
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSterically demanding Csp2(ortho-substitution)–Csp3(tertiary) bond formation via carboxylate-directed Mizoroki-Heck reaction under extra-ligand-free conditions†
Wei
Zeng‡
ab,
Ai-Wen
Chen‡
ab,
Ming-Jie
Yan
ab and
Jie
Wang
 *abc
*abc
aSchool of Chinese Materia Medica, Nanjing University of Chinese Medicine, Nanjing 210023, China. E-mail: jiewang@simm.ac.cn
bState Key Laboratory of Chemical Biology, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China
cUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 22nd November 2023
Abstract
Construction of the sterically demanding Csp2(oS)–Csp3(T) bond was achieved by carrying out the Pd-catalyzed carboxylate-directed Mizoroki-Heck reaction under extra-ligand-free aqueous conditions. The cooperative role of the presence of water with the absence of phosphine ligand was proposed to accelerate the migratory insertion process considerably, delivering a broad substrate scope.
Due to the importance of sp3 carbon atoms1 in accessing a diversified chemical space for drug discovery, there has been much recent interest in developing new methods to construct Csp2–Csp3 bonds.2 There are extra challenges when targeting the sterically demanding Csp2(oS)–Csp3(T) bonds characterized by an ortho-substituted sp2 carbon attached to a tertiary sp3 carbon center, which are often found in complex natural products. Over the past decade, although a plethora of interesting new approaches have emerged allowing for versatile access to tertiary Csp2–Csp3 bonds intermolecularly, only a few of them could tolerate ortho-substitution on the sp2 coupling partner, including the Friedel–Crafts reaction,3 directed Mizoroki-Heck reaction4 and C–H functionalization,5 redox-relay Heck reaction,6 and the widely studied cross-coupling reactions.7 Of these approaches, the directing group strategy is particularly highly reliable and shows adequate regio- and stereo-chemical control.8 For example, the amine-directed intermolecular Mizoroki-Heck reaction developed by Hallberg was used to construct a Csp2(oS)–Csp3(T) bond efficiently in a highly regio- and enantio-selective manner (Fig. 1A).4 Aniline derivatives and aryl iodide are effective directing groups in ruthenium-catalyzed and electrochemically catalyzed C–H alkylation reactions, securing a versatile construction of Csp2(oS)–Csp3(T) bonds.5 In this context, we are interested in constructing Csp2(oS)–Csp3(T) bonds using the directivity of carboxylic acid, particularly due to the ubiquitous nature, good stability, and remarkable transformational versatility of this functional group in organic synthesis. Although the directivity of free carboxylic acid was previously well documented in palladium-catalyzed C–H functionalizations,9 it has hardly been used at all in Mizoroki-Heck reactions.10 While this strategy has been studied by White,10a Zhao10b and Chou10c for Csp2–Csp2 bond formation, the method reported by Shenvi and coworkers is, to the best of our knowledge, the only example for constructing a Csp2–Csp3(T) bond with a sterically hindered quaternary carbon center (Fig. 1B).10d–e However, ortho-substituted and electron deficient aryl bromides were reported to be incompatible as commented in their article and supporting information. Here, in this work, we present the development of a new extra-ligand-free aqueous condition allowing access to the extremely challenging Csp2(oS)–Csp3(T) bonds. Aryl bromides or iodides of different electronic characteristics and other substitution patterns were well tolerated.
 | ||
| Fig. 1 (A) Directing group strategy for intermolecular Csp2(oS)–Csp3(T) bond formation. (B) Construction of the Csp2–Csp3 bond via the directivity of carboxylic acid: precedent and this work. | ||
Our reaction conditions were optimized with γ,δ-unsaturated carboxylic acid 1 and aryl halide 2 as model substrates (Table 1). Initially, the conditions reported by Shenvi and coworkers were attempted with 2-bromoanisole as the aryl coupling partner. However, the desired product 3 was not detected even in the slightest amount (entry 1).10e To our delight, by simply changing 2-bromoanisole to 2-iodoanisole (2a), 1 was delivered in 30% yield under the otherwise same conditions (entry 2). Ligand screening revealed that inclusion of PPh3 and DavePhos increased the yields slightly (entry 3) while P(o-Tol)3 and JohnPhos provided product in less than 10% yield (entry 4). Bidentate phosphine ligands were also tested and proved to be ineffective for this chemistry (see ESI† for details). Interestingly, we found that the reaction also proceeded to some extent in the absence of phosphine ligand, with a low yield of 15% obtained (entry 5). Under these extra-ligand-free conditions, other single solvents such as DMA, 1,4-dioxane, and DMSO all performed poorly (entry 6). Gratifyingly, a thorough solvent screen led us to discover H2O to be a crucial co-solvent for increasing the reaction yield significantly (entries 7–9), and the optimal DMF/H2O ratio was identified to be 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, providing 3 in 60% yield (entry 9). The H2O promotion phenomenon was also observed to be general, for several other solvents tested (entries 10–12 vs. entry 6). DMSO/H2O gave the same yield as did DMF/H2O (entry 12 vs. entry 9) and showed comparable efficiencies for other substrates except for strongly electron-deficient ones (see ESI† for a detailed list of substrates tested). Under the newly developed conditions, addition of phosphine ligands exhibited a deleterious effect on the reaction yields (entries 13 and 14). Finally, changing the temperature (entry 15), palladium source (entry 16) and base (entry 16) all delivered inferior results.
:1, providing 3 in 60% yield (entry 9). The H2O promotion phenomenon was also observed to be general, for several other solvents tested (entries 10–12 vs. entry 6). DMSO/H2O gave the same yield as did DMF/H2O (entry 12 vs. entry 9) and showed comparable efficiencies for other substrates except for strongly electron-deficient ones (see ESI† for a detailed list of substrates tested). Under the newly developed conditions, addition of phosphine ligands exhibited a deleterious effect on the reaction yields (entries 13 and 14). Finally, changing the temperature (entry 15), palladium source (entry 16) and base (entry 16) all delivered inferior results.
|
|
|||
|---|---|---|---|
| Entry | Ligand | Solvent | Yieldb |
| a Reaction conditions: 1 (0.1 mmol), 2a (1.5 equiv.), Pd2(dba)3·CHCl3 (3 mol%), ligand (12 mol%) if used, and K2CO3 (2.5 equiv.) were dissolved in solvent (0.6 mL) and heated to 100 °C under an Ar atmosphere. b Yield determined from a crude 1H NMR analysis using 1,3,5-trimethoxybenzene as an internal standard. c 2-Bromoanisole was used instead of 2a. d DMF (0.3 mL) and H2O (0.3 mL). e DMF (0.4 mL) and H2O (0.2 mL). f Isolated yield. g Reaction temperature was 90 or 110 °C. h Pd(OAC)2, PdCl2, Pd(PPh3)2Cl2, or Pd(PPh3)4 was used instead of Pd2(dba)3·CHCl3. i NaHCO3, Na2CO3, Cs2CO3, tBuONa, or Et3N was used instead of K2CO3. See ESI† for more details. | |||
| 1c | TrixiePhos | DMF | 0% |
| 2 | TrixiePhos | DMF | 30% |
| 3 | PPh3/DavePhos | DMF | 39%/33% |
| 4 | P(o-Tol)3, JohnPhos | DMF | <10% |
| 5 | — | DMF | 15% |
| 6 | — | DMA, 1,4-dioxane, DMSO | <5% |
| 7d | — | DMF/H2O (1/1) | 44% |
| 8e | — | DMF/H2O (2/1) | 52% |
| 9 | — | DMF/H 2 O (5/1) | 60% (58%) |
| 10 | — | DMA/H2O (5/1) | 32% |
| 11 | — | 1,4-dioxane/H2O (5/1) | 30% |
| 12 | — | DMSO/H2O (5/1) | 60% |
| 13 | TrixiePhos | DMF/H2O (5/1) | 36% |
| 14 | PPh3/DavePhos | DMF/H2O (5/1) | 39%/39% |
| 15g | — | DMF/H2O (5/1) | 47–51% |
| 16h | — | DMF/H2O (5/1) | 12–45% |
| 17i | — | DMF/H2O (5/1) | 15–45% |

With the optimized conditions in hand, we set out to investigate the substrate scope. It turned out that the new conditions developed above were generally effective across a range of substrates (>40 examples) as shown in Scheme 1. The aryl coupling partner scope was first investigated (Scheme 1A). Various ortho-substituents ranging from electron-rich to electron-poor ones including MeO (3, 4, 5, 7), Me (6), F (8), and carbonyl (9) were well tolerated. Even a highly hindered bis-ortho-substituted substrate provided product in 33% yield (7). Commercial aryl halides with other substitution patterns and different electronic characteristics were then evaluated. While a similar efficiency was observed for electron-rich and electron-neutral arenes (11–20) as was observed for the Shenvi conditions, a significant improvement in yields for electron-poor ones was obtained.10e For example, aryl bromides with strongly electron-withdrawing groups in the para position were previously reported to be unreactive, while that in the meta position was delivered in low yield. In contrast, under our conditions, these substrates all provided products in moderate to good yields (21–23). Moderately electron-deficient F- and Cl-substituted arenes also worked well, providing 24 and 25 in 54% and 71% yields, respectively. Various heteroaromatic substrates were also tested and showed improved efficiency. In this regard, electron-rich heterocycles such as benzothiophene (26), benzofuran (27), and either 2- or 3-thiophene with or without ortho-substitution (28–30) all gave moderate yields. Electron-poor ones such as quinoline (31) and chromone (32) also proved to be viable substrates under our conditions.
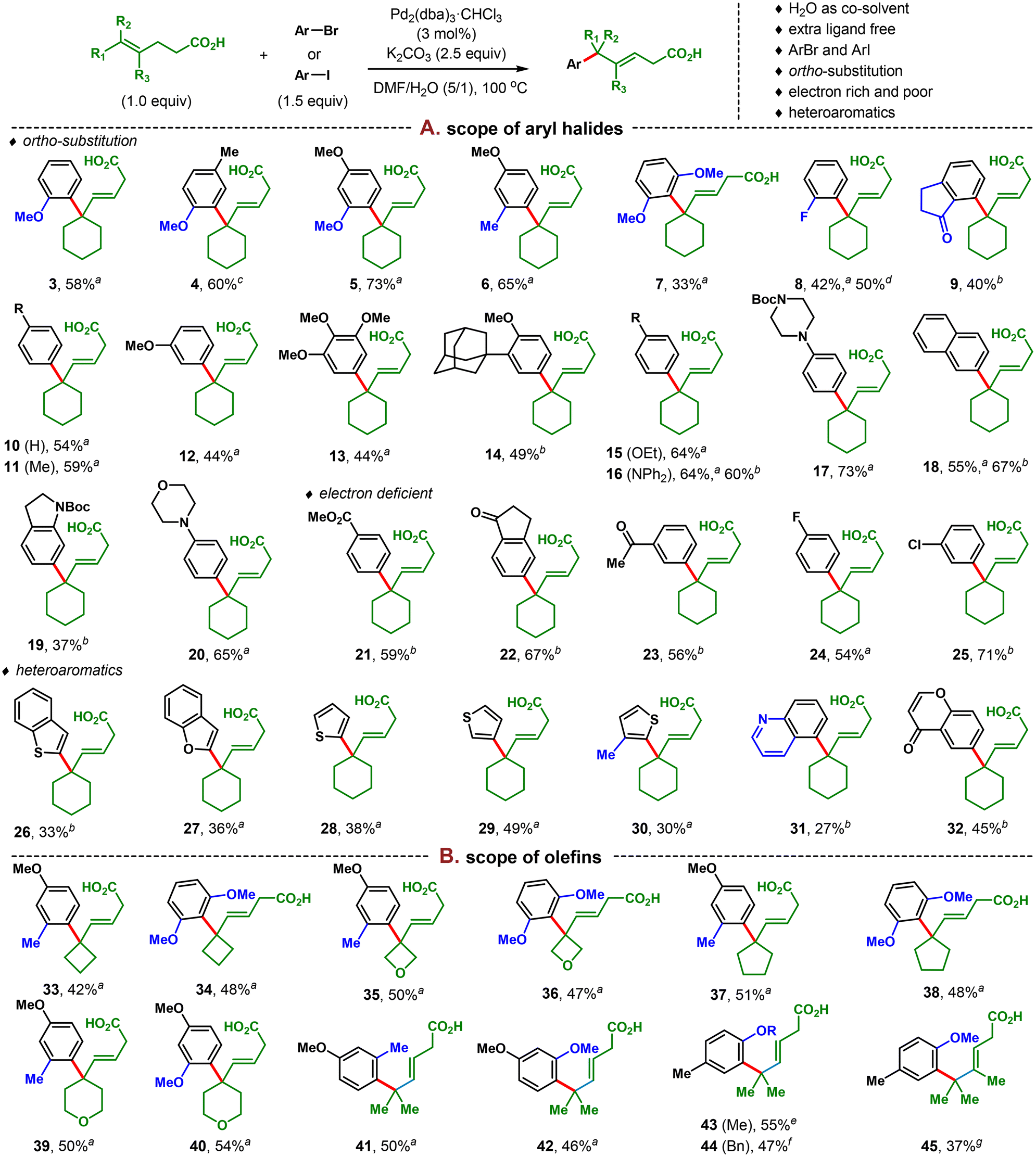 | ||
| Scheme 1 Reaction conditions: olefin (0.1 mmol, 1.0 equiv.), aryl halide (1.5 equiv.), Pd2(dba)3·CHCl3 (3 mol%), and K2CO3 (2.5 equiv.) were dissolved in DMF/H2O (5/1, 0.6 mL) and stirred at 100 °C under an argon atmosphere for 5–12 hours. Yield referred to isolated yield unless otherwise noted. aArI was used. bArBr was used. cDMF/H2O (5/1, 1.2 mL) was used. dOlefin (0.5 mmol), ArI (1.5 equiv.), DMF/H2O (5/1, 6.0 mL). e1.0 mmol scale. fArI (1.0 mmol), olefin (1.5 equiv.). gArI (3.0 equiv.), Pd2(dba)3·CHCl3 (10 mol%). | ||
Next, several other γ,δ-unsaturated carboxylic acids were evaluated with o-OMe- or bis-o-OMe-substituted aryl coupling partners as shown in Scheme 1B. Yields of 37–55% were obtained consistently for all the 4–6-membered cyclic, heterocyclic, and noncyclic substrates (33–45). The ortho-benzyl ether protecting group was also tolerated, providing slightly lower yield than provided by the ortho-OMe counterpart (47% vs. 55% for 44 and 43). With a more hindered tetrasubstituted olefin (45), a useful yield of 37% was also obtained after slight modification to the original conditions including use of 10 mol% catalyst.
The successful implementation of this newly developed condition also relied largely on the judicious choice of aryl bromide or iodide precursors. Empirically, aryl iodides outperformed the corresponding aryl bromides for electron-rich and neutral substrates (1, 5, 6, 12, 13, 15, 17, 20, and 30), while aryl bromides worked better for electron-deficient ones (8, 23, 25). The generally moderate yields obtained throughout the investigation of substrate scope mainly stemmed from byproduct where the double bond migrated to be conjugated with the carboxylic acid through chain walking (typically 1/5–1/10 with regards to the shown products) and, in some cases, incomplete conversions.
While reactions with most of the substrates shown in Scheme 1 were performed on a 0.1 mmol scale, scale-up proved uneventful and was exemplified by substrates 8 and 44 on 0.5 and 1.0 mmol scales, respectively. They were then easily converted to known respective intermediates and natural products containing Csp2(oS)–Csp3(T) bonds, further demonstrating the utility of this new method (Schemes 2A and B). To this end, compound 47, a synthetic intermediate for S1P receptor modulator,11 was straightforwardly accessed from 8 in two steps and 64% overall yield by successive reduction of the alkene and carboxylic acid functional group. As another example, the sesquiterpenoid natural product himasecolone (49)12 was synthesized simply from 44 by carrying out a one-step Bn deprotection/alkene hydrogenation followed by transforming the carboxylic acid group to methyl ketone in 43% overall yield. In both cases, the strategic construction of the synthetically challenging Csp2(oS)–Csp3(T) moiety in an early stage provided extreme immediacy and simplicity for subsequent functional group manipulations.
 | ||
| Scheme 2 (A) Synthesis of drug intermediate 47 and (B) natural product himasecolone (49). (C) Proposed role of H2O and absence of bulky ligand. | ||
Based on previous reports of palladium-catalysed Mizoroki-Heck reactions in aqueous media,13 we arrived at an understanding of the mechanistic advantages of the current conditions (Scheme 2C). Oxidative addition and migratory insertion have often been regarded as rate-determining steps in Heck reactions.14 In the carboxylate-directed Heck reaction, we assumed the ArPd(II) carboxylate formation, coordination to alkene, and subsequent migratory insertion to be a relatively slow process, but one accelerated under our H2O-containing and ligand-free conditions.15 The H2O co-solvent not only increased the solubility of the inorganic base K2CO3 and the effective concentration of palladium carboxylate,16 but also promoted the migratory insertion to follow cationic mechanism by displacing the poisonous iodide or bromide anion from the coordination shell of palladium, the effect of which could be more pronounced under extra-ligand-free conditions.17 On the other hand, the omission of a bulky phosphine ligand also contributed significantly to this process by allowing for a free coordination site on the palladium center, reducing the steric congestion and thus increasing the rate of migratory insertion.18 While the sluggish migratory insertion step was accelerated substantially, the reaction showed better tolerance for the rate of oxidative addition, that is to say, a wide range of both aryl bromides and iodides with varied electronic characteristics could be used. In the case of electron-deficient substrates (9, 21–23, 31–32), our aqueous conditions free of phosphine ligand might have led to slower oxidative addition of aryl bromides,19 which probably better matched the accelerated downstream migratory insertion step and prevented the Ar-Pd(II) accumulation and deactivation process proposed by Shenvi and coworkers.10e
Conclusions
In summary, the construction of sterically demanding Csp2(oS)–Csp3(T) bonds (>20 examples) was achieved by carrying out palladium-catalyzed carboxylate-directed Mizoroki-Heck reactions. The extra-ligand-free aqueous conditions were suitable for aryl halides with various substitution patterns and electronic characteristics, and the utility of this method was further demonstrated through a few-step synthesis of the Csp2(oS)–Csp3(T)-bond-containing S1PR modulator and sesquiterpenoid natural product himasecolone. Mechanistically, we proposed that the high substrate scope arose from the rapid migratory insertion, which was significantly accelerated by the use of H2O as co-solvent and the absence of phosphine ligand acting cooperatively.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this work was provided by the National Natural Science Foundation of China (Grant No. 22101290), Chinese Academy of Sciences and Shanghai Institute of Materia Medica. We thank Professor Hui-Xiong Dai for helpful discussions.References
- (a) F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS; (b) W. P. Walters, J. Green, J. R. Weiss and M. A. Murcko, J. Med. Chem., 2011, 54, 6405–6416 CrossRef CAS.
- For selected reviews on Csp2−Csp3 cross-couplings, see: (a) R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417–1492 CrossRef CAS; (b) G. C. Fu, ACS Cent. Sci., 2017, 3, 692–700 CrossRef CAS PubMed; (c) M. L. Neidig, S. H. Carpenter, D. J. Curran, J. C. DeMuth, V. E. Fleischauer, T. E. Iannuzzi, P. G. N. Neate, J. D. Sears and N. J. Wolford, Acc. Chem. Res., 2018, 52, 140–150 CrossRef; (d) R. Shi, Z. Zhang and X. Hu, Acc. Chem. Res., 2019, 52, 1471–1483 CrossRef CAS PubMed; (e) A. Guérinot and J. Cossy, Acc. Chem. Res., 2020, 53, 1351–1363 CrossRef; (f) W. Xue, X. Jia, X. Wang, X. Tao, Z. Yin and H. Gong, Chem. Soc. Rev., 2021, 50, 4162–4184 RSC.
- S. Zhang, M. Vayer, F. Noël, V. D. Vuković, A. Golushko, N. Rezajooei, C. N. Rowley, D. Lebœuf and J. Moran, Chem, 2021, 7, 3425–3441 CAS.
- P. Nilsson, M. Larhed and A. Hallberg, J. Am. Chem. Soc., 2003, 125, 3430–3431 CrossRef CAS.
- (a) S. Bauri, S. Kumar and A. Rit, Adv. Synth. Catal., 2023, 365, 2385–2391 CrossRef CAS; (b) Z. Deng, W. Fan, J. Liu, G. Tu, P. Huang, X. Xu, J. Tan, S. J. Ji and Y. Zhao, Chem. – Eur. J., 2023, 29 DOI:10.1002/chem.202300905; (c) X. Lv, Y. Cheng, Y. Zong, Q. Wang, G. An, J. Wang and G. Li, ACS Catal., 2023, 13, 7310–7321 CrossRef CAS; (d) X. Zheng, P. Peng, C. Huang and Q. Lu, CCS Chem., 2023, 5, 1086–1095 CrossRef CAS.
- (a) T.-S. Mei, H. H. Patel and M. S. Sigman, Nature, 2014, 508, 340–344 CrossRef CAS PubMed; (b) C. C. Oliveira, A. Pfaltz and C. R. D. Correia, Angew. Chem., Int. Ed., 2015, 54, 14036–14039 CrossRef CAS.
- (a) A. Joshi-Pangu, C.-Y. Wang and M. R. Biscoe, J. Am. Chem. Soc., 2011, 133, 8478–8481 CrossRef CAS PubMed; (b) X. Wang, G. Ma, Y. Peng, C. E. Pitsch, B. J. Moll, T. D. Ly, X. Wang and H. Gong, J. Am. Chem. Soc., 2018, 140, 14490–14497 CrossRef CAS; (c) T. G. Chen, H. Zhang, P. K. Mykhailiuk, R. R. Merchant, C. A. Smith, T. Qin and P. S. Baran, Angew. Chem., Int. Ed., 2019, 58, 2454–2458 CrossRef CAS PubMed; (d) B. Li, J.-L. Shi and Y. Xia, Org. Lett., 2023, 25, 2674–2679 CrossRef CAS.
- For selected reviews, see: (a) M. Oestreich, Eur. J. Org. Chem., 2005, 783–792 CrossRef CAS; (b) K. Murali, L. A. Machado, R. L. Carvalho, L. F. Pedrosa, R. Mukherjee, E. N. Da Silva Junior and D. Maiti, Chem. – Eur. J., 2021, 27, 12453–12508 CrossRef CAS PubMed; (c) J. Das, W. Ali and D. Maiti, Trends Chem., 2023, 5, 551–560 CrossRef CAS.
- For selected reviews, see: (a) K. M. Engle, T. S. Mei, M. Wasa and J. Q. Yu, Acc. Chem. Res., 2012, 45, 788–802 CrossRef CAS PubMed; (b) G. Shi and Y. Zhang, Adv. Synth. Catal., 2014, 356, 1419–1442 CrossRef CAS; (c) M. P. Drapeau and L. J. Goossen, Chem. – Eur. J., 2016, 22, 18654–18677 CrossRef.
- (a) J. H. Delcamp, A. P. Brucks and M. C. White, J. Am. Chem. Soc., 2008, 130, 11270–11271 CrossRef CAS; (b) S. Yang, L. Liu, Z. Zhou, Z. Huang and Y. Zhao, Org. Lett., 2021, 23, 296–299 CrossRef CAS PubMed; (c) S. Y. Tsai, Y. W. Huang and C. M. Chou, Adv. Synth. Catal., 2023, 365, 699–703 CrossRef CAS; (d) J. J. Roach, Y. Sasano, C. L. Schmid, S. Zaidi, V. Katritch, R. C. Stevens, L. M. Bohn and R. A. Shenvi, ACS Cent. Sci., 2017, 3, 1329–1336 CrossRef CAS; (e) T. R. Huffman, Y. Wu, A. Emmerich and R. A. Shenvi, Angew. Chem., Int. Ed., 2019, 58, 2371–2376 CrossRef CAS PubMed.
- P. X. Nguyen, T. M. Heidelbaugh and J. R. Cappiello, (Allergan Inc.), WO 2014/093253A1, 2014.
- (a) P. K. Agarwal and R. P. Rastogi, Phytochemistry, 1981, 20, 1319–1321 CrossRef CAS; (b) S. V. Trivedi and V. R. Mamdapur, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 1986, 25, 1160 Search PubMed.
- (a) L.-M. Tao, Q.-G. Li, W.-Q. Liu, Y. Zhou and J.-F. Zhou, Water-Promoted Palladium-Catalysed Heck Cross-Coupling Reactions of Aryl Halides with Alkenes in TBAB, J. Chem. Res., 2011, 35, 154–156 CrossRef CAS; (b) F. Christoffel and T. R. Ward, Palladium-Catalyzed Heck Cross-Coupling Reactions in Water: A Comprehensive Review, Catal. Lett., 2017, 148, 489–511 CrossRef; (c) S. N. Jadhav and C. V. Rode, An efficient palladium catalyzed Mizoroki–Heck cross-coupling in water, Green Chem., 2017, 19, 5958–5970 RSC.
- C. S. Consorti, F. R. Flores and J. Dupont, J. Am. Chem. Soc., 2005, 127, 12054–12065 CrossRef CAS.
- L. Xu, M. J. Hilton, X. Zhang, P.-O. Norrby, Y.-D. Wu, M. S. Sigman and O. Wiest, J. Am. Chem. Soc., 2014, 136, 1960–1967 CrossRef CAS PubMed.
- F. Zhao, M. Shirai and M. Arai, J. Mol. Catal. A: Chem., 2000, 154, 39–44 CrossRef CAS.
- I. P. Beletskaya and A. V. Cheprakov, Chem. Rev., 2000, 100, 3009–3066 CrossRef CAS PubMed.
- (a) J. H. Groen, J. G. P. Delis, P. W. N. M. van Leeuwen and K. Vrieze, Organometallics, 1997, 16, 68–77 CrossRef CAS; (b) V. H. Menezes da Silva, N. H. Morgon, C. R. D. Correia and A. A. C. Braga, J. Organomet. Chem., 2019, 896, 5–15 CrossRef CAS.
- (a) F. Barrios-Landeros, B. P. Carrow and J. F. Hartwig, J. Am. Chem. Soc., 2009, 131, 8141–8154 CrossRef CAS; (b) B. Noverges-Pedro, M. Medio-Simón and A. Jutand, ChemCatChem, 2017, 9, 2136–2144 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ob01784b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |