
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTetraarylammonium salts – synthesis, properties and emerging use
Alexander
Marriott
and
Götz
Bucher
 *
*
School of Chemistry, University of Glasgow, Joseph Black Building, University Avenue, Glasgow G12 8QQ, UK. E-mail: goetz.bucher@glasgow.ac.uk
First published on 28th October 2024
Abstract
The chemistry of tetraarylammonium salts, first explored in the 1960s Soviet Union, has long been a dormant field of research. This is owing to the inherent difficulty in adding a fourth benzene ring to the nitrogen atom of the sterically demanding and low-nucleophilicity triphenylamine molecule. Only recently have new developments in synthetic methodology made access to tetraarylammonium salts less of a tour de force, and first applications are beginning to emerge. As a consequence, the number of publications in this field of research is growing. The review covers the complete field of research, from the beginnings to the present day.
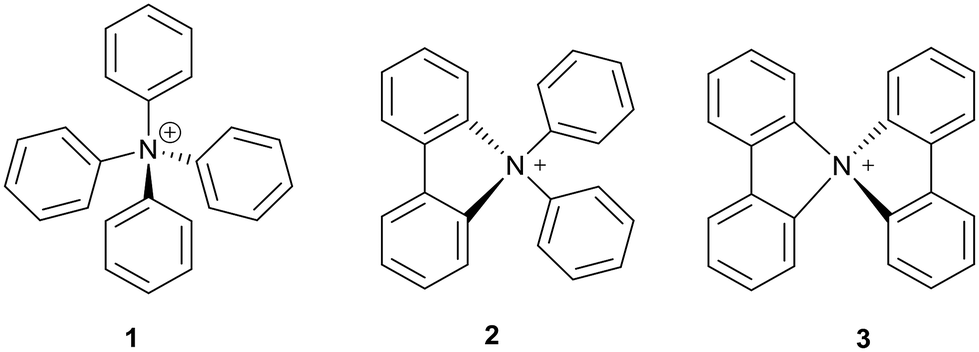
Tetraarylammonium salts (TAASs) are a relatively new class of ionic compounds consisting of cationic nitrogen species with four aromatic substituents bonded to the central nitrogen atom, with a range of counter-anions.1,2 Examples include salts of cations like tetraphenylammonium 1, N,N-diphenylcarbazolium 2, and spiro-dicarbazolium 3 (Scheme 1). TAASs are related to the far more widely known tetraarylphosphonium salts (Ar4P+ X−), which find application in biomedical research,3,4 as phase transfer catalysts,5 and more recently also in battery research.6
 | ||
| Scheme 1 Some tetraarylammonium cations. | ||
Quaternary ammonium salts (QASs) have a variety of industrial uses such as antimicrobials, surfactants, and phase transfer catalysts. Perhaps most crucial to the needs of modern society is their potential to be used as cationic functional groups in the membranes of anion exchange-membrane fuel cells (AEMFCs) and water electrolysers (AEM-WEs). However, the harsh alkaline conditions of such applications lead to rapid degradation via reaction with hydroxide.7 TAASs tend to exhibit better thermo- and hydrolytic stability than their alkyl counterparts,2 hence the importance of developing synthetic routes and studying the effect of functionalised aryl substituents on the properties of QASs.
Synthetic strategies
Tetraarylammonium cations are significantly harder to synthesize than most other per-aryl-X-onium ions, such as the easily attainable tetraarylphosphonium ions. This is attributed to the short C–N bonds and mostly planar structure of triaryl amines.8 Furthermore, the nitrogen lone pair is partially dispersed through the aromatic systems and thus less available as a nucleophile than it is in other amines. The combination of this steric crowding and low nucleophilicity presents a challenge to the synthesis of tetraaryl ammonium cations.8 Due to this lack of nucleophilicity of triphenylamine, nucleophilic aromatic substitution (SN2Ar) as an arylation strategy fails. This was demonstrated by Makarova and Nesmeyanov, in an early unsuccessful attempt (see Scheme 2) at the synthesis of tetraphenylammonium via the reaction of triphenylamine 4 with diphenyliodonium salts 5.9 | ||
| Scheme 2 An early unsuccessful approach towards tetraarylammonium cations. | ||
A successful synthesis must overcome the disinclination of triaryl amines to react via their nitrogen lone pairs, which therefore necessitates the use of extremely electrophilic and reactive arylating agents – aryl cations. The use of extremely reactive aryl cations as reaction partners, however, comes at the cost of drastically reduced selectivity. Singlet aryl cations, such as the parent phenyl cation, are known to be extremely unselective, reacting with little or no barrier with whatever nucleophile is present. To force the arylation reaction to take place on the triarylamine nitrogen atom (and not in the periphery, on one of the benzene rings), the final N–C bond forming step is normally designed to be an intramolecular cyclization reaction, to make the reaction as entropically favourable as possible.
There have been two common ways to generate the aryl cations required, such as 8: first by the decomposition of a diazonium substituent (in 7) to produce an aryl cation proximal to the N-centre, and more recently by the silyl cation-promoted activation of an aryl C–F bond (in 9) with similar regiochemistry.8 It has been shown that both methods produce similar byproducts, indicating that they both proceed via the same phenyl cation-type intermediate 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 8 as mapped in Scheme 3. The arylation of lone pair-containing heteroatoms in this manner is facilitated by the formation of a 5-membered ring.
8 as mapped in Scheme 3. The arylation of lone pair-containing heteroatoms in this manner is facilitated by the formation of a 5-membered ring.
 | ||
| Scheme 3 Preparation of N,N-diphenylcarbazolium 2via a phenyl cation intermediate 8. | ||
This diazonium salt decomposition strategy was first demonstrated by Nesmeyanov, who used it to prepare N,N-diphenylcarbazolium 2 from 2′-diphenylaminobiphenyl-2-diazonium 7.10 Hellwinkel and Seifert then applied this method to the formation of 9,9′-spirobicarbazolium 3 and detailed the pathway to the diazonium precursor (Scheme 4).11,12 2,2′-Dinitrobiphenyl 10 was formed via Ullman coupling of 2-iodonitrobenzene. Partial reduction of this product by sodium polysulfide, followed by a Sandmeyer reaction, afforded 2-iodo-2′-nitrobiphenyl 12. This aryl halide was used to arylate carbazole through copper catalysis, yielding a triaryl amine 13 with a proximal nitro group ready for reduction and diazotisation.
 | ||
| Scheme 4 Hellwinkel's synthesis of spirodicarbazolium 3. | ||
Hellwinkel and Seifert found the yields of these intramolecular cyclisations to be somewhat lacking, so conducted an examination of the byproducts formed. Chromatography suggested that they were crystalline with triarylamine-like properties.12 It was postulated that the aryl cation intermediate was not only reacting with the nitrogen lone pair but also with the π-system of either aromatic ring, forming a bond with the carbon ortho to the triarylamine nitrogen to yield a cyclohexadienyl-type carbocation 15 as shown in Scheme 5. The resulting azepine 16 (and azepino carbazole in the case of a spirobicarbazolium target) was confirmed with mass spectrometry.12 It was found that the ratio of tetraarylammonium product to such a byproduct was consistent across a range of conditions and was markedly higher for formation of 9,9′-spirobicarbazoliums than of N,N-diphenylcarbazolium. This was rationalised by the presence of a bridge between aromatic rings that limits the number of ortho positions left vulnerable to this side reaction.12
 | ||
| Scheme 5 Formation of azepine derivatives by addition of a phenyl cation intermediate to an aromatic ring. | ||
Subsequent syntheses of TAASs have mostly been variations of the same strategy with alternative pathways to a diazonium salt, the decomposition of which allows a phenyl cation to react intramolecularly with a triaryl amine. Unsatisfied with the many-step synthetic pathway previously established, Gjineci et al. recently developed a 3-step synthesis from a 2,2′-dinitrobiphenyl 10 starting material (Scheme 6).13 This was reduced using zinc in acidic conditions to 2,2′-diaminobiphenyl 17, which was coupled with two equivalents of iodobenzene in an Ullmann arylation reaction. It was observed that after arylation of one primary amino group, the second arylation occurs preferentially with the same (now secondary) amine.14 This regioselectivity was exploited to reliably produce 2′-diphenylaminobiphenyl-2-amine 18. Finally, diazotisation and subsequent decomposition afforded N,N-diphenyl carbazolium 2via the same intramolecular ring-closing reaction as previous syntheses.
 | ||
| Scheme 6 Synthesis of N,N-diphenylcarbazolium 2 according to Gjineci et al. | ||
This simpler approach has allowed larger-scale preparation of TAASs, but the overall yield is still not entirely satisfactory (12% over three steps).14 It should also be noted that one of the key intermediates (17) is a regioisomer of benzidine, a known carcinogen which has been heavily restricted.15 Variation of the aryl halide in this pathway has afforded a range of substituents on the phenyl rings of N,N-diphenyl carbazolium, and the use of a biphenyl dihalide created 9,9′-spirobicarbazolium.13 However, this arylation reaction took 7 days to complete, due to the challenging steric hindrance at the ortho position of the halide.
More recently, a synthesis has been demonstrated to reach halogenated spirobicarbazolium salts via Suzuki coupling between an N-iodoaryl carbazole 18 and an aminoaryl pinacolyl boronic ester (Scheme 7), yielding an N-biphenylamine carbazole 19 with the correct regiochemistry to undergo a ring-closing diazonium decomposition as before.2 This strategy provided modest yields that varied widely in relation to the electron-withdrawing nature of halide substituents on the starting materials. Synthesis of unsubstituted spirobicarbazolium via Suzuki coupling to 2-fluorophenylboronic acid (Scheme 7) had been reported earlier and had a good yield of fluorobiphenyl carbazole 20,8 however the process utilised a C–F activation step rather than the more popular diazonium salt decomposition strategy. This involves the use of silyl cations and microwave radiation, and this synthetic pathway also required the preparation of the aryl carbazole starting material 18.8
 | ||
| Scheme 7 Access to precursors to spirobicarbazolium via Suzuki coupling. | ||
The synthetic achievements outlined above have all relied on the intramolecular ring-closing reaction of a phenyl cation to circumvent the poor reactivity of a triaryl amine group. This has meant that the products all contain, by necessity, at least one bridge moiety between aromatic rings. A simpler, non-bridged tetraphenyl ammonium cation may serve as an important benchmark for the characterisation of TAASs but has historically been hard to come by.16 With this goal in mind, Fujita et al. developed a novel synthetic strategy for the arylation of a triaryl amine. Instead of an intramolecular cyclisation of an aryl closed-shell cation with a neutral tertiary amine, an intermolecular radical coupling reaction was performed between a neutral aryl radical and a triarylammonium radical cation.16
Quantum calculations had shown that in a triphenyl ammonium radical cation, the SOMO is spread across the phenyl groups as well as the central nitrogen.16 Thus, the main hurdle in the development of this synthesis was dimerisation through the para positions of the phenyl rings leading to an undesired neutral closed-shell byproduct. To combat this, a triaryl amine was developed with bulky tert-butyl groups in the meta positions and bromides in the para positions, which could be removed after coupling. This steric hindrance of the aromatic rings allowed an intermolecular radical coupling via the central ammoniumyl radical cation (Scheme 8).16 It is noted that this coupling reaction is facilitated by the persistent radical effect, based on the kinetic stability of the triarylamine radical cation.
 | ||
| Scheme 8 Synthesis of a tetraarylammonium cation via radical coupling reaction. | ||
This protected triaryl amine 21 was activated using silver tetrafluoroborate for a one-electron oxidation reaction. The resulting triarylammoniumyl radical cation 22 was coupled with an aryl radical 24, which also had tert-butyl blocking groups and was produced in situ from a dibenzoyl peroxide derivative 23.16 Although the yield of the desired product of this radical coupling was extremely low, it allowed the isolation of a non-bridged tetraaryl ammonium cation 25. Removal of the bromine substituents via lithium – halogen exchange using n-BuLi followed by protonation gave tert-butylated cation 26, which, after removal of the tert-butyl groups by heating in trifluoromethane sulfonic acid, resulted in the first extensive characterisation of a tetraphenyl ammonium salt 1.16
Superheavy hydrogen 3H (or tritium T) decays to 3He via β− decay, with a half-life of t½ = 12.32 years. This radioactive decay process has been used to generate highly reactive phenyl cations 29 from hexatritiobenzene 27, as the initially formed phenyl cation-helium complex 28 dissociates readily. The tritiated phenyl cations 29 thus formed can be generated in unpolar solvents like benzene, are extremely reactive, and are readily trapped even by very weak nucleophiles. Due to radiological hazards and the high cost of tritium, these reactions can only be performed on a very small scale (typically using μL amounts of C6T6), and as reaction times of several years (corresponding to a significant portion of the half-life of tritium) are impracticable, only extremely small degrees of conversion are achievable. Hence, any characterization of ionic products formed by necessity is limited to an assay of allocating the activity of the remaining tritium to different fractions of the product mixture, and in some cases TLC or HPLC analysis, thus providing indirect evidence only. This method has been used to generate elusive diarylfluoronium cations,17,18 but could also be used to generate parent tetraphenylammonium 1-T5 from triphenylamine, albeit in a small yield (ca. 3% of the remaining activity) (Scheme 9).19
 | ||
| Scheme 9 Formation of a TAAS 1-T5 from hexatritiobenzene 27 and triphenylamine. | ||
Crystallographic and spectroscopic characterisation
Single-crystal X-ray diffraction allowed Fujita et al. to confirm the tetrahedral structure of the tetraphenyl ammonium cations 1 and 26 that they had produced, as well as derive the bond lengths within and compare them to similar compounds.16 It was found that the N–C(sp2) bond at the centre of the structure had a length of 1.529 Å, which was longer than the corresponding bond length in other methylphenyl ammonium cations 30–32 as well as in N,N-diphenylcarbazolium 2.16 From this comparison (Fig. 1) it was deduced that the tetraphenyl environment provided a high level of steric hindrance around the ammonium centre. Other tetraphenyl examples, such as tetraphenylmethane 33 or tetraphenylborate anion 34, exhibit a longer bond from the central atom to the aromatic carbon than their ammonium counterpart. This indicated that tetraphenyl ammonium is the most sterically congested of the class, which may be an explanation for its elusiveness.16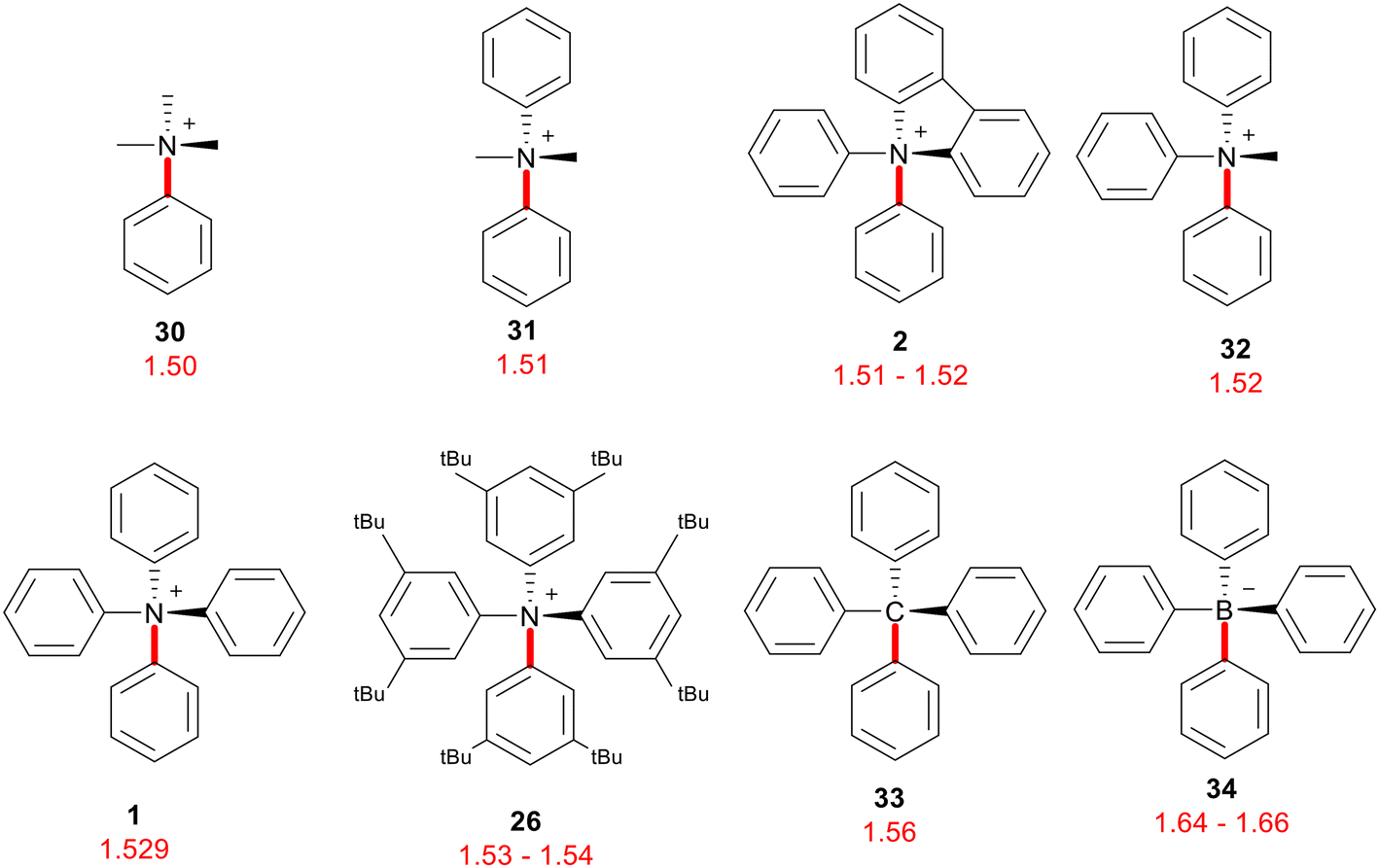 | ||
| Fig. 1 N–C(sp2) bond lengths (Å) of various aryl ammonium cations, and corresponding lengths in tetraphenyl analogues. | ||
Also measured was the shortest distance between the ortho-hydrogen and ipso carbon of neighbouring phenyl groups at 2.46 Å, which is shorter than the combined van der Waals radii of carbon and hydrogen thus steric repulsion is generated.16 Once again, other tetraphenyl analogues 33, 34 have a longer H–C distance and thus a less significant amount of steric repulsion (Fig. 2). The octa-tert-butylated tetraphenyl ammonium cation 26 was also analysed, and it was found that both the N–C(sp2) bond length and H–C distance were slightly longer than in the simple tetraphenyl ammonium case. This was taken to suggest that these meta-tert-butyl groups generated steric and electronic effects on the ammonium system.16
 | ||
| Fig. 2 o-H–i′-C distances (Å) in tetraphenyl ammonium and analogues. | ||
The X-ray crystallography of bridged N,N-diaryl carbazolium salts had been reported previously by Gjineci et al., who found that the N–C(sp2) bond length for the freely-rotating phenyl ring was 1.516 Å (shorter than that of tetraphenyl ammonium, but longer than other common arylammonium salts) and for the carbazole moiety was ca. 1.51 Å.13 This discrepancy in N–C bond length between the phenyl rings and the carbazole moiety was observed both with and without alkyl substitution on the phenyl rings. Furthermore, it was found that the N–C(sp2) bond length in 9,9′-spirobicarbazolium was shorter still, ranging from 1.498–1.506 Å.13
Although Hellwinkel and Seifert did not conduct crystallographic studies on diaryl carbazolium salts, they did publish spectroscopic data obtained by UV and NMR methods.12 It was found that while anilinium and methylphenyl ammonium compounds exhibited UV spectra that were similar to benzene, the spectra of the group of carbazolium-based cations resembled those of the fluorene class.14 In these carbazolium ions, a trend was observed where successive substitution of methyl groups with phenyl groups (35, 36, 2) induced a bathochromic shift in the UV spectrum of each member of the series as listed in Fig. 3, with spirobicarbazolium 3 having its longest wavelength absorption band at 301 nm.14 This trend is also present in the fluorene series, where the progression from 9,9-dimethylfluorene to 9,9′-spirobifluorene is accompanied by bathochromic shift. Hellwinkel and Seifert attributed this trend to a conjugative interaction between perpendicular π-systems separated by a tetrahedral centre.12
 | ||
| Fig. 3 Long-wavelength absorption maxima [nm] in the UV spectra of carbazolium cations. | ||
Hellwinkel and Seifert noted that the 1H NMR spectrum of spirobicarbazolium was surprisingly simple, with aromatic proton signals that could be trivially assigned by comparison with the spectrum of the tetramethylated spirobicarbazolium that was also recorded.12 It was observed that the position ortho to the ammonium centre was farthest upfield while the meta position (ortho to the bridge moiety) was furthest downfield. By comparison with other carbazolium systems with a range of N-substituents, it was established that the charge and degree of substitution on the nitrogen were the primary factors influencing the chemical shift of the ortho position. In neutral cases such as unsubstituted and monosubstituted N-methyl carbazoles, this resonance is comparable to other aromatic resonances, but coordination of another methyl substituent to form an ammonium cation resulted in substantial deshielding and thus a downfield shift.12 In contrast, the case of spirobicarbazolium saw that the deshielding effect of the positively charged central nitrogen is outweighed by the shielding effect of the second carbazole moiety caused by the geometric conditions of the cation. The result of this is that the chemical shift of the ortho hydrogens in spirobicarbazolium 3, listed in Fig. 4, is further upfield than in other carbazolium cations.12
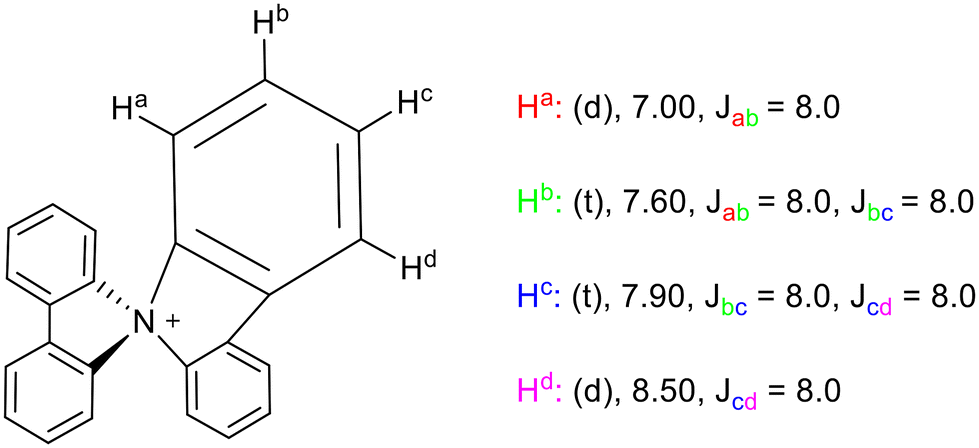 | ||
| Fig. 4 1H NMR signals of spirobicarbazolium 3, assigned to specific protons with (multiplicity), chemical shift δ (ppm), and J values (Hz). | ||
Alongside their crystallographic data, Fujita et al. also published 1H and 13C NMR spectra of tetraphenyl ammonium 1, which showed that the resonances were all shifted downfield in comparison to triphenylamine.16 This was attributed to the strong electron withdrawing effect of the positively charged nitrogen in tetraphenyl ammonium as well as the loss of lone pair resonance present in triphenylamine. It was also observed that the proton resonances at the meta and para positions of tetraphenyl ammonium 1 appeared further upfield than the corresponding signals from tetraphenyl phosphonium 37 (Fig. 5).16 It was postulated that this was due to strong anisotropic effects in tetraphenyl ammonium as a result of being smaller than the phosphonium counterpart. The same relation was observed when comparing the spectrum of tetraphenyl methane 33 with that of tetraphenyl silane 38.16
 | ||
| Fig. 5 Chemical shifts δ (ppm) of meta and para1H NMR resonances in tetraphenyl compounds 37, 1, 38 and 33. | ||
Alkaline stability
The development of water electrolysers and hydrogen fuel cells for the storage of electrical energy is a highly active and competitive field in current research. A leading technological framework in this field has been the proton exchange membrane water electrolyser (PEM-WE) or fuel cell (PEMFC).7 Devices such as these involve the transfer of protons between the electrodes via an electrolyte. Since the anode and cathode cell compartments must be kept separate to prevent the mixing of gases, a PEM (proton exchange membrane) is used that will allow the passing of protons but nothing else. These membranes are often based on fluorinated polymers which ultimately have a negative environmental impact.20 Furthermore, PEMFCs and PEM-WEs both use platinum catalysts at the electrodes, which is expensive and in short supply.20 To tackle these limitations, alternate technology has been explored in the form of AEM (anion exchange membrane) systems.20AEMFCs and AEM-WEs do not involve the passage of protons across a membrane, but rather of hydroxide anions. Importantly, they allow the use of non-platinum-group elements as electrocatalysts and fluorine-free polymers as the backbone for the AEM, improving both their cost and environmental impact.20 AEMFCs use a solid electrolyte, granting them a comparatively simple operation and improved performance stability. AEMs have been found to allow very high hydroxide conductivity, in some cases exceeding the proton conductivity of PEMs.20 However, current AEM technology faces a major hurdle: the stability of cationic functional groups in harsh alkaline conditions. While ex situ stability in heated alkaline conditions can be achieved, in situ stability has proven much more challenging.7 This has been attributed to the water gradient in the cell, which is produced by the water consumption at the relevant electrode. This results in an anhydrous environment around the membrane, where the unsolvated hydroxide anions have increased nucleophilicity and thus rapidly degrade the cationic functional groups, neutralising the membrane and halting conductivity.7,20 This occurs through a variety of degradation mechanisms, including Hoffmann elimination, deprotonation, and in some cases even a single-electron-transfer (SET) radical reaction.20
AEM research has largely focused on QASs since they tend to exhibit superior stability in an anhydrous alkaline environment, although they still leave a lot to be desired. To be useful in an industrial setting, the membrane material would have to be stable at high current densities for thousands of hours.7 QASs have been found to be stable in ex situ alkaline solutions with fully solvated hydroxides, but they tend to fail rapidly in situ. An ex situ testing protocol to predict the in situ stability of QASs has been developed, whereby a crown ether is used to capture potassium cations and leave the hydroxides ‘naked’, before being mixed with the QAS of interest in a dry DMSO solution.7 By this method, it was found that even leading QAS-functionalised polymers degrade rapidly in anhydrous alkaline conditions. Since these QASs typically include one or more alkyl groups, it has been understood that QAS degradation primarily occurs by two mechanisms: abstraction of an acidic β-hydrogen leading to Hoffmann elimination, and classic nucleophilic attack of an α-carbon leading to an SN2 reaction.13
With the goal of impeding these degradation mechanisms, research has been conducted into the stability of TAASs, as they consist of a central nitrogen connected to aromatic sp2-hybridised carbons rather than aliphatic sp3-carbons. It is expected that in tetraphenyl ammonium, the steric hindrance of the aromatic rings slows down an SN2 reaction, and β-hydrogen abstraction produces a strained benzyne intermediate.13 Indeed, Nesmeyanov observed that N,N-diphenylcarbazolium proved remarkably stable and decomposed only when heated in 50% aq. potassium hydroxide.10 However, it was later determined that diphenylcarbazolium degraded rapidly in the severe anhydrous conditions expected in an electrochemical cell.22 Analysis of the degradation products indicated preferential attack on the freely-rotating phenyl rings rather than the carbazole moiety but did not help to identify the principal degradation mechanism.13
To this end, a N,N-di(p-tolyl)carbazolium salt 39 was prepared, and the degradation products were once again analysed to provide insight on the regiochemistry of hydroxide attack. Results indicated that the predominant mechanism is an SN2 or addition–elimination attack on the ipso carbon, but this did not seem likely due to the steric buffer provided by the aryl rings.13 The observation of preferential attack on the freely-rotating rings led to the testing of an ammonium without such rings; namely, 9,9′-spirobicarbazolium 3. This salt proved to be perfectly stable in heated aqueous potassium hydroxide, and NMR analysis alongside mass spectrometry revealed a hydrogen/deuterium exchange with the solvent at the ortho positions of the carbazolium rings without decomposition.13 This cast further doubt on the mechanism of hydroxide attack on the ipso carbon.
An alternative mechanism was proposed in order to rationalise these findings. This proposal suggested that hydroxide was acting as a reducing agent, causing a single electron transfer (SET) to the carbazolium ion 39 which would provide the observed degradation products 41 and 42via the mechanism shown in Scheme 10.13 Cyclic voltammetry analysis suggested that a discrete SET from hydroxide to diphenyl carbazolium was improbable, and so it was postulated that this mechanism was additionally driven by a subsequent radical coupling with the remaining hydroxyl radical, yielding hydroxycyclohexadiene 40. It was concluded that the alkaline degradation mechanism of N,N-diaryl carbazolium salts 39 is an inner-sphere SET, followed by radical coupling and elimination of the carbazole group 41 to restore aromaticity.13
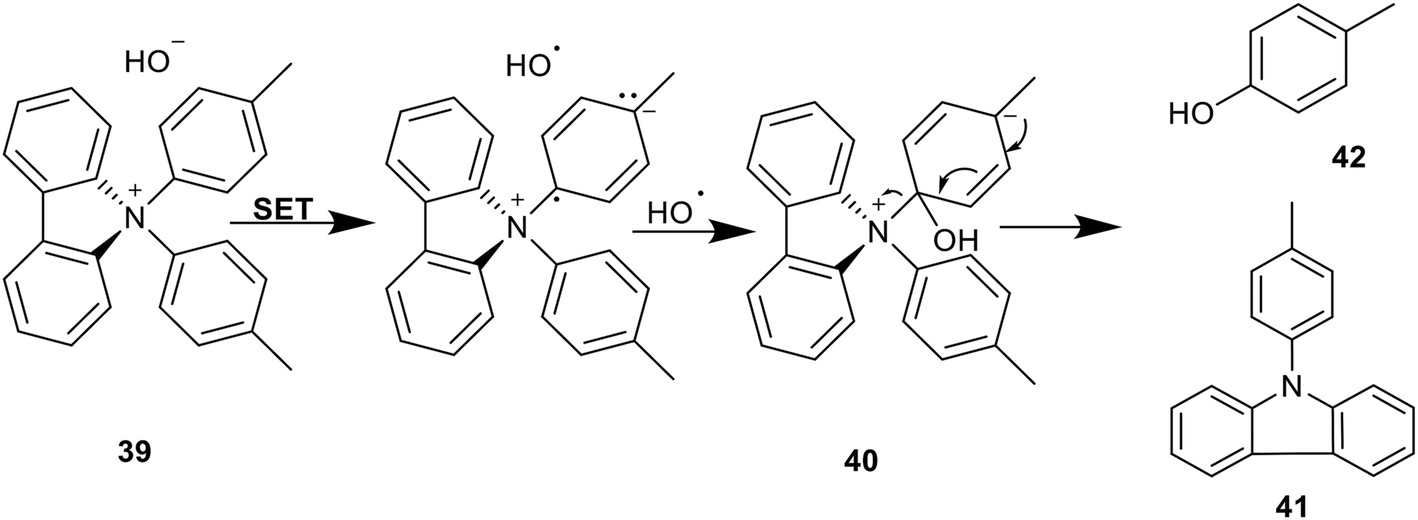 | ||
| Scheme 10 Degradation of N,N-di(p-tolyl)carbazolium 39via SET and subsequent radical coupling. | ||
The verification of this degradation mechanism allowed a new way to tune the alkaline stability of diaryl carbazolium cations. It was hypothesised that substituting electron-donating groups or electron-withdrawing groups onto the phenyl rings would have substantial effects on their reactivity with hydroxide, and this was found to be the case.22 A particularly exceptional result was the enhanced stability of N,N-di-p-hydroxyphenyl carbazolium 43 depicted in Scheme 11, which demonstrated a half-life of 138.6 hours under anhydrous alkaline conditions.22 This was taken to prove the concept that increasing electron-density in the aryl rings of a diaryl carbazolium will hinder the SET mechanism by which it degrades in the presence of hydroxide.
 | ||
| Scheme 11 N,N-4,4′-Dihydroxyphenylcarbazolium. | ||
It was later found that, in heated aqueous potassium hydroxide solution, tetraphenyl ammonium showed superior alkaline stability to diphenyl carbazolium,16 although this was not tested in anhydrous conditions. Octa-tert-butylated tetraphenyl ammonium exhibited an even higher stability, attributed to the steric protection on the phenyl rings.16 These steric and electronic effects on the stability of TAASs may be combined for improved results, upon functionalisation with appropriate groups in appropriate positions.
Phenyllithium is another very strong base that has been reacted with a tetraarylammonium salt. Hellwinkel and Seifert investigated the reaction of spirodicarbazolium 3 (as iodide salt) with PhLi, aiming at the synthesis of a pentacoordinate nitrogen compound, 44.23 While they were unable to synthesise 44, they did observe a very slow degradation of 3, occurring over the course of two days in refluxing ether. The reaction yielded a product mixture, with products 46 or 47 formed indicative of a mechanism running via ortho-didehydrobenzene (o-benzyne) intermediates such as 45 (Scheme 12).
 | ||
| Scheme 12 Reaction of 3 with phenyllithium. | ||
Deuterium labelling experiments were employed to support this mechanism. With methyllithium as base or nucleophile, analogous products were observed, and evidence was also obtained for deprotonation of 3 occurring when KOtBu or concentrated (50%) KOH in water was used as base.23
Functionalisation
Typically, functionalisation of tetraaryl ammonium cations has been achieved by positioning of substituents on the starting materials rather than direct functionalisation of an otherwise unsubstituted case. Alongside synthesis of spirobicarbazolium, Hellwinkel and Seifert also demonstrated a synthesis, shown in Scheme 13, of a tetramethylated derivative 48 by combining 2,7-dimethylcarbazole 49 with a similarly dimethylated iodonitrobiphenyl derivative 50.12 | ||
| Scheme 13 Synthesis of tetramethyl-spirodicarbazolium. | ||
Similarly, derivatives of N,N-diphenyl carbazolium with various substituents have been synthesised by Gjineci et al. by inclusion of para-substituents on the aryl iodides used to arylate diaminobiphenyl.12,21 The particularly stable para-hydroxy derivative 43 was synthesised by a post-carbazolium-formation functional group interchange (FGI), where a methoxy group was demethylated using boron tribromide.21 The synthesis of tetraphenyl ammonium via intermolecular radical coupling necessitated the synthesis of a tetraphenyl ammonium with para-bromo groups as well as meta-tert-butyl groups, the latter of which proved to have a positive effect on alkaline stability.15
Recently, a class of mono-, di-, tri-, and tetra-halogenated spirobicarbazoliums, including chiral examples, have been synthesised by the positioning of various halide substituents in the meta or para positions of the carbazole and aryl halide starting materials prior to Suzuki coupling with an aminoaryl boronic ester, also carrying halide substituents (Scheme 7 and 14).2 The resulting meta- or para-bromo substituted spirobicarbazolium salts like 51 were further functionalised through a subsequent Suzuki reaction with phenyl boronic acid to replace any bromo groups with phenyl rings, as in 52. The reaction was found to proceed faster and at lower temperatures than the corresponding reaction of spirobifluorene analogues.2 This relation was explained by the electron deficient nature of positively charged spirobicarbazolium salts in comparison to neutral spirobifluorenes, which facilitates the oxidative addition step of the Suzuki catalytic cycle. It was also shown to work with substituted phenyl boronic acids to provide spirobicarbazolium complexes with a range of functional groups connected through a phenyl ring.2
 | ||
| Scheme 14 Functionalization of a spiro-dicarbazolium salt via Suzuki reaction. | ||
Conclusion and outlook
The chemistry of tetraarylammonium salts (TAASs) has come some way from being a largely neglected laboratory curiosity, accessible only via lengthy syntheses requiring rather forcing conditions. Nowadays, modern palladium-catalysed coupling chemistry affords carbazole-based TAASs in a few synthetic steps only. Due to the extraordinary base stability of some TAASs, applications in membrane technology are beginning to emerge, and the research field can be expected to develop more rapidly.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.References
- A. N. Nesmeyanov, T. P. Tolstaya and A. N. Kost, Diarylhalonium, Triaryloxonium, and Tetraarylammonium Salts, Probl. Org. Khim., 1970, 21–41 Search PubMed.
- M. Lu, J. Xu, K. Baldridge and J. Siegel, Propeller, Linear, Cruciform and Stellate Spiro-bicarbazolium Salts, Chem. – Eur. J., 2022, 29(6), e202203035, DOI:10.1002/chem.202203035.
- M. F. Ross, G. F. Kelso, F. H. Blaikie, A. M. James, H. M. Cochemé, A. Filipovska, T. Da Ros, T. R. Hurd, R. A. J. Smith and M. P. Murphy, Lipophilic Triphenylphosphonium Cations as Tools in Mitochondrial Bioenergetics and Free Radical Biology, Biochemistry, 2005, 70, 222–230, DOI:10.1007/s10541-005-0104-5.
- J.-J. Min, S. Biswal, C. Deroose and S. S. Gambhir, Tetraphenylphosphonium as a Novel Probe for Imaging Tumors, J. Nucl. Med., 2004, 45, 636–643 CAS.
- M. Sefkow, N. Borsuk and M. O. Wolff, Chim. Oggi, 2001, 19, 19–22 CAS.
- G. Mandouma, J. Collins and D. Williams, Synthesis, Crystal Structure, and Conductivity of a Weakly Coordinating Anion/Cation Salt for Electrolyte Application in Next-Generation Batteries, Acc. Chem. Res., 2023, 56, 1223–1230, DOI:10.1021/acs.accounts.2c00584.
- S. Willdorf-Cohen, A. Mondal, D. Dekel and C. Diesendruck, Chemical stability of pol(phenylene oxide)-based ionomers in an anion exchange-membrane fuel cell environment, J. Mater. Chem. A, 2018, 6, 22234–22239, 10.1039/c8ta05785k.
- M. Lu, O. Allemann, J. Xu, A. Linden, K. Baldridge and J. Siegel, Peraryl-X-onium ions of nitrogen and oxygen, Org. Chem. Front., 2019, 6, 2640–2646, 10.1039/c9qo00633h.
- L. G. Makarova and A. N. Nesmeyanov, The decomposition and formation of onium salts and the synthesis of organoelemental compounds through onium compounds. I. Two types of decomposition of diphenyliodonium salts, Izv. Akad. Nauk. USSR, 1945, 6, 617–626 Search PubMed.
- A. Nesmeyanov, T. Tolstaia and A. Grib, Diphenyl-o, o’-diphenyleneammonium salts, Dokl. Akad. Nauk SSSR, 1963, 153(3), 608–611 CAS.
- D. Hellwinkel and H. Seifert, 2,2′-Biphenylyleneammonium salts, Chem. Commun., 1968, 24, 1683–1684, 10.1039/C19680001683.
- D. Hellwinkel and H. Seifert, Ring Closure of 2′-Heterosubstituted Biphenyl-2-diazonium Salts to (Spiro)cyclic Tetraarylammonium Salts and Tribenz[b.d.f]azepines, Chem. Ber., 1972, 105(3), 880–906, DOI:10.1002/cber.19721050320.
- N. Gjineci, S. Aharonovich, S. Willdorf-Cohen, D. Dekel and C. Diesendruck, The Reaction Mechanism Between Tetraarylammonium Salts and Hydroxide, Eur. J. Org. Chem., 2020, 3161–3168, DOI:10.1002/ejoc.202000435.
- S. Aharonovich, N. Gjineci, D. Dekel and C. Diesendruck, An Effective Synthesis of N,N-Diphenyl Carbazolium Salts, Synlett, 2018, 1314–1318, DOI:10.1055/s-003601591848.
- G. Choudhary, Human health perspectives on environmental exposure to benzidine: A review, Chemosphere, 1996, 32(2), 267–291, DOI:10.1016/0045-6535(95)00338-X.
- H. Fujita, O. Sasamoto, S. Kobayashi, M. Kitamura and M. Kunishima, Synthesis and characterization of tetraphenylammonium salts, Nat. Commun., 2022, 13, 2537, DOI:10.1038/s41467-022-30282-y.
- N. E. Shchepina, G. A. Badun, V. D. Nefedov, M. A. Toropova, V. M. Fedoseev, V. V. Avrorin and S. B. Lewis, Synthesis of arylhalonium compounds [including (4-methylphenyl) phenylfluoronium] by the nuclear–chemical method, Tetrahedron Lett., 2002, 43, 4123–4124, DOI:10.1016/S0040-4039(02)00717-7.
- N. E. Shchepina, V. D. Nefedov, M. A. Toropova, V. V. Avrorin, S. B. Lewis and B. Mattson, Ion-molecular reactions of free phenylium ions, generated by tritium β-decay with bidentate arenes, Tetrahedron Lett., 2000, 41, 25–27, DOI:10.1016/S0040-4039(00)00854-6.
- W. D. Nefedov, M. A. Toropova, N. E. Shchepina, V. V. Avrorin, W. E. Shuravleva and N. I. Trofimova, Radiokhimya, 1980, 5, 69–71 Search PubMed.
- C. Santoro, A. Lavacchi, P. Mustarelli, V. Noto, L. Elbaz, D. Dekel and F. Jaouen, What is Next in Anion-Exchange Membrane Water Electrolyzers? Bottlenecks, Benefits, and Future, ChemSusChem, 2022, 15(8), e202200027, DOI:10.1002/cssc.202200027.
- K. Aggarwal, N. Gjineci, A. Kaushansky, S. Bsoul, J. Douglin, S. Li, I. Salam, S. Aharonovich, J. Varcoe, D. Dekel and C. Diesendruck, Isoindolinium Groups as Stable Anion Conductors for Anion-Exchange Membrane Fuel Cells and Electrolyzers, ACS Mater. Au, 2022, 2(3), 367–373, DOI:10.1021/acsmaterialsau.2c00002.
- N. Gjineci, S. Aharonovich, D. Dekel and C. Diesendruck, Increasing the Alkaline Stability of N,N-Diaryl Carbazolium Salts Using Substituent Electronic Effects, ACS Appl. Mater. Interfaces, 2020, 12(44), 49617–49625, DOI:10.1021/acsami.0c14132.
- D. Hellwinkel and H. Seifert, Zur Frage des pentakoordinierten Stickstoffs: Reaktionen von (spiro)cyclischen Tetraarylammoniumsalzen mit Nucleophilen, Liebigs Ann. Chem., 1972, 762, 29–54, DOI:10.1002/jlac.19727620105.
| This journal is © The Royal Society of Chemistry 2024 |