
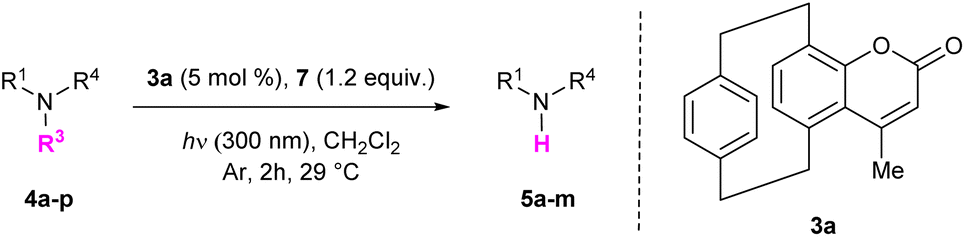
[2.2]Paracyclophane-based coumarins: effective organo-photocatalysts for light-induced desulfonylation processes†‡
Jules
Brom
a,
Antoine
Maruani
 a,
Serge
Turcaud
a,
Sonia
Lajnef
a,
Fabienne
Peyrot
ab,
Laurent
Micouin
a and
Erica
Benedetti
*a
a,
Serge
Turcaud
a,
Sonia
Lajnef
a,
Fabienne
Peyrot
ab,
Laurent
Micouin
a and
Erica
Benedetti
*a
aUniversité Paris Cité, CNRS, Laboratoire de Chimie et de Biochimie Pharmacologiques et Toxicologiques, F-75006 Paris, France. E-mail: erica.benedetti@u-paris.fr
bSorbonne-Université, Institut National Supérieur du Professorat et de l'Education (INSPE) de l'Académie de Paris, F-75016 Paris, France
First published on 23rd November 2023
Abstract
Herein, we demonstrate for the first time that coumarins derived from [2.2]paracyclophane (pCp) can act as effective organo-photocatalysts and promote the reductive cleavage of sulfonamides under light-irradiation. In the presence of these original compounds, photodesulfonylation reactions occur under mild conditions at low catalyst loadings in the presence of Hantzsch ester. Theoretical and experimental investigations are described, which elucidate the reaction mechanism and the nature of the active species involved in the photocatalytic process. This proof-of-concept study paves the way for further application of pCps in the field of photocatalysis.
[2.2]Paracyclophane (pCp) is an original organic molecule that possesses a unique three-dimensional structure. Described for the first time by Brown and Farthing in 1949,1 this compound incorporates two benzene rings stacked in a face-to-face geometry and linked together by two ethylene bridges at their para position.2 pCp displays unusual optoelectronic properties due to the proximity of its aromatic cores that favours through-space and through-bond interactions.3 As a result, this substrate and its derivatives have been extensively used in material science as building-blocks for the development of various luminescent objects.4 These compounds have also frequently served as precursors to obtain aromatic polymers or functionalized surfaces using chemical vapor deposition (CVD) methods.5 In addition, substituted pCps can show planar chirality6 and have been increasingly employed over the years as ligands or catalysts in stereoselective synthesis7 or as scaffolds for the development of chiroptical dyes.8
Despite their interesting photophysical and catalytic behaviours that have led to such a wide range of applications, [2.2]paracyclophanes have rarely been considered as useful precursors for the development of novel photocatalysts. This possibility was nonetheless anticipated by Hopf as early as 1991.9 Recently, an example of a heterobimetallic photoredox complex based on a pCp backbone has been reported by Bräse and co-workers.10 However, to the best of our knowledge, no examples of organo-photocatalysts derived from [2.2]paracyclophanes have been described to date.
Herein, we demonstrate for the first time that pCp-fused coumarin systems can play the role of photoredox catalysts and promote the reductive cleavage of sulfonamides under light-irradiation.
Sulfonamides display several advantageous properties including general inertia to acids, bases, or electrophiles and stability under oxidizing or mild reducing conditions. The difficult cleavage of these compounds, however, still greatly limits the use of such functions as amine protecting groups in organic synthesis. Indeed, removal of N-sulfonyl moieties usually involve treatments with strong reducing agents, acids or bases.11 Electrochemical methods or procedures relaying on SmI2-promoted electron transfer reactions have also been described in the literature.12 Recently, photocatalytic processes have unlocked new possibilities for performing light-driven desulfonylation reactions under milder conditions, and few examples have been reported.13 These include, for instance, the use of extremely potent acridine radical photoreductants.14 Methods involving different organophotocatalysts in the presence of hydride donors15 or expensive transition metal complexes in combination with Hantzsch esters (HE) have also been developed.16
Coumarin dyes are known to be able to mimic powerful reductant [Ir(III)] complexes in various light-promoted transformations, such as the radical coupling of carbonyl compounds and imines, the trifluoromethylation of alkenes or the reductive protonation of bromoketones.17
On the basis of our ongoing work on the synthesis and spectroscopic characterization of small three-dimensional dyes derived from [2.2]paracyclophane,18 we envisaged the possibility to employ luminescent pCp-based coumarins instead of iridium-based catalysts to promote the reductive photocleavage of sulfonamides (Scheme 1).
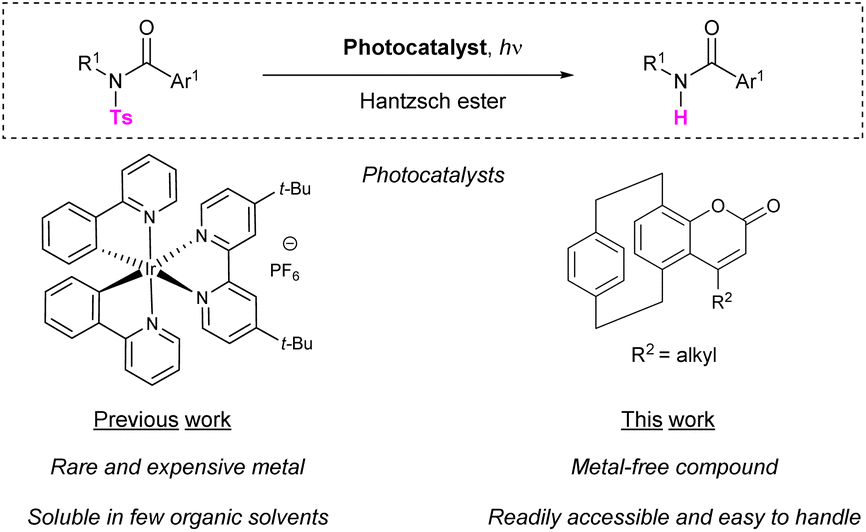 | ||
| Scheme 1 Examples of catalytic photodesulfonylation reactions. | ||
We began our investigation by synthesizing differently substituted pCp-based coumarins. These compounds were rapidly obtained on synthetically useful scales (2 mmol) starting from 4-hydroxy[2.2]paracyclophane19 and following a procedure previously developed in our laboratory (Scheme 2).18b Esterification of pCp 1 with different propiolic acid derivatives and subsequent gold-catalysed cyclization of esters 2a–c allowed us to isolate products 3a–c incorporating a methyl, an isopropyl or a phenyl substituent on their heterocyclic moiety. Spectroscopic characterization revealed that these coumarin dyes can absorb light at 300–330 nm in CH2Cl2 (Table 1). In this solvent, compounds 3a–c also display broad emission bands with maxima around 435 nm or 460 nm depending on the nature of their substituents (Table 1). We next set out to study the ability of pCp-based coumarins 3a–c to promote the photocleavage of N–S bonds in combination with the Hantzsch ester (7, Table 2). To this end, an N-sulfonylated model compound (4a, Table 2) was prepared starting from benzylamine via a tosylation and subsequent benzoylation reaction (see the ESI‡ for more information).
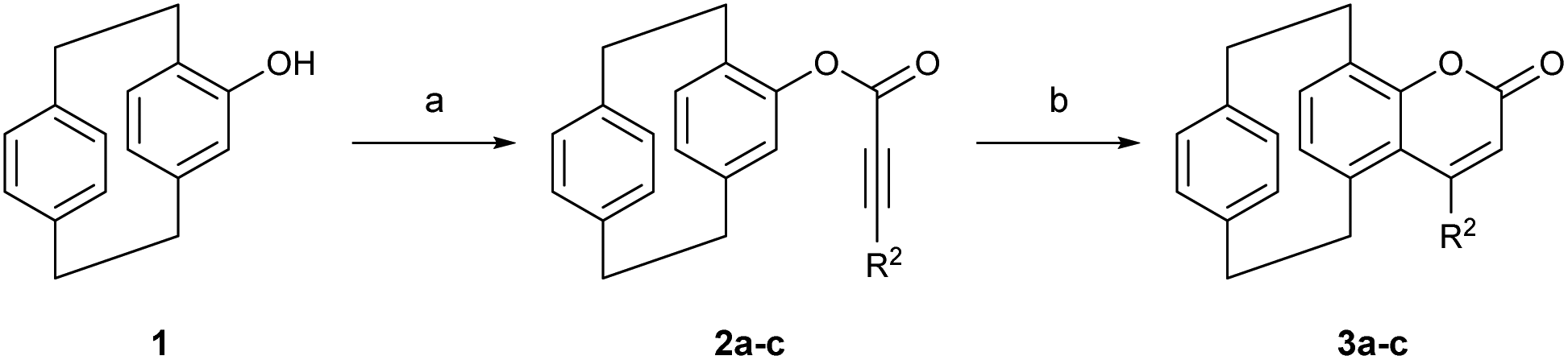 | ||
| Scheme 2 Synthesis and spectroscopic properties of pCp-based coumarins. Reaction conditions. (a) Propiolic acid (1.5 equiv.), DCC (1.5 equiv.), DMAP (0.15 equiv.), CH2Cl2, rt, 4 h; (b) Echavarren's catalyst (5 mol%), DCE, Ar, 80 °C, MW, 30 min. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | Photocat. (mol%) | Deviation from standard conditionsa | [H] | t (h) | Conv.b (%) |
| a Standard conditions: reactions were performed in a Rayonet photochemical reactor equipped with eight 300 nm lamps under argon atmosphere (T = 29 °C, c = 0.05 M, solvent = CH2Cl2). b Determined by 1H NMR analysis of the crude reaction mixture. | |||||
| 1 | 3a (20) | — | 7 | 2 | 66 |
| 2 | 3a (20) | — | 7 | 16 | 71 |
| 3 | 3a (15) | — | 7 | 2 | 63 |
| 4 | 3a (5) | — | 7 | 2 | 65 |
| 5 | 3a (5) | c = 0.025 M | 7 | 2 | 64 |
| 6 | 3a (5) | Solvent = DCE | 7 | 2 | 45 |
| 7 | 3a (5) | Solvent = PhCl | 7 | 2 | 28 |
| 8 | 3a (5) | Solvent = toluene | 7 | 2 | — |
| 9 | 3a (5) | Solvent = DMSO | 7 | 2 | — |
| 10 | 3a (5) | λ = 350 nm | 7 | 2 | 25 |
| 11 | 3b (5) | — | 7 | 2 | 56 |
| 12 | 3c (5) | — | 7 | 2 | 26 |
| 13 | 3a (5) | In the dark | 7 | 2 | — |
| 14 | 3a (5) | — | — | 2 | — |
| 15 | — | — | 7 | 2 | 9 |
| 16 | 3a (5) | O2 (1 atm.) | 7 | 2 | 19 |
| 17 | 3a (5) | TEMPO (1.2 equiv.) | 7 | 2 | — |
| 18 | 3a (5) | BHT (1.2 equiv.) | 7 | 2 | 18 |
| 19 | 3a (5) | — | 8 | 2 | — |
| 20 | 3a (5) | — | 9 | 2 | — |
| 21 | 3a (5) | — | n-Bu4NBH4 | 2 | — |
| 22 | 6 (5) | — | 7 | 2 | 14 |
| 23 | pCp (5) | — | 7 | 2 | 9 |
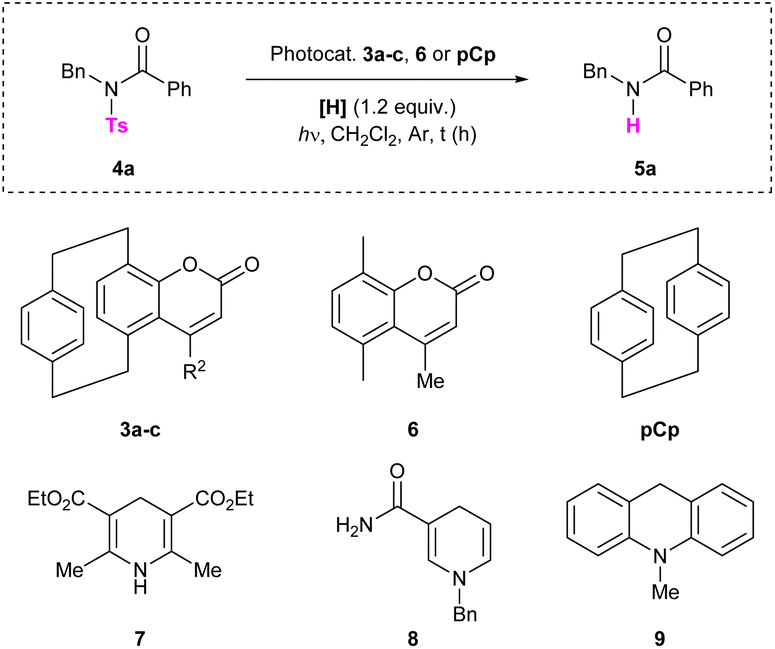
With this compound in hand, we first tested the photodesulfonylation under light irradiation at 300 nm in the presence of catalyst 3a (20 mol%) and 7 (1.2 equiv.), using dichloromethane as the solvent. Under these conditions, we were pleased to observe that the reaction reached 66% conversion after 2 h (Table 2, entry 1). Note that a longer reaction time (16 h) only slightly improved the reaction outcome (Table 2, entry 2). The catalytic charge could be reduced to 5 mol% without any significant loss in the reaction efficiency (Table 2, entries 3 and 4). Performing the transformation under more diluted conditions did not have a remarkable impact on the outcome (Table 2, entry 5). Different solvents were then tested. In dichloroethane (DCE) and chlorobenzene (PhCl) the reaction reached 45% and 28% conversion respectively (Table 2, entries 6 and 7). The cleavage however failed to occur in toluene (Table 2, entry 8) and in dimethyl sulfoxide (DMSO, Table 2, entry 9) due to the poor solubility of the reaction partners in these media. Low conversion was observed when the reaction medium was irradiated at 350 nm for 2 h (Table 2, entry 10). Notably, compound 3b, incorporating an isopropyl group on its coumarin moiety, also prove to catalyse the photodeprotection as 56% conversion could be reached after 2 h (Table 2, entry 11). On the contrary, derivative 3c, displaying an aromatic substituent on its core, delivered product 5a in a less efficient fashion (Table 2, entry 12). Control reactions were performed and proved the utility of all reaction partners. Light irradiation was found to be essential for the reaction to work, as no product 5a was formed when the detosylation was performed in the dark (Table 2, entry 13). Compound 4a did not undergo the photodeprotection in the absence of the Hantzsch ester (Table 2, entry 14), and only traces of product 5a were observed in the absence of the catalyst (Table 2, entry 15). This last result presumably arises from the fact that both the Hantzsch ester and the model compound 4a can absorb light at 300 nm. Note that the background reaction between the substrate and 7 was previously reported in the literature.16 In the absence of the catalyst, however, the photodesulfonylation does not allow to isolate the desired product in a good yield even after irradiating for 16 h. The reaction efficiency was significantly decreased when performing the transformation under an oxygen atmosphere (Table 2, entry 16). Very low conversions were observed when radical scavengers (TEMPO or BHT, 1.2 equiv., Table 2, entries 17 and 18) were added to the reaction mixture, suggesting that the N–S bond cleavage occurs via the formation of radical intermediates. Different hydrogen donors were also screened, including 1,4-dihydronicotinamide 8, acridine derivative 9 and n-Bu4NBH4, but all failed to deliver product 5a (Table 2, entries 19–21). Finally, the paracyclophane-deprived coumarin 6 and commercially available pCp were tested as catalysts and proved to be unreactive. Indeed, in these cases, low conversions were observed (Table 2, entries 22 and 23), comparable to that obtained in the absence of the catalyst. These last results confirm the necessity to employ a pCp-based coumarin to promote the transformation. The optimized detosylation of 4a (Table 2, entry 4) was also performed on the millimolar scale. In this case, product 5a was isolated in 57% yield by irradiating the reaction mixture for 16 h (Scheme 3).
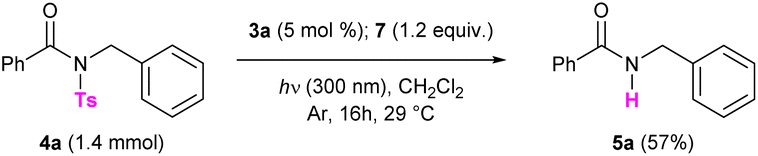 | ||
| Scheme 3 Scale-up of the optimized reaction. | ||
We next turned our attention to the reaction scope (Table 3). The cleavage of different sulfonyl groups (Ts, SO2Ph, SO2Mes, Ms) was tested in the presence of 3a (5 mol%). Analogously to the Ts group (Table 3, entry 1), a phenyl sulfonyl moiety could easily be removed to afford product 5a in 59% yield (Table 3, entry 2). Mesityl and mesyl groups, however, could not be removed under the same conditions as only traces of product 5a were isolated after the reactions (Table 3, entries 3 and 4). Tosylated precursors bearing various substituents were well-tolerated, including differently substituted (hetero)aromatic motifs, alkyl chains and even a Boc group (Table 3, entries 5–12). Compounds incorporating different aroyl moiety reacted as well (Table 3, entries 13 and 14), even if a modest yield was obtained in the presence of an electron-donating group (Table 3, entry 14). Starting from enantiopure compounds 4o and 4p, products 5l and 5m were isolated in 36% and 30% yields respectively without any loss in their enantiopurity, thus demonstrating that base-sensitive stereogenic centers did not racemize under the optimized reaction conditions (Table 3, entries 15 and 16).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | SM | R1 | R4 | R3 | Pa | Yield (%)a |
| Reactions were performed in a Rayonet photochemical reactor equipped with eight 300 nm lamps (T = 29 °C, c = 0.05 M).a Isolated yields.b The reaction was irradiated overnight.c The starting material was recovered after the reaction. | ||||||
| 1 | 4a | Bn | COPh | Ts | 5a | 65 |
| 2 | 4b | Bn | COPh | SO2Ph | 5a | 59 |
| 3b | 4c | Bn | COPh | SO2Mes | 5a | 11 |
| 4b | 4d | Bn | COPh | Ms | 5a | 3 |
| 5 | 4e |
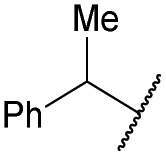
|
COPh | Ts | 5b | 72 |
| 6b | 4f |
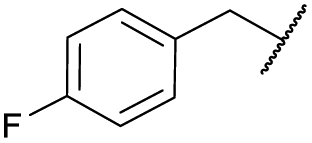
|
COPh | Ts | 5c | 60 |
| 7b | 4g |
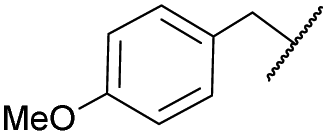
|
COPh | Ts | 5d | 61 |
| 8 | 4h |
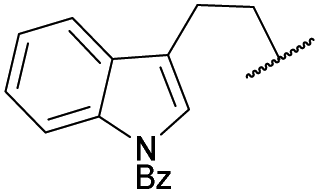
|
COPh | Ts | 5e | 54 |
| 9 | 4i | Me | COPh | Ts | 5f | 58 |
| 10 | 4j | n-Bu | COPh | Ts | 5g | 56 |
| 11 | 4k | Boc | COPh | Ts | 5h | 84 |
| 12 | 4l | allyl | COPh | Ts | 5i | 78 |
| 13b | 4m | Bn |
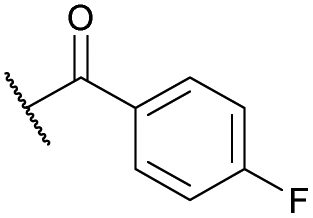
|
Ts | 5j | 54 |
| 14b | 4n | Bn |
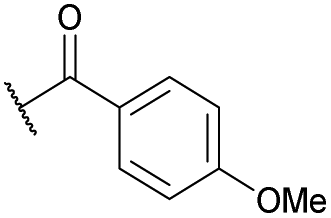
|
Ts | 5k | 39 |
| 15b | 4o |
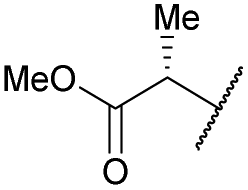
|
COPh | Ts | 5l | 36 |
| 16b | 4p |
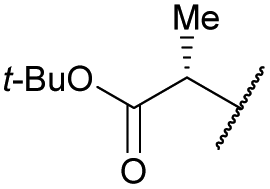
|
COPh | Ts | 5m | 30 |
| 17 | 4q | Bn | Boc | Ts | 5n | —c |
| 18 | 4r | Bn | COMe | Ts | 5o | —c |
| 19 | 4s | Bn | Me | Ts | 5p | —c |
Note that the presence of an aroyl moiety on the sulfonylated precursors was required for the reaction to work. Indeed, acetyl, Boc- or Me-substituted sulfonamides 4q–s proved to be unreactive (Table 3, entries 17–19). Based on this observation, the possibility to perform a chemoselective cleavage of sulfonamides was investigated. Starting from compound 4t, which incorporates two different tosyl groups, we were happy to observe the formation of the mono-deprotected product 5q in 61% yield (Scheme 4).
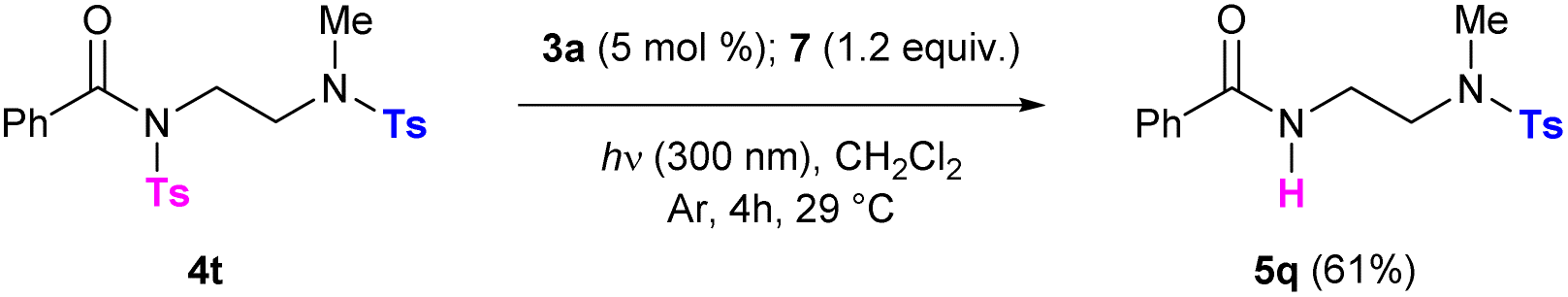 | ||
| Scheme 4 Chemoselective photocleavage of differently substituted sulfonamides. | ||
We finally wished to gain an insight into the mechanism of the photodesulfonylation promoted by the pCp-based coumarins. Theoretical calculations revealed that an energy transfer from the excited states of catalyst 3a to the lowest excited state of substrate 4a is endergonic and can therefore be ruled out (see the ESI‡ for more details). By analogy with the previously described method relying on the use of an Iridium-based catalyst in combination with the Hantzsch ester,16 the observed photocleavage of sulfonamides was thus believed to proceed via an electron transfer (ET) pathway. Considering the redox potentials of 3a, 4a and 7 as well as the excitation energy (E*) associated with 3a, the ET process seems to be thermodynamically favourable only when it occurs between the catalyst and the Hantzsch ester (ΔGET = −0.63 eV, see the ESI‡ for more details). On the contrary, direct electron transfer between 3a and 4a appears to be unfavourable (ΔGET = 0.13 eV, see the ESI‡ for more details). The formation of electron donor–acceptor (EDA) complexes between the reaction partners was excluded by UV-vis analysis of catalyst 3a in the presence of increasing quantities of either 4a or 7.
In addition, we confirmed that the reaction stops as soon as the light irradiation is turned off, therefore excluding the possibility for the photocleavage to happen through a self-sustaining radical process.
Electron paramagnetic resonance (EPR) spectroscopy allowed us to observe the formation of a radical species when the photodesulfonylation was performed in the presence of all the reaction partners. The signal appeared as a single line centered at g = 2.0055 (Fig. 1). A similar but significantly less intense signal was observed by monitoring a light-promoted process between the Hantzsch ester 7 and 4a, thus confirming that a low-yielding background reaction can occur between these two reagents. No EPR signal was recorded in the absence of light, or by mixing only the catalyst and the substrate or the catalyst and the Hantzsch ester. Interestingly, a different radical species was detected while performing a room temperature photolysis of p-toluene sulfonyl chloride in the presence of di-t-butyl peroxide and triethyl silane. In this case, the observed signal was centered at g = 2.0046, in good agreement with data previously reported for arylsulfonyl radicals (ArSO2˙).20 Note that hyperfine splitting of this EPR signal could be observed, as previously described,20,21 when generating the radical in toluene at low temperature (193 K, see the ESI‡ for more details). This result allowed us to exclude that ArSO2˙ radicals may be the species observed by EPR in our reaction conditions.
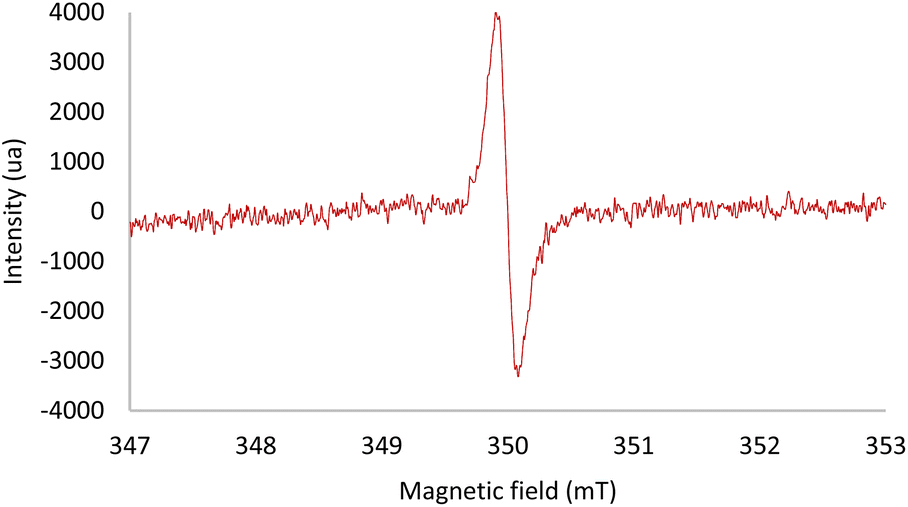 | ||
| Fig. 1 EPR spectroscopy of a solution of 3a (5 mol%), 4a (1 equiv.) and 7 (1.2 equiv.) in CH2Cl2 (0.05 M) under irradiation at 300 nm in a quartz tube under an argon atmosphere. | ||
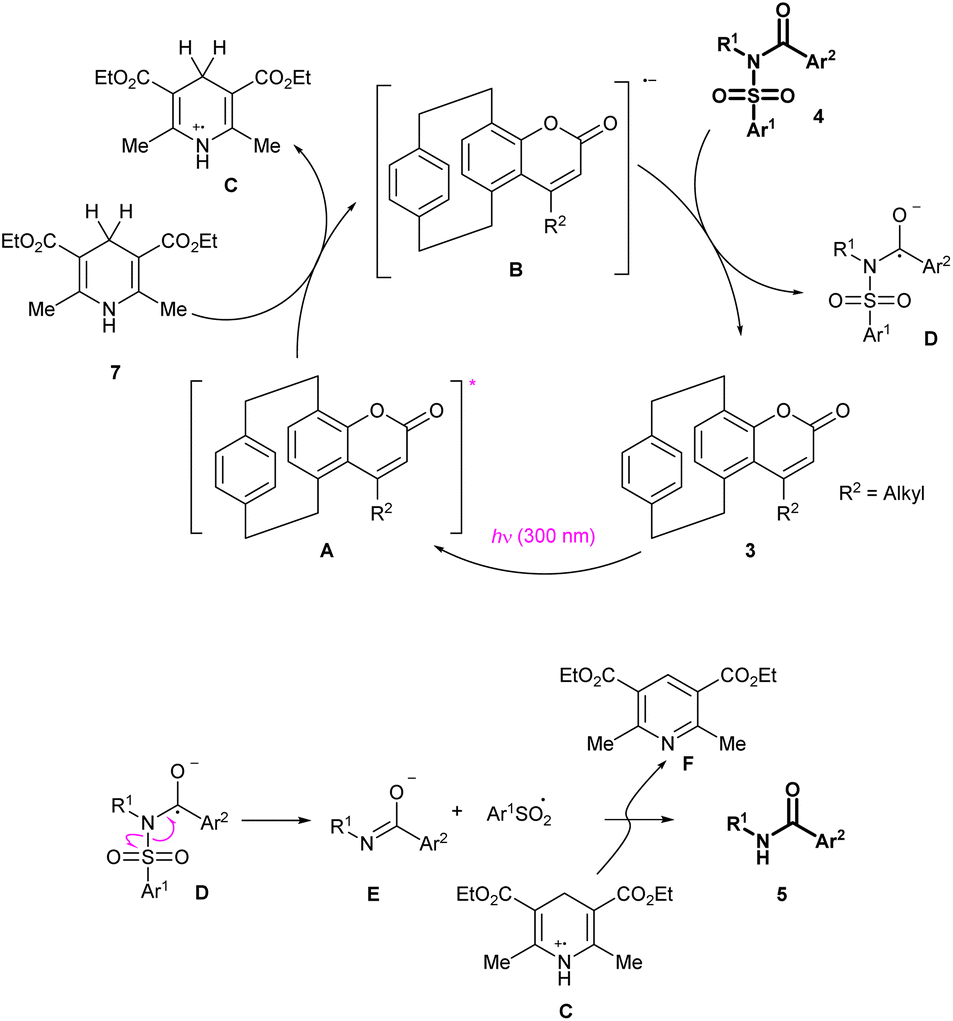
Considering all this experimental evidence, the observed transformation is supposed to occur through the mechanism depicted in Scheme 5. The pCp-based catalyst 3 is excited by light irradiation at 300 nm. The resulting excited species (A) reacts with the Hantzsch ester 7. A single electron transfer occurs at this stage, leading to the formation of a dihydropyridine radical cation (C) and a coumarin-derived radical anion (B).
 | ||
| Scheme 5 Proposed reaction mechanism. | ||
The latter interacts with the reaction substrate 4 to form radical D and regenerate the photocatalyst. Note that formation of the reaction intermediate D was previously proposed for analogous electrochemical transformations,12g and is supported by the fact that the reaction does not occur in the absence of an aroyl moiety on the starting material. The reactive species D decomposes through an homolytic cleavage of its N–S bond to afford a carboxamide anion (E) and an aryl sulfonyl radical (Ar1SO2˙). Quenching reactions occurring between these species and the dihydropyridine radical C finally afford the detosylated product 5.
DFT calculations were performed to elucidate further this putative mechanism and obtain a better understanding of this novel reaction. First, as previously anticipated, an energy transfer pathway was ruled out as it was found to be thermodynamically unfavoured (ΔGEnT = +1.13 eV, see ESI‡ for details). The free energy of each intermediate was then computed to verify that each proposed step was exergonic (see ESI‡ for details). In an attempt to rationalize the yield discrepancy observed between the photodesulfonylations performed with catalysts 3a, 6 and 3c (Table 2, entry 4 vs. entries 22 and 12), the difference between the ΔG of the two diverse reactions yielding intermediate D (Scheme 4) was calculated. It was found that the photocleavage catalysed by 3c (compound incorporating a phenyl substituent) has a free energy 5.5 kcal mol−1 higher than the one promoted by 3a (compound incorporating a methyl substituent), thus providing a possible explanation for the yield difference. Additionally, TD-DFT calculations were conducted, and it was found that the first excited state of 6 is 9.2 kcal mol−1 higher than that of 3a, which could explain the yield discrepancy between these catalysts.
Calculations were finally performed to try and clarify the nature of the radical species observed by EPR spectroscopy. Starting from the optimized geometries of the different intermediates, the EPR parameters of all potential radicals were calculated (see ESI‡ for details). The results obtained after this study were in accordance with published g values for the known radical TsSO2˙ (gmeasured = 2.0046; gcalculated = 2.0048).20 Unfortunately, D was the only intermediate with a calculated g that would correspond to the observed signal (gmeasured = 2.0055; gcalculated = 2.0050) but its calculated nitrogen isotropic hyperfine coupling (HFC) constant of Acalc = 0.14 mT is incompatible with the experimental spectrum obtained by EPR analysis, which does not display any hyperfine structure. The HFCs calculations being known to be quite sensitive to the geometry of the molecules, further studies are ongoing to precisely determine the identity and structure of the radical observed. These will be reported in due course.
Conclusions
In conclusion, we have reported for the first time that [2.2]paracyclophane-fused coumarins can be employed as organophotocatalysts to promote light-induced desulfonyation reactions in combination with the Hantzsch ester. The transformation tolerates various substituents on the starting materials. An aroyl moiety is, however, required on the sulfonylated precursors to observe the formation of the desired products. Experimental mechanistical investigations and theoretical calculations suggest that this reaction occurs via an electron transfer pathway which leads to the formation of a key radical intermediate that requires further characterization. This proof-of-concept study opens new horizons for expanding the range of applications of paracyclophanes in the field of photocatalysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully thank the Agence Nationale de la Recherche (ANR PhotoChiraPhane), CNRS, IdEx Université Paris Cité, and the Ministère de l'Enseignement Supérieur et de la Recherche for financial support. This work was granted access to the HPC resources of IDRIS under the allocation 2022-AD010813916 made by GENCI. P. Gerardo is kindly acknowledged for his help with mass analysis. We are grateful to B. Colasson for the fruitful discussions.References
- C. J. Brown and A. C. Farthing, Nature, 1949, 164, 915 CrossRef CAS.
- H. Hope, J. Bernstein and K. N. Trueblood, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1972, 28, 1733 CrossRef CAS.
- (a) I. Majerz and T. Dziembowska, J. Phys. Chem. A, 2016, 120, 8138 CrossRef CAS PubMed; (b) S. E. Galembeck, R. P. Orenha, R. M. Madeira, L. B. Peixoto and R. L. T. Parreira, J. Braz. Chem. Soc., 2021, 32, 1447 CAS; (c) D. J. Cram, N. L. Allinger and H. Steinberg, J. Am. Chem. Soc., 1954, 76, 6132 CrossRef CAS; (d) D. Klee, N. Weiss and J. Lahann, in Modern Cyclophane Chemistry, ed. R. Gleiter and H. Hopf, Wiley-VCH, Weinheim, 2004 Search PubMed.
- (a) J. P. Chen, K. Ueno and K. Suzuki, US6869698, 2005; (b) H. Goromaru, N. Shigeiwa, M. Murata and S. Maeda, JP4918803, 2012; (c) G. P. Bartholomew, I. Ledoux, S. Mukamel, G. C. Bazan and J. Zyss, J. Am. Chem. Soc., 2002, 124, 13480 CrossRef CAS PubMed; (d) J. Zyss, I. Ledoux, S. Volkov, V. Chernyak, S. Mukamel, G. P. Bartholomew and G. C. Bazan, J. Am. Chem. Soc., 2000, 122, 11956 CrossRef CAS.
- H. Nandivada, H.-Y. Chen, L. Bondarenko and J. Lahann, Angew. Chem., Int. Ed., 2006, 45, 3360 CrossRef CAS.
- (a) D. J. Cram and N. L. Allinger, J. Am. Chem. Soc., 1955, 77, 6289 CrossRef CAS; (b) R. S. Cahn, C. K. Ingold and V. Prelog, Angew. Chem., Int. Ed. Engl., 1966, 5, 385 CrossRef CAS.
- (a) S. Felder, S. Wu, J. Brom, L. Micouin and E. Benedetti, Chirality, 2021, 33, 506 CrossRef CAS PubMed; (b) J. Paradies, Synthesis, 2011, 3749 CrossRef CAS; (c) S. E. Gibson and J. D. Knight, Org. Biomol. Chem., 2003, 1, 1256 RSC.
- (a) Y. Morisaki and Y. Chujo, Bull. Chem. Soc. Jpn., 2019, 92, 265 CrossRef CAS; (b) Z. Hassan, E. Spuling, D. M. Knoll, J. Lahann and S. Bräse, Chem. Soc. Rev., 2018, 47, 6947 RSC; (c) S. Felder, M.-L. Delcourt, R. Rodríguez, L. Favereau, J. Jeanne Crassous, L. Micouin and E. Benedetti, J. Mater. Chem. C, 2023, 11, 2053 RSC.
- H. Hopf, T. Laue and M. Zander, Angew. Chem., Int. Ed. Engl., 1991, 30, 432 CrossRef.
- D. M. Knoll, C. Zippel, P. Weis, M. M. Kappes, Z. Hassan, M. Nieger and S. Bräse, Dalton Trans., 2019, 48, 17704 RSC.
- (a) H. R. Snyder and R. E. Heckert, J. Am. Chem. Soc., 1952, 74, 2006 CrossRef CAS; (b) D. I. Weisblat, B. J. Magerlein and D. R. Myers, J. Am. Chem. Soc., 1953, 75, 3630 CrossRef CAS; (c) E. Wellner, H. Sandin and L. Pääkkönen, Synthesis, 2003, 0223 CrossRef CAS; (d) B. A. Merrill and E. LeGoff, J. Org. Chem., 1990, 55, 2904 CrossRef CAS; (e) N. K. Garg, R. Sarpong and B. M. Stoltz, J. Am. Chem. Soc., 2002, 124, 13179 CrossRef CAS PubMed; (f) S. Ji, L. B. Gortler, A. Waring, A. J. Battisti, S. Bank, W. D. Closson and P. A. Wriede, J. Am. Chem. Soc., 1967, 89, 5311 CrossRef CAS.
- (a) T. Ankner and G. Hilmersson, Org. Lett., 2009, 11, 503 CrossRef CAS; (b) E. Vedejs and S. Lin, J. Org. Chem., 1994, 59, 1602 CrossRef CAS; (c) H. Knowles, A. Parsons and R. Pettifer, Synlett, 1997, 271 CrossRef CAS; (d) K. L. Jensen, P. T. Franke, L. T. Nielsen, K. Daasbjerg and K. A. Jørgensen, Angew. Chem., Int. Ed., 2010, 49, 129 CrossRef CAS PubMed; (e) G. Blay, L. Cardona, E. Climent and J. R. Pedro, Angew. Chem., Int. Ed., 2008, 47, 5593 CrossRef CAS; (f) M. Kuriyama, T. Soeta, X. Hao, Q. Chen and K. Tomioka, J. Am. Chem. Soc., 2004, 126, 8128 CrossRef CAS PubMed; (g) P. Viaud, V. Coeffard, C. Thobie-Gautier, I. Beaudet, N. Galland, J.-P. Quintard and E. Le Grognec, Org. Lett., 2012, 14, 942 CrossRef CAS PubMed.
- B. Paul, H. Paul and I. Chatterjee, Synthesis, 2022, 5409 CrossRef CAS.
- I. A. MacKenzie, L. Wang, N. P. R. Onuska, O. F. Williams, K. Begam, A. M. Moran, B. D. Dunietz and D. A. Nicewicz, Nature, 2020, 580, 76 CrossRef CAS PubMed.
- (a) E. Hasegawa, T. Tanaka, N. Izumiya, T. Kiuchi, Y. Ooe, H. Iwamoto, S. Takizawa and S. Murata, J. Org. Chem., 2020, 85(6), 4344 CrossRef CAS PubMed; (b) J. Brom, A. Maruani, L. Micouin and E. Benedetti, J. Org. Chem., 2023, 88, 5923 CrossRef CAS PubMed.
- J. Xuan, B.-J. Li, Z.-J. Feng, G.-D. Sun, H.-H. Ma, Z.-W. Yuan, J.-R. Chen, L.-Q. Lu and W.-J. Xiao, Chem. – Asian J., 2013, 8, 1090 CrossRef CAS.
- (a) A. Gualandi, G. Rodeghiero, E. Della Rocca, F. Bertoni, M. Marchini, R. Perciaccante, T. P. Jansen, P. Ceroni and P. G. Cozzi, Chem. Commun., 2018, 54, 10044 RSC; (b) A. Gualandi, A. Nenov, M. Marchini, G. Rodeghiero, I. Conti, E. Paltanin, M. Balletti, P. Ceroni, M. Garavelli and P. G. Cozzi, ChemCatChem, 2021, 13, 981 CrossRef CAS.
- (a) E. Benedetti, M.-L. Delcourt, B. Gatin-Fraudet, S. Turcaud and L. Micouin, RSC Adv., 2017, 7, 5047 RSC; (b) M.-L. Delcourt, C. Reynaud, S. Turcaud, L. Favereau, J. Crassous, L. Micouin and E. Benedetti, J. Org. Chem., 2019, 84, 888 CrossRef CAS PubMed; (c) S. Felder, M.-L. Delcourt, M. H. E. Bousquet, D. Jacquemin, R. Rodríguez, L. Favereau, J. Crassous, L. Micouin and E. Benedetti, J. Org. Chem., 2022, 87, 147 CrossRef CAS PubMed.
- C. J. Friedmann, S. Ay and S. Bräse, J. Org. Chem., 2010, 75, 4612 CrossRef CAS PubMed.
- (a) M. Griesser, J.-P. R. Chauvin and D. A. Pratt, Chem. Sci., 2018, 9, 7218 RSC; (b) J. E. Bennett, G. Brunton, B. C. Gilbert and P. E. Whittall, J. Chem. Soc., Perkin Trans. 2, 1988, 1359 RSC; (c) A. G. Davies, B. P. Roberts and B. R. Sanderson, J. Chem. Soc., Perkin Trans. 2, 1973, 626 RSC.
- M. C. R. Symons, J. Am. Chem. Soc., 1969, 91, 5924 CrossRef CAS.
Footnotes |
| † A preprint of this manuscript has been posted on ChemRxiv (https://doi.org/10.26434/chemrxiv-2023-n4ds3). |
| ‡ Electronic supplementary information (ESI) available: Synthetic procedures for the preparation of all products, UV-vis absorption spectra and cyclic voltammograms of the reaction partners, details on the photochemical apparatus, characterization data, theoretical data, copies of 1H NMR and 13C NMR spectra for all the synthesized compounds. See DOI: https://doi.org/10.1039/d3ob01711g |
| This journal is © The Royal Society of Chemistry 2024 |