Quaternary ammonium fluorides and difluorosilicates as nucleophilic fluorination reagents†
Received
18th November 2023
, Accepted 21st December 2023
First published on 25th December 2023
Abstract
TBAT (tetrabutylammonium difluorotriphenylsilicate) is an excellent homogeneous nucleophilic fluorination reagent, but a high excess of the reagent was reported to be essential. We hence optimized the reaction conditions and compared its nucleophilic fluorination reactivity with that of other common commercial nucleophilic fluorination reagents, such as anhydrous TBAF and TASF (tris(dimethylamino)sulfonium difluorotrimethylsilicate). As the substrates, we employed a standard set of primary and secondary octyl substrates under identical conditions. To eliminate the possibility of hydrogen fluoride elimination in the above reagents, we prepared four quaternary ammonium fluorides lacking β-elimination possibility in the hydrocarbon chain, transformed them to the corresponding difluorotriphenylsilicates, and compared their reactivity with that of the commercial reagents. Furthermore, attempts to isolate analogous tetrabutylammonium difluoromethyldiphenylsilicate or difluorodimethylphenylsilicate failed, as was confirmed by comparison of the published experimental data with computed 19F NMR spectra. Finally, we studied the transition states of decomposition of various tetramethylammonium methylphenyldifluorosilicates by DFT methods and found that their relative energies increase with an increasing number of phenyl groups. The formation of difluorosilicates is a nearly barrierless process.
Introduction
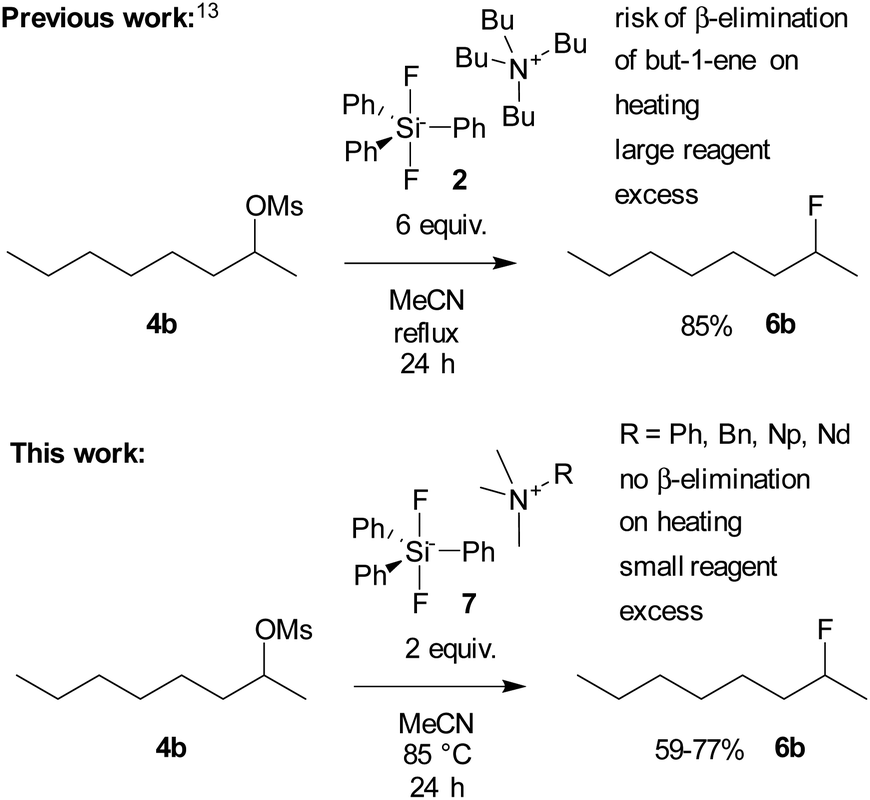
Fluorinated compounds play an indispensable role in industrial chemistry,1 medicinal chemistry2–4 and agrochemistry.5 Among plentiful variants of their preparation methods, nucleophilic fluorination is the most traditional and still extensively applied approach.6,7 Two basic versions of nucleophilic fluorination are available, with deoxyfluorination of alcohols or phenols being probably the most popular approach.8 However, haloalkanes are common substrates in organic chemistry, and so far, methods for converting them to fluorides are limited and insufficient. The main drawback of cheap and therefore frequently used reagents such as KF is their poor solubility and high basicity, which can be solved by the use of ionic liquids, crown ethers or cryptands7,9 Combination of crown ethers with an appropriate sterically hindered alcohol improved the selectivity of nucleophilic fluorination of primary substrates and was studied both experimentally and by DFT methods, but was not tested on critical secondary substrates.10–13 Promising results were obtained with KF and highly sophisticated calixcrown ethers in combination with sterically hindered alcohols as solvents even for one selected secondary bromide.14,15 Unfortunately, no standard secondary substrates were tested. The most common way to cope with the solubility problem is the use of tetrabutylammonium fluoride (TBAF, 1).16 However, its basicity still remains a critical issue and the elimination side reaction is the main obstacle in nucleophilic substitution reactions. To cope with that, a sterically hindered hydrogen donor, tert-butylalcohol, was successfully employed to control TBAF basicity, which significantly improved substitution/elimination ratio for primary substrates.17 Unfortunately, this system was again not tested for critical secondary substrates. Fluorination selectivity and the structure of such complexes were further studied both experimentally18 and by DFT methods.19 Moreover, NMR spectroscopy was employed to understand better the coordination of naked F− anions with ureas.20 Probably the best recent solution for fluorination of secondary substrates is the use of modified phenanthrene complexed CuHF2.21 Another problem of TBAF is that it crystallizes with three molecules of water, and attempts to remove the water by heating result in its decomposition.22 Synthesis of anhydrous TBAF was developed by DiMagno et al.,23 and this solution is commercially available. To avoid the decomposition of the tetrabutylammonium cation and the presence of water, hypervalent silicon compounds have been introduced as a stable source of anhydrous fluoride anions. However, a large excess of tetrabutylammonium difluorotriphenylsilicate (TBAT, 2) has been reported to be used to achieve satisfactory yield in fluorination of octan-2-yl mesylate (4b). Moreover, only low yield of the fluorinated product was obtained from 2-bromooctane (5b).24 Tris(dimethylamino)sulfonium difluorotrimethylsilicate (TASF, 3) has been developed and proved to be highly efficient in fluorination of sterically hindered reagents such as protected saccharides25 or amino acids (Fig. 1).26 With the original aim of synthesizing hypervalent inorganic anions using quaternary ammonium fluorides lacking β-hydrogen and hence not prone to β-elimination, trimethylneopentylammonium fluoride has been synthesized.27
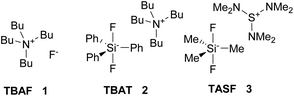 |
| | Fig. 1 Nucleophilic fluorination reagents used for comparison. | |
Herein, we wish to report a systematic study comparing the reactivity of quaternary ammonium fluorides lacking β-hydrogen and the corresponding difluorosilicates with that of the abovementioned fluorination reagents. Furthermore, a computational DFT study dealing with the stability of various tetramethylammonium methylphenylsilicates and the transition states of their decomposition to the corresponding fluorosilanes and TBAF was performed (Scheme 1).
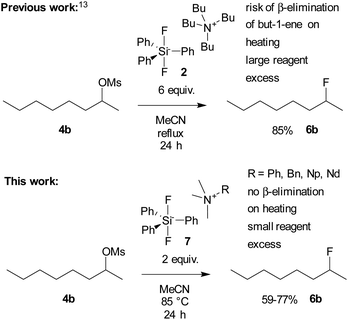 |
| | Scheme 1 Nucleophilic fluorination with difluorosilicates. | |
Results and discussion
Quaternary ammonium fluorides 8
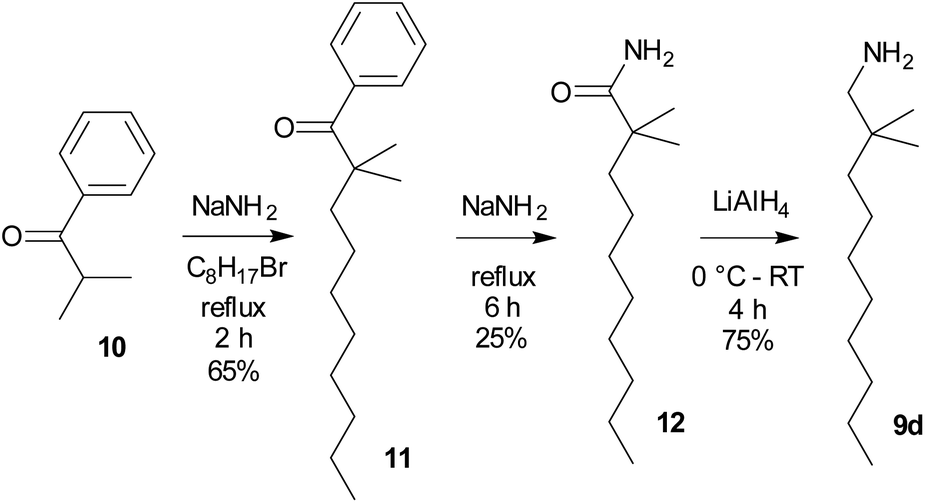
As the starting compounds, the corresponding primary amines 9 were employed. While amines 9a–9c are commercially available, 2,2-dimethyldecan-1-amine (9d) was prepared from 2-methyl-1-phenylpropan-1-one (isobutyrophenone, 10) by a three step procedure in analogy to ref. 28 (Scheme 2). This long-chained amine, 9d, was synthesized with the aim of improving the solubility of both fluoride 8d and difluorosilicate 7d in organic solvents.
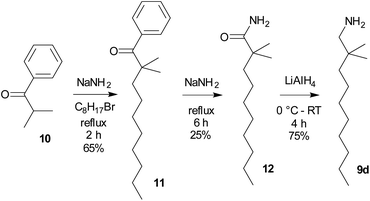 |
| | Scheme 2 Preparation of the starting long chain branched amine 9d. | |
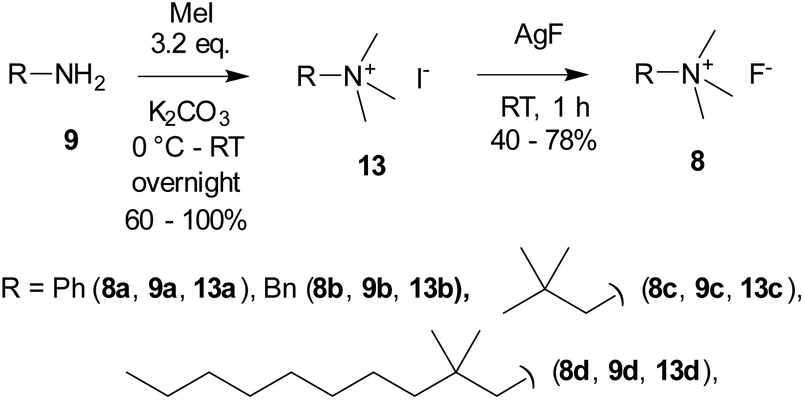
Quaternary ammonium fluorides 8 were obtained from the starting amines 9 by quaternization with methyl iodide, followed by iodine–fluorine exchange with AgF in analogy to ref. 27 (Scheme 3).
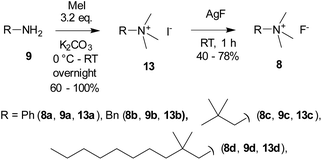 |
| | Scheme 3 Preparation of quaternary ammonium fluorides 8. | |
Quaternary ammonium difluorosilicates 7
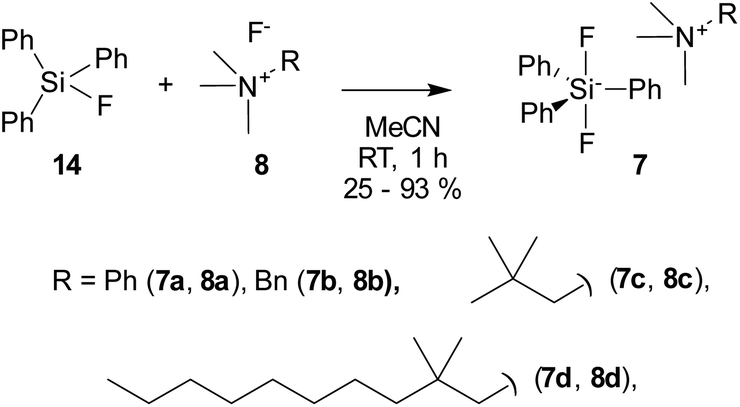
The reaction of fluorides 8 with triphenylsilyl fluoride (14) at room temperature in acetonitrile afforded the target difluorosilicates 7 in poor to good yields as white solids (Scheme 4).
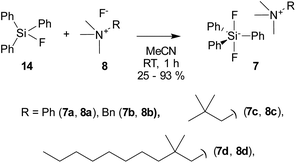 |
| | Scheme 4 Preparation of quaternary ammonium difluorosilicates 7. | |
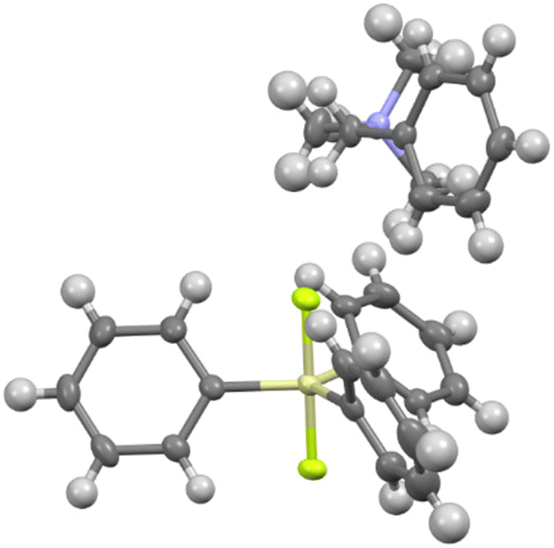
A single crystal of benzyltrimethylammonium difluorotriphenylsilicate (7b) was obtained and its X-ray structure is shown in Fig. 2. The ammonium cation is coordinated to one fluorine and two phenyl rings of the silicate anion. Although the X-ray structure of TBAT has been reported,29 the position of the ammonium cation is not shown and hence the structures could not be compared. On the other hand, the X-ray structure of potassium difluorotriphenylsilicate complexed with [2.2.2]cryptand shows no such coordination due to the bulky complexed cation.30 The details of crystallographic analysis are given in the ESI.†
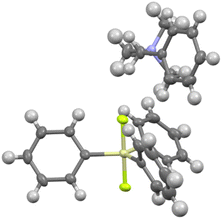 |
| | Fig. 2 Single crystal structure of benzylsilicate 7b. Crystallographic details are given in the ESI.† | |
Nucleophilic fluorinations with fluorides 8 and difluorosilicates 7
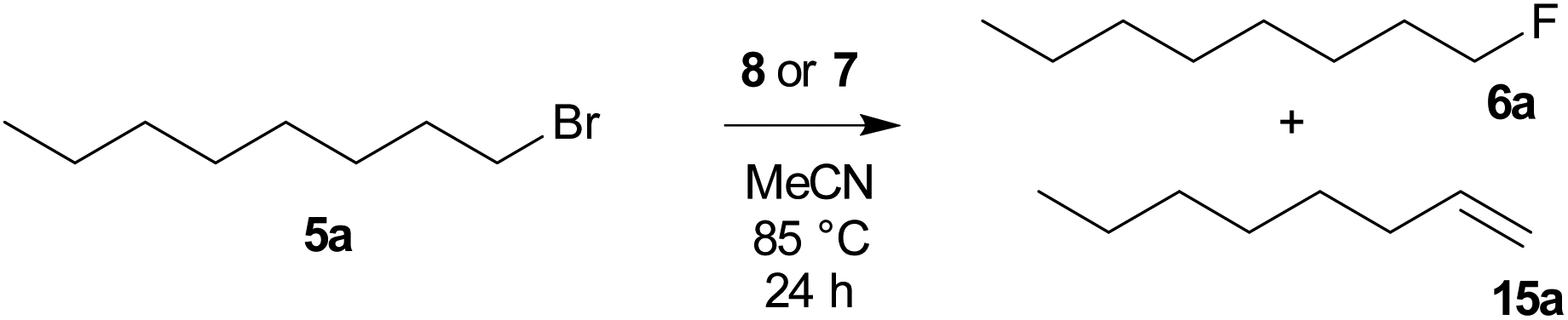
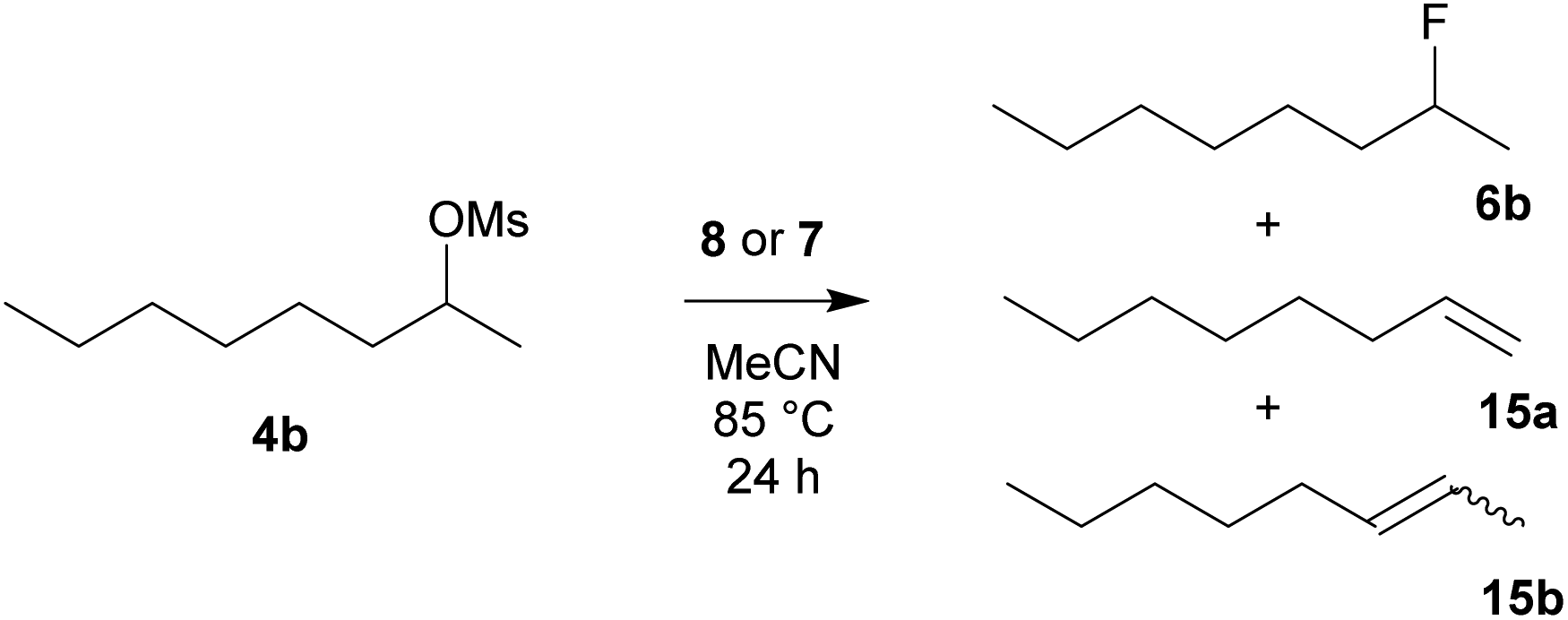
To compare the activity of various fluorination reagents, two primary and two secondary substrates were chosen. In analogy to the paper devoted to TBAT,24 we employed octyl mesylate (4a) and 1-bromooctane (5a) as the primary substrates, while octan-2-yl mesylate (4b) and 2-bromooctane (5b) were used as the secondary substrates. All reactions were performed in sealed Schlenk flasks in CD3CN at 85 °C for 24 h using a two-fold excess of the reagent. In all cases, the average of two fluorination runs is given.
We first tested whether the published24 high excess of 6 equivalents of reagent 2 is really needed. To our surprise, 2 equivalents were sufficient to obtain nearly quantitative conversion of octyl mesylate (4a) (Table 1) and we hence used it in all fluorinations. Lower temperatures gave significantly lower conversions.
Table 1 Optimization of reaction conditions for fluorination of octyl mesylate (4a) with TBAT (2)
| TBAT equiv. |
Time (h) |
Temp. (°C) |
Conversion (%) |
|
High excess of reagent employed in ref. 24.
|
| 1.1 |
24 |
85 |
62 |
| 1.1 |
48 |
85 |
71 |
| 2 |
24 |
85 |
95 |
| 2 |
24 |
40 |
∼3 |
| 2 |
24 |
RT |
0 |
| 6a |
24 |
85 |
96 |
Fluorination of 2-bromooctane (5b).
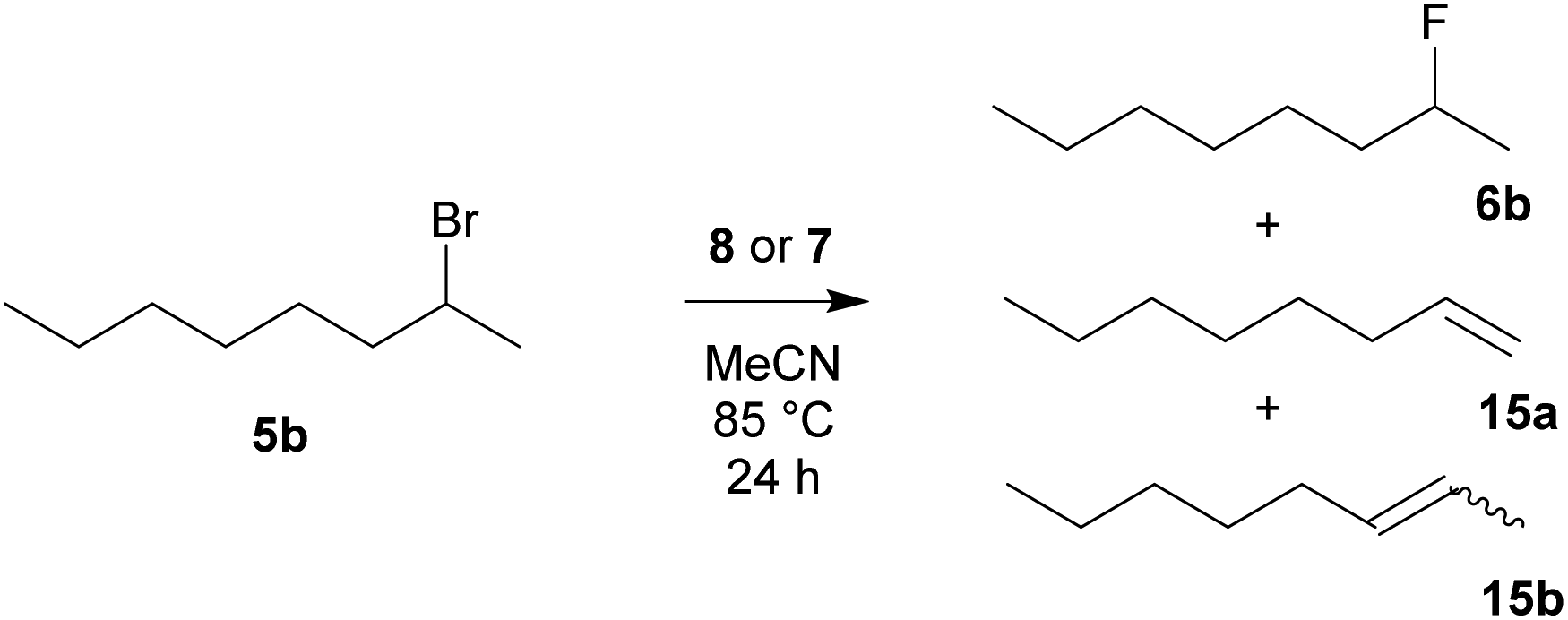
The results of fluorination of 2-bromooctane (5b) (Scheme 8) are summarized in Table 5. As expected, 2-bromooctane (5b) proved to be the most demanding substrate for nucleophilic fluorination reactions. Again, TBAT (2) was the most successful reagent, closely followed by benzyl-based difluorosilicate 7b. TASF reagent (3) again showed the worst selectivity with complete elimination. Among the prepared reagents, difluorosilicates 7 gave better selectivities than fluorides 8.
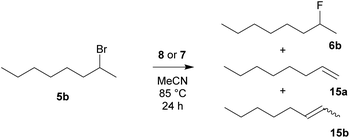 |
| | Scheme 8 Fluorination of 2-bromooctane (5b) with reagent 8 or 7. | |
Table 5 Results of fluorination of 2-bromooctane (4b) with quaternary ammonium fluorides 8 and difluorotriphenylsilicates 7
| Reagent |
Conversion |
Product ratio 6b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15b:15a :15b:15a |
|
Np = 2,2-dimethylpropyl (neopentyl).
Nd = 2,2-dimethyldecyl (neododecyl).
|
| TBAF (1) |
99% |
28:62:10 |
| TBAT (2) |
92% |
39:53:8 |
| TASF (3) |
99% |
0:81:19 |
| PhN+Me3 F− (8a) |
69% |
12:77:11 |
| BnN+Me3 F− (8b) |
99% |
14:72:14 |
| NpN+Me3 F− (8c)a |
90% |
19:69:12 |
| NdN+Me3 F− (8d)b |
99% |
19:71:10 |
| PhN+Me3 Ph3SiF2− (7a) |
65% |
33:59:8 |
| BnN+Me3 Ph3SiF2− (7b) |
92% |
36:56:8 |
| NpN+Me3 Ph3SiF2− (7c)a |
82% |
34:57:9 |
| NdN+Me3 Ph3SiF2− (7d)b |
41% |
20:72:8 |
The main advantage of the newly prepared reagents 8 and 7 is that their synthesis avoids highly corrosive conditions such as HF, as well as the use of expensive anhydrous TBAF. Moreover, while the use of TBAT (2) at higher temperatures cannot fully exclude that the actual fluorinating agent is tetrabutylammonium hydrogen difluoride formed by decomposition of TBAT, this is not possible for reagents 8 and 7. Among the new reagents, benzyltrimethylammonium difluorotriphenylsilicate (7b) was probably the most efficient for difficult secondary substrates with results close to those of the commercial TBAT reagent.
Attempted synthesis of difluoromethylphenylsilicates 16 and 17.
Synthesis of tetrabutylammonium difluoromethyldiphenylsilicate (16) and difluorodimethylphenylsilicate (17) has been reported about twenty years before.31,32 However, the reported 19F NMR shifts (−140 to −150 ppm) do not correspond to the 19F NMR shift of TBAT (−96.0 ppm24 ) and potassium difluoromethyldiphenylsilicate complexed with [2.2.2]cryptand (−85.6 ppm).30 The previously reported data are thus erroneous and correspond to the HF2− anion. When we mixed a solution of fluoromethyldiphenylsilane (18) in CD3CN with “anhydrous” TBAF in an NMR tube and immediately recorded the 19F NMR spectrum at −40 °C, two major signals in a 1:3 ratio were observed (Scheme 9). The minor signal (−86.4 ppm, bs, 24%) corresponds to the target difluorosilicate in good agreement with the computed value (see Table 6), while the major signal (−151.0 ppm, bd, 1JH–F = 128 Hz, 76%) is the signal of the HF2− anion (see Fig. S37 in the ESI†). After stirring the mixture for 1 h at RT, only the signal of the HF2− anion was observed.
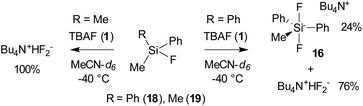 |
| | Scheme 9 Attempted synthesis of difluoromethylphenylsilicates 16 and 17. | |
Table 6 Comparison of computed and reported experimental 19F NMR shifts for difluoromethylphenylsilicates 2, 16, 17, 20–23 and the fluorosilane–tetramethylammonium fluoride complexes 24–27
| R |
Silicate |
Exp. δ |
Silicate |
Comp. δ |
Silane–F− complex |
Comp. δ |
|
Erroneous values.
−86.4 ppm was observed by us for 16 in good agreement with computations.
|
| Ph3 |
2
|
−96.0 (ref. 24) |
20
|
−100.2 |
24
|
−203.9 |
| Ph2Me |
16
|
−157.3 (ref. 31)a,b |
21
|
−88.6 |
25
|
−204.9 |
| PhMe2 |
17
|
−150.9 (ref. 31)a |
22
|
−86.6 |
26
|
−196.3 |
| Me3 |
—
|
−144.2 (ref. 31)a |
23
|
−76.7 |
27
|
−189.3 |
In an analogous experiment with fluorodimethylphenylsilane (19) and “anhydrous” TBAF (Scheme 9), only the signal of the HF2− anion was observed in the 19F NMR spectrum (see Fig. S38 in the ESI†).
Computations
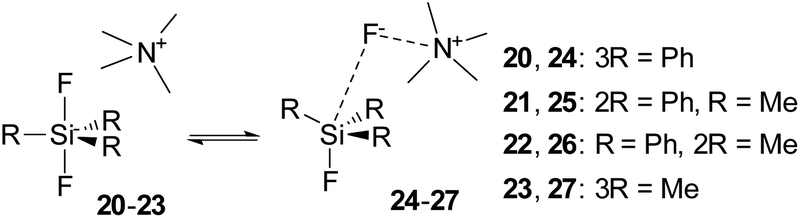
Both previous and our failures to synthesize difluorosilicates modified with methyl groups resulted in our aim to study the stability of these compounds and the connected fluorosilane–quaternary ammonium fluoride complexes by DFT methods. To simplify our calculations, we substituted the tetrabutylammonium cation in TBAT (2), and in 16 and 17, the tetramethylammonium cation. The computational results imply that all difluorosilicates 20–23 should be stable compounds in contrast to the experimental data. For the obtained minima of difluorosilicates 20–23 and the corresponding fluorosilane–ammonium fluoride complexes 24–27 (Scheme 10), chemical shieldings of fluorine atoms and the respective computed chemical shifts obtained by comparison with CCl3F standard shielding are listed in Table 6.
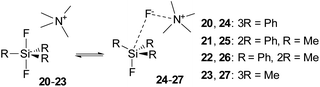 |
| | Scheme 10 Equilibrium among difluorosilicates 20–23 and fluorosilane–TMAF complexes 24–27. | |
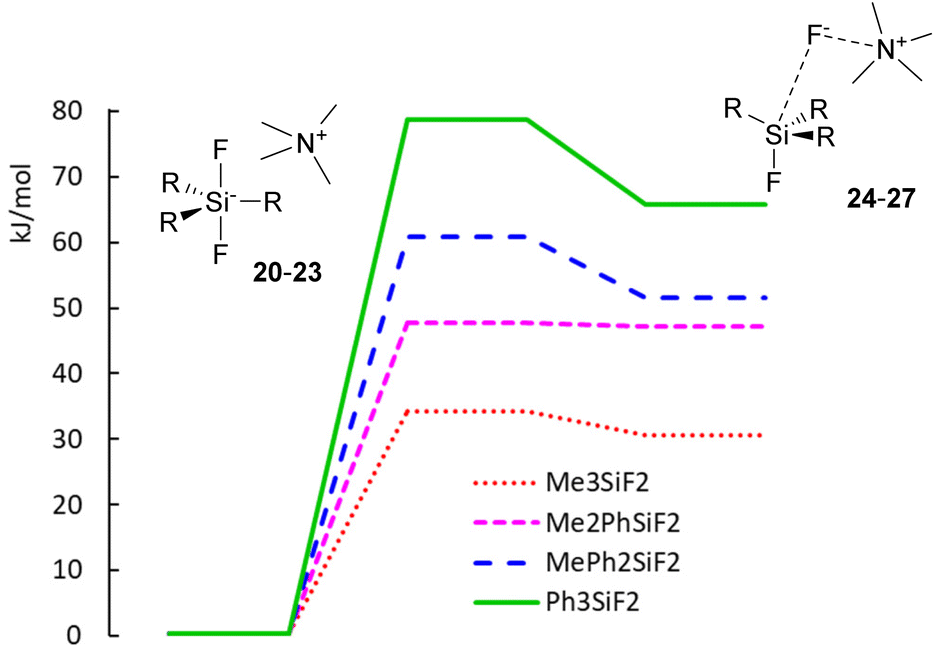
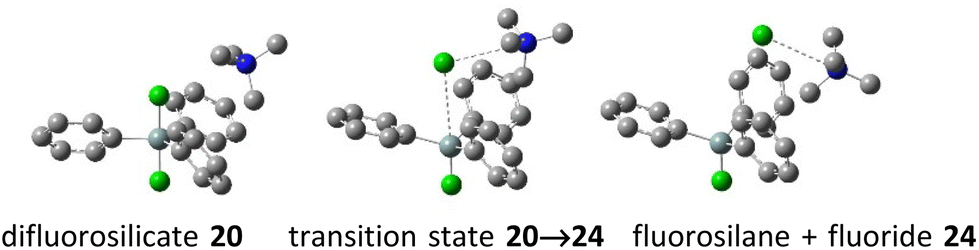
While the computed results agree well with the experimental chemical shifts for TBAT (2), they strongly differ from the erroneously reported shifts for methylated difluorosilicates 16 and 17. Furthermore, we were interested in how high is the energy barrier of decomposition of difluorosilicates 20–23 to fluorosilane–fluoride complexes 24–27. We hence started a DFT study of the corresponding potential energy surfaces. The results show that the transition state Gibbs free energy gradually increases with an increasing number of phenyls in the corresponding difluorosilicates (see Fig. 3 for the individual barriers and Fig. 4 for critical structures). Interestingly, the computational results show that the formation of difluorosilicates from fluorosilane and fluoride anions is a nearly barrierless process.
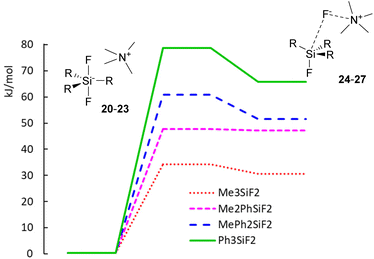 |
| | Fig. 3 Potential energy surfaces of decomposition of difluorosilicates 20–23 to fluorosilane–ammonium fluoride complexes 24–27. | |
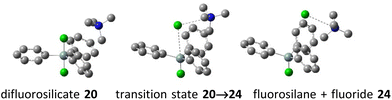 |
| | Fig. 4 Examples of computed critical structures – decomposition of tetramethylammonium difluorotriphenylsilicate (20) to fluorosilane–fluoride complex 24. | |
Experimental
Materials and methods
All reactions were performed under an argon atmosphere in oven dried flasks using a standard inert technique, unless otherwise noted. Fluorinations were performed in sealed vials. 1H NMR spectra were recorded with an Agilent 400-MR DDR2 spectrometer at working frequencies of 399.94 MHz for 1H NMR, 376.29 MHz for 19F NMR and 100.58 MHz for 13C NMR or with a JEOL-ECZL400G spectrometer at working frequencies of 399.78 MHz for 1H NMR, 376.17 MHz for 19F NMR and 100.53 MHz for 13C NMR, in deuterated solvents. Chemical shifts (δ) are reported in parts per million (ppm) with reference to the residual solvent peak. Signals are described as s = singlet, d = doublet, t = triplet, m = multiplet, bs = broad singlet. Coupling constants (J) are reported in Hz. Mass spectra were recorded on an LTW Orbitrap XL (Thermo Fisher Scientific) instrument. Octyl mesylate (4a),33 octan-2-yl mesylate (4b),34 fluoromethyldiphenylsilane (18) and fluorodimethylphenylsilane (19)32 were prepared according to the published procedures.
2,2-Dimethyl-1-phenyldecan-1-one (11).
To a flask containing NaNH2 (0.92 g; 24 mmol; 1.4 equiv.) equipped with a mechanical stirrer, dry toluene (10 mL) and isobutyrophenone (10, 3.0 g; 20 mmol; 1.2 equiv.) were added and the mixture was heated to reflux for 2 h. Then a solution of octyl bromide (3.2 g; 17 mmol; 1.0 equiv.) in anhydrous toluene (10 mL) was added and heated to reflux for 6 h. The mixture was cooled to r.t., quenched with water and the organic layer was washed with water (3 × 20 mL), brine (3 × 20 mL) and dried with anh. MgSO4. The solvent was evaporated to give ketone 11 (2.8 g, 65%). 1H NMR (399.94 MHz, CDCl3): δ 0.86 (t, 3JH–H = 6.9 Hz, 3H, CH3); 1.18–1.33 (m, 14H, CH2); 1.29 (s, 6H, C(CH3)2); 7.35–7.42 (m, 1H, Ar–H); 7.42–7.48 (m, 2H, Ar–H); 7.63–7.68 (m, 2H, Ar–H) ppm. 13C NMR (100.58 MHz, CDCl3): δ 14.2 (1C, CH3); 22.8 (1C, CH2); 24.9 (1C, CH2); 26.3 (2C, C(CH3)2); 29.3 (1C, CH2); 29.5 (1C, CH2); 30.3 (1C, CH2); 32.0 (1C, CH2); 41.2 (1C, CH2); 48.0 (1C, C(CH3)2); 127.7 (2C, Ar–CH); 128.2 (2C, Ar–CH); 130.9 (1C, Ar–CH); 139.4 (1C, Ar–C); 209.5 (1C, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) ppm. HRMS (ESI): calcd for C18H29O [M + H]+ 261.2213, found 261.2214.
O) ppm. HRMS (ESI): calcd for C18H29O [M + H]+ 261.2213, found 261.2214.
2,2-Dimethyldecanamide (12)
To a flask containing NaNH2 (0.84 g; 22 mmol; 2.0 equiv.), a solution of ketone 11 (2.8 g, 11 mmol; 1.0 equiv.) in anhydrous toluene (15 mL) was added and the mixture was heated to reflux. After 6 h, the reaction mixture was cooled to r.t., quenched with water and the organic layer was washed with water (3 × 20 mL), brine (3 × 20 mL), and dried with anh. MgSO4. Then the solvent was evaporated to give amide 12 (0.53 g, 25%). 1H NMR (399.94 MHz, CDCl3): δ 0.87 (t, 3JH–H = 7.0 Hz, 3H, CH3); 1.18 (s, 6H, C(CH3)2); 1.26 (m, 10H, CH2); 1.49 (m, 2H, CH2C); 5.21 (s, 1H, H2N); 5.56 (s, 1H, H2N) ppm. 13C NMR (100.58 MHz, CDCl3): δ 14.3 (1C, CH3); 22.8 (1C, CH2); 25.0 (1C, CH2); 25.7 (2C, C(CH3)2); 29.4 (1C, CH2); 29.7 (1C, CH2); 30.3 (1C, CH2); 32.0 (1C, CH2); 41.6 (1C, CH2); 42.2 (1C, C(CH3)2); 180.7 (1C, CO) ppm. HRMS (ESI): calcd for C12H25NNaO [M + Na]+ 222.1828, found 222.1829.
2,2-Dimethyldecan-1-amine (9d)
Amide 12 (0.92 g; 4.6 mmol; 1.0 equiv.) was dissolved in anhydrous Et2O (25 mL) and the mixture was cooled to 0 °C in an ice bath. To the reaction mixture, LiAlH4 (4.6 mL; 19 mmol; 4.0 equiv., 4 M sol. in Et2O) was slowly added. After 4 h, the reaction mixture was cooled down in an ice bath and was slowly quenched with water. To the mixture, NaOH was added (10 mL, 10% aqueous solution) and the mixture was stirred for 30 min. The organic layer was separated and washed with water (3 × 20 mL), brine (2 × 20 mL), and dried with anh. MgSO4. After evaporation, amine 9d (0.65 g, 75%) was obtained. 1H NMR (399.94 MHz, CDCl3): δ 0.79 (s, 6H, C(CH3)2); 0.85 (t, 3JH–H = 7.0 Hz, 3H, CH3); 1.07–1.32 (m, 14H, CH2); 2.39 (s, 2H, H2NCH2) ppm. 13C NMR (100.58 MHz, CDCl3): δ 14.3 (1C, CH3); 22.8 (1C, CH2); 24.0 (2C, C(CH3)2); 24.8 (1C, CH2); 29.5 (1C, CH2); 29.8 (1C, CH2); 30.7 (1C, CH2); 32.0 (1C, CH2); 34.5 (1C, C(CH3)2); 39.6 (1C, CH2); 52.9 (1C, H2NCH2) ppm. HRMS (ESI): calcd for C12H28N [M + H]+ 186.2216, found 186.2217.
Preparation of ammonium iodides 13
General procedure.
To a suspension of K2CO3 (1.2 equiv.) in anhydrous ethanol, amine 9 (1.0 equiv.) was added and the mixture was cooled to 0 °C in an ice bath. Then MeI (3.2 equiv.) was slowly added to it, and the mixture was allowed to warm to r.t. and stirred overnight. Solids were filtered off and the solvent was evaporated. The crude mixture was dissolved in a small amount of methanol, then Et2O was added to it and white precipitate was collected, washed with Et2O and dried under vacuum.
Trimethylphenylammonium iodide (13a).
From aniline (9a, 0.51 g; 5.0 mmol), K2CO3 (0.92 g; 6.4 mmol), and MeI (2.4 g; 17 mmol), ammonium iodide 13a (1.4 g, 60%) was obtained according to the general procedure. 1H NMR (399.94 MHz, DMSO-d6): δ 3.61 (s, 9H, N(CH3)3); 7.22–8.26 (m, 5H, Ar–H) ppm.35
Benzyltrimethylammonium iodide (13b).
From benzylamine (9b, 0.52 g; 4.6 mmol), K2CO3 (0.80 g; 5.6 mmol), and MeI (2.1 g; 15 mmol), ammonium iodide 13b (1.3 g, 100%) was obtained according to the general procedure. 1H NMR (399.94 MHz, DMSO-d6): δ 3.04 (s, 9H, N(CH3)3); 4.58 (s, 2H, CH2); 7.53 (5H; Ar–H) ppm.36
Trimethylneopentylammonium iodide (13c).
From neopentylamine (9c, 5.0 g; 57 mmol), K2CO3 (9.5 g; 68 mmol) and MeI (26 g; 0.18 mol), ammonium iodide 13c (10.7 g, 73%) was obtained according to the general procedure. 1H NMR (399.94 MHz, DMSO-d6): δ 1.12 (s, 9H, C(CH3)3); 3.14 (s, 2H, CH2); 3.28 (s, 9H, N(CH3)3) ppm.37
(2,2-Dimethyldecyl)trimethylammonium iodide (13d).
From neododecylamine 9d (3.3 g; 18 mmol), K2CO3 (3.3 g; 22 mmol) and MeI (8.1 g; 57 mmol), ammonium iodide 13d (4.0 g, 66%) was obtained according to the general procedure. 1H NMR (399.94 MHz, DMSO-d6): δ 0.85 (t, 3JH–H = 7.0 Hz, 3H, CH3); 1.10 (s, 6H, C(CH3)2); 1.26 (m, 14H, CH2); 3.15 (s, 9H, N(CH3)3); 3.28 (s, 2H, NCH2) ppm. 13C NMR (100.58 MHz, DMSO-d6): δ 14.0 (1C, CH3); 22.1 (1C, CH2); 23.0 (1C, CH2); 26.6 (2C, C(CH3)2); 28.7 (1C, CH2); 28.9 (1C, CH2); 29.7 (1C, CH2); 31.3 (1C, CH2); 35.5 (1C, C(CH3)2); 42.3 (1C, CH2); 54.6 (t, 1JC–N = 4.0 Hz, 3C, (CH3)3N+); 74.1 (1C, NCH2) ppm. HRMS (ESI): calcd for C15H34N [M − I]+ 228.2686, found 228.2687.
Preparation of ammonium fluorides 8
General procedure.
Ammonium iodide 13 (1.0 equiv.) was dissolved in anhydrous methanol (10 mL) and slowly added to a suspension of AgF (1.1 equiv.) in anhydrous methanol (10 mL). The suspension was stirred for 1 h, and then the solution was filtered first through Celite and then through filter paper to yield a clear solution. After evaporation of the solvent, solid ammonium fluoride 8 was dried under vacuum (0.1 kPa) for 14 days while heated to 50 °C. Dry fluorides 8 were stored in a glove box.
Trimethylphenylammonium fluoride (8a).
From ammonium iodide 13a (1.0 g; 3.8 mmol) and AgF (0.53 g; 4.2 mmol), ammonium fluoride 8a (0.41 g, 61%) was obtained according to the general procedure. 1H NMR (399.94 MHz, DMSO-d6): δ 3.64 (s, 9H, N(CH3)3); 7.22–8.27 (m, 5H, Ar–H) ppm. 19F NMR (376.29 MHz, DMSO-d6): δ −101.6 (bs, 1F, F−) ppm. 13C NMR (100.58 MHz, CD3OD): δ 57.7 (3C, (CH3)3N); 121.0 (1C, Ar–CH); 131.7 (2C, Ar–CH); 131.7 (2C, Ar–CH); 148.6 (1C, Ar–C) ppm. HRMS (ESI): calcd for C9H14N [M − F]+ 136.1121, found 136.1121.
Benzyltrimethylammonium fluoride (8b).
From ammonium iodide 13b (1.0 g; 3.6 mmol) and AgF (0.50 g; 3.9 mmol), ammonium fluoride 8b was obtained (0.25 g, 40%) according to the general procedure. 1H NMR (399.94 MHz, DMSO-d6): δ 3.02 (s, 9H, N(CH3)3); 4.54 (s, 2H, CH2); 7.53 (5H, Ar–H) ppm. 19F NMR (376.29 MHz, DMSO-d6): δ −108.24 (s, 1F, F−) ppm. 13C NMR (100.58 MHz, CD3OD): δ 53.1 (t, 1JC–N = 4.2 Hz, 3C, N(CH3)3); 70.5 (t, 1JC–N = 2.8 Hz, CH2, 1C); 129.1 (1C, Ar–C); 130.4 (2C, Ar–CH); 132.0 (1C, Ar–CH); 134.0 (2C, Ar–CH) ppm.
Trimethylneopentylammonium fluoride (8c).
From ammonium iodide 13c (1.0 g; 3.9 mmol) and AgF (0.54 g; 4.3 mmol), ammonium fluoride 8c was obtained (0.45 g, 78%) according to the general procedure. 1H NMR (399.94 MHz, DMSO-d6): δ 1.12 (s, 9H, C(CH3)3); 3.14 (s, 2H, CH2); 3.28 (s, 9H, N(CH3)3) ppm. 19F NMR (376.29 MHz, DMSO-d6): δ −102.11 (bs, 1F, F−) ppm.38
(2,2-Dimethyldecyl)trimethylammonium fluoride (8d).
From ammonium iodide 13d (1.0 g; 2.8 mmol) and AgF (0.41 g; 3.3 mmol), ammonium fluoride 8d was obtained (0.35 g, 48%) according to the general procedure. 1H NMR (399.94 MHz, CD3OD): δ 0.89 (t, 3JH–H = 7.0 Hz, 3H, CH3); 1.22 (s, 6H, C(CH3)2); 1.37 (m, 14H, CH2); 3.25 (s, 9H, N(CH3)3); 3.37 (s, 2H, NCH2) ppm. 19F NMR (376.29 MHz, CD3OD): δ −152.09 (s, 1F, F−) ppm. 13C NMR (100.58 MHz, CD3OD): δ 14.5 (1C, CH3); 23.7 (1C, CH2); 24.7 (1C, CH2); 27.4 (2C, C(CH3)2); 30.5 (1C, CH2); 30.7 (1C, CH2); 31.4 (1C, CH2); 33.1 (1C, CH2); 37.1 (1C, CH2); 44.0 (1C, C(CH3)2); 56.1 (t, 1JC–N = 4.0 Hz, 3C, (CH3)3N); 77.0 (t, 1JC–N = 2.2 Hz, 1C, NCH2) ppm. HRMS (ESI): calcd for C15H34N [M − F]+ 228.2686, found 228.2687.
Preparation of difluorosilicates 7
General procedure.
To a solution of ammonium fluoride 8 (1.0 equiv.) in anhydrous MeCN (2 mL), a solution of fluorotriphenylsilane (14) (1.0 equiv.) in anhydrous MeCN (2 mL) was added and the mixture was stirred for 1 h. Then the solvent was evaporated to yield crude difluorosilicate 7, which was purified by dissolving the white powder in CH2Cl2, precipitating it with hexane and drying in vacuo.
Trimethylphenylammonium difluorotriphenylsilicate (7a).
From ammonium fluoride 8a (100 mg; 0.36 mmol) and Ph3SiF (14, 56 mg; 0.36 mmol), difluorosilicate 7a was obtained according to the general procedure (145 mg, 93%). 1H NMR (399.94 MHz, DMSO-d6): δ 7.04–7.16 (m, 9H, Ar–H); 7.51–7.69 (m, 5H, Ar–H); 7.83–8.02 (m, 6H, Ar–H) ppm. 19F NMR (376.29 MHz, DMSO-d6): δ −97.85 (s, 2F, Si–F2) ppm. 13C NMR (100.58 MHz, DMSO-d6) δ 56.4 (3C, N(CH3)3); 120.4 (2C, Ar–CH); 126.0 (6C, Ar–CH); 126.3 (6C, Ar–CH); 130.1 (3C, Ar–CH); 137.1 (3C, Ar–CH); 147.3 (1C, Ar–CH); 150.5 (3C, Ar–CH) ppm. HRMS (ESI+): calcd for C9H14N [M]+ 136.1121, found 136.1121. HRMS (ESI−): calcd for C18H15F2Si [M]− 297.0917, found 297.0918.
Benzyltrimethylammonium difluorotriphenylsilicate (7b).
From ammonium fluoride 8b (209 mg; 0.75 mmol) and Ph3SiF (14, 127 mg; 0.75 mmol), difluorosilicate 7b was obtained according to the general procedure (244 mg, 73%). 1H NMR (399.94 MHz, DMSO-d6): δ 2.99 (s, 9H, CH3); 4.49 (s, 2H, CH2); 7.05–7.16 (m, 10H, Ar–H); 7.49–7.57 (m, 5H, Ar–H); 7.85–7.89 (m, 5H, Ar–H) ppm.3119F NMR (376.29 MHz, DMSO-d6): δ −97.82 (s, F, Si−F2) ppm.3113C NMR (100.58 MHz, DMSO-d6) δ 51.7 (t, 1JC–N = 3.6 Hz, 3C, N(CH3)3); 67.8 (t, 1JC–N = 2.6 Hz, 1C, NCH2); 126.1 (6C, Ar–CH); 126.3 (6C, Ar–CH); 128.3 (1C, Ar–C); 129.0 (2C, Ar–CH); 130.3 (2C, Ar–CH); 132.8 (1C, Ar–CH); 137.0 (3C, Ar–CH); 150.5 (t, 3JC–F = 41.1 Hz, 3C, Ar–C) ppm. HRMS (ESI+): calcd for C10H16N [M]+ 150.1277, found 150.1278. HRMS (ESI−): calcd for C18H15F2Si [M]− 297.0917, found 297.0919.
Trimethylneopentylammonium difluorotriphenylsilicate (7c).
From ammonium fluoride 8c (107 mg; 0.63 mmol) and Ph3SiF (14, 176 mg; 0.63 mmol), difluorosilicate 7c was obtained according to the general procedure (216 mg, 80%). 1H NMR (399.94 MHz, DMSO-d6): δ 1.10 (s, 9H, C(CH3)3); 3.11 (s, 9H, N(CH3)3); 3.24 (s, 2H, CH2); 7.09 (m, 9H, Ar–H); 7.80–7.89 (m, 6H, Ar–H) ppm.2719F NMR (376.29 MHz, DMSO-d6): δ −97.31 (bs, 2F, Si–F2) ppm.2713C NMR (100.58 MHz, DMSO-d6): δ 29.3 (3C, C(CH3)3); 32.9 (1C, C(CH3)3); 54.5 (t, 1JC–N = 3.8 Hz, 3C, N(CH3)3); 75.5 (t, 1JC–N = 2.3 Hz, 1C, NCH2); 126.1 (6C, Ar–CH); 126.3 (6C, Ar–CH); 137.0 (3C, Ar–CH); 150.8 (m, 3C, Ar–C) ppm. HRMS (ESI+): calcd for C8H20N [M]+ 130.1590, found 130.1591. HRMS (ESI−): calcd for C18H15F2Si [M]− 297.0917, found 297.0915.
(2,2-Dimethyldecyl)trimethylammonium difluorotriphenylsilicate (7d).
From ammonium fluoride 8d (107 mg; 0.63 mmol) and Ph3SiF (14, 176 mg; 0.63 mmol), difluorosilicate 7d was obtained according to the general procedure (216 mg, 80%). 1H NMR (399.94 MHz, DMSO-d6): δ 0.86 (t, 3JH–H = 6.8 Hz, 3H, CH3); 1.09 (s, 6H, C(CH3)2); 1.19–1.37 (m, 14H, CH2); 3.13 (s, 9H, N(CH3)3); 3.26 (s, 2H, NCH2); 7.01–7.16 (m, 10H, Ar–H); 7.84–7.89 (m, 5H, Ar–H) ppm. 19F NMR (376.29 MHz, DMSO-d6): δ −97.83 (bs, 2F, Si–F2) ppm. 13C NMR (100.58 MHz, DMSO-d6): δ 14.0 (1C, CH3CH2); 22.1 (1C, CH2); 23.0 (1C, CH2); 26.7 (1C, CH2); 28.7 (2C, C(CH3)2); 29.0 (1C, CH2); 29.8 (1C, CH2); 31.3 (1C, CH2); 35.5 (1C, CH2); 42.4 (1C, C(CH3)2); 54.7 (t, 1JC–N = 4.2 Hz, 3C, N(CH3)3); 74.3 (1C, NCH2); 126.0 (s, 6C, Ar–CH); 126.3 (s, 6C, Ar–CH); 137.0 (3C, Ar–CH) ppm. HRMS (ESI+): calcd for C15H34N [M]+ 228.2686, found 228.2686. HRMS (ESI−): calcd for C18H15F2Si [M]− 297.0917, found 297.0918.
Fluorinations
General procedure.
A 5 mL Schlenk flask was charged with the fluorination reagent (2.0 equiv.), substrate (1.0 equiv., 20 mg) and CD3CN (0.7 mL). The flask was sealed and heated on a metallic block to 85 °C for 24 h. After cooling, the samples were measured by NMR (16 scans, 20 s relaxation delay) and the conversions were determined from the characteristic peaks of the products 14 and 15 given below.
1-Fluorooctane (6a).
1H NMR (399.94 MHz, CD3CN): δ 4.44 (dt, 2JH–F = 47.5 Hz, 3JH–H = 6.2 Hz, 2H, CH2F) ppm.
2-Fluorooctane (6b).
1H NMR (399.94 MHz, CD3CN): δ 4.64 (ddqd, 2JH–F = 49.3 Hz, 3JH–H = 7.5 Hz, 3JH–H = 6.2 Hz, 3JH–H = 4.7 Hz, 1H, CHF) ppm.
Oct-1-ene (15a).
1H NMR (399.94 MHz, CD3CN): δ 5.85 (ddt, 3JH–H = 17.0 Hz, 3JH–H = 10.2, 3JH–H = 6.7 Hz, 1H, CHCH2) ppm.
Oct-2-ene (15b).
1H NMR (399.94 MHz, CD3CN): δ 5.33–5.53 (m, 2H, CHCH) ppm.
Attempted preparation of tetrabutylammonium difluoromethyldiphenylsilicate (16).
An NMR tube was charged with fluoromethylphenylsilane 18 (30 mg, 27 μL, 0.14 mmol) and CD3CN, followed by the addition of a solution of “anhydrous” TBAF in anhydrous THF (1 M, 140 μL, 0.14 mmol). The mixture was stirred for 1 h at RT and the 19F NMR spectrum was recorded at RT and at −40 °C. At −40 °C, the minor signal of silicate 16 was observed (−86.4 ppm, bs, 24%), while the major signal corresponded to the signal of the HF2− anion (−151.0, bd, 1JH–F = 128 Hz, 76%).
Attempted preparation of tetrabutylammonium difluorodimethylphenylsilicate (17).
An NMR tube was charged with fluoromethylphenylsilane 19 (20 mg, 27 μL, 0.14 mmol) and anhydrous CD3CN, followed by the addition of a solution of “anhydrous” TBAF in anhydrous THF (1 M, 140 μL, 0.14 mmol). The mixture was stirred for 1 h at RT and the 19F NMR spectrum was recorded at RT and at −40 °C.
Computational details
Preliminary computations were performed using the Gaussian1639 program suite using the pure M-06L functional,40 which enabled the use of the RI (resolution of identity) approach,41 together with the double-ζ def2-SVP basis set,42 which greatly accelerated the initial calculations. To better describe the anionic structures, we also used the double-ζ def2-SVPD basis set with additional diffuse functions.43 The solvent (MeCN) was simulated using the SMD variant of the IEF-PCM method.44 The results of preliminary computations are given in the ESI.† Higher level productive calculations were accomplished using the ORCA computational program,45 which uses the efficient RIJCOSX approximation46 to accelerate hybrid functional computation. We employed the M06-2X hybrid functional40 together with the minimally augmented ma-def2-TZVP basis set.47 Weigend's universal auxiliary basis set was used for the RI approximation calculations.48 The MeCN solvent was simulated with the CPCM method49 and the description of the non-covalent interactions was improved by the dispersion correction.50
Results of the preliminary computations, copies of NMR spectra and xyz files of all computed structures are given in the ESI.†
Conclusions
We synthesized a series of new quaternary ammonium fluorides 8 and difluorotriphenylsilicates 7 resistant against β-elimination and compared their reactivity with that of standard nucleophilic fluorination reagents, TBAF (1), TBAT (2) and TASF (3), in the reaction with standard C8 primary and secondary substrates, mesylates and bromides. We found that the reported24 high excess of the TBAT reagent is not essential and a two-fold excess of the reagent is fully sufficient for successful fluorination. TBAF and TASF displayed significantly worse selectivity than TBAT. Newly prepared difluorosilicates 7 were more selective reagents than fluorides 8, with benzyltrimethylammonium difluorotriphenylsilicate 7b showing reactivity and selectivity close to those of TBAT (2). As an advantage, the synthesis of the newly prepared difluorosilicates 7 avoids the use of corrosive HF or expensive anhydrous TBAF. Although we were not able to isolate alkylated difluorosilicates, we observed the signal of tetrabutylammonium difluoromethyldiphenylsilicate (16) in the low temperature 19F NMR spectra together with that of the HF2− anion. Analogous formation of tetrabutylammonium difluorodimethylphenylsilicate (17) was not observed. In contrast to that, DFT calculations showed that methylated difluorosilicates should be stable compounds and the discrepancy between theoretical and experimental data will be further studied. Transition state energies of the decomposition of difluorosilicates 20–23 increase with an increasing amount of phenyl substituents.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors gratefully acknowledge the Czech Science Foundation for financial support of the Project No. 21-29531K (Advanced reagents for (asymmetric) nucleophilic fluorination).
References
- A. Harsanyi and G. Sandford, Green Chem., 2015, 17, 2081–2086 RSC.
- M. Inoue, Y. Sumii and N. Shibata, ACS Omega, 2020, 5, 10633–10640 CrossRef CAS PubMed.
- J. He, Z. Li, G. Dhawan, W. Zhang, A. E. Sorochinsky, G. Butler, V. A. Soloshonok and J. Han, Chin. Chem. Lett., 2023, 34, 107578 CrossRef CAS.
- S. Caron, Org. Process Res. Dev., 2020, 24, 470–480 CrossRef CAS.
- Y. Ogawa, E. Tokunaga, O. Kobayashi, K. Hirai and N. Shibata, iScience, 2020, 23, 101467 CrossRef CAS PubMed.
- A. M. Sorlin, F. O. Usman, C. K. English and H. M. Nguyen, ACS Catal., 2020, 10, 11980–12010 CrossRef CAS.
- P. A. Champagne, J. Desroches, J.-D. Hamel, M. Vandamme and J.-F. Paquin, Chem. Rev., 2015, 115, 9073–9174 CrossRef CAS PubMed.
- T. Aggarwal, Sushmita and A. K. Verma, Org. Chem. Front., 2021, 8, 6452–6468 RSC.
- M. Khandelwal, G. Pemawat and R. K. Khangarot, Asian J. Org. Chem., 2022, 11, e202200325 CrossRef CAS.
- S. L. Silva, M. S. Valle and J. R. Pliego Jr., J. Org. Chem., 2020, 85, 15457–15465 CrossRef CAS PubMed.
- A. S. Melo, M. S. Valle and J. R. Pliego, J. Fluorine Chem., 2023, 269, 110146 CrossRef CAS.
- S. L. Silva, M. S. Valle and J. R. Pliego, J. Mol. Liq., 2020, 319, 114211 CrossRef CAS.
- E. V. Dalessandro and J. R. Pliego, Mol. Syst. Des. Eng., 2020, 5, 1513–1523 RSC.
- S. M. Kang, C. H. Kim, K. C. Lee and D. W. Kim, Org. Lett., 2019, 21, 3062–3066 CrossRef CAS PubMed.
- V. H. Jadhav, W. Choi, S.-S. Lee, S. Lee and D. W. Kim, Chem. – Eur. J., 2016, 22, 4515–4520 CrossRef CAS PubMed.
- D. P. Cox, J. Terpinski and W. Lawrynowicz, J. Org. Chem., 1984, 49, 3216–3219 CrossRef CAS.
- D. W. Kim, H.-J. Jeong, S. T. Lim and M.-H. Sohn, Angew. Chem., Int. Ed., 2008, 47, 8404–8406 CrossRef CAS PubMed.
- K. M. Engle, L. Pfeifer, G. W. Pidgeon, G. T. Giuffredi, A. L. Thompson, R. S. Paton, J. M. Brown and V. Gouverneur, Chem. Sci., 2015, 6, 5293–5302 RSC.
- F. M. Lisboa and J. R. Pliego, J. Mol. Model., 2022, 28, 159 CrossRef CAS PubMed.
- F. Ibba, G. Pupo, A. L. Thompson, J. M. Brown, T. D. W. Claridge and V. Gouverneur, J. Am. Chem. Soc., 2020, 142, 19731–19744 CrossRef CAS PubMed.
- Y. Liu, C. Chen, H. Li, K.-W. Huang, J. Tan and Z. Weng, Organometallics, 2013, 32, 6587–6592 CrossRef CAS.
- R. K. Sharma and J. L. Fry, J. Org. Chem., 1983, 48, 2112–2114 CrossRef CAS.
- H. Sun and S. G. DiMagno, J. Am. Chem. Soc., 2005, 127, 2050–2051 CrossRef CAS PubMed.
- A. S. Pilcher, H. L. Ammon and P. DeShong, J. Am. Chem. Soc., 1995, 117, 5166–5167 CrossRef CAS.
- W. A. Szarek, G. W. Hay and B. Doboszewski, J. Chem. Soc., Chem. Commun., 1985, 663–664 RSC.
- W. Qu, Z. Zha, K. Ploessl, B. P. Lieberman, L. Zhu, D. R. Wise, C. B. Thompson and H. F. Kung, J. Am. Chem. Soc., 2011, 133, 1122–1133 CrossRef CAS PubMed.
- A. R. Mahjoub, X. Zhang and K. Seppelt, Chem. – Eur. J., 1995, 1, 261–265 CrossRef CAS.
- K. E. Hamlin and M. Freifelder, J. Am. Chem. Soc., 1953, 75, 369–373 CrossRef CAS.
- A. F. Hill, J. S. Ward and Y. Xiong, Organometallics, 2015, 34, 5057–5064 CrossRef CAS.
- S. Yamaguchi, S. Akiyama and K. Tamao, Organometallics, 1999, 18, 2851–2854 CrossRef CAS.
- M. Penso, D. Albanese, D. Landini and V. Lupi, J. Mol. Catal. A: Chem., 2003, 204–205, 177–185 CrossRef CAS.
- J. Kvíčala, P. Mysík and O. Paleta, Synlett, 2001, 547–549 CrossRef.
- Y.-R. Hong and C. B. Gorman, J. Org. Chem., 2003, 68, 9019–9025 CrossRef CAS PubMed.
- J. J. Tufariello, H. Meckler, K. Pushpananda and A. Senaratne, Tetrahedron, 1985, 41, 3447 CrossRef CAS.
- Z. Kokan, M. Duskova-Smrckova and V. Sindelar, Chem, 2021, 7, 2473–2490 CAS.
- C. I. Nwachukwu, T. P. McFadden and A. G. Roberts, J. Org. Chem., 2020, 85, 9979–9992 CrossRef CAS PubMed.
- J. Rickmeier and T. Ritter, Angew. Chem., Int. Ed., 2018, 57, 14207–14211 CrossRef CAS PubMed.
- U. Groß, S. Rüdiger, A.-R. Grimmer and E. Kemnitz, J. Fluorine Chem., 2002, 115, 193–199 CrossRef.
-
J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2007, 120, 215–241 Search PubMed.
- O. Vahtras, J. Almlöf and M. W. Feyereisen, Chem. Phys. Lett., 1993, 213, 514–518 CrossRef CAS.
- D. Rappoport and F. Furche, J. Chem. Phys., 2010, 133, 134105 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS PubMed.
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98–109 CrossRef CAS.
- J. Zheng, X. Xu and D. Truhlar, Theor. Chem. Acc., 2011, 128, 295–305 Search PubMed.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Kateřina
Kučnirová
a,
Kateřina
Kučnirová