
Synthetic access to diverse thiazetidines via a one-pot microwave assisted telescopic approach and their interaction with biomolecules†
Ramdas Nishanth
Rao
a,
Soumyadip
Das
a,
Kezia
Jacob
a,
Mohammed Mujahid
Alam
c,
M. M.
Balamurali
 *d and
Kaushik
Chanda
*ab
*d and
Kaushik
Chanda
*ab
aDepartment of Chemistry, School of Advanced Sciences, Vellore Institute of Technology, Vellore 632014, India. E-mail: chandakaushik1@gmail.com
bDepartment of Chemistry, Rabindranath Tagore University, Hojai 782435, Assam, India
cDepartment of Chemistry, College of Science, King Khalid University, P.O. Box 9004, Abha 61413, Saudi Arabia
dChemistry Division, School of Advanced Sciences, Vellore Institute of Technology, Chennai, Tamil Nadu 600027, India. E-mail: mmbala@gmail.com
First published on 26th March 2024
Abstract
A one-pot microwave assisted telescopic approach is reported for the chemo-selective synthesis of substituted 1,3-thiazetidines using readily available 2-aminopyridines/pyrazines/pyrimidine, substituted isothiocyanates and 1,2-dihalomethanes. The procedure involves thiourea formation from 2-aminopyridines/pyrazines/pyrimidine with the substituted isothiocyanates followed by a base catalysed nucleophilic attack of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S bond on the 1,2-dihalomethane. Subsequently, a cyclization reaction occurs to yield substituted 1,3-thiazetidines. These four membered strained ring systems are reported to possess broad substrate scope with high functional group tolerance. The above synthetic sequence for the formation of four membered heterocycles is proven to be a modular and straightforward approach. Further the mechanistic pathway for the formation of 1,3-thiazetidines was supported by computational evaluations and X-ray crystallography analyses. The relevance of these thiazetidines in biological applications is evaluated by studying their ability to bind bio-macromolecules like proteins and nucleic acids.
S bond on the 1,2-dihalomethane. Subsequently, a cyclization reaction occurs to yield substituted 1,3-thiazetidines. These four membered strained ring systems are reported to possess broad substrate scope with high functional group tolerance. The above synthetic sequence for the formation of four membered heterocycles is proven to be a modular and straightforward approach. Further the mechanistic pathway for the formation of 1,3-thiazetidines was supported by computational evaluations and X-ray crystallography analyses. The relevance of these thiazetidines in biological applications is evaluated by studying their ability to bind bio-macromolecules like proteins and nucleic acids.
Introduction
Heterocyclic compounds have been extensively utilized in the development of numerous pharmaceuticals, agrochemicals, dyes and plastics.1 The versatile nature of these heterocycles permits possible modifications for various applications via simple chemical reactions. Almost one third of the marketed drugs are heterocyclic that serve as important pharmacophore.2 They also possess excellent photophysical properties that lead to their utilization for bio-imaging and other interdisciplinary applications.3 Most importantly, they are basic components of nucleic acids as purines and pyrimidines, and can significantly influence a wide range of biological activities as modulators and inhibitors in the form of bicyclic,4 polycyclic5 and organometallic complexes.6 Recently, various chemical methodologies for the synthesis of heterocyclic compounds were reviewed for their potential biological interest such as nanoparticle catalyzed synthesis,7 ionic liquid mediated synthesis,8 microwave and water assisted methodologies,9etc.Among the various heterocyclic compounds, either natural or synthetic, four membered heterocyclic rings have their own significance. They belong to the ‘small ring’ group with a wide range of biological activities including anti-depressant,10 analgesic,11 ACP reductase12 activity, etc. Well known anti-bacterials like penicillin and cephalosporin contain a four membered β-lactam ring as their key structural element.13 The strained internal angle of 90° in four membered rings deviates significantly from the ideal bond angle of 109.5° for sp3 hybridized carbon. Conformational changes in the ring system induced by the observed steric repulsions of various substituents result in them possessing ‘wing-shaped’ orientations. Regarding their potency as anti-bacterial agents, most of the four membered rings suffer the drawbacks of anti-bacterial resistance and bond angle conformations that make them less stable molecules. In this regard, investigations are required to identify new four membered rings with hybrid modifications that can further add to the library of already existing stable structures. One such moiety is 1,3-thiazetidine containing one sulphur atom in addition to a nitrogen atom at the 1st and 3rd positions of the ring. The four-membered 1,3-thiazetidines also serve as important precursors with a broad range of therapeutic activities like anti-viral (A), pesticidal (B) and anti-bacterial (C) as shown in Fig. 1.14
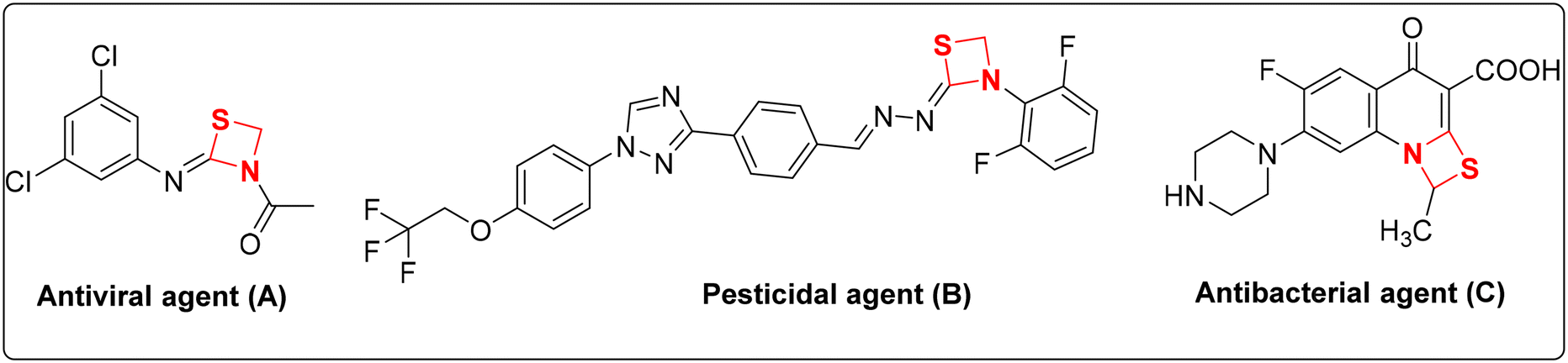 | ||
| Fig. 1 Pharmacologically important 1,3-thiazetidine derivatives. | ||
Although thiazetidines are known for their significant roles in numerous chemical or pharmacological applications, their synthesis techniques are restricted. Unlike the case of 3-membered aziridines, addition of a sulphur atom to azetidines in place of a carbon atom would relieve the strain and the resulting thiazetidines are expected to display better reactivity. Recently, the updated synthetic advancement and reactivity of strain driven azetidine heterocycles was reported by Szostak's group.15Fig. 2 demonstrates the most recent methods employed for the synthesis of 1,3-thiazetidine rings. In 1970, Field et al. performed the 1,2-thiazetidine ring synthesis by chlorinating 2-tosylamidoethyl disulfide to form the corresponding sulfenyl chloride followed by its treatment with triethyl amine (Fig. 2, a).16 Further in 1990, Goto et al. demonstrated the synthesis of 2-imino-1,3-thiazetidines from CS2, dialkylcyanamide and benzylideneaniline under high pressure conditions (Fig. 2, b).17 Subsequently, in 2011, Yadav et al. performed the coupling of various aldehydes with O,O-diethyl hydrogen phosphorodithioate and aldimines in THF using sodium hydride as base to yield 1,3-thiazetidines (Fig. 2, c).18 Recently, the solvent effect on the synthesis of 1,3-thiazetidine derivatives was demonstrated by Shao et al. Using a polar protic solvent like ethanol, the reaction between phenylthioureas and 1,1-dichloro-2-nitroethene yielded 1,3-thiazetidines while the same reaction in the presence of a polar solvent like chloroform yielded 1,4,2-dithiazolidines (Fig. 2, d).19 In the same year, Sun et al. reported the NaH catalyzed synthesis of thiazetidines linked with benzimidazole moieties in 10 min under one-pot conditions in good yield. Subsequently, benzimidazole fused thiadiazines in appreciable yields were generated when Et3N was used at room temperature for 30 min in acetone solvent via a stepwise protocol using thiourea as an intermediate (Fig. 2, e).20
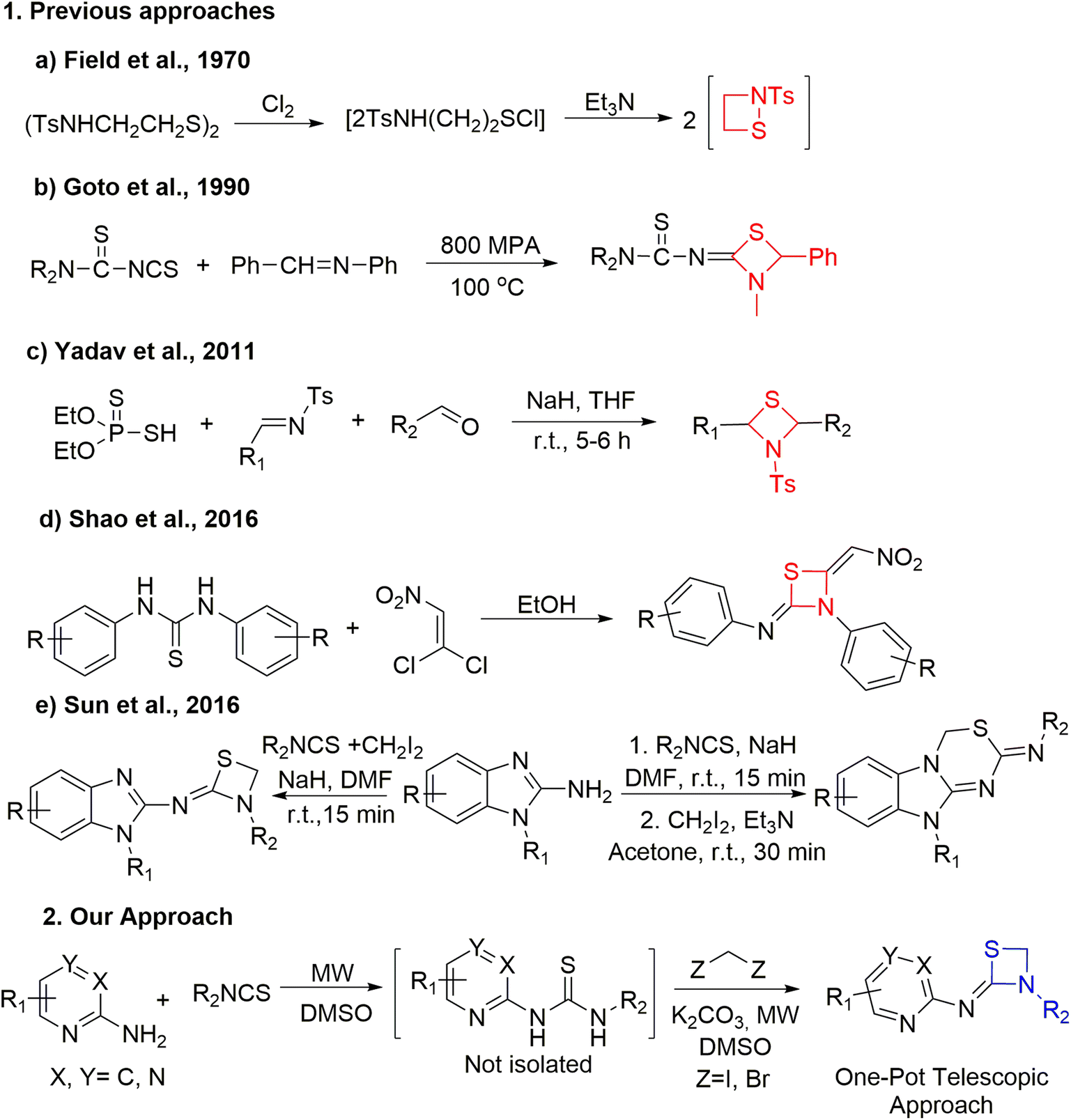 | ||
| Fig. 2 Various approaches for the synthesis of 1,3-thiazetidines. | ||
Moreover, most of the reported synthetic routes used high pressure, toxic gases and longer reaction times, and lack functional diversity. Herein the development of a new generation of heterocyclic scaffolds employing various synthetic techniques starting from 2-aminopyridine21 is reported for the first time for 2-aminopyridine/pyrimidine/pyrazine derived 1,3-thiazetidines. Further to tune the intrinsic properties of the above mentioned bioactive thiazetidines, a one-pot telescopic approach was employed to generate 1,3-thiazetidines using different electron donating and electron withdrawing groups at various positions and the products were investigated for their ability to interact with biomolecules.
The versatility of the one-pot telescopic approach has been well established for the synthesis of heterocyclic moieties and drugs over the past decades.22 This one-pot method avoids separation and purification of intermediates and provides high yields in a time efficient manner.23 Recently, there has been huge demand for mild and eco-friendly reactions that use microwaves to avoid multistep synthetic sequences and longer reaction times. We have recently reported a one-pot telescopic approach for the chemo-selective synthesis of substituted benzo[e]pyrido/pyrazino/pyridazino[1,2-b][1,2,4]thiadiazine dioxides.24 Herein, the use of dihalomethanes as one carbon units for the production of structurally diverse 1,3-thiazetidines via a one-pot microwave assisted telescopic approach is reported.
Results and discussion
Initially, a benchmark reaction was performed with 2-aminopyridine (1a) and phenyl isothiocyanate (2a) to obtain thiourea (3a) that could serve as a precursor for the synthesis of four membered 1,3-thiazetidines as depicted in Scheme 1. As expected, thiourea product (3a) was obtained in 95% yield under microwave irradiation for 4 min in DMSO solvent at 120 °C.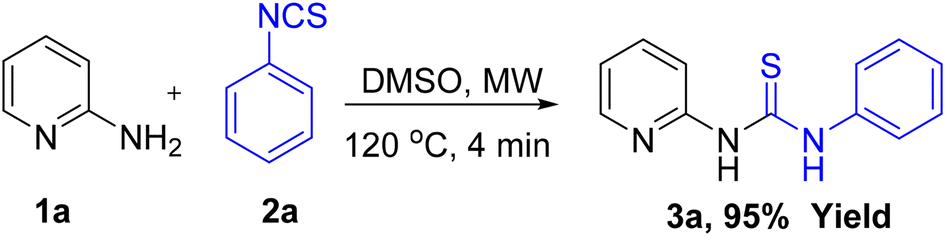 | ||
| Scheme 1 Synthesis of thiourea intermediate 3a by microwave irradiation. | ||
Following the formation of the above thiourea intermediate (3a), further steps in the synthetic sequence involved the formation of the four membered 1,3-thiazetidine framework using a one carbon fragment electrophile, 1,2-diiodoomethane, as template for the reaction with the thiourea intermediate (3a). Further evaluation of various key factors and optimization of the reaction conditions were carried out. Initially, the effect of base and solvent conditions was examined and the relevant data are compiled in Table 1. Particularly, the thiourea intermediate (3a) skeleton was oriented in such a way that there is a possibility of base mediated nucleophillic attack of the CS bond on 1,2-diiodoomethane followed by intramolecular ring closure reaction via the pyridine N atom or the aniline N atom generating two 1,3-thiazetidines 4a and 4a′ as depicted in Fig. 3.
 | ||
| Fig. 3 Two different modes of 1,3-thiazetidine ring formation. | ||
| Entry | Solvent | Base | Temperature | Time (h) | Yieldc (%) |
|---|---|---|---|---|---|
| a Reaction was performed using 3a (1 mmol), 1,2-diiodomethane (1.2 mmol), and base (1.1 mmol). b Microwave reactions were carried out in microwave model no. CATA R (Catalyst Systems, Pune) at a power of 28 watts. c Yield of the isolated product. d Reaction was carried out with 1,2-dibromomethane. | |||||
| 1 | Neat | — | r.t. | 5 | 0 |
| 2 | Neat | — | 100 °C | 5 | 0 |
| 3 | EtOH | Et3N | Reflux | 5 | 0 |
| 4 | EtOH | K2CO3 | Reflux | 5 | 20 |
| 5 | DMF | K2CO3 | 100 °C | 5 | 25 |
| 6 | CH3CN | K2CO3 | Reflux | 5 | 35 |
| 7 | DMSO | K2CO3 | 100 °C | 5 | 65 |
| 8 | DMSO | K2CO3 | MWb, 120 °C | 0.12 | 90 |
| 9 | DMSO | NaOH | MWb, 120 °C | 0.12 | 65 |
| 10 | DMSO | Et3N | MWb, 120 °C | 0.12 | 0 |
| 11 | DMSO | Piperidine | MWb, 120 °C | 0.12 | 0 |
| 12 | DMSO | K2CO3 | MWb, 120 °C | 0.12 | 90d |
| 13 | DMSO | KOtbu | 100 °C | 5 | 60 |
| 14 | DMSO | KOtbu | MWb, 120 °C | 0.12 | 75 |
| 15 | DMSO | Cs2CO3 | 100 °C | 5 | 0 |
| 16 | DMSO | Cs2CO3 | MWb, 120 °C | 0.12 | 0 |
When compound 3a was treated with 1,2-diiodomethane under neat conditions at room temperature, it did not yield the cyclised product 4a or 4a′ (Table 1, entry 1). Moreover, heating the reaction mixture to 100 °C for 5 h or using Et3N as base under refluxing EtOH solution also did not yield any cyclised product (Table 1, entries 2 and 3 respectively). However, upon changing the base to K2CO3, the same reaction under refluxing EtOH for 5 h resulted in the formation of product 4a or 4a′ in 20% yield (Table 2, entry 4). Further to increase the yield of the desired 1,3-thiazetidines, the reaction was attempted in polar aprotic solvent (DMF) at 100 °C or in refluxing acetonitrile solution (Table 1, entries 5 and 6 respectively). However, no substantial increment in the product yield was observed. But interestingly, the yield of the product increased to 65% when the same reaction was performed in DMSO at 100 °C for 5 h (Table 1, entry 7). Further increase in the yield of the product to 90% was observed when the reaction was carried out with microwave irradiation at 120 °C for 7 min (Table 1, entry 8). However, upon using NaOH as the base, under the same conditions (microwave irradiation), no significant increase in the yield of the product was noted (Table 1, entry 9). Moreover, the application of mild bases such as triethylamine and piperidine also did not yield any product formation (Table 1, entries 10 and 11, respectively).

The subsequent use of 1,2-dibromomethane as the electrophile in the heterocyclization reaction did not lead to any significant changes in the outcome of the reaction (Table 1, entry 12) which clearly demonstrated the advantages of microwave irradiation in ring closure reactions. The use of strong bases such as KOtBu did not cause any substantial changes in the yield of the reaction (Table 1, entries 13 and 14, respectively) whereas the reaction didn't proceed at all in the presence of Cs2CO3 as base both under the conventional and microwave conditions (Table 1, entries 15 and 16, respectively). Further to ascertain the formation of compound 4a over 4a′, the newly formed product was isolated and subjected to spectroscopic analysis including regular proton NMR spectrometry. It was noticed that peaks at 14.5 and 10.2 ppm correspond to two NH protons of thiourea intermediate 3a in spectrum A of Fig. 4. Along with the emergence of five aromatic protons in the phenyl isothiocyanate moiety, the chemical shift of these two protons moved further downfield. Intermediate 3a upon reacting with 1,2-diiodomethane resulted in the formation of the thiazetidine 4a or 4a′ which could not be confirmed through 1H NMR spectra. In spectrum B (Fig. 4), the loss of two NH protons and the emergence of one singlet at 4.9 ppm confirms the formation of the desired four membered thiazetidine 4a or 4a′.
 | ||
| Fig. 4 Stepwise monitoring of (A) thiourea and (B) 1,3-thiazetidine ring formation 4a or 4a′ formation by 1H NMR spectroscopy. | ||
Since it was not possible to ascertain the formation of 4a or 4a′ by regular proton NMR spectroscopy, computational investigations were attempted to get mechanistic details on the reaction process. The computational approaches involved the optimization of geometry using density functional theory and the B3LYP hybrid functional with the 6-311G++** basis set in their ground states.25 The potential energy profile was created using the same technique to follow the mechanistic pathways (concerted/path 1/path 2) that result in the generation of product 4a or 4a′ (Fig. 5).
 | ||
| Fig. 5 Geometry optimized structures of intermediates, transition states and corresponding products. | ||
The results of geometry optimization revealed that the strain induced by the approach of 1,2-dibromomethane increased the dihedral angle between the pyridine and the phenyl ring from 24.6° to 44.5° upon deprotonation of the pyridyl amine and to 41.9° for the aniline moiety. Further, upon cyclization to form product 4a or 4a′, the dihedral angle between >CS and the pyridine ring was reduced from 36.8° to 1.6° and for the phenyl ring it was decreased to 9.9°. The above observations revealed that the reaction proceeds via geometry alterations to overcome the steric strain.
In order to ascertain the attainment of minimum energy structures, frequency calculations were carried out to ensure the absence of any negative frequencies. Further to identify the high energy structures of the transition states, force constants were evaluated applying the same functional and basis set as were used for geometry optimizations. The possible formation of products 4a or 4a′ can be proposed to proceed through two different mechanisms – (i) a concerted mechanism wherein the abstraction of the proton from the pyridine amine/aniline amine occurs simultaneously with the attack of CH2–Br, and (ii) sequential abstraction of protons followed by the attack of CH2–Br. The concerted mechanism with a trimolecular process involving simultaneous deprotonation and SN2 substitution is entropically not likely. Therefore, a sequential reaction is expected. In this, the intermediate 3a1′ was found to possess 10.05 kcal mol−1 lower energy than 3a1 while the two transition states, ts and ts′ (0.20 kcal mol−1) were revealed to possess almost similar energies. Moreover in the sequential mechanism, two pathways can be proposed – abstraction from the pyridine amine –NH (path 1) and the aniline –NH (path 2). As shown in Fig. 6, path 1 involves the nucleophilic attack of the CS bond on 1,2-dihalomethane to produce the transition state ts, which is accomplished by the abstraction of the proton by the base from the pyridine amine nitrogen. While in path 2, the nucleophilic attack of the CS bond on the 1,2-dibromomethane, followed by the nucleophilic abstraction of the proton from the aniline nitrogen results in the formation of intermediate ts′.
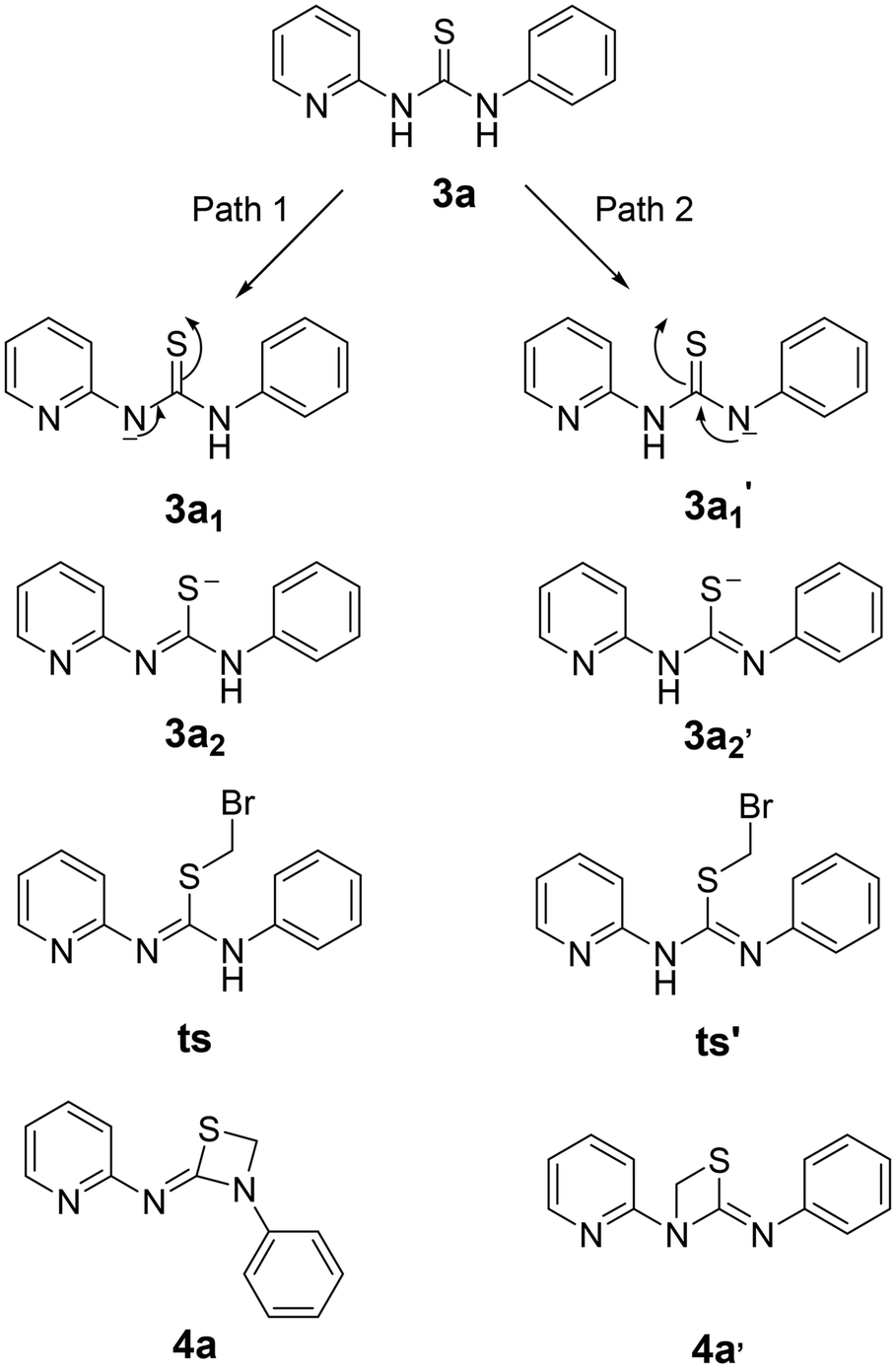 | ||
| Fig. 6 Mechanistic investigation for the possible formation of products 4a and 4a′. | ||
The energy profile for the reaction is depicted in Fig. 7. The energy barrier for the conversion of 3a1 to 4a is 30.94 kcal mol−1 and for 3a1′ to 4a′ it is 40.79 kcal mol−1. This shows that the attainment of equilibrium is energetically more favourable in the former case (3a1 to 4a). Moreover, the energy required for the reverse reaction is 35.41 and 32.73 kcal mol−1, respectively. Further, to ascertain the preference for pyridine amine proton abstraction over aniline amine proton abstraction and vice versa, in the sequential mechanism the abstraction of the aniline amine proton is energetically more favoured by 8.25 kcal mol−1 than that of the pyridine amine proton (18.30 kcal mol−1). It is known that pyridine >N–H is more acidic than aniline >N–H. In fact, aniline >N–H is more nucleophilic than pyridine >N–H. In this case, the chemoselectivity is controlled by the acidity of pyridine >N–H and higher nucleophilicity of aniline >N–H. The calculated energetics revealed the possibility for the formation of product 4a over 4a′. Finally it has been established that the 1,3-thiazetidine ring formation by the attack on the aniline nitrogen is favoured rather than on the pyridyl amine nitrogen as depicted in Fig. 7 because the amidic resonance of anilides is much higher than that of pyridyl-amides.26 The formation of product 4a was further confirmed with the crystal structure data obtained for pyrazine derivative 4g (Fig. 8 and the ESI†).
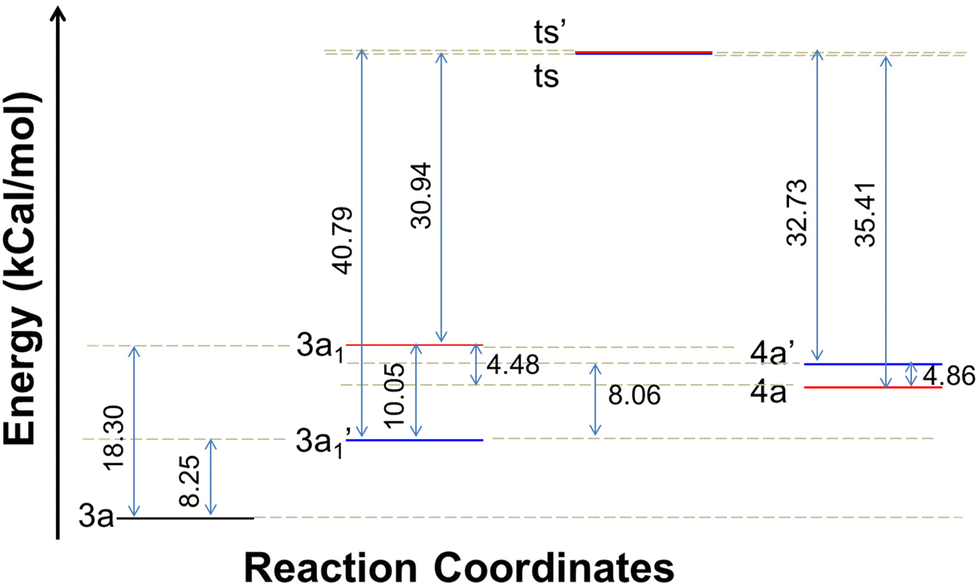 | ||
| Fig. 7 Energy profile depicting the reaction coordinate for the conversion of intermediates to products. | ||
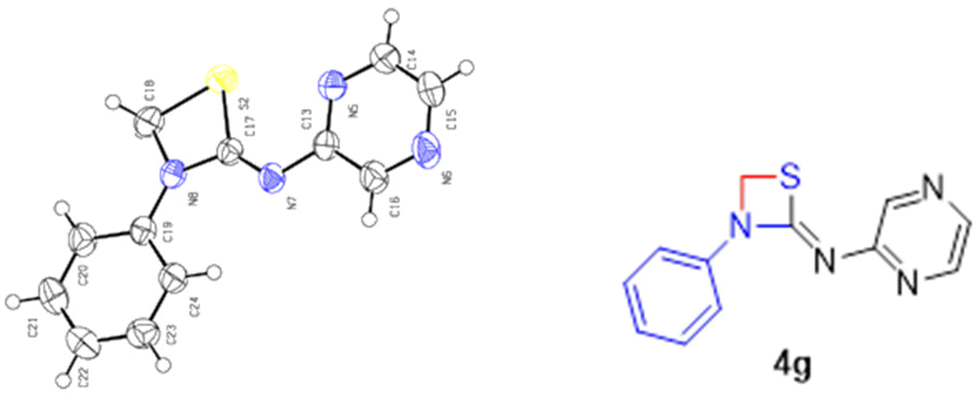 | ||
| Fig. 8 ORTEP diagram of compound 4g with 40% thermal ellipsoid probability. | ||
To further confirm the formation of 1,3-thiazetidine 4a, the X-ray crystallographic study of compound 4g was carried out. Fig. 8 depicts the ORTEP diagram of compound 4g (X-ray crystallographic data given in the ESI†). The X-ray crystal structure of compound 4g indicates exclusively the formation of the 1,3-thiazetidine ring that is favoured by the attack on the aniline nitrogen rather than on the pyridyl amine nitrogen atom, which unequivocally confirms its structure.27
After successfully executing the 1,3-thiazetidine ring 4 formation, efforts to achieve the chemo-selective synthetic manipulation were attempted. For this reason, a one-pot telescopic reaction was performed, where the reaction of substituted 2-aminopyridines 1 with substituted isothiocyanates 2 under microwave irradiation for 4 min at 120 °C in DMSO solvent resulted in the formation of thioureas 3. Without isolating the intermediate products 3, the same reaction mixtures were subjected to microwave irradiation for 7 to 10 min at 120 °C with in situ addition of 1,2-diiodomethane and K2CO3 to obtain the desired 1,3-thiazetidine derivatives as depicted in Scheme 2.
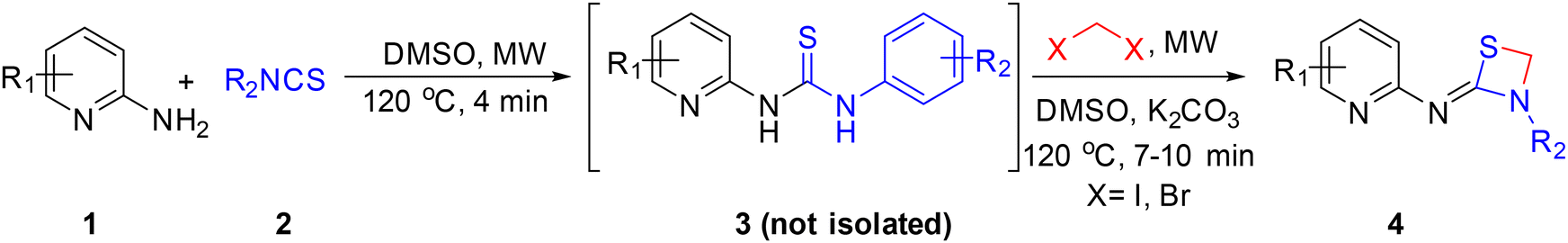 | ||
| Scheme 2 One-pot telescopic approach to synthesize substituted 1,3-thiazetidenes via a chemo-selective pathway. | ||
Upon completion of the reaction, the 1H NMR spectra of the synthesized compounds indicated the formation of pure 1,3-thiazetidines 4 in excellent yields. Further efforts to expand the substrate scope, to include 2-amino pyrimidine and 2-aminopyridazine as appropriate substrates were attempted in order to assess the effectiveness of the one-pot telescopic technique and to swiftly increase the size of the unique chemical library. As depicted in Table 2, a smooth transformation of 2-aminopyrimidine and 2-aminopyridazine to unique four membered 1,3-thiazetidine moieties was observed in excellent yields. Interestingly, using aniline as one of the components in the one-pot telescopic approach did not yield any 1,3-thiazetidine compound.
Finally, the corresponding 1,3-thiazetidine derivatives 4 were successfully produced through the telescopic reaction in excellent yields. This was followed by straightforward work-up procedures that involved the removal of solvent under reduced pressure, extraction and solvent evaporation procedures. Finally, the pure products were obtained after purifying through column chromatography, which was followed by the spectroscopic analysis of the pure compounds using 1H NMR and 13C NMR spectroscopy, mass spectrometry (MS) and IR spectroscopy. The above model reaction was also successfully attempted for gram scale synthetic applicability following this one-pot telescopic method. According to Scheme 3, the reaction produced 2.05 g of 4a in 85% yield with no appreciable loss in efficiency, highlighting the potential use of the current technique for large-scale synthesis of the scaffold 1,3-thiazetidine derivatives.
 | ||
| Scheme 3 Gram-scale synthesis of 1,3-thiazetidine via a chemo-selective pathway. | ||
Interaction studies of various 1,3-thiazetidine derivatives with bio-macromolecules
In order to evaluate the biological significance and potential of various synthesized thiazetidine derivatives, their interaction with calf thymus DNA (ct-DNA) and bovine serum albumin (BSA) protein was evaluated by following their electronic absorption and fluorescence spectra, respectively.28 The equilibrium binding constant, number of binding sites and Stern–Volmer quenching constants were calculated following their interaction profiles.29The fluorescence quenching interactions can be described by the Stern–Volmer equation (eqn (1)).
| I0/I = 1 + KSV[Q] | (1) |
log[(I0 − I)/I] = log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K + nlog[Q] K + nlog[Q] | (2) |
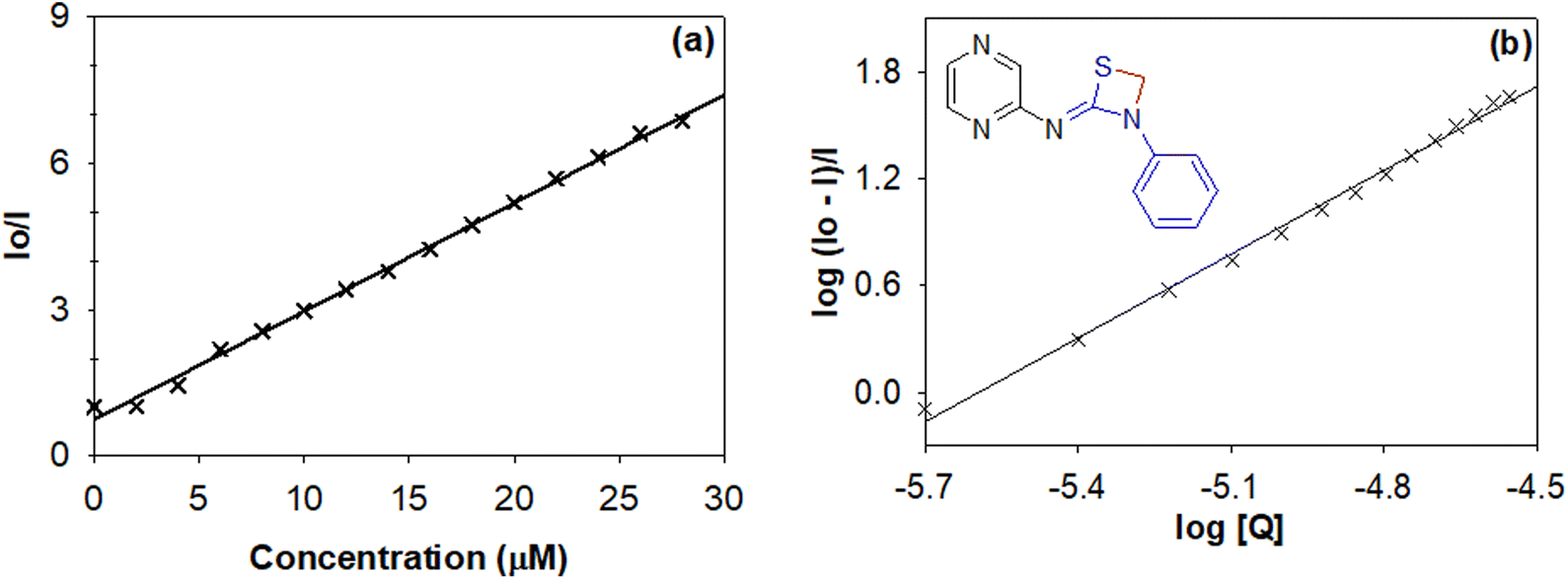 | ||
| Fig. 9 (a) Stern–Volmer and (b) Scatchard plots to evaluate the quenching constant and binding constant for 1,3-thiazetidine derivative 4g. | ||
| Entry | K SV × 105 (M−1) | K (M−1) | n |
|---|---|---|---|
| 4a | 0.768 | 5.20 × 106 | 1.3 |
| 4b | 0.943 | 9.29 × 106 | 1.3 |
| 4c | 0.571 | 9.28 × 105 | 1.2 |
| 4d | 0.231 | 2.04 × 104 | 0.9 |
| 4e | 0.360 | 5.23 × 105 | 1.2 |
| 4f | 0.336 | 1.25 × 105 | 1.0 |
| 4g | 2.087 | 6.05 × 10 8 | 1.3 |
| 4h | 0.233 | 3.30 × 104 | 1.0 |
| 4i | 0.329 | 2.80 × 104 | 0.9 |
| 4j | 0.176 | 4.09 × 106 | 1.4 |
| 4k | 0.429 | 3.64 × 106 | 1.3 |
| 4l | 0.249 | 1.54 × 105 | 1.1 |
| 4m | 0.106 | 4.72 × 105 | 1.3 |
| 4n | 0.053 | 6.58 × 105 | 1.3 |
| 4o | 0.161 | 2.65 × 104 | 1.0 |
| 4p | 0.642 | 8.05 × 105 | 1.1 |
| 4q | 0.300 | 0.02 × 106 | 0.97 |
| 4r | 0.243 | 0.06 × 106 | 0.99 |
Where I0 and I are the fluorescence intensities in the absence and presence of quencher, K is the equilibrium binding constant, [Q] is the concentration of the quencher and n is the number of equivalent binding sites on the macromolecule.
The data in Table 3 reveal that thiazetidine derivative 4g binds more strongly to the protein with a binding constant of 6.05 × 108 M−1. In general it could be observed that electron donating groups like –CH3 on the pyridyl/pyrimidyl/pyrazine ring lead to strong interaction with the protein while electron withdrawing groups like –Br lead to a relatively weaker interaction. In addition to that, phenyl substitution on the thiazetidine nitrogen leads to a stronger binding interaction when compared to benzyl or alkyl substituents. Therefore, it could be the size or the steric effect that stabilizes the interactions in addition to the electronic effects as discussed above. Moreover, the quenching constant values indicate that the intrinsic fluorescence from the amino acids including tryptophan, tyrosine and phenylalanine is quenched either by their exposure to solvent or through energy transfer to quencher molecules. The value of n indicates that there exists only one binding site offered by the protein for its interaction with thiazetidine derivatives.
 | ||
| Fig. 10 UV-Vis absorption spectra depicting the interaction of calf thymus DNA with 1,3-thiazetidine derivatives 4b and 4p. | ||
The binding strength (Kb) values of complexes with ct-DNA were calculated by using the following equation.36
| [DNA]/(εa − εf) = [DNA]/(εb − εf) + 1/Kb(εb − εf) | (3) |
Where [DNA] is the concentration of the DNA in base pairs, the apparent absorption coefficients εa, εf and εb correspond to Aobsd/[complex], the extinction coefficient for the complex in its free form and the extinction coefficient for the complex fully bound with DNA, respectively. The intrinsic binding constant (Kb) is obtained from the ratio of the slope to the intercept and the relevant data are given in Table 4.
| Entry | K b (M−1) | Entry | K b (M−1) | Entry | K b (M−1) | Entry | K b (M−1) |
|---|---|---|---|---|---|---|---|
| 4a | 3.20 × 104 | 4e | 4.46 × 104 | 4i | 7.02 × 104 | 4m | 8.29 × 104 |
| 4b | 1.42 × 10 5 | 4f | 7.46 × 103 | 4j | 4.13 × 103 | 4n | 2.48 × 104 |
| 4c | 1.41 × 104 | 4g | 2.45 × 104 | 4k | 4.78 × 104 | 4o | 3.36 × 104 |
| 4d | 3.97 × 104 | 4h | 5.13 × 103 | 4l | 2.00 × 104 | 4p | 1.35 × 105 |
| 4q | 1.60 × 104 | 4r | 2.70 × 104 |
From the data reported in Table 4, it is evident that derivatives 4b and 4p display exceptionally higher binding affinity as compared to the others (Table 5). In most of the cases, a stronger interaction was observed when an alkyl substituent was present on the pyridine ring. Not limited to the above, the geometry and orientation of the molecule in the binding cavity of the macromolecule also plays a key role in determining the binding interactions. All these results indicate that the herein reported 1,3-thiazetidine derivatives possess significant nucleic acid interaction potential and hence can be utilized for their applications as biomolecular probes. Further development of this type of unique four membered ring compound might be of interest to medicinal chemists.

Conclusions
A microwave-assisted one-pot telescopic synthesis of diverse 1,3-thiazetidines via thiourea formation followed by the base catalysed nucleophilic addition of 1,2-diiodomethane with subsequent intramolecular cyclization is reported for the first time in the literature. With this protocol it is possible to synthesize efficiently, various diverse four membered 1,3-thiazetidines by taking advantage of the broad substrate scope and short reaction times with good to excellent yields. Further the mechanistic pathway was supported by DFT evaluations and X-ray crystallography. The biological significance of the synthesized 1,3-thiazetidines was evaluated by investigating their ability to bind biomacromolecules including proteins and nucleic acids. Compound 4g exhibited efficient binding affinity with BSA protein while 4b and 4p revealed a relatively stronger interaction with ct-DNA as compared to all the other derivatives. Further, this study suggested that these unique four membered 1,3-thiazetidines might serve as potent candidates for biomolecular probes for drug discovery applications.Experimental section
General methods
Unless otherwise indicated all common reagents and solvents were used as obtained from commercial suppliers without further purification. 1H NMR (400 MHz) and 13C NMR (100 MHz) spectra were recorded on a Bruker DRX400 spectrometer. Chemical shifts are reported in ppm relative to the internal solvent peak. Coupling constants, J, are given in Hz. Multiplicities of peaks are given as: d (doublet), m (multiplet), s (singlet), and t (triplet). Mass spectra were recorded on a PerkinElmer Calrus 600 GC-MS spectrometer. HRMS spectra were recorded on an LC-QTOF-HRMS. IR spectra were recorded on a Bomen DA8 3FTS spectrometer. Microwave assisted reactions were carried out in a Catalyst Scientific Microwave oven system (Model No: CATA R (Catalyst System, Pune) operating at 2450 MHz equipped with glass vial extension by a condenser was used for performing the reaction. The microwave was equipped with a temperature control system (external probe). All the starting materials such as 2-aminopyridine, 2-aminopyrimidine, 2-aminopyridazine and substituted isothiocyanates were purchased from either Sigma-Aldrich or Avra Scientific Limited.Yield = 0.216 g, 90%; white solid; mp: 63–65 °C; Rf = 0.75 (10% EtOAc/n-hexane); 1H NMR (400 MHz, CDCl3) δ 8.33 (d, J = 4 Hz, 1H), 7.62 (dd, J = 7.8, 6.7 Hz, 3H), 7.37 (t, J = 7.8 Hz, 2H), 7.10–7.04 (m, 2H), 6.92 (t, J = 6.2 Hz, 1H), 4.92 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 159.9, 156.0, 146.6, 140.6, 137.8, 129.1, 122.9, 119.8, 118.4, 116.0, 50.9; MS (GC-MS) 241; HRMS (EI, m/z) calcd for C13H11N3S: m/z 241.0674; found 241.0670; IR (cm−1, KBr) 3049, 2875, 1550, 740, 680.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Chancellor and Vice Chancellor of Vellore Institute of Technology for providing the opportunity to carry out this study. Further, the authors wish to thank the management of this institute for providing seed money as a research grant. Thanks are also due to the Central Instrumentation Facility VIT for recording the spectra and for use of the single-crystal X-ray diffractometer. Mohammed Mujahid Alam is also grateful to the Deanship of Scientific Research, King Khalid University, for support through the Large Research Group Project number R.G.P. 2/210/1444. KC thanks the Vice Chancellor of Rabindranath Tagore University, Hojai for his keen interest in this work.References
- D. Ye, H. Lu, Y. He, J. Zheng, J. Wu and W. Hao, Nat. Commun., 2022, 13, 3337 CrossRef PubMed
and references cited therein.
-
(a) N. T. A. Dawoud, E. M. A. El-Fakharany, A. E. Hamada, E. I. Gendi and D. R. Lotfy, Sci. Rep., 2022, 12, 3424 CrossRef CAS PubMed
-
(a) K. Chanda and M. M. Balamurali, Chem. Soc. Rev., 2021, 50, 3706–3719 RSC
- R. Kaur and K. Kumar, Eur. J. Med. Chem., 2021, 215, 113220 CrossRef CAS PubMed
- L. M. De Coen, T. S. A. Heugebaert, D. García and C. V. Stevens, Chem. Rev., 2016, 116, 80–139 CrossRef CAS PubMed
-
(a) R. L. Panchangam, R. N. Rao, V. Manickam, M. M. Balamurali and K. Chanda, Inorg. Chem., 2021, 60, 17593 CrossRef CAS PubMed
- S. Das and K. Chanda, ChemNanoMat, 2022, 8, e202200375 CrossRef CAS
- M. G. Ahmad and K. Chanda, Coord. Chem. Rev., 2022, 472, 214769 CrossRef CAS
- R. N. Rao, S. Jena, M. Mukherjee, B. Maiti and K. Chanda, Environ. Chem. Lett., 2021, 19, 3315 CrossRef
- P. Melloni, A. D. Torre, M. Meroni, A. Ambrosini and A. C. Rossi, J. Med. Chem., 1979, 22, 183–191 CrossRef CAS PubMed
- D. C. Bishop, J. F. Cavalla, I. M. Lockhart, M. Wright, C. V. Winder, A. Wong and M. Stephens, J. Med. Chem., 1968, 11, 466–470 CrossRef CAS PubMed
- M. Takhi, K. Sreenivas, C. K. Reddy, M. Munikumar, K. Praveena, P. Sudheer, B. N. V. M. Rao, G. Ramakanth, J. Sivaranjani, S. Mulik, Y. R. Reddy, K. N. Rao, R. Pallavi, A. Lakshminarasimhan, S. K. Panigrahi, T. Antony, I. Abdullah, Y. K. Lee, M. Ramachandra, R. Yusof, N. A. Rahman and S. Hosahalli, Eur. J. Med. Chem., 2014, 84, 382–394 CrossRef CAS PubMed
- K. Tahlan and S. E. Jensen, J. Antibiot., 2013, 66, 401–410 CrossRef CAS PubMed
-
(a)
Q. Ding, N. Jiang and R. J. Weikert, Ativiral Compounds, WO2014135472A1, 2014
- H. Mughal and M. Szostak, Org. Biomol. Chem., 2021, 19, 3274–3286 RSC
- N. E. Heimer and L. Field, J. Org. Chem., 1970, 35, 1668–1670 CrossRef CAS
- T. Tohru, M. Yasumoto, I. Shibuya, Y. Taguchi, K. Yonemoto and M. Goto, Chem. Lett., 1990, 19, 1423–1426 CrossRef
- R. Ankita and L. D. S. Yadav, Tetrahedron Lett., 2011, 52, 3933–3936 CrossRef
- Y. Feng, M. Zou, R. Song, X. Shao, Z. Li and X. Qian, J. Org. Chem., 2016, 81, 10321–10327 CrossRef CAS PubMed
- M. Selvaraju, S. Dhole and C. M. Sun, J. Org. Chem., 2016, 81, 8867–8875 CrossRef CAS PubMed
-
(a) R. N. Rao and K. Chanda, Chem. Commun., 2022, 58, 343–382 RSC
- H. Ishikawa, T. Suzuki and Y. Hayashi, Angew. Chem., Int. Ed., 2009, 48, 1304–1307 CrossRef CAS PubMed
- S. Das and K. Chanda, Synthesis, 2024, 56, 693–699 CrossRef CAS
- R. D. Padmaja, M. M. Balamurali and K. Chanda, J. Org. Chem., 2019, 84, 11382–11390 CrossRef CAS PubMed
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, revision B.01, Gaussian, Inc., Wallingford, 2010 Search PubMed
-
(a) S. Shi and M. Szostak, Org. Lett., 2017, 19, 4656–4659 CrossRef PubMed
- Final product 4g was crystallized by slow evaporation of its ethyl acetate–hexane (1:1, v/v) solution at room temperature. Crystal data: C12H10N4S, M = 242.30, triclinic, space group P1. The crystal data has been deposited at Cambridge Crystallographic Data Centre [CCDC no. 2130702†].
- S. U. Parsekar, K. Paliwal, P. Haldar, P. K. S. Antharjanam and M. Kumar, ACS Omega, 2022, 7, 2881–2896 CrossRef CAS PubMed
- F. Hashemi-Shahraki, B. Shareghi and S. Farhadian, Int. J. Biol. Macromol., 2023, 227, 1151–1161 CrossRef CAS PubMed
- R. Lange, P. Anzenbacher, S. Muller, L. Maurin and C. Balny, Eur. J. Biochem., 1994, 226, 963–970 CrossRef CAS PubMed
- F. I. Khan, M. T. Rehman, F. Sameena, T. Hussain, M. F. AlAjmi, D. Lai and M. K. A. Khan, J. Mol. Recognit., 2022, 35, e2958 CrossRef CAS PubMed
- M. Makarska-Bialokoz, Spectrochim. Acta, Part A, 2018, 193, 23–32 CrossRef CAS PubMed
- G. Vignesh, S. Arunachalam, S. Vignesh and R. A. James, Spectrochim. Acta, Part A, 2012, 96, 108–116 CrossRef CAS PubMed
- S. L. Zhang, H. Yao, C. Wang and K. Y. Tam, Bioorg. Med. Chem. Lett., 2014, 24, 4963–4968 CrossRef CAS PubMed
- E. Lissi and E. Abuin, J. Fluoresc., 2011, 21, 1831–1833 CrossRef CAS PubMed
- M. Feizi-Dehnayebi, E. Dehghanian and H. Mansouri-Torshizi, J. Iran. Chem. Soc., 2022, 19, 3155–3175 CrossRef CAS
- T. Göktürk, E. S. Çetin, T. Hökelek, H. Pekel, Ö. Şensoy, E. N. Aksu and R. Güp, ACS Omega, 2023, 8, 31839–31856 CrossRef PubMed
- B. C. Yallur, U. Katrahalli, P. M. Krishna and M. D. Hadagal, Spectrochim. Acta, Part A, 2019, 222, 117192 CrossRef CAS PubMed
- T. A. Farghaly, A. M. A. Alnaja, H. A. El-Ghamry and M. R. Shaaban, Bioorg. Chem., 2020, 102, 104103 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR, 13C NMR, IR and HRMS spectra of compounds 4. ct-DNA and BSA binding assays for compounds 4, and X-ray data of compound 4g. CCDC 2130702. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ob00075g |
| This journal is © The Royal Society of Chemistry 2024 |