
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Palladium-catalyzed C–C bond cleavage of N-cyclopropyl acylhydrazones†
Hiroki
Fujioka
a,
Motohiro
Yasui
 *a,
Shohei
Hamada
b,
Kohei
Fukumi
a,
Norihiko
Takeda
a,
Yusuke
Kobayashi
b,
Takumi
Furuta
b and
Masafumi
Ueda
*a
*a,
Shohei
Hamada
b,
Kohei
Fukumi
a,
Norihiko
Takeda
a,
Yusuke
Kobayashi
b,
Takumi
Furuta
b and
Masafumi
Ueda
*a
aKobe Pharmaceutical University, Motoyamakita, Higashinada, Kobe 658-8558, Japan. E-mail: masa-u@kobepharma-u.ac.jp
bDepartment of Pharmaceutical Chemistry, Kyoto Pharmaceutical University, Yamashina-ku, Kyoto 607-8414, Japan. E-mail: furuta@mb.kyoto-phu.ac.jp
First published on 1st April 2024
Abstract
Despite their utility as directing groups, the C–C bond cleavage of cyclopropanes utilizing hydrazones has not been explored. Herein, Pd-catalyzed C–C bond cleavage reaction of N-cyclopropyl acylhydrazones, followed by cycloisomerization to yield pyrazoles, has been developed. The protocol enables the synthesis of various α-pyrazole carbonyl compounds, which have a potential of biological activity. Control experiments and DFT calculations suggest that β-carbon elimination of a stable 6-membered chelate palladium complex occurs, generating a conjugated azine as a reaction intermediate for the following cycloisomerization.
Introduction
Deconstructive strategy via C–C bond cleavage is valuable for synthesizing complex compounds.1–3 Since cyclopropanes have 27.5 kcal mol−1 of strain energy, the strain-released ring-opening reaction of cyclopropanes is thermodynamically advantageous to the C–C bond cleavage process.4 Therefore, cyclopropanes are often used as a synthetic intermediate for producing complex compounds.4,5 As the most typical examples, Lewis acid-mediated ring-opening reactions of donor–acceptor cyclopropanes, which have an electronically-biased C–C bond, have been reported.6 For cyclopropanes bearing a less electronically-biased C–C bond, ring-opening reactions mostly proceed via oxidative addition of a transition metal complex to a C–C bond.7 Among them, cyclopropanes having an adjacent electron-withdrawing group, such as ketones and imines or alkylidene cyclopropanes, have been employed. In contrast, the treatment of cyclopropanes linked to an electron-donating group (EDG), such as the hydroxy, siloxy, amino, and thio groups with appropriate transition metals, undergoes β-carbon cleavage8 or EDG-assisted ring-opening.9 Moreover, indirect C–C bond cleavage via palladium-catalyzed C(sp3)–H activation of aminocyclopropane derivatives has been also reported.10,11The proximity of transition metals to a reaction site is important for the activation of inactive bonds such as C–H or C–C bonds.12 In the cleavage of C–C bonds in aminocyclopropane derivatives, only examples of covalent proximity effects via oxidative addition to C–X bond (X = Cl, Br, or I) have been reported (Scheme 1a).10,11 In contrast, a widely used activation method without C–X bond is the use of a directing group.13 Hydrazones have been used for various C–H functionalizations,14 first reported on C(sp2)–H functionalization by Inamoto's group in 2007 (Scheme 1b).14a More recently, C(sp3)–H functionalization using 2,6-dimethoxyphenyl groups as a substituent on the hydrazone carbon has also been reported by Dong's group.14b However, C–C bond cleavage of cyclopropanes utilizing hydrazones has not been explored. Thus, as part of our continuous research into the reactivity of hydrazones and the synthesis of nitrogen-containing heterocycles,15 we expected that the C–C bond cleavage reaction of N-cyclopropyl hydrazones would lead a development of new methodology in the field of organic synthetic chemistry. Herein, we report palladium-catalyzed sequential reaction involved in C–C bond cleavage of cyclopropanes followed by cycloisomerization, along with the elucidation of the reaction pathway by a series of control experiments and DFT calculations. The protocol enables the synthesis of α-pyrazole carbonyl compounds, which have a potential of biological activity.16,17
 | ||
| Scheme 1 Pd-catalyzed C–C or C–H bond activation of amine derivatives. | ||
Results and discussion
At the beginning of this study, the reaction conditions were optimized using cyclopropylhydrazone 1aa in xylene, expecting C–C bond cleavage via C(sp3)–H activation (Table 1).11 The use of Rh(III) or Ni(II) catalysts, which were reported in directing group-accelerated C(sp2)–H activation or C(sp3)–H activation, did not yield pyrazole 2aa efficiently (entries 1 and 2).18,19 In contrast, the yield improved to 49% when Pd(OAc)2 was used (entry 3). To accelerate the reaction, Pd(TFA)2 was examined; however, the yield decreased (entry 4). Subsequently, additives were examined. The addition of AgOAc, which was expected to act as a promoter for the regeneration of Pd(OAc)2, did not improve the yield (entry 5).20 When hexafluoro-2-propanol (HFIP) was added to promote C–C bond cleavage via C–H activation,21 a higher yield was observed to produce the desired pyrazole (entry 6). In contrast, the use of HFIP as a solvent was not effective for the sequential reaction (entry 7). Finally, when tert-amyl alcohol was used as a solvent, the yield improved to 80% (entry 8).22|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Additive (equiv.) | Solvent | T (h) | Yield (%) |
| a Conditions: 1aa (0.09–0.17 mmol), catalyst (10 mol%), MS4A (79–100 mg) in solvent (0.10 M) at reflux. b Yields were determined by 1H NMR using triphenylmethane as an internal standard. | |||||
| 1 | [RhCp*Cl2]2 | Cu(OAc)2 (1.0) | Xylene | 16 | 13b |
| K2CO3 (1.0) | |||||
| 2 | Ni(OTf)2 | PivOH (1.0) | xylene | 16 | N.D. |
| 3 | Pd(OAc)2 | — | xylene | 14 | 49b |
| 4 | Pd(TFA)2 | — | xylene | 16 | 42b |
| 5 | Pd(OAc)2 | AgOAc (2.0) | xylene | 16 | 39b |
| 6 | Pd(OAc)2 | HFIP (2.0) | xylene | 15 | 69 |
| 7 | Pd(OAc)2 | — | HFIP | 21 | N.D. |
| 8 | Pd(OAc)2 | — | t-AmylOH | 4 | 80 |

With the optimized conditions established, the substrate scope was investigated (Scheme 2). First, a substrate bearing aryl groups on the hydrazone carbon was examined. When various 4-substituted aryl groups were examined, pyrazoles were obtained with good yields except for the hydrazone 1ah bearing bromo arene moiety (2ab–2aj). It is noted that although the synthesis of 2ab has been reported by N–H insertion with pyrazole,17b–d an alternative reliable synthetic method was provided. Moreover, we confirmed that the E/Z geometry of hydrazones did not affect the yield (2ac), which suggests that E/Z isomerization through azo-hydrazone tautomerism driven by acyl group occurs during the reaction.23 To confirm the steric effect of the substituents, substrates having a methyl or methoxy group at the 2-position on the benzene ring were tested. This resulted in pyrazoles 2ak and 2al exhibiting nearly identical yields as the para-substituted substrates. The yield of pyrazole 2am having a methyl group at the 3-position on the benzene ring decreased, while a methoxy or cyano group was not affected, giving pyrazole 2an or 2ao a good yield. A substrate 1ap bearing a methylenedioxy group at the 3- and 4-positions could also be applied to the reaction; however, a higher reaction temperature was required. When 1-naphtylhydrazone 1aq was used, the reaction proceeded smoothly. The substrates linked to a heterocycle, such as thiophene and benzofuran, were applicable to the reaction (2ar, 2as). Subsequently, the reaction of substrate 1at bearing a phenyl group at the cyclopropane moiety produced 5-aryl pyrazole in moderate yield although the reaction necessitated an elevated temperature and more catalyst.24
 | ||
Scheme 2 Scope for aryl groups of N-cyclopropy acylhydrazones. Conditions: 1 (0.067–0.20 mmol), Pd(OAc)2 (10 mol%), MS4A (60–168 mg) in t-AmylOH (0.10 M) at reflux for 5–28 h. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1ac (2.8 mmol) was used. b(E)-Isomer was used. cAfter 19 h, stirred at 150 °C in a sealed tube for 9 h. dAfter stirred at 140 °C in a sealed tube for 21 h, Pd(OAc)2 (10 mol%) was added and stirred for 4 h. 1ac (2.8 mmol) was used. b(E)-Isomer was used. cAfter 19 h, stirred at 150 °C in a sealed tube for 9 h. dAfter stirred at 140 °C in a sealed tube for 21 h, Pd(OAc)2 (10 mol%) was added and stirred for 4 h. | ||
Next, substrates bearing an alkyl group on the hydrazone carbon were studied. When methyl, benzyl, and cyclopentyl groups, as well as a non-substituted substrate (R1 = H), were examined, the reaction proceeded smoothly (Scheme 3, 2au–2ax). Hydrazone bearing a tert-butyl group required a higher temperature and more catalyst, producing pyrazole 2ay in low yield. Finally, different acyl groups on the hydrazone carbon were examined. Pyrazole 2az was obtained in high yield from lactone having an α-dimethyl group, which might fix orientation of the carbonyl group accelerated the C–C bond cleavage process. In addition to phenyl ketones 1ba and 1bb, the desired products were also obtained from hydrazone 1bc, which was derived from camphorquinone. Furthermore, amide 1ca was also applicable to the reaction.
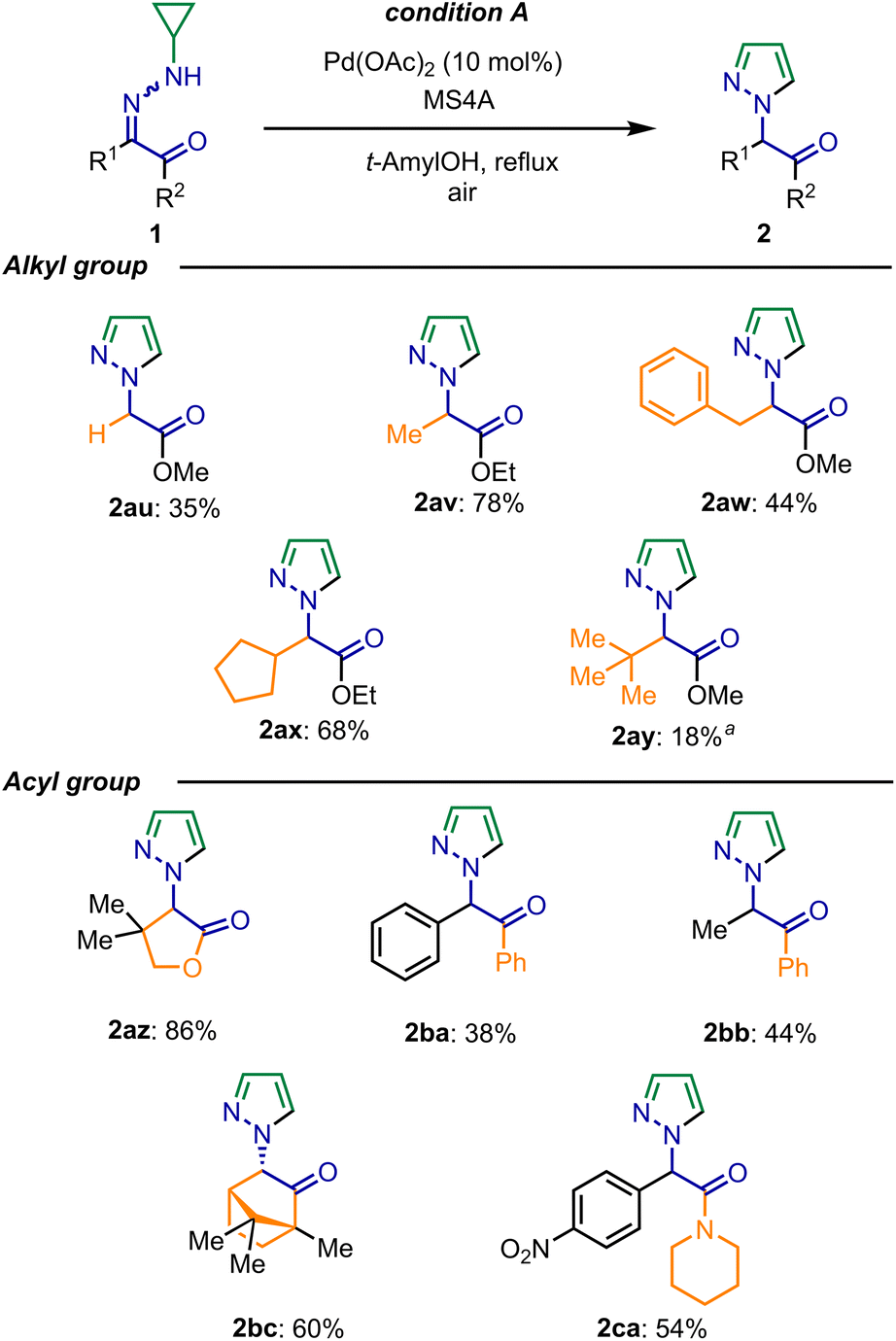 | ||
| Scheme 3 Scope for alkyl and acyl groups of N-cyclopropyl acylhydrazones. Conditions: 1 (0.068–0.18 mmol), Pd(OAc)2 (10 mol%), MS4A (60–118 mg) in t-AmylOH (0.10 M) at reflux for 5–32 h. aAfter stirred at 130 °C in a sealed tube for 13 h, Pd(OAc)2 (10 mol%) was added and stirred for 11 h. | ||
Subsequently, we conducted several studies to obtain knowledge of the reaction mechanism. To identify the reaction intermediate, hydrazone 1ac was treated under condition A, and its transformation was observed at any particular time (Fig. 1a). As a result, the formation of conjugated azine 3ac was observed before the disappearance of the starting material, while the existence of 3ac was not confirmed at 17 h. Furthermore, when azine 3ac was subjected to condition A, pyrazole 2ac was obtained in high yield. These results suggest that conjugated azine is the reaction intermediate. In addition, pyrazole 2ac was obtained in a slightly higher yield when the reaction was conducted without a palladium catalyst, supporting the notion that a palladium catalyst is not involved in the transformation from azine to pyrazole. When α-siloxyhydrazone 1da was employed under the optimized condition, conjugated azine and pyrazole were not formed (Fig. 1b). The result suggests that the acyl group is essential for the C–C bond cleavage and implies that it works as a directing group for the palladium complex. Finally, control experiments were performed under optimized conditions using hydrazone 1ab (Fig. 1c). First, regarding the palladium catalyst, the exchange from Pd(OAc)2 to PdCl2 did not produce pyrazole (entries 1 and 2). In addition, pyrazole was not obtained with BF3·OEt2 or without a catalyst (entries 3 and 4). These results suggest that the C–C bond cleavage reaction occurs rather than vinylcyclopropane rearrangement.25 The yield decreased without MS4A a little (entry 5). Finally, an argon atmosphere decreased the yield, which suggests that the Pd(0) species formed in situ are regenerated into Pd(II) species by oxygen molecules (entry 6).
 | ||
| Fig. 1 Mechanistic studies. aYields were determined by 1H NMR using triphenylmethane as an internal standard. | ||
Control experiments could not determine definitively whether C–C bond cleavage proceeds directly8a or is mediated by C–H activation.11 Therefore, to elucidate further detail process in the sequential reaction, an energy profile of the C–C bond cleavage/cycloisomerization reaction was estimated by DFT studies (Fig. 2). The calculations were first performed at the B97D/Lanl2DZ (for Pd) and B97D/6-31G+(d,p)-def2TZV (for the other atoms) level. Then, single-point energy calculations in the presence of solvent (chlorobenzene) were performed. Initially, hydrazone 1 is transformed to 6-membered chelate palladium complex A, which is more stable than 5-membered complex A′. Subsequently, β-carbon elimination of A proceeds in 19.6 kcal mol−1 of activation energy to produce imino-palladacycle B along with enamino-tautomer B′ in equilibrium. Next, conjugated azine C is formed from enamine B′ through the elimination of palladium hydride species. Then, the palladium hydride species undergo reductive elimination without any energy barriers. Pd(0) species are oxidized to Pd(OAc)2 by oxygen molecules to complete the catalytic cycle.26,27 Finally, cycloisomerization proceeds under heating conditions to produce pyrazole 2.28 The possibility of C–C bond cleavage via C–H activation is ruled out due to the high energy level of 5-membered chelate palladium complex A′ and the subsequent transition state in A′ to palladacycle D.11
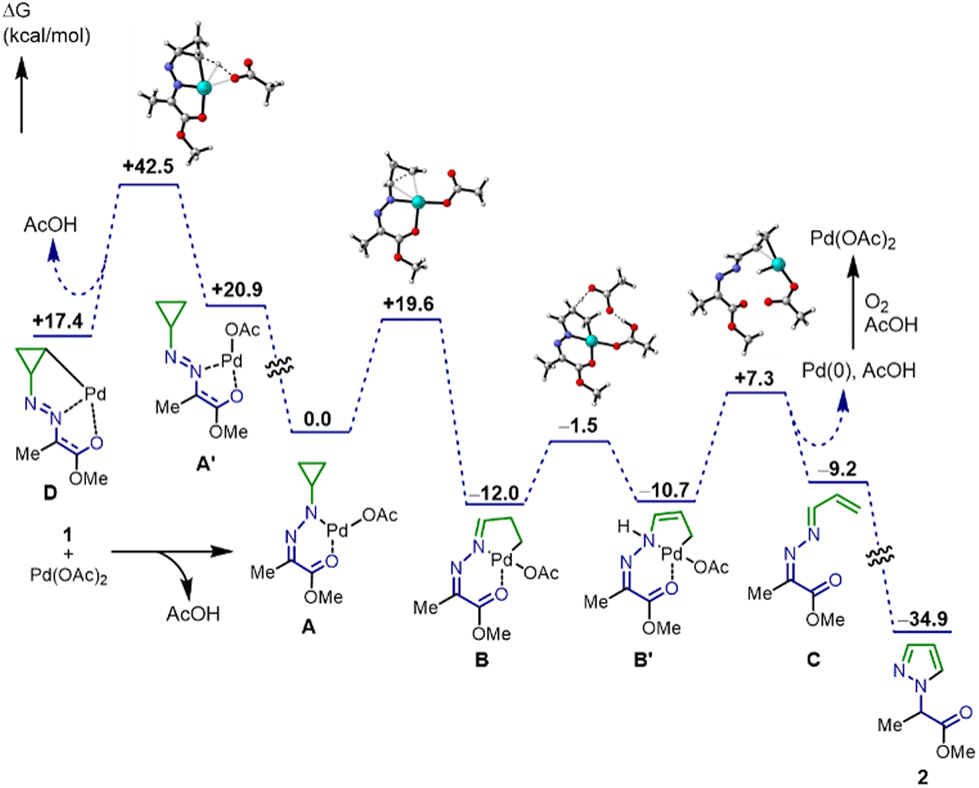 | ||
| Fig. 2 Gibbs free energy profile. | ||
Conclusions
In conclusion, we have developed Pd-catalyzed C–C bond cleavage of N-cyclopropyl acylhydrazones through β-carbon elimination of cyclopropane, followed by cycloisomerization to produce pyrazoles. Regarding substrate scope, various aryl and alkyl groups on hydrazone carbons can be utilized. Furthermore, various carbonyl functional groups such as esters, lactones, ketones, and amides can also be applied to the sequential reaction. A series of control experiments and DFT calculations suggest that C–C bond cleavage proceeds via β-carbon elimination followed by tautomerization/depalladation, resulting in the formation of conjugated azines. We expect the protocol leads to atom-economical skeletal editing, including C–H bond and C–C bond scissions as well as incorporating directing groups. This synthetic strategy can introduce sterically hindered secondary and tertiary alkyl groups under weakly acidic conditions. In addition, it enables the synthesis of 1-alkyl-5-aryl pyrazole, which is difficult to obtain with chemo/regioselectivity, as a single product.29,30 These α-pyrazole carbonyl compounds have a potential of biological activity. Therefore, this synthetic strategy might be one way to enrich compound library in medicinal chemistry. A more efficient synthetic method for 1,5-disubstituted pyrazoles using cyclopropyl hydrazones through the deconstructive strategy is currently under development.Author contributions
HF and KF conducted the experiments. HF wrote the original draft. MY conceptualized and managed the work, and were involved in review and editing. SH and YK conducted the computational study. NT and TF reviewed the work. MU supervised and review the work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by a Grant-in-Aid from JSPS KAKENHI, the MEXT Leading Initiative for Excellent Young Researchers grant, and Nagai Memorial Research Scholarship from the Pharmaceutical Society of Japan (N-235304, H. F.).References
- (a) J. B. Roque, Y. Kuroda, L. T. Göttemann and R. Sarpong, Science, 2018, 361, 171–174 CrossRef CAS PubMed; (b) Y. Xia, G. Lu, P. Liu and G. Dong, Nature, 2016, 539, 546–550 CrossRef CAS PubMed.
- M. Murakami and N. Chatani, in Cleavage of Carbon–Carbon Single Bonds by Transition Metals, Wiley-VCH, Weinheim, 2015, DOI:10.1002/9783527680092.
- Y. Xia and G. Dong, Acc. Chem. Res., 2022, 55, 2341–2354 CrossRef PubMed.
- H. N. C. Wong, M.-Y. Hon, C.-W. Tse, Y.-C. Yip, J. Tanko and T. Hudlicky, Chem. Rev., 1989, 89, 165–198 CrossRef CAS.
- P. Tang and Y. Qin, Synthesis, 2012, 2969–2984 CAS.
- T. F. Schneider, J. Kaschel and D. B. Werz, Angew. Chem., Int. Ed., 2014, 53, 5504–5523 CrossRef CAS PubMed.
- (a) L. Souillart and N. Cramer, Chem. Rev., 2015, 115, 9410–9464 CrossRef CAS PubMed; (b) L. Marek, A. Masarwa, P.-O. Delaye and M. Leibeling, Angew. Chem., Int. Ed., 2015, 54, 414–429 CrossRef PubMed.
- (a) S.-B. Park and J. K. Cha, Org. Lett., 2000, 2, 147–149 CrossRef CAS PubMed; (b) D. Rosa and A. Orellana, Org. Lett., 2011, 13, 110–113 CrossRef CAS PubMed.
- (a) A. D. Santos, L. E. Kaim, L. Grimaud and R. Ramozz, Synlett, 2012, 438–442 Search PubMed; (b) S. Ponra, A. Nyadanu, N. Pan, E. Martinand-Lurin, A. Savy, M. Vitale, L. E. Kaim and L. Grimaud, Org. Process Res. Dev., 2020, 24, 827–834 CrossRef CAS.
- O. O. Sokolova and J. F. Bower, Chem. Rev., 2021, 121, 80–109 CrossRef CAS PubMed.
- (a) S. Rousseaux, B. Liegault and K. Fagnou, Chem. Sci., 2012, 3, 244–248 RSC; (b) C. Ladd, D. S. Roman and A. B. Charette, Tetrahedron, 2013, 69, 4479–4487 CrossRef CAS; (c) K. Saint-Jacques, C. L. Ladd and A. B. Charette, Chem. Commun., 2022, 58, 7550–7553 RSC.
- (a) L. N. Lewis and J. F. Smish, J. Am. Chem. Soc., 1986, 108, 2728 CrossRef CAS; (b) S. Murai, F. Kakiuchi, S. Sekine, Y. Tanaka, A. Kamatani, M. Sonoda and N. Chatani, Nature, 1993, 366, 529 CrossRef CAS.
- (a) Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107–1295 RSC; (b) R. Logeswaran and M. Jeganmohan, Adv. Synth. Catal., 2022, 364, 2113–2139 CrossRef CAS; (c) H. Amistadi-Revol, S. Liu and S. Prévost, Eur. J. Org. Chem., 2023, e202300582 CrossRef CAS; (d) M. S. Ahmad and K. Meguellati, ChemistrySelect, 2022, 7, e202103716 CrossRef CAS; (e) B. Liu, A. M. Romine, C. Z. Rubel, K. M. Engle and B.-F. Shi, Chem. Rev., 2021, 121, 14957–15074 CrossRef CAS PubMed; (f) S. Rej, A. Das and N. Chatani, Coord. Chem. Rev., 2021, 431, 213683 CrossRef CAS; (g) C. Sambiagio, D. Sch önbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaa, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnürch, Chem. Soc. Rev., 2018, 47, 6603–6743 RSC; (h) J. Zhang, X. Lu, C. Shen, L. Xu, L. Dinga and G. Zhong, Chem. Soc. Rev., 2021, 50, 3263–3314 RSC; (i) U. Dutta and D. Malti, Acc. Chem. Res., 2022, 55, 354–372 CrossRef CAS PubMed.
- (a) K. Inamoto, T. Saito, M. Katsuno, T. Sakamoto and K. Hiroya, Org. Lett., 2007, 9, 2931–2934 CrossRef CAS PubMed; (b) Z. Huang, C. Wang and G. Dong, Angew. Chem., Int. Ed., 2016, 55, 5299–5303 CrossRef CAS PubMed; (c) A. Ros, R. López-Rodríguez, B. Estepa, E. Álvarez, R. Fernández and J. M. Lassaletta, J. Am. Chem. Soc., 2012, 134, 4573–4576 CrossRef CAS PubMed; (d) A. Park, K.-S. Jeong, H. Lee and H. Kim, ACS Omega, 2021, 6, 6498–6508 CrossRef CAS PubMed; (e) L. Zhang, J. Chen, X. Chen, X. Zheng, J. Zhou, T. Zhong, Z. Chen, Y.-F. Yang, X. Jiang, Y.-B. She and C. Yu, Chem. Commun., 2020, 56, 7415–7418 RSC; (f) M. Asamdi, M. M. Shaikh, P. M. Chauhan and K. H. Chikhalia, Tetrahedron, 2018, 74, 3719–3727 CrossRef CAS; (g) D. S. Deshmukh and B. M. Bhanage, Org. Biomol. Chem., 2018, 16, 4864–4873 RSC; (h) P. Xu, G. Wang, Z. Wu, S. Li and C. Zhu, Chem. Sci., 2017, 8, 1303–1308 RSC.
- (a) Y. Ito, M. Ueda, N. Matsuda, Y. Nishida and O. Miyata, Heterocycles, 2014, 89, 963–969 CrossRef CAS; (b) Y. Ito, M. Ueda, N. Takeda and O. Miyata, Chem. – Eur. J., 2016, 22, 2616–2619 CrossRef CAS PubMed; (c) H. Matsuzaki, N. Takeda, M. Yasui, Y. Ito, K. Konishi and M. Ueda, Org. Lett., 2020, 22, 9249–9252 CrossRef CAS PubMed; (d) H. Matsuzaki, N. Takeda, M. Yasui and M. Ueda, Chem. Commun., 2021, 57, 12187–12190 RSC; (e) N. Takeda, Y. Kobori, M. Yasui, K. Matsumoto, K. Orihara, Y. Kido and M. Ueda, Tetrahedron Lett., 2021, 73, 153098 CrossRef CAS; (f) M. Yasui, M. Hasegawa, K. Konishi, N. Takeda and M. Ueda, Heterocycles, 2021, 103, 661–669 CrossRef PubMed; (g) M. Yasui, H. Fujioka, N. Takeda and M. Ueda, Org. Lett., 2022, 24, 43–47 CrossRef CAS PubMed.
- (a) R. L. jinamatada and C. Pandit, WO2016185342A1, Nov. 24, 2016; (b) J. D. Rodgers, S. Shepard, T. P. Maduskuie, H. Wang, N. Falahatpisheh, M. Rafalski, A. G. Arvanitis, L. Storace, R. K. Jalluri, J. S. Fridman and K. Vaddi, US20070135461A1, June 14, 2007; (c) M. Zak, P. Gibbons, Y.-X. Cheng and S. C. Goodacre, WO2019139714A1, July 18, 2019; (d) H. Uneme, O. Ujigawa, H. Ishizuka and T. Okauchi, JP06287171A, Oct. 11, 1994.
- For recent reports on the synthesis of α-pyrazole carbonyl compounds: (a) S. Dhanju, A. C. Caravana and R. J. Thomson, Org. Lett., 2020, 22, 8055–8058 CrossRef CAS PubMed; (b) J. Miller, W. Zhao, J. D. Herr and A. T. Radosevich, Angew. Chem., Int. Ed., 2012, 51, 10605–10609 CrossRef PubMed; (c) M. L. Stivanin, A. A. G. Fernandes, A. F. Da Sliva, C. Y. Okada Jr. and I. D. Jurberg, Adv. Synth. Catal., 2020, 362, 1106–1111 CrossRef CAS; (d) W. Zhao and A. T. Radosevich, Org. Synth., 2015, 92, 267–276 CrossRef CAS PubMed.
- P. Xu, G. Wang, Z. Wu, S. Li and C. Zhu, Chem. Sci., 2017, 8, 1303–1308 RSC.
- (a) Y. Aihara and N. Chatani, J. Am. Chem. Soc., 2014, 136, 898–901 CrossRef CAS PubMed; (b) M. Li, J. Dong, X. Huang, K. Li, Q. Wu, F. Song and J. You, Chem. Commun., 2014, 50, 3944–3946 RSC.
- R. Padmavathi, R. Sankar, B. Gopalakrishnan, R. Parella and S. A. Babu, Eur. J. Org. Chem., 2015, 3727–3742 CrossRef CAS.
- (a) S. K. Sinha, T. Bhattacharya and D. Maiti, React. Chem. Eng., 2019, 4, 244–253 RSC; (b) T. Bhattacharya, A. Ghosha and D. Maiti, Chem. Sci., 2021, 12, 3857–3870 RSC.
- N. Dastbaravardeh, T. Toba, M. E. Farmer and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 9877–9884 CrossRef CAS PubMed.
- (a) S. M. Landge, E. Tkatchouk, D. Benitez, A. A. Lanfranchi, M. Elhabri, W. A. Goddard III and I. Aprahamian, J. Am. Chem. Soc., 2011, 133, 9812–9823 CrossRef CAS PubMed; (b) H. Y. Lee, X. Song, H. Park, M.-H. Baik and D. Lee, J. Am. Chem. Soc., 2010, 132, 12133–12144 CrossRef CAS PubMed; (c) A. Mitchell and D. C. Nonhebel, Tetrahedron, 1979, 35, 2013–2019 CrossRef CAS.
- Unfortunately, trisubstituted cyclopropanes have not been investigated because the substrate could not be prepared.
- M. Meazza, H. Guo and R. Rios, Org. Biomol. Chem., 2017, 15, 2479–2490 RSC.
- (a) Y. Izawa, I. Shimizu and A. Yamamoto, Bull. Chem. Soc. Jpn., 2004, 77, 2033–2045 CrossRef CAS; (b) C. Wang, Z. Zhang, Y. Tu, Y. Li, J. Wu and J. Zhao, J. Org. Chem., 2018, 83, 2389–2394 CrossRef CAS PubMed.
- M. Hu, W. Wu and H. Jiang, ChemSusChem, 2019, 12, 2911–2935 CrossRef CAS PubMed.
- T. A. Albright, S. Evans, C. S. Kim, C. S. Labaw, A. B. Russiello and E. E. Schweizer, J. Org. Chem., 1977, 42, 3691–3697 CrossRef CAS.
- J. S. Uber, Y. Vogels, D. van den Helder, I. Mutikainen, U. Turpeinen, W. T. Fu, O. Roubeau, P. Gamez and J. Reedijk, Eur. J. Inorg. Chem., 2007, 4197–4206 CrossRef CAS.
- (a) M. L. Stivanin, A. G. Fernandes, A. F. Da Silva, C. Y. Okada Jr. and I. D. Jurberg, Adv. Synth. Catal., 2020, 362, 1106–1111 CrossRef CAS; (b) A. Beladhria, K. Beydoun, H. B. Ammar, R. B. Salem and H. Doucet, Synthesis, 2011, 2553–2560 CAS; (c) F. Bellina, M. Lessi and C. Manzini, Eur. J. Org. Chem., 2013, 5621–5630 CrossRef CAS; (d) D. Xu, L. Frank, T. Nguyen, A. Stumpf, D. Russell, R. Angelaud and F. Gosselin, Synlett, 2020, 595–599 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ob00349g |
| This journal is © The Royal Society of Chemistry 2024 |