
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn electrochemical access to 2-amino-2,3-dihydro-1,4-benzodioxanes derived from hydroxytyrosol†
Anne
Neudorffer
 *,
Patrick
Deschamps
,
Martine
Largeron
and
Brigitte
Deguin
*,
Patrick
Deschamps
,
Martine
Largeron
and
Brigitte
Deguin
Université Paris Cité, CNRS, CiTCoM, F-75006 Paris, France. E-mail: anne.neudorffer@u-paris.fr
First published on 2nd January 2024
Abstract
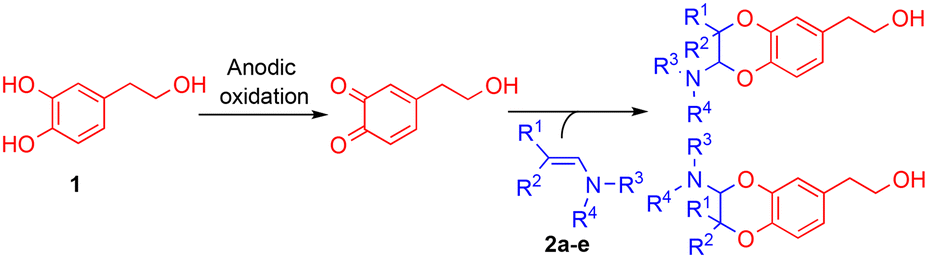
The anodic oxidation of a natural antioxidative catechol, hydroxytyrosol, was developed in an acetonitrile/dimethylsulfoxide (or acetonitrile/water) solvent mixture to produce in a stable way the resulting non-activated o-quinone and generate structural analogues. 2-Amino-2,3-dihydro-1,4-benzodioxane derivatives were obtained as two regioisomers in good to high overall yields (65–90%) and 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratios, through an inverse electron demand Diels–Alder (IEDDA) reaction between the electrogenerated o-quinone and tertiary enamines. The insertion of an electron withdrawing (or electron donating) group on the catechol modified their relative proportions, so that the reaction became regiospecific. With some aliphatic enamines, a competitive 1,6-Michael addition took place, affording 2-hydroxy-1,2,4,5-tetrahydrobenzo[d]oxepine compounds.
:3 ratios, through an inverse electron demand Diels–Alder (IEDDA) reaction between the electrogenerated o-quinone and tertiary enamines. The insertion of an electron withdrawing (or electron donating) group on the catechol modified their relative proportions, so that the reaction became regiospecific. With some aliphatic enamines, a competitive 1,6-Michael addition took place, affording 2-hydroxy-1,2,4,5-tetrahydrobenzo[d]oxepine compounds.
Introduction
The Mediterranean diet induces various health benefits, in particular a lower incidence of cancers, diabetes, and cardiovascular and Alzheimer's diseases.1 These effects attributed to the large consumption of fruits and vegetables are also correlated with the abundant intake of olives and extra virgin olive oil (EVOO), rich in fatty unsaturated acids and secoiridoids2 such as oleuropein, ligstroside, oleacein and oleocanthal (Fig. 1).3–5 Hydroxytyrosol 1, a catechol compound present in high concentration in EVOO and constitutive of oleuropein aglycon, was found to contain strong radical scavenging properties as well as inhibiting activities on the inflammatory pathways.6 Structural modifications have been carried out to improve its pharmacological properties.7,8 Most of them consisted of the esterification of the alcohol chain with lipophilic acids9 or protecting the catechol moiety responsible for the antioxidant properties with acetonide10 or sulphate11 groups. Other functionalisations of hydroxytyrosol 1 were realised on the 6-position of the aromatic ring, through nitration,12 arylation13 (Suzuki–Miyaura reaction), or cyclisation in isochroman14 in the presence of carbonyl compounds.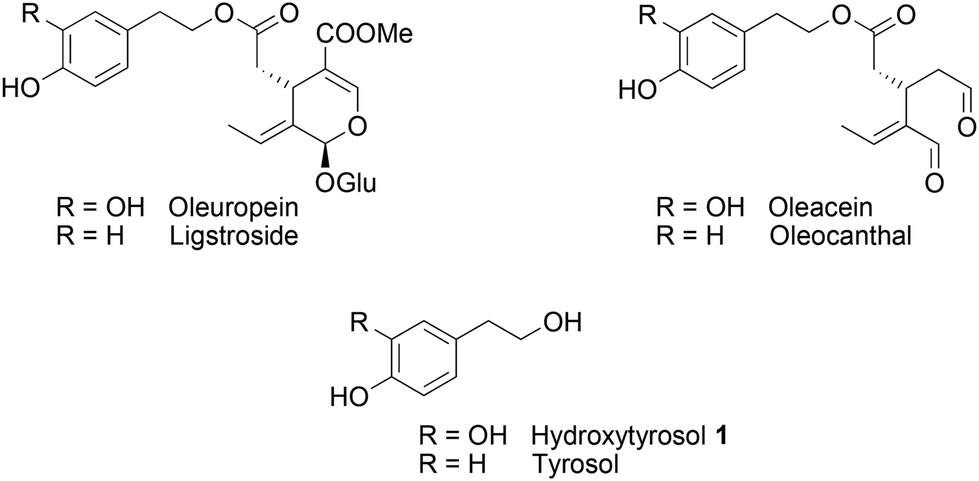 | ||
| Fig. 1 Chemical structures of the major phenolic compounds in EVOO. | ||
During the past decades, the construction of 2,3-dihydro-1,4-benzodioxane rings from catechols was extensively developed, due to their occurrence in natural products and pharmaceutical compounds.15 However, very few methods led to 2-amino-2,3-dihydro-1,4-benzodioxanes, although similar structures were identified in compounds isolated from insects16–20 and marine organisms.21 Such natural products isolated as pairs of two trans regioisomers, except in the case of orthidines A–D (Fig. 2), possess noticeable antioxidant and anti-inflammatory activities, including inhibition of cyclooxygenases. Therefore, we envisaged the synthesis of 2-amino-2,3-dihydro-1,4-benzodioxane derivatives starting from hydroxytyrosol 1.
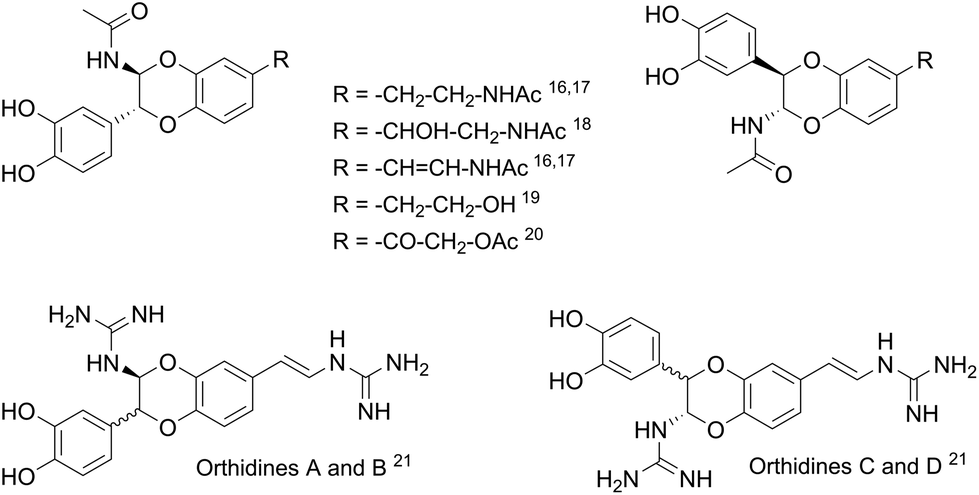 | ||
| Fig. 2 Some natural 2-amino-2,3-dihydro-1,4-benzodioxanes. | ||
With the exception of a biomimetic oxidative dimerisation of N-acetyl-dehydrodopamine,22 the principal route to 2-amino-2,3-dihydro-1,4-benzodioxanes consists of inverse electron-demand Diels–Alder reactions (IEDDA) between enamines and o-quinone heterodienes. The o-quinones used then were commercially available halogenated o-quinones, such as o-chloranil and o-bromanil,23 or o-quinones obtained through the oxidation of monophenols by IBX (Scheme 1).24 A few years ago, we reported that the electrochemical oxidation of o-diphenols was an efficient tool for producing o-quinones under environmentally friendly conditions. These unstable species could be involved in further Michael reactions,25,26 intramolecular cyclisation27 or IEDDA reactions with enamines,28 through diverse one-pot processes. Until now, [4 + 2] cycloadditions with enamine dienophiles were possible only from electron-poor electrogenerated o-quinone heterodienes (or electron-poor o-azaquinones).29 Compared to the anodic oxidation of monophenols,30 the electrochemical oxidation of non-activated catechols was less investigated, even if the resulting o-quinones engaged in subsequent reactions gave various compounds, such as catechol thioethers,31,32 dihydroxy-phenyl-indolin-2-ones,33 benzofurans,34 or a dimethyl-fulvene coupling product.35 Previously, two enzymatic oxidations of hydroxytyrosol 1 were attempted. None of them produced the expected o-quinone under stable conditions. The first led to a benzodioxan type dimer thanks to a catechol-quinone intermolecular 1,4-Michael addition,36 while the second afforded, after the suroxidation of the transient o-quinone species, an hydroxylated quinonoid-phenyl dimer.37
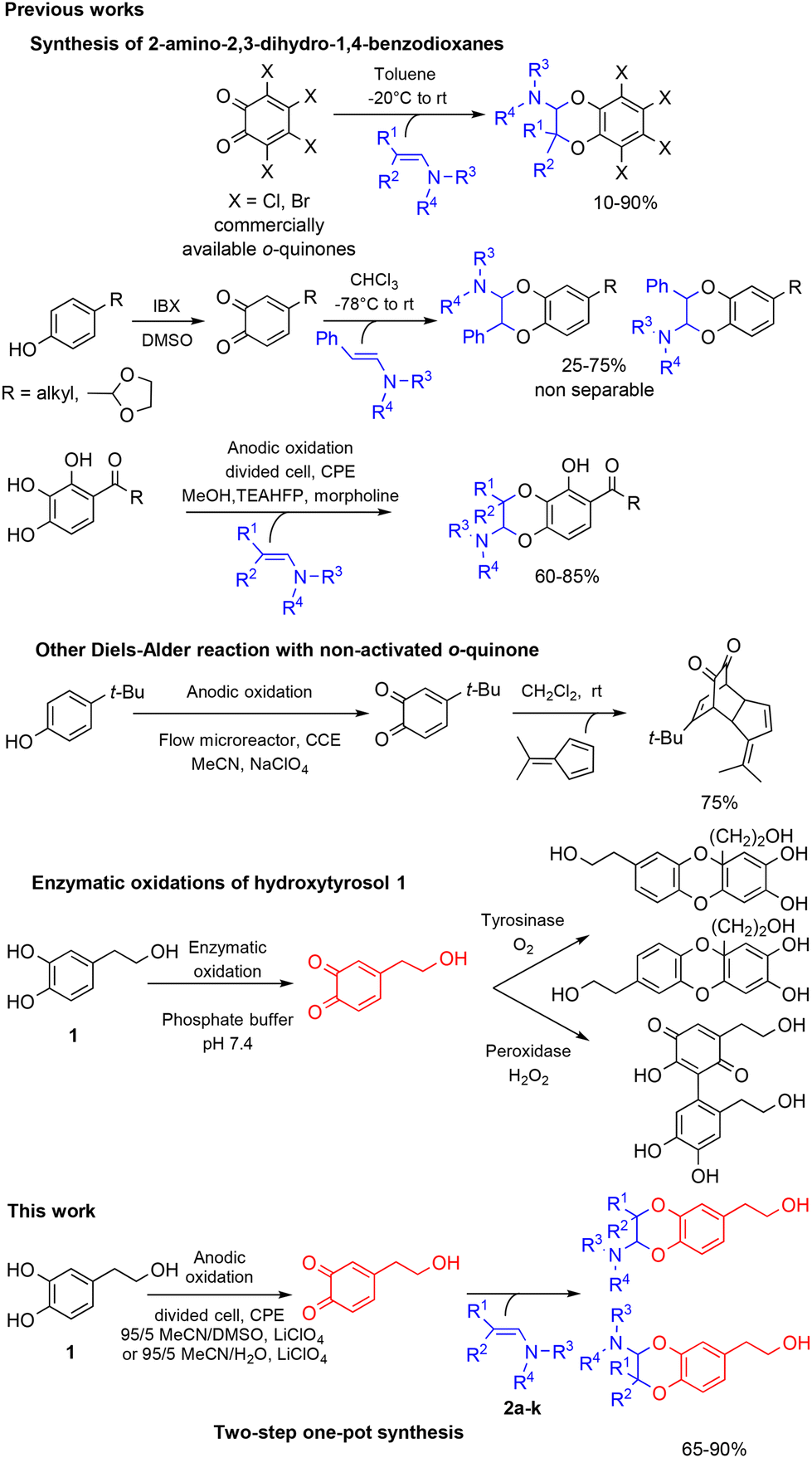 | ||
| Scheme 1 Reactivity of various o-quinone species. | ||
Therefore, we describe herein an electrochemical process to generate in a stable way the expected o-quinone(s) and obtain, through an IEDDA reaction with enamines, 2-amino-2,3-dihydro-1,4-benzodioxanes derived from hydroxytyrosol 1. The parameters that influence the [4 + 2] cycloaddition are further explored in terms of heterodienes (non-activated/activated electrogenerated o-quinones) and dienophiles (aromatic/aliphatic enamines).
Results and discussion
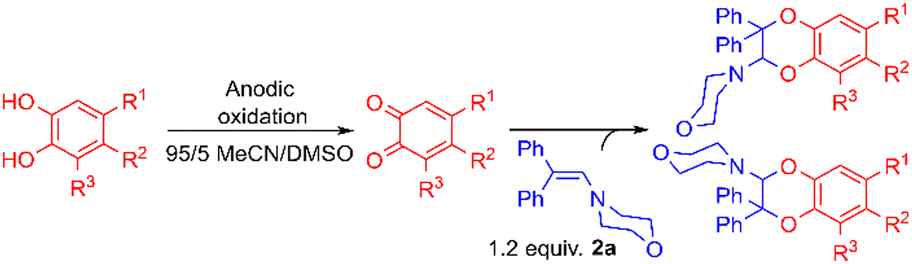
Synthesis of compounds 3 and 4
For this purpose, the solvent of the electrolysis had to be compatible both with the two-electron oxidation of the catechol and with the good stability of the enamine species involved in the [4 + 2] cycloaddition reaction. While the use of acetonitrile or dimethylformamide is usually preferred to generate radical intermediates such as phenoxyls, buffered aqueous solutions, methanol or water/acetonitrile solvent mixtures are often used for the oxidation of catechols into o-quinones. In aqueous buffered solutions, the pH of the solution and the concentration of the enamine strongly influence the rate of hydrolysis of the dienophile.38 Therefore to start, a 50/50 (v/v) phosphate buffer pH 8.0/acetonitrile solvent was used for the electrochemical oxidation of hydroxytyrosol by controlled potential electrolysis. Under these conditions, the cyclic voltammogram of 1, recorded at a platinum working electrode in the presence of 0.05 M LiClO4 as the supporting electrolyte, showed a broad anodic peak Pa around +1.8 V vs. Ag/AgCl, without any cathodic peak on the reverse sweep (0.1 V s−1 scan rate). When the controlled potential of the electrolysis was fixed at +1.8 V vs. Ag/AgCl, a coulometric value of 2.0 ± 0.1 F was found for the number of electrons (n) involved in the oxidation of one mole of 1. The monitoring of the UV-visible absorption spectrum in the course of the electrolysis showed a decrease in the absorption band due to the neutral form of 1 at λ = 281 nm, while new bands developed at λ = 257 and 395 nm. The presence of four isosbestic points confirmed the existence of a simple equilibrium between the two species (Fig. 3), hydroxytyrosol 1 and the o-quinone species.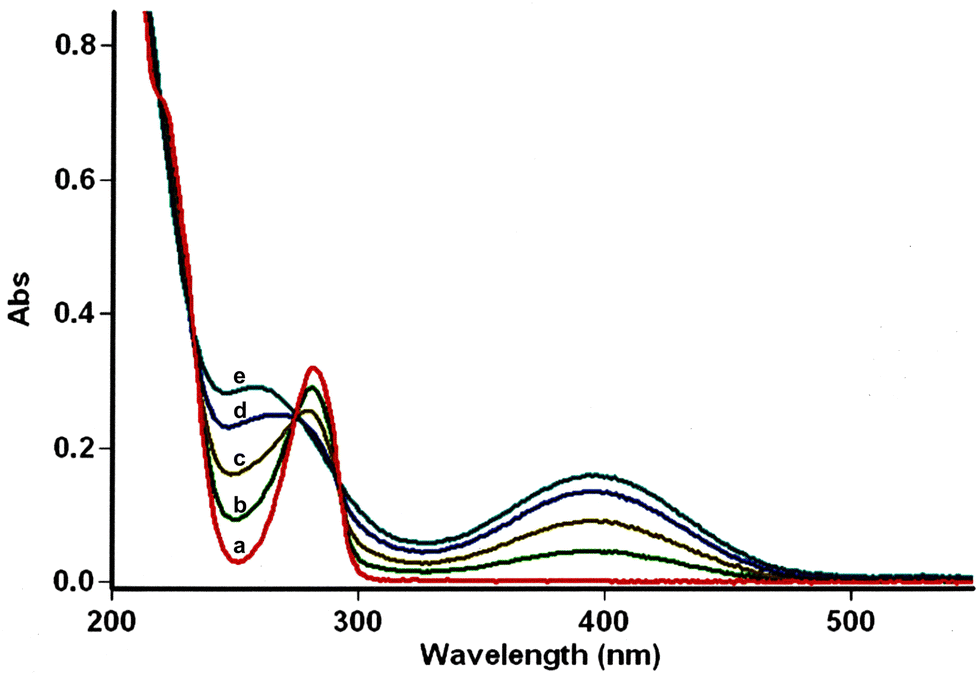 | ||
| Fig. 3 UV-visible absorption spectra in the course of the anodic oxidation of 1. Hydroxytyrosol 1 (1.25 mM), 50/50 (v/v) phosphate buffer pH 8.0/acetonitrile solution containing LiClO4 (0.05 M) as the supporting electrolyte, platinum anode, Eox = +1.8 V vs. Ag/AgCl, stirring for 40 min under Ar at room temperature. (a) Before electrolysis, (b) 0.5 F mol−1, (c) 1.0 F mol−1, (d) 1.5 F mol−1, and (e) 2.0 F mol−1. | ||
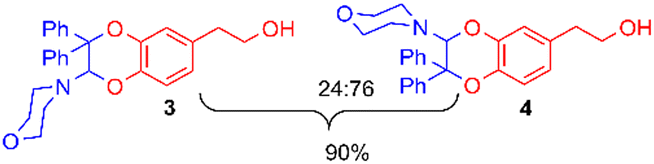
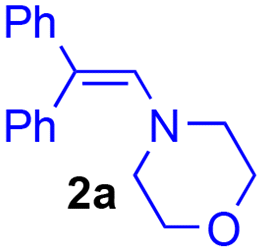
The addition of 1-(4-morpholino)-2,2-diphenylethene 2a (5 equiv.) to the exhaustively oxidised solution induced the slow discolouration of the yellow colour characteristic of the o-quinone (30 min). Two 1,4-benzodioxane regioisomers 3 and 4, separable by flash chromatography, could then be isolated in 61% overall yield, with 27/73 ratio (Table 1, entry 1).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Potential | Equiv. of enamine | Yieldc | Ratiod3/4 |
| a Electrosynthesis: [1] = 1.25 mM, 0.05 M LiClO4 as the supporting electrolyte, divided cell, platinum grid anode, platinum plate cathode,40 oxidation potential Eox referred to Ag/AgCl. b Diels–Alder reaction: addition of enamine 2a to a solution of o-quinone. c Overall isolated yield of both regioisomers 3 and 4. d 1H NMR ratio of 3/4. e Aqueous phase = 3 mM phosphate buffer pH 8.0. f 0.05 M LiClO4 replaced with 0.02 M NEt4PF6. g +5 equiv. of morpholine. | |||||
| 1 | 50/50 H2O/MeCNe | +1.8 V | 5 | 61% | 27/73 |
| 2 | MeCN | +1.4 V | 5 | — | — |
| 3 | MeOHf | +1.0 V | 5 | — | — |
| 4 | MeOHf,g | +1.0 V | 5 | — | — |
| 5 | 95/5 MeCN/DMSO | +1.0 V | 5 | 71% | 24/76 |
| 6 | 95/5 MeCN/DMSO | +1.0 V | 2.5 | 90% | 24/76 |
| 7 | 95/5 MeCN/DMSO | +1.0 V. | 1.2 | 90% | 25/75 |
| 8 | 95/5 MeCN/H2O | +1.0 V | 5 | 63% | 24/76 |
| 9 | 95/5 MeCN/H2O | +1.0 V | 1.2 | 27% | 24/76 |
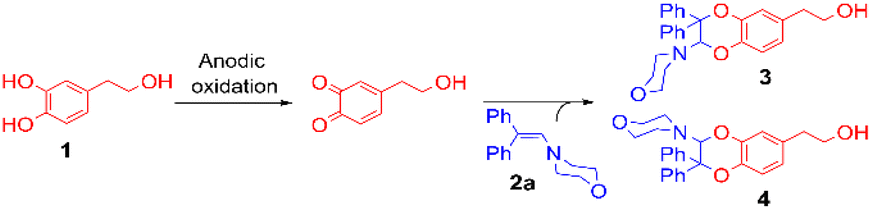
The structural identification of the major regioisomer 4 was first established thanks to the HMBC correlation of the single hydrogen of the dihydrodioxin ring (δH = 5.70 ppm) with C-3 (Fig. S1†). As confirmed by X-ray crystallographic data (Fig. 4), the point of fixation of the morpholino group faced the 3-position. Comparatively, the hydrogen of the dihydrodioxin ring of the other 1,4-benzodioxane product correlated with C-4. Compound 3 was therefore identified as the second regioisomer that could be formed through the [4 + 2] cycloaddition between the enamine and the electrogenerated o-quinone (see the equation in Table 1).
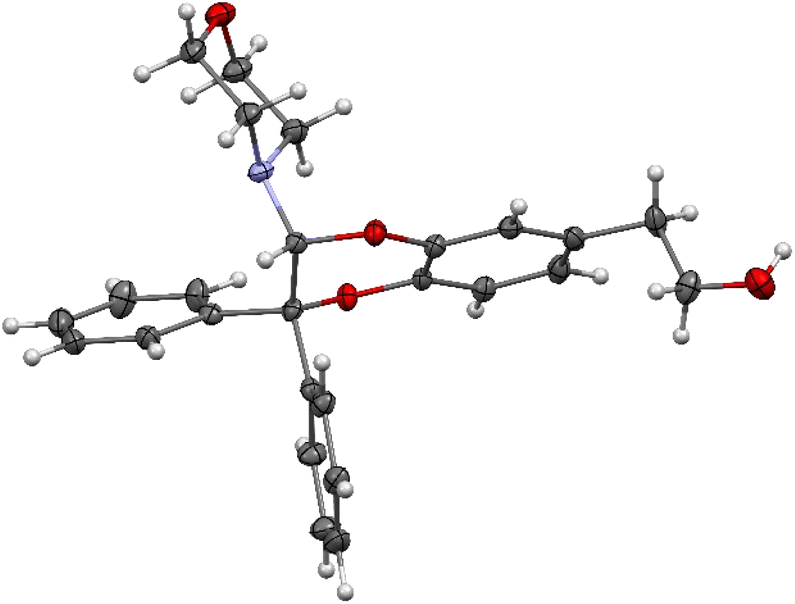 | ||
| Fig. 4 ORTEP view of compound 4. Displacement ellipsoids are drawn at the 50% probability level. | ||
Optimisation of reaction conditions
Usually, the yields of the IEDDA reactions markedly depend on the nature of the solvent.39 Here, the replacement of 50/50 water/acetonitrile solvent with pure acetonitrile or methanol induced strong modifications in the evolution of UV-visible spectra, together with the loss of isosbestic points (Table 1, entries 2–4). No formation of stable o-quinone was possible, whatever the ionisation level under which the hydroxytyrosol catechol was oxidised: neutral species (entry 3) or monoanionic form obtained through morpholine addition (entry 4).28To limit the hydrolysis of enamine without affecting the rate of the cycloaddition, we replaced the large amount of water (50% of aqueous buffer pH 8.0) with 5% of dimethylsulfoxide. The potential of the platinum anode could then be fixed at a lower potential (+1.0 V vs. Ag/AgCl), since the cyclic voltammogram exhibited an anodic peak at +0.9 V vs. Ag/AgCl (scan rate 0.1 V s−1). A small cathodic peak appeared on the reverse sweep at 0 V. vs. Ag/AgCl corresponding to the reduction of the electrogenerated o-quinone (Fig. S2†). As previously observed in the water/acetonitrile solvent, a coulometric value of 2.0 ± 0.1 F was necessary for the exhaustive oxidation of one mole of hydroxytyrosol 1. Under these optimised reaction conditions, the yield of 3 and 4 reached 90%, even in the presence of only 1.2 equiv. of enamine (entries 5–7).
The reaction efficiency was still suitable in 95/5 acetonitrile/water mixtures with 5 equiv. of enamine (entry 8), with the ratio of regioisomers 3 and 4 remaining close to 25/75. However, decreasing the amount of enamine to 1.2 equiv. was no more possible without a severe diminution of the yield (entry 9).
The importance of the order of addition of the diene and the dienophile should be underlined. The dienophile (enamine) had to be added to the diene solution (o-quinone). When MeCN/water solution of o-quinone was added in fractions to MeCN solution of 5 equiv. of enamine, no 1,4-benzodioxane formed. In the presence of water, the partial hydrolysis of the enamine induced the liberation of an excess of base (morpholine) and the polymerisation of a small fraction of o-quinone (dark purple colour).
Variation of the catechol substrate and ratio of the regioisomers
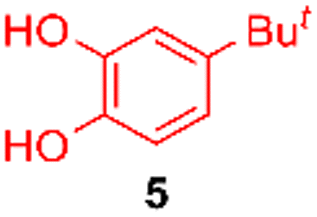
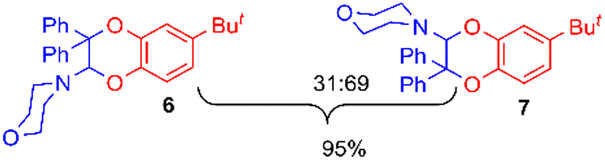
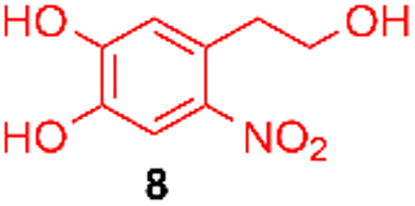
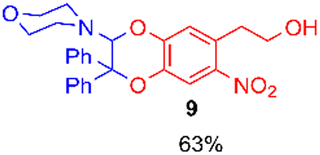
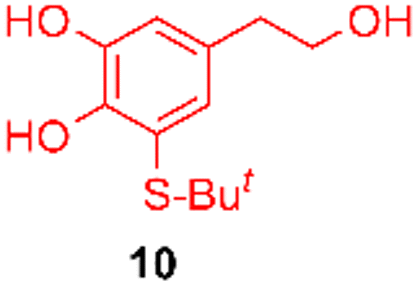
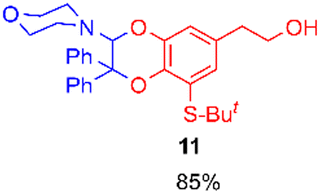
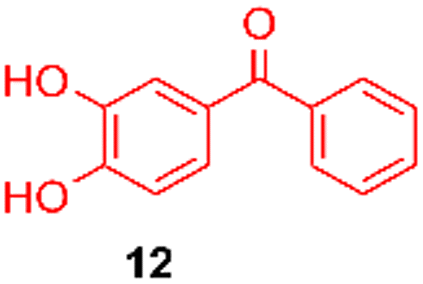
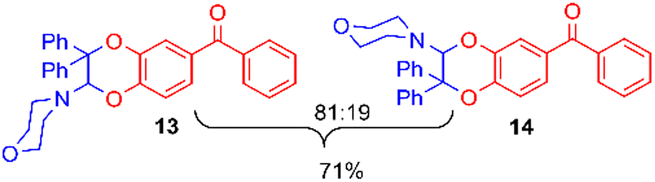
Using the conditions providing the best yield with enamine 2a (95/5 MeCN/DMSO, 1.2 equiv. of enamine), we turned to other catechol substrates to evaluate the regioselectivity of the [4 + 2] cycloaddition (Table 2). With 4-tert-butyl-catechol 5, another source of non-activated o-quinone, two regioisomers 6 and 7 could be synthesised in 95% overall yield. As expected for an IEDDA reaction, in the presence of compound 8, for the hydroxytyrosol homologue bearing an electron-withdrawing nitro group in position 6, compound 9 was the sole regioisomer formed (63% yield). With compound 10 substituted in position 5 by the –S-tBu hindered thiol group, the reaction afforded also only one regioisomer 11 (85% yield). In contrast, with 3,4-dihydroxybenzophenone 12, bearing an electron-withdrawing group in position 1, the concerted attack of the enamine is favoured on the oxo in position 3, leading to compounds 13/14 in 81/19 ratio and 71% overall yield.|
|
||
|---|---|---|
| Catechol | 2-Amino-2,3-dihydro-1,4-benzodioxanesc | |
| a Electrosynthesis: [catechol] = 1.25 mM, [LiClO4] = 0.05 M, divided cell, platinum grid anode, platinum plate cathode, Eox = +1.4 V vs. Ag/AgCl (except for compound 1, Eox = +1.0 V vs. Ag/AgCl), 95/5 MeCN/DMSO. b Diels–Alder reaction: addition of 1.2 equiv. of enamine 2a to a solution of o-quinone. c Isolated yields of regioisomers separable by flash chromatography (except for regioisomer 14). | ||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Variation of the dienophile part
|
|
||
|---|---|---|
| Enamine | 2-Amino-2,3-dihydro-1,4-benzodioxanesd | |
| a [Catechol] = 1.25 mM, [LiClO4] = 0.05 M, divided cell, platinum grid anode, platinum plate cathode, Eox = + 1.0 V vs. Ag/AgCl. Solvent of electrolysis and quantity of enamine added for the Diels–Alder reaction:b 95/5 MeCN/DMSO, 1.2 equiv. of enamine.c 95/5 MeCN/H2O, 5 equiv. of enamine.d Isolated yields of regioisomers separable by flash chromatography. | ||
|
|
|
|
|
|
|
|
|
|
|
|
|
— | |
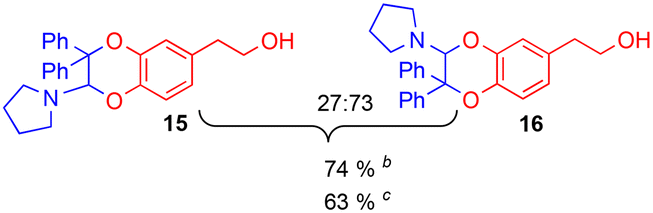
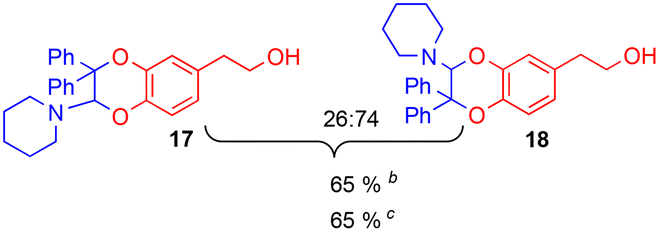
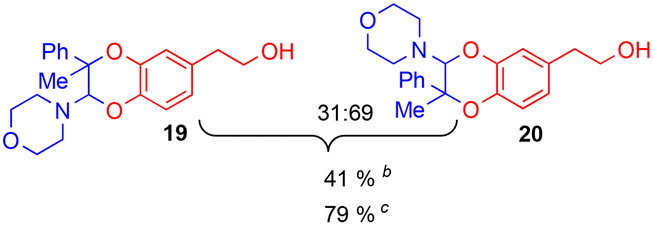
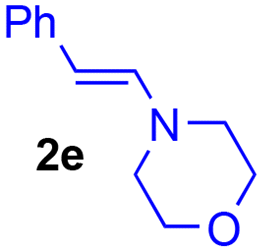
The improving impact of water on Diels–Alder reactions usually reported41 appeared more clearly with the enamine 2d (R1 = Ph, R2 = Me), since the yield almost doubled from 41% to 79%, with 5% of water instead of 5% of DMSO. However, no stable product could be isolated with secondary enamine 2e (R1 = Ph, R2 = H), whatever the solvent mixture used.
Since the enamine 2d had different substituents R1 = Ph and R2 = Me, regioisomers 19 and 20 were obtained as two pairs of diastereoisomers 19′,19′′ and 20′,20′′, in 18.5:12/44.5:25 ratio (Fig. 5). In the case of compounds 20′ and 20′′, the proton of the dihydrodioxin ring Hdioxin-20′ was located at 5.02 ppm while Hdioxin-20′′ was located at 4.88 ppm. Both of them presented an HMBC correlation with C-3. NOESY experiments revealed that the methyl group of 20′ was on the same face as CH2-O and CH2-N of the morpholine group. Comparatively, in compound 20′′ the morpholine faced the phenyl and methylene CH2-N correlating with aromatic protons. In the same way, HMBC and NOESY correlations showed that compound 19′ resulted from the trans coupling mode, with compound 19′′ corresponding to the cis one. The 1H NMR spectral evolution of a 6.25 × 10−3 M solution of crystallised trans enamine 2d42 in 95/5 CD3CN/D2O confirmed that no isomerisation of the dienophile occurred under these conditions.
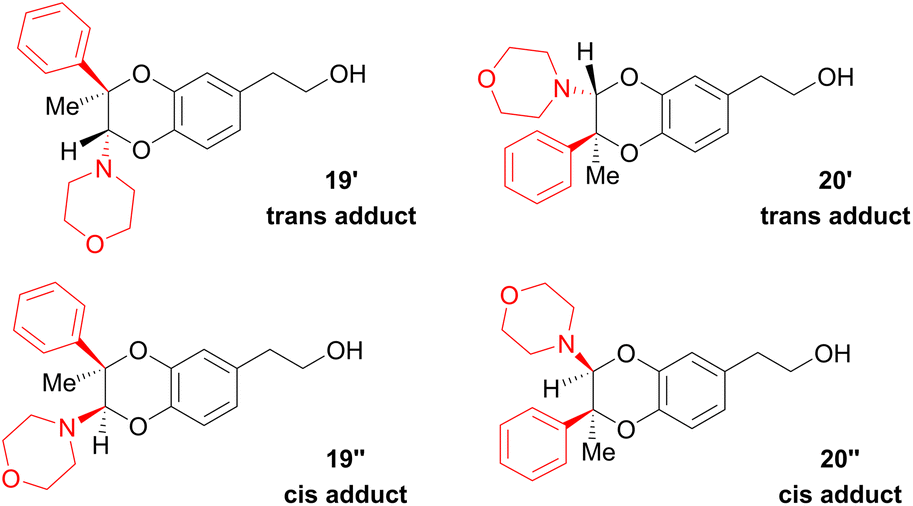 | ||
| Fig. 5 Relative configuration of the four regioisomeric products 19′,19′′/20′,20′′ obtained through the IEDDA reaction between the o-quinone and enamine 2d. | ||
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Enamine | Benzodioxanes | Yieldb | Benzoxepine | Yield | |||
| R1 | R2 | HN (R3,R4) | ||||||
| a Conditions of electrosynthesis: [catechol] = 1.25 mM, divided cell, platinum grid anode, platinum plate cathode, Eox = + 1.0 V vs. Ag/AgCl, [LiClO4] = 0.05 M, 95/5 MeCN/H2O, 5 equiv. of extemporaneously prepared enamine. b Overall yield after flash chromatography, 21/22 separable by HPLC. | ||||||||
| 1 | 2f | Et | Et |
|
21/22 | 65% | — | — |
| 2 | 2g | Et | Et |
|
23/24 | 87% | — | — |
| 3 | 2h | Et | Et |
|
25/26 | 85% | — | — |
| 4 | 2i | Et | Et |
|
27/28 | 26% | 33 | 18% |
| 5 | 2j | Me | Me |
|
29/30 | 41% | 33 | 41% |
| 6 | 2k | Cyclohexyl |
|
31/32 | 27% | 34 | 49% | |
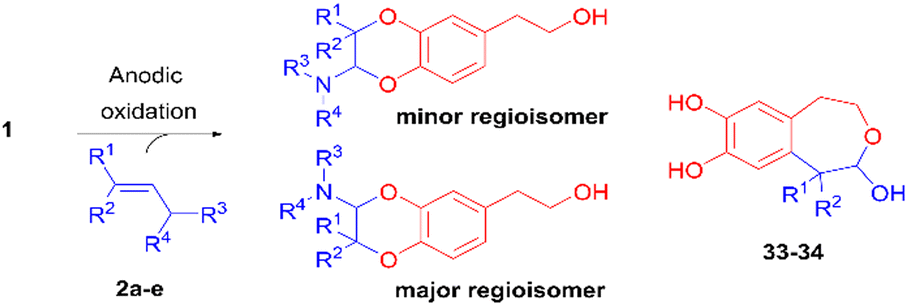
With enamines bearing methyl or cyclohexyl groups, a competitive reaction occurred which generated original 1,2,4,5-tetrahydrobenzo[d]oxepines 33 and 34 (entries 4–6). Such compounds, hydroxylated in the 2-position, were in solution in equilibrium with their aldehyde open forms 33′ and 34′, as characterised by the 1H NMR spectrum in acetone-d6. They probably resulted from a 1,6-Michael addition of the enamine on the electrogenerated o-quinone (Scheme 2), followed, in the presence of water, first by the elimination of the amine and then by the intramolecular cyclisation of the resulting transient aldehyde. In the specific case of hindered enamine 2k, this reaction became predominant, so that compound 34 was isolated as the major product in 49% yield.
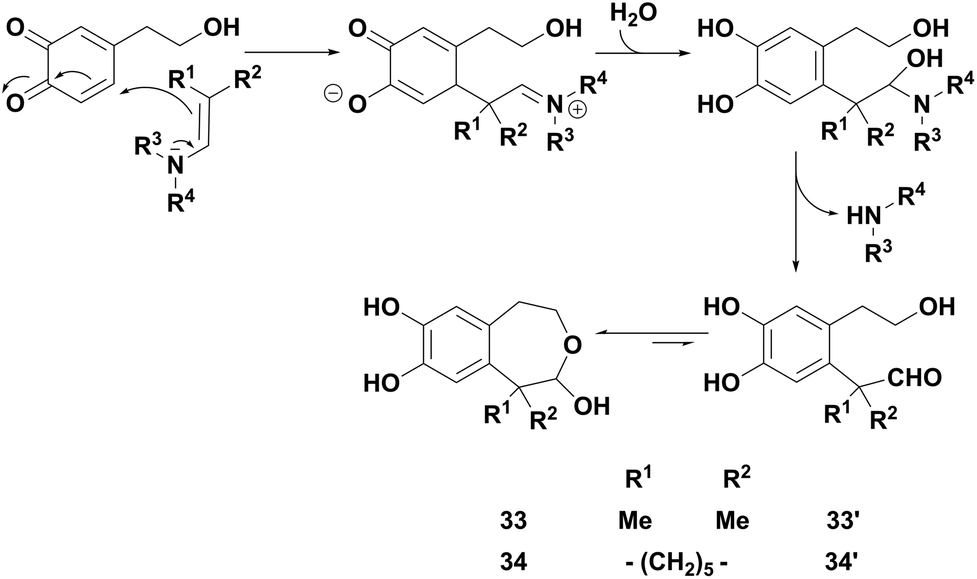 | ||
| Scheme 2 Proposed mechanism for the formation of 1,2,4,5-tetrahydrobenzo[d]oxepines 33 and 34. | ||
Conclusion
In summary, we have developed an electrochemical method to modify the catechol moiety of hydroxytyrosol 1 (or analogues) and obtained 2-amino-2,3-dihydro-1,4-benzodioxane derivatives, through the cycloaddition of enamines with the corresponding o-quinone(s). The use of the 95/5 MeCN/DMSO (or 95/5 MeCN/H2O) mixture as the solvent, combined with the anodic oxidation of hydroxytyrosol under the neutral form, turned out to be compatible both with the bi-electronic oxidation of the catechol into o-quinone and with the enamine cycloaddition. As expected, the regiospecificity of the reaction is determined by the presence of electron-withdrawing (or electron-donating) substituents on the o-quinone, favouring the cycloaddition of the enamine on one of the other oxo groups of the diene. With non-activated o-quinone, two regioisomers were obtained, whose substituents in the 3-position of the 1,4-benzodioxin ring could be aromatic or aliphatic. Our objective in the future will be to apply this method to other catechols abundant in olive oil, such as oleuropein.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank CNRS and Université Paris Cité for financial support. The authors would like to thank Cyril Colas from the “Fédération de Recherche” ICOA/CBM (FR2708) for HRMS analysis and Regis Guillot from ICCMO – Université Paris Saclay for collecting crystallographic data.References
- (a) M. Guasch-Ferré and W. C. Willett, J. Intern. Med., 2021, 290, 549 CrossRef PubMed; (b) R. J. Widmer, A. J. Flammer, L. O. Lerman and A. Lerman, Am. J. Med., 2015, 128, 229 CrossRef PubMed.
- (a) H. Lemoine, D. Marković and B. Deguin, J. Org. Chem., 2014, 79, 4358 CrossRef CAS PubMed; (b) K. Vougogiannopoulou, C. Lemus, M. Halabalaki, C. Pergola, O. Werz, A. B. Smith III, S. Michel, L. Skaltsounis and B. Deguin, J. Nat. Prod., 2014, 77, 441 CrossRef CAS PubMed.
- S. Charoenprasert and A. Mitchell, J. Agric. Food Chem., 2012, 60, 7081 CrossRef CAS PubMed.
- V. Francisco, C. Ruiz-Fernández, V. Lahera, F. Lago, J. Pino, L. Skaltsounis, M. A. González-Gay, A. Mobasheri, R. Gómez, M. Scotece and O. Gualillo, J. Agric. Food Chem., 2019, 67, 3845 CrossRef CAS PubMed.
- A. Angelis, D. Michailidis, L. Antoniadi, P. Stathopoulos, V. Tsantila, J.-M. Nuzillard, J.-H. Renault and L. A. Skaltsounis, Sep. Purif. Technol., 2021, 255, 117692 CrossRef CAS.
- T. Hu, X.-W. He, J.-G. Jiang and X.-L. Xu, J. Agric. Food Chem., 2014, 62, 1449 CrossRef CAS PubMed.
- M. Oliverio, M. Nardi, M. L. Di Gioia, P. Costanzo, S. Bonacci, S. Mancuso and A. Procopio, Nat. Prod. Rep., 2021, 38, 444 RSC.
- A. Procopio, S. Alcaro, M. Nardi, M. Oliverio, F. Ortuso, P. Sacchetta, D. Pieragostino and G. Sindona, J. Agric. Food Chem., 2009, 57, 11161 CrossRef CAS PubMed.
- (a) M. Trujillo, R. Mateos, L. Collantes de Teran, J. L. Espartero, R. Cert, M. Jover, F. Alcudia, J. Bautista, A. Cert and J. Parrado, J. Agric. Food Chem., 2006, 54, 3779 CrossRef CAS PubMed; (b) A. Procopio, C. Celia, M. Nardi, M. Oliverio, D. Paolino and G. Sindona, J. Nat. Prod., 2011, 74, 2377 CrossRef CAS PubMed.
- A. Gambacorta, D. Tofani, R. Bernini and A. Migliorini, J. Agric. Food Chem., 2007, 55, 3386 CrossRef CAS PubMed.
- P. Begines, D. Biedermann, K. Valentová, L. Petrásková, H. Pelantová, I. Maya, J. G. Fernández-Bolaños and V. Křen, J. Agric. Food Chem., 2019, 67, 7281 CrossRef CAS PubMed.
- M. Trujillo, E. Gallardo, A. Madrona, L. Bravo, B. Sarriá, J. A. González-Correa, R. Mateos and J. L. Espartero, J. Agric. Food Chem., 2014, 62, 10297 CrossRef CAS PubMed.
- R. Bernini, S. Cacchi, G. Fabrizi and E. Filisti, Org. Lett., 2008, 10, 3457 CrossRef CAS PubMed.
- M. Guiso, C. Marra and C. Cavarischia, Tetrahedron Lett., 2001, 42, 6531 CrossRef CAS.
- For examples, see: (a) S. F. Campbell, M. J. Davey, J. D. Hardstone, B. N. Lewis and M. J. Palmer, J. Med. Chem., 1987, 30, 49 CrossRef CAS PubMed; (b) Y. Liu and W. Bao, Org. Biomol. Chem., 2010, 10, 2700 RSC; (c) J. Habermann, S. V. Ley, J. J. Scicinski, J. S. Scott, R. Smits and A. W. Thomas, J. Chem. Soc., Perkin Trans. 1, 1999, 2425 RSC; (d) M. Massacret, P. Lhoste, R. Lakhmiri, T. Parella and D. Sinou, Eur. J. Org. Chem., 1999, 2665 CrossRef CAS; (e) N. R. Guz and F. R. Stermitz, J. Nat. Prod., 2000, 63, 1140 CrossRef CAS PubMed; (f) T. Song, B. Zhou, G.-W. Peng, Q.-B. Zhang, L.-Z. Wu, Q. Liu and Y. Wang, Chem. – Eur. J., 2014, 20, 678 CrossRef CAS PubMed; (g) K. C. Nicolaou, J. Wang and Y. Tang, Angew. Chem., Int. Ed., 2008, 47, 1432 CrossRef CAS PubMed.
- Y.-N. Shi, Z.-C. Tu, X.-L. Wang, Y.-M. Yan, P. Fang, Z.-L. Zuo, B. Hou, T.-H. Yang and Y.-X. Cheng, Bioorg. Med. Chem. Lett., 2014, 24, 5164 CrossRef CAS PubMed.
- Y.-M. Yan, L.-J. Li, X.-C. Qin, Q. Lu, Z.-C. Tu and Y.-X. Cheng, Bioorg. Med. Chem. Lett., 2015, 25, 2469 CrossRef CAS PubMed.
- L. Yang, G. Y. Li, H. Y. Wang, K. Zhang, Y. Zhu, W. Zhao, H. Wang and J. H. Wang, Phytochem. Lett., 2016, 16, 97 CrossRef CAS.
- X.-Q. Pang, X.-M. Wu, Q. Wang, D. Meng, Y.-M. Huang, J.-L. Xu, Y. Li, H. Liu, H. Xiao and Z.-T. Ding, Nat. Prod. Commun., 2022, 17, 1 CrossRef.
- P. Thapa, Y. Gu, Y.-S. Kil, S. C. Baek, K. H. Kim, A.-R. Han, E. K. Seo, H. Choi, J.-H. Chang and J.-W. Nam, Bioorg. Chem., 2020, 102, 104095 CrossRef CAS PubMed.
- A. N. Pearce, E. W. Chia, M. V. Berridge, E. W. Maas, M. J. Page, J. L. Harper, V. L. Webb and B. R. Copp, Tetrahedron, 2008, 64, 5748 CrossRef CAS.
- Y. S. Cheah, S. Santhanakrishnan, M. B. Sullivan, K. G. Neoh and C. L. L. Chai, Tetrahedron, 2016, 72, 6543 CrossRef CAS.
- W. Ried and E. Torok, Justus Liebigs Ann. Chem., 1965, 187 CrossRef CAS.
- J. Zhang, C. Taylo, E. Bowman, L. Savage-Low, M. W. Lodewyk, L. Hanne and G. Wu, Tetrahedron Lett., 2013, 54, 6298 CrossRef CAS.
- C.-M. Martinez, A. Neudörffer and M. Largeron, Org. Biomol. Chem., 2012, 10, 3739 RSC.
- A. Felim, A. Urios, A. Neudörffer, G. Herrera, M. Blanco and M. Largeron, Chem. Res. Toxicol., 2007, 20, 685 Search PubMed.
- K. Cottet, A. Neudörffer, M. Kritsanida, S. Michel, M.-C. Lallemand and M. Largeron, J. Nat. Prod., 2015, 78, 2136 CrossRef CAS PubMed.
- D. Xu, A. Chiaroni and M. Largeron, Org. Lett., 2005, 7, 5273 CrossRef CAS PubMed.
- M. Largeron, A. Neudorffer, M. Vuilhorgne, E. Blattes and M.-B. Fleury, Angew. Chem., Int. Ed., 2002, 41, 824 CrossRef CAS PubMed.
- (a) S. Yamamura and S. Nishiyama, Synlett, 2002, 533 CrossRef CAS; (b) T. Yamamoto, T. Saitoh, Y. Einaga and S. Nishiyama, Chem. Rec., 2021, 21, 1 CrossRef; (c) S. R. Waldvogel and B. Janza, Angew. Chem., Int. Ed., 2014, 53, 7122 CrossRef CAS PubMed; (d) B. Elsler, D. Schollmeyer and S. R. Waldvogel, Faraday Discuss., 2014, 172, 413 CAS.
- D. Nematollahi and E. Tammari, J. Org. Chem., 2005, 70, 7769 CrossRef CAS PubMed.
- C.-C. Zeng, D.-W. Ping, S.-C. Zhang, R.-G. Zhong and J. Y. Becker, J. Electroanal. Chem., 2008, 622, 90 CrossRef CAS.
- D. Nematollahi, S. S. H. Davarani and P. Mirahmad, ACS Sustainable Chem. Eng., 2014, 2, 579 CrossRef CAS.
- D. Nematollahi, D. Habibi, M. Rahmati and M. Rafiee, J. Org. Chem., 2004, 69, 2637 CrossRef CAS PubMed.
- K. Tanaka, H. Yoshizawa and M. Atobe, Synlett, 2019, 30, 1194 CrossRef CAS.
- D. Vogna, A. Pezzella, L. Panzella, A. Napolitano and M. d'Ischia, Tetrahedron Lett., 2003, 44, 8289 CrossRef CAS.
- M. De Lucia, L. Panzella, A. Pezzella, A. Napolitano and M. d'Ischia, Tetrahedron, 2006, 62, 1273 CrossRef CAS.
- (a) P. Y. Sollenberger and R. B. Martin, J. Am. Chem. Soc., 1970, 92, 4261 CrossRef CAS; (b) W. Maas, M. J. Janssen, E. J. Stamhuis and H. Wynberg, J. Org. Chem., 1967, 32, 1111 CrossRef CAS.
- C. Cativiela, J. I. Garcia, J. A. Mayoral and L. Salvatella, Chem. Soc. Rev., 1996, 209 RSC.
- Similar results were obtained when replacing in entry 1 LiClO4 with NaCl or in entry 2 LiClO4 with TEAHFP. The platinum anode could be replaced with carbon graphite, and the platinum cathode with carbon felt (Table S1†).
- A. Kumar, Chem. Rev., 2001, 101, 1 CrossRef CAS PubMed.
- L. Duhamel, P. Duhamel, S. Combrisson and P. Siret, Tetrahedron Lett., 1972, 34, 3603 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, crystallographic data in CIF and spectra of novel compounds. CCDC 2294269. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ob01858j |
| This journal is © The Royal Society of Chemistry 2024 |