
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydrogenation of functionalised pyridines with a rhodium oxide catalyst under mild conditions†
Sydney
Williams
 a,
Leiming
Qi
a,
Robert J.
Cox
b,
Prashant
Kumar
a and
Jianliang
Xiao
*a
a,
Leiming
Qi
a,
Robert J.
Cox
b,
Prashant
Kumar
a and
Jianliang
Xiao
*a
aDepartment of Chemistry, University of Liverpool, Crown Street, L69 7ZD, Liverpool, UK. E-mail: jxiao@liverpool.ac.uk
bChemical Development, AstraZeneca, Silk Road Business Park, SK10 2NA, Macclesfield, UK
First published on 3rd January 2024
Abstract
Piperidines are one of the most widely used building blocks in the synthesis of pharmaceutical and agrochemical compounds. The hydrogenation of pyridines is a convenient method to synthesise such compounds as it only requires reactant, catalyst, and a hydrogen source. However, this reaction still remains difficult for the reduction of functionalised and multi-substituted pyridines. Here we report the use of a stable, commercially available rhodium compound, Rh2O3, for the reduction of various unprotected pyridines. The reaction only requires mild conditions, and the substrate scope is broad, making it practically useful.
Introduction
Piperidines are saturated heterocyclic amine compounds and have been identified as one of the most common heterocyclic structures present in FDA approved drugs.1–3 These biologically active compounds can be synthesised or occur naturally. Examples of naturally occurring, biologically active piperidine-based compounds are shown in Fig. 1. Piperine and pseudodistomin A are derived from black pepper and marine life, respectively.2,4 Examples of active drugs that contain a piperidine functional group are also shown in Fig. 1. Mesoridazine is a neuroleptic drug used in the treatment of schizophrenia, while raloxifene is used for the treatment of osteoporosis in post-menopausal-women.5,6 Paroxetine is an antidepressant that contains an unprotected piperidine.7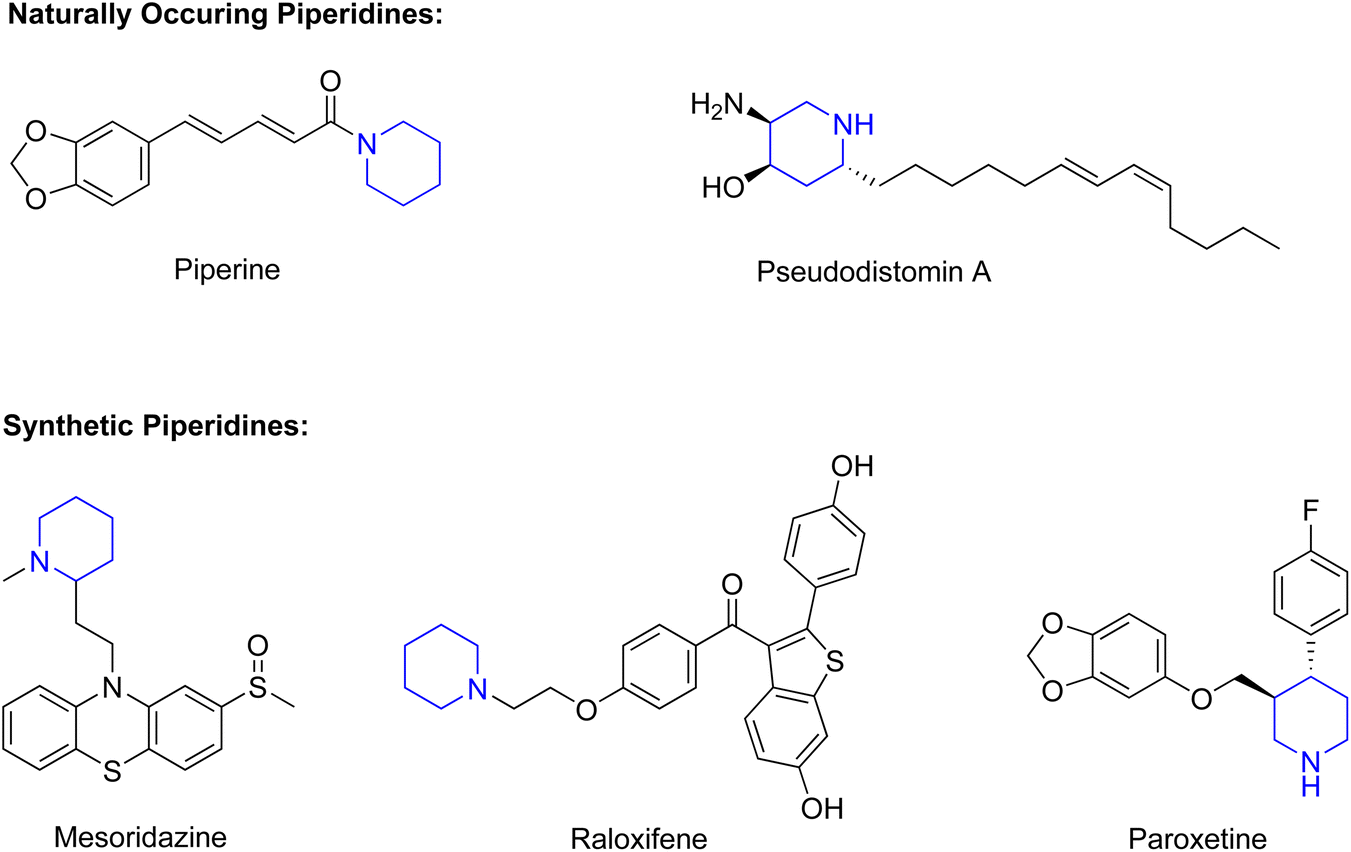 | ||
| Fig. 1 Examples of biologically active compounds that contain a piperidine unit. | ||
There are multiple approaches for the synthesis of piperidines. Examples include cycloadditions, reductive amination, nucleophilic substitutions, radical cyclisation and reduction of pyridines.8–21 In principle, hydrogenation is a favourable method as it only requires a pyridine substrate, a catalyst and a hydrogen source. However, this remains a challenging reaction and the current reported catalytic methods tend to require harsh conditions and are generally substrate specific.17–19
Hydrogenation of N-heterocycles has been extensively studied using a variety of hydrogen sources and catalysts.18–21 Although using molecular hydrogen carries risks with the use of specialised high pressure equipment, previous research has shown that this type of hydrogenation can effectively reduce activated N-heterocycles with both high yield and high stereoselectivity.21 Issues can arise when reducing unactivated or neutral N-heterocycles, such as pyridines, as the nitrogen can bind to the metal centre and poison the catalyst in addition to the reduced reactivity of the ring toward metal hydride.16 These issues can be alleviated by creating a quarternised pyridinium salt.22,23 Although the hydrogenation of unprotected pyridines is less common with homogeneous catalysts, the use of heterogenous catalysts have been reported.10–12,18,24–29 Some recent examples are shown in Fig. 2. The most commonly used catalysts include palladium on carbon, rhodium on carbon and platinum oxide.12,27–29 However, these reactions tend to require harsh conditions, high catalyst loading and/or long reaction times with varying degrees of success. The substrate scope also tends to be narrow, featuring few functional groups.
 | ||
| Fig. 2 Recent work on the hydrogenation of pyridines. | ||
Herein we report the use of rhodium(III) oxide (Rh2O3) for the hydrogenation of a wide range of unprotected pyridines with H2 under mild conditions using low catalyst loading. Rh2O3 is one of the commercially most easily available rhodium compounds, and it is stable and easy to handle and store. Whilst rhodium compounds are one of the most widely used catalysts in hydrogenation, it is surprising somewhat that Rh2O3 has rarely been explored in any catalytic reactions, and our literature search found no reported use of Rh2O3 for the hydrogenation of pyridines. Only a few studies of using rhodium oxide as catalysts are available, e.g. for the reduction of carbon monoxide and the hydroformylation of alkenes.30–32
Results and discussion
Previous work within our group has involved the use of rhodium pincer complexes for the reduction of aromatic rings.33 It soon became apparent that the hydrogenation relied on the transformation of the pincer complex to an insoluble black precipitate, suggesting the formation of an active heterogeneous catalyst, as is often observed with other molecular rhodium complexes.10 We therefore decided to screen a range of commercially available heterogeneous rhodium compounds, as the pincer complexes could not reduce functionalised pyridines. When using the conditions previously established within our group, we found three catalysts that were active, completely reducing 2,6-lutidine (1e) to the corresponding piperidine (2e) in hexafluoroisopropanol (HFIP). These catalysts were rhodium on carbon, rhodium on alumina and rhodium oxide (Table 1, entries 1–3). Although Rh/C and Rh/Al2O3 have already been established as catalysts for the hydrogenation of pyridines,18,28 we were surprised to find Rh2O3 was also active and with slightly higher diastereoselectivity (entry 3), as there are no reports of using commercially available Rh2O3 for the hydrogenation of pyridines. Considering the harsh conditions usually seen in reduction with other catalysts (e.g.Fig. 2) and the dearth of studies of Rh2O3 as hydrogenation catalyst, we therefore decided to focus on Rh2O3 and optimise the reaction conditions.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | H2 pressure (bar) | Temperature (°C) | Conversionb (%) |
cis![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :trans :trans |
| a Reaction conditions (unless otherwise stated): 2,6-lutidine (0.79 mmol.) and catalyst in solvent (1 mL). b Conversion determined by 1H NMR spectroscopy using an internal standard (1,3,5-trimethoxybenzene). c Reaction time 1.5 hours. | ||||||
| 1 | Rh/C (2 mol%) | HFIP | 50 | 45 | 100 | 96:4 |
| 2 | Rh/Al2O3 (2 mol%) | HFIP | 50 | 45 | 100 | 96:4 |
| 3 | Rh2O3 (1 mol%) | HFIP | 50 | 45 | 100 | 97:3 |
| 4 | Rh2O3 (1 mol%) | HFIP | 10 | 45 | 100 | 95:5 |
| 5 | Rh2O3 (1 mol%) | H2O | 10 | 45 | 0 | N/A |
| 6 | Rh2O3 (1 mol%) | THF | 10 | 45 | 0 | N/A |
| 7 | Rh2O3 (1 mol%) | DCE | 10 | 45 | 17 | N.D |
| 7 | Rh2O3 (1 mol%) | 1,4-Dioxane | 10 | 45 | 0 | N/A |
| 9 | Rh2O3 (1 mol%) | Cyclohexane | 10 | 45 | 7 | N.D |
| 10 | Rh2O3 (1 mol%) | MeOH | 10 | 45 | 100 | 94:6 |
| 11 | Rh2O3 (1 mol%) | TFE | 10 | 45 | 100 | 96:4 |
| 12 | Rh2O3 (1 mol%) | TFE | 5 | 45 | 100 | 94:6 |
| 13 | Rh2O3 (1 mol%) | MeOH | 5 | 45 | 14 | N.D |
| 14 | Rh2O3 (1 mol%) | TFE | 1 | 45 | 6 | N.D |
| 15 | Rh2O3 (1 mol%) | TFE | 5 | 40 | 100 | 97:3 |
| 16 | Rh2O3 (0.5 mol%) | TFE | 5 | 40 | 100 | 97:3 |
| 17 | Rh2O3 (0.5 mol%)c | TFE | 5 | 40 | 60 | 97:3 |
| 18 | Nishimura‘s catalyst (1 mol% Rh)c | TFE | 5 | 40 | 54 | 98:2 |

Results from the optimisation are also shown in Table 1, testing a variety of parameters, such as solvent, hydrogen pressure, and temperature. Reducing the pressure from 50 bar to 10 bar did not affect the reaction yield (entry 4). A number of other solvents were screened (entries 5–11), among which trifluoroethanol (TFE) showed the best result in terms of activity and selectivity (entry 11). Reducing the pressure to 5 bar did not affect the activity but there was a slight reduction in the selectivity between cis and trans isomers (entry 12). Although the reaction in methanol also showed excellent activity at 10 bar (entry 10), the drop to 5 bar dramatically reduced the activity (entry 13). It was also possible to reduce the temperature and catalyst loading to 40 °C and 0.5 mol%, respectively (entry 16). We compared Rh2O3 to the commercially available Nishimura‘s Catalyst, which is a combination of Rh2O3 and PtO2 and has been reported for the hydrogenation of aromatic compounds.34–37 Interestingly, when using the same amount of rhodium, Rh2O3 outperformed the Nishimura‘s Catalyst in activity (entries 17 and 18).34 The conditions in entry 16 represent the now optimised reaction conditions for the substrate scope investigations, i.e. 5 bar of H2 with 0.5 mol% of Rh2O3 at 40 °C in TFE.
We went on to examine the substrate scope of the hydrogenation. Although the reduction of 2,6-lutidine (1e) showed 100% conversion within 3 hours, some substrates with substituents at the 3 and 4 positions required a longer time. It was therefore decided that it was more efficient to expand the substrate scope using an overnight reaction. Due to the volatility of most piperidines, we used 1H NMR in the presence of an internal standard to determine the product yield. However, a few compounds were isolated, aimed to validate the NMR yield. The optimal conditions were highly successful in reducing a variety of alkyl pyridines, as shown in Fig. 3. Notably, the reaction was also successful in reducing pyridines with bulky groups that sterically hinder the nitrogen, e.g. 2,4,6-trimethylpyridine (1k) and 2,6-di-tert-butylpyridine (1m). The majority of multisubstituted pyridines were reduced to give mainly the cis product, which is expected for heterogeneous arene hydrogenation (see ESI† for details).38 The diastereoselectivity ratio is included in Fig. 3 for the multisubstituted piperidines that exhibit both cis and trans isomers. The trans isomer would be expected to be the thermodynamically favoured configuration for 2,3- and 2,5-dimethylpiperidine (2f, h) when considering the possible chair conformations of disubstituted piperidines. This may explain why the selectivity is lower when compared to 2,6- (2e) and 2,4-dimethylpiperidine (2g) and indicates that thermodynamics plays a role in determining the product selectivity.39 Although a variety of multi-substituted alkyl pyridines were fully reduced, a lower yield was encountered when the fourth position is sterically hindered, as observed in the case of 3,4- and 3,5-lutidine (2i–j) and 4-tert-butylpyridine (c.f.2r and 2q). This provides some insight into the potential mechanism of the reaction, which was investigated further (vide infra). An issue of chemoselectivity was observed when reducing 2-vinylpyridine (1p) and 4-vinylpyridine (1q), where the alkene was also reduced.
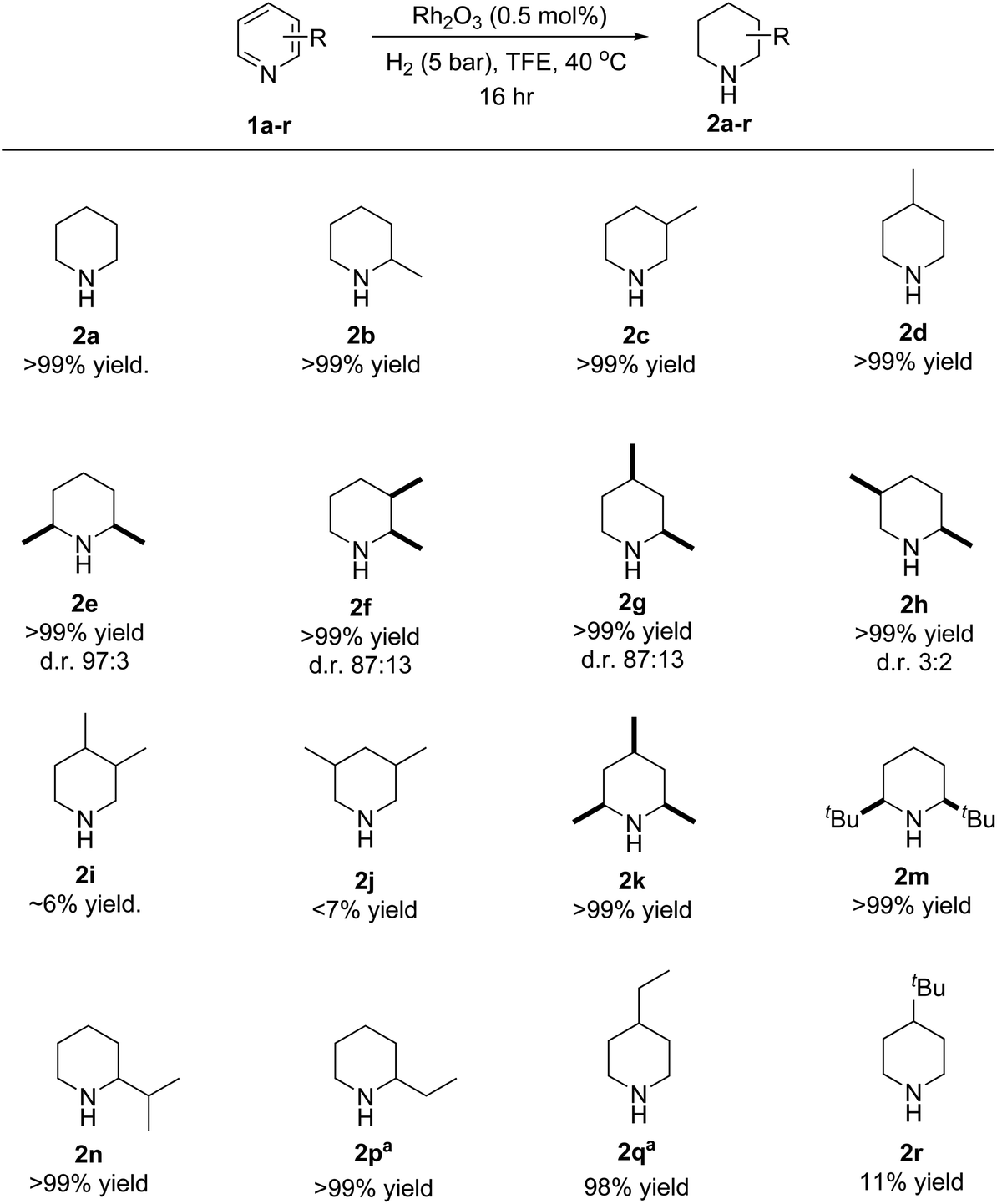 | ||
| Fig. 3 Hydrogenation of alkyl pyridines with Rh2O3 catalyst. Reaction conditions: substrate (0.8 mmol.), Rh2O3 (1 mg, 0.5 mol%) and TFE (1 mL) with molecular hydrogen (5 bar) at 40 °C for 16 hours. Yield determined using NMR spectroscopy with an internal standard (1,3,5-trimethoxybenzene; a yield of >99% is assigned when the substrate is invisible in the spectrum). aSubstrate R group is an alkene. | ||
The hydrogenation of pyridines with alcohol groups was also achieved, shown in Fig. 4. The optimised conditions were effective in reducing pyridines with alcohol groups attached directly to the ring but also separated by a carbon chain. It was found that the hydrogenation of 2-hydroxypyridines (3a–c) formed δ-lactams rather than 2-hydroxypiperidines. This was expected as similar occurrences have been reported in the reduction of 2-hydroxypyridine as a result of amide-iminol tautomerization.27,40,41 The reduction of 2-pyridinemethanol (3e) was achieved with a lower yield (76%). This is believed to be caused by possible coordination of the pyridine or the product to the rhodium metal to give 5-membered ring metal species, poisoning the catalyst. An increase in time from 16 to 24 hours did not improve the conversion, neither did an increase in pressure to 10 bar. Further supporting our surmise, 3-pyridinemethanol gave 86% conversion (4f), with the rest of the product subject to an elimination reaction to afford 3-methylpiperidine (2c), and pyridines 3g and 3h were fully reduced during the reaction to give the desired products 4g and 4h. These results indicate that it is not the alcohol functional group itself that affects the hydrogenation.
 | ||
| Fig. 4 Hydrogenation of alcohol pyridines with Rh2O3 catalyst. Reaction conditions: substrate (0.8 mmol.), Rh2O3 (1 mg, 0.5 mol%) and TFE (1 mL) with molecular hydrogen (5 bar) at 40 °C for 16 hours. Yield determined using NMR spectroscopy with an internal standard (1,3,5-trimethoxybenzene). Isolated yield for 4b in parentheses. aSubstrate R group is a hydroxyl group. | ||
It was also possible to reduce pyridines with primary and tertiary amines at the 2-position (Fig. 5). For the hydrogenation of substrates with an amino unit at the 2-position, 1H NMR indicated the formation of 3,4,5,6-tetrahydopyridin-2-amines, rather than 2-aminopiperidines. This observation for 2-aminopyridine (5a) is expected as the substrate exhibits tautomerism and the resulting 3,4,5,6-tetrahydopyridin-2-amine (6a) product shows some resistance towards further reduction under the slightly acidic conditions (pKa of TFE: 12.5).42,43 The same is true with pyridines 5b and 5c. These observations are also reported within the literature regarding tertiary amine at the second position of the pyridine,44,45 and are reminiscent of that observed with 3a. However, an elimination side reaction was observed when there was an amine attached to the pyridine ring (e.g.6a and 6b);46 improved yields were obtained at a shorter reaction time. In the case of 2-picolylamine (5d) the conversion is low. This is similar to the reduction of 2-pyridine methanol (3e) and the lower conversion can be explained by the stronger coordinating capability of a free amine. In line with this, complete reduction of Boc-protected 2-picolylamine (5e) was achieved. Unfortunately, 2-nicotinonitrile was not tolerated and the 4-dimethylamino-substituted pyridine (5f) afforded only a low yield, reminiscent of 1r.
 | ||
| Fig. 5 Hydrogenation of amine-functionalised pyridines with Rh2O3 catalyst. Reaction conditions: substrate (0.8 mmol.), Rh2O3 (1 mg, 0.5 mol%) and TFE (1 mL) with molecular hydrogen (5 bar) at 40 °C for 16 hours. Yield determined using NMR spectroscopy with an internal standard (1,3,5-trimethoxybenzene). Isolated yield for 6e in parentheses. | ||
The reaction was successful in reducing the pyridine ring when a carboxylic acid, ester or amide is directly attached to the ring (Fig. 6). Both 2- and 3-substituted carbonyl compounds were well tolerated, and di-substituted carbonyl compounds also showed excellent activity to give the corresponding cis product as the major isomer. Pipecolic acid (8a), piperidine-3-carboxamide (8g), 5-(methoxycarbonyl)piperidine-2-carboxylic acid (8h) and dimethyl-piperidine-2,6-dicarboxylate (8j) were isolated with excellent yield, supporting the use of an NMR internal standard to determine yield. There was an issue with the chemoselectivity when reducing 2-acetylpyridine (7d), in which the ketone was also reduced along with the pyridine ring to give 8d.
 | ||
| Fig. 6 Hydrogenation of carbonyl pyridines with Rh2O3 catalyst. Reaction conditions: substrate (0.8 mmol.), Rh2O3 (1 mg, 0.5 mol%) and TFE (1 mL) with molecular hydrogen (5 bar) at 40 °C for 16 hours. Yield determined using NMR spectroscopy with an internal standard (1,3,5-trimethoxybenzene or maleic acid). Isolated yield for 8a, 8g and 8j in parentheses. aSubstrate is 2-acetylpyridine. bSubstrate is 6-hydroxypyridine-2-carboxylic acid. | ||
Poor reduction of phenyl pyridines was observed when the benzene ring is directly attached to the pyridine (Fig. 7). This has been previously seen in the literature.47,48 We were unsure whether this was caused by issues with the individual substrate or catalyst poisoning. As there is low conversion for both 2-phenylpyridine (9a) and 3-phenylpyridine (9b), the issue does not appear to be caused by potential coordination to the rhodium via e.g. C–H activation. The issue could be due to interactions of the aromatic rings with the rhodium atoms, as 2- and 3-phenylpyridines are planar molecules. This is to some degree supported by the complete reduction of 2-benzylpyridine 9d and 2-benzoylpyridine 9c. Although 9c is also a planar molecule, the molecule would be non-planar if the ketone is reduced first. We also attempted the hydrogenation of halogenated pyridines. Unfortunately, the hydrogenation conditions employed resulted in dehalogenation, regardless of halogen or position. This has also been observed within the literature.40,44,45,49
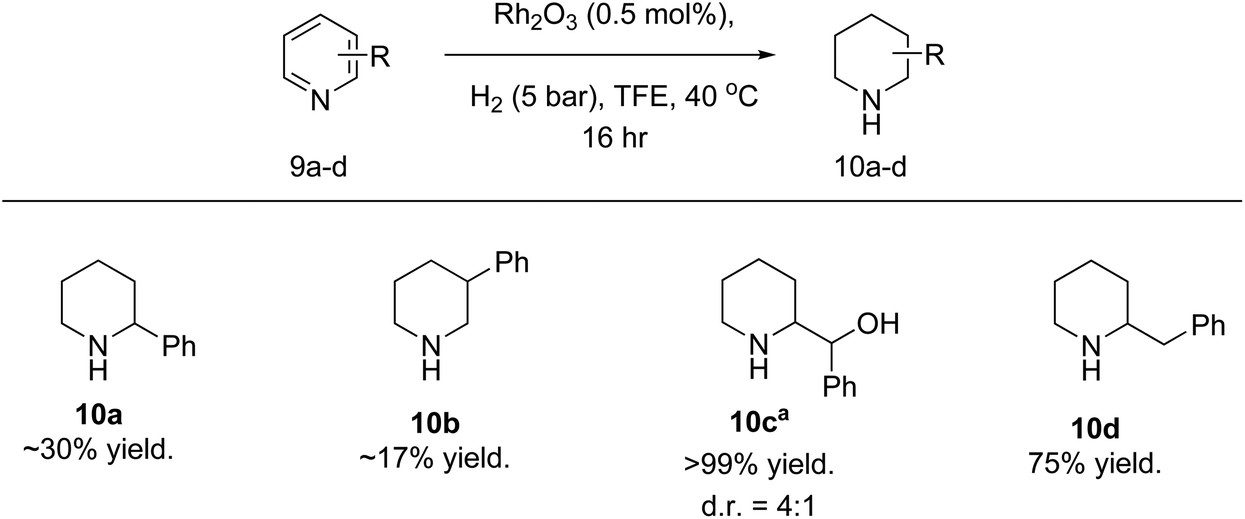 | ||
| Fig. 7 Hydrogenation of phenyl pyridines using Rh2O3 catalyst. Reaction conditions: substrate (0.8 mmol.), Rh2O3 (1 mg, 0.5 mol%) and TFE (1 mL). Yield determined using NMR spectroscopy with an internal standard (1,3,5-trimethoxybenzene). aSubstrate is 2-benzoylpyridine. | ||
The reduction of pyridines where the 4th position was sterically hindered was difficult, evident from the low yields of 2i, 2j and 2r. This is an indication that the reduction may proceed via hydrogen addition at the 4th position. To shed more light on this conjecture, a few 4-substituted pyridines with different electronic effects were examined in a short reaction time (Fig. 8). As expected, the reduction of 4-methoxypyridine (11a) gave the desired product (12a) in a low yield, presumably because the 4th position has a strong electron donor which would disfavour the hydride addition. When a less electron-donating methyl group was at the same position, a higher product yield was observed (2d), and more remarkably, the electron withdrawing trifluoromethyl analogue afforded full conversion (12b), although sterically 11a is less demanding. Together with the observations made with 1m and 1r, these results suggest that the hydrogenation starts with hydrogen transfer to the 4th position of pyridines. This is common with the reduction of pyridines, pyridinium salts and quinolines under the conditions of homogeneous catalysis.21
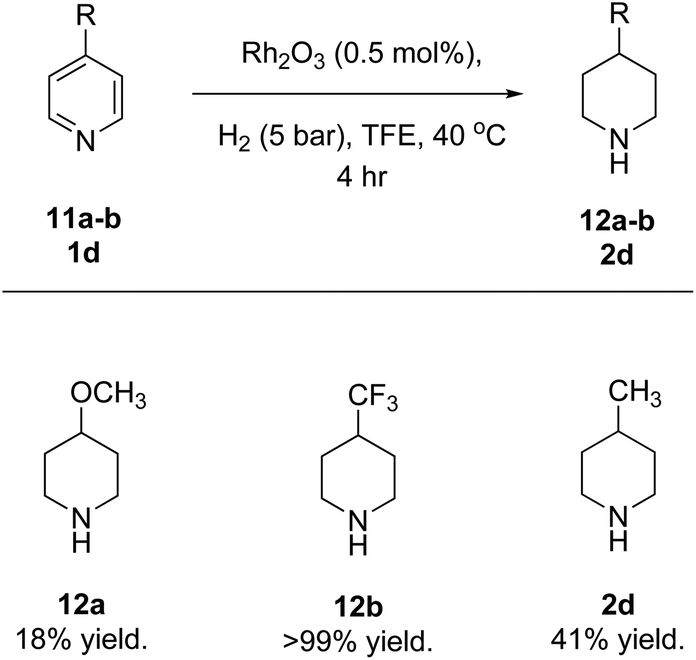 | ||
| Fig. 8 Hydrogenation of pyridines with an EDG or EWG at the 4-position. Reaction conditions: substrate (0.8 mmol.), Rh2O3 (1 mg, 0.5 mol%) and TFE (1 mL) with molecular hydrogen (5 bar) at 40 °C for 4 hours. Yield determined using NMR spectroscopy with an internal standard (1,3,5-trimethoxybenzene). | ||
Conclusion
We have identified the commercially available rhodium oxide to be a highly active catalyst for the hydrogenation of a wide variety of unprotected pyridines under mild conditions. The optimal conditions tolerate a number of functional groups, including alcohols, amines and carbonyls. The reaction can also reduce multisubstituted pyridines to give the corresponding cis piperidines as the major product. Issues can arise with chemoselectivity when the functional group attached to the pyridine ring can also be reduced, for example olefins, nitro groups and ketones, which were also reduced during the reaction. We suspect that the hydrogenation proceeds via initial hydride transfer to the 4th position of pyridine ring, as electron donating and/or sterically bulky groups hinder the reaction. Study is ongoing in our lab to explore further the potential of this easily available, easy-to-handle catalyst.Experimental section
A pyridine (0.79 mmol) and rhodium(III) oxide (1.0 mg, 0.5 mol%) were added to a glass vial equipped with a stirrer bar and degassed. Trifluoroethanol (1 mL) was introduced and the mixture briefly flushed with nitrogen. The vial was placed in an autoclave and purged with hydrogen three times. Once the autoclave had been charged with hydrogen gas (5 bar), the reaction mixture was heated to 40 °C and stirred for the allocated amount of time. The autoclave was removed from the heat source and the pressure carefully released in a fumehood at room temperature. An internal standard (1,3,5-trimethoxybenzene, 0.33 equiv. or maleic acid, 0.5 equiv.) was added to the crude mixture, which was then partially concentrated in vacuo. CD3Cl was added to the mixture which was filtered through Celite prior to NMR analysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the EPSRC/AstraZeneca for financial support (PhD studentship for SW). We also thank the China Scholarship Council (CSC)/University of Liverpool for a PhD scholarship (LQ), the Royal Society (UK) for an International Collaboration Award (IC170044) and financial support (PK), and the Analytical Services of the Department of Chemistry of the University of Liverpool for product analysis.References
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- M. M. Abdelshaheed, I. M. Fawzy, H. I. El-Subbagh and K. M. Youssef, Future J. Pharm. Sci., 2021, 7, 188 CrossRef.
- P. Goel, O. Alam, M. J. Naim, F. Nawaz, M. Iqbal and M. I. Alam, Eur. J. Med. Chem., 2018, 157, 480–502 CrossRef CAS PubMed.
- A. J. Freyer, A. D. Patil, L. Killmer, N. Troupe, M. Mentzer, B. Carte, L. Faucette and R. K. Johnson, J. Nat. Prod., 1997, 60, 986–990 CrossRef CAS PubMed.
- J. R. Bagley, L. V. Kudzma, N. L. Lalinde, J. A. Colapret, B. S. Huang, B. S. Lin, T. P. Jerussi, M. J. Benvenga, B. M. Doorley, M. H. Ossipov, T. C. Spaulding, H. K. Spencer, F. G. Rudo and R. L. Wynn, Med. Res. Rev., 1991, 11, 403–436 CrossRef CAS PubMed.
- T. Elavarasan, D. P. Bhakiaraj and M. Gopalakrishnan, ISRN Org. Chem., 2014, 2014, 1–9 CrossRef PubMed.
- M. S. Yu, I. Lantos, Z. Q. Peng, J. Yu and T. Cacchio, Tetrahedron Lett., 2000, 41, 5647–5651 CrossRef CAS.
- M. G. P. Buffat, Tetrahedron, 2004, 60, 1701–1729 CrossRef CAS.
- F. X. Felpin and J. Lebreton, Eur. J. Org. Chem., 2003, 3693–3712 CrossRef CAS.
- S. Kim, F. Loose, M. J. Bezdek, X. Wang and P. J. Chirik, J. Am. Chem. Soc., 2019, 141, 17900–17908 CrossRef CAS PubMed.
- K. Murugesan, V. G. Chandrashekhar, C. Kreyenschulte, M. Beller and R. V. Jagadeesh, Angew. Chem., Int. Ed., 2020, 59, 17408–17412 CrossRef CAS PubMed.
- R. Sreenivasulu, K. V. S. Ranganath and R. R. Raju, Asian J. Chem., 2015, 27, 4358–4360 CrossRef CAS.
- L. Lückemeier, M. Pierau and F. Glorius, Chem. Soc. Rev., 2023, 4996–5012 RSC.
- L. Hegedus, V. Háda, A. Tungler, T. Máthé and L. Szepesy, Appl. Catal., A, 2000, 201, 107–114 CrossRef CAS.
- B. Barwinski, P. Migowski, F. Gallou, G. Franciò and W. Leitner, J. Flow Chem., 2017, 7, 41–45 CrossRef CAS.
- F. Glorius, Org. Biomol. Chem., 2005, 3, 4171–4175 RSC.
- Y. G. Zhou, Acc. Chem. Res., 2007, 40, 1357–1366 CrossRef CAS PubMed.
- A. Gualandi and D. Savoia, RSC Adv., 2016, 6, 18419–18451 RSC.
- R. Gunasekar, R. L. Goodyear, I. Proietti Silvestri and J. Xiao, Org. Biomol. Chem., 2022, 20, 1794–1827 RSC.
- A. N. Kim and B. M. Stoltz, ACS Catal., 2020, 10, 13834–13851 CrossRef CAS PubMed.
- D. S. Wang, Q. A. Chen, S. M. Lu and Y. G. Zhou, Chem. Rev., 2012, 112, 2557–2590 CrossRef CAS PubMed.
- B. Balakrishna, J. L. Núñez-Rico and A. Vidal-Ferran, Eur. J. Org. Chem., 2015, 5293–5303 CrossRef CAS.
- J. Wu, Z. Chen, J. H. Barnard, R. Gunasekar, C. Pu, X. Wu, S. Zhang, J. Ruan and J. Xiao, Nat. Catal., 2022, 5, 982–992 CrossRef CAS.
- A. Kumar, V. Goyal, N. Sarki, B. Singh, A. Ray, T. Bhaskar, A. Bordoloi, A. Narani and K. Natte, ACS Sustainable Chem. Eng., 2020, 8, 15740–15754 CrossRef CAS.
- T. Wagener, A. Heusler, Z. Nairoukh, K. Bergander, C. G. Daniliuc and F. Glorius, ACS Catal., 2020, 10, 12052–12057 CrossRef CAS PubMed.
- N. Tanaka and T. Usuki, Eur. J. Org. Chem., 2020, 2020, 5514–5522 CrossRef CAS.
- C. Cheng, J. Xu, R. Zhu, L. Xing, X. Wang and Y. Hu, Tetrahedron, 2009, 65, 8538–8541 CrossRef CAS.
- M. Freifelder, R. M. Robinson and G. R. Stone, J. Org. Chem., 1962, 27, 284–286 CrossRef CAS.
- G. van der Heijden, T. B. van Schaik, V. Mouarrawis, M. J. M. de Wit, C. M. L. V. Velde, E. Ruijter and R. V. A. Orru, Eur. J. Org. Chem., 2019, 2019, 5313–5325 CrossRef CAS.
- P. R. Watson and G. A. Somorjai, J. Catal., 1981, 72, 347–363 CrossRef CAS.
- J. Gustafson, R. Westerström, A. Resta, A. Mikkelsen, J. N. Andersen, O. Balmes, X. Torrelles, M. Schmid, P. Varga, B. Hammer, G. Kresse, C. J. Baddeley and E. Lundgren, Catal. Today, 2009, 145, 227–235 CrossRef CAS.
- C. Botteghi and P. Pino, Org. Synth., 1977, 57, 11 CrossRef.
- P. Kumar, L. Qi, S. Williams, R. A. Dop, Y. Liu, T. Zhang, C. Li and J. Xiao, J. Organomet. Chem., 2023, 997, 122795 CrossRef CAS.
- S. Nishimura, Bull. Chem. Soc. Jpn., 1960, 33, 566–567 CrossRef CAS.
- S. Nishimura, Bull. Chem. Soc. Jpn., 1961, 34, 10–27 Search PubMed.
- S. Nishimura, Bull. Chem. Soc. Jpn., 1961, 34, 32–36 CrossRef CAS.
- A. Kaithal, H. S. Sasmal, S. Dutta, F. Schäfer, L. Schlichter and F. Glorius, J. Am. Chem. Soc., 2023, 145, 4109–4118 CrossRef CAS PubMed.
- M. Wollenburg, A. Heusler, K. Bergander and F. Glorius, ACS Catal., 2020, 10, 11365–11370 CrossRef CAS PubMed.
- Y. Takeuchi, H. Suzuki, K. Ogawa and Y. Nomura, J. Am. Chem. Soc., 1984, 106, 831–841 CrossRef.
- T. Wagener, L. Lückemeier, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2021, 60, 6425–6429 CrossRef CAS.
- Y. Wei, B. Rao, X. Cong and X. Zeng, J. Am. Chem. Soc., 2015, 137, 9250–9253 CrossRef CAS PubMed.
- T. Grave, J. Am. Chem. Soc., 1924, 46, 1460–1470 CrossRef.
- A. D. Dwivedi, R. K. Rai, K. Gupta and S. K. Singh, ChemCatChem, 2017, 9, 1930–1938 CrossRef CAS.
- F. Berardi, N. A. Colabufo, G. Giudice, R. Perrone, V. Tortorella, S. Govoni and L. Lucchi, J. Med. Chem., 1996, 39, 176–182 CrossRef CAS PubMed.
- V. Smout, A. Peschiulli, S. Verbeeck, E. A. Mitchell, W. Herrebout, P. Bultinck, C. M. L. Vande Velde, D. Berthelot, L. Meerpoel and B. U. W. Maes, J. Org. Chem., 2013, 78, 9803–9814 CrossRef CAS PubMed.
- Y. Kamochi and T. Kudo, Heterocycles, 1993, 36, 2383–2396 CrossRef CAS.
- R. A. Murphy, A. Y. Chen, S. K. Nair, G. M. Gallego, N. W. Sach and G. Smith, Tetrahedron Lett., 2016, 57, 5588–5591 CrossRef CAS.
- F. Martinez-Espinar, P. Blondeau, P. Nolis, B. Chaudret, C. Claver, S. Castillón and C. Godard, J. Catal., 2017, 354, 113–127 CrossRef CAS.
- H. Prokopcová, S. D. Bergman, K. Aelvoet, V. Smout, W. Herrebout, B. Van Der Veken, L. Meerpoel and B. U. W. Maes, Chem. – Eur. J., 2010, 16, 13063–13067 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ob01860a |
| This journal is © The Royal Society of Chemistry 2024 |