic degradation and to denaturation which can in turn cause precipitation under certain conditions. In contrast, helical aromatic oligoamides are not degraded by proteases and, provided they are sufficiently hydrophilic, their clear solutions are unchanged after months of storage at 4 °C. However, designing a foldamer that specifically recognizes a given protein surface remains a challenging task and only multiple such endeavours will reveal the general suitability of the approach. Here, we sought to estimate the chances of a given proteomorphous aromatic foldamer to have affinity for any of the proteins present in a cell lysate. Using pull-down experiments and proteomic analysis, we observed significant enrichments and subsequently determined submicromolar dissociation constants (KD), in the absence of any specific design. The capacity of a single foldamer to interact with proteins hints at a potential to interfere with their functions, although this was not investigated here. These results thus bode well for future developments and encourage further screening of various foldamers from different backbones and with different side chain arrangements, not only for binding, but also for their ability to modify interesting cellular phenotypes.
Foldamer 2 and its biotinylated analogue 1 consist of a dodecaamide of 8-amino-2-quinoline carboxylic acid (Fig. 1) bearing side chains in position 4. In water, such oligomers fold into stable aromatic helices having 2.5 units per turn and a pitch equal to the thickness of one aromatic ring.8 Since each δ-amino acid monomer is equivalent in size to a dipeptide, the helices of 1 and 2 reach the size of a 24mer peptide. These sequences, including final biotinylation, were synthesized on solid support using recently optimized protocols (Fig. S1 in the ESI†) and purified by reversed-phase HPLC.12 The anionic, cationic, polar neutral, or hydrophobic nature of the side chains and their location on the helix surface were chosen among the building blocks available at the time to promote various types of intermolecular interactions but without any prejudice of which protein could be recognized. The arrangement of the side chains in space avoids that different parts of the helix surface resemble and thus enhance the chances that some proteins bind one area of the helix or another via shape recognition and complementary hydrophobic and electrostatic contacts. Sequences 1 and 2 contain several instances of contiguous residues that are identical or have similar polar or hydrophobic features. Yet, due to the high helix curvature – a monomer spans 0.4 helix turn – contiguous residues project their side chains towards different sides of the helix (Fig. 1b). Putting two identical residues contiguous in the sequence in fact contributes to the diversity of environments at the surface of the foldamer. The presentation of diverse arrays of side chains on the different sides of the helix is highlighted by the helix wheel in Fig. 1b.
 | ||
| Fig. 1 (a) Foldamer aromatic building blocks colour coded according to their side chain nature: hydrophobic (black), polar neutral (green), cationic (blue) and anionic (red). (b) Sequence of dodecamer 2 used for X-ray crystallographic studies and its biotinylated version 1 used in the pull-down assay. The five-pointed star helix wheel representation depicts the side-chain positioning around the oligoquinoline backbone. | ||
High-quality crystals of compound 2 were obtained and diffracted at atomic resolution, allowing for the elucidation of its solid state structure (Fig. 2a). The top view of the structure illustrates the distribution of side chains. It also reveals a slight deviation of helix curvature from the usual 2.5 units per turn, possibly due to crystal packing interactions (Fig. S2†). The electrostatic charge potential shown on the solvent accessible surface of 2 (Fig. 2b) provides a graphical illustration of its proteomorphous nature. Despite the quinoline monomers being coarse, i.e. twice as large as an α-amino acid, the display of the side chains at the surface of the helix makes it look protein-like. Its size remains small, comparable to that of a 24mer peptide. If required, substantially larger (as well as smaller) foldamers would be accessible through solid phase synthesis.
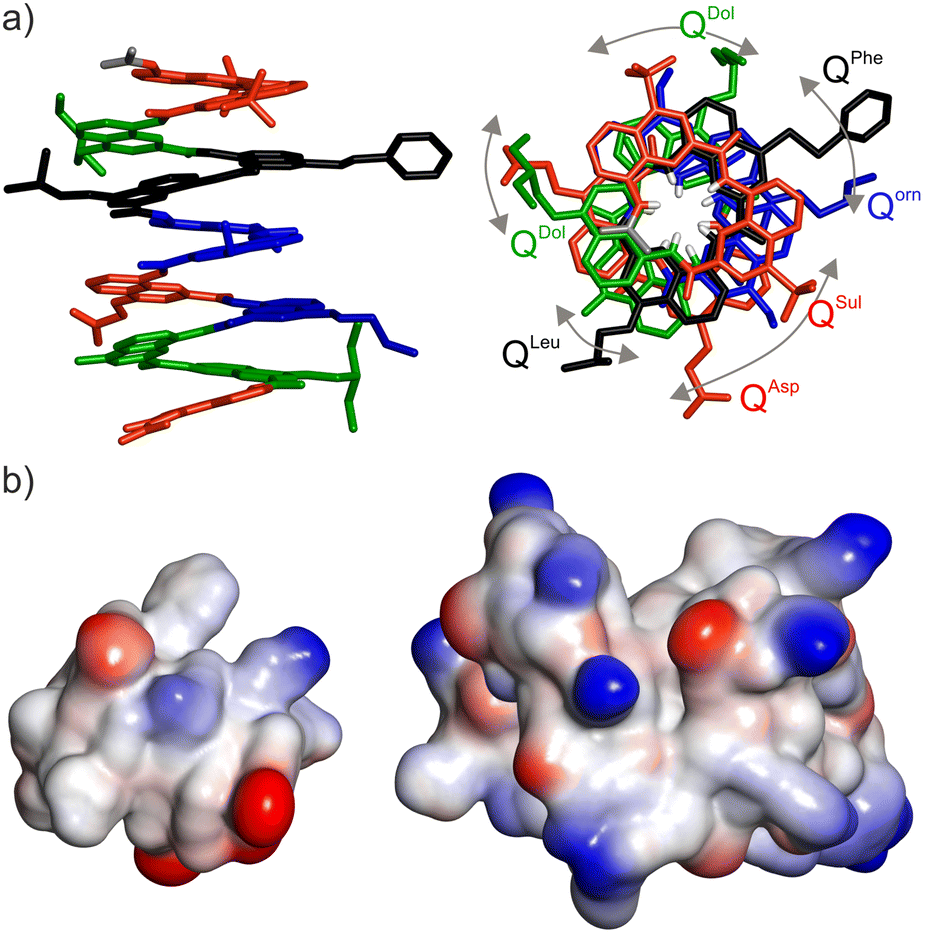 | ||
| Fig. 2 (a) Solid state structure of foldamer 2 determined by X-ray crystallography. Each monomer in stick representation is colour-coded as in Fig. 1a. Included solvent molecules and hydrogen atoms are omitted for clarity, except in the top view at right where amide NH protons are shown in white at the inner rim of the helix. Bent arrows indicate side chains that should be aligned in the top view if curvature was exactly 2.5 units per turn (see Fig. 1b). (b) Comparison of the size, shape and electrostatic potential (blue: cationic, red: anionic, white: neutral) of foldamer 2 (left) and a small protein shown at the same scale (Sac7D, PDB #1AZQ, 66 residues)14 illustrating the mini-protein-like aspect of the foldamer. | ||
For the pull-down experiments, biotinylated foldamer 1 was immobilized on streptavidin-coated magnetic beads and used as a bait.13 Since the main chain helix of 1 does not contain stereogenic centers, it exists as a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of right-handed (P) and left-handed (M) conformers. It is thus two, mirror-imaged helices that were used as baits. The beads were then incubated in a solution of cell lysate of S. cerevisiae (Fig. 3) to allow the foldamer to interact with putative prey proteins. After a thorough wash with phosphate buffered saline, bound prey proteins were eluted off from the beads (Fig. S3†) and subsequently digested with trypsin. Extracted peptides were then subjected to LC-MS/MS proteomic analysis. To ensure high fidelity of LC-MS/MS and assess statistical significance, the solution of extracted peptides was divided into three portions and analysed separately. In addition, the whole triplicate pull-down assay was repeated in three independent experiments. Proteomic analysis by LC-MS/MS spectrometry identified approximately 2000 proteins (Fig. S4 and ESI†). The abundance of proteins was compared to that obtained from control experiments with non-modified magnetic beads (i.e. streptavidin without foldamer as a bait). Identified prey proteins were then sorted based on criteria such as fold change and confidence. Across the three experiments, 153, 166 and 214 proteins with a fold change >2.0 were identified. Among those, 74 proteins were consistently observed with a fold change >2.0 in all three independent experiments, and another 75 proteins in two out of three experiments, highlighting the reproducibility of both protein pull-down and relative abundance (Fig. S5†). It should be pointed here that the conditions of the pull-down assay were not stringent, as reflected in the total number of proteins identified with high confidence and still large number of proteins with a fold change >2.0. Our priority was to establish the reproducibility of the assay and we started without knowing what the outcome would be. In future experiments, one may consider using wash buffers more efficient at disrupting weak interactions than the phosphate buffered saline used here.
:1 mixture of right-handed (P) and left-handed (M) conformers. It is thus two, mirror-imaged helices that were used as baits. The beads were then incubated in a solution of cell lysate of S. cerevisiae (Fig. 3) to allow the foldamer to interact with putative prey proteins. After a thorough wash with phosphate buffered saline, bound prey proteins were eluted off from the beads (Fig. S3†) and subsequently digested with trypsin. Extracted peptides were then subjected to LC-MS/MS proteomic analysis. To ensure high fidelity of LC-MS/MS and assess statistical significance, the solution of extracted peptides was divided into three portions and analysed separately. In addition, the whole triplicate pull-down assay was repeated in three independent experiments. Proteomic analysis by LC-MS/MS spectrometry identified approximately 2000 proteins (Fig. S4 and ESI†). The abundance of proteins was compared to that obtained from control experiments with non-modified magnetic beads (i.e. streptavidin without foldamer as a bait). Identified prey proteins were then sorted based on criteria such as fold change and confidence. Across the three experiments, 153, 166 and 214 proteins with a fold change >2.0 were identified. Among those, 74 proteins were consistently observed with a fold change >2.0 in all three independent experiments, and another 75 proteins in two out of three experiments, highlighting the reproducibility of both protein pull-down and relative abundance (Fig. S5†). It should be pointed here that the conditions of the pull-down assay were not stringent, as reflected in the total number of proteins identified with high confidence and still large number of proteins with a fold change >2.0. Our priority was to establish the reproducibility of the assay and we started without knowing what the outcome would be. In future experiments, one may consider using wash buffers more efficient at disrupting weak interactions than the phosphate buffered saline used here.
 | ||
| Fig. 3 Schematic representation of the pull-down assay and proteomic analysis. | ||
Among several candidates of prey proteins that were a priori amenable to simple recombinant expression in E. coli and were devoid of posttranslational modification, we chose to express four different proteins and assess their affinity for 1. The DNA repair and homologous recombination protein Rad5215 showed reproducible, moderate enrichment and high confidence. Through the three pull-down assays, the fold changes of Rad52 were ranked in 20th, 22nd, and 22nd place among all identified proteins. The same stands for the RNA binding protein SGN116 (ranked at the 31st, 25th and 43rd positions in the three pull-downs), the coenzyme Q9 homolog (COQ9) protein17 (ranked at the 50th, 5th and 32nd positions) and the splicing factor Mud2 (ranked at the 14th, 27th and 6th positions).18 All four proteins showed calculated p-values from Student's T-test that were below the threshold of 0.05, indicating a nominal statistical significance (Table 1).
| Exp. number | Fold change (rank)a | T-testb |
|---|---|---|
| a Enrichment in abundance when the bait was replaced from streptavidin to foldamer 1. b T-test uses the two-tailed distribution for samples with unequal variances. | ||
| Rad52 | ||
| 1st | 7.57 (20th) | 4.45 × 10−3 |
| 2nd | 6.44 (22nd) | 8.63 × 10−3 |
| 3rd | 11.39 (22nd) | 1.98 × 10−6 |
| SGN1 | ||
| 1st | 5.63 (31st) | 2.31 × 10−3 |
| 2nd | 5.89 (25th) | 9.65 × 10−3 |
| 3rd | 6.51 (43rd) | 2.92 × 10−6 |
| COQ9 | ||
| 1st | 4.01 (50th) | 4.71 × 10−3 |
| 2nd | 17.85 (5th) | 8.43 × 10−3 |
| 3rd | 8.51 (32nd) | 1.36 × 10−3 |
| Mud2 | ||
| 1st | 10.19 (14th) | 9.69 × 10−4 |
| 2nd | 5.38 (27th) | 3.99 × 10−5 |
| 3rd | 27.89 (6th) | 1.64 × 10−4 |
All four proteins were recombinantly expressed in E. coli. However, Mud2 proved to be somewhat problematic (poor overexpression, propensity to precipitate) and was not considered further. Binding studies using biolayer interferometry (BLI) were performed with the three remaining proteins to measure both binding kinetics and dissociation constants (KD). This technique was preferred because it allowed us to immobilize the biotinylated foldamer on the sensor and overcome problems associated with its aggregation or even poor solubility at concentrations that would be relevant for KD determination in solution. Isothermal titration calorimetry, for example, was attempted but gave poor results, on top of requiring a lot of material. To assess binding selectivity, we set to measure binding to P-1 and M-1 separately. These two compounds may in principle serve as ideal controls for one another in that their physical properties are essentially identical – thus alleviating non-specific effects – but the projection of their side chains are mirror images and may thus not lead to identical recognition properties. We thus endeavoured in the separation of P-1 and M-1 by chiral HPLC. Note that the handedness of helices of such length is kinetically inert in water at 25 °C: despite being conformational isomers, P-1 and M-1 do not interconvert to a detectable extent.19
In previous studies, we have demonstrated the possibility to separate the P- and M-helical conformers of organo-soluble aromatic oligoamides of moderate sizes (from hexamer to hexadecamer) by chiral HPLC using a Chiralpak™ IA stationary phase under normal phase conditions (i.e. an n-hexane/chloroform mixture).20 Here, we sought for a stationary phase and eluting conditions allowing for a separation in reversed-phase mode. We selected the Chiralpak™ QN-AX, a quinine-based stationary phase initially developed for the separation of chiral α-amino acids.21 Finding elution conditions took some screening and optimization but we eventually found that a solvent mixture composed of 30% acetonitrile in triethylammonium acetate buffer (150 mM, pH 7.21) resulted in an impressive separation of the two helical conformers with a separation factor of 3.7 between M-1 and P-1. The assignment of the handedness was made using a circular dichroism (CD) detector connected after the UV/Vis detector (Fig. S6†) and based on previously published absolute handedness assignment of such aromatic helices.22 Thanks to the remarkable peak separation, we could directly use the analytical column to successfully isolate both helix conformers from the M-1/P-1 mixture. We finally further confirmed the chiral purity by reinjecting the collected pure fractions on the same column (Fig. S7†). Even an octamer shows no detectable handedness inversion after long incubation in water.19,20,23
The biotinylated P- and M-helices of 1 were next independently immobilized on BLI streptavidin sensors tips. Loading was performed at a concentration of 2 μg mL−1. The sensors were then exposed to a range of protein concentrations for time course monitoring of the association before being dipped in a buffer solution to record the dissociation. The interaction between Rad52 and between P-1 and M-1 was assessed in multiple runs with a protein concentration ranging from 125 nM to 7.75 nM. Real-time BLI sensorgrams fit very well to a 1:1 kinetic binding model. Rapid association (large ka) and slow dissociation (low kd) were calculated by curve fitting (Fig. 4, Table 2) yielding a remarkably low nanomolar KD. The values obtained for M-1 and P-1 differed marginally (1.3 and 1.5 nM, respectively), suggesting that the chiral features of 1 are not critical for binding. The lack of selectivity came as a surprise given the low Kd values. The exact binding mode was not further investigated. Nevertheless, one may recall that yeast Rad52 is a 490 amino acid protein. A 90-residue domain near the N-terminus is known to form a stable decamer15a with an inner cavity of 32 nm. Most of the remaining sequence is not seen experimentally by cryo-electron microscopy15a and is predicted by AlphaFold not to belong to a stable tertiary structure (Fig. S8†). In solution, a 10:1 foldamer binding ratio of the foldamer to the 10mer may be considered unless the foldamer binds in the central cavity of the 10mer. This is precluded in the BLI experiments where the foldamer is immobilized on the sensor. One may still envisage that a certain degree of multivalency (or “avidity”), e.g. two or more Rad52 subunits of the 10mer binding to two or more foldamers on the sensor, contribute to the low KD values. We performed additional BLI experiments decreasing the loading of foldamer on the sensor to prevent multivalency and observed no change in the shape of the response curve, which suggests the binding observed is indeed 1:1.
 | ||
| Fig. 4 Distinct BLI experiments report on the 1:1 binding affinity of protein Rad52 to P-1 (left graphs) and M-1 (right graphs) yielding KD = 1.5 and 1.3 nM, respectively. Both biotinylated chiral helices were immobilized onto the BLI SA sensors tips (3 × P-helix and 3 × M-helix tips at 2 μg ml−1 for the loading and two reference tips were used). The BLI tips were then dipped into solutions containing varying concentrations of Rad52 in buffer (20 mM Hepes (pH 7.4), 150 mM NaCl, 0.02% Tween-20®) to record the concentration-dependent association over 240 s: (a) 125–31.25 nM and (b) 31–7.75 nM. Next the sensors were dipped in pure buffer solution to record dissociation events over 300 s. Experimental curves are shown in blue and calculated curves according to a 1:1 binding isotherm are shown in red. | ||
| Protein/P-1 or M-1 | k a (103 M−1 s−1) | k d (10−6 s−1) | K D (nM) |
|---|---|---|---|
|
a The KD values have been calculated assuming an equilibrium level of the SGN1 binding to the sensor tips (Langmuir's equation).
b Curve fitting to a 1:1 binding model was not ideal. We thus choose not to report precise KD values but rather an order of magnitude (see ESI†).
|
|||
| Rad52 | |||
| P-1 | 125 ± 0.36 | 187 ± 1.72 | 1.49 ± 0.015 |
| M-1 | 124 ± 0.31 | 163 ± 1.51 | 1.31 ± 0.013 |
| SGN1 | |||
| P-1 | — | — | 104.9 ± 10.7a |
| M-1 | — | — | 115.5 ± 9.2a |
| COQ9 | |||
| P-1 | 4.10 ± 0.033 | 2034 ± 15.22 | ≈500b |
| M-1 | 3.38 ± 0.026 | 1695 ± 12.88 | ≈500b |
The binding of SGN1 was estimated in a similar manner. Rapid association and dissociation were recorded but, after the initial binding event when a steady state should be reached, a gradual linear climbing of the association step was noticed as if SGN1 was further associating to itself on top of its binding to 1 on the sensor. Adding 2% Bovine Serum Albumin (BSA) to the buffer reduced this phenomenon but did not completely remove it, making the curve fitting to a 1:1 kinetic binding model inaccurate. Consequently, the KD values of SGN1 binding to M-1 and P-1 were calculated with the Langmuir's equation and assuming that equilibrium was reached after the initial climb of the signal (steady state). Binding in the three-digit nanomolar range was observed and again revealed similar association to M-1 and P-1 (KD of 105 and 115 nM, respectively, Fig. S9†). Unlike for Rad52, not much is known about the structure of yeast SGN1 other than that it contains an RNA binding domain.16 Alphafold predicts that only a small part of its 250 residues belong to a stable tertiary fold (Fig. S8†) but such a prediction may be taken with caution. In contrast, the structure of the 228-residue yeast COQ9 is not known but AlphaFold predicts a fully folded protein similar to the structure of human COQ9 which contains ten α-helices (Fig. S8†). For BLI measurement with COQ9, we screened a range of buffers with variable amounts of BSA (1–2%) and/or Tween-20® as a detergent (0.05–0.1%) to reduce protein aggregation and eventually obtained a good, albeit not ideal, fit to a 1:1 kinetic binding model. KD is therefore provided as an order of magnitude (Table 2). The best binding behaviour was again obtained with HEPES buffer. Binding of COQ9 to 1 was weaker than for the two other proteins, although still in the submicromolar range, with again no preference for the P- or M-helix (Table 2 and Fig. S10†).
Conclusions
A single helical aromatic foldamer, not resulting from a specific design but simply displaying different side chains at its surface, was shown to possess nanomolar affinity for certain proteins identified from a cell lysate. These results bode well for the use of such medium-sized molecules to interfere with cellular functions. While efforts towards the structure-based design of helical foldamers to recognize protein surfaces are already under way,9–11 these new results suggest that simple screening approaches may also yield promising results. The current study focused on the detection of binding and led to the identification of protein binders that do not necessarily play an important role in diagnostics or therapeutic intervention. For that reason, further investigations on the selectivity of binding, on the thermodynamics involved – are associations entropy or enthalpy driven – on the structural elucidation of the interactions involved, or on whether these interactions result in interference with protein function were not undertaken. Future efforts will instead focus on screening the effects of various foldamers for relevant cell phenotypes ensuring that what is detected is not solely binding but also function.Author contributions
SK, VM, LW, PKM, SC, and CD performed the experiments. IH supervised the research. All authors contributed to the writing, reviewing and editing of the manuscript and approved its final version.Data availability
The data supporting this article have been included as part of the ESI. Crystallographic data for 2 has been deposited at the CCDC under # 2280177 and can be obtained from https://www.ccdc.cam.ac.uk.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) through project CRC1309-C7 (project ID 325871075) to I. H. We would like to thank Prof. T. Sugiyama for having kindly offered us the Rad52 construct.References
- (a) A. A. Vinogradov, Y. Yin and H. Suga, J. Am. Chem. Soc., 2019, 141, 4167 CrossRef CAS PubMed; (b) T. A. F. Cardote and A. Ciulli, ChemMedChem, 2016, 11, 787 CrossRef CAS PubMed; (c) P. Timmerman, J. Beld, W. C. Puijk and R. H. Meloen, ChemBioChem, 2005, 6, 821 CrossRef CAS; (d) C. Heinis, T. Rutherford, S. Freund and G. Winter, Nat. Chem. Biol., 2009, 5, 502 CrossRef CAS PubMed; (e) K. Maola, J. Wilbs, J. Touati, M. Sabisz, X.-D. Kong, A. Baumann, K. Deyle and C. Heinis, Angew. Chem., 2019, 131, 11927 CrossRef; (f) Z. C. Adams, A. P. Silvestri, S. Chiorean, D. T. Flood, B. P. Balo, Y. Shi, M. Holcomb, S. I. Walsh, C. A. Maillie, G. K. Pierens, S. Forli, K. J. Rosengren and P. E. Dawson, ACS Cent. Sci., 2023, 9, 648 CrossRef CAS PubMed; (g) A. Krzyzanowski, L. M. Esser, A. Willaume, R. Prudent, C. Peter, P. Hart and H. Waldmann, J. Med. Chem., 2022, 65, 15300 CrossRef CAS PubMed.
- (a) J. Helma, M. C. Cardoso, S. Muyldermans and H. Leonhard, J. Cell Biol., 2015, 209, 633 CrossRef CAS PubMed; (b) D. Schumacher, J. Helma, A. F. L. Schneider, H. Leonhardt and C. P. R. Hackenberger, Angew. Chem., Int. Ed., 2018, 57, 2314 CrossRef CAS PubMed; (c) F. Y. Frejd and K.-T. Kim, Exp. Mol. Med., 2017, 49, e306 CrossRef CAS PubMed; (d) S. Schlehuber and A. Skerra, Expert Opin. Biol. Ther., 2005, 5, 1453 CrossRef CAS PubMed.
- (a) P. S. Pendergrast, H. N. Marsh, D. Grate, J. M. Healy and M. Stanton, J. Biomol. Tech., 2005, 16, 224 Search PubMed; (b) M. Takahashi, Biochimie, 2018, 145, 63 CrossRef CAS PubMed; (c) Z. Zhang, J. Li, R. Amini, A. Mansfield, J. Gu, J. Xia, J. D. Brennan and Y. Li, Analysis Sensing, 2023, 3, e202300001 CrossRef CAS.
- L.-G. Milroy, T. N. Grossmann, S. Hennig, L. Brunsveld and C. Ottmann, Chem. Rev., 2014, 114, 4695 CrossRef CAS PubMed.
- W. S. Horne and T. N. Grossmann, Nat. Chem., 2020, 12, 331 CrossRef CAS PubMed.
- (a) D.-W. Zhang, X. Zhao, J.-L. Hou and Z.-T. Li, Chem. Rev., 2012, 112, 5271 CrossRef CAS PubMed; (b) I. Huc, Eur. J. Org. Chem., 2004, 17 CrossRef CAS.
- (a) D. Mazzier, S. De, B. Wicher, V. Maurizot and I. Huc, Angew. Chem., Int. Ed., 2020, 59, 1606 CrossRef CAS PubMed; (b) S. De, B. Chi, T. Granier, T. Qi, V. Maurizot and I. Huc, Nat. Chem., 2018, 10, 51 CrossRef CAS PubMed.
- X. Hu, S. J. Dawson, P. K. Mandal, X. de Hatten, B. Baptiste and I. Huc, Chem. Sci., 2017, 8, 3741 RSC.
- (a) J. M. Alex, V. Corvaglia, X. Hu, S. Engilberge, I. Huc and P. B. Crowley, Chem. Commun., 2019, 55, 11087 RSC; (b) P. S. Reddy, B. Langlois d'Estaintot, T. Granier, C. D. Mackereth, L. Fischer and I. Huc, Chem. – Eur. J., 2019, 25, 11042 CrossRef CAS PubMed; (c) M. Vallade, M. Jewginski, L. Fischer, J. Buratto, K. Bathany, J.-M. Schmitter, M. Stupfel, F. Godde, C. D. Mackereth and I. Huc, Bioconjugate Chem., 2019, 30, 54 CrossRef CAS PubMed; (d) J. Buratto, C. Colombo, M. Stupfel, S. J. Dawson, C. Dolain, B. Langlois d'Estaintot, L. Fischer, T. Granier, M. Laguerre, B. Gallois and I. Huc, Angew. Chem., Int. Ed., 2014, 53, 883 CrossRef CAS PubMed.
- (a) S. Kumar, M. Birol, D. E. Schlamadinger, S. P. Wojcik, E. Rhoade and A. D. Miranker, Nat. Commun., 2016, 7, 11412 CrossRef CAS PubMed; (b) S. Kumar, A. Henning-Knechtel, I. Chehade, M. Magzoub and A. D. Hamilton, J. Am. Chem. Soc., 2017, 139, 17098 CrossRef CAS PubMed; (c) J. Ahmed, T. C. Fitch, C. M. Donnelly, J. A. Joseph, T. D. Ball, M. M. Bassil, A. Son, C. Zhang, A. Ledreux, S. Horowitz, Y. Qin, D. Paredes and S. Kumar, Nat. Commun., 2022, 13, 2273 CrossRef CAS PubMed.
- (a) K. Ziach, C. Chollet, V. Parissi, P. Prabhakaran, M. Marchivie, V. Corvaglia, P. P. Bose, K. Laxmi-Reddy, F. Godde, J. M. Schmitter, S. Chaignepain, P. Pourquier and I. Huc, Nat. Chem., 2018, 10, 511 CrossRef CAS PubMed; (b) V. Corvaglia, D. Carbajo, P. Prabhakaran, K. Ziach, P. K. Mandal, V. D. Santos, C. Legeay, R. Vogel, V. Parissi, P. Pourquier and I. Huc, Nucleic Acids Res., 2019, 47, 5511 CrossRef CAS PubMed; (c) V. Kleene, V. Corvaglia, E. Chacin, I. Forne, D. B. Konrad, P. Khosravani, C. Douat, C. F. Kurat, I. Huc and A. Imhof, Nucleic Acids Res., 2023, 51, 9629 CrossRef CAS PubMed.
- (a) V. Corvaglia, F. Sanchez, F. S. Menke, C. Douat and I. Huc, Chem. – Eur. J., 2023, e202300898 CrossRef CAS PubMed; (b) B. Baptiste, C. Douat-Casassus, K. Laxmi-Reddy, F. Godde and I. Huc, J. Org. Chem., 2010, 75, 7175 CrossRef CAS PubMed.
- J. Maryas, J. Faktor, L. Čapkova, P. Müller, P. Skladal and P. Bouchal, Cancer Genomics Proteomics, 2018, 15, 395 CrossRef CAS PubMed.
- S. Su, Y.-G. Gao, H. Robinson, Y.-C. Liaw, S. P. Edmondson, J. W. Shriver and A. H.-J. Wang, J. Mol. Biol., 2000, 303, 395 CrossRef CAS PubMed.
- (a) J. Deveryshetty, R. Chadda, J. R. Mattice, S. Karunakaran, M. J. Rau, K. Basore, N. Pokhrel, N. Englander, J. A. J. Fitzpatrick, B. Bothner and E. Antony, Nat. Commun., 2023, 14, 6215 CrossRef CAS PubMed; (b) N. V. Khade and T. Sugiyama, PLoS One, 2016, 11, e0158436 CrossRef PubMed.
- A. Gaspary, R. Laureau, A. Dyatel, G. Dursuk, Y. Simon and L. E. Berchowitz, J. Cell Biol., 2023, 222, 1 CrossRef PubMed.
- C. H. He, D. S. Black, T. P. T. Nguyen, C. Wang, C. Srinivasan and C. F. Clarke, Biochim. Biophys. Acta, 2015, 1851, 1227 CrossRef CAS PubMed.
- (a) J.-C. Rain and P. Legrain, EMBO J., 1997, 16, 1759 CrossRef CAS PubMed; (b) R. Minocha, V. Popova, D. Kopytova, D. Misiak, S. Hüttelmaier, S. Georgieva and K. Sträßer, Nucleic Acids Res., 2018, 46, 9749 CrossRef CAS PubMed.
- T. Qi, V. Maurizot, H. Noguchi, T. Charoenraks, B. Kauffmann, M. Takafuji, H. Ihara and I. Huc, Chem. Commun., 2012, 48, 6337 RSC.
- N. Delsuc, T. Kawanami, J. Lefeuvre, A. Shundo, H. Ihara, M. Takafuji and I. Huc, ChemPhysChem, 2008, 9, 1929 CrossRef PubMed.
- M. Lämmerhofer and W. Lindner, J. Chromatogr. A, 1996, 741, 33 CrossRef.
- A. M. Kendhale, L. Poniman, Z. Dong, K. Laxmi-Reddy, B. Kauffmann, Y. Ferrand and I. Huc, J. Org. Chem., 2011, 76, 195 CrossRef CAS PubMed.
- S. J. Dawson, Á. Mészáros, L. Petho, C. Colombo, M. Csékei, A. Kotschy and I. Huc, Eur. J. Org. Chem., 2014, 4265 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary figures and data, detailed experimental protocols and characterisation of new compounds. CCDC 2280177. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ob01436g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |