
Synthesis of the aminocyclopentenediol fragment of queuosine by way of the stereoselective addition of an organometallic reagent to a N-t-butanesulfinyl glycosylamine†
Floriane
Heis
 ,
Estelle
Gallienne
and
Olivier R.
Martin
*
,
Estelle
Gallienne
and
Olivier R.
Martin
*
Institute of Organic and Analytical Chemistry, UMR 7311, University of Orleans and CNRS, Rue de Chartres, 45067 Orleans, France. E-mail: olivier.martin@univ-orleans.fr
First published on 28th November 2023
Abstract
An innovative, concise synthesis of the aminocyclopentenediol fragment of queuosine is reported. The synthesis is based on the stereocontrolled addition of a vinylGrignard·LiCl reagent to a t-butanesulfinyl L-ribofuranosylamine, followed by dehydrodeoxygenation to generate a second vinyl group and ring-closing metathesis to form the five-membered ring scaffold of the natural product. This approach has the potential for the development of a larger scale synthesis.
Introduction
Queuosine (nucleoside Q) is a hypermodified, non-canonical RNA nucleoside found in many eukaryotic and bacterial species, usually present in the anticodon loop of certain tRNAs.1 In addition to being a 7-deazaguanosine analog, queuosine carries a unique 4,5-dihydroxycyclopentenylamine substituent branched at the 7-position of the nucleobase, the relative stereochemistry of which was established in 1976 by the pioneering work of Goto et al.2 (Fig. 1). In higher vertebrates and humans, the nucleoside queuosine also exists as O-mannosylated or O-galactosylated derivatives in the carbocyclic diol, the precise glycosylation site and anomeric configuration of which have been recently elucidated by Carell and coworkers.3,4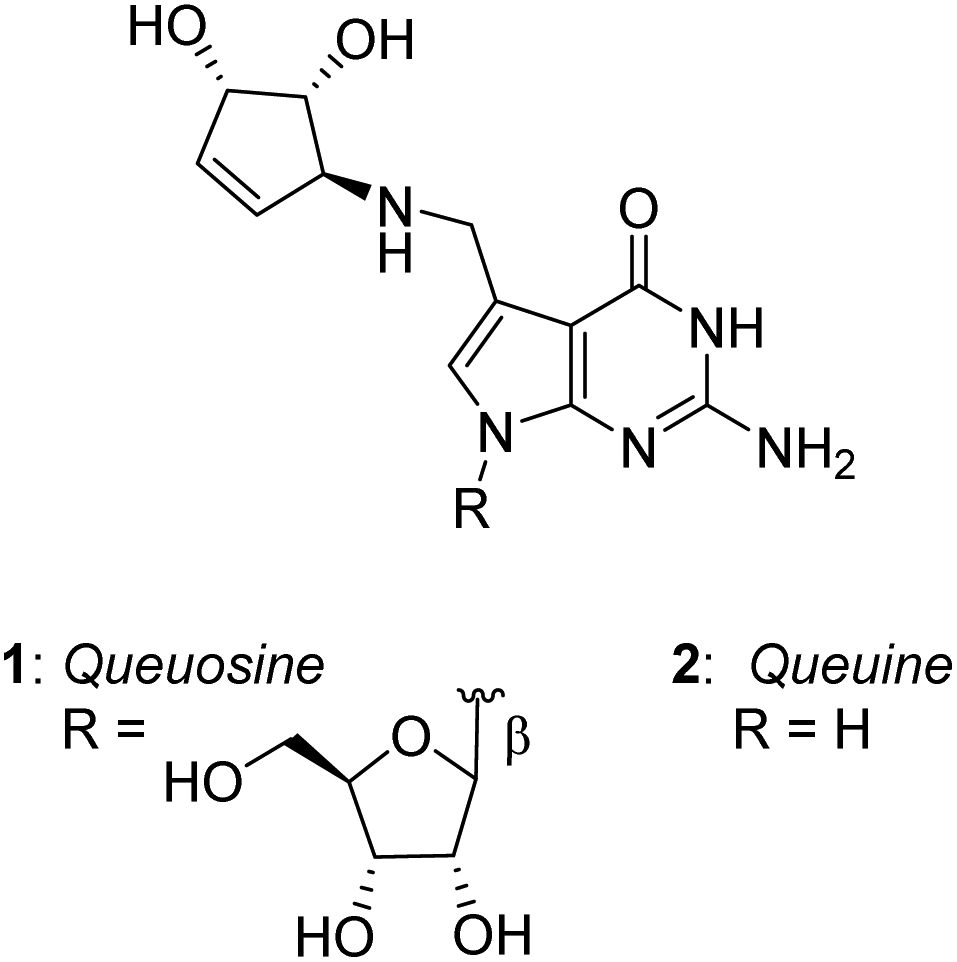 | ||
| Fig. 1 Structure of queuosine (nucleoside Q) and queuine. | ||
The nucleobase component of queuosine, known as queuine 2, has been shown very recently to exhibit remarkable biological activities: it has a protective effect in in vitro models of neurodegeneration.5 As queuine itself is exclusively produced by gut bacteria, it appears that this compound could be a key mediator of the link between microbiome imbalance and several neurodegenerative disorders including Parkinson's and Alzheimer's diseases, and as it is enriched in the brain, exerts a neuroprotective effect.6
The absolute configuration of the carbocyclic component of queuine has been established following the first synthesis of queuosine reported as early as 1979 by Goto et al.7 The first asymmetric synthesis of this component was reported by Ogasawara,8 and the total syntheses of the glycosylated nucleosides Q were reported recently by Carell.3,4
In view of the considerable therapeutic potential of queuine, a concise and efficient synthetic route to this compound is required. Previous synthetic efforts have concentrated on a convergent approach involving the preparation of a protected cyclopentenylamine such a 3 and its efficient coupling with a 7-formyl deazaguanosine derivative 4 by reductive amination as reported by Showalter9 and by Carell in the case of queuosine,10 according to the retrosynthetic scheme (Scheme 1).
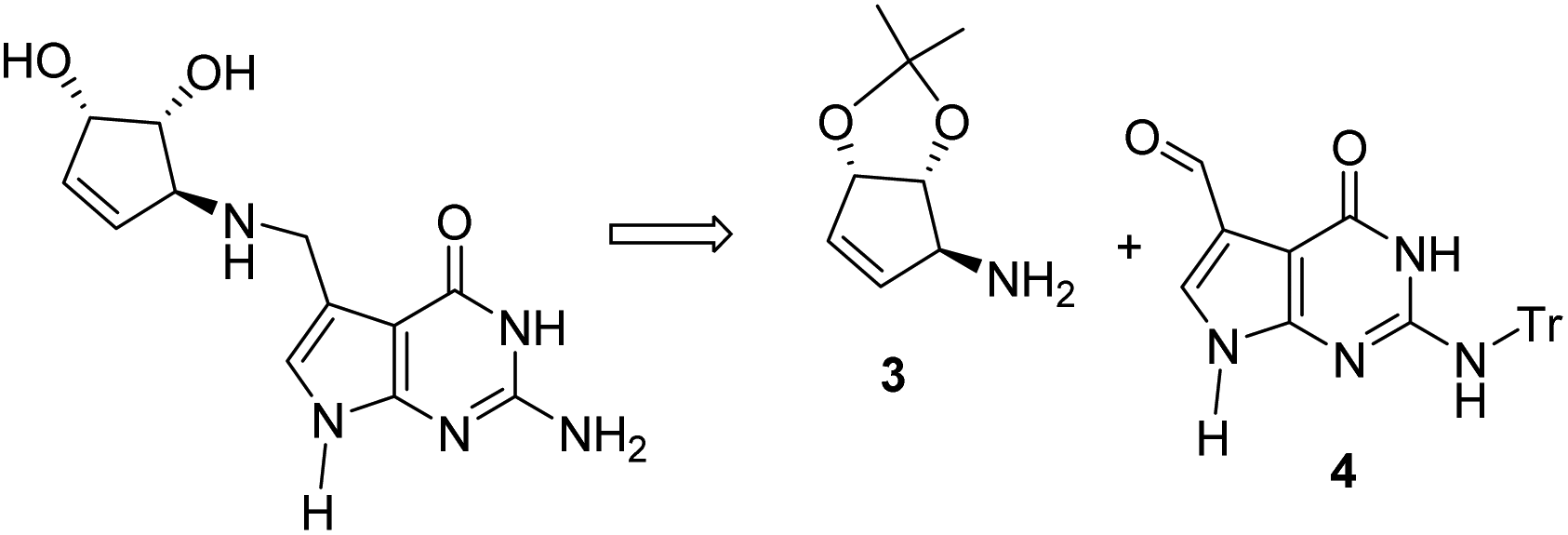 | ||
| Scheme 1 Retrosynthesis of the nucleobase of queuosine. | ||
The bottleneck of the synthesis of the modified nucleobase is the access to the functionalized, stereochemically well defined cyclopentene derivative. Two main approaches to the five-membered carbocyclic structure have been investigated.
(A) Syntheses from cyclopentadiene derivatives, in which the five-membered ring is present in the starting material
(B) Formation of the 5-membered ring in the course of the synthesis by a ring-closing metathesis (RCM)
Stategy (A) was implemented in the first synthesis of queuosine,7 and later in the first asymmetric synthesis of the cyclopentenyl derivative by way of a (−)-3-endo-hydroxy-dicyclopentadiene.8 These lengthy syntheses were considerably shortened by the work of Trost et al.,11 in which the enantiomerically pure target compound was generated in 5 steps from racemic cyclopentadiene epoxide. In a related approach, Miller et al.12 reported a synthesis based on the cycloaddition of cyclopentadiene with an amino acid-derived acyl nitroso compound, thus introducing simultaneously a hydroxyl and an amino group.
Strategy (B) was pioneered by van Boom and coworkers13,14 (Scheme 2). Starting from a hexose (D-mannose), a multistep process including a Wittig chain extension leads to an open-chain triol carrying 2 vinyl groups A, substrate for the RCM reaction. The latter leads to a protected cyclopentenetriol B which is then submitted to an Overmann rearrangement by way of a trichloroacetimidate C. Cyclopentenetriol derivatives such as B are key precursors of carbocyclic nucleosides.15
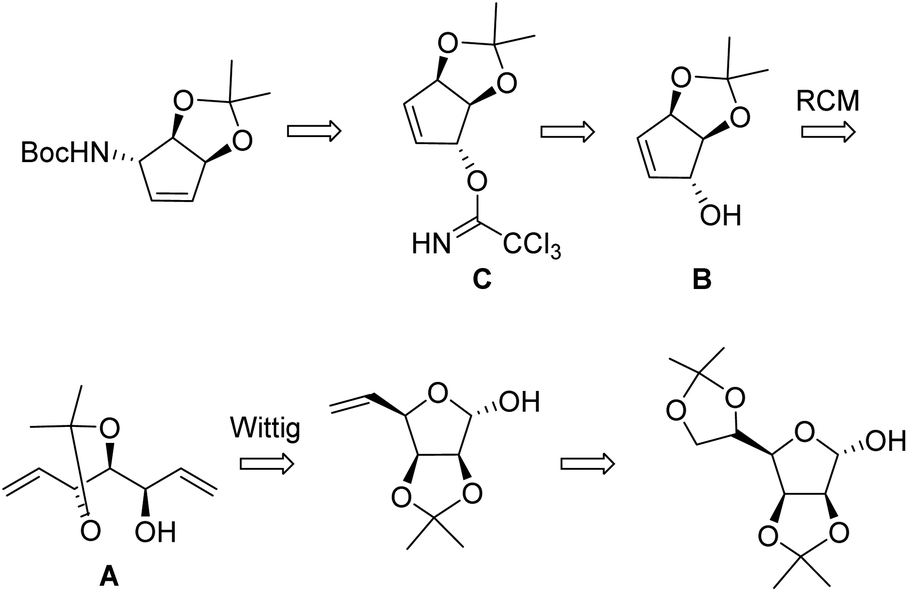 | ||
| Scheme 2 van Boom's strategy from D-mannose.13 | ||
This process is fully stereocontrolled but requires 11 steps from D-mannose. A related strategy from D-ribose was implemented by Carell,10 the cyclisation being achieved by an intramolecular Wittig–Horner process. These syntheses require however lengthy multistep processes and are difficult and costly to scale up.
We have developed recently an efficient methodology leading to aminoalditols of well-defined stereochemistry by the addition of organometallic reagents to N-t-butanesulfinylglycosylamines.16 We propose herein an innovative synthesis of the carbocyclic component of queuine based on this methodology. As outlined in Scheme 3, this methodology would provide a more concise approach to the carbocyclic component of queuine.
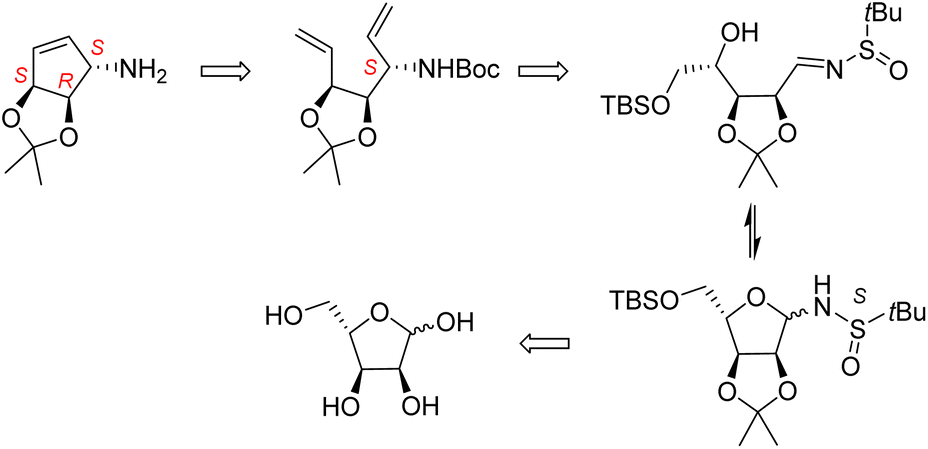 | ||
| Scheme 3 Proposed retrosynthetic scheme to form the cyclopentenyl-amine derivative. | ||
The key step is the stereocontrolled chain extension of a pentose-derived glycosylamine to give an open chain aminoalditol. As shown by our group, the stereoselectivity of such additions to (R) or (S)-t-butanesulfinylglycosylamines can be tuned to provide the desired configuration, usually with very high de. Thus, our plan was to perform a 2C chain extension by the addition of a vinyl group to an L-ribofuranosylamine, generating an aminoalditol having the appropriate configuration at the amino-carrying carbon, followed by the dehydrodeoxygenation of the terminal diol function in the resulting intermediate. Starting from an L-ribo substrate, this sequence is expected to lead to the required diene precursor carrying, unlike other syntheses, all 3 stereogenic centers of the final product with the appropriate functions and configurations. The synthesis is then completed by a RCM. We report in this article the implementation of this strategy.
Results and discussion
The synthesis that was developed according to these plans is outlined in Scheme 4. Both L-ribose or D-lyxose could be considered as starting materials for the synthesis, both of which having the appropriate configurations at C-2 and C-3. The C-4 configuration is not important as this stereocenter is lost in the course of the synthesis. The development work was done from L-ribose, since preliminary work indicated that the stereoselectivity of the addition of the organometallic reagent to the corresponding D-lyxofuranosylamines was, unexpectedly, very poor.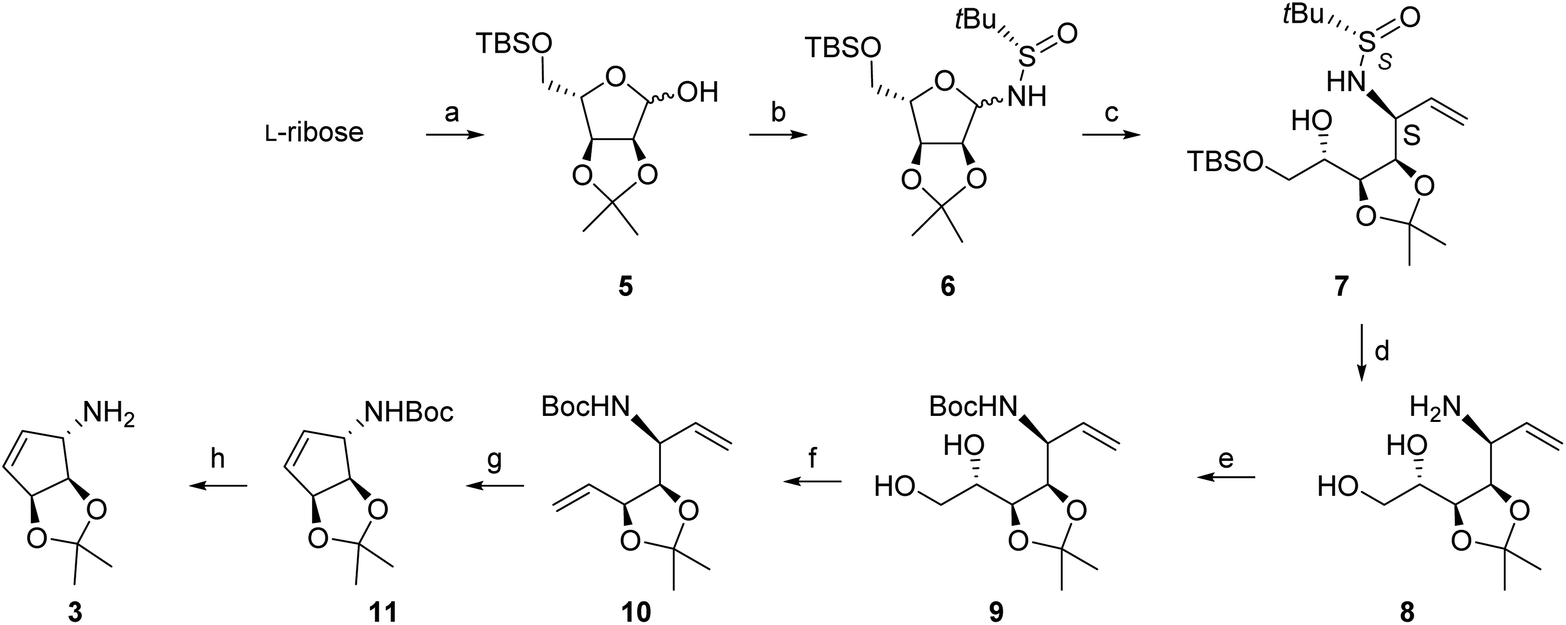 | ||
Scheme 4 Synthesis of 3 from L-ribose. Reagents and conditions: (a) acetone, conc. H2SO4, 1.5 h, 0 °C → rt then TBSCl, pyridine, DCM 0 °C to RT, overnight, 54% (2 steps). (b) (S)-t-Butanesulfinamide, Cs2CO3, MS 4 Å, DCM, reflux 24 h, 64%. (c) VinylMgBr, LiCl, THF, −60 to −20 °C, 83%. (d) HCl (from AcCl), MeOH, 0 °C → rt. (e) Boc2O, DMAP, MeCN/H2O (8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2), 82% (2 steps). (f) 2,4,5-Triiodoimidazole, triphenylphosphine, imidazole, toluene, reflux 1.5 h, 75%. (g) Grubbs 2nd generation (5 mol%), DCM, 35 °C, 4 h, 87%. (h) TFA/DCM, 3 h, 50%. :2), 82% (2 steps). (f) 2,4,5-Triiodoimidazole, triphenylphosphine, imidazole, toluene, reflux 1.5 h, 75%. (g) Grubbs 2nd generation (5 mol%), DCM, 35 °C, 4 h, 87%. (h) TFA/DCM, 3 h, 50%. | ||
The protected D-ribofuranose derivative D-5 (enantiomer of 5) has been used extensively as starting material in a diversity of syntheses and is commercially available. It is readily prepared from D-ribose and the procedure has been described by several groups with minor variations in the protocols,15,17 in particular by the Carell's group,18 with yield varying from 50% to 72% for the 2 steps (25–500 g scale). The L-ribose derivative 5 was reported for the first time in 2010,19 but no detailed procedure was provided: the authors claim to have used the conditions generally reported for the D-ribose derivative. We have also used the reported D-ribose conditions to convert L-ribose into 5 (54% overall yield).
From our previous work,16 we anticipated that the reaction of an (S)-configured t-butanesulfinyl-L-ribofuranosylamine would lead predominantly to the desired (S)-configuration of the new chiral center carrying the amino group (1,2-syn in the open-chain product), as required for the final cyclopentenylamine. Thus, the corresponding glycosylamine 6 was prepared by the reaction of L-ribofuranose 5 with (S)-t-butanesulfinamide in the presence of cesium carbonate in DCM, conditions that we had previously developed for acid-sensitive substrates.16 The product was submitted to a rapid purification by column chromatography which afforded very clean glycosylamine 6 as a 3:1 mixture of anomers (β-anomer, 1,2-trans probably major) in 64% yield.
We then investigated the addition of a vinylmagnesium reagent to glycosylamine 6 in THF at low temperature, without additive. The addition worked quite efficiently to give isomers 7S and 7R in 84% isolated yield, but with modest diastereoselectivity (de = 68%) (see NMR spectra below, lowest trace). The addition was then investigated in the presence of additives, especially lithium chloride. We had observed previously a remarkable effect of this salt on the diastereoselectivity of related reactions,16a due apparently to the formation of a highly reactive LiCl-RMgX reagent (Knochel's Turbo Grignard reagent).20 This reagent was prepared from strictly dry LiCl and the mixed organometallic species was reacted with glycosylamine 6: this change led to an impressive improvement of the diastereoselectivity, with less than 3% of the second isomer being formed (de > 95%), the minor isomer becoming hardly detectable by NMR (Fig. 2).
 | ||
| Fig. 2 Crude NMR spectra of 7 in CDCl3, 300 MHz, 298 K. With 3.5 eq. of LiCl (top); without LiCl (bottom). | ||
Further elaboration of 7 required the removal of the N-sulfinyl group and of the silyl group at O-5. Preliminary investigations involving the stepwise cleavage of the silyl group with fluoride ion before or after the cleavage of the sulfinyl group did not give satisfactory yields. We found that the treatment of 7S with methanolic HCl for a short period promoted the cleavage of both the silyl group and the sulfinyl group without affecting the isopropylidene acetal. This excellent selectivity provided a simple access to the free amine and diol 8 in essentially quantitative yield. The crude amino compound was directly converted into a t-butyl carbamate (Boc group). The product was submitted to a short silica gel column for purification, to give the product 9 in 82% yield over 2 steps.
The configuration at C-1 was determined by preparing a cyclic derivative of 7: pyrrolidine 12 was obtained by mesylation of the C-4 alcohol function followed by an internal SN. In the cyclic product, it is expected that both H-1 and H-4 are in cis relationship if C-1 has an S configuration. This was confirmed by the NOESY analysis of compound 12 (Fig. 3): a correlation is observed between these two signals, thus confirming the absolute configuration at C-1.
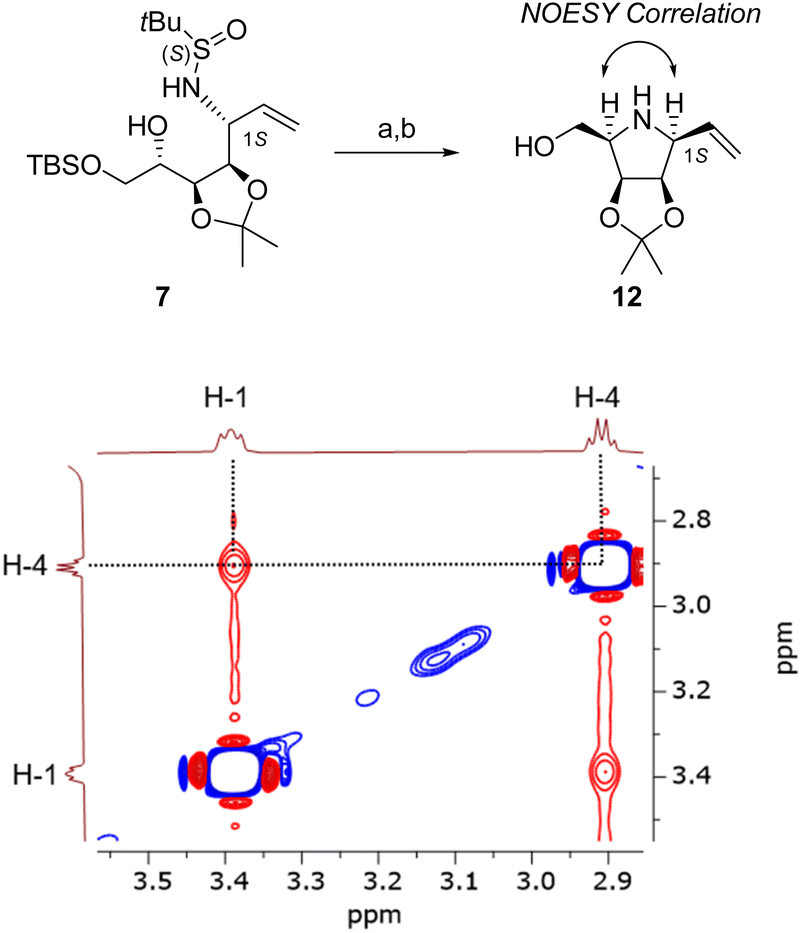 | ||
| Fig. 3 Synthesis of 12 (a) MsCl, Et3N, DCM, 0 °C → rt; (b) HCl (from AcCl), MeOH, rt, 1.5 h, 75%. NOESY of 12 showing correlation between H-1 and H-4 (400 MHz, CDCl3, 298 K). Compound 12 has a ‘β-D-lyxo’ configuration. | ||
The next step of the synthesis, namely the conversion of the terminal dihydroxyethyl group into a vinyl group required extensive investigations. Tipson-Cohen-type processes21 were unsuccessful as the presence of a leaving group in 1,4-relationship with respect to the protected amine led readily to the formation of the pyrrolidine. The conditions reported by Sarma (Me3SiCI-NaI in MeCN)22 gave intractable mixtures. More promising results were obtained using variations of the Garegg–Samuelsson conditions.23,24 The conditions involve a phosphine source, an iodine source and imidazole. Under standard conditions, the first attempt (100 mg-scale) worked brilliantly, giving very clean divinyl compound 10 in 53% yield (and triphenylphosphine oxide). This reaction was, however, not reproducible. The conditions were therefore further investigated. While no satisfactory results could be obtained using PPh3 or ClPPh2, the reaction leading to mixtures of products and/or intermediates such as the 5-iodo derivative, we turned our attention to using 2,3,5-triiodoimidazole as an iodine source.25 This reagent has the favourable advantage of not producing HI in the course of the reaction, the formation of this acid being possibly a cause of the degradation observed with iodine. The 2,4,5-triiodoimidazole was synthesized following a published procedure,26 but it is also commercially available. Performing the reaction in 2 stages (apparently by forming first the 5-iodo derivative), at 80 °C for 30 min, until all SM was consumed, then heating under reflux for 2 h gave the desired product in reproducible yield up to 85%. The reaction performed well at the gram-scale level; a chromatography purification was needed to clean the dark-coloured product obtained from this reaction. NMR data were fully consistent with the structure, showing clearly the presence of the 2 vinyl groups.
From literature precedents,13–15 the formation of the cyclopentene ring by a ring-closing metathesis was expected to work well. Indeed, exposing the diene 10 to Grubbs II ruthenium catalyst (5 mol%) in degassed DCM gave the desired cyclopentene derivative 11 cleanly and in high yield (87%, 500 mg-scale), after filtration through a short column of silica gel and through a short pad of active charcoal to remove traces of the catalyst. The catalyst load could eventually be reduced to 2 mol%. The structure and the configuration of 11 was unambiguously established from NMR data and by comparison with literature data. The same compound has been prepared in 1996 by Ogasawara et al.8 by a totally different approach. Remarkably, the NMR data reported by this author (300 MHz, CDCl3, data in ESI†) matched exactly those measured for our compound 11.
This excellent agreement constitutes the final proof that the structure of compound 11 is the expected one, and that the stereoselective addition of the vinyl group to glycosylamine 6 gave the desired S configuration, validating the entire process for the synthesis of compound 3.
The selective deprotection of the Boc group was achieved using TFA in dichloromethane, leading to the free amine 3 in 50% yield. This process was not optimized but our preliminary investigations clearly showed that the Boc group can be cleaved selectively using anhydrous TFA, without affecting the isopropylidene group. The synthesis of queuine could be completed from the amine 3 thus obtained under the conditions reported.9
Conclusions
The new strategy developed for the synthesis of the carbocyclic component of queuine has definite advantages over previously reported syntheses: it does not require the difficult reactions (SN or 3,3-sigmatropic rearrangements) needed to introduce the amino group on the cyclopentenetriol scaffold, nor does it require an atom-expensive Wittig-type reaction. It is significantly shorter than the van Boom method as the target compound can be reached in 6 steps from a 5-O-protected L-ribofuranose derivative, in an 25% (84%/step) overall yield, unoptimized. This approach has the potential for the development of a larger scale synthesis.Experimental
General
All solvents were dried prior to use. Dichloromethane (DCM) was distilled over P2O5. THF and toluene were dried using a solvent drying machine and the water content regularly analyzed by K.-F. analysis titration. All the reactions at room temperature (rt) were conducted at 25 °C. Molecular sieves (4 Å, powder) were activated in an oven overnight prior to use. Flash silica gel chromatography was performed on Silica gel 60 (0.040–0.063 mm, Merck, Darmstadt). The reactions were monitored by TLC on aluminum-coated sheets (silica gel 60 GF254, Merck), and spots were detected by charring with sulfuric acid (5% in ethanol) or Cerium Ammonium Molybdenum (CAM) revelator. 1H and 13C NMR spectra were recorded at 298 K with a Bruker NanoBay 400 MHz instrument in CDCl3 unless otherwise indicated. High-resolution mass spectra (HRMS) were recorded on a Bruker Q-TOF MaXis spectrometer. PE = Petroleum ether; EA = Ethyl acetate. sm = starting material.:3) to afford compound 6 as a white solid (6.34 g, 64%).
1H NMR (400 MHz, CDCl3, mixture of anomers labeled as maj = major and min = minor) δ 5.35 (dd, J = 11.5, 4.1 Hz, 1H, H-1 maj), 5.25 (d, J = 10 Hz, 0.3H, H-1 min), 4.92 (d, J = 10.0 Hz, 0.3H, N–H min), 4.74 (d, J = 6.0 Hz, 1H, H-3 maj), 4.67 (d, J = 6.1 Hz, 0.3H, H-3 min), 4.61 (dd, J = 6.1, 1.6 Hz, 0.3H, H-2 min), 4.54 (dd, J = 6.0, 4.2 Hz, 1H, H-2 maj), 4.37 (d, J = 11.5 Hz, 1H, N–H maj), 4.22 (br s, 0.3H, H-4 min), 4.11 (br s, 1H, H-4 maj), 3.79 (dd, J = 10.8, 2.7 Hz, 1H, H-5B maj), 3.77–3.70 (m, 0.6H, H-5A, 5B min), 3.65 (dd, J = 10.8, 1.9 Hz, 1H, H-5A maj), 1.48 (m, 3.7H, CH3 isop min + maj), 1.33 (s, 3H, CH3 isop maj), 1.30 (s, 0.9H, CH3 isop min), 1.21 (s, 9H, tBu maj), 1.19 (s, 2.9H, tBu min), 0.90 (s, 2.7H, tBu min), 0.86 (s, 9H, tBu maj), 0.10 (2s, 1.8H, Si-CH3 min), 0.05 (2s, 6H, Si-CH3 maj); 13C NMR (101 MHz, CDCl3) δ 112.6 (CIV isop min), 112.4 (CIV isop maj), 93.2 (C-1 min), 89.0 (C-1 maj), 87.5 (C-2 min), 86.0 (C-4 min), 82.4 (C-4 maj), 82.1 (C-3 min), 82.0 (C-3 maj), 80.7 (C-2 maj), 65.7 (C-5 maj), 65.0 (C-5 min), 56.2 (CIV S-tBu min), 56.2 (CIV S-tBu maj), 26.8 (CH3 isop min), 26.2 (CH3 isop maj), 26.0 (tBu-S maj), 25.8 (tBu-S min), 25.2 (CH3 isop min), 24.7 (CH3 isop maj), 22.5 (Si-tBu min), 22.46 (Si-tBu maj), 18.5 (CIV-Si min), 18.0 (CIV-Si maj), −5.26 and −5.37 (Si-CH3 min), −5.55 and −5.72 (Si-CH3 maj). ESI + HRMS [M + H]+ m/z 408.2234 calcd for C18H38NO5SSi found: 408.2230.
:5. A silica gel chromatography was performed (PE/EA 8:2) to remove the minor isomer and to afford compound 7S as a white amorphous solid (4 g, 83%).
1H NMR (400 MHz, CDCl3) δ 6.00 (ddd, J = 16.8, 10.4, 6.0 Hz, 1H, H-6), 5.41 (dt, J = 17.3, 1.7 Hz, 1H, H-7 trans), 5.24 (dt, J = 10.4, 1.7 Hz, 1H, H-7 cis), 4.73 (d, J = 5.0 Hz, 1H, N–H), 4.36 (t, J = 5.8 Hz, 1H, H-2), 4.21 (m, 1H, H-1), 4.13 (dd, J = 9.7, 5.9 Hz, 1H, H-3), 4.00 (m, 1H, H-4), 3.79 (dd, J = 10.2, 3.0 Hz, 1H, H-5B), 3.67 (dd, J = 10.2, 4.8 Hz, 1H, H-5A), 2.91 (br s, 1H, OH), 1.47 (s, 3H, CH3 isop), 1.32 (s, 3H, CH3 isop), 1.24 (s, 9H, S-tBu), 0.89 (s, 9H, Si-tBu), 0.07 (s, 6H, Si-CH3). 13C NMR (101 MHz, CDCl3) δ 136.4 (C-6), 117.4 (C-7), 108.4 (CIV isop), 79.2 (C-2), 76.8 (C-3), 68.0 (C-4), 64.4 (C-5), 56.9 (C-1), 55.7 (CIV, S-tBu), 27.1 (CH3 isop), 25.9 (S-tBu), 25.0 (CH3 isop), 22.8 (tBu-Si), 18.33 (CIVtBu-Si), −5.38 (CH3-Si), −5.46 (CH3-Si). ESI + HRMS [M + H]+m/z C20H42NO5SSi calc. 436.2547 found 436.2539.
:1) was submitted to flash chromatography (8:2 to 7:3 PE/EA) which afforded compounds 7S (265 mg, 70%) and 7R (52 mg, 14%).
7R : 1H NMR (400 MHz, CDCl3) δ 6.07 (ddd, J = 17.3, 10.5, 6.6 Hz, 1H, H-6), 5.45 (d, J = 17.3 Hz, 1H, H-7trans), 5.35 (d, J = 10.6 Hz, 1H, H-7cis), 5.02 (d, J = 5.1 Hz, 1H, N–H), 4.47 (dd, J = 8.2, 5.2 Hz, 1H, H-2), 4.17 (dd, J = 9.7, 5.2 Hz, 1H, H-3), 4.11 (br q, 1H, H-1), 3.88 (dd, J = 9.9, 3.3 Hz, 1H, H-5B), 3.81 (m, 1H, H-4), 3.62 (dd, J = 9.9, 7.2 Hz, 1H, H-5A), 3.10 (br d, J = 3.4 Hz, 1H, OH), 1.40 (s, 3H, CH3 isop), 1.35 (s, 3H, CH3 isop), 1.21 (s, 9H, S-tBu), 0.92 (s, 9H, Si-tBu), 0.10 (s, 6H, Si-CH3). 13C NMR (101 MHz, CDCl3) δ 136.0 (C-6), 118.9 (C-7), 108.6 (CIV isop), 79.9(C-2), 77.3 (C-3), 68.8 (C-4), 64.5 (C-5), 57.3 (C-1), 55.9 (CIV, S-tBu), 29.7 (CH3 isop), 25.9 (S-tBu), 25.6 (CH3 isop), 22.8 (tBu-Si), 18.33 (CIVtBu-Si), −5.37 (CH3-Si), −5.40 (CH3-Si). ESI + HRMS [M + H]+m/z C20H42NO5SSi calc. 436.2547 found 436.2540.
1H NMR (400 MHz, CDCl3) δ 6.08 (ddd, J = 17.1, 10.4, 6.8 Hz, 1H, H-6), 5.20 (d, J = 17.2 Hz, 1H, H-7 trans), 5.14 (d, J = 10.3 Hz, 1H, H-7 cis), 4.16 (br dd, J = 5.6, 2.3 Hz, 1H, H-2), 3.97 (dd, J = 8.8, 5.4 Hz, 1H, H-3), 3.79–3.86 (m, 2H, H-4, H-5B), 3.76 (br d, J = 6.7 Hz, 1H, H-1), 3.61 (dd, J = 12.2, 7.4 Hz, 1H, H-5A), 3.3–3.5 (br s, 4H, OH, NH2), 1.45 (s, 3H, CH3 isop), 1.32 (s, 3H, CH3 isop). 13C NMR (101 MHz, CDCl3) δ 140.3 (C-6), 114.9 (C-7), 107.9 (CIV isop), 79.1(C-2), 77.9 (C-3), 68.8 (C-4), 64.3 (C-5), 52.8 (C-1), 27.8 (CH3 isop), 25.4 (CH3 isop). ESI + HRMS [M + H]+m/z C10H20NO4 calc. 218.1387 found: 218.1382.
:2) and Boc2O (4.36 g, 20 mmol) and DMAP (611 mg, 5 mmol) were added. The mixture was stirred at rt until TLC showed complete conversion of the sm. The mixture was diluted with ethyl acetate (100 mL) and a saturated ammonium chloride solution was added (100 mL). The layers were separated and the aqueous layer was extracted twice with ethyl acetate (2 × 50 mL). The combined organic layers were washed with H2O and then dried over MgSO4 and concentrated. The residue was purified over a short silica gel column (DCM/MeOH 9:1) to give 9 as a colourless oil (2.61 g, 82% over two steps). 1H NMR (400 MHz, CDCl3) δ 5.91 (ddd, J = 16.3, 10.4, 5.0 Hz, 1H, H-6), 5.21 (d, J = 17.1 Hz, 1H, H-7trans), 5.18 (br s, 1H, NH), 5.16 (d, J = 10.3 Hz, 1H, H-7cis), 4.71 (br s, 1H, H-1), 4.37 (br s, 1H, OH), 4.31 (br d, J = 7.1 Hz, 1H, H-2), 4.11 (dd, J = 9.7, 7.1 Hz, 1H, H-3), 3.86 (br, 1H, H-4), 3.81 (br d, J = 12 Hz, 1H, H-5B), 3.58 (br, 1H, H-5A), 3.17 (br s, 1H, OH), 1.45 (s, 12H, CH3 isop, tBu), 1.33 (s, 3H, CH3 isop). 13C NMR (101 MHz, CDCl3) δ 156.2 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 137.3 (C-6), 115.1 (C-7), 108.2 (CIV isop), 80.3 (CIV, O-tBu), 78.2 (C-2), 76.5 (C-3), 69.3 (C-4), 65.2 (C-5), 51.1 (C-1), 28.4 (tBu), 26.7 (CH3 isop), 24.1 (CH3 isop). ESI + HRMS [M + H]+m/z C15H28NO6 calc. 318.1911 found: 318.1916.
:2 PE/EA). The mixture was cooled down to rt and the tarry residue was washed with toluene (3 × 20 mL). The toluene layer was washed with water (2 × 50 mL), dried over MgSO4 and concentrated. A silica gel chromatography was performed (8:2 PE/EA) to afford the diene 10 as a white waxy solid (754 mg, 75%). 1H NMR (400 MHz, CDCl3) δ 5.94 (ddd, J = 17.4, 10.3, 7.4 Hz, 1H, H-4), 5.83 (ddd, J = 17.4, 10.4, 5.5 Hz, 1H, H-6), 5.37 (d, J = 17.2 Hz, 1H, H-5trans), 5.29 (d, J = 10.3 Hz, 1H, H-5cis), 5.24 (d, J = 17.1 Hz, 1H, H-7trans), 5.18 (d, J = 10.3 Hz, 1H, H-7cis), 4.89 (br, 1H, NH), 4.66 (t, J = 7.5 Hz, 1H, H-3), 4.24 (br d, J = 6.7 Hz, 1H, H-2), 4.17 (br s, 1H, H-1), 1.55 (s, 3H, CH3 isop), 1.46 (s, 9H, OtBu), 1.39 (s, 3H, CH3 isop). 13C NMR (101 MHz, CDCl3) δ 154.9 (CO), 136.9 (C-6), 133.5 (C-4), 119.3 (C-5), 115.7 (C-7), 108.6 (OC(CH3)2), 79.6 (C-2), 79.4 (OC(CH3)3), 79.2 (C-3), 52.1 (C-1), 28.4 (OC(CH3)3), 26.9, 24.36 (2C, CH3 isop). ESI + HRMS [M + H]+m/z C15H26NO4 calc: 284.1856 found: 284.1859.
:2). The mixture was concentrated and a short silica gel chromatography was performed (PE/EA 9:1). The product was then diluted in EA and filtered through a short pad of activated carbon to remove traces of catalyst, and the adsorbent rinsed with EA to afford 11 as a colourless oil (415 mg, 87%). 1H NMR (400 MHz, CDCl3) δ 5.98 (d, J = 5.7 Hz, 1H, H-1), 5.77 (d, J = 5.7 Hz, 1H, H-2), 5.23 (d, J = 5.6 Hz, 1H, H-5), 4.6 (br s, 2H, H-3, NH), 4.49 (d, J = 5.8 Hz, 1H, H-4), 1.46 (s, 9H, tBu), 1.41 (s, 3H, CH3 isop), 1.34 (s, 3H, CH3 isop). 13C NMR (101 MHz, CDCl3) δ 155.1 (CO), 135.1 (C-1), 132.5 (C-2), 111.5 (CIV isop), 84.9 (C-4), 84.3 (C-5), 79.8 (CIV, O-tBu), 62.5 (C-3), 28.4 (tBu), 27.4, 25.7 (2C, CH3 isop). ESI + HRMS [M + Na]+m/z C13H21NNaO4 calc. 278.1362 found: 278.1365.
:1). The mixture was stirred for 2 h at 0 °C then quenched carefully with saturated aqueous NaHCO3. After stirring for 10 min, the layers were separated and the aqueous layer was extracted twice with DCM (2 × 10 mL). The combined organic layers were dried with Na2SO4 then concentrated in vacuo. The crude compound was obtained as a yellowish oil which was purified by chromatography (DCM/MeOH 95:5) to afford pure 3 (47 mg, 50%), 1H NMR (400 MHz, CDCl3) δ 5.87 (br d, AB, J = 5.7 Hz, 1H, H-1), 5.83 (br d, AB, J = 5.7 Hz, 1H, H-2), 5.25 (br d, J = 5.6 Hz, 1H, H-5), 4.36 (d, J = 5.6 Hz, 1H, H-4), 3.97 (br s, 1H, H-3), 1.68 (br s, 2H, NH2), 1.39 (s, 3H, CH3 isop), 1.32 (s, 3H, CH3 isop). 13C NMR (101 MHz, CDCl3) δ 155.1 (CO), 136.7 (C-2), 132.9 (C-1), 111.1 (CIV isop), 87.3 (C-4), 84.3 (C-5), 63.0 (C-3), 27.5, 25.8 (2C, CH3 isop). ESI + HRMS [M + Na]+ ESI + HRMS [M + H]+ m/z C8H14NO2 calc. 156.1019 found: 156.1019.
O), 137.3 (C-6), 115.1 (C-7), 108.2 (CIV isop), 80.3 (CIV, O-tBu), 78.2 (C-2), 76.5 (C-3), 69.3 (C-4), 65.2 (C-5), 51.1 (C-1), 28.4 (tBu), 26.7 (CH3 isop), 24.1 (CH3 isop). ESI + HRMS [M + H]+m/z C15H28NO6 calc. 318.1911 found: 318.1916.
:2 PE/EA). The mixture was cooled down to rt and the tarry residue was washed with toluene (3 × 20 mL). The toluene layer was washed with water (2 × 50 mL), dried over MgSO4 and concentrated. A silica gel chromatography was performed (8:2 PE/EA) to afford the diene 10 as a white waxy solid (754 mg, 75%). 1H NMR (400 MHz, CDCl3) δ 5.94 (ddd, J = 17.4, 10.3, 7.4 Hz, 1H, H-4), 5.83 (ddd, J = 17.4, 10.4, 5.5 Hz, 1H, H-6), 5.37 (d, J = 17.2 Hz, 1H, H-5trans), 5.29 (d, J = 10.3 Hz, 1H, H-5cis), 5.24 (d, J = 17.1 Hz, 1H, H-7trans), 5.18 (d, J = 10.3 Hz, 1H, H-7cis), 4.89 (br, 1H, NH), 4.66 (t, J = 7.5 Hz, 1H, H-3), 4.24 (br d, J = 6.7 Hz, 1H, H-2), 4.17 (br s, 1H, H-1), 1.55 (s, 3H, CH3 isop), 1.46 (s, 9H, OtBu), 1.39 (s, 3H, CH3 isop). 13C NMR (101 MHz, CDCl3) δ 154.9 (CO), 136.9 (C-6), 133.5 (C-4), 119.3 (C-5), 115.7 (C-7), 108.6 (OC(CH3)2), 79.6 (C-2), 79.4 (OC(CH3)3), 79.2 (C-3), 52.1 (C-1), 28.4 (OC(CH3)3), 26.9, 24.36 (2C, CH3 isop). ESI + HRMS [M + H]+m/z C15H26NO4 calc: 284.1856 found: 284.1859.
:2). The mixture was concentrated and a short silica gel chromatography was performed (PE/EA 9:1). The product was then diluted in EA and filtered through a short pad of activated carbon to remove traces of catalyst, and the adsorbent rinsed with EA to afford 11 as a colourless oil (415 mg, 87%). 1H NMR (400 MHz, CDCl3) δ 5.98 (d, J = 5.7 Hz, 1H, H-1), 5.77 (d, J = 5.7 Hz, 1H, H-2), 5.23 (d, J = 5.6 Hz, 1H, H-5), 4.6 (br s, 2H, H-3, NH), 4.49 (d, J = 5.8 Hz, 1H, H-4), 1.46 (s, 9H, tBu), 1.41 (s, 3H, CH3 isop), 1.34 (s, 3H, CH3 isop). 13C NMR (101 MHz, CDCl3) δ 155.1 (CO), 135.1 (C-1), 132.5 (C-2), 111.5 (CIV isop), 84.9 (C-4), 84.3 (C-5), 79.8 (CIV, O-tBu), 62.5 (C-3), 28.4 (tBu), 27.4, 25.7 (2C, CH3 isop). ESI + HRMS [M + Na]+m/z C13H21NNaO4 calc. 278.1362 found: 278.1365.
:1). The mixture was stirred for 2 h at 0 °C then quenched carefully with saturated aqueous NaHCO3. After stirring for 10 min, the layers were separated and the aqueous layer was extracted twice with DCM (2 × 10 mL). The combined organic layers were dried with Na2SO4 then concentrated in vacuo. The crude compound was obtained as a yellowish oil which was purified by chromatography (DCM/MeOH 95:5) to afford pure 3 (47 mg, 50%), 1H NMR (400 MHz, CDCl3) δ 5.87 (br d, AB, J = 5.7 Hz, 1H, H-1), 5.83 (br d, AB, J = 5.7 Hz, 1H, H-2), 5.25 (br d, J = 5.6 Hz, 1H, H-5), 4.36 (d, J = 5.6 Hz, 1H, H-4), 3.97 (br s, 1H, H-3), 1.68 (br s, 2H, NH2), 1.39 (s, 3H, CH3 isop), 1.32 (s, 3H, CH3 isop). 13C NMR (101 MHz, CDCl3) δ 155.1 (CO), 136.7 (C-2), 132.9 (C-1), 111.1 (CIV isop), 87.3 (C-4), 84.3 (C-5), 63.0 (C-3), 27.5, 25.8 (2C, CH3 isop). ESI + HRMS [M + Na]+ ESI + HRMS [M + H]+ m/z C8H14NO2 calc. 156.1019 found: 156.1019.
(b) A methanolic solution of HCl was prepared by adding AcCl (82 μL, 1.15 mmol) to MeOH (5 mL) and stirring for 30 min. This solution was added to the mesylate of 7S (118 mg, 0.23 mmol) and the mixture was stirred for 1.5 h. The solution was neutralized with [OH−]-ion exchange resin, the suspension filtered and the solvent evaporated. The residue was submitted to a flash chromatography (9:1 DCM/MeOH) which afforded pyrrolidine 12 as an oily material (34 mg, 75% for 2 steps). 1H NMR (400 MHz, CDCl3) δ 5.98 (ddd, J = 17.3, 10.5, 6.6 Hz, 1H, H-6), 5.38 (narrow dt, J = 17.3, 2 × 1.5 Hz, 1H, H-trans), 5.30 (narrow dt, J = 10.6, 2 × 1.3 Hz, 1H, H-7cis), 4.77 (dd, J = 5.6, 4.3 Hz, 1H, H-3), 4.66 (dd, J = 5.6, 4.1 Hz, 1H, H-2), 4.05 (dd, J = 11.9, 3.9 Hz, 1H, H-5B), 3.94 (dd, J = 11.9, 4.8 Hz, 1H, H-5A), 3.43 (m, 1H, H-1), 2.95 (br q, J = 3 × 4.3 Hz, 1H, H-4), 1.51 (s, 3H, CH3 isop), 1.33 (s, 3H, CH3 isop). 13C NMR (101 MHz, CDCl3) δ 133.1 (C-6), 118.1 (C-7), 111.5 (CIV isop), 83.7 (C-2), 83.4 (C-3), 64.7 (C-1), 62.5 (C-4), 60.9 (C-5), 25.5, 23.7 (2C, CH3 isop). ESI + HRMS [M + H]+m/z C10H18NO3 calc. 200.1281 found 200.1279.
Author contributions
F. H. investigation, writing; E.G. supervision, reviewing; O.M. conceptualization, funding acquisition, writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding from Stellate Therapeutics for the support of this project is gratefully acknowledged. The authors also thank the SALSA platform for spectroscopic measurements and spectrometric analyses. The projects CHemBio (FEDER-FSE 2014-2020-EX003677), Valbiocosm (FEDER-FSE 2014-2020-EX003202), Techsab (FEDER-FSE 2014-2020-EX011313), QUALICHIM (APR-IA-PF 2021-00149467), the RTR Motivhealth (2019-00131403) and the Labex programs SYNORG (ANR-11-LABX-0029) and IRON (ANR-11-LABX-0018-01) are also gratefully acknowledged for laboratory equipment funding.References
- (a) H. M. Goodman, J. Abelson, A. Landy, S. Benner and J. D. Smith, Nature, 1968, 217, 1019–1024 CrossRef CAS PubMed; (b) H. Kasai, Z. Ohashi, F. Harada, S. Nishimura, N. J. Oppenheimer, P. F. Crain, J. C. Liehr, D. L. von Minden and J. A. McCloskey, Biochemistry, 1975, 14, 4198–4208 CrossRef CAS PubMed; (c) H. Kasai, K. Nakanishi, R. D. Macfarlane, D. F. Torgerson, Z. Ohashi, J. A. McCloskey, H. J. Gross and S. Nishimura, J. Am. Chem. Soc., 1976, 98, 5044–5046 CrossRef CAS.
- T. Ohgi and T. Goto, Tetrahedron Lett., 1976, 367–370 CrossRef CAS.
- P. Thumbs, T. T. Ensfelder, M. Hillmeier, M. Wagner, M. Heiss, C. Scheel, A. Schön, M. Müller, S. Michalakis, S. Kellner and T. Carell, Angew. Chem., Int. Ed., 2020, 59, 12352–12356 CrossRef CAS.
- M. Hillmeier, M. Wagner, T. T. Ensfelder, E. Korytiakova, P. Thumbs, M. Müller and T. Carell, Nat. Commun., 2021, 12, 7123–7129 CrossRef CAS.
- P. Richard, L. Kozlowski, H. Guillorit, P. Garnier, N. C. McKnight, A. Danchin and X. Manière, PlosONE, 2021, 16, e0253216 CrossRef CAS.
- N. McKnight, P. Richard, X. Manière, A. Danchin and P. Garnier, WO2020EP75581, 2020.
- T. Ohgi, T. Kondo and T. Goto, J. Am. Chem. Soc., 1979, 101, 3629–3633 CrossRef CAS.
- (a) K. Tanaka and K. Ogasawara, Synthesis, 1995, 1237–1239 CrossRef CAS; (b) K. Tanaka and K. Ogasawara, Synthesis, 1996, 219–221 CrossRef CAS.
- A. F. Brooks, G. A. Garcia and H. D. Hollis Showalter, Tetrahedron Lett., 2010, 51, 4163–4165 CrossRef CAS.
- F. Klepper, E.-M. Jahn, V. Hickmann and T. Carell, Angew. Chem., Int. Ed., 2007, 46, 2325–2327 CrossRef CAS PubMed.
- B. M. Trost and M. T. Sorum, Org. Process Res. Dev., 2003, 7, 432–435 CrossRef CAS.
- K.-H. Kim and M. J. Miller, Tetrahedron Lett., 2003, 44, 4571–4573 CrossRef CAS.
- H. Ovaa, J. D. C. Codée, B. Lastdrager, H. S. Overkleeft, G. A. van der Marel and J. H. van Boom, Tetrahedron Lett., 1998, 39, 7987–7900 CrossRef CAS.
- H. Ovaa, B. Lastdrager, J. D. C. Codée, G. A. van der Marel, H. S. Overkleeft and J. H. van Boom, J. Chem. Soc., Perkin Trans. 1, 2002, 2370–2377 RSC.
- H. R. Moon, W. J. Choi, H. O. Kim and L. S. Jeong, Tetrahedron: Asymmetry, 2002, 13, 1189–1193 CrossRef CAS.
- (a) C. Cocaud, C. Nicolas, A. Bayle, T. Poisson, X. Pannecoucke and O. R. Martin, Eur. J. Org. Chem., 2015, 4330–4334 CrossRef CAS; (b) C. Cocaud, C. Nicolas, T. Poisson, X. Pannecoucke, C. Legault and O. R. Martin, J. Org. Chem., 2017, 82, 2753–2763 CrossRef CAS; (c) D. Kamzol, M. Neuville, C. Cocaud, K. Tiger, B. Taffoureau, K. Lewiński, J. Jaszczyk, O. R. Martin, S. Baś, S. Routier, I. Gillaizeau, F. Buron and C. Nicolas, Eur. J. Org. Chem., 2023, 26, e202300762 CrossRef CAS.
- B. Kaskar, G. L. Heise, R. S. Michalak and B. R. Vishnuvajjala, Synthesis, 1990, 1031–1032 CrossRef CAS.
- T. Bruckl, F. Klepper, K. Gutsmiedl and T. Carell, Org. Biomol. Chem., 2007, 3821–3825 RSC.
- M. Shiozaki, T. Tashiro, H. Koshino, R. Nakagawa, S. Inoue, T. Shigeura, H. Watarai, M. Taniguchi and K. Mori, Carbohydr. Res., 2010, 345, 1663–1684 CrossRef CAS PubMed.
- D. S. Ziegler, B. Wie and P. Knochel, Chem. – Eur. J., 2019, 25, 2695–2703 CrossRef CAS.
- R. S. Tipson and A. Cohen, Carbohydr. Res., 1965, 1, 338–340 CrossRef CAS.
- J. C. Sarma, N. C. Barua, R. P. Sharma and J. N. Barua, Tetrahedron, 1983, 39, 2843–2846 CrossRef CAS.
- (a) P. J. Garegg and B. Samuelsson, Synthesis, 1979, 469–470 CrossRef CAS; (b) P. J. Garegg and B. Samuelsson, Synthesis, 1979, 813–814 CrossRef CAS; H. Ohrui, K. Nagihashi and H. Meguro, Biosci., Biotechnol., Biochem., 1992, 56, 1721–1724 Search PubMed.
- Z. Liu, B. Classon and B. Samuelsson, J. Org. Chem., 1990, 55, 4273–4275 CrossRef CAS.
- P. J. Garegg and B. Samuelsson, J. Chem. Soc., Perkin Trans. 1, 1980, 2866–2869 RSC; P. J. Garegg and B. Samuelsson, J. Chem. Soc., Perkin Trans. 1, 1982, 681–683 RSC; See also; J. Singleton, K. Sahteli and J. O. Hoberg, Synthesis, 2008, 3682–3686 Search PubMed.
- R. D. C. Gallo, I. M. Ferreira, G. A. Casagrande, L. Pizzuti, D. Oliveira-Silva and C. Raminelli, Tetrahedron Lett., 2012, 53, 5372–5375 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Comparison of NMR data of compound 11 with the data reported in 1996 by Ogasawara; copies of NMR spectra. See DOI: https://doi.org/10.1039/d3ob01713c |
| This journal is © The Royal Society of Chemistry 2024 |