
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An assay for aryl radicals using BHAS coupling†‡
Kenneth F.
Clark
a,
Seb
Tyerman
a,
Laura
Evans
b,
Craig M.
Robertson
c and
John A.
Murphy
 *a
*a
aDepartment of Pure and Applied Chemistry, University of Strathclyde, 295 Cathedral Street, Glasgow, G1 1XL, UK. E-mail: john.murphy@strath.ac.uk
bMedicinal Chemistry, Research and Early Development, Oncology R&D, AstraZeneca, Cambridge, CB10 1XL, UK
cGSK Medicines Research Centre, Gunnels Wood Road, Stevenage, Herts SG1 2NY, UK
First published on 23rd December 2023
Abstract
Aryl radicals are intermediates in many reactions, but determining their presence unambiguously is often challenging. As we recently reported, reaction of 2-iodo-1,3-dimethylbenzene (7) in benzene with KOtBu and a suitable organic additive, leads to a base-induced homolytic aromatic substitution (BHAS) coupling reaction giving 2,6-dimethylbiphenyl (12) and biphenyl (3) as coupled products, together with xylene (13). In this case, biphenyl arises from a radical translocation and is the major coupling product. This paper now quantitatively investigates that reaction, which shows a very similar ratio for 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :12 [ca. 4:1] when using different sources of radical initiation. Deuterium isotope studies provide detailed mechanistic support for the proposed mechanism; when carried out in C6D6vs. C6H6, the reaction is characterised by a strong isotope effect for formation of 3-d10vs. 3, but not for formation of 12-d5vs. 12. These distinctive properties mean that the transformation can act as an assay for aryl radicals. An advantage of such a BHAS process is its sensitivity, since it involves a chain reaction that can amplify radical activity.
:12 [ca. 4:1] when using different sources of radical initiation. Deuterium isotope studies provide detailed mechanistic support for the proposed mechanism; when carried out in C6D6vs. C6H6, the reaction is characterised by a strong isotope effect for formation of 3-d10vs. 3, but not for formation of 12-d5vs. 12. These distinctive properties mean that the transformation can act as an assay for aryl radicals. An advantage of such a BHAS process is its sensitivity, since it involves a chain reaction that can amplify radical activity.
Introduction
A wide range of reactions of redox-active metals achieve coupling of organic moieties and are brought about in the presence of alkali metal tert-butoxides and other basic alkali metal salts.1–9 In many instances, discussions about whether key steps involve organometallic intermediates or radical intermediates have arisen. This has led to the concept of hybrid organometallic/organic radical species.10–15 In these cases, an organometallic intermediate might undergo reversible cleavage of the metal–carbon bond to form a carbon radical. It can be challenging to show the presence of radicals when a substrate is converted to a product structure that could plausibly arise from an organometallic or a radical or indeed a hybrid pathway. Aryl radicals are so reactive that they cannot be directly assayed in routine organic reaction conditions such as ours by methods such as EPR.16 Strong evidence for radicals can emerge when a radical is directly trapped by TEMPO or other radical traps to form a direct adduct that is then fully characterised, but reactions are often inhibited by TEMPO and related molecules without products of direct trapping being detected. In these cases, caution is needed before interpreting the inhibition as due to trapping of organic radicals by TEMPO since the inhibition could be due to other reasons, e.g. direct reaction of TEMPO with metal complexes.17,18 Our aim is to provide a diagnostic test for aryl radicals that are produced in the presence of base.19Aryl radicals are intermediates in Base-promoted Homolytic Aromatic Substitution (BHAS, Scheme 1).20–22 In these reactions, an aryl halide 1 couples with an arene 2 to form a biaryl. In detail, the substrate is converted into an aryl radical 4 that then adds to an arene 2 or heteroarene to afford radical 5. Treatment of 5 with KOtBu leads to radical anion 6 which is transformed into coupled product 3 by transferring an electron to another molecule of the starting aryl halide 1, thereby establishing a BHAS cycle. BHAS reactions are generally efficient in producing high yields of products.23
 | ||
| Scheme 1 BHAS coupling. | ||
The reactions are initiated by the formation of aryl radicals, usually with the help of an organic additive; a common mechanism of action is by reaction of the additive with alkali metal tert-butoxide to form an organic electron donor that initiates the BHAS cycle (i.e. converts substrate 1 to radical 4).22–25 However, regardless of the method of initiation, BHAS chemistry operates when aryl radicals are produced in the presence of arenes under basic conditions. Our recent report22 introduced substrate 7 as a useful mechanistic tool to identify BHAS chemistry and now, in this paper, we characterise the reactions of special substrate 7 through product ratio studies, and we use deuterium isotope studies to show that the reaction shows the same characteristics even when different sources of initiation are used. The reaction can thus be used as an assay for aryl radicals; in papers based on these findings, we use the assay to indicate when (i) Pd-catalysed reactions of aryl halides afford radicals in ground-state reactions26 and (ii) Ni complexes are involved in aryl radical formation.27
Aryl iodide 7 is a hindered substrate, and this gives it a special advantage in mechanistic studies. Instead of providing a single product, this substrate provides a defined mixture of three products 12, 13 and 3 that act as a signature of the radical process (Scheme 2). An advantage of this substrate 7 is that it cannot be converted to a benzyne by reaction with KOtBu. (Benzynes have been proposed as an alternative initiation source for BHAS chemistry; in the case of 7, this pathway cannot interfere.22 The chemistry is not confined to substrate 7; we will shortly disclose the reactivity of 8 and a range of other substrates.)
 | ||
| Scheme 2 BHAS chemistry of substrate 7. | ||
In our proposed mechanism, BHAS chemistry is triggered by conversion of substrate 7 into xylyl radical 9. Radical 9 can add to benzene in the normal way to afford 2,6-dimethylbiphenyl 12 but this is not the major coupled product – instead, the major coupled product is biphenyl 3. We proposed that the steric effect of the two methyl groups inhibits the m-xylyl radical 9 from easily adding to the π-system of benzene, so that, instead, 9 primarily abstracts a hydrogen atom from benzene, leading to a phenyl radical 14 and the volatile m-xylene 13. This phenyl radical can then add to benzene to form the cyclohexadienyl radical 5, which can be swiftly deprotonated by tert-butoxide anion, forming the cyclohexadienyl radical anion 6. This intermediate then transfers an electron to another molecule of the starting haloarene 7 and, in so doing, generates biphenyl 3.
Previous experiments22 had shown that, on reaction under various conditions of temperature and concentrations with a tetraazafulvalene organic electron donor and with phenanthroline or with 5,6-dimethylphenanthroline for different durations, biphenyl 3 and dimethylbiphenyl 12 had been produced in a ratio of approximately 4:1 as determined using 1H NMR with an internal standard. For NMR, samples needed to be concentrated in vacuo prior to analysis and this meant that one of the products, m-xylene, was lost. In this paper, the reaction products are more fully analysed by GC(FID), GCMS and NMR. By calibrating GC(FID) with authentic samples of the substrate and products in advance, the yields of products are accurately determined. Samples do not need to be concentrated in vacuo prior to analysis, allowing characterisation of the m-xylene 13 that is formed, which turns out to be very revealing.
Results and discussion
Our reaction conditions [2 equiv. KOtBu, 130 °C, 24 h using phenanthroline 15 (40 mol%) as the organic additive] gave an approximate ratio of ca. 4:1 for biphenyl 3:dimethylbiphenyl 12. The amount of additive used was based on previous experiments in the literature.28,29 (Reaction of phenanthroline with base is known to afford strong electron donors to initiate the formation of aryl radicals from haloarenes.22,28–30) Repeating the reaction with analysis by GC(FID) now confirmed the results (Table 1, entry 1) with a ratio of 3.97:1 after 24 h.31 These results underline the fact that biphenyl 3 is the major coupling product arising from reaction of this substrate 7 in benzene, in line with our mechanistic proposal that hydrogen atom abstraction from benzene leading to phenyl radical 14 (and thereafter, biphenyl 3) is faster than addition to benzene to give radical 10 (and thereafter, biaryl 12).
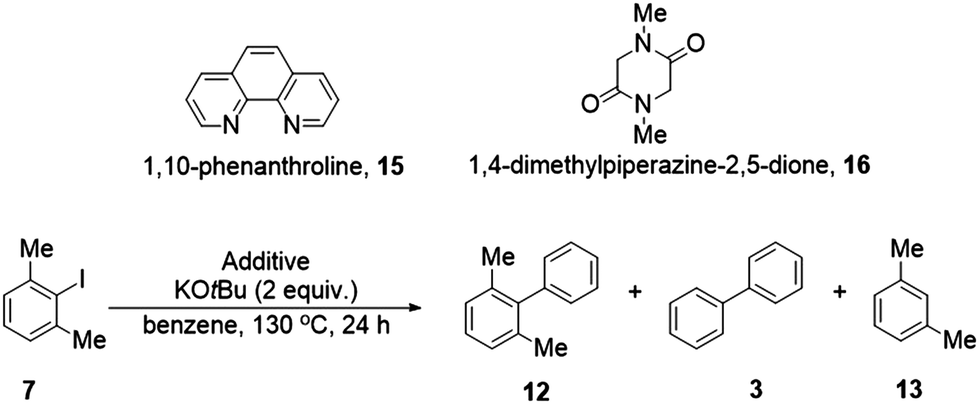
If this BHAS reaction is to be used as an assay for aryl radical formation from different sources, then it is important to test how the product profile alters in the presence of quite different reagents. For this reason, a completely different additive, the piperazinedione 16, was chosen. This additive is among those that are converted into a strong electron donor in the presence of KOtBu.23 In contrast to phenanthroline, this additive has really reactive C–H bonds in the NCH2C(O) methylene groups – abstraction of such a hydrogen by a reactive radical would afford a highly stabilised captodative radical, and so this additive might be expected to contribute to quenching any reactive aryl radicals arising from the substrate 7. Repeating the reaction with piperazinedione 16 (20 mol%) as additive gave a similar ratio of biphenyl 3:dimethylbiphenyl 12 (4.17:1, Table 1, entry 2). Once again, the amount of additive used was decided by following prior literature examples.23
Thus, for these very different additives, 15 and 16, the BHAS experiments provide similar ratios for the coupled products with biphenyl 3 as the major coupled product, giving confidence that the method can be applied widely to aryl radicals generated by other reagents. For both additives, the variation in the ratio dimethylbiphenyl 12:biphenyl 3 is likely caused by reaction of some of the additive molecules with radical intermediates. It is also noted that the conditions used for additive 16 resulted in some unchanged starting substrate 7. This likely means that fewer radical chains are maintained under the conditions of that reaction.
The detail of Scheme 2 was now scrutinised, through comparison of reactions in C6H6 and C6D6 as elaborated in Scheme 3. Previous studies by other authors of BHAS reactions with routine unhindered substrates have shown from deuterium labelling experiments with C6D6vs. C6H6 that the deprotonation reactions 5 → 6 (Scheme 1) are not rate-determining, so that no isotope effect is seen in the formation of products from the reaction.29,31 By contrast, the proposed hydrogen atom transfer step of an (sp2 C–H) hydrogen atom in benzene 2 to xylyl radical 9 is unique to hindered substrates like 7, and looks to be a challenging reaction and a good candidate for the rate-determining step. To explore this, the experiments of Table 1 were repeated, but using C6D6 as solvent in place of C6H6. It was expected that the amount of biphenyl (3-d10) formed in this reaction would decrease relative to the amount of 3 that routinely forms from C6H6, due to the more difficult abstraction of a deuterium atom from C6D6 compared with a hydrogen atom from C6H6. It was also thought that the yields of dimethylbiphenyl (12 and 12-d5 respectively) in the experiments with C6D6 and C6H6 where the rate determining step is C–C bond formation should remain relatively constant.
 | ||
| Scheme 3 Considering BHAS reactions with C6D6. | ||
Accordingly, the experiment was repeated in C6D6 as solvent. Before analysis of the yields of products produced from the reaction, the mass spectra of the three products biphenyl (3), dimethylbiphenyl (12) and m-xylene (13) were examined to verify that the expected deuterated isotopologues were formed. Products 3-d10, and 12-d5 were indeed formed, but the m-xylene 13 was predominantly not deuterated as discussed below.
Table 2, (entry 1) reproduces the data from the reaction in C6H6 previously shown in (Table 1, entry 1). The yields of biaryl products from the reaction in C6D6 (Table 2, entry 2) demonstrated that our expectations were correct, and use of C6D6 as solvent clearly affects the yield. The amount of biphenyl 3-d10 was greatly decreased compared to 3 in the respective experiment, from ∼32% down to 4%, highlighting the significant isotope effect associated with the formation of that compound. Dimethylbiphenyl 12-d5 (11%) was formed in the experiment, which represents a slight rise on the 8.1% of 12 formed from the control reaction in benzene. A rise can be in-line with expectations, since a greater proportion of xylyl radicals 9 will add to C6D6 rather than abstract a D atom from it, when compared to the analogous events in C6H6.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Additive (mol%) | Base | Residual 7 (%) | 12 (%) | 3 (%) | 13 (%) |
| a Reactions were conducted in duplicate. b Product identities verified by GCMS, NMR; ratios determined by calibrated GCFID. | ||||||
| 1 | 15 (40) | KOtBu, C6H6 | 0.0 | 8.1 | 32.2 | 58.0 |
| 2 | 15 (40) | KOtBu, C6D6 | 12.0 | 11.0 | 4.0 | 45.0 |
| 3 | 15 (40) | KOtBu-d9, C6D6 | 31.0 | 10.7 | 3.5 | 30.4 |
| 4 | 16 (20) | KOtBu, C6H6 | 35.3 | 3.0 | 12.5 | 47.9 |
| 5 | 16 (20) | KOtBu, C6D6 | 43.9 | 2.1 | 0.9 | 25.5 |
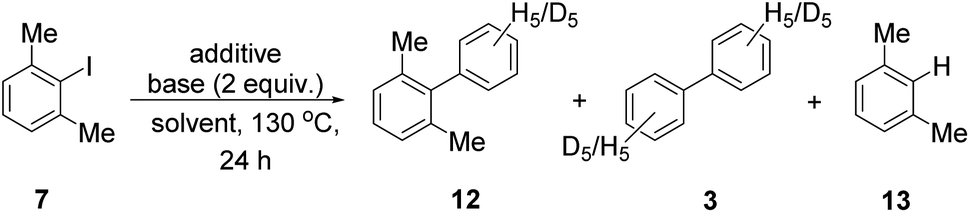
Unlike in the reaction with C6H6, unreacted aryl halide 7 (12%) remained from the reaction in C6D6. This can be explained by less propagation of the BHAS cycle, with slower formation of the cyclohexadienyl radical anion 6-d10 in C6D6 compared to its counterpart in unlabelled benzene, due to the deuterium isotope effect.32
The xylene formed in this experiment was predominantly undeuterated, (the estimated deuteration level was 12%). Clearly, the xylyl radicals 9 that form, predominantly abstract H atoms from available sites rather than D atoms from C6D6. The most likely sites targeted are the benzylic C–H sites of xylyl substrate 7 (and compounds 12 and 13 derived therefrom), as these would represent the weakest C–H bonds.
Table 2, entry 3 shows the same experiment with KOtBu-d9 and C6D6, showing very similar amounts of 12 and 3.
The experiment was repeated with piperazinedione additive 16 in C6D6 (Table 2, entry 5). For easy comparison, the results with C6H6 which were shown as Table 1, entry 2 are now reproduced as Table 2, entry 4. The reactions with piperazinedione additive 16 displayed the same expected trend as for phenanthroline, with similar quantities of dimethylbiphenyl-12-d5 being formed compared to the reaction of its unlabelled counterpart in C6H6 (3.0% → 2.1%) while the amount of biphenyl 3-d10 was greatly decreased relative to its counterpart (12.5% → 0.9%). The level of labelling of the xylene product 13 was so low that it could not be accurately estimated, and this was consistent with the low level of biphenyl-d10 produced.
To probe the source of the transferred H-atom in the formation of unlabelled xylene in experiments in C6D6, we considered preparing deuterated iodoxylene, 7-d6 (i.e. labelled in the methyl groups) and using it in place of 7 in this experiment. However, it was more convenient to prepare iodomesitylene-d9, 17 (Scheme 4) as shown in ESI.‡ To detect the formation of the Ar–D bond in a mesitylene product, 2H NMR was used. This gave rise to a peak at 6.87 ppm (Fig. S1‡), which is essentially the same chemical shift as the Ar–H of mesitylene (Fig. S4‡). Thus, the CD3 groups can act as the ultimate source of the Ar–D bond.33
 | ||
| Scheme 4 Reaction of iodomesitylene-d9. | ||
Conclusions
In summary, deuterium isotope effects are consistent with the proposed mechanisms of formation of products from 2,6-dimethyliodobenzene 7, involving a normal BHAS route to the formation of 2,6-dimethylbiphenyl 12 and an anomalous HAT from benzene to a xylyl radical leading to biphenyl 3.The reaction shows a very similar ratio of yields for 3:12 [ca. 4:1] when using different sources of radical initiation. Deuterium isotope studies provide detailed mechanistic support for the proposed mechanism; when carried out in C6D6vs. C6H6 the reaction is characterised by a strong isotope effect for formation of 3-d10vs.3. In combination, these characteristics allow us to propose this system as an assay for aryl radicals under basic conditions, as we now report in our related papers.26,27
Author contributions
KC and ST performed the experiments, analysed the results and drafted the paper, LE, CR and JM supervised the research, analysed the data and edited the draft paper and JM conceived of the research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank AstraZeneca and the University of Strathclyde for funding a studentship to KFC and EPSRC and GlaxoSmithKline for funding an iCASE studentship to ST (grant number EP/W522260/1). The authors thank Craig Irving, Patricia Keating, Jessica Bame and Graeme Anderson for assistance with spectroscopy.References
- J. Canivet, J. Yamaguchi, I. Ban and K. Itami, Org. Lett., 2009, 11, 1733–1736 CrossRef CAS PubMed
.
- M. Li and R. Hua, Tetrahedron Lett., 2009, 50, 1478–1481 CrossRef CAS
- M. M. Heravi, Z. Kheilkordi, V. Zadsirjan, M. Heydari and M. Malmir, J. Organomet. Chem., 2018, 861, 17–104 CrossRef CAS
- R. Dorel, C. P. Grugel and A. M. Haydl, Angew. Chem., Int. Ed., 2019, 58, 17118–17129 CrossRef CAS PubMed
- M. M. Heravi, V. Zadsirjan, M. Malmir and L. Mohammadi, Monatsh. Chem., 2021, 152, 1127–1171 CrossRef CAS
- F. Vallée, J. J. Mousseau and A. B. Charette, J. Am. Chem. Soc., 2010, 132, 1514–1516 CrossRef PubMed
- Y. Huang, M. E. Moret and R. J. M. K. Gebbink, Eur. J. Org. Chem., 2014, 3788–3793 CrossRef CAS
- R. Zhang, C.-X. Miao, S. Wang, C. Xia and W. Sun, ChemCatChem, 2012, 4, 192–195 CrossRef CAS
- H. Li, C.-L. Sun, M. Yu, D.-G. Yu, B.-J. Li and Z.-J. Shi, Chem. – Eur. J., 2011, 17, 3593–3597 CrossRef CAS PubMed
- K. S. Bloome, R. L. McMahen and E. J. Alexanian, J. Am. Chem. Soc., 2011, 133, 20146–20148 CrossRef CAS PubMed
- P. Chuentragool, M. Parasram, Y. Shi and V. Gevorgyan, J. Am. Chem. Soc., 2018, 140, 2465–2468 CrossRef CAS PubMed
- P. Chuentragool, D. Kurandina and V. Gevorgyan, Angew. Chem., Int. Ed., 2019, 58, 11586–11598 CrossRef CAS PubMed
- W.-J. Zhou, G.-M. Cao, Z.-P. Zhang and D.-G. Yu, Chem. Lett., 2019, 48, 181–191 CrossRef CAS
- Q. Liu, X. Dong, J. Li, J. Xiao, Y. Dong and H. Liu, ACS Catal., 2015, 5, 6111–6137 CrossRef CAS
- D. P. Curran, T. M. Morgan, C. E. Schwartz, B. B. Snider and M. A. Dombroski, J. Am. Chem. Soc., 1991, 113, 6607–6617 CrossRef CAS
- For low-temperature measurement of EPR spectra of phenyl radicals in cryomatrices, see:
(a) F. P. Sargent and E. M. Gardy, J. Chem. Phys., 1977, 67, 1793–1795 CrossRef CAS
- D. G. H. Hetterscheid, J. Kaiser, E. Reijerse, T. P. J. Peters, S. Thewissen, A. N. J. Blok, J. M. M. Smits, R. de Gelder and B. de Bruin, J. Am. Chem. Soc., 2005, 127, 1895–1905 CrossRef CAS PubMed
- D. Isrow and B. Captain, Inorg. Chem., 2011, 50, 5864–5866 CrossRef CAS PubMed
- This paper appeared in preprint form K. F. Clark, S. Tyerman, L. Evans, C. M. Robertson and J. A. Murphy, ChemRxiv preprint 2023. https://chemrxiv.org/engage/chemrxiv/article-details/6460e271fb40f6b3ee94c5df.
- S. Yanagisawa, K. Ueda, T. Taniguchi and K. Itami, Org. Lett., 2008, 10, 4673–4676 CrossRef CAS PubMed
- A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2011, 50, 5018–5022 CrossRef CAS PubMed
- S. Zhou, G. M. Anderson, B. Mondal, E. Doni, V. Ironmonger, M. Kranz, T. Tuttle and J. A. Murphy, Chem. Sci., 2014, 5, 476–482 RSC
- S. Zhou, E. Doni, G. M. Anderson, R. G. Kane, S. W. MacDougall, V. M. Ironmonger, T. Tuttle and J. A. Murphy, J. Am. Chem. Soc., 2014, 136, 17818–17826 CrossRef CAS PubMed
- G. Nocera and J. A. Murphy, Synthesis, 2020, 52, 327–336 CrossRef CAS
- A. J. Smith, D. L. Poole and J. A. Murphy, Sci. China: Chem., 2019, 62, 1425–1438 CrossRef CAS
- K. F. Clark, S. Tyerman, L. Evans, C. M. Robertson, D. J. Nelson, A. R. Kennedy and J. A. Murphy, J. Am. Chem. Soc., 2023, 145, 20849–20858 CrossRef CAS PubMed
- See accompanying paper, Org. Biomol. Chem., 2024 10.1039/d3ob01745a
- C.-L. Sun, H. Li, D.-G. Yu, M. Yu, X. Zhou, X.-Y. Lu, K. Huang, S.-F. Zheng, B.-J. Li and Z.-J. Shi, Nat. Chem., 2010, 2, 1044–1049 CrossRef CAS PubMed
- E. Shirakawa, K. I. Itoh, T. Higashino and T. Hayashi, J. Am. Chem. Soc., 2010, 132, 15537–15539 CrossRef CAS PubMed
- H. Yi, A. Jutand and A. Lei, Chem. Commun., 2015, 51, 545–548 RSC
- In these experiments, phenanthroline derivatives are not detected by GCMS. We have reported22 a study of the effect of KOtBu on phenanthroline which, under optimal conditions, led to the isolation of dimer in 38% yield. In fact, this is the basis of the electron transfer nature of KOtBu + phenanthroline. The initial dimeric anionic structure is very electron-rich and, by behaving as an electron donor, forms the neutral dimer that we isolated. This dimer is then susceptible to further reaction in situ, leading to oligomers and polymers.
- A point about Table 2, entry 2 that deserves comment relates to mass balance. In this experiment under deuterium conditions (and the same holds for Table 2, entries 3 and 5, which are the other experiments under deuterium conditions), the mass balance comes to approximately 75%, whereas with the non-deuterated experiments, the mass balance approaches 100%. This means that more xylyl radicals are diverted to undetected products in the deuterated experiments. Our prior study of the reaction of KOtBu with phenanthroline22 showed that base-induced dimerisation and oligomerisation of phenanthroline occur. Indeed, the isolated electron donor in that study derived from a dimerisation of phenanthroline. These higher order compounds do not appear on GCMS traces. As the reactions progress, the concentrations of the oligomeric phenanthrolines rise and, as polycyclic aromatic structures, these may be more susceptible to addition by xylyl radicals, and, with extended conjugation, they should be more susceptible compared to phenanthroline itself. Our lower mass balances in experiments with C6D6 would be explained by this difference in reaction speed being responsible for the greater loss of xylyl radicals in the deuterated conditions due to trapping by these oligomers.
- However, we cannot state definitively whether Ar–Me groups is the exclusive source of H atom transfer to xylyl radicals. As an alternative, we contemplated H/D-atom transfer reaction to xylyl radical 9 from phenanthroline, although the C–H/D bonds in phenanthroline should be a lot stronger than the C–H/D of the xylyl methyl groups. It turns out that KOtBu, acting as a base, promotes H+/D+ transfer between phenanthroline and ArMe groups. We demonstrated this through D+ exchange between mesitylene-d9 and phenanthroline, as well as between perdeuterated phenanthroline and the methyl groups of mesitylene (see ESI‡). Accordingly, efforts to distinguish between phenanthroline and xylyl methyl groups being involved in quenching of xylyl radicals 9, cannot rely on the use of deuterated precursors.
Footnotes |
| † This paper is dedicated to Professor Shigeru Yamago on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ob01743e |
| This journal is © The Royal Society of Chemistry 2024 |