
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Radical coupling of aryl halides to arenes facilitated by Ni(COD)(DQ) and other nickel sources†‡
Seb
Tyerman
a,
Craig M.
Robertson
b and
John A.
Murphy
 *a
*a
aDepartment of Pure and Applied Chemistry, University of Strathclyde, 295 Cathedral Street, Glasgow G1 1XL, UK. E-mail: John.murphy@strath.ac.uk
bGSK Medicines Research Centre, Gunnels Wood Road, Stevenage, Herts SG1 2NY, UK
First published on 23rd December 2023
Abstract
The air-stable complex Ni(COD)(DQ) (COD = 1,5-cyclooctadiene, DQ = duroquinone) promotes the coupling of aryl halides to arenes in the presence of KOtBu. This complex has recently been shown to perform coupling reactions based on organonickel intermediates, but in this case the coupling reactions proceed via aryl radicals as shown by our newly developed assay for aryl radicals. Coupling with this nickel source is more efficient than with Ni(COD)2, Ni(PPh3)4 and Ni(acac)2, all of which we also show to operate through aryl radical pathways.
Introduction
Nickel complexes have been used to couple aryl halides with arenes through C–H functionalisation of the arene. (For reviews, see references.1–3) Itami coupled azoles to haloarenes with Ni(OAc)2 and 2,2′-bipyridine or dppf in the presence of LiOtBu4,5 proposing a standard mechanism involving a Ni(0)/Ni(II) cycle and organonickel intermediates. Simultaneously, Miura coupled azoles to haloarenes with NiBr2 in the presence of phenanthroline, LiOtBu and zinc powder and also proposed a classic Ni(0)/Ni(II) pathway (Scheme 1).6 | ||
| Scheme 1 Cross-coupling between aryl halides and unactivated arenes, facilitated by nickel salts and complexes. | ||
Meanwhile, in 2009 Yamakawa et al. used Cp2Ni (Cp = cyclopentadienyl), BEt3 and KOtBu to couple iodo- and bromoarenes with unactivated arenes such as benzene.7 With naphthalene or pyridine as the arene, the product regioisomer ratios suggested a complicated reaction mechanism, as acknowledged by the authors. In 2011 Mao et al. utilised Ni(OAc)2, 1,10-phenanthroline and KOtBu to carry out similar transformations and cited evidence for a radical mechanism.8 However, prior and subsequent reports showed that 1,10-phenanthrolines form strong electron donors in situ with KOtBu9–16 in the absence of nickel salts, and these electron donors are known to initiate coupling of haloarenes with arenes via base-assisted homolytic aromatic substitution (BHAS).17–19 In support of this, Mao reported a reaction (4-iodoanisole coupling with toluene in the presence of KOtBu (2 eq.) at 110 °C for 24 h) in the absence of Ni(OAc)2 that gave only slightly decreased yield of 37% [vs. 54% when Ni(OAc)2 was added]. Since this coupling proceeds in the absence of a nickel source, the importance and role of nickel needs clarification. Thus, the nickel-promoted mechanism of coupling of haloarenes to arenes like benzene has been unclear for some time. As the accompanying paper shows, we have recently developed a stringent assay for aryl radicals, and the mechanism of coupling of haloarenes to arenes was a clear case where the assay could be applied. This paper now details our investigation into the cross-coupling of aryl halides with unactivated arenes using nickel sources.
Results and discussion
Recently, the Engle lab reported that bench-stable 18-electron complex Ni(COD)(DQ) (COD = 1,5-cycloctadiene; DQ = duroquinone) serves as an active pre-catalyst in Ni-mediated reactions, including Suzuki–Miyaura reactions and Buchwald–Hartwig aminations.20,21 However, coupling of haloarenes to arenes was not reported. Inspired by the simple synthesis and remarkable stability of this Ni(0) complex, it was employed in our system. Ni(COD)(DQ) promoted the coupling of iodoarenes 1a,b in excellent yields (Table 1, entries 1 and 2), with near quantitative coupling of 1a (95%). Other nickel sources, Ni(acac)2 (acac = 2,4-pentanedionate), Ni(PPh3)4 and Ni(COD)2 (COD = 1,5-cyclooctadiene) were less effective (63%–69%, entries 3–8). A blank reaction featuring no nickel source gave a low yield of products. (entries 9 and 10) which, under these conditions, is proposed to arise via a benzyne-initiated BHAS mechanism.12 Ni(OAc)2 provided only marginally improved yields over the blank (entries 11 and 12).
To demonstrate the synthetic utility of Ni(COD)(DQ), examples of substrates were now employed in the coupling reaction with benzene under our standard conditions (Scheme 2). Unactivated aryl iodides and those bearing electron-donating para-substituents coupled in excellent yields (5a–c, 95–86%), with the corresponding aryl bromides giving slightly lower yields (84–78%). The reactions yielding 5a and 5b were repeated at a shorter reaction time of 4 h and, while good yields were obtained, the reactions were not complete; thus, it was decided to retain a 16 h reaction time to allow all substrates to react fully. Aryl chlorides were not amenable to coupling, yielding only 13% of the biphenyl product 5c. Substrates with ortho- or meta-substituents (5d–f) coupled satisfactorily (78–66%). Products 5g–5h, where substrates had electron-withdrawing substituents, afforded moderate to good yields. When 1-(allyloxy)-2-iodobenzene was employed, product 5i was obtained. Here the allyl group is rapidly isomerised to the Z-vinyl ether by KOtBu,22 which cannot undergo unfavourable 5-endo-trig or 4-exo-trig cyclisations and thus the intermolecular coupling product is obtained (67%). Pyridine-based heteroaryl-derived products 5j–k were obtained from the corresponding bromides in moderate yields (51–49%), expanding the potential applications of this system to heteroaryl halides.
 | ||
Scheme 2 Substrate scope. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) reaction run for 4 h. bp-Terphenyl also obtained in 16% yield. reaction run for 4 h. bp-Terphenyl also obtained in 16% yield. | ||
To determine the mechanism, our assay was then applied to the coupling reactions. The hindered 2,6-dimethyliodobenzene (6) was employed as the mechanistic test substrate.12,23 The first part of our assay is based on ratio of products when aryl radicals are formed from 6; two products are formed, namely 5c and 7 (Scheme 3) in a ratio approaching 4:1 (this ratio results from the relative rate constants for the two reactions that occur between aryl radical 9 and benzene 4, namely hydrogen atom abstraction (kC–H) and addition to benzene (kC–C) (for details of the rationale, see the accompanying paper)). Table 2 shows that nickel sources that proved active in the reaction (entries 1–4) all gave ratios 5c:7 that were in line with BHAS chemistry involving aryl radicals (for full screen including control reactions, see ESI‡).
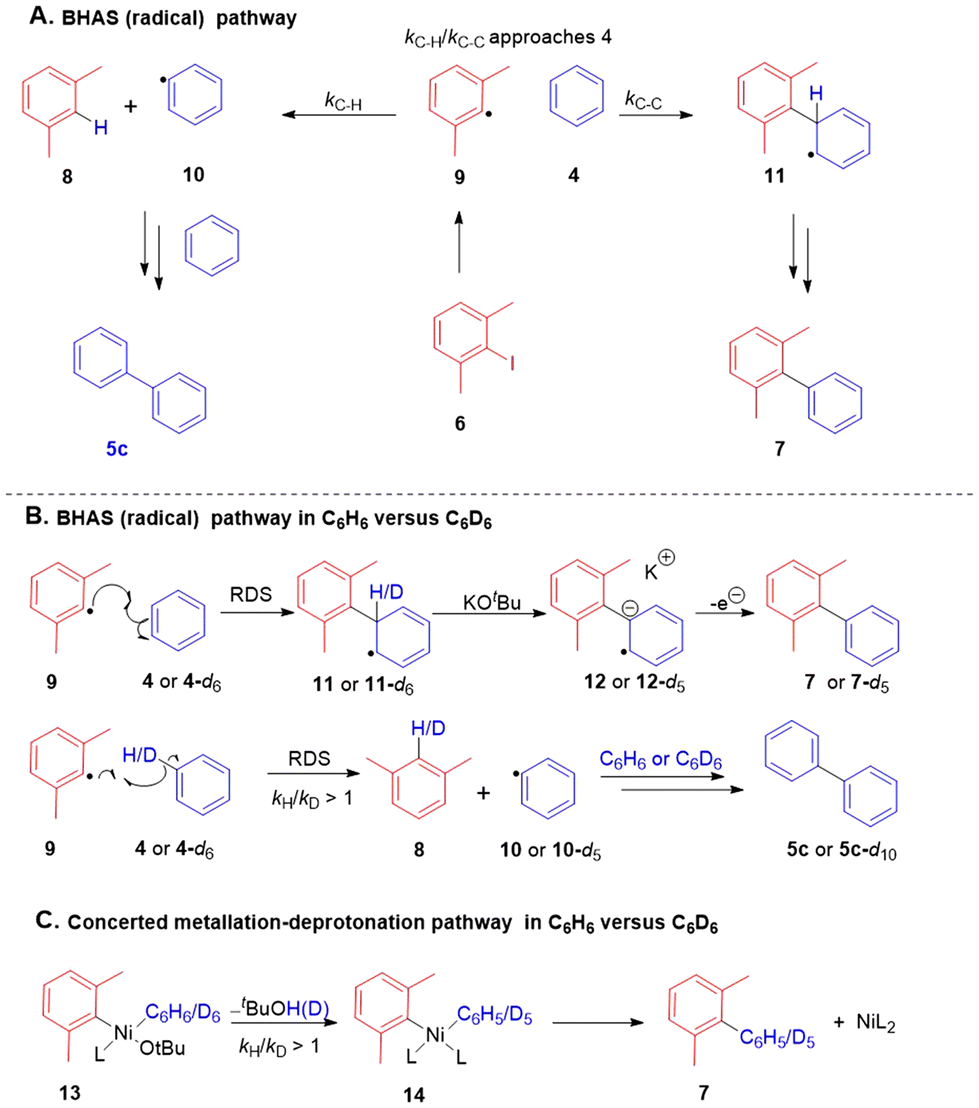 | ||
| Scheme 3 Expected KIE effects via organometallic or radical reaction routes. | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Entrya | Ni source | 6 (yield %) | 7 | 5c | 8 | Ratio 5c:7 |
| a Measured using GC-FID with n-dodecane as internal standard. | ||||||
| 1 | Ni(COD)(DQ) | Trace | 6.1 | 22.1 | 75.9 | 3.6 |
| 2 | Ni(COD)2 | 49.0 | 1.2 | 3.6 | 31.0 | 3.1 |
| 3 | Ni(acac)2 | 66.6 | 1.5 | 4.8 | 13.9 | 3.2 |
| 4 | Ni(PPh3)4 | 43.0 | 1.3 | 4.7 | 15.9 | 3.6 |
| 3 | Ni(OAc)2 | 86.9 | Trace | Trace | 0.8 | — |
| 6 | NiCl2 | 87.7 | Trace | Trace | Trace | — |
| 7 | — | 88.3 | Trace | Trace | Trace | — |

The second part of the assay examines the effect of employing deuterated benzene, C6D6. In the formation of 7 in a standard BHAS reaction in C6H6vs. C6D6, no primary kinetic isotope effect (KIE) would be observed, as the deprotonation step (11 → 12) in all BHAS reactions examined to date is known not to be the rate-determining step (RDS).10,24,25 However, formation of m-xylene (8) involves a challenging hydrogen atom abstraction by radical 9 from benzene 4 as the RDS, which would therefore incur a primary KIE (Scheme 3A). Thus, the rate of formation of biphenyl (5c-d10) would also decrease versus5c, as fewer phenyl radicals 10-d5versus10 would be formed in the respective experiments. Alternatively, if the coupling were proceeding through an organometallic pathway, for example through concerted metalation–deprotonation (CMD) of 13 to give 14, a primary KIE would be expected (Scheme 3C).26
The outcomes of reactions in C6H6vs. C6D6 (Table 3) show a significant decrease in the yields of the 5c-d10 relative to 5c in the C6H6 experiment, as expected from a hydrogen atom abstraction pathway. The yield of 8, which interestingly was not significantly deuterated (we estimate 3% from mass spectra), also declined. The non-deuteration points towards an alternative hydrogen atom source in the reaction mixture for abstraction by radical 9 that outcompetes abstraction from d6-benzene to form phenyl radical 10-d5 (see ESI‡ for this investigation; this outcome was mirrored in the accompanying paper under nickel-free conditions23). This could also explain why there was a small drop in the yield of 7-d5 relative to 7, as abstraction of hydrogen atoms by radical 9 from sources other than benzene would not easily lead to the radical anion required to propagate the chain. Substrate 6 was not fully consumed in this reaction (entry 2), supporting this explanation.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | 6 | 7/7-d5 | 5c/5c-d10 | 8 |
| 1 | C6H6 | Trace | 6.1 | 22.1 | 75.9 |
| 2 | C6D6 | 30.4 | 4.2 | 1.5 | 48.4 |

This result rationalises the formation of large quantities of m-xylene (8) in reactions employing substrate 6. Radical scavengers were employed to probe for radical intermediates, and 2,2,6,6-tetramethyl-1-piperidinyloxyl (TEMPO) was added to the reaction of 6 in both 0.5 and 3.0 equivalents (see ESI‡). When 0.5 equivalents of TEMPO were added the yields of 7, 5c and 8 decreased significantly (2.9%, 6.8% and 30.0% respectively), and when 3.0 equivalents were added the reaction was completely inhibited. This supports radical intermediates, although the result should be interpreted with caution, as it should be noted that TEMPO can affect reactions due to alternative effects, e.g. TEMPO has been shown to coordinate to Ni0 centres as a ligand.27
Conclusions
In summary, it is shown that, under ground-state conditions, air-stable Ni(COD)(DQ) and KOtBu promote the coupling of a range of simple aryl halides to benzene in high yields. Mechanistic studies support aryl radicals and radical anions as intermediates, and it is proposed that this nickel system acts as an initiator to produce aryl radicals, which then go onto react via a base-assisted homolytic aromatic substitution (BHAS) radical chain reaction. While Ni(COD)(DQ) was found to be by far the most effective complex employed, other nickel sources also facilitate this reaction, with evidence that they may also proceed via radical intermediates in a BHAS pathway.Author contributions
ST performed the experiments, analysed the results and drafted the paper. CMR and JAM supervised the research, analysed the data and edited the draft paper and JAM conceived of the research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank GSK and EPSRC for funding an i-CASE studentship to ST (grant number EP/W522260/1). The authors thank Craig Irving, Patricia Keating, Jessica Bame and Graeme Anderson for assistance with spectroscopy.References
- P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192–2452 CrossRef CAS PubMed.
- J. Yamaguchi, K. Muto and K. Itami, Top. Curr. Chem., 2016, 374, 55, DOI:10.1007/s41061-016-0053-z.
- S. M. Khake and N. Chatani, Chem, 2020, 6, 1056–1081 CAS.
- J. Canivet, J. Yamaguchi, I. Ban and K. Itami, Org. Lett., 2009, 11, 1733–1736 CrossRef CAS PubMed.
- T. Yamamoto, K. Muto, M. Komiyama, J. Canivet, J. Yamaguchi and K. Itami, Chem. – Eur. J., 2011, 17, 10113–10122 CrossRef CAS PubMed.
- H. Hachiya, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2009, 11, 1737–1740 CrossRef CAS PubMed.
- O. Kobayashi, D. Uraguchi and T. Yamakawa, Org. Lett., 2009, 11, 2679–2682 CrossRef CAS PubMed.
- G. Xie, T. Li, X. Qu and J. Mao, J. Mol. Catal. A: Chem., 2011, 340, 48–52 CrossRef CAS.
- E. Shirakawa, K. I. Itoh, T. Higashino and T. Hayashi, J. Am. Chem. Soc., 2010, 132, 15537–15539 CrossRef CAS PubMed.
- C. L. Sun, H. Li, D. G. Yu, M. Yu, X. Zhou, X. Y. Lu, K. Huang, S. F. Zheng, B. J. Li and Z. J. Shi, Nat. Chem., 2010, 2, 1044–1049 CrossRef CAS PubMed.
- H. Li, C. L. Sun, M. Yu, D. G. Yu, B. J. Li and Z. J. Shi, Chem. – Eur. J., 2011, 17, 3593–3597 CrossRef CAS PubMed.
- S. Zhou, G. M. Anderson, B. Mondal, E. Doni, V. Ironmonger, M. Kranz, T. Tuttle and J. A. Murphy, Chem. Sci., 2014, 5, 476–482 RSC.
- Z. Xu, L. Gao, L. Wang, M. Gong, W. Wang and R. Yuan, ACS Catal., 2015, 5, 45–50 CrossRef CAS.
- S. Zhou, E. Doni, G. M. Anderson, R. G. Kane, S. W. MacDougall, V. M. Ironmonger, T. Tuttle and J. A. Murphy, J. Am. Chem. Soc., 2014, 136, 17818–17826 CrossRef CAS PubMed.
- H. Yi, A. Jutand and A. Lei, Chem. Commun., 2015, 51, 545–548 RSC.
- M. Patil, J. Org. Chem., 2016, 81, 632–639 CrossRef CAS.
- A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2011, 50, 5018–5022 CrossRef CAS PubMed.
- G. Nocera and J. A. Murphy, Synthesis, 2020, 52, 327–336 CrossRef CAS.
- A. J. Smith, D. L. Poole and J. A. Murphy, Sci. China: Chem., 2019, 62, 1425–1438 CrossRef CAS.
- V. T. Tran, Z. Q. Li, O. Apolinar, J. Derosa, M. V. Joannou, S. R. Wisniewski, M. D. Eastgate and K. M. Engle, Angew. Chem., Int. Ed., 2020, 59, 7409–7413 CrossRef CAS PubMed.
- For other forefront developments in nickel-catalysed coupling reagents, see: L. Nattmann, R. Saeb, N. Nöthling and J. Cornella, Nat. Catal., 2020, 3, 6–13 CrossRef CAS.
- M. Shi, L. Wang, Q. Chen, M. He, M. Shen and Z. H. Zhang, Tetrahedron Lett., 2020, 61, 152278 CrossRef CAS.
- See accompanying K. F. Clark, S. Tyerman, L. Evans, C. M. Robertson and J. A. Murphy, An Assay for Aryl Radical using BHAS coupling, Org. Biomol. Chem., 2024 10.1039/d3ob01743e.
- Z. Xu, L. Gao, L. Wang, M. Gong, W. Wang and R. Yuan, ACS Catal., 2015, 5, 45–50 CrossRef CAS.
- E. Shirakawa, K. I. Itoh, T. Higashino and T. Hayashi, J. Am. Chem. Soc., 2010, 132, 15537–15539 CrossRef CAS PubMed.
- S. I. Gorelsky, D. Lapointe and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 10848–10849 CrossRef CAS PubMed.
- D. Isrow and B. Captain, Inorg. Chem., 2011, 50, 5864–5866 CrossRef CAS PubMed.
Footnotes |
| † A draft of this paper has been pre-published in ChemRxiv, 2023: https://chemrxiv.org/engage/chemrxiv/article-details/6460f4bbf2112b41e98e610c. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ob01745a |
| This journal is © The Royal Society of Chemistry 2024 |