
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Carbones (–C2−–), carbenes (–C:–) and carbodications (–C2+–) on the magnetic criterion†
Erich
Kleinpeter
 * and
Andreas
Koch
* and
Andreas
Koch
Universität Potsdam, Institut für Chemie, Karl-Liebknecht-Str. 24-25, D-14476 Potsdam(Golm), Germany. E-mail: ekleinp@uni-potsdam.de; Andreas.Koch@uni-potsdam.de
First published on 30th January 2024
Abstract
The spatial magnetic properties, particularly the through-space NMR shieldings (TSNMRSs, the anisotropy effect in 1H NMR spectroscopy) of carbenes, carbones and carbodication (carbo2+) compounds (with and without stabilization by NMe2 π-donation) and those of a number of carbo2+ analogues have been calculated using the GIAO perturbation method, employing the nucleus-independent chemical shift (NICS) concept, and visualized as iso-chemical-shielding surfaces (ICSS) of various sizes and directions. TSNMRSs prove the electronic structure of carbo2+ compounds to be completely different from those of carbenes and carbones, preferring both the π-electron distribution and the structure of allenes/cumulenes despite the central carbon atom being the most electrophilic centre.
Introduction
Carbenes are a class of bivalent carbon species with six valence electrons (one σ-type lone pair and one vacant pz orbital). In N-heterocyclic carbenes (NHCs) and cyclic(alkyl)(amino)-carbenes (CAACs), the extreme stabilization by one or two nitrogen atom(s) adjacent to the carbene electron-deficient centre was already emphasized in the initial reports1,2 [the electronegativity of the nitrogen atom(s) (−I substituent effect) and the electron donation of the N-lone pair(s) (+M substituent effect) stabilize the carbenes via ylide mesomeric contributor(s) (Scheme 1)]:The π-donation of the nitrogen lone pair(s) into the formerly empty pz orbital of the ylide carbon atom is proved by dynamic NMR studies of the restricted rotation about the partial C,N double bonds (Scheme 1) in non-cyclic bis(dialkylamino)carbenes (NCACs): dependent on the substituents at nitrogen, the C,N bonds have variable but substantial double bond character (ΔG# = 10.7 to 19.35 kcal mol−1).3,4
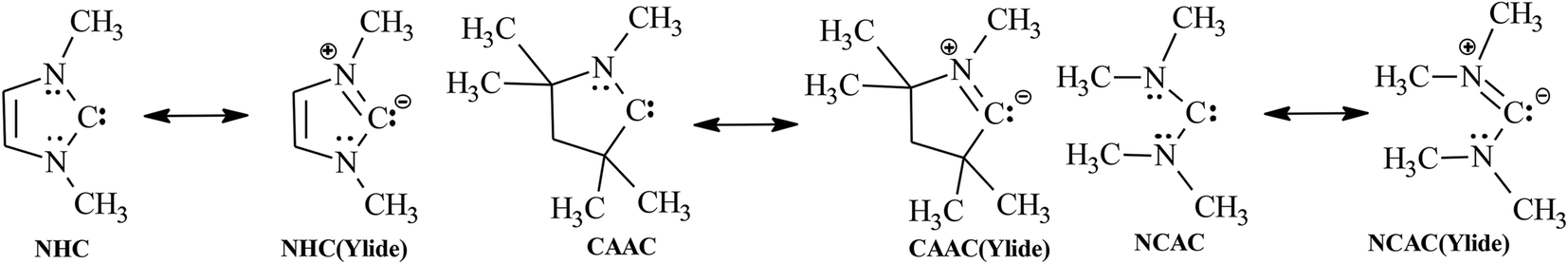 | ||
| Scheme 1 Mesomeric contributors of NHCs, CAACs and NCACs. | ||
Carbones bear comparable allene-like, carbene-like and carbone-like resonance contributors, among them the one with the central carbon atom carrying two negative charges (Scheme 2).5–9
 | ||
| Scheme 2 Mesomeric contributors of carbones. | ||
Besides allene-like and carbene-like mesomeric contributors (the latter in phosphorus allenes), a multiplicity of carbone-like compounds (bent allenes, carbodiphosphoranes and chalcogen-stabilized carbones) can be identified.
Finally, another carbon species was very recently discovered by Bertrand and coworkers10 with a mesomeric contributor of two positive charges on the central carbon atom (carbo2+, doubly oxidized carbene); however, it can stabilize itself in a number of allene-like, cumulene-like canonical structures, and even a higher degree of multiple bond character on the central carbon atom is suspected (Scheme 3).10
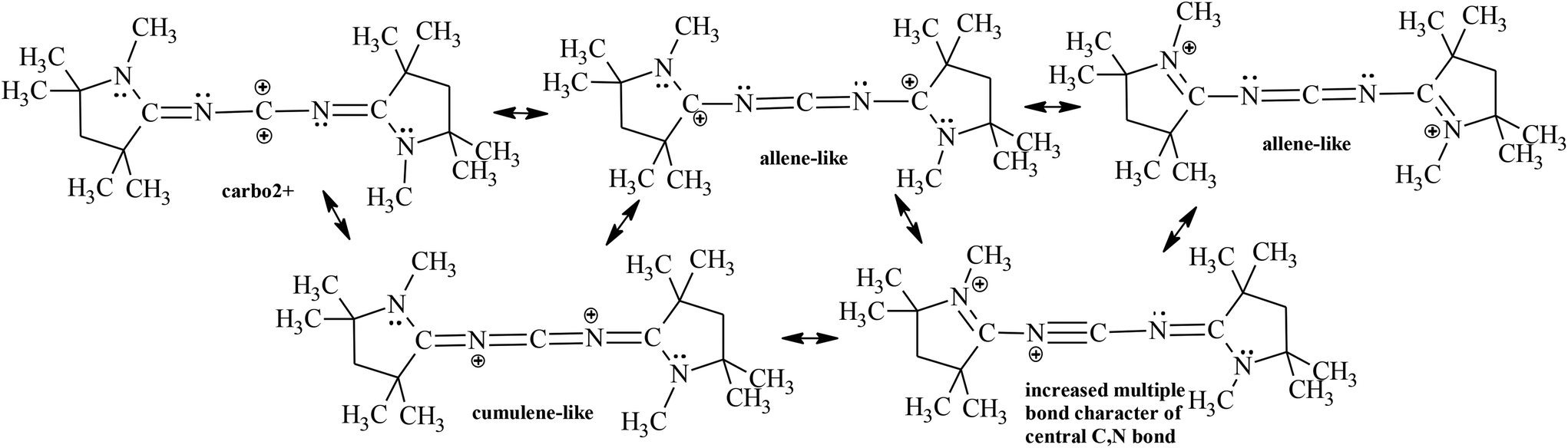 | ||
| Scheme 3 Mesomeric contributors of carbo2+ compounds. | ||
After unequivocally identifying the dominant mesomeric contributors in carbenes (ylide structures)11 and carbones (bent allenes, carbodiphosphoranes and chalcogen-stabilized carbones) on the magnetic criterion,12 we were strongly interested in how the extremely conjugated carbo2+ structure (Scheme 3) behaves under the same criteria. The main goal hereby was to identify the predominant mesomeric contributor and, from this, to assign the existing π-electron distribution and thus the electronic structure of the novel carbo2+ compound.10 This is the main object of this paper.
We employed our through-space NMR shielding (TSNMRS) concept13–15 to qualify the spatial magnetic properties (actually, the anisotropy effects in 1H NMR spectroscopy) of the studied species. Along this concept, the NICS values were calculated for a grid of ghost atoms surrounding the molecules in order to locate diatropic and paratropic regions around the structures. The TSNMRSs were visualized as iso-chemical-shielding surfaces (ICSS) and employed to qualify and quantify the anisotropy effects of the studied compounds. While the normally employed specifications of NICS values to quantify e.g. (anti)aromaticity are theoretical items, the experimental Δδ/ppm in proton NMR spectra are the molecular response properties of TSNMRS values.16
Results and discussion
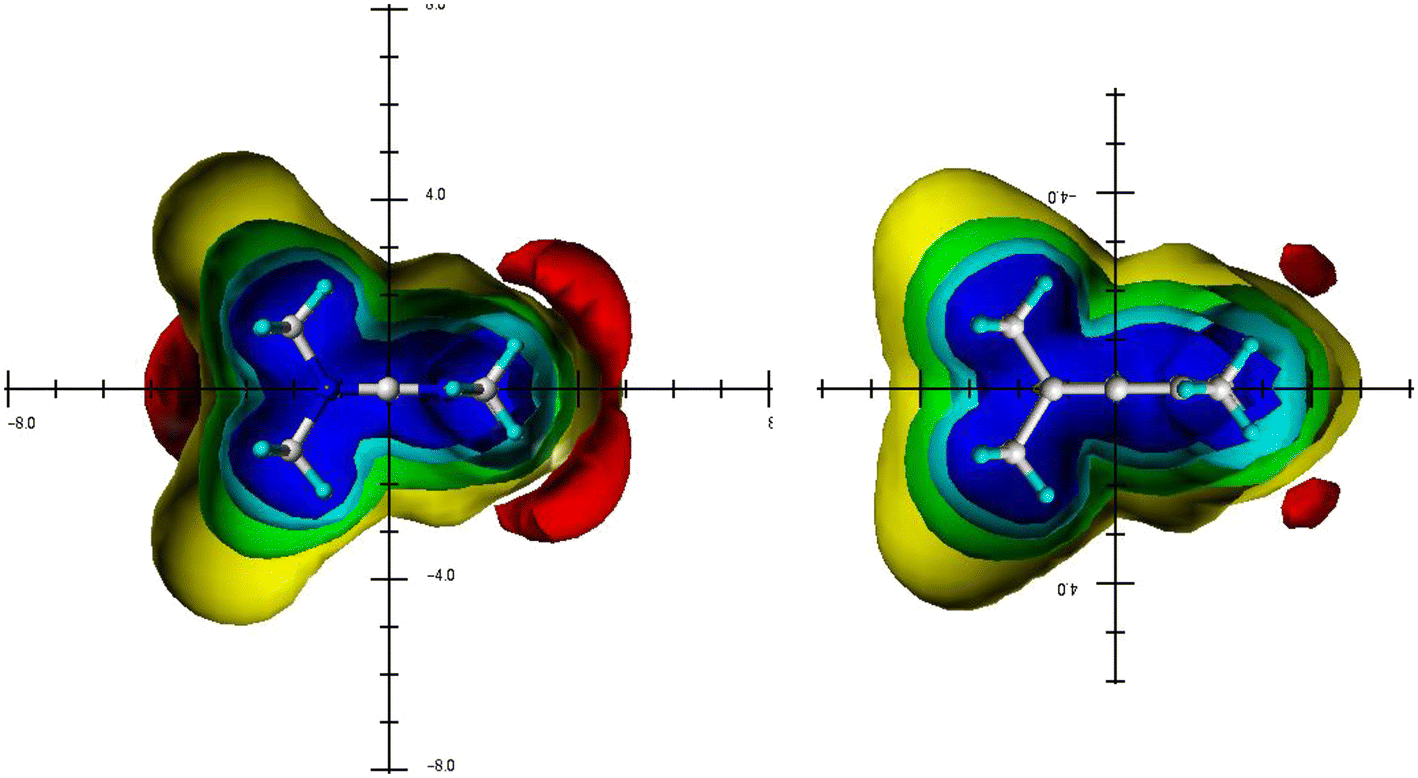
Introducing the topic, the various ICSSs of the TSNMRS of singly non-conjugated carbenes 1 (Me2C:), carbones 2 (Me2C− −) and the carbo2+ relative 3 (Me2C+ +) have been calculated (Scheme 4)17 and are shown in Fig. 1. While anisotropy effects of the central carbon atom of carbenes and carbones are comparable in size and extension, the corresponding anisotropy effect in the comparable carbon2+ compound 3 proves to be completely different: the carbon2+ compound 3 is linear (see also Table 1); the distinct shielding anisotropy effect above/below plane in the angled carbenes and carbones disappeared and are replaced by a paratropic area located at the central carbon atom; the shielding ICSSs (familiar from carbenes and carbones, Fig. 1) are shifted to the methyl carbon atoms.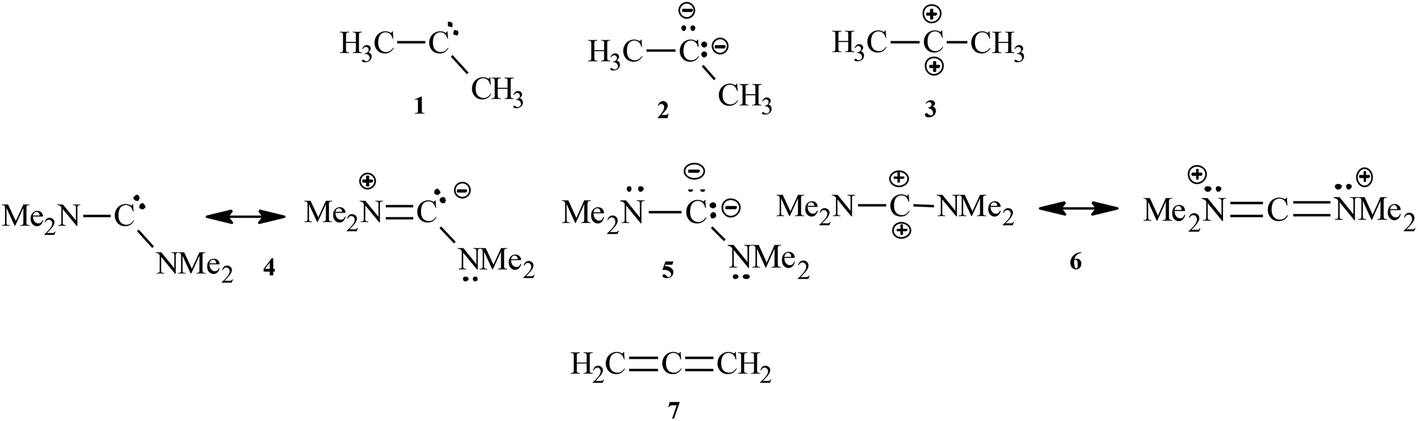 | ||
| Scheme 4 Dis-methyl- (1–3), dis-dimethylamino-carbene, -carbone and -carbo2+ (4–6) and allene (7). | ||
 | ||
| Fig. 1 Visualisation of the spatial magnetic properties (TSNMRSs) of dimethylcarbene 1, dimethylcarbone 2 and dimethylcarbon2+ 3 by different ICSS of −0.1 ppm (red) deshielding and 5 ppm (blue), 2 ppm (cyan), 0.5 ppm (green) and 0.1 ppm (yellow) shielding. | ||
| No. | Geometry | NMR parameters | ||
|---|---|---|---|---|
| Bond length (Å) | Bond angle | δ(13C)/ppm | ICSS (+5, +2 and 0.5)/(Å) | |
| dC–C/C–N | C–C–C/N−C−N | |||
| Me2C# | ||||
| 1 | 1.481 | 111.6° | 820.0 | 2.0, 2.7 and 4.25 |
| 2 | 1.454 | 103.55° | 152.0 | 1.8, 2.7 and 4.2 |
| 3 | 1.346 | Linear | 325.7 | paratropic hole |
| (NMe2)2C# | ||||
| 4 | 1.354 | 116.1° | 283.4 | 1.6, 2.1 and 3.0 |
| 5 | 1.339 | 145.0° | 145.1 | 1.3, 1.7 and 2.3 |
| 6 | 1.245 | Linear | 189.8 | 1.0, 1.4 and 2.1 |
CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CCH2 CCH2 |
||||
| 7 | 1.430 | Linear | 203.5 | 1.2, 1.5 and 2.1, but above centers of CC bonds: 1.5, 1.8 and 2.3 |
For the dis-dimethylamino analogues in Fig. 2, stabilization by conjugation proves to be an alternative: the conjugated carbenes 4 stabilize themselves essentially,11 if not exclusively,5,6via ylide structures and thereby significantly reduce the anisotropy effect of the central electron-deficient center; due to the angled structure of the ylide, the dimethylamino substituents are slightly twisted from the common resonance plane (Table 1). Conjugated carbone structures 5 also significantly reduce the anisotropy effect but retain the characteristic ball-like anisotropy effect of the central carbon atom (–C2−–).12 The carbone molecule is also angled (Table 1), as the Me2N+C−–NMe2 carbene analogue 4 is perfectly symmetric with the NMe2 moieties exactly orthogonal to each other, but the NMe2 groups are structurally pyramidal; because of the already existing doubly negatively charged central atom in carbones, the π-donor effect of the amino groups can only develop to an extent reduced accordingly.
 | ||
| Fig. 2 Visualisation of the spatial magnetic properties (TSNMRSs) of dis-dimethylamino-carbene 4, dis-dimethylamino-carbone 5 and dis-dimethylamino-carbon2+ 6 by different ICSS of −0.1 ppm (red) deshielding and 5 ppm (blue), 2 ppm (cyan), 0.5 ppm (green) and 0.1 ppm (yellow) shielding. | ||
The carbon2+ structure 6, finally, exhibits a completely different behavior on the magnetic criterion: the distinctive but not very strong deshielding zone around the central carbon atom in 3 disappeared, but the linearity in 6 remained. An allene-conform structure can be seen (see also Table 1), which has CN double bonds in-plane of the C(NMe2)2 fragments, with the CC(NMe2)2 moieties exactly orthogonal to each other. In-plane of the completely planar C(sp)C(NMe2)2 fragments, the anisotropic effect of the CN double bonds emerges clearly as in real allene structures (vide infra).
These initial results of the model carbenes, carbones and carbo2+ structures brings us significantly forward in answering the initial question: as Guy Bertrand and coworkers suggested,10 due to the higher electronegativity of nitrogen compared to carbon and the distinctive lone pair donation (+M effect) of the amino groups, the positive charges are not mainly located on the central carbon of the carbon2+ compound 6 but on the neighboring nitrogen atoms in exact allene structures; the direct comparison of the spatial magnetic properties with the allene CH2CCH27 in Fig. 3 proves to be unequivocal. Especially crucial proves to be, at this point, the characteristic anisotropy effect of the CN and CC double bonds, respectively, of the two CX(NMe2)2 (X = C, N) fragments.
 | ||
| Fig. 3 Visualisation of the spatial magnetic properties (TSNMRSs) of dis-dimethylamino-carbon2+ (4) and allene CH2CCH2 (7) by different ICSS of −0.1 ppm (red) deshielding and 5 ppm (blue), 2 ppm (cyan), 0.5 ppm (green) and 0.1 ppm (yellow) shielding. | ||
Now to the recently synthesized carbo2+ structure 8,10 two analogues (9, 10) and [4]cumulene 11 that are intended to support the generality of the former conclusions from the model compounds 1–7 on the magnetic criterion: the TSNMRS values as ICSSs of different size and direction are given in Fig. 4., and structural and magnetic data for 8–11 are given in Table 2. The occurrence of allene-like structures of the carbon2+ compounds 8–10 compared with the [4]cumulene structure 11 is confirmed. This is particularly demonstrated by the orthogonality of the terminal moieties. However, only 9 (as 6) is still linear; 8 and 11 deviate from linearity, with N–C–N at 168.5° and 163.2°, respectively, and C–N–C at 168.0° for both. On the other hand, the spatial magnetic properties (TSNMRSs) are even more convincing. As in [4]cumulene 11, the various ICSSs of 8–10 are spread more or less evenly over the allene(cumulene)-like double bonds and rise slightly towards the end of the molecules [see ICSS (+0.1 ppm) yellow]; perpendicular to this (the other side of the molecule due to sp hybridization of the central carbon atom), they fall off quickly. All in all, the present allene(cumulene)-like structure is convincingly depicted. The TSNMRSs of the CAAC molecule 8 are somewhat confusing due to the highly substituted 5-membered rings, but despite the extensive substitution, the present allene(cumulene)-like structure and corresponding π-electron distribution proves to be predominant (Scheme 5).
 | ||
| Fig. 4 Visualisation of the spatial magnetic properties (TSNMRSs) of carbodication 12+ (8), two analogues (9 and 11) and tetramethyl-[4]cumulene (10) (from left) by different ICSS of −0.1 ppm (red) deshielding and 5 ppm (blue), 2 ppm (cyan), 0.5 ppm (green) and 0.1 ppm (yellow) shielding. | ||
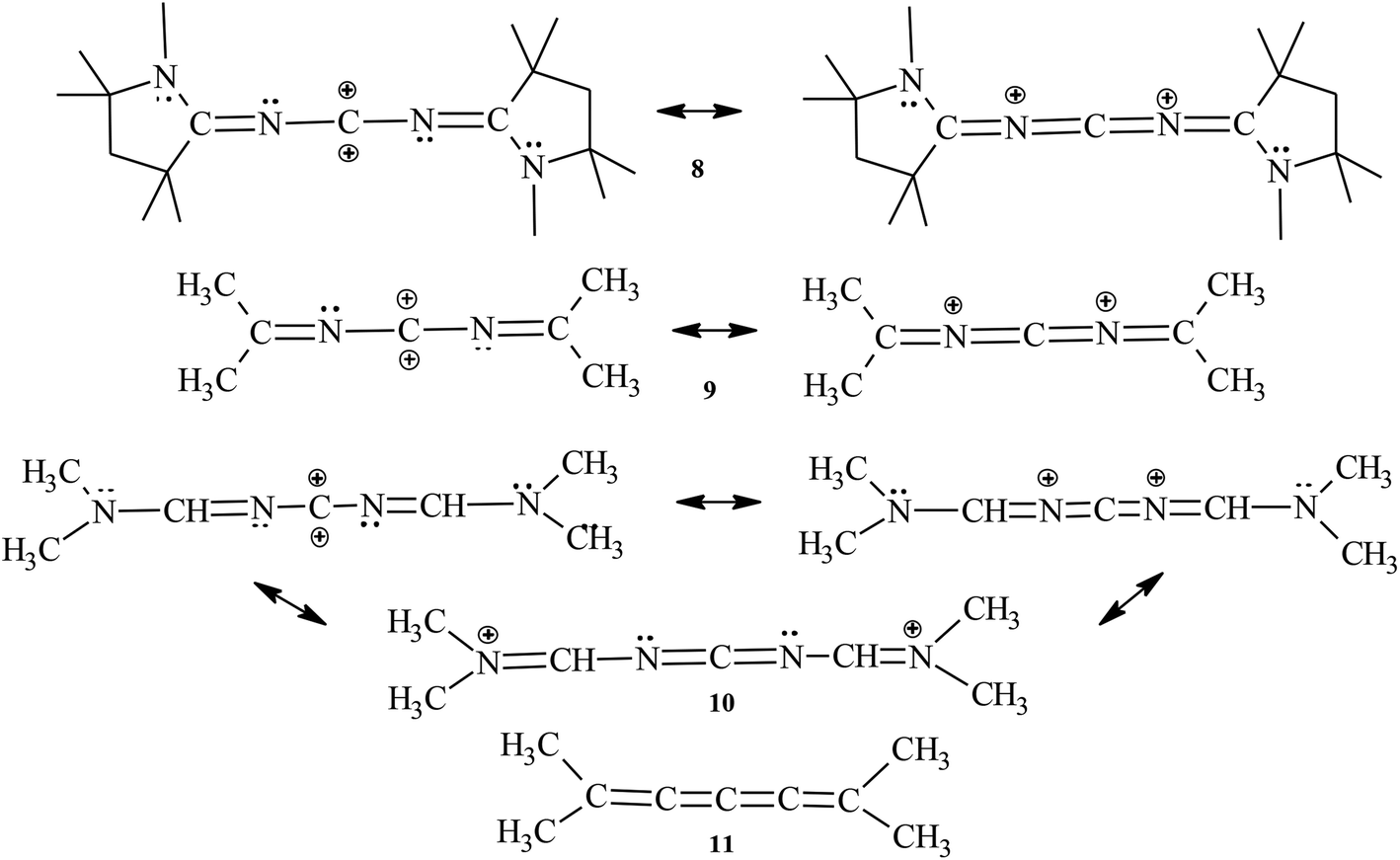 | ||
| Scheme 5 Structures of the carbo2+ family 8–10 and [4]cumulene 11. | ||
| No. | Geometry | NMR parameters | |||
|---|---|---|---|---|---|
| Bond lengths (Å) | Bond angles | δ(13C)/ppm | Others | ||
| dC1–N (Å) | dC2–N(Å) | N–C–—N/C2–N–C1 | |||
| CCCCC |
|||||
| 10 | 1.284 | 1.326 | Linear | 126.5 | C2 (189.0); C3 (114.5) |
| CNCNC |
|||||
| 8 | 1.225; 1.197 | 1.368; 1.349 | 168.5°;163.2° | 138.6; 119.8 | C3 (183.0); CAAC units almost orthogonal (79.6°) |
| 1.361°;147.8° | |||||
| 9 | 1.210 | 1.311 | Linear | 245.4 | CMe2 units (85.2°) |
| 11 | 1.223 | 1.336; 136.8° | 168.0° | 135.2 | C3 (157.9); –CHNMe2 units (83.4°) |
| 4 | 1.245 | — | Linear | 189.8 | –NMe2 units orthogonal |
The choice between cumulative (suggested by Bertrand et al.)10 and allene resonance contributors (Scheme 3) cannot be decided only from the spatial magnetic properties: Wiberǵs bond orders in 8 [C1–N1 (1.92), N1 –C2 (1.29) and C2–N2 (1.63)]10 and in the CAAC (ylide) [(1.53)11b (Scheme 1)] point more urgently to a predominant allene resonance contributor. The dominating, if not complete, CN double bond of the ylide structure of carbenes has been proven.3,4 Furthermore, the 13C chemical shifts of the central carbon atom in 8, 9 and 11 also demonstrate the clear dependence on the presence of the CAAC nitrogen in 8 (and of the terminal nitrogen atom in 11) and supports the presence of the allene-like resonance contributor. Without the terminal nitrogen atoms, the 13C chemical shift of the central carbon atom is much further down field (Scheme 6).
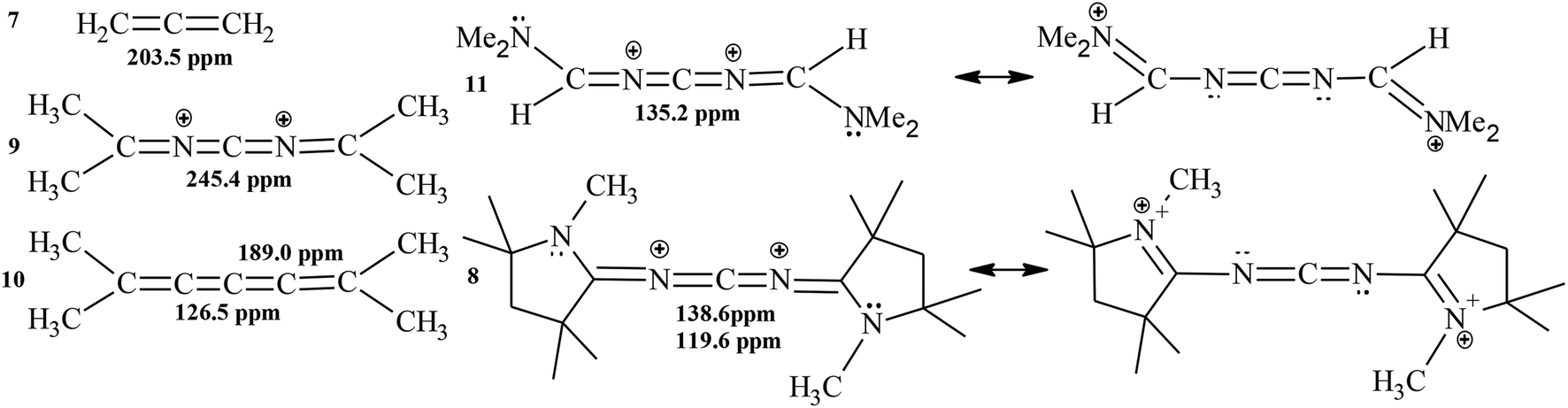 | ||
| Scheme 6 13C Chemical shifts of the central carbon atom in 7–11. | ||
Despite the by-far predominant allene (cumulene)-like mesomeric contributor, the canonical structure of the carbo2+ compound 8 with the corresponding vacant orbitals must be detectably present, because the central C atom is chemically confirmed (double bond formation, successful mono- and di-nucleophilic attack)10 as the dominant electrophilic site of the molecule.
Conclusion
The spatial magnetic properties, through space NMR shieldings (TSNMRSs, the anisotropy effects in 1H NMR spectroscopy) of carbenes, carbones and the recently synthesized new class of carbo2+ compounds have been calculated and, together with geometry and electronic structure, compared on the magnetic criterion. While carbenes prefer the ylide (N+C−–), carbones, the carbone (C–C2−–C) and not the allene (CCC) resonance contributor, carbo2+ compounds unequivocally occur as allene (N+CN+) canonical structure, confirmed by geometry and 13C chemical shift data.
Author contributions
The authors declare equal participation.Conflicts of interest
The authors declare no conflicts of interest.References
- A. Igau, H. Grützmacher, A. Baceiredo and G. Bertrand, Analogous α,α′-bis- carbenoid triply bonded species: synthesis of a stable λ3-phosphinocarbene–λ3- phosphaacetylene, J. Am. Chem. Soc., 1988, 110, 6463–6466 CrossRef CAS.
- A. J. Arduengo III, R. L. Harlow and M. Kline, A stable crystalline carbene, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef.
- W. R. Roger, W. Alder, P. R. Allen, M. Murray and A. Orpen, Bis(diisopropy1- amino)-carbene, Angew. Chem., Int. Ed. Engl., 1996, 35, 1121–1132 CrossRef.
- R. W. Alder, M. E. Blakem and J. M. Oliva, Diaminocarbenes; calculation of barriers to rotation about Ccarbene-N bonds, barriers to dimerization, proton affinities, and 13C NMR shifts, J. Phys. Chem. A, 1999, 103, 11200–11211 CrossRef CAS.
- M. Alcarazo, C. W. Lehmann, A. Anoop, W. Thiel and A. Fürstner, Coordination chemistry at carbon, Nat. Chem., 2009, 1, 295–2301 CrossRef CAS PubMed.
- (a) R. Tonner, F. Öxler, B. Neumüller, W. Petz and G. Frenking, Carbodiphosphoranes: The chemistry of divalent carbon(0), Angew. Chem., Int. Ed., 2006, 45, 8038–8042 CrossRef CAS PubMed; (b) R. Tonner and G. Frenking, C(NHC)2: Divalent carbon(0) compounds with N-heterocyclic carbene ligands − Theoretical evidence for a class of molecules with promising chemical properties, Angew. Chem., Int. Ed., 2007, 46, 8695–8698 CrossRef CAS PubMed.
- H. Schmidbaur, A Rélique: A new concept for bonding in carbodiphosphoranes?, Angew. Chem., Int. Ed., 2007, 46, 2984–2985 CrossRef CAS PubMed , and references therein.
- M. Alcarazo, On the metallic nature of carbon in allenes and heterocumulenes, Dalton Trans., 2011, 40, 1839–1845 RSC.
- T. Morosaki and T. Fujii, Recent advances in heteroatom stabilized carbones and their metal complexes, Adv. Organomet. Chem., 2017, 68, 137–196 CrossRef.
- Y. K. Loh, M. Melaimi, M. Gembicky, D. Munz and G. Bertrand, A crystalline doubly oxidized carbene, Nature, 2023, 623, 66–70 CrossRef CAS PubMed.
- (a) E. Kleinpeter and A. Koch, Stable carbenes or betaines?, Eur. J. Org. Chem., 2018, 3114–3121 CrossRef CAS; (b) E. Kleinpeter and A. Koch, Is the term “Carbene” justified for remote N-heterocyclic carbenes (r-NHCs) and abnormal N-heterocyclic carbenes (a-NHCs/MICs)?, Tetrahedron, 2019, 75, 1549–1554 Search PubMed; (c) E. Kleinpeter and A. Koch, The 13C chemical shift and the anisotropy effect of the carbene electron-deficient centre: Simple means to characterize the electron distribution of carbenes, Magn. Reson. Chem., 2020, 58, 280–292 CrossRef CAS PubMed; (d) E. Kleinpeter and A. Koch, Bent allenes or di-1,3- betaines − An answer given on the magnetic criterion, J. Phys. Chem. A, 2020, 124, 3180–3190 CrossRef CAS PubMed; (e) E. Kleinpeter and A. Koch, Intramolecular carbene stabilization via, 3c,2e bonding on basis of the magnetic criterion, Tetrahedron, 2021, 95, 132357 CrossRef CAS; (f) E. Kleinpeter and A. Koch, Quantification of σ-acceptor and π-donor stabilization in O, S and Hal analogues of N-heterocyclic carbenes (NHCs) on the magnetic criterion, J. Phys. Chem. A, 2021, 125, 7235–7245 CrossRef CAS PubMed; (g) E. Kleinpeter and A. Koch, Phosphorus stabilization of the carbene function in P-analogues of non-cyclic carbenes, N-heterocyclic carbenes and cyclic(alkyl)-(amino)-carbenes − An assessment on basis of geometry, 13C, 31P chemical shifts and the anisotropy effects of the carbene electron deficient centres, Tetrahedron, 2022, 121, 132923 CrossRef CAS.
- E. Kleinpeter and A. Koch, Carbones − a classification on the magnetic criterion, Chem. – Asian J., 2023, e202300826 Search PubMed , and references therein.
- S. Klod and E. Kleinpeter, Ab initio, calculation of the anisotropy effect of multiple bonds and the ring current effect of arenes−application in conformational and configurational analysis, J. Chem. Soc., Perkin Trans. 2, 2001, 1893–1898 CAS.
- E. Kleinpeter, S. Klod and A. Koch, Visualization of through space NMR shieldings of aromatic and anti-aromatic molecules and a simple means to compare and estimate aromaticity, J. Mol. Struct. (THEOCHEM), 2007, 811, 45–60 CrossRef CAS and references cited therein.
- E. Kleinpeter, Quantification and Visualization of the Anisotropy Effect in NMR Spectroscopy by Through-Space NMR Shieldings, in Annual Reports on NMR Spectroscopy, ed. G. A. Webb, Elsevier, 2014, ch. 3, vol. 82 Search PubMed.
- E. Kleinpeter and A. Koch, Antiaromaticity proved by the anisotropic effect in 1H NMR spectra, J. Phys. Chem. A, 2012, 116, 5674–5680 CrossRef CAS PubMed.
- The quantum chemical calculations were performed using the Gaussian 09 program package18 and carried out on LINUX clusters. The studied structures were fully optimized at the MP2/6-311G(d,p) level of theory without constraints.19 The obtained structures have been confirmed as local minima by performing harmonic frequency calculations at the optimized geometries.20 NICS values21,22 were computed on the basis of the MP2/6-311G(d,p) geometries using the gauge-including atomic orbital (GIAO) method23,24 at the B3LYP/6-311G(d,p)25–27 theory level.28 Variation of the basis set was found to be of non-significant influence on the NICS values. To calculate the spatial NICS, ghost atoms were placed on a lattice of −10 Å to +10 Å with a step size of 0.5 Å in the three directions of the Cartesian coordinate system. The zero points of the coordinate system were positioned at the centers of the studied structures. The resulting 68
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 921 NICS values, thus obtained, were analyzed and visualized by the SYBYL 7.3 molecular modeling software;29 different iso-chemical-shielding surfaces (ICSS) of −0.5 ppm (orange) and −0.1 ppm (red) deshielding, and 5 ppm (blue), 2 ppm (cyan), 0.5 ppm (green) and 0.1 ppm (yellow) shielding were used to visualize the TSNMRSs of the studied structures in the various figures. ICSSs are a quantitative indication of the anisotropy effect in 1H NMR spectroscopy;3–5 the computed shielding(deshielding) ICSSs quantify the corresponding anisotropy effect in 1H NMR spectroscopy subject to the distance from the center of the molecules (in Å).13–15.
921 NICS values, thus obtained, were analyzed and visualized by the SYBYL 7.3 molecular modeling software;29 different iso-chemical-shielding surfaces (ICSS) of −0.5 ppm (orange) and −0.1 ppm (red) deshielding, and 5 ppm (blue), 2 ppm (cyan), 0.5 ppm (green) and 0.1 ppm (yellow) shielding were used to visualize the TSNMRSs of the studied structures in the various figures. ICSSs are a quantitative indication of the anisotropy effect in 1H NMR spectroscopy;3–5 the computed shielding(deshielding) ICSSs quantify the corresponding anisotropy effect in 1H NMR spectroscopy subject to the distance from the center of the molecules (in Å).13–15. - M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, et al., Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- C. Møller and M. S. Plesset, Note on an approximation treatment for many-electron systems, Phys. Rev., 1934, 46, 618–622 CrossRef.
- W. J. Hehre, L. Radom, P. V. R. Schleyer and J. A. Pople, Ab initio Molecular Orbital Theory, Wiley, New York, 1986 Search PubMed.
- P. V. R. Schleyer, C. Maerker, A. Dransfield, H. Jiao and N. J. van Eikema Hommes, Nucleus-Independent Chemical Shifts: A simple and efficient aromaticity probe, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS PubMed.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. V. R. Schleyer, Nucleus-Independent Chemical Shifts (NICS) as an aromaticity criterion, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS PubMed.
- R. Ditchfield, Self-consistent perturbation theory of diamagnetism I. A gauge- invariant LCAO method for N.M.R. chemical shifts, Mol. Phys., 1974, 27, 789–807 CrossRef CAS.
- G. W. Cheeseman, T. A. Trucks and M. J. A. Keith, A comparison of models for calculating nuclear magnetic shielding tensors, J. Chem. Phys., 1996, 104, 5497–5509 CrossRef.
- A. D. Becke, Density-functional thermochemistry III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- B. Miehlich, A. Savin, H. Stoll and H. Preuss, Results obtained with the correlation energy density functionals of Becke and Lee, Yang and Parr, Chem. Phys. Lett., 1989, 157, 200–206 CrossRef CAS.
- The lattice points (“ghost atoms”) should be sensor points only without energy contribution in the present calculations. Only if DFT or HF calculations are applied this is true; in case of electron correlation calculations, the “ghost atoms” get their own electron density and show some influence on the energy of the studied molecule. In these cases the TSNMRS surfaces are heavily distorted.
- SYBYL 7.3 Tripos Inc., 1699 South Hanley Road, St. Louis, Missouri 63144, USA, 2007.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ob00063c |
| This journal is © The Royal Society of Chemistry 2024 |