
α/β-Stereo- and diastereoselective glycosylation with n-pentenyl glycoside donors, promoted by N-iodosuccinimide and catalyzed by chiral Brønsted acid†
Maniyamma
Aswathy
ab,
Mohammed Sadik
N. K.
c,
Purushothaman C.
Harikrishnan
a,
Sasikumar
Parameswaran
a,
Kokkuvayil Vasu
Radhakrishnan
 *ab and
Ravi S.
Lankalapalli
*ab
*ab and
Ravi S.
Lankalapalli
*ab
aChemical Sciences and Technology Division, CSIR-National Institute for Interdisciplinary Science and Technology (CSIR-NIIST), Thiruvananthapuram-695019, India. E-mail: radhu2005@gmail.com; ravishankar@niist.res.in
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad-201002, India
cDepartment of Applied Chemistry, CUSAT, Kochi-22, India
First published on 21st November 2023
Abstract
A method involving stereoselective glycosylation catalyzed by (+)-isomenthol ester of pentacarbomethoxycyclopentadiene as a chiral Brønsted acid, with n-pentenyl glycosides in the presence of N-iodosuccinimide as the promoter is described; this method offered a chiral recognition of racemic substrates.
Hydrolysis of the glycosidic bond by glycosidases and glycosylation by glycosyltransferases are governed by acid-base catalysis, with the amino acids Asp, Glu, and Tyr as the catalytic residues in the active site.1,2 Brønsted acids (BAs) can act as enzymes when used with appropriate glycosyl donors for synthetic glycosylation. While the activation of glycosyl donors by Lewis acids is dominant in glycosylation, BA-based promoters are generally employed for 1-hydroxy and trichloroacetimidate glycosyl donors.3–10 Sulfuric acid immobilized on silica as BA in combination with N-iodosuccinimide (NIS) was utilized to activate a thioglycoside for the synthesis of tetrasaccharide fragments.11–13 Furthermore, attempts at achieving α/β-stereoselectivity in glycosylation were made with chiral BAs (CBAs)activation. This involved the use of a tetrazole-amino acid ionic liquid,9 chiral binaphthol (BINOL) phosphoric acids,14–16 and peptides bearing carboxylic acid groups in combination with an MgBr2 Lewis acid.17
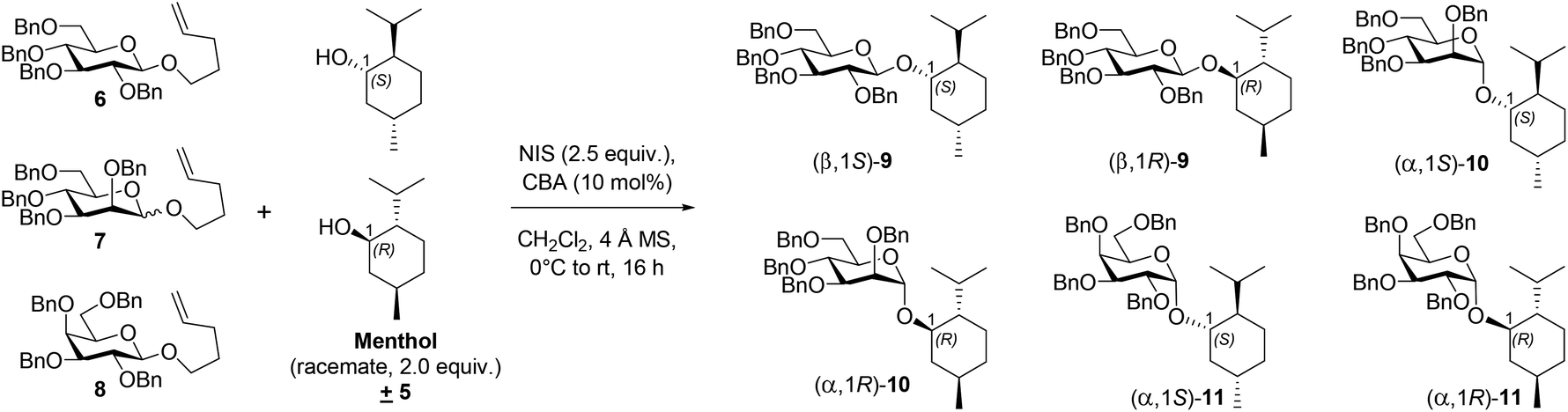
Recently, we reported the catalytic utility of another BA, namely pentacarbomethoxycyclopentadiene (PCCP), for stereoselective glycosylation with n-pentenyl orthoesters (NPOEs) of D-glucose and D-galactose in the presence of NIS.18 Oxidative hydrolysis of n-pentenyl glucoside with N-bromosuccinimide to form a hemiacetal was an outcome of a serendipitous observation made by Fraser-Reid,19 which led to the inception of n-pentenyl glycoside (NPG) and NPOE glycosyl donors. BA and PCCP are defined in the initial paragraphs was a pioneering contribution by Lambert et al., who prepared the chiral version of this BA by appending to it a naturally occurring (−)-menthol as an ester and showed the utility of this BA in Mukaiyama–Mannich and oxocarbenium aldol reactions in an enantioselective manner.20 Toshima et al. utilized chiral BINOL phosphoric acid as a BA, resulting in an enantioselective catalysis for glycosylation with trichloroacetimidate donors and racemic acceptors, thus affording excellent α/β-stereo- and diastereoselectivity.14 Inspired by these two reports, we herein investigated stereoselective glycosylation with an NPG donor in the presence of NIS, using the efficient chiral catalyst from (+)-isomenthol ester of PCCP; this process offered chiral recognition of racemic aglycones, namely menthol, benzylic alcohols, and Boc-protected serine.
CBA 1 (Fig. 1) was synthesized from PCCP and (+)-isomenthol as per the reported procedure.20 Racemic menthol 5 was chosen as the acceptor for investigating the chiral recognition in a glycosylation reaction catalyzed by CBA 1. The initial glycosylation attempt was performed with benzyl-protected glucosyl-NPGs 6 and 5 in the presence of NIS and CBA 1 (10 mol%), under the optimized reaction conditions of our previous report with BA-PCCP,18 which afforded β-glycoside 9 at 92% yield (entry 1, Table 1). The relative amounts of (β, 1S)-9 and (β, 1R)-9, determined by computing the area percentages of both peaks in the chromatogram of high-performance liquid chromatography and by comparing the retention times of the respective glycosides of each menthol enantiomer (see ESI†), were found to be 86.83% and 13.17%, respectively (entry 1, Table 1). Thus, in addition to β-selectivity with CBA 1, chiral recognition of the racemic menthol favoring glycosylation of (+)-menthol afforded (β,1S)-9 with a diastereomeric excess of 74%. Glycosylation with mannosyl-NPG 7 (α, β mixture) in the presence of CBA 1 afforded glycoside 10 with the expected α-stereoselectivity (entry 2, Table 1). Note that glycosylation with the less studied galactosyl-NPG 8 produced glycoside 11 with α-stereoselectivity (entry 3, Table 1). Once again, chiral recognition favored glycosylation of (+)-menthol to afford diastereoselective products (α,1S)-10 and (α,1S)-11, with each of them showing a diastereomeric excess of 69% (entries 2 and 3, Table 1). The results of glycosylation with CBA 1 motivated us to compare its role in α/β-stereo- and diastereoselectivity with other chiral and nonchiral BAs under optimized glycosylation conditions.18 Galactosyl-NPG 8 has hardly been investigated for glycosylation reactions, much less so compared to NPGs 6 and 7. Hence, glycosylation with NPG 8 and BINOL-derived chiral phosphoric acid catalysts (R)-2 and (S)-2 (Fig. 1) were attempted and afforded α-glycoside 11 as did with CBA 1; however, the diastereoselectivity was poor with a slightly favored glycosylation of (−)-menthol (entries 4 and 5, Table 1). A glycosylation attempt with NPG 8 in the presence of Lewis acid activator Sc(OTf)3, a conventional activator for NPG donors,21 and BAs such as achiral PCCP and triflic acid afforded neither stereoselectivity nor diastereoselectivity (entries 6–8, Table 1). In all three attempts, we observed the formation of the R/S mixture of both α and β glycosides of the corresponding NPGs. To assess the importance of the selection of (+)-isomenthol in CBA synthesis, CBA 3 and CBA 4 (Fig. 1) were synthesized from (+)-menthol and (−)-menthol, respectively. NPG 7 was chosen to study the effect of steric hindrance of the β-face of the oxocarbenium ion on glycosylation in the presence of CBA 3 and CBA 4 catalysts; α-glycosides 10 were produced as an outcome with no diastereoselectivity.
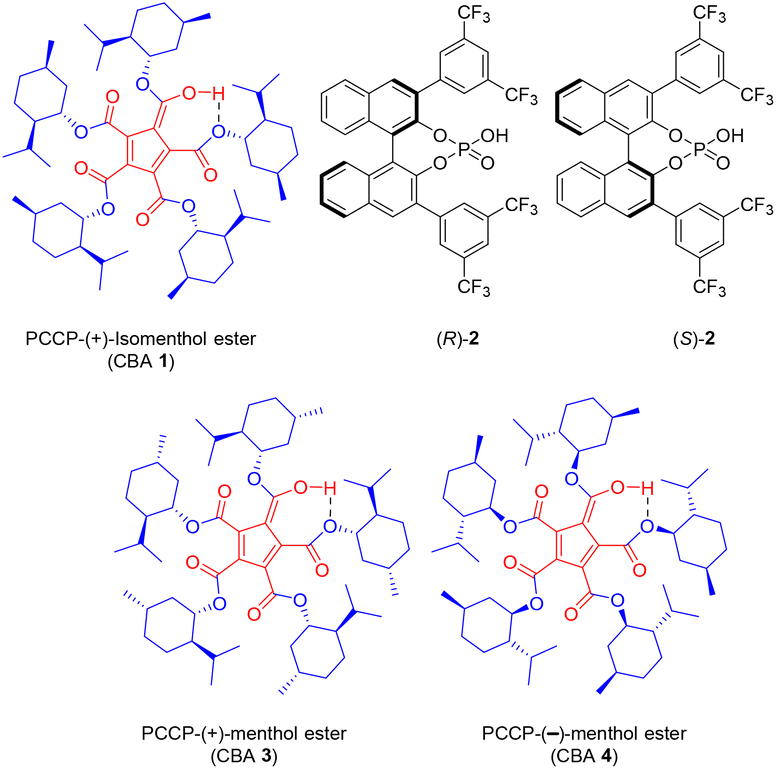 | ||
| Fig. 1 Structures of chiral Brønsted acids (CBAs). | ||
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Donor | Activator | Yieldb (%) | Relative amounts of diastereomeric glycosidesc (%) | |||||
| (β,1S)-9 | (β,1R)-9 | (α,1S)-10 | (α,1R)-10 | (α,1S)-11 | (α,1R)-11 | ||||
| a Reaction conditions: NIS (2.5 equiv.) and CBA (10 mol%) activators in addition to CBA (15 mol%) were added to each of mixtures of 6, 7, or 8 (1.0 equiv.) with ± 5 (2.0 equiv.) in CH2Cl2 at 0 °C, under 4 Å MS, and the resulting mixtures were slowly warmed to room temperature. b Isolated yields from column chromatography analysis. c Relative amounts of diastereomeric glycosides obtained from the area% of an HPLC chromatogram. | |||||||||
| 1 | 6 | 1 | 92 | 86.8% | 13.2% | — | — | — | — |
| 2 | 7 | 1 | 85 | — | — | 84.7% | 15.3% | — | — |
| 3 | 8 | 1 | 94 | — | — | — | — | 84.5% | 15.5% |
| 4 | 8 | (R)-2 | 90 | — | — | — | — | 40.1% | 59.9% |
| 5 | 8 | (S)-2 | 83 | — | — | — | — | 45.7% | 54.3% |
| 6 | 8 | Sc(OTf)3 | 86 | Neither stereoselective nor diastereoselective | |||||
| 7 | 8 | PCCP | 73 | Neither stereoselective nor diastereoselective | |||||
| 8 | 8 | CF3SO3H | 35 | Neither stereoselective nor diastereoselective | |||||
| 9 | 7 | 3 | 90 | — | — | 54.2% | 45.8% | — | — |
| 10 | 7 | 4 | 85 | — | — | 45.9% | 54.1% | — | — |
The benzyl protection of NPGs 6–8 was intended to avoid any participation effect during glycosylation, similar to the approach by Toshima et al.14 Glycosylation, either in the absence of CBA or NIS, did not occur, suggesting a role for the CBA catalyst in the activation of NIS. Next, we treated one of the obtained products, (β,1S)-9, with CBA 1 and NIS under the optimized glycosylation conditions without the acceptor ±5. The product (β,1S)-9 was quantitatively recovered without any isomerization even after 24 h of reaction, indicating that the stereo- and diastereoselectvities of the reaction were under kinetic control. The mechanistic rationale behind the stereoselectivity, shown in Scheme 1, involved the positioning of the oxocarbenium ion, generated from NPG activation,22 and menthol on the chiral cyclopentadienyl anion platform, in line with the model proposed by Lambert et al. for enantioselective addition onto oxocarbenium with the necessary organizational element of H-bonding.20 From the proposed mechanistic rationale, the H-bonding interactions of the cyclopentadienyl anion with certain C atoms—specifically with the C1 carbon, and with the C5 carbons adjacent to the oxygen of the oxocarbenium ion—were posited to be conventional CH/π H bonding interactions (Scheme 1a).23 During glycosylation, menthol was suggested to be positioned in the hydrophobic pocket formed by the (+)-isomenthyl groups and held by H-bonding with the carbonyl of the catalyst. The organization of the donor and acceptor on the cyclopentadienyl ring, prior to glycosylation, was concluded to be the key to the observed stereo- and diastereoselectivity. The α/β-stereoselectivity of the glycosylation was modeled to be governed by the steric hindrance by C2-OBn and C4-OBn groups for the incoming nucleophilic attack on the oxocarbenium ion. Accordingly, in the case of glucosyl-NPG 6, the less-hindered β-face was found to be preferred for nucleophilic attack by the menthol oxygen atom; in the case of galactosyl-NPG 8, the α-face was preferred. Mannosyl-NPG 7 exhibited conventional α-selectivity. The observation of β-selectivity with NPG 6 and α-selectivity with NPG 7 and 8 reinforced the proposed model in Scheme 1 as a highly-conserved catalytic platform with organizational elements to promote stereoselectivity. The diastereoselectivity was indicated, according to the developed rationale, to be governed by the sterics of the interaction of the all-equatorial conformation of the menthol with the axial methyl group of (+)-isomenthyl of the catalyst. (−)-Menthol 5 was posited to exhibit van der Waals repulsion in the proposed model (Scheme 1b), favoring (+)-menthol 5 in the glycosylation (Scheme 1a). Thus, based on the proposed model (Scheme 1), α/β-stereo- and diastereoselectivity with NPGs 6–8 and chiral recognition of racemic menthol 5 (entries 1–3, Table 1) can be rationalized. The choice of ester in the catalyst was found to be essential—as demonstrated with menthyl esters of CBA 3 and 4 that have all three groups in the equatorial position of their most stable conformation, causing a lack of diastereoselectivity (entries 9 and 10, Table 1). Similarly, the PCCP CBA catalyst with (−)-isomenthyl group can offer opposite chiral recognition; however, due to the lack of commercial availability of (−)-isomenthol, we could not pursue synthesis of the catalyst. The importance of CBA 1 catalysis was obvious in glycosylation experiments—as attempts with chiral phosphoric acid catalysts (R)-2 and (S)-2, though having offered stereoselectivity, showed no appreciable chiral recognition of racemic menthol (entries 4 and 5, Table 1).
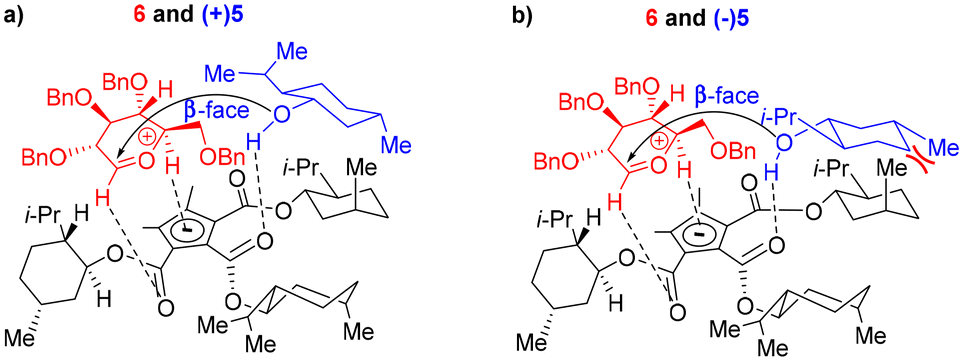 | ||
| Scheme 1 Rationale for stereo- and diastereocontrol in glycosylation with CBA 1. | ||
We performed density functional theory (DFT) calculations to establish a correlation between the suggested mechanism responsible for the oxocarbenium intermediate-assisted transition state, presence of H-bonding interactions in the three systems, and electron transitions from the acceptor to the oxocarbenium intermediate. Our experimental analysis aligned seamlessly with the DFT assessments, confirming the presence of three significant H-bonding interactions within optimized geometries. A visual depiction of the DFT analysis, illustrated in Fig. S1 (see ESI†), unveiled precise interactions: with a hydrogen bond formed between the hydrogen atom (H39) of the hydroxyl group of the acceptor moiety and the carbonyl oxygen (O11) of the catalyst with an approximate length of 1.44 Å. Another hydrogen bond interaction between the carbonyl oxygen (O3) of the catalyst and the hydrogen atom (H54) of the oxocarbenium intermediate, measured at approximately 1.47 Å, was discerned. Furthermore, a π-hydrogen bond was observed between the catalyst and the oxocarbenium ion (H67) by positioning a dummy atom (O84) at the centre of cyclopentadienyl system, with an approximate bond length of 1.96 Å. The consistency of these parameters with findings from the literature further substantiated the proposed mechanism.23 Moreover, the electron transition between the oxocarbenium intermediate and the acceptor groups were illustrated by calculating the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) energy levels for the optimized geometry. As given in Table S1 (in ESI†), these calculations indicated the HOMO energy level (6.40 eV) to be concentrated over the acceptor moiety, whereas the LUMO energy level (3.04 eV) to be concentrated over the oxocarbenium intermediate. These results confirmed the nucleophilic and electrophilic natures of the acceptor and intermediate systems, respectively.
To assess the generality of the developed process, we investigated the chiral recognition with racemic substrates 12, 13, and 14 for the CBA 1-catalyzed glycosylation reaction with NPGs 6–8 (Scheme 2). Exclusive diastereocontrol in chiral recognition of the S-enantiomers of benzylic alcohols 12 and 13 in the glycosylation reaction afforded 15–20 in excellent yields. Glucosyl-NPG 6 afforded β-anomers 15 and 18 selectively; however, mannosyl-NPG 7 afforded anomeric mixtures 16 and 19, with the α-anomer being predominant, and galactosyl-NPG 8 produced anomeric mixtures 17 and 20, with the β-anomer being predominant. Interestingly, glycosylation attempts with racemic Boc-protected serine 14, with a free carboxylic acid, also afforded chiral recognition of the S-enantiomer in good yields; herein, glucosyl-NPG 6 afforded α-anomer 21 selectively, and mannosyl-NPG 7 afforded an anomeric mixture of 22, with the β-anomer being predominant. However, galactosyl-NPG 8 produced α-anomer 23 selectively and did not afford chiral recognition, unlike its congeners. In line with the mechanistic rationale outlined in Scheme 1, a preference for the S-enantiomers of benzylic alcohols 12 and 13 in the glycosylation reaction was identified, as shown in Scheme S3 (ESI†). However, in the case of racemic 14, intricate secondary interactions arising from the Boc and COOH groups in the transition state for the glycosylation reaction apparently dictated the stereo- and diastereoselectivities, a feature difficult to comprehend.
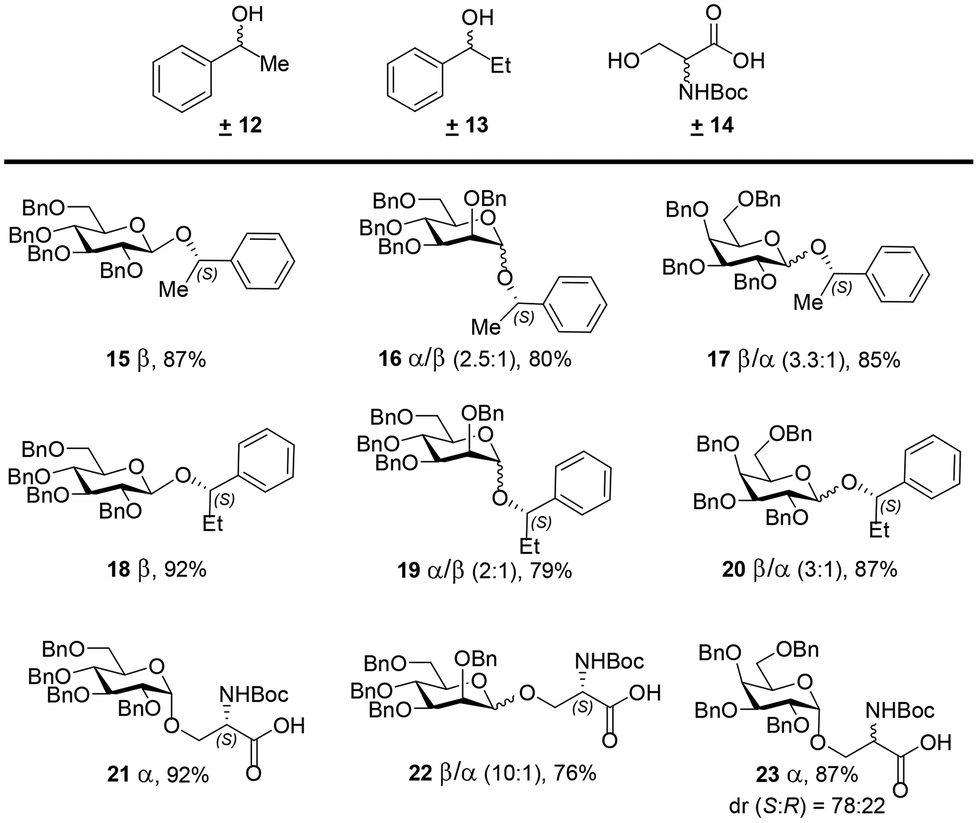 | ||
| Scheme 2 Diastereocontrol in glycosylation of racemic acceptors 12, 13, and 14 with CBA 1. Reaction conditions: NIS (2.5 equiv.) and CBA 1 (10 mol%) were added to each of mixtures of 6, 7, or 8 (1.0 equiv.) with ±12, ±13, or ±14 (2.0 equiv.) in CH2Cl2 at 0 °C, under 4 Å MS, and the resulting mixtures were slowly warmed to room temperature. The reported yields are isolated yields determined using column chromatography. The ratios were determined from integration of the anomeric protons in 1H NMR spectra, identified by their respective chemical shifts of the glycosides with pure enantiomers and HMQC spectra (see ESI†). Relative amounts of diastereomeric glycosides(%). | ||
In conclusion, as noted in the pioneering contribution by Lambert et al., chiral PCCP BA catalysis can be made more efficient by optimizing the catalyst structure; herein, we modified their original CBA 4 with (−)-menthol to CBA 1 with (+)-isomenthol and evaluated its catalytic role in stereoselective glycosylation with NPG donors in the presence of NIS as the promoter. Apart from α/β-stereoselectivity with NPGs, the glycosylation reaction in the presence of CBA 1 was efficient in chiral recognition of the racemic menthol, favoring (+)-menthol, with excellent diastereoselectivity. The stereo- and diastereocontrols observed here with CBA 1 and NPGs were superior to those of chiral BINOL phosphoric-acid-catalyzed glycosylation with trichloroacetimidate donors.14 The mechanistic model behind CBA 1 catalysis by H-bonding organization of the oxocarbenium ion, generated from NPG activation and menthol with the chiral cyclopentadienyl anion platform, assisted in providing the rationale behind the lack of diastereocontrol with other chiral, BINOL-derived and menthyl CBA esters, and nonchiral BAs. The present investigation with CBA 1 catalyst motivated us to investigate its utility with other racemic aglycons, and the resulting stereo- and diastereocontrol observed in the glycosylation with NPGs suggested the potential benefits of CBA in a myriad BA-catalyzed synthetic organic transformations.
Author contributions
K. V. R. conceived and supervised this project. M. A. performed majority of the synthesis. M.S. performed theoretical calculations. P. C. H. and S. P. performed a minor part of the synthesis. R. S. L. supervised this project and wrote the original draft of the manuscript, which was edited by all the authors.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
AcSIR student M. A. thanks CSIR for the fellowship, Mr Billu Abraham and Ms C. P. Devika for their help with HPLC, and Ms Deepa Sebastian for the help in interpreting theoretical calculations. Financial support from DST, Science, and Engineering Research Board, India (Grant Number: CRG/2020/004993) is gratefully acknowledged.References
- J.-L. Viladot, E. de Ramon, O. Durany and A. Planas, Biochemistry, 1998, 37, 11332–11342 CrossRef CAS PubMed.
- L. L. Lairson, B. Henrissat, G. J. Davies and S. G. Withers, Annu. Rev. Biochem., 2008, 77, 521–555 CrossRef CAS PubMed.
- A. Kumar, V. Kumar, R. T. Dere and R. R. Schmidt, Org. Lett., 2011, 13, 3612–3615 CrossRef CAS PubMed.
- H. Hinou, N. Saito, T. Maeda, M. Matsuda, Y. Kamiya and S.-I. Nishimura, J. Carbohydr. Chem., 2011, 30, 575–586 CrossRef CAS.
- A. A. Chaugule, A. R. Jadhav and H. Kim, RSC Adv., 2015, 5, 104715–104724 RSC.
- Y. Kobayashi, Y. Nakatsuji, S. Li, S. Tsuzuki and Y. Takemoto, Angew. Chem., Int. Ed., 2018, 57, 3646–3650 CrossRef CAS PubMed.
- T. Yamanoi, Y. Oda, S. Matsuda, I. Yamazaki, K. Matsumura, K. Katsuraya, M. Watanabe and T. Inazu, Tetrahedron, 2006, 62, 10383–10392 CrossRef CAS.
- T. Yamanoi, S. Matsuda, I. Yamazaki, R. Inoue, K. Hamasaki and M. Watanabe, Heterocycles, 2006, 68, 673–677 CrossRef CAS.
- S. Delacroix, J.-P. Bonnet, M. Courty, D. Postel and A. N. van Nhien, Carbohydr. Res., 2013, 381, 12–18 CrossRef CAS.
- S. Shirakawa and S. Kobayashi, Org. Lett., 2007, 9, 311–314 CrossRef CAS PubMed.
- S. Dasgupta and B. Mukhopadhyay, Eur. J. Org. Chem., 2008, 2008, 5770–5777 CrossRef.
- G. Karki and P. K. Mandal, ARKIVOC, 2019, 2019, 196–210 Search PubMed.
- G. Karki, H. Kumar, R. Rajan and P. K. Mandal, Synlett, 2016, 2581–2586 CAS.
- T. Kimura, M. Sekine, D. Takahashi and K. Toshima, Angew. Chem., Int. Ed., 2013, 52, 12131–12134 CrossRef CAS PubMed.
- D. Liu, S. Sarrafpour, W. Guo, B. Goulart and C. S. Bennett, J. Carbohydr. Chem., 2014, 33, 423–434 CrossRef CAS.
- D. J. Cox, M. D. Smith and A. J. Fairbanks, Org. Lett., 2010, 12, 1452–1455 CrossRef CAS PubMed.
- N. D. Gould, C. Liana Allen, B. C. Nam, A. Schepartz and S. J. Miller, Carbohydr. Res., 2013, 382, 36–42 CrossRef CAS.
- M. Aswathy, B. Abhijith, R. S. Lankalapalli and K. V. Radhakrishnan, Carbohydr. Res., 2022, 522, 108684 CrossRef CAS PubMed.
- B. Fraser-Reid and J. C. López, in Top Curr Chem, 2011, vol. 301, pp. 1–29 Search PubMed.
- C. D. Gheewala, B. E. Collins and T. H. Lambert, Science, 2016, 351, 961–965 CrossRef CAS PubMed.
- K. N. Jayaprakash and B. Fraser-Reid, Synlett, 2004, 301–305 CAS.
- B. Fraser-Reid, G. Anilkumar, M. R. Gilbert, S. Joshi and R. Kraehmer, in Carbohydrates in Chemistry and Biology, Wiley-VCH Verlag GmbH, Weinheim, Germany, 2000, pp. 135–154 Search PubMed.
- M. Nishio, Phys. Chem. Chem. Phys., 2011, 13, 13873–13900 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C and 2D NMR, experimental procedures, computational studies, NMR spectral data, HPLC chromatograms. See DOI: https://doi.org/10.1039/d3ob01633a |
| This journal is © The Royal Society of Chemistry 2024 |