
Synthesis of 2-amidoindenone derivatives through an ynamide carbosilylation/Houben–Hoesch cyclization 2-step sequence†
Pierre
Hansjacob‡
,
Célia
Schwoerer‡
,
Frédéric R.
Leroux
 and
Morgan
Donnard
*
and
Morgan
Donnard
*
University of Strasbourg, University of Haute-Alsace, CNRS, UMR 7042-LIMA, ECPM, 67000 Strasbourg, France. E-mail: donnard@unistra.fr
First published on 29th November 2023
Abstract
Herein we report the efficient and selective two-step synthesis of various 3-silyl-2-amidoindenones from easily accessible ynamides. This sequence involves a regio- and stereo-selective silylcyanation followed by a Houben–Hoesch type cyclization. Thanks to post-transformations, various 3-substituted 2-amidoindenones could be obtained.
1-Indenone derivatives can be frequently found in natural molecules and represent an important scaffold in pharmaceuticals and agrochemicals.1 Therefore, general synthetic routes towards such molecular structures have been intensively investigated over the last decades.2 However, when we consider the synthesis of indenones bearing a heteroatom at position 2, the number of available methods drops dramatically and the reported strategies are often very limited in terms of functionalization at position 3. Quite surprisingly, given the prevalence of nitrogen-based functionalities in bioactive molecules, the number of available methods to access 2-aminoindenones is remarkably low.14 Among the reported syntheses we can cite the reaction of o-alkynylbenzoyl cyanides with carboxylic acids developed by the group of Murakami3 (Fig. 1A) and the gold-catalyzed oxidation of 1-(2-alkynylphenyl)ketones investigated by Liu and coworkers4 (Fig. 1B). Recently, two direct annulation strategies using ynamides as partners have been reported. The first one, described by the Sahoo group, involves a key step of cobalt-catalyzed C–H activation at the ortho position of arylthioamides5 (Fig. 1C). The second one, reported by our group, relies on a palladium-catalyzed Larock-type annulation of o-iodo-benzaldehydes6 (Fig. 1D). Notably, both methods, although efficient and practical, appeared to be limited to aryl-ynamides and so ending in 3-aryl-2-amidoindenones. In summary, although the reported methods to access 2-aminoindenone derivatives present various synthetic possibilities, they also show limitation regarding the substituent diversity on the final indenones, especially at position 3, and, in some cases, in terms of regioselectivity. To address both issues, we have designed a new strategy to access selectively versatile 3-silyl-2-amidoindenones (Fig. 1E). This two-step approach involves the silylcyanation of various ynamides functionalized with a nucleophilic ring followed by a cyclization through an intramolecular Houben–Hoesch type reaction. The resulting 2-amidoindenones are substituted in position 3 by a TMS group that can be then converted into other functional groups.
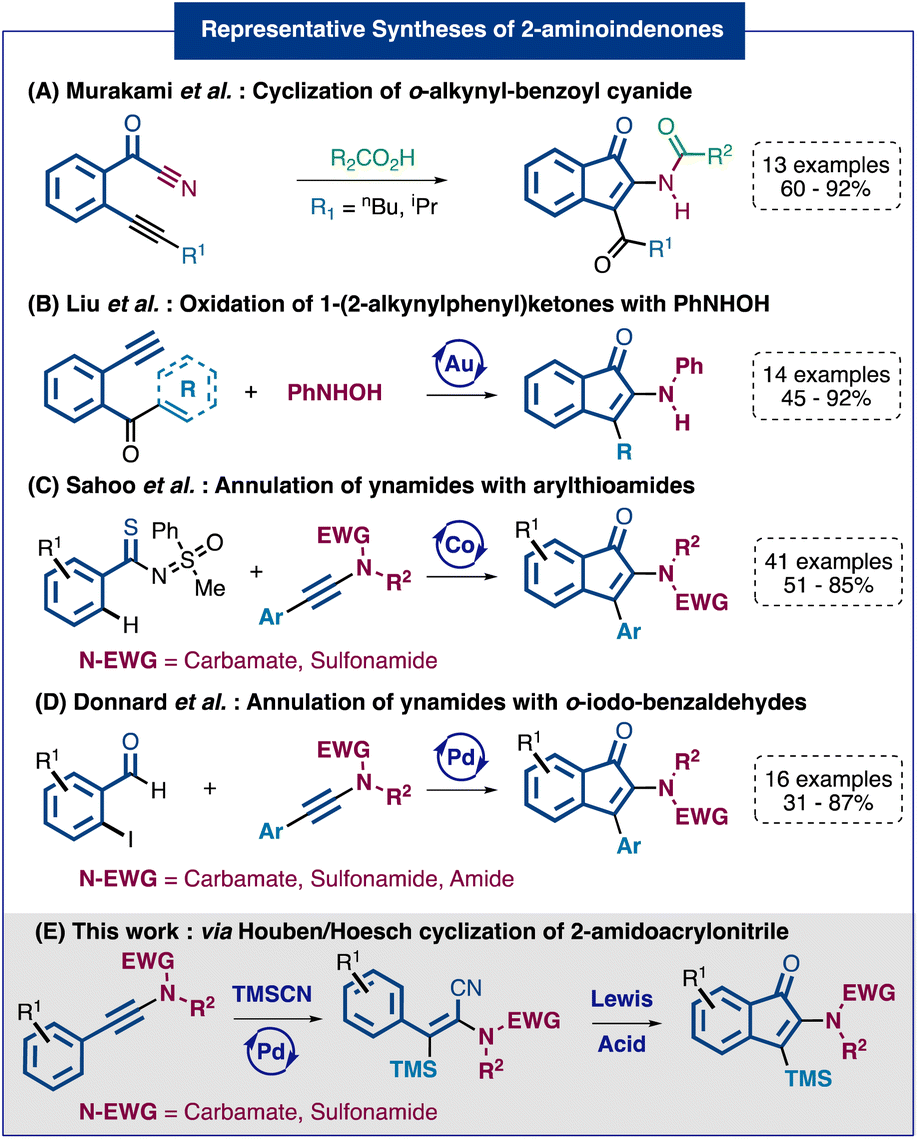 | ||
| Fig. 1 Selected synthetic routes to access 2-aminoindenones. | ||
As a starting point for our research, using an ynamide silylcyanation reaction previously developed in our group,7 we synthesized variously substituted 2-amido-acrylonitrile derivatives bearing nucleophilic aromatic rings at the 3-position (Fig. 2). Overall, a total of 12 products 2a–2l were obtained in good to very good yields and high stereoselectivities (Z/E ratios of around 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10). In the case of heteroaromatic-substituted ynamides (2e–2h) a slight erosion of the yields (41–66%) could be observed and, in the case of the furan substituted product 2e, a decrease in the stereoselectivity was observed. In the same way, ynamides bearing an N-methylsulfonamide substituent (2d, 2h, 2i, and 2l) were obtained in lower yields ranging from 49% to 66%. In most cases, the products were obtained in an inseparable Z/E mixture which was used as such for the following cyclization step. Considering that only the Z isomer can lead to the desired ring closure, we envisaged that isomerization could potentially take place during the cyclization step, leading to the complete conversion of the mixture to the desired indenones. With this set of substrates in hand, we began the optimization of the Houben–Hoesch type cyclization step, using compound 2a as a substrate, by studying the influence of several parameters such as the solvent, temperature, time of reaction and the nature of the Lewis acid (Table 1).
:10). In the case of heteroaromatic-substituted ynamides (2e–2h) a slight erosion of the yields (41–66%) could be observed and, in the case of the furan substituted product 2e, a decrease in the stereoselectivity was observed. In the same way, ynamides bearing an N-methylsulfonamide substituent (2d, 2h, 2i, and 2l) were obtained in lower yields ranging from 49% to 66%. In most cases, the products were obtained in an inseparable Z/E mixture which was used as such for the following cyclization step. Considering that only the Z isomer can lead to the desired ring closure, we envisaged that isomerization could potentially take place during the cyclization step, leading to the complete conversion of the mixture to the desired indenones. With this set of substrates in hand, we began the optimization of the Houben–Hoesch type cyclization step, using compound 2a as a substrate, by studying the influence of several parameters such as the solvent, temperature, time of reaction and the nature of the Lewis acid (Table 1).
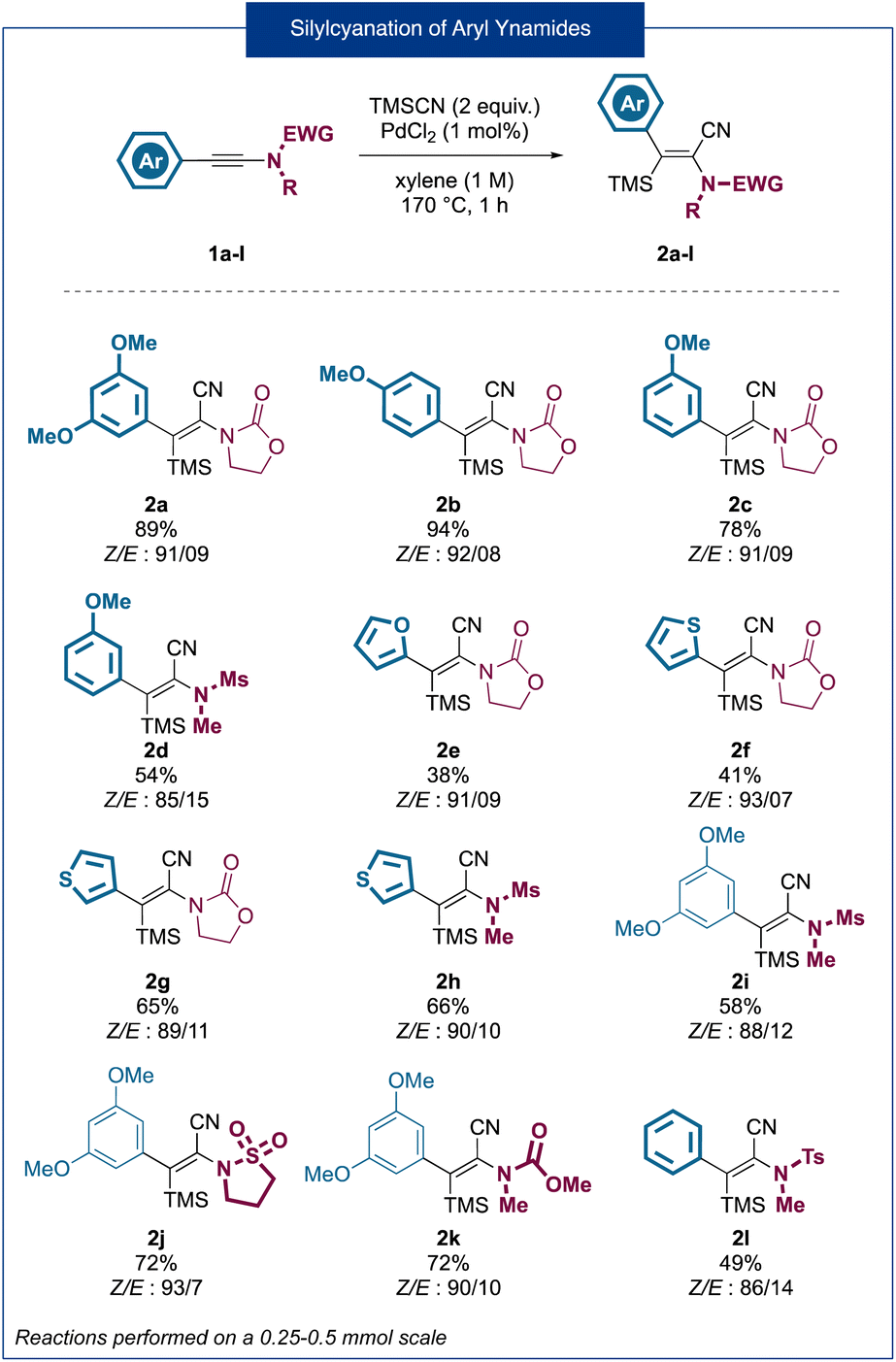 | ||
| Fig. 2 Silylcyanation of ynamides bearing an electron-rich aromatic substituent. | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Lewis acid (equiv.) | Solv. | t (h) | Conv. Z-2a (%) | 1H NMR ratioa3a/4a | 1H NMR yieldb (%) 4a |
| Reactions were performed on a 0.1 mmol scale.a 1H NMR ratio before SiO2 mediated hydrolysis.b 4a1H NMR yield with respect to the Z isomer of the Z/E mixture, after SiO2 mediated hydrolysis, with CH2BrCl as the internal standard.c Isolated yield in parentheses.d Reaction performed at 25 °C.e 2nd equiv. added at t = 5 h.f Reaction performed at 50 °C. |
||||||
| 1 | BF3·OEt2 (0.5) | Tol. | 15 | 66 | 74/26 | 58 |
| 2 | BF3·OEt2 (1.0) | Tol. | 15 | 81 | 73/27 | 75 (72)c |
| 3 | AlCl3 (1.0) | Tol. | 15 | 0 | — | 0 |
| 4 | BiCl3 (1.0) | Tol. | 15 | 66 | 92/8 | 38 |
| 5d | TiCl4 (1.0) | Tol. | 15 | 84 | 97/3 | 62 |
| 6 | FeCl3 (1.0) | Tol. | 15 | 66 | 62/38 | 52 |
| 7 | BF3·OEt2 (1.0) | THF | 15 | 66 | 97/3 | 63 |
| 8 | BF3·OEt2 (1.0) | ACN | 15 | 62 | 96/4 | 47 |
| 9 | BF3·OEt2 (1.0) | DCE | 15 | 67 | 94/6 | 50 |
| 10e | BF3·OEt2 (2.0) | Tol. | 15 | 85 | 100/0 | 84 |
| 11 | BF 3 ·OEt 2 (2.0) | Tol. | 24 | 100 | 100/0 | 91 (91) |
| 12f | BF3·OEt2 (2.0) | Tol. | 24 | 33 | 100/0 | 24 |
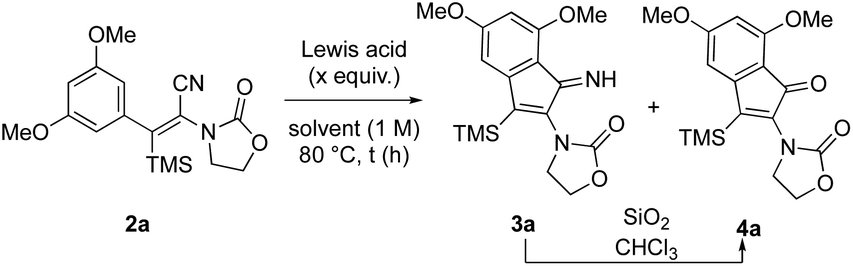
Inspired by the work of Campaigne and Mais,8 the reaction was first carried out with 50 mol% of BF3·OEt2 in toluene at 80 °C for 15 h. Gratifyingly, the expected product was obtained as a 74:26 mixture of indenimine 3a and indenone 4a. A subsequent hydrolysis of the mixture in a suspension of silica in chloroform led to the desired product 4a in a decent 58% yield (entry 1). Notably, unlike our expectations, no isomerization of the starting acrylonitrile was observed as the E isomer was completely recovered at the end of the reaction in the initial proportion. The use of 1 equivalent of the Lewis acid allowed an increase in the isolated yield to 72%, but the conversion remained incomplete (81%) (entry 2). Other Lewis acids (entries 3–6) gave lower yields with various 3a/4a ratios, except for aluminum trichloride that showed no conversion of the starting material (entry 3). A slight decrease in the yield was observed when carrying out the reaction in other solvents (entries 7–9). Finally, the addition of a second equivalent of BF3·OEt2 after 5 h of reaction was required to achieve full conversion and, after hydrolysis, indenone 4a was afforded in a 91% isolated yield after 24 h as 15 h led to an incomplete conversion (entries 10 and 11). Notably, the use of 2 equivalents of BF3·OEt2 directly from the beginning of the reaction gave significantly lower yields. An attempt to reduce the temperature of the reaction led to a significant decrease in the conversion (33% after 24 h).
With these optimized conditions defined, we then turned our attention to the scope of the cyclization reaction (Fig. 3A). Notably, similar to 3a, some of the indenimine intermediates could be isolated and characterized. When required, the latter were hydrolyzed quantitatively to the corresponding indanones. The higher stability of indenimine in some cases can potentially be explained by the higher electron density coming from richer aromatic rings and/or more electron-donating amido groups (sulfonamides). Predictably, no conversion of substrate 2b, bearing a less nucleophilic p-methoxyphenyl, was observed. While the mono-methoxylated m-methoxyphenyl acrylonitrile 2c, bearing an oxazolidinone as a nitrogen moiety, led to a modest 27% yield and a 70:30 mixture of regioisomers, the analogous substrate 2d bearing a sulfonamide led to a significant increase in both yield and selectivity. This trend could be observed in most examples involving sulfonamide substrates (4h–4j) and could be explained by the more electron-donating nature of sulfonamides compared to that of carbamates. A substrate bearing a furan as a nucleophilic ring did not lead to the targeted cyclopenta[b]furan-6-one 4e but only to the isomerized product E-2e. The electron depletion of the furan moiety, possibly caused by coordination to the Lewis acid, could explain this observation. Remarkably, substrates 2f–2h bearing a thiophene all led to the corresponding products 4f–4h in good to very good yields (62–86%), providing access to unprecedented compounds with scaffolds very rarely described in the literature5,9 (especially cyclopenta[b]thiophen-6-one derivatives such as 4g and 4h). These results are all the more interesting as they represent, as far as we know, the first examples of the Houben–Hoesch reaction involving thiophene rings. Moreover, the reaction was compatible with various substitutions on the nitrogen moiety of the starting material leading to the targeted indenones (4i–4k) in very good yields (77%–93%). Although substituted by a sulfonamide, an acrylonitrile lacking a nucleophilic aryl substituent (2l) did not undergo the cyclization reaction under our conditions. In an attempt to reach dibenzotropone derivatives using our strategy, we synthesized 2-amido-acrylonitrile 2m bearing an electron-rich biaryl substituent. Unfortunately, this substrate appeared completely inert under our cyclization conditions (Fig. 3B). Interestingly, the desilylated amido-acrylonitrile 2a′ did not react under our Houben–Hoesch conditions. A potential explanation for this difference in the reactivity with the analogous silylated substrate could be the stabilizing silicon effect of the arenium intermediate formed during the cyclization (Fig. 3C).10
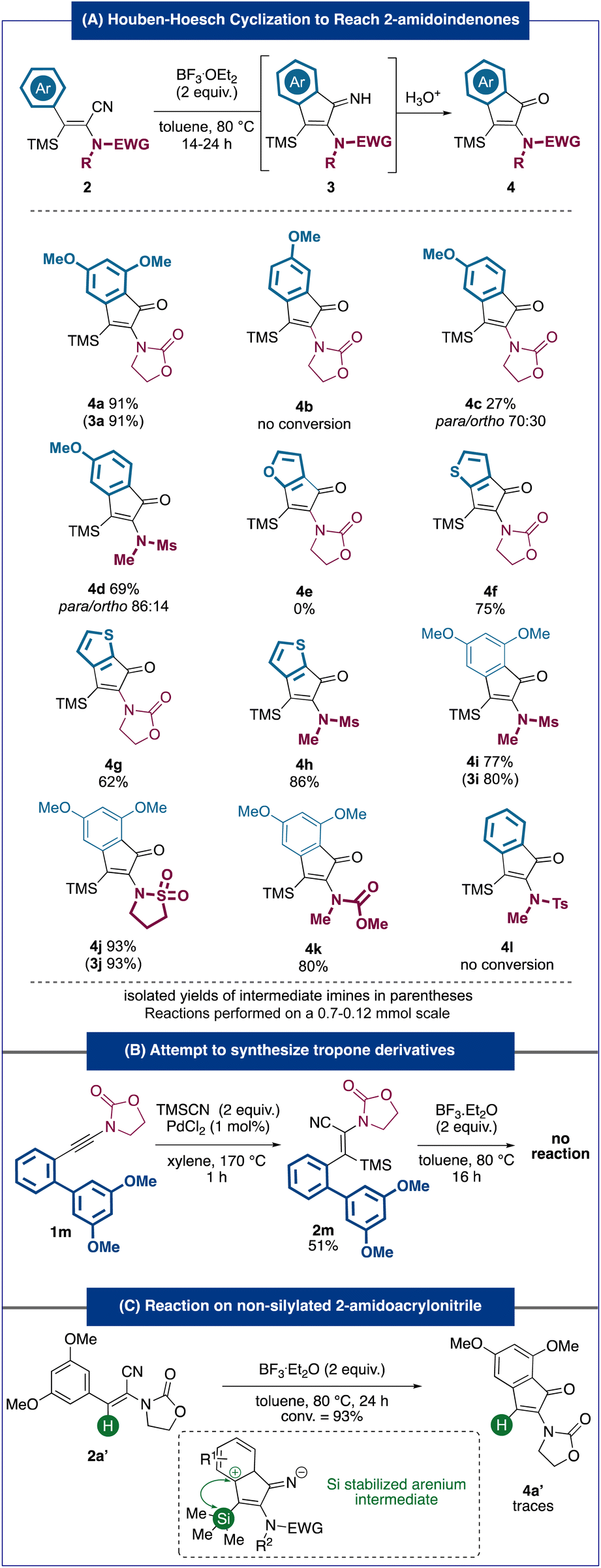 | ||
| Fig. 3 Scope and limitations of the methodology to access 2-amidoindenones. | ||
Finally, the resulting 2-amido-indenones were then valorised through various post-functionalization reactions (Fig. 4).
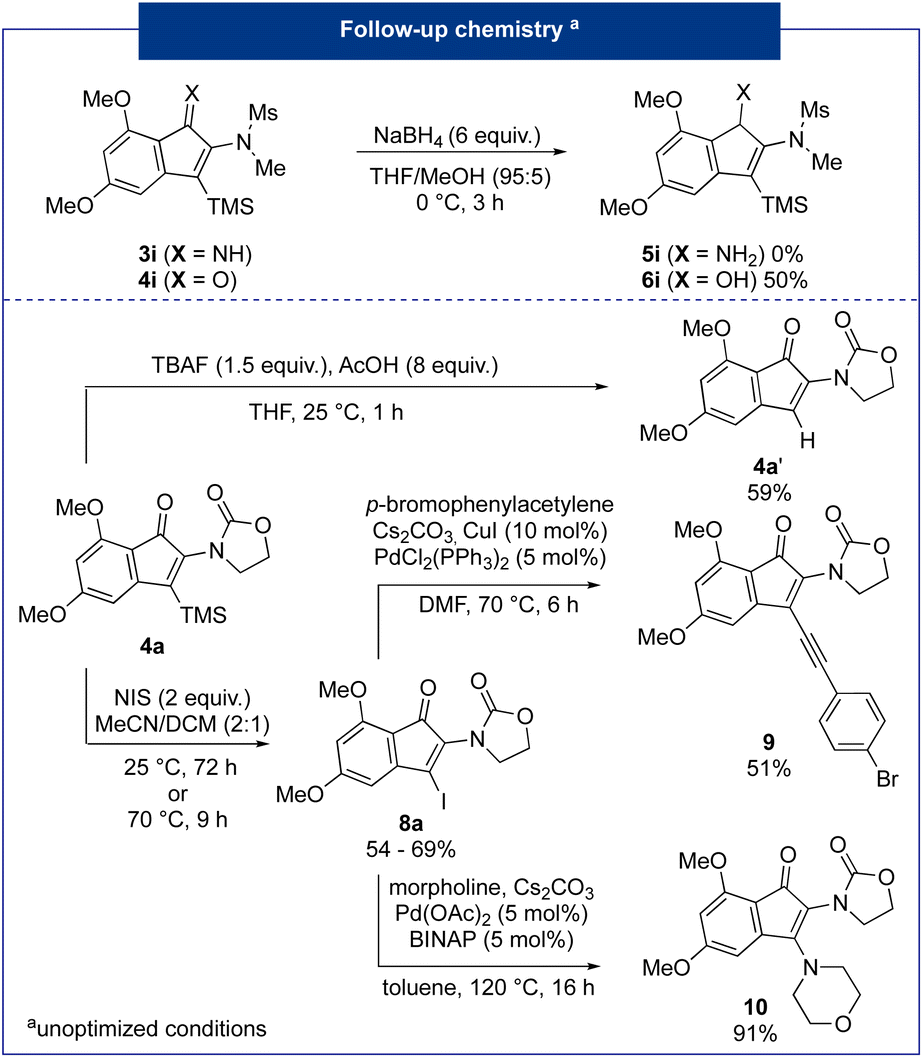 | ||
| Fig. 4 Follow-up chemistry on 3-silyl-2-aminoindenones 4a and 4i. | ||
First, carbonyl reduction of indenone was carried out using sodium borohydride as the reducing agent11 and the corresponding alcohol 6i was afforded in a good 50% yield. Conversely, an attempt to reduce the imine 3i led only to the desilylation of the starting substrate. Transformations of the silicon group were then investigated. First, a protodesilylation reaction using TBAF in THF with an excess of acetic acid led to the indenone 4a′ in 56% yield. Then, an iododesilylation reaction was envisaged to access a versatile 3-iodo-indenone 8a that could then be functionalized through various cross-coupling reactions.12 Using NIS as an electrophilic source of iodine, the desired product was obtained in a good 69% yield after 72 h at 25 °C. Notably, increasing the temperature to 70 °C allowed the formation of the desired product in only 9 h, but in a significantly lower 54% yield. The iodinated derivative 8a was then engaged in two typical palladium-catalyzed cross-coupling reactions, namely an alkynylation and an amination. Firstly, a Sonogashira reaction in the presence of p-bromophenylacetylene was carried out.13 Product 9 was obtained in a moderate 51% yield. This can be explained by a portion of the over-reaction leading to a second coupling on the bromo-aryl moiety linked to the alkynyl moiety. Secondly, a Buchwald–Hartwig coupling with morpholine was carried out and the desired 2,3-diaminoindenone 10 was yielded in an excellent 91% yield.14 In conclusion, we have been able to regioselectively access versatile 3-silyl-2-amido-indenones through a 2-step sequence involving a silylcyanation of ynamides followed by an intramolecular Houben–Hoesch reaction. In some cases, 2-amido-indenimine intermediates were isolated and indenones bearing a heteroatom such as thiophene were obtained in very good yields. The resulting products were finally valorised through a reduction of the carbonyl function. The vinylsilane function was also valorised through protodesilylation and iododesilylation reactions. Finally, an iodinated derivative was subjected to Sonogashira and Buchwald–Hartwig cross-coupling reactions, affording good to excellent yields.
Conflicts of interest
There are no conflicts to declare.References
- (a) R. Nigam, K. R. Babu, T. Ghosh, B. Kumari, D. Akula, S. N. Rath, P. Das, R. Anindya and F. A. Khan, Bioorg. Med. Chem., 2018, 26, 4100–4112 CrossRef CAS; (b) P.-C. Lv, M. S. A. Elsayed, K. Agama, C. Marchand, Y. Pommier and M. Cushman, J. Med. Chem., 2016, 59, 4890–4899 CrossRef CAS; (c) J. H. Ahn, M. S. Shin, S. H. Jung, S. K. Kang, K. R. Kim, S. D. Rhee, W. H. Jung, S. D. Yang, S. J. Kim, J. R. Woo, J. H. Lee, H. G. Cheon and S. S. Kim, J. Med. Chem., 2006, 49, 4781–4784 CrossRef CAS; (d) M. A. Ernst-Russell, C. L. L. Chai, J. H. Wardlaw and J. A. Elix, J. Nat. Prod., 2000, 63, 129–131 CrossRef CAS PubMed; (e) R. E. McDevitt, M. S. Malamas, E. S. Manas, R. J. Unwalla, Z. B. Xu, C. P. Miller and H. A. Harris, Bioorg. Med. Chem. Lett., 2005, 15, 3137–3142 CrossRef CAS PubMed; (f) H. Kim, I. Yang, S.-Y. Ryu, D. H. Won, A. G. Giri, W. Wang, H. Choi, J. Chin, D. Hahn, E. Kim, C. Han, J. Lee, S.-J. Nam, W.-K. Ho and H. Kang, J. Nat. Prod., 2015, 78, 363–367 CrossRef CAS; (g) T. Ito, T. Tanaka, M. Iinuma, K. Nakaya, Y. Takahashi, R. Sawa, J. Murata and D. Darnaedi, J. Nat. Prod., 2004, 67, 932–937 CrossRef CAS; (h) S. A. Patil, R. Patil and S. A. Patil, Eur. J. Med. Chem., 2017, 138, 182–198 CrossRef CAS PubMed.
- (a) L. E. Hutchinson and D. J. Wilger, Adv. Synth. Catal., 2022, 364, 3441–3465 CrossRef CAS; (b) X. Yan, S. Zou, P. Zhao and C. Xi, Chem. Commun., 2014, 50, 2775–2777 RSC; (c) P. Zhao, Y. Liu and C. Xi, Org. Lett., 2015, 17, 4388–4391 CrossRef CAS PubMed; (d) M. B. Floyd and G. R. Allen, J. Org. Chem., 1970, 35, 2647–2653 CrossRef CAS; (e) B. Suchand and G. Satyanarayana, J. Org. Chem., 2017, 82, 372–381 CrossRef CAS PubMed.
- H. Shimizu and M. Murakami, Synlett, 2008, 1817–1820 CAS.
- B. D. Mokar, D. B. Huple and R. Liu, Angew. Chem., Int. Ed., 2016, 55, 11892–11896 CrossRef CAS.
- S. Sau, A. Ghosh, M. Shankar, M. P. Gogoi and A. K. Sahoo, Org. Lett., 2022, 24, 9508–9513 CrossRef CAS PubMed.
- S. Golling, P. Hansjacob, N. Bami, F. R. Leroux and M. Don-nard, J. Org. Chem., 2022, 87, 16860–16866 CrossRef CAS.
- P. Hansjacob, F. R. Leroux, V. Gandon and M. Donnard, Angew. Chem., Int. Ed., 2022, 61, e202200204 CrossRef CAS PubMed.
- E. Campaigne and D. E. Mais, J. Heterocycl. Chem., 1975, 12, 267–271 CrossRef CAS.
- For selected examples see: (a) Ref. 5; ; (b) M. Rostami, A. Khosropour, V. Mirkhani, M. Moghadam, S. Tangestaninejad and I. Mohammadpoor-Baltork, Tetrahedron Lett., 2011, 52, 7149–7152 CrossRef CAS; (c) H. M. F. Madkour, Heterocycl. Commun., 2002, 8, 501–508 CAS.
- D. D. Roberts and M. G. McLaughlin, Adv. Synth. Catal., 2022, 364, 2307–2332 CrossRef CAS.
- G. Liu, X. Wang, X.-H. Xu, X. Lu, E. Tokunaga, S. Tsuzuki and N. Shibata, Org. Lett., 2013, 15, 1044–1047 CrossRef CAS.
- D. P. Stamos, A. G. Taylor and Y. Kishi, Tetrahedron Lett., 1996, 37, 8647–8650 CrossRef CAS.
- A. Reding, P. G. Jones and D. B. Werz, Org. Lett., 2018, 20, 7266–7269 CrossRef CAS.
- C. Meyers, B. U. W. Maes, K. T. J. Loones, G. Bal, G. L. F. Lemière and R. A. Dommisse, J. Org. Chem., 2004, 69, 6010–6017 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ob01787g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |