DOI:
10.1039/D3OB01679J
(Review Article)
Org. Biomol. Chem., 2024,
22, 37-54
Recent advances in ligand-enabled palladium-catalyzed divergent synthesis
Received
15th October 2023
, Accepted 27th November 2023
First published on 27th November 2023
Abstract
Developing efficient and straightforward strategies to rapidly construct structurally distinct and diverse organic molecules is one of the most fundamental tasks in organic synthesis, drug discovery and materials science. In recent years, divergent synthesis of organic functional molecules from the same starting materials has attracted significant attention and has been recognized as an efficient and powerful strategy. To achieve this objective, the proper adjustment of reaction conditions, such as catalysts, solvents, ligands, etc., is required. In this review, we summarized the recent efforts in chemo-, regio- and stereodivergent reactions involving acyclic and cyclic systems catalyzed by palladium complexes. Meanwhile, the reaction types, including carbonylative reactions, coupling reactions and cycloaddition reactions, as well as the probable mechanism have also been highlighted in detail.
 Yue Wang | Yue Wang was born in Henan Province, China. He received his B.S. and Ph.D. degrees in organic chemistry from Zhengzhou University (ZZU) in 2015 and 2022, respectively, under the supervision of Prof. Zheng Duan and Prof. Er-Qing Li. His doctoral project was focused on transition metal catalysis and organic synthesis methodologies. In 2022, he joined Prof. Teck-Peng Loh's team at the College of Advanced Interdisciplinary Science and Technology, Henan University of Technology. His research interests include the chemistry of biological coupling and green catalytic methodologies. |
 Jinzan Feng | Jinzan Feng was born in Henan, China. She is currently a graduate student at Henan University of Technology. She graduated from the Department of Chemistry and Chemical Engineering of Jilin University of Chemical Technology with a Bachelor's degree. In 2022, she joined the College of Advanced Interdisciplinary Science and Technology at Henan University of Technology, under the mutual supervision of Prof. Teck-Peng Loh and Dr Yue Wang. Her research interests include the chemistry of biological coupling. |
 Er-Qing Li | Prof. Er-Qing Li was born in 1987 in Henan province, China. He received his PhD in 2015 from Nankai University under the guidance of Prof. You Huang. Subsequently, he worked as a postdoctoral fellow with Prof. Lutz F. Tietze at Göttingen University, Germany. He then joined Zhengzhou University at the end of 2016 and started his independent research. His major research interests are asymmetric catalysis, design and synthesis of P-stereogenic phosphine ligands and transition metal catalysis. |
 Zhenhua Jia | Zhenhua Jia is a full professor at the College of Advanced Interdisciplinary Science and Technology, Henan University of Technology. He received his BS in 2004 from Tianjin University and PhD in 2013 from Sun Yat-sen University and joined McGill University under the supervision of Prof. Albert S. C. Chan and Prof. Chao-Jun Li. Then he continued to pursue his postdoctoral research at Marquette University, Chinese University of Hong Kong and the University of Alberta with Prof. Chae S. Yi, Prof. Henry N. C. Wong, and Prof. Frederick G. West, respectively. In 2018, he joined Prof. Teck-Peng Loh's team and held his current position. His research interests include green and sustainable synthesis and organic transformations under biocompatible conditions. |
 Teck-Peng Loh | Teck-Peng Loh is a distinguished university professor of Chemistry at Nanyang Technological University, Singapore. Under the tutelage of Professor E. J. Corey, he obtained his PhD (1994) from Harvard University. He has been awarded outstanding researcher awards from both the National University of Singapore and Nanyang Technological University. In 2017, he received the Yoshida Prize (Japan) and the prestigious President's Science Award (individual) in Singapore. He has been a Fellow of Academia of Sciences, Singapore (2018) and a Fellow of the Academia of Sciences, Malaysia since 2010. His research work mainly focuses on the development of new synthetic methodologies, green chemistry, and the synthesis of natural and unnatural products. |
1. Introduction
As a result of the specificity of living organisms, high-throughput screening of compound libraries is required in drug discovery, these libraries mainly consist of a large number of structurally similar molecules.1 Therefore, precise synthesis of organic molecules is one of the fundamental tasks in synthesis chemistry. Organic chemists have been concerned with the synthesis of structurally diverse “privileged” molecular scaffolds with high yields and high selectivity. Normally, different starting materials and reaction types are used to separately prepare structurally similar molecules by organic chemists, which makes the process time-consuming and laborious. Recently, a simpler strategy was developed: using the same starting material and small variations in reaction conditions realizes the synthesis of diverse isomers with different chemo-, regio- or stereoselectivities. Due to the advantage of diversity-oriented synthesis, divergent synthesis from the same starting materials has attracted increasing attention in worldwide labs.
The concept of divergent synthesis considers reactions which proceed through different reaction routes or transition states to generate different isomers starting from the same substrate, just by changing usually the catalysts, ligands, solvents or other reaction conditions. Besides, dual catalysis, pioneered by Carreira in 2013,2 has emerged as a hot research topic to realize the stereodivergent synthesis of acyclic and cyclic frameworks (including dual metal, metal/organocatalysis and dual organocatalysis).3 Among them, transition metal-catalyzed diverse divergent synthetic strategies have evolved and remarkable advances have been achieved in recent years. Apparently, the electronic and steric properties of the ligands are predominant for the activity and selectivity of metal complex catalysts. In other words, ligands, much like a chemist's hands, manipulate the reactivity and selectivity in metal catalysis.4 Therefore, the ligand-enabled divergent synthesis strategy has attracted more and more attention, and several reviews and literature reports involving divergent synthesis have been reported by organic chemists.5 For example, Kong and co-workers recently reported advances in Ni-catalyzed ligand-controlled divergent and selective synthesis.6 Meanwhile, these reviews have covered partial aspects of metal-catalyzed divergent synthesis. Surprisingly, to the best of our knowledge, there has still not been a comprehensive review involving palladium-catalyzed divergent synthesis. Given the importance of the palladium catalysis system, catalyzed divergent synthesis will provide organic chemists with a systematic picture of this research field, leading to further development in this new reaction mode.
In this review, we focus on the recent advances in palladium-catalyzed divergent synthesis. The main emphasis of this review is on showing some synthetic potential, exhibiting a new type of catalyst and reaction mechanism, or helping us to understand the origin of selectivity. The review is divided into three major parts: (1) chemodivergent synthesis; (2) regiodivergent synthesis; (3) stereodivergent synthesis. The diverse reactivities, various reaction modes and the mechanistic studies in representative works will be described in detail in this review. We hope that the comprehensive summary will inspire chemists to explore and discover more types of reactions for palladium-catalyzed divergent synthesis.
2 Chemodivergent synthesis
2.1 Ligand-controlled chemodivergent carbonylative reactions
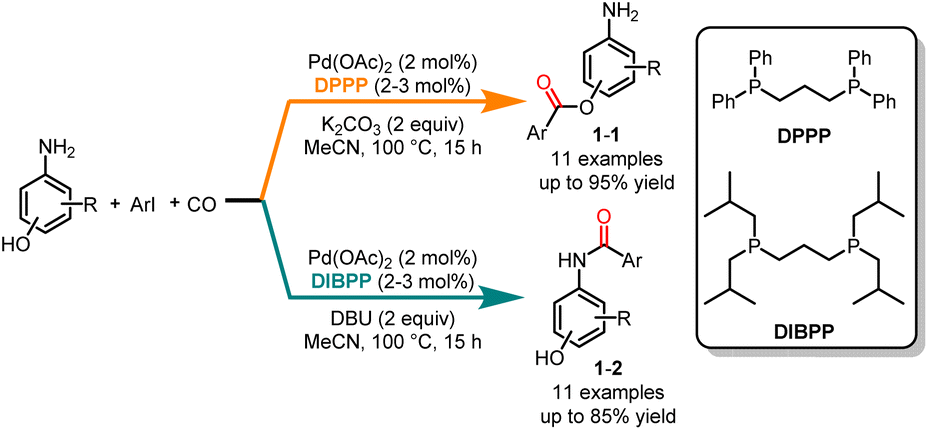
Esters and amides play crucial roles in natural products, drug intermediates, pesticides and materials.7 With its efficiency in constructing carbonyl-containing skeletons, palladium-catalyzed carbonylation has emerged as a versatile tool for the preparation of the corresponding amides and esters.8 In 2014, Alper and co-workers published a groundbreaking study on a ligand-controlled chemoselective carbonylation of aminophenols using iodoarenes (Scheme 1).9 The phosphine ligand played a key role in the selectivity of the carbonylation reaction. The reaction, facilitated by a palladium catalyst, employed 1,3-bis-(diphenylphosphino)propane (DPPP) as the ligand and K2CO3 as the base, and carbonylation occurred on the hydroxyl group, yielding esters 1-1 with high yields and excellent selectivities. In comparison, amides 1-2 were selectively obtained when 1,3-bis-(diisobutylphosphino)-propane (DIBPP) was used as the ligand and DBU as the base. Because the amino group is a stronger nucleophile than the hydroxyl group, electron-rich ligands could more easily react with the amino group to form amide compounds. Remarkably, the exclusive formation of amide products from 2-aminophenols under both reaction conditions suggested coordination at both the nitrogen and oxygen atoms.
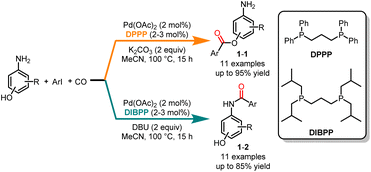 |
| | Scheme 1 Pd-catalyzed chemoselective carbonylation of iodoarenes with aminophenols. | |
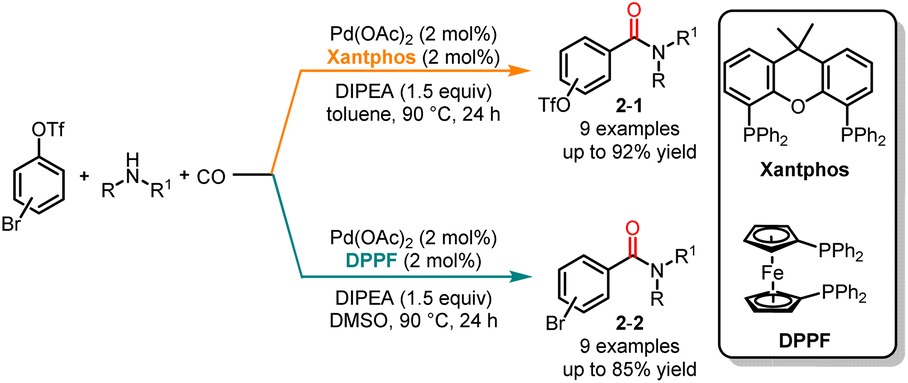
Since the pioneering work of Tsuji and Heck, the chemoselectivity of palladium-catalyzed carbonylation, specifically when a nucleophile possesses two nucleophilic sites, has been extensively investigated by chemists.10 In contrast to the extensive research on chemoselective carbonylation using nucleophiles with multiple nucleophilic sites, there are only a few reported instances of selective carbonylation of dihaloaromatic compounds. In 2017, Wu and co-workers developed palladium-catalyzed ligand- and solvent-controlled chemoselective carbonylation of bromoaryl triflates in high selectivities and yields (Scheme 2).11 The use of 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene (Xantphos) as the ligand in toluene promoted the selective carbonylation of the C-Br bond and produced the products 2-1; while employing 1,1′-bis(diphenylphosphino)ferrocene (DPPF) in DMSO facilitated the carbonylation of the C–O bond yielding the products 2-2. Density functional theory (DFT) calculations revealed that the lower activation barrier for the oxidative addition to the C–Br bond was 25.4 kJ mol−1 compared to the C–O bond when Xantphos was used in toluene. Similarly, using DPPF in DMSO resulted in a slightly higher activation barrier for the C–Br bond compared to that for the C–O bond. The calculated results were consistent with the experimental results.
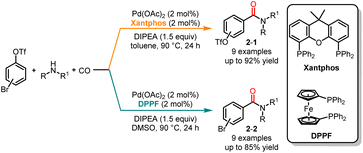 |
| | Scheme 2 Pd-catalyzed chemoselective carbonylation of bromoaryl triflates with amines. | |
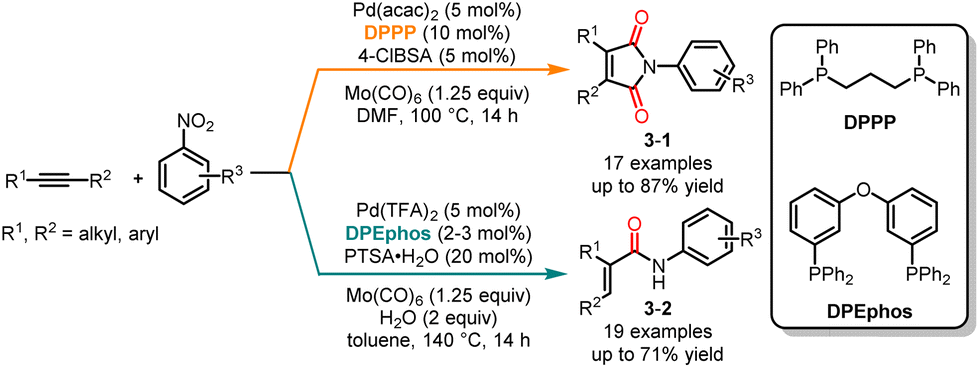
In 2019, Wu and co-workers investigated a ligand-controlled palladium-catalyzed aminocarbonylation of alkynes and nitroarenes (Scheme 3).12 This study resulted in the selective synthesis of a variety of α,β-unsaturated amides and maleimides with moderate to good yields. The combination of Pd(acac)2 and DPPP as the catalyst led to the selective formation of maleimide derivatives 3-1, while using Pd(TFA)2 and DPEphos as the catalysts yielded α,β-unsaturated amides 3-2. A diverse array of nitrobenzenes with electron-donating groups and electron-withdrawing groups could be used in the coupling or cyclization reactions.
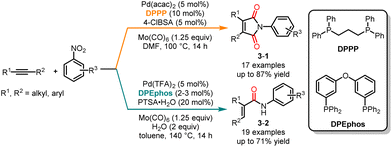 |
| | Scheme 3 Pd-catalyzed chemoselective carbonylation of alkynes and nitroarenes. | |
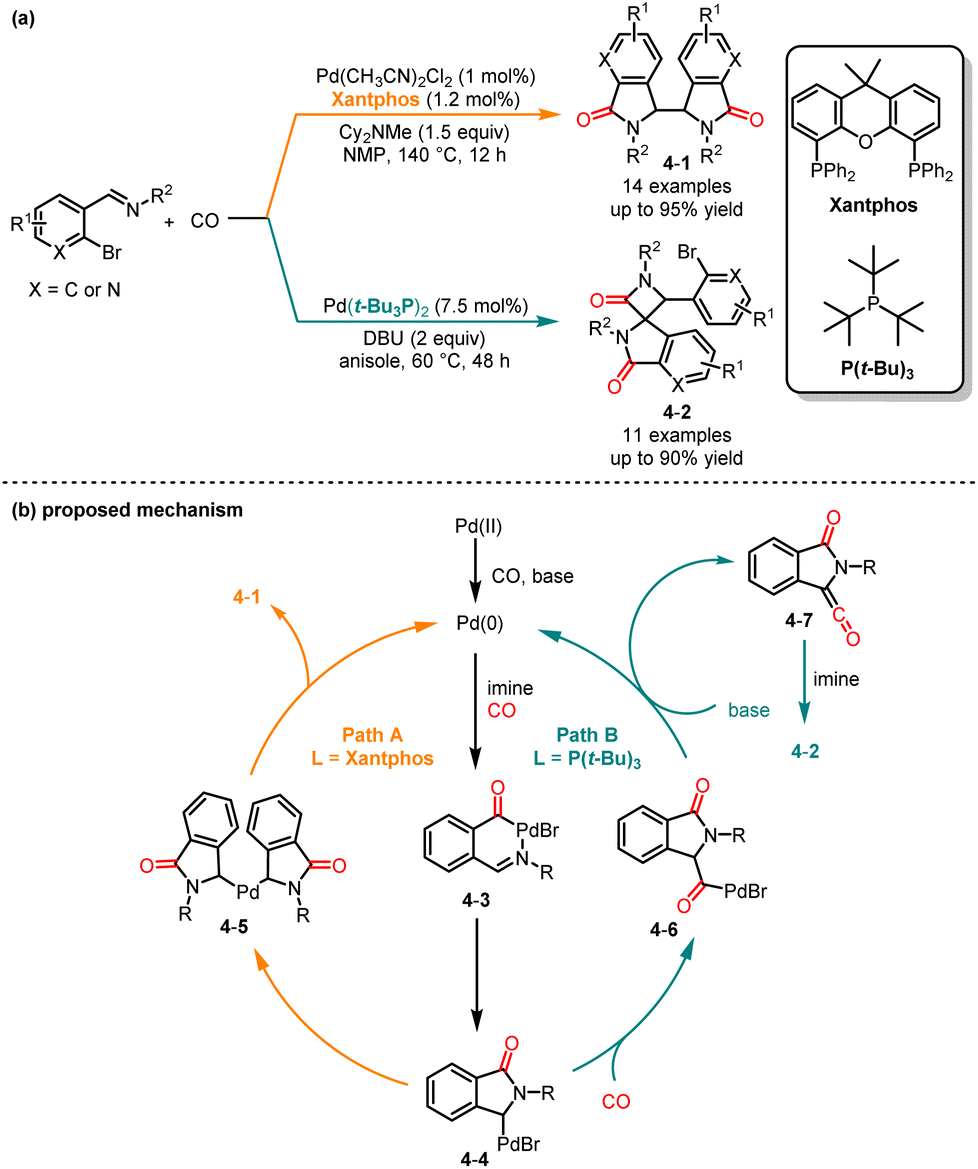
Although various catalytic methods have been established for the synthesis of isoindolinone,13 there is still a lack of methods for the synthesis of biisoindolinones. In 1997, Shim and co-workers introduced a novel carbonylative cyclization reaction to produce biisoindolinone.14 In 2009, Iwasawa and co-workers revealed a molybdenum-catalyzed carbonylative cyclization of β-haloalkenylimines to yield biisoindolinones.15 Recently, Huang and co-workers developed a palladium-catalyzed ligand- and temperature-controlled chemoselective carbonylative cyclization reaction of o-bromoarylimines, providing an efficient method for the synthesis of functionalized biisoindolinones and spirocyclic isoindolinones (Scheme 4a).16 In the presence of Xantphos in anisole at 140 °C, biisoindolinones 4-1 were obtained with good yields and low diastereoselectivity. Alternatively, spirocyclic isoindolinones 4-2 were obtained with high selectivity when Pd(t-Bu3P)2 was used as the catalyst at 60 °C. Based on the experimental results and previous literature reports, two reaction paths were proposed as shown in Scheme 4b. The in situ formed Pd(0) from Pd(II) undergoes oxidative addition with o-bromoarylimine and then insertion of CO to afford intermediates 4-3 and 4-4. At higher temperatures, 4-4 dimerizes to form intermediate 4-5; the biisoindolinone was obtained followed by reductive elimination. At lower temperatures, a second CO insertion and β-hydride elimination occurred to afford intermediates 4-6 and 4-7. Finally, [2 + 2] cycloaddition occured to deliver spirocyclic isoindolinone.
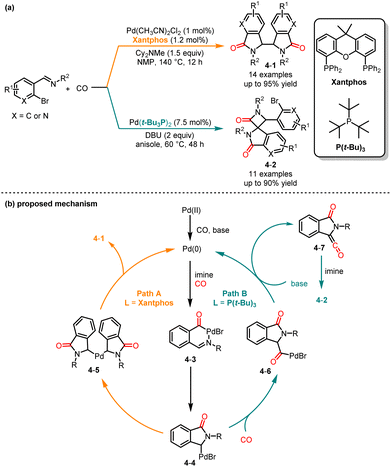 |
| | Scheme 4 Pd-catalyzed chemoselective carbonylation cyclization reaction of o-bromoarylimines. | |
2.2 Ligand-controlled chemodivergent Suzuki coupling reactions
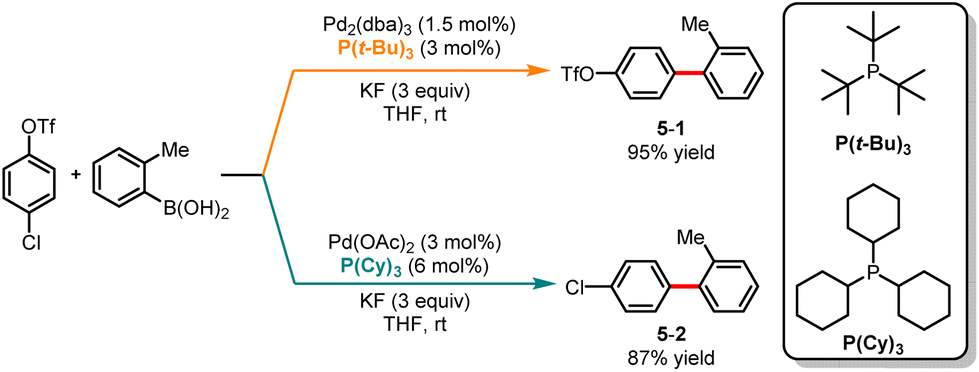
The palladium-catalyzed Suzuki cross-coupling reaction is a powerful synthetic method widely used in organic chemistry for coupling aryl triflates or aryl halides with arylboronic acids.17 As a result, in the past decades, the Suzuki cross-coupling reaction has witnessed exponential growth.18 However, achieving precise control over site selectivity in cross-coupling reactions can be challenging, especially in the presence of multiple (pseudo)halides. In 2010, Fu and co-workers disclosed a palladium-catalyzed ligand-controlled chemoselective Suzuki cross-coupling reaction of boronic acid with 4-chlorophenyltriflate (Scheme 5).19 Using P(t-Bu)3 as a ligand and KF as an additive, cross-coupling occurred at the C–Cl bond leading to the formation of 5-1 with excellent selectivity, while employing PCy3 as a ligand facilitated the cross-coupling of the C–OTf bond generating 5-2 in good yield.
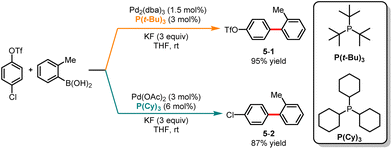 |
| | Scheme 5 Pd-catalyzed chemoselective Suzuki cross-coupling reaction of 4-chlorophenyltriflate. | |
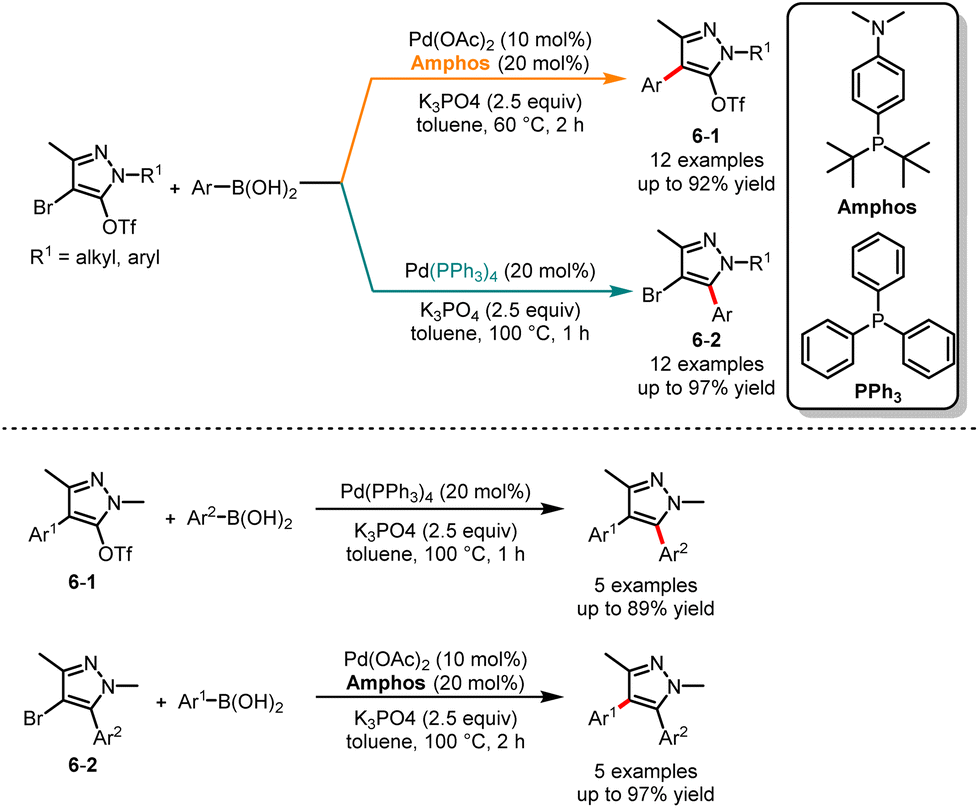
Fujii and co-workers in 2019 described an interesting report on palladium-catalyzed site-selective Suzuki cross-coupling of 4-bromopyrazol-5-yl triflates (Scheme 6).20 It was observed that the selectivity of the arylation reaction at the C4 or C5 position in pyrazole can be guided by the choice of phosphine ligands. The experimental results indicated that the use of a large ligand such as Amphos, resulted in the formation of the C4 coupling products 6-1. Alternatively, a small ligand such as PPh3 afforded the C5 coupling products 6-2. In addition, with the application of each catalytic condition, the obtained triflate 6-1 and bromide 6-2 can be subjected to additional arylation, resulting in two distinct aryl-substituted products.
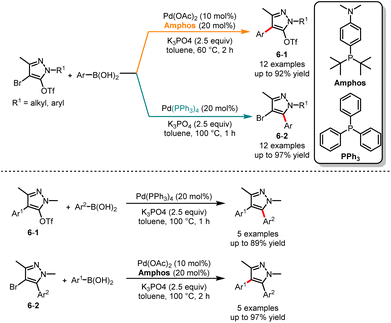 |
| | Scheme 6 Pd-catalyzed chemoselective Suzuki cross-coupling reaction of 4-bromopyrazol-5-yl triflates. | |
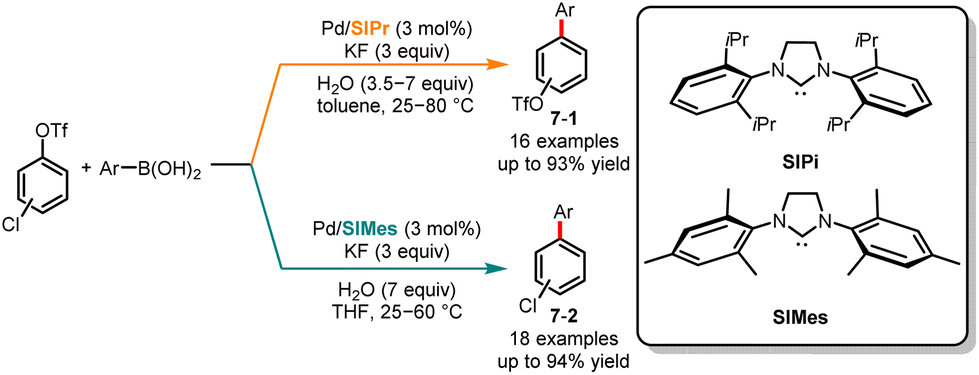
Contrary to the method suggested by Fu's and Fujii's group for selectively controlling Suzuki cross-coupling of multiple (pseudo)halides using phosphine ligands, in the same year, Neufeldt and co-workers revealed an N-heterocyclic carbene ligand-controlled chemoselective Suzuki cross-coupling of chloroaryl triflates (Scheme 7).21 Selective cross-coupling at chloride and triflate is achieved by utilizing 1,3-bis(2,6-diisopropylphenyl)-4,5-dihydroimidazol-2-ylidene (SIPr) and 1,3-bis (2,4,6-trimethylphenyl)-4,5-dihydroimidazol-2-ylidene (SIMes) as ligands, respectively. Through the computational and experimental results, the author concluded that the monoligated [Pd(SIPr)] transition state promoted oxidative addition preferentially at the C–Cl bond, while the oxidative addition of C–OTf might be attributed to bisligated [Pd(SIMes)2] or [Pd(SIMes)X]−.
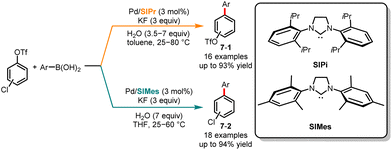 |
| | Scheme 7 Pd-catalyzed chemoselective Suzuki cross-coupling reaction of chloroaryl triflates. | |
2.3 Ligand-controlled chemodivergent cycloaddition reaction
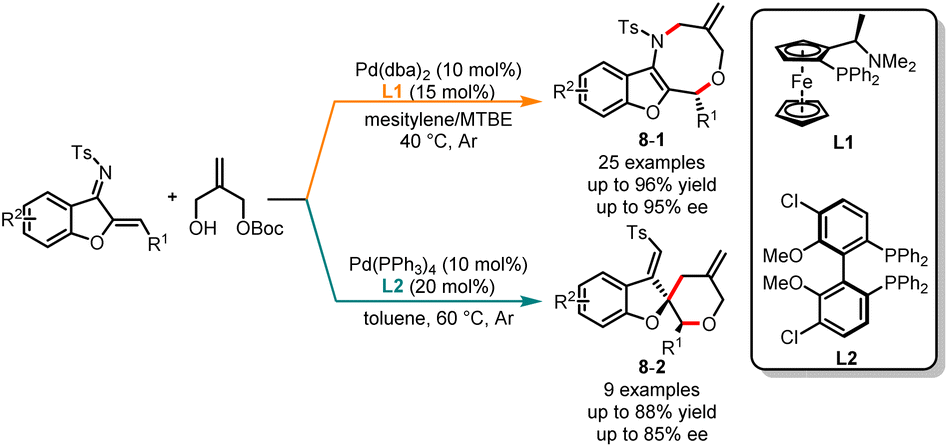
Palladium-catalyzed asymmetric cycloaddition reactions are crucial methods in organic synthesis for the construction of chiral carbon–carbon and carbon–heteroatom bonds, allowing chemists to efficiently create complex molecular structures.22 Different ligands can modulate the electronic and steric properties of the palladium catalyst, thereby influencing its reactivity and ability to control stereochemistry to achieve high regio- and stereoselectivity. Despite the well-established nature of transition metal-mediated asymmetric divergent synthesis reactions, it remains difficult to achieve chemo-, regio-, and enantioselectivity simply by choosing different chiral ligands. In 2021, Lin and co-workers devised a novel ligand-controlled, palladium-catalyzed asymmetric [4 + 4] and [2 + 4] cycloaddition protocol, enabling the coupling of benzofuran-derived azadienes with allyl carbonate (Scheme 8).23 When a chiral P,N-ligand such as (S,Rp)-PPFA (L1) was used, the chemoselective [4 + 4] cycloaddition reactions were observed to deliver the products 8-1 in good yields with high stereo- and chemoselectivities. In contrast, when the chiral P,P-ligand (S)-Cl-MeO-BIPHEP (L2) was employed, the [2 + 4] cyclization products 8-2 were obtained with good yields and moderate stereoselectivities.
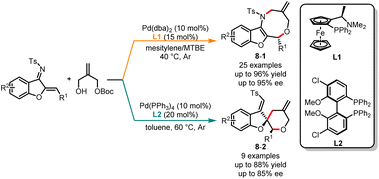 |
| | Scheme 8 Pd-catalyzed chemoselective cycloaddition reaction of benzofuran-derived azadienes. | |
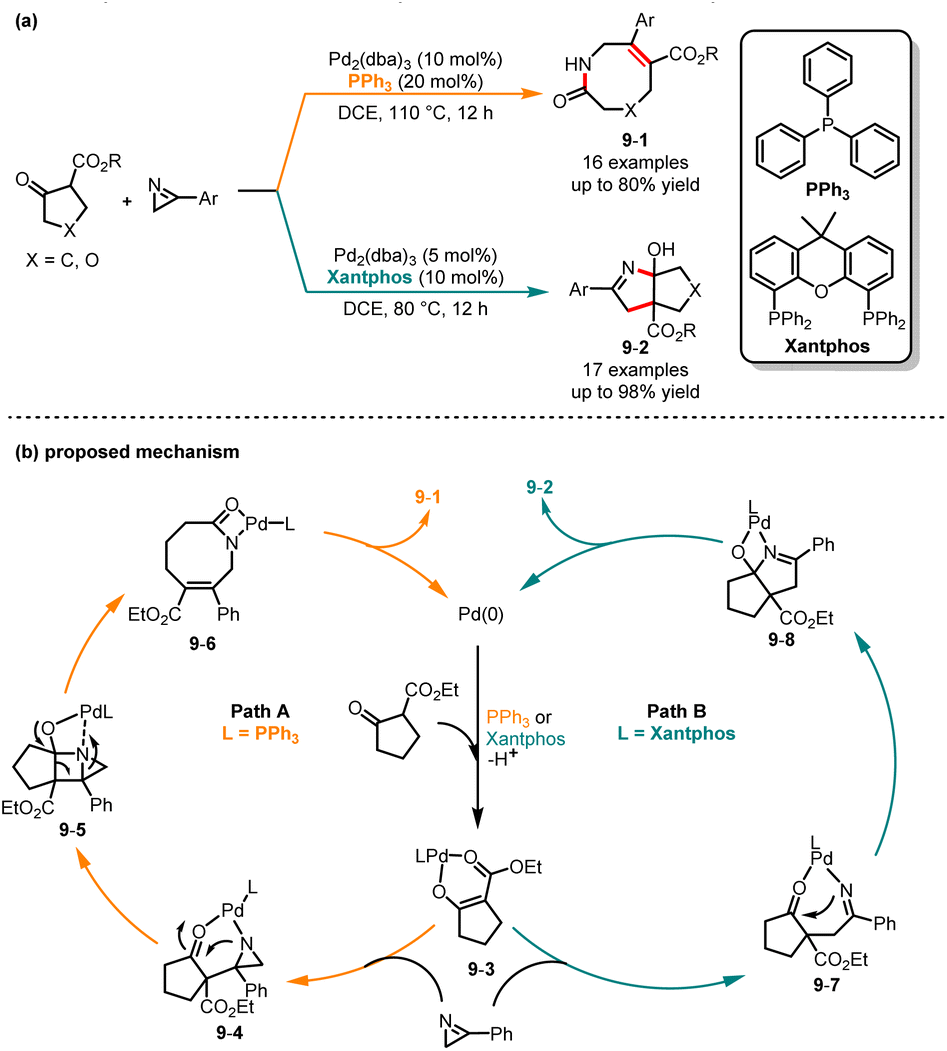
Later, Xie and co-workers reported a ligand-dependent chemodivergent synthesis of eight-membered ring lactams and bicyclic 3,4-dihydro-2H-pyrrole derivatives from cyclic β-keto esters with 3-aryl-2H-azirines (Scheme 9).24 Interestingly, ligand optimization experiments show that the utilization of different ligands can produce entirely distinct products from the same substrate. It was demonstrated that when substrates were treated at 110 °C under the catalysis of Pd2(dba)3/PPh3, a range of eight-membered ring products 9-1 were yielded in moderate to good yields. In contrast, when Pd2(dba)3/Xantphos was used as a catalyst, bicyclic derivatives 9-2 were afforded with good efficiency. A proposed mechanism is outlined in Scheme 9b. Initially, β-ketoester undergoes oxidative addition with the Pd/L complex to afford intermediate 9-3. In path A, intermediate 9-3via a nucleophile attack on the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C3 bond of azirine generates intermediate 9-4. Then, an intramolecular nucleophilic attack and intramolecular ring expansion form intermediate 9-6. Finally, protonation of intermediate 9-6 affords product 9-1. In path B, intermediate 9-3via a nucleophile attack on the NC2 bond gives intermediate 9-7. Subsequent intramolecular nucleophilic addition and protonation afford product 9-2.
C3 bond of azirine generates intermediate 9-4. Then, an intramolecular nucleophilic attack and intramolecular ring expansion form intermediate 9-6. Finally, protonation of intermediate 9-6 affords product 9-1. In path B, intermediate 9-3via a nucleophile attack on the NC2 bond gives intermediate 9-7. Subsequent intramolecular nucleophilic addition and protonation afford product 9-2.
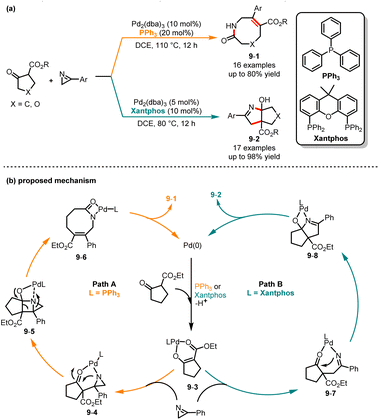 |
| | Scheme 9 Pd-catalyzed chemoselective cycloaddition reaction of β-keto esters with azirines. | |
2.4 Other ligand-controlled chemodivergent reactions
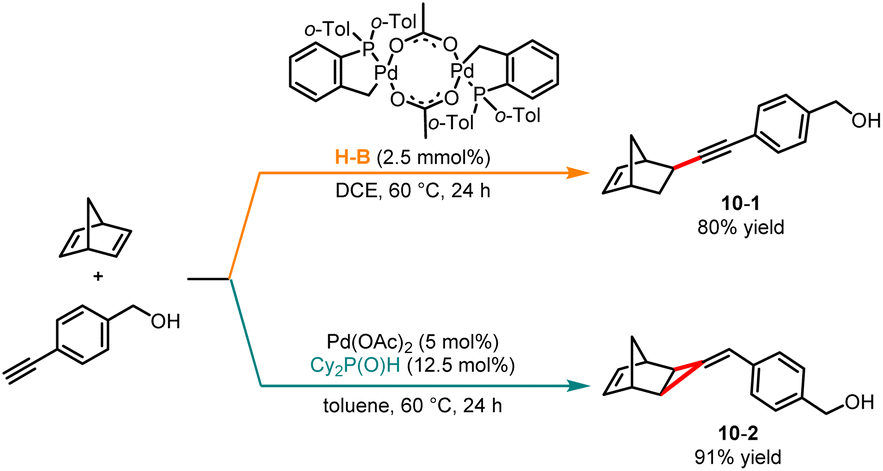
In 2015, Humbel and co-workers disclosed a palladium-catalyzed ligand-controlled hydroalkynation and [2 + 1] cycloaddition of norbornadiene with terminal alkynes (Scheme 10).25 In the presence of Herrmann–Beller phosphapalladacycle as a catalyst, the C–C coupling reaction generated product 10-1 in overall good yields. Contrarily, when phosphinous Cy2P(O)H was employed as a ligand, a [2 + 1] cycloaddition reaction occurred selectively to give the methylenecyclopropane product 10-2. The authors discovered that incorporating AcOH into the reaction system enhanced the catalytic activity. Density functional theory (DFT) calculations revealed that the distance between the proton and the ligand influences the ligand on the interaction with the metal center, resulting in the reaction proceeding through two distinct pathways.
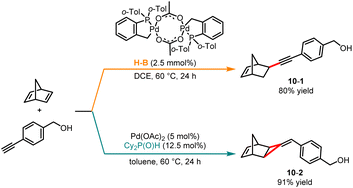 |
| | Scheme 10 Pd-catalyzed divergent hydroalkynation and cycloaddition. | |
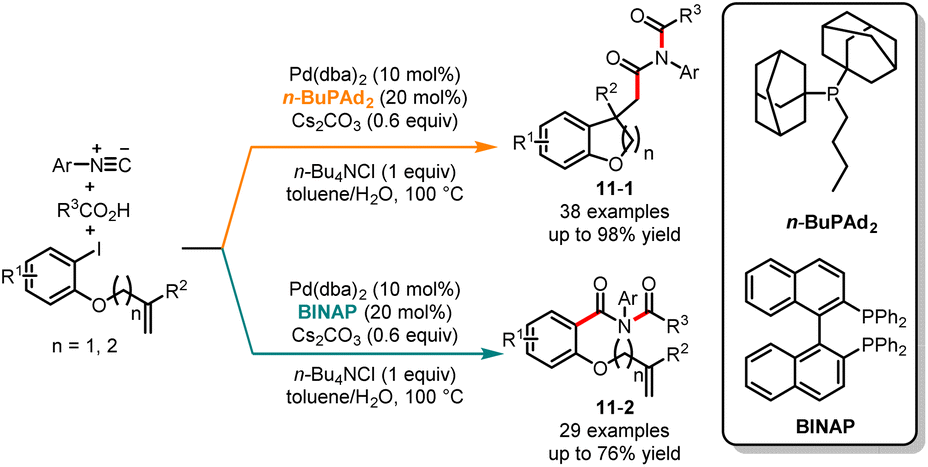
In recent years, the palladium-catalyzed tandem cyclization or coupling reaction has garnered considerable attention due to its ability to efficiently construct functionalized carbocycles and heterocycles.26 This synthetic strategy offers great potential for the synthesis of diverse and complex organic molecules. In 2020, Yao, Fang and co-workers revealed an interesting palladium-catalyzed chemoselective tandem cyclization reaction of aryl iodides, carboxylic acids and isocyanides to access valuable imides (Scheme 11).27 Interestingly, the detailed optimization studies revealed that the ligands play a crucial role in controlling the chemoselectivity by tuning the steric and electronic properties of the catalytic center. When the reaction was treated with the palladium-catalytic system derived from bulky and electron-rich phosphine n-BuPAd2 as a ligand, the cyclization products 11-1 were obtained in good yields with excellent selectivity. On the other hand, employing the bidentate phosphine ligand BINAP resulted in the direct imidation of aryl iodides, leading to the formation of aryl imides 11-2. Very recently, Peng and co-workers reported the ligand-controlled chemoselective synthesis of dihydro-2H-pyrans and γ-dienyl-α-ketoesters through the reaction of β,γ-unsaturated α-ketoesters and alkyl cyclopropanes. When using n-BuPAd2 as the ligand, the 1,4-addition products were generated, while with PPh3 as the ligand, the cyclization products were obtained as the main product.28
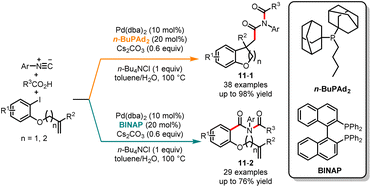 |
| | Scheme 11 Pd-catalyzed chemoselective tandem cyclization reaction of aryl iodides, carboxylic acids and isocyanides. | |
3 Regiodivergent synthesis
3.1 Ligand-controlled regiodivergent carbonylative reactions
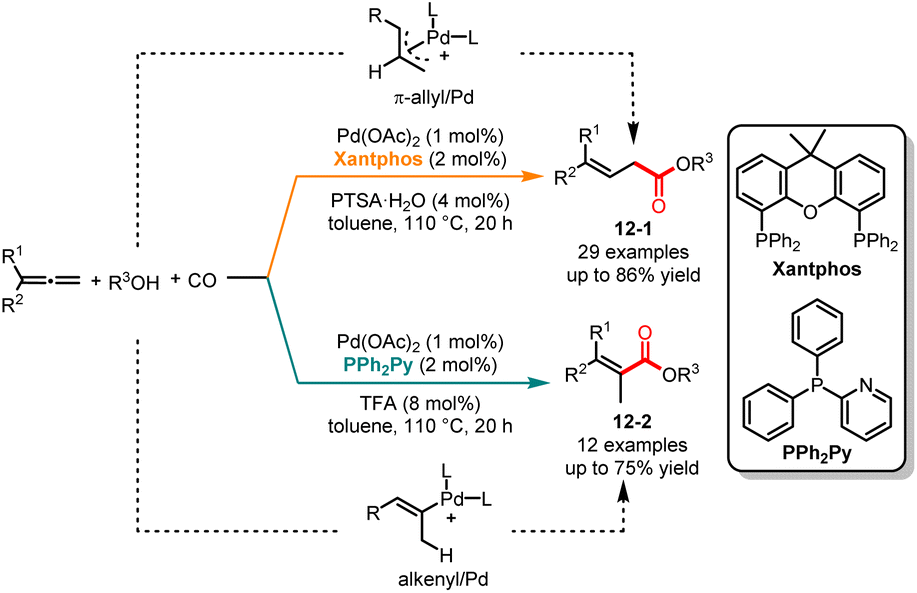
Carbonylation reactions are widely applied in various industries, including industrial production, pharmaceuticals, and functional materials, due to their ability to efficiently introduce multifunctional carbonyl groups into compound skeletons and facilitate the expansion of carbon chains.29 Transition metal-catalyzed alkoxycarbonylation for the synthesis of α,β- and β,γ-unsaturated carboxylic acid derivatives has garnered significant attention in recent years.30 However, there are still some challenges in the following areas, including the substrate scope, atom economy and regioselectivity. In 2015, Beller and co-workers revealed a ligand-controlled, palladium-catalyzed regioselective alkoxycarbonylation of aliphatic alcohols with allenes (Scheme 12).31 It was demonstrated that ligands play a key role in controlling the regioselectivity in this reaction. It was noted that in the presence of Pd(OAc)2/Xantphos as the catalyst and PTSA·H2O as the acid additive, a series of β,γ-unsaturated esters 12-1 were obtained in good yields with excellent selectivities. When the reaction was treated with PPh2Py as a ligand and trifluoroacetic acid as an acid co-catalyst, the corresponding α,β-unsaturated esters 12-2 were selectively obtained in good yields. The deuterium labeling experiment indicated that the C2–C3 double bond of allene can selectively be inserted into the Pd–H bond. A mechanism study showed that the use of Xantphos as a ligand could form a π-allyl/Pd intermediate, resulting in the selective formation of β,γ-unsaturated esters. Conversely, when using PPh2Py as a ligand, the reaction via the alkenyl/Pd intermediate leads to the generation of α,β-unsaturated esters.
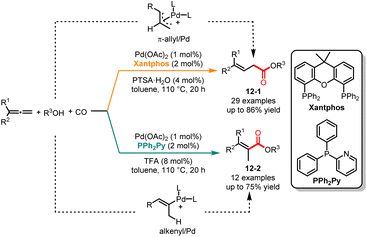 |
| | Scheme 12 Pd-catalyzed regioselective alkoxycarbonylation of aliphatic alcohols with allenes. | |
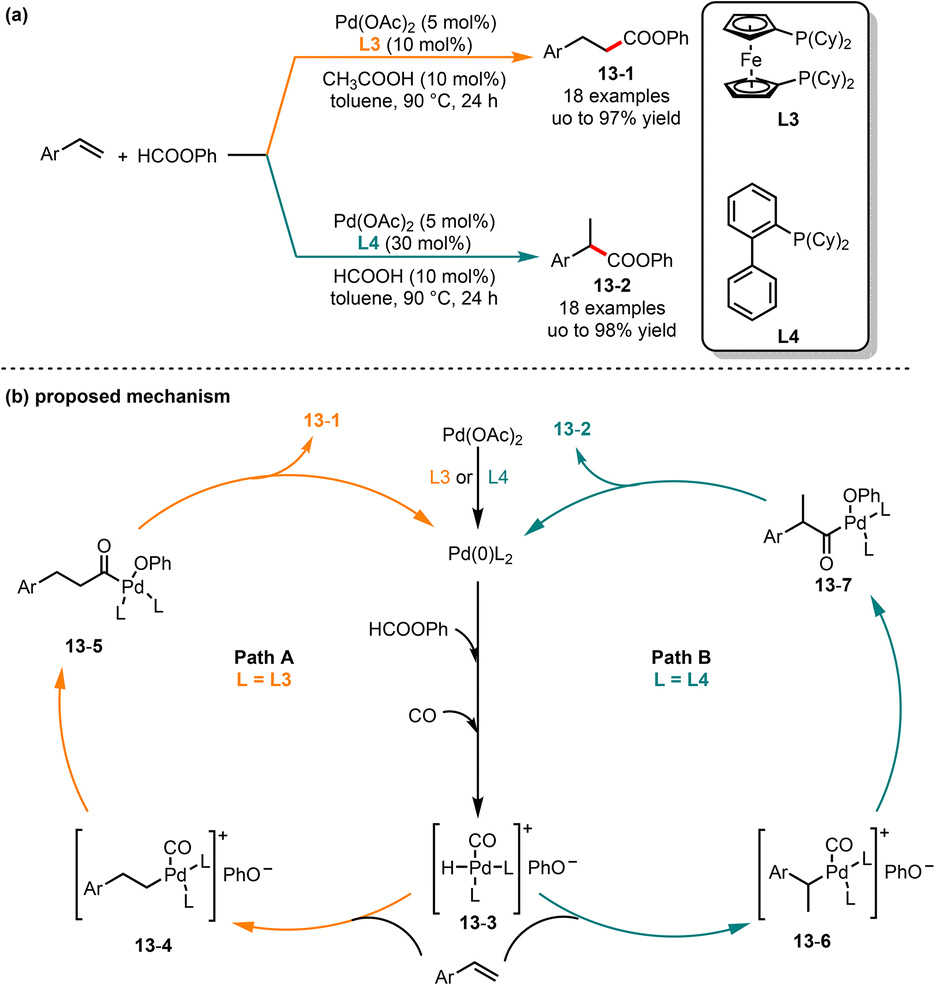
In traditional transition-metal catalyzed hydroesterification, CO was used as the starting material, and the reaction typically requires high pressure and temperature.32 Over the past few decades, chemists have increasingly directed their attention towards the utilization of formate as a substitute for CO in the process of alkoxycarbonylation.33 In 2015, Shi and co-workers introduced a ligand-controlled palladium-catalyzed regioselective hydroesterification of aryl olefins (Scheme 13).34 In this reaction, linear or branched phenyl arylpropanoates could be obtained by choosing different phosphorus ligands, respectively. In the case of bisphosphine ligand L3, linear phenyl arylpropanoates 13-1 were much more favored in good yields. Interestingly, when the monophosphine ligand L4 was employed, the regioselectivity was reversed and produced the branched phenyl arylpropanoates 13-2 in moderate to good yields. Mechanistically, the in situ formed Pd(0) from Pd(II) underwent oxidative addition with phenyl, followed by rearrangement to offer the intermediate 13-3. Then, the intermediate 13-3via a migratory insertion with aryl olefin gave the intermediates 13-6 and 13-7. Finally, linear and branched esters were obtained by reductive elimination of intermediates 13-6 and 13-7, respectively.
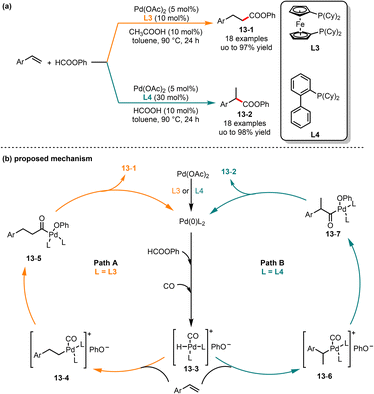 |
| | Scheme 13 Pd-catalyzed regioselective hydroesterification of aryl olefins. | |
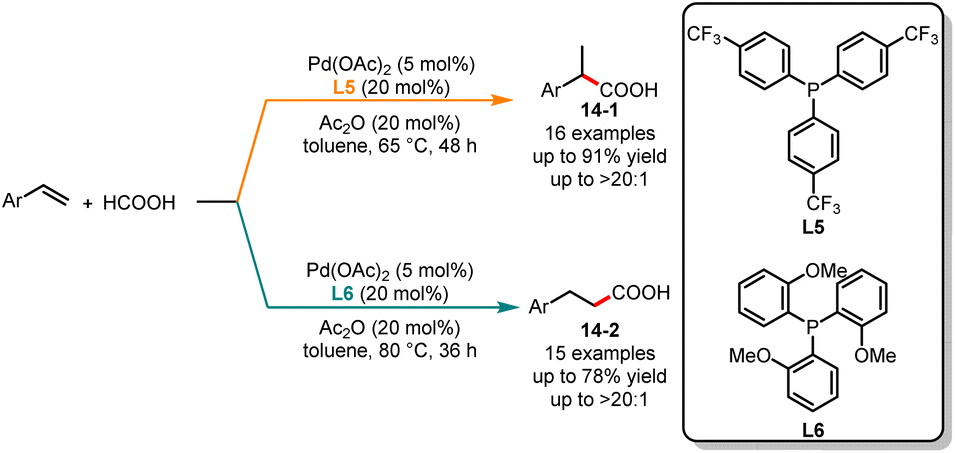
Later, the same group expanded a palladium-catalyzed ligand-controlled regioselective hydrocarboxylation of aryl olefins with formic acid (Scheme 14).35 In this reaction, the authors used formic acid as a CO source. When the reaction proceeded with tris[4-(trifluoromethyl)phenyl]-phosphine (L5) as a ligand, it gave the branched acids 14-1 with excellent yields. In contrast, the linear acids 14-2 were obtained with high selectivity when substrates were treated under the catalysis of Pd(OAc)2/L6.
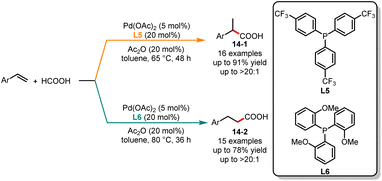 |
| | Scheme 14 Pd-catalyzed regioselective hydrocarboxylation of aryl olefins with formic acid. | |
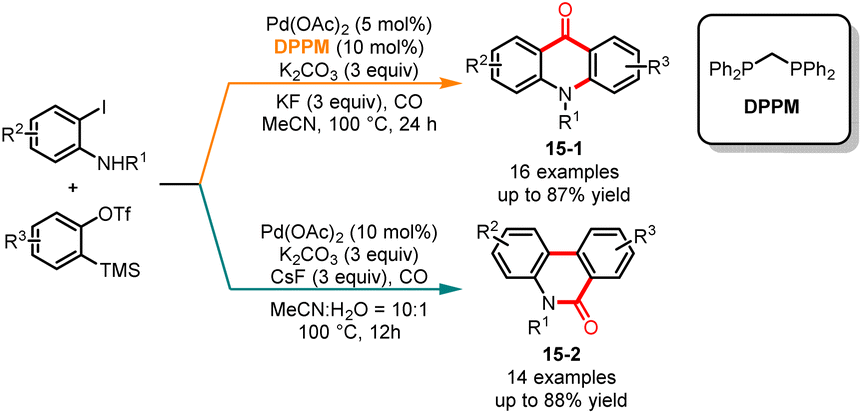
In 2015, Jiang and co-workers published a palladium-catalyzed ligand-controlled regioselective carbonylation of 2-iodoaniline and aryne (Scheme 15).36 In this multi-component reaction, the utilization of ligands enabled the regioselective synthesis of phenanthridinones and acridones. In the presence of the electron-abundant bisphosphine ligand bis(diphenylphosphino)methane (DPPM), the acridones 15-1 were obtained as sole products in moderate to good yields. On the other hand, when the substrates were treated without the ligand, the corresponding phenanthridinones 15-2 were selectively afforded with excellent regioselectivity. The reaction demonstrated extensive substrate versatility and the corresponding compounds could be efficiently synthesized from both electron-deficient and electron-rich iodoanilines. The control experiments showed that the incorporation of CsF and TBAI in the reaction mixture enhanced both the rate of the reaction and the regioselectivity. To further validate the practicality of this research methodology, the authors successfully employed this approach for synthesizing the natural product 2,3-methylenedioxy-10-methyl-9-acridanone.
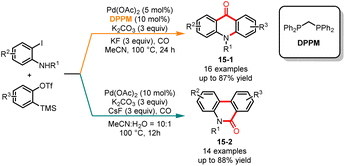 |
| | Scheme 15 Pd-catalyzed regioselective carbonylation of 2-iodoaniline and aryne. | |
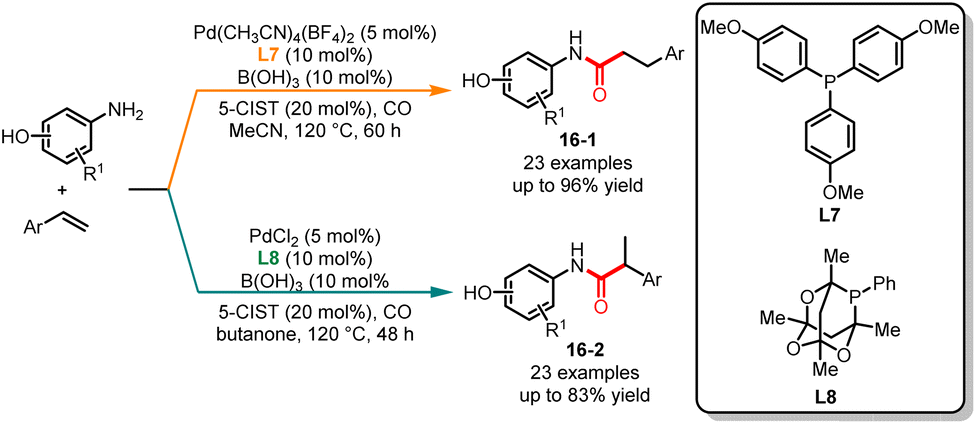
In the previous section, we reviewed the ligand- and base-controlled chemoselective carbonylation of aminophenols reported by Alper's group. The same group published a ligand-controlled regioselective carbonylation of aminophenols with tyrenes in 2016 (Scheme 16).37 In this paper, by employing tris(4-methoxyphenyl)phosphine (L7) as the ligand and 5-chlorosalicylic acid and B(OH)3 as the additives, the corresponding linear amides 16-1 were obtained in good yields and moderate to high selectivities. By changing the ligand to 1,3,5,7-tetramethyl-2,4,8-trioxa-6-phenyl-6-phosphaadamantane (L8) in butanone branched amides were obtained in good yields and excellent selectivities. In addition, ligand screening experiments showed that the utilization of electron-rich ligands, while maintaining the cone angle, imparted greater selectivity for the synthesis of linear amides. Using electron-rich ligands with a bulky structure selectively forms branched amides.
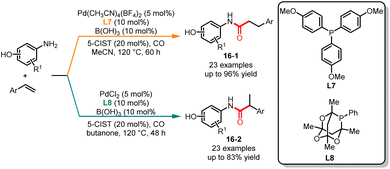 |
| | Scheme 16 Pd-catalyzed regioselective carbonylation of aminophenols with tyrenes. | |
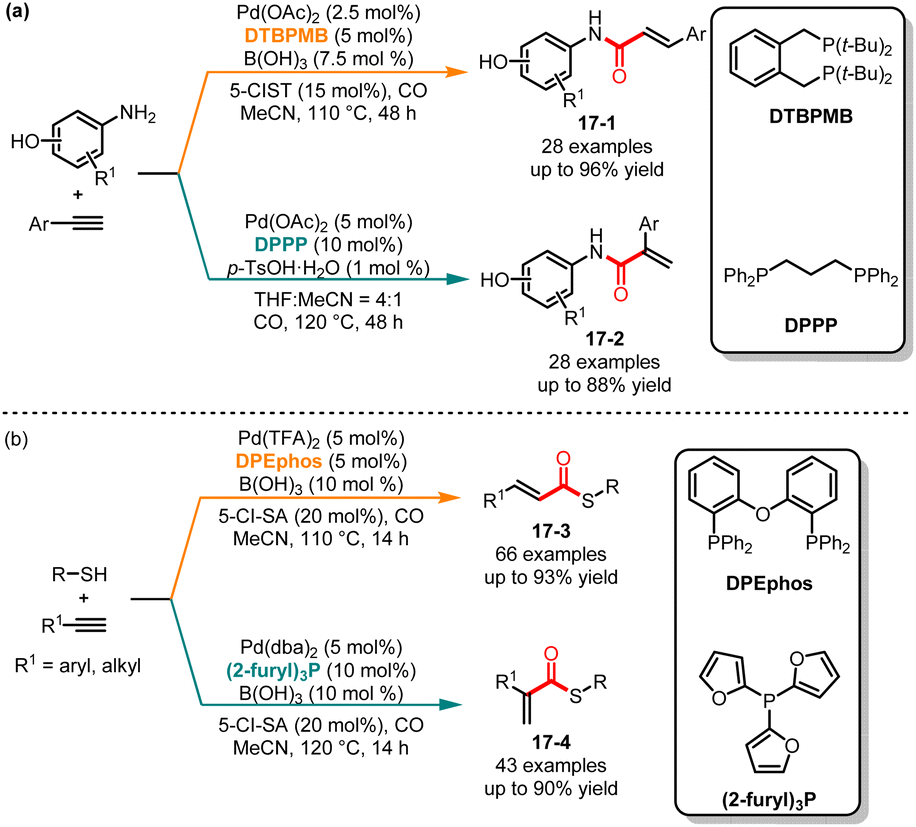
Compared to alkenes, alkynes generally exhibit higher reactivity due to their higher carbon-carbon triple bond energy. This higher reactivity of alkynes often makes it more challenging to control the selectivity when reacting with aminophenols. In 2017, Alper and co-workers demonstrated a ligand-controlled regioselective aminocarbonylation of aminophenols and alkynes (Scheme 17a).38 By simply selecting different additives and ligands, a variety of branched or linear α,β-unsaturated amides could be obtained with high yields. In the case of 1,2-bis(di-tert-butylphosphinomethyl)-benzene (DTBPMB) as a ligand, and 5-chlorosalicylic acid and B(OH)3 as the additives, the linear products 17-1 were selectively obtained. However, branched α,β-unsaturated amides 17-2 could be achieved by utilizing p-TsOH·H2O as the additive and DPPP as the ligand. This method demonstrated excellent substrate versatility, and arylamines containing hydroxyl, carboxyl and vinyl substituents could be efficiently obtained. It was important to note that mechanistic studies have indicated that the intermediates LnPd(CO)NHAr and LnPd(CO)–H controlled two distinct reaction pathways, respectively. The additive functioned as a proton donor, facilitating the formation of the LnPd-H intermediate, which promoted the selective generation of linear products. Later, in 2021, Wu and co-workers disclosed a ligand-controlled regioselective thiocarbonylation of alkynes. When using DPEphos as the ligand, the linear α,β-unsaturated thioesters 17-3 were formed in moderate to good yields. On the other hand, when the substrates were treated with tri(2-furyl)phosphine as the ligand, the corresponding branched products 17-4 were selectively approached with excellent regioselectivity (Scheme 17b).39
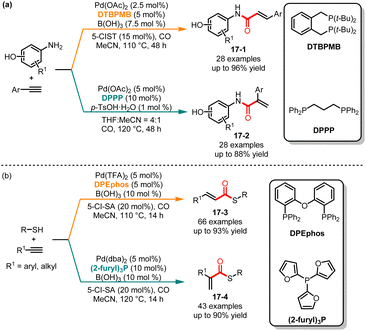 |
| | Scheme 17 Pd-catalyzed regioselective aminocarbonylation or thiocarbonylation of alkynes. | |
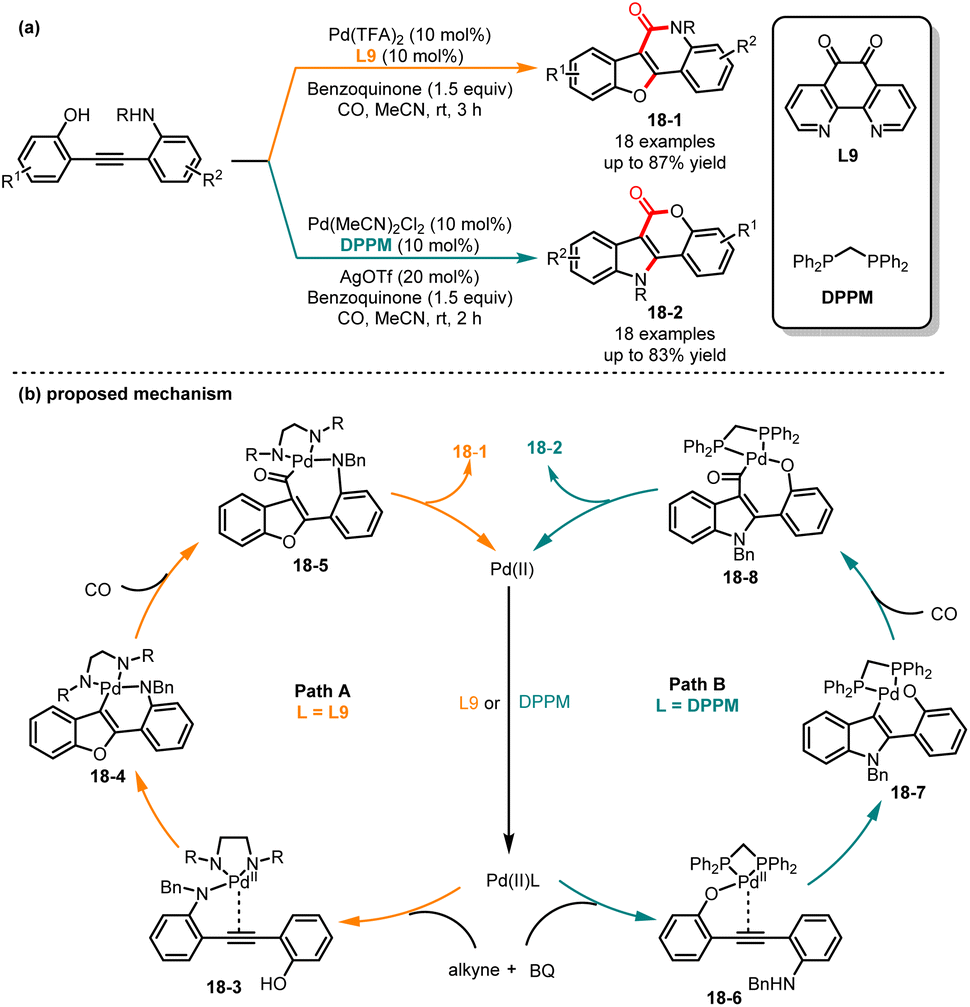
In 2018, Jiang and co-workers discovered an interesting ligand-dependent divergent synthesis of polycyclic compounds indolo[3,2-c]coumarins 18-1 and benzofuro-[3,2-c]quinolinones 18-2 by sequential cyclization and carbonylation of diphenylethyne derivatives (Scheme 18).40 In the presence of the electron-deficient nitrogen-containing ligand 1,10-phenanthroline-5,6-dione (L9) and benzoquinone as a reoxidant, the O-attack/N-carbonylation cyclization compounds 18-1 were generated as the sole product in high yields and selectivities. Alternatively, when the electron-donating bidentate phosphine ligand DPPM was used, the N-attack/O-carbonylation cyclization compounds 18-2 were successfully obtained with high yields. Subsequently, the authors investigated nucleophilic reagents besides phenols, such as amines, carboxylic acids, and aromatic rings, which were also found to be applicable in the reaction system and yielded the corresponding polycyclic products with excellent yields. The control experiments confirmed that the reaction proceeded through an intramolecular cyclization followed by a CO insertion sequence. Mechanistically, the in situ formation of palladium complex 18-3 by Pd(II) and L9 made it more electrophilic and allowed for coordination with the amino group, resulting in the formation of intermediate 18-4. Furthermore, the intermediate 18-4 underwent intramolecular nucleophilic cyclization and subsequent CO insertion to form intermediate 18-5. Finally, the product 18-1 was generated through reductive elimination of intermediate 18-5. In contrast, when dppm was used, the chemoselectivity showed a transformation. The Pd(II)/DPPM catalyst coordinated with the hydroxy group to generate the intermediate 18-6, which experienced sequential nucleophilic attack and CO insertion, yielding intermediate 18-8. The product 18-2 was produced through reductive elimination. Later, in 2019, Chen and co-workers investigated the factors contributing to the chemo- and regioselectivity of the reaction using DFT calculations and confirmed the feasibility of the proposed mechanism.41
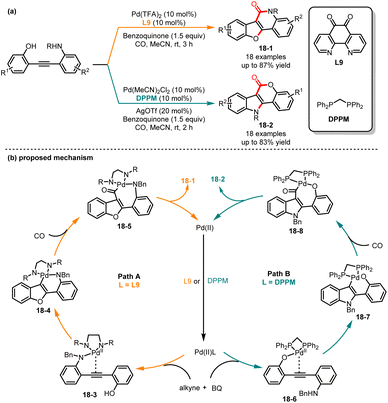 |
| | Scheme 18 Trifluoromethylation reaction of aliphatic alcohols. | |
3.2 Ligand-controlled regiodivergent coupling reactions
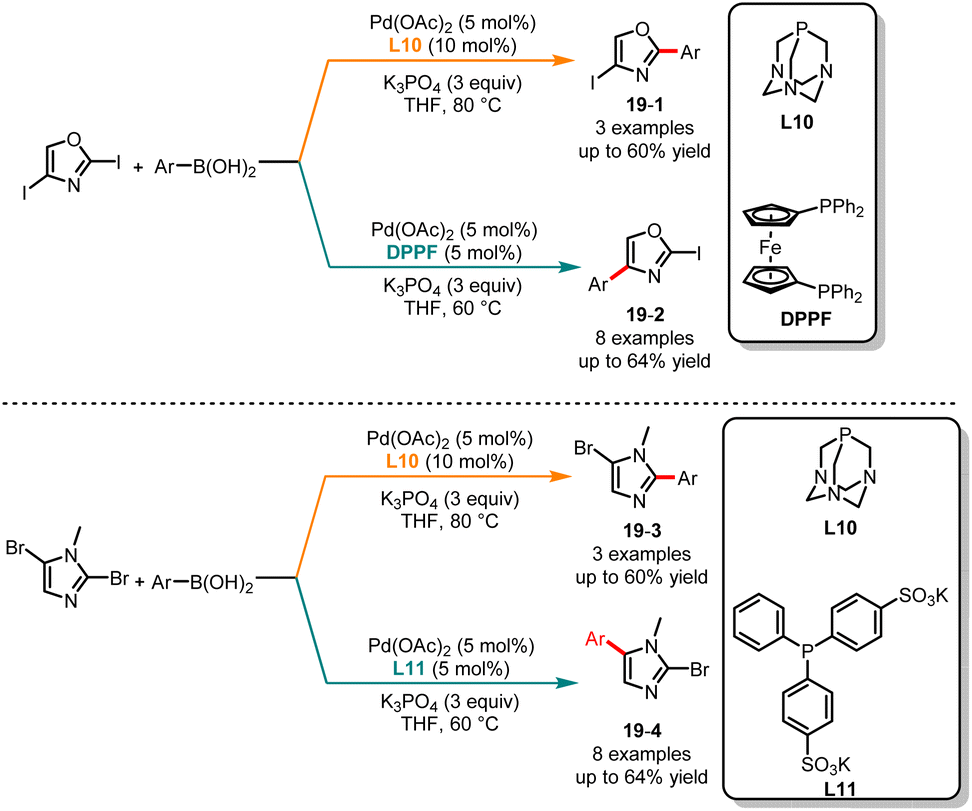
Since the pioneering work of Suzuki and Miyaura in 1979, the Suzuki–Miyaura cross-coupling reaction has gained significant interest in the field of heteroarene chemistry, owing to its broad substrate compatibility and the ability to introduce diverse functional groups into the heterocyclic framework.42 Based on the studies by Greaney and Miura, it has been shown that the C2 position in oxazole has a higher pKa, thereby exhibiting higher reactivity compared to the C4 position.43 In 2010, Strotman and co-workers successfully realized a ligand-controlled regioselective Suzuki coupling reaction of 2,4-diiodooxazole (Scheme 19a).44 In the case of the electron-rich ligand 1,3,5-triaza-7-phosphaadamantane (L10) and THF as the solvent, a series of monoarylated products 19-1 with high selectivity at the C2 position were obtained, whereas the treatment of 2,4-diiodooxazole with a Pd(OAc)2/DPPF catalyst system in THF afforded monoarylated products 19-2 at the C4 position with medium yields and selectivities. The authors discovered that this modular approach is also applicable to monoarylation of dibromoimidazoles. The utilization of L10 yielded arylated products 19-3 at the C2 position, while bis(p-sulfonatophenyl)-phenylphosphine dihydrate dipotassium salt (L11) facilitated coupling at the C4 position giving the appropriate products 19-4. Subsequently, Buchwald and co-workers published a ligand-controlled regioselective Suzuki coupling reaction of (hetero)aryl halides with allylboronates, and a range of α- and γ-selective allylation products were obtained in good yields and excellent selectivity.45
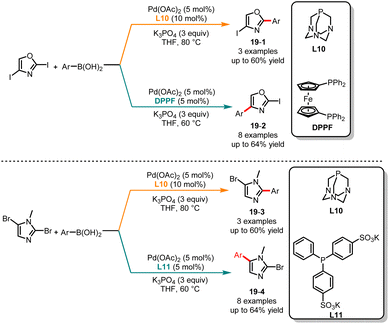 |
| | Scheme 19 Pd-catalyzed regioselective carbonylation of dihaloimidazoles or dihalooxazoles. | |
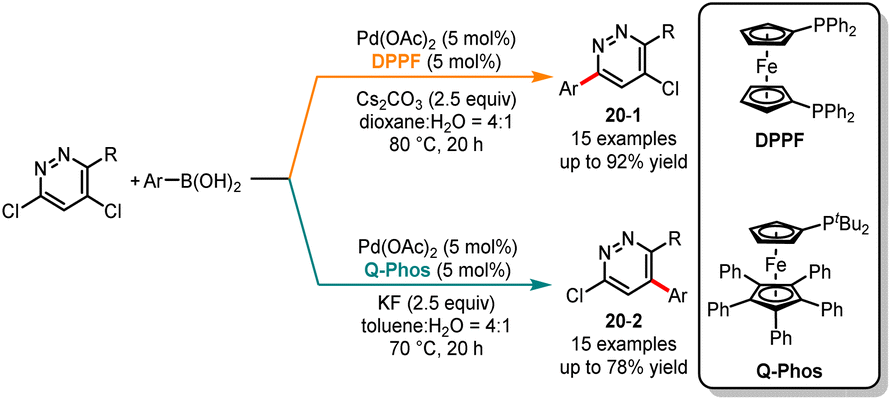
Selective cross-couplings can be achieved for substrates with different halogens based on their reactivity disparities, but it becomes challenging for polyhalogenated heteroarenes with identical halogens. In 2013, Dai and co-workers achieved ligand-controlled regioselective Suzuki monocoupling at the C3 or C5 positions of 3,5-dichloropyridazines, resulting in a series of arylated and heteroarylated products with high yields and selectivities (Scheme 20).46 It was noted that the ligand played a crucial role in determining the efficiency and selectivity of the reaction. In the presence of Pd(OAc)2/DPPF as a catalyst and Cs2CO3 as a base, a series of C3-coupled products 20-1 were obtained in good yields. In contrast, the use of the monodentate ligand Q-Phos favored coupling at the C5 position, affording 20-2 as major products. To further expand the applicability of this modular approach, the authors accomplished selective monoarylation at the C2 and C4 positions of 2,4-dichloropyridine. Employing DPPF and Q-Phos ligands separately, the C2-coupled product was obtained in a high yield of 90% and the C4-coupled product in a yield of 36%. Later, in 2020, Manabe and co-workers published a ligand-controlled regioselective arylation reaction at the C2 or C3 positions of 2,5-diaryl-1H-pyrroles. When using JohnPhos as the ligand, 2,2,5-triaryl-2H-pyrroles were produced with high yields and selectivities. The different isomers of C3-arylation products can be generated when bulky monophosphine t-BuBrettPhos was used.47
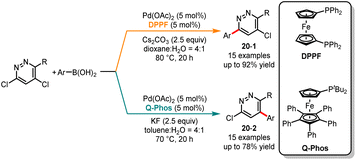 |
| | Scheme 20 Pd-catalyzed regioselective carbonylation of 3,5-dichloropyridazines. | |
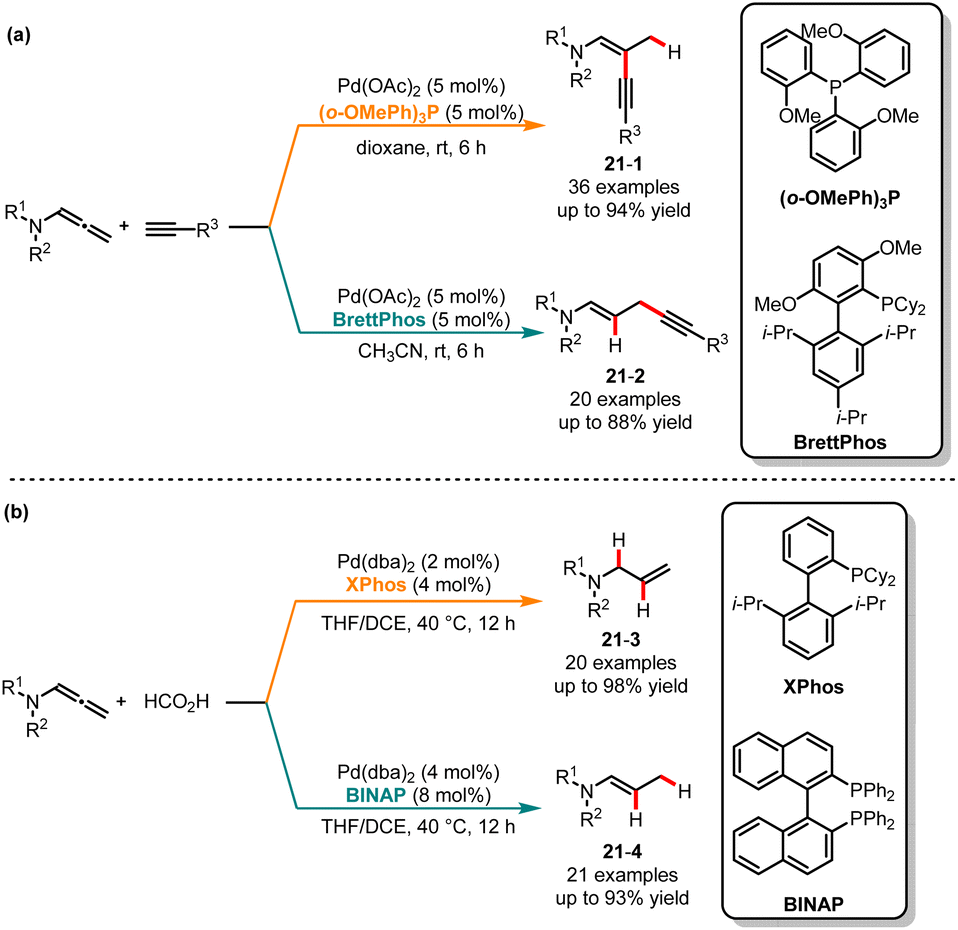
Chemists have shown renewed interest in 1,n-enynes as precursors for the construction of bioactive cyclic compounds.48 The field of utilizing alkynes as substrates for hydroalkynylation to construct 1,3- and 1,4-enynes has seen significant and rapid exploration in recent decades.49 Despite successful advancements in regional and stereoselective hydroalkynylation, the issues of regiodivergent and stereoselectivity persist as neighboring reactive π-systems are susceptible to isomerization, potentially yielding mixtures containing both regio- and stereoisomeric products. In 2018, Park and co-workers published a regioselective and stereospecific cross-coupling method for the synthesis of cis-1,3- and trans-1,4-ynenamides, utilizing two distinct ligands (Scheme 21a).50 With Pd(OAc)2/(o-OMePh)3P as the catalytic system in dioxane, β-branched ynenamides 21-1 were obtained in excellent yields with complete regioselectivity. Whereas BrettPhos was used as a ligand in CH3CN, an excellent regioselectivity switch was observed, yielding the skipped ynenamides 21-2. The kinetic studies revealed a higher reaction rate for the β-alkynylation compared to the γ-alkynylation. When the BrettPhos ligand was employed, the steric effect causes an alkyne attack on the γ-position of allene through carbopalladation processes, facilitating the formation of linear products. Contrary to expectations, based on the Curtin–Hammett principle, using a small-sized ligand facilitates the attack of the H atom on the γ-position of the allene, leading to the branched products. Recently, Cui and co-workers released a ligand-controlled regiodivergent semireduction of allenamides. When the monodentate ligand XPhos was used, allylic amides with high chemo-, regio- and stereoselectivity were obtained through a 1,2-semireduction. Conversely, the different isomers of (E)-enamide derivatives can be generated with high selectivity when BINAP was used (Scheme 21b).51
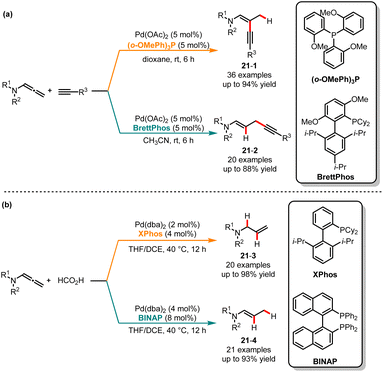 |
| | Scheme 21 Pd-catalyzed regioselective cross-coupling. | |
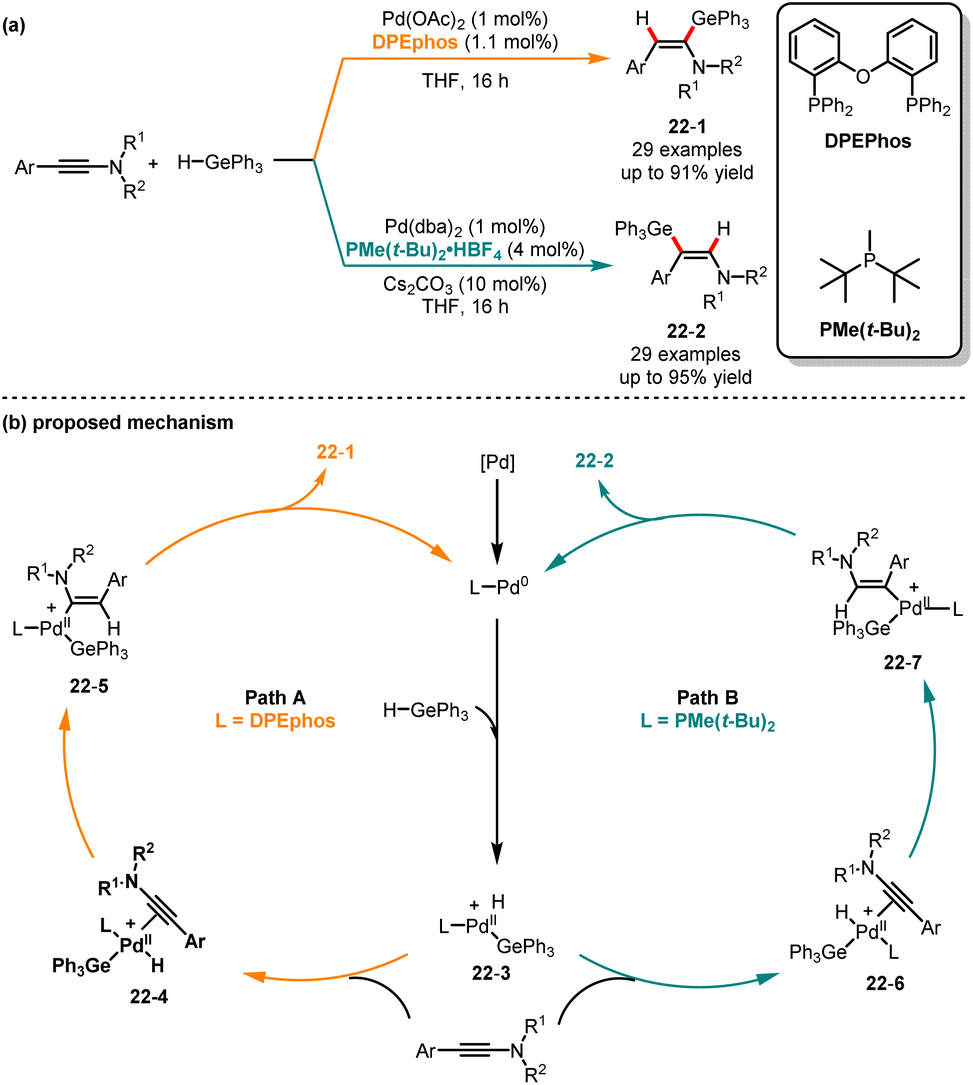
Despite the reported hydrogermylation reactions of alkynes since the 1950s, there are only a few reports on the regio- and stereoselective synthesis of vinylgermanes.52 In 2001, Cintrat and co-workers reported the α-regioselective palladium-catalyzed hydrostannylation of alkynes.53 Subsequently, the Perez-Luna group demonstrated that the β,E-germylated enamides were synthesized through hydrogermylation of alkynes under zinc catalysis.54 Later in 2020, Bizet and co-workers presented a regiodivergent hydrometalation of ynamides by employing different ligands, generating selectively α,E- and β,E-germylated and stannylated enamides (Scheme 22).55 When bidentate phosphorus ligand DPEphos was used, leading to the α-germylated enamides 22-1 in good yields. In contrast, the different isomers of β-germylated enamides 22-2 can be generated with high selectivity when bulky monophosphine (t-Bu)2PMe was used. In this palladium catalysis, two different pathways were proposed. Initially, an oxidative addition of the palladium(0) catalyst with triphenylgermanium hydride generates the palladium(II) intermediate 22-3. By employing DPEphos as a ligand, the palladium(II) complex 22-3 coordinates with ynamide, followed by a hydropalladation step leading to intermediate 22-5, which could undergo reductive elimination and deliver the α,E-enamides 22-1. Conversely, when switching DPEphos to bulky monophosphine (t-Bu)2PMe, the intermediate 22-7 was generated via hydropalladation and produced the β,E-enamides 22-2. DFT calculations provide evidence for a hydropalladation mechanism and demonstrate that the regioselectivity can be influenced by the steric properties of the phosphine ligands.
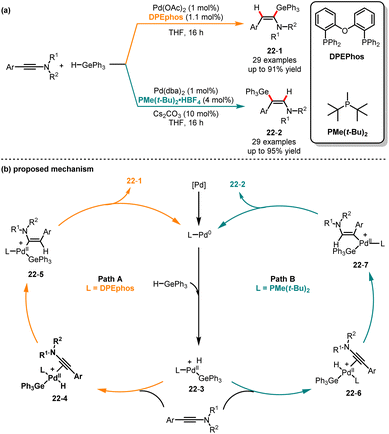 |
| | Scheme 22 Pd-catalyzed regioselective hydrometalation of ynamides. | |
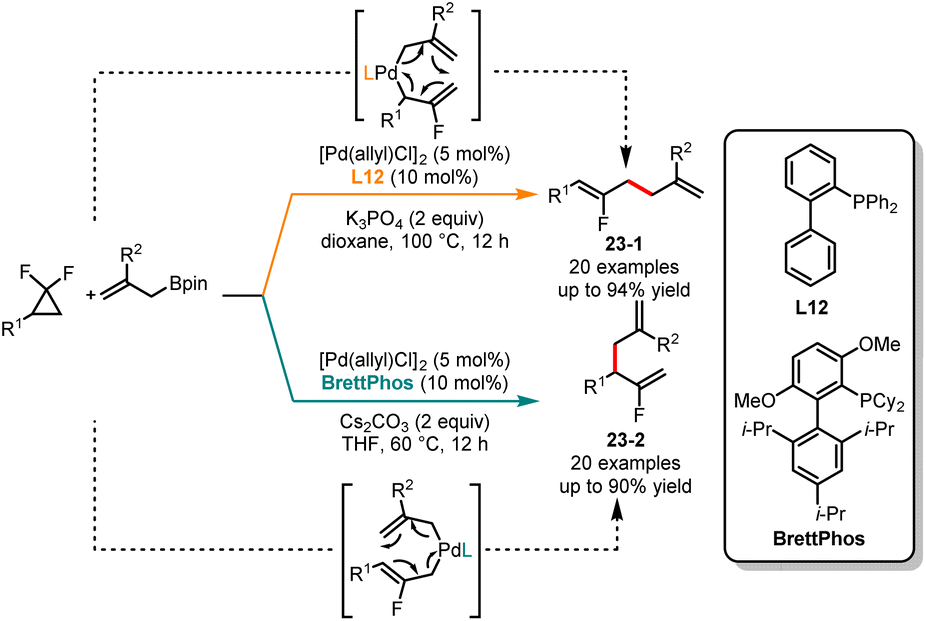
Fluorinated compounds have been widely applied in the fields of refrigeration, pharmaceuticals, and materials science.56 In 2020, Hartwig and coworkers achieved stereoselective monofluoroalkene formation by utilizing 3,3-difluoropropenes as allylic electrophiles in reaction with carbon nucleophiles.57 Xia and coworkers in 2021 described a rhodium-catalyzed coupling of gem-difluorocyclopropanes with arenes, affording a range of linear fluorinated allylic derivatives.58 Subsequently, Li and coworkers introduced a highly regioselective coupling of gem-difluorocyclopropanes, promoted by palladium catalysis to provide branched fluorinated allylic derivatives.59 Very recently, Shi and coworkers developed a ligand-dependent divergent synthesis of linear and branched fluorinated allylic derivatives from gem-difluorocyclopropanes bearing allylboronates (Scheme 23).60 The use of biarylphosphine (L12) as a ligand and K3PO4 as a base in dioxane provided the linear coupling products 23-1, whereas the employment of BrettPhos as a ligand and Cs2CO3 as a base in THF led to the formation of branched products 23-2 with high selectivity. The deuterium labeling experiments and DFT calculations confirmed that the regioselectivity of the reaction was influenced by the steric effect of the ligands.
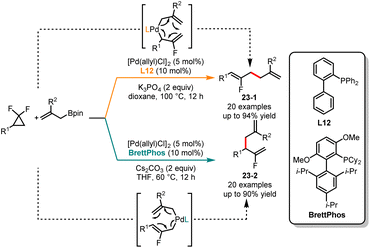 |
| | Scheme 23 Pd-catalyzed regioselective coupling of gem-difluorocyclopropanes. | |
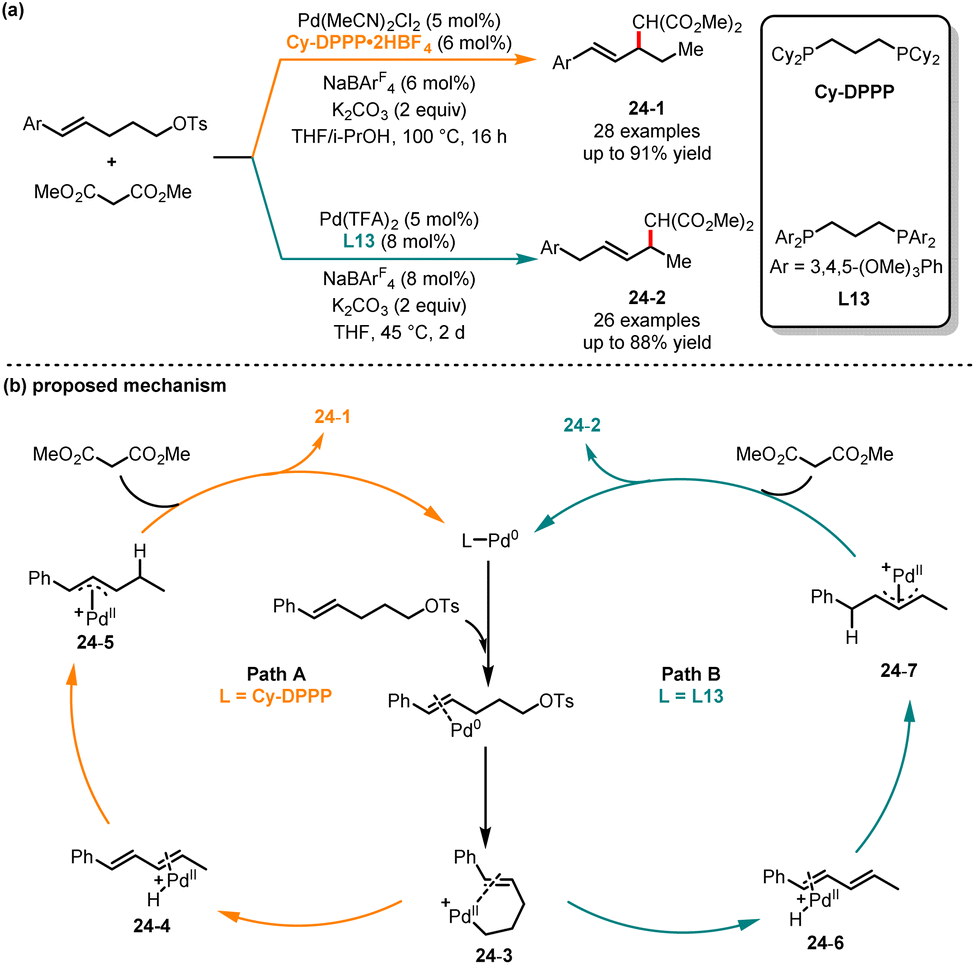
Traditional palladium-catalyzed allylic substitution reactions commonly result in the formation of η3-π metal intermediates.61 In 2020, Liu and co-workers developed a ligand-controlled regioselective diamination of alkenes. In this reaction system, the quinox ligand promoted the formation of 6-endo products, while the use of the pyox ligand yielded 5-exo products.62 However, generating these intermediates for alkenes with distal leaving groups was rarely reported, as it is unlikely for thermodynamically stable η3-allylmetal species to be formed. In 2023, Lin, He and coworkers reported a ligand-controlled regioselective allylic functionalization reaction of styrene bearing a remote OTs group with malonates (Scheme 24).63 In the presence of Pd(MeCN)2Cl2/Cy-DPPP as a catalyst and THF/i-PrOH as solvents, the 4,3-addition products 24-1 were achieved in good yields with excellent regioselectivities. Conversely, the 1,4-addition products 24-2 were obtained as major isomers when electron-rich bisphosphine ligand L13 was used in THF. Under two different catalytic pathways, the transformation exhibited high functional group tolerance. It is noteworthy that the distance between the leaving group and the olefin unit in the electrophilic reagent should not exceed three linear carbon atoms. Based on kinetic experiments and deuterium studies, the authors have proposed two different pathways that are associated with the ligands. Initially, the reaction involved the oxidation addition of the OTs group in styrene to the Pd(0) catalyst, generating the Pd(II) intermediate 24-3. Subsequently, the isomerization afforded the conjugated diene intermediate coordinated with the Pd(II) catalyst (intermediate 24-4 or 24-6). Then, the conjugated diene intermediate treated with the PdH catalyst through 4,3-addition or 1,4-addition produced the η3-allylpalladium intermediate (24-5 or 24-7) by employing different ligands. Finally, the corresponding product was obtained through turnover-limiting allylic substitution with malonate.
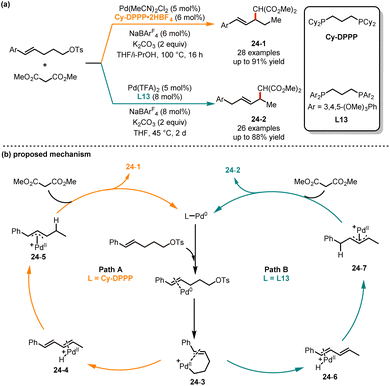 |
| | Scheme 24 Pd-catalyzed regioselective allylic functionalizations of styrene with malonates. | |
3.3 Ligand-controlled regiodivergent cycloaddition reactions
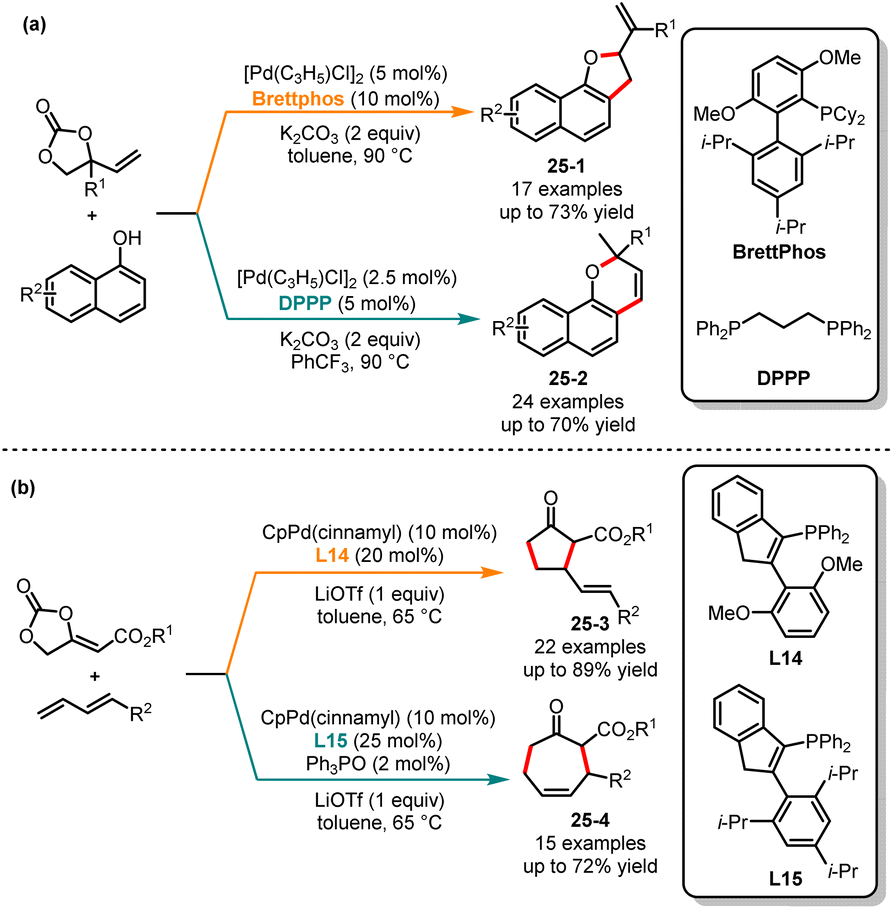
Since the 1990s, there has been extensive research interest in transition metal-stabilized oxyallyl cation-mediated cyclization reactions.64 In 2018, Trost and co-workers reported Pd-oxyallyl-mediated [3 + 2] cycloaddition reactions of 1,3-dienes with bifunctional precursors, yielding a series of tetrahydrofuran derivatives.65 Subsequently, DFT calculations confirmed that the C–O reductive elimination was the preferred pathway.66 It still poses a challenge to generate cyclic ketones through C–C reductive elimination of Pd-oxyallyl species. In 2019, Liang and co-workers introduced ligand-controlled cycloaddition reactions of vinylethylene carbonates and naphthols. When using Brettphos and DPPP as ligands, the corresponding [3 + 2] and [3 + 3] cycloaddition products are obtained in medium yields and selectivities, respectively (Scheme 25a).67 Recently, Zi and co-workers have successfully achieved Lewis acid- and ligand-controlled cycloaddition reactions of methylene ethylidene carbonates with 1,3-dienes (Scheme 25b).68 By employing L14 as a ligand and LiOTf as the Lewis acid in toluene, [3 + 2] cycloaddition products 25-1 were generated as the sole product in medium to good yields. In contrast, when the steric bulk ligand L15 was used, the corresponding [4 + 3] cycloaddition products 25-2 were produced with good yields and medium regioselectivities. Mechanistically, the coordination between the lithium ion and the alkoxide group interrupts the C–O reductive elimination process, resulting in a Li-enolate tethered π-allyl-Pd species. Then, under the steric influence of the ligand, the enolate moiety underwent intramolecular allylic attack at the C5 or C7 position of the π-allyl-Pd moiety.
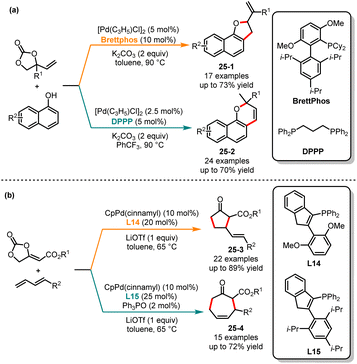 |
| | Scheme 25 Pd-catalyzed regioselective cycloaddition reactions of vinylethylene carbonates. | |
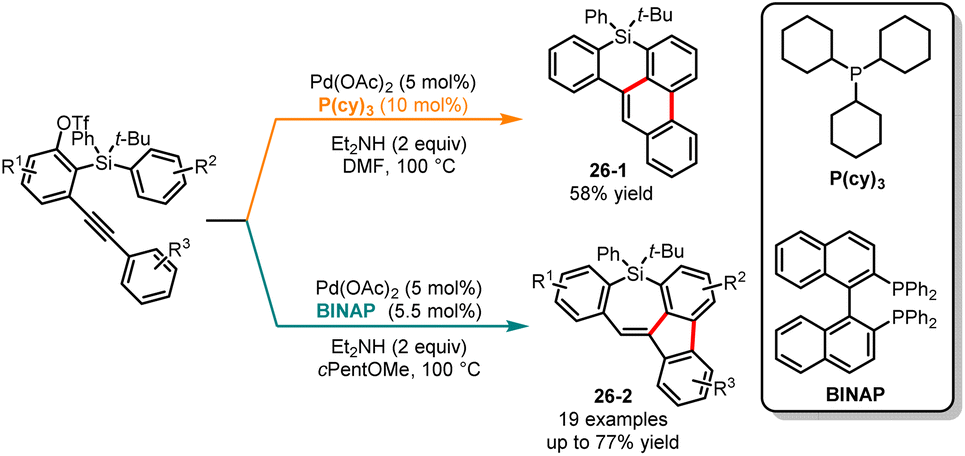
The activation of challenging C–H bonds, which are typically difficult to directly functionalize, can be achieved through 1,n-metal migration, enabling the synthesis of complex organic molecules from simple precursors.69 In 2020, Shintani and co-workers developed a palladium-catalyzed ligand-free catalytic system for the synthesis of 8H-benzo[e]-phenanthro[1,10-bc]silines through a 1,4-palladium migration coupling.70 Subsequently, in 2021, the same group discovered a palladium-catalyzed ligand-controlled 1,n-palladium migration coupling for the selective synthesis of benzophenanthrosiline 26-1 and 5H-dibenzo-[b,f]silepins 26-2 from silicon-tethering triflates (Scheme 26). When using the monophosphine ligand (PCy3 or P(t-Bu)3), the 1,4-palladium migration product 26-1 was obtained in moderate yield. Conversely, by using the diphosphine ligand BINAP in the reaction system, other novel products 5H-dibenzo-[b,f]silepins 26-2 were obtained through 1,5-palladium migration.71 Recently, Yang and co-workers elucidated the reaction mechanisms through two pathways and factors influencing regioselectivity through DFT calculations.72 The computational results indicated that alkyne insertion is the determining step for regioselectivity. In the absence of the ligand, the barrier for 1,2-alkyne insertion is lower by 4.2 kcal mol−1 compared to that of 2,1-alkyne insertion, favoring the formation of 6,6-fused benzophenanthrolines. In the case of BINAP ligand, the barrier for 2,1-alkyne insertion is even lower, favoring the generation of 5,7-fused benzofluorenosilepins.
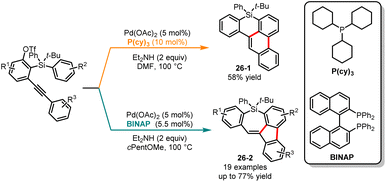 |
| | Scheme 26 Pd-catalyzed regioselective cycloaddition reactions of silicon-tethering triflates. | |
4 Stereodivergent synthesis
4.1 Ligand-controlled stereodivergent coupling reactions
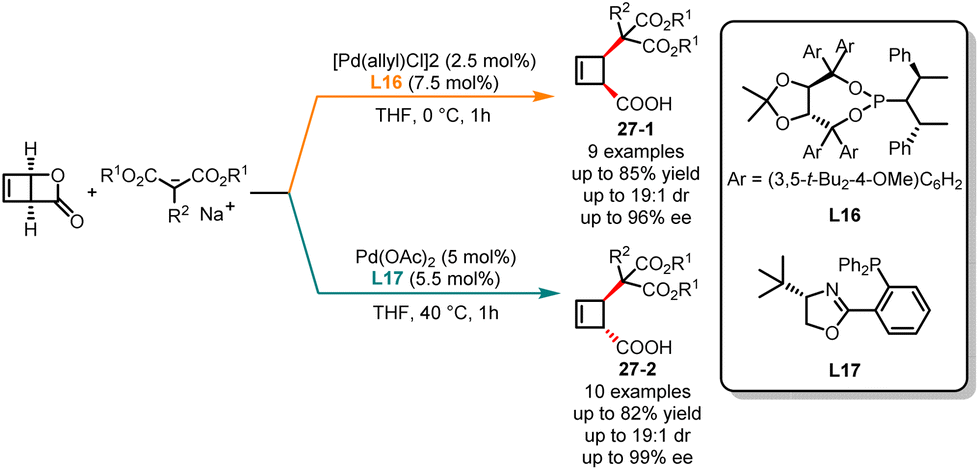
With the advancement and continuous development of asymmetric catalysis, deracemization has become a pivotal chemical transformation. The deracemization of racemates provides increased possibilities and choices for synthesizing chiral compounds and studying chirality in chemistry. Palladium-catalyzed alkene allylation reactions allowed the introduction of alkyl groups onto alkene molecules, expanding the allyl frameworks through the construction of C–C bonds.73 This strategy found widespread application in the synthesis of pharmaceuticals, natural products and materials, effectively building diverse and complex molecular structures. In 2011, Maulide and co-workers published an innovative palladium-catalyzed allylic alkylation reaction, where diastereodivergent deracemization of racemic lactone with malonate nucleophiles was controlled by the ligand (Scheme 27).74 In the presence of phosphoramidite L16 as the ligand and THF as solvents, the highly cis-selective products 27-1 were achieved in good yields with excellent enantioselectivities. Conversely, the trans-disubstituted cyclobutenes 27-2 were obtained as a major isomer when phosphine oxazoline L17 was used in THF at 40 °C. In both reaction pathways, multiple functional groups were found to be tolerated.
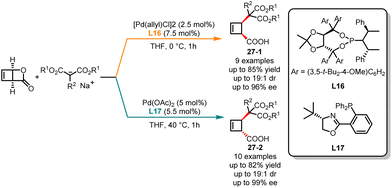 |
| | Scheme 27 Trifluoromethoxylation–halogenation of arynes. | |
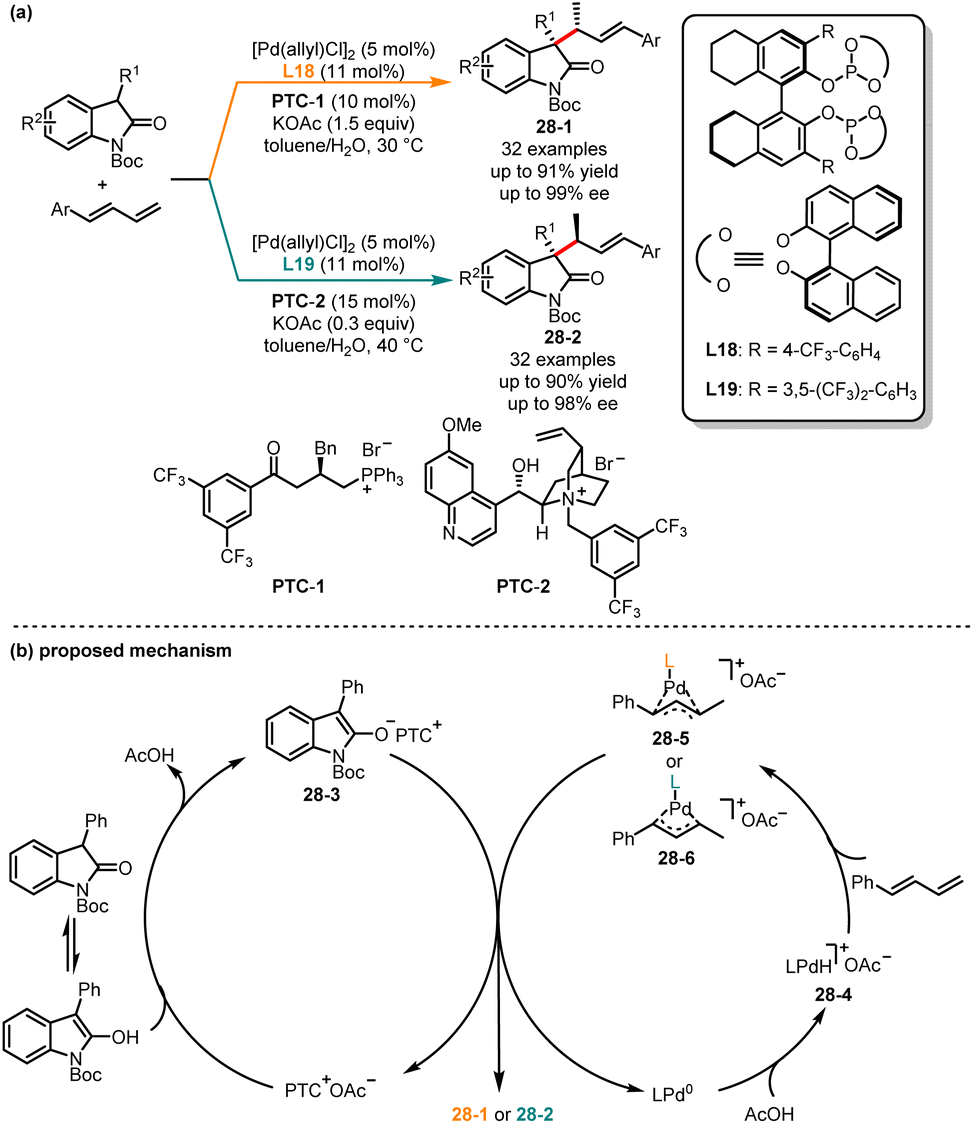
The selective generation of a single diastereomer has been extensively reported in the literature, but the stereoconvergent synthesis of all diastereomers still poses certain challenges, because it requires precise control over the absolute and relative configurations of multiple stereocenters within the molecule. In 2022, Chen and co-workers revealed an interesting example of palladium-catalyzed decarboxylative dienylation reaction of (E, E)-2,4-dienoic acids and imines. By switching different ligands, a series of diene derivatives with both (E,E) and (Z,E) configurations were obtained.75 Recently, Zi and co-workers described a palladium/phase-transfer synergistic catalytic approach for diastereodivergent C–C coupling of oxindoles with 1,3-dienes (Scheme 28).76 It was revealed that the steric and electronic properties of ligands coordinated to the Pd(0) center and both syn and anti-coupling products could be obtained with good yields and excellent diastereo- and enantioselectivities. When bidentate bisphosphite ligand L18 and phase-transfer catalysts PTC-1 were employed, the (αS,βR)-oxindoles 28-1 were provided, whereas (αR,βR)-oxindoles 28-2 were selectively obtained when the chiral ligand L19 and phase-transfer catalysts PTC-2 were utilized. Mechanistically, PTC+ combined with OAc− and transferred it to the organic phase. Then the ion pair intermediate 28-3 was obtained by deprotonation of oxindole. In the palladium catalytic cycle, the LPd(0) complexed with AcOH to form the intermediate 28-4. Next, under the influence of the ligands, Pd–H underwent migratory insertion into 1,3-diene, resulting in the formation of intermediates 28-5 and 28-6. Finally, intermediate 28-3via C–C coupling of either intermediate 28-5 or intermediate 28-6 generated the desired stereoisomer.
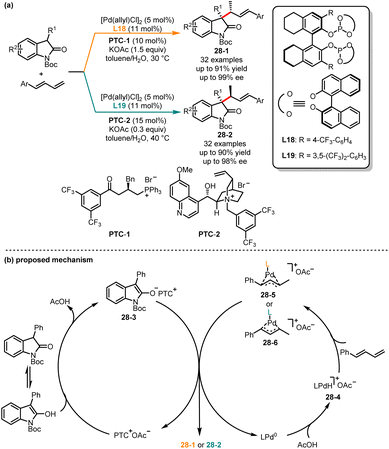 |
| | Scheme 28 Pd-catalyzed diastereoselective C–C coupling of oxindoles with 1,3-dienes. | |
Very recently, Fang and co-workers explored an interesting stereodivergent synthesis of various trisubstituted acrylonitriles from the hydrocyanation of propiolamides, using palladium catalysts with different ligands (Scheme 29).77 The use of DPPF as the ligand selectively afforded E-cyanation products 29-1 with good yields and high stereoselectivities. In contrast, when employing L20 as the ligand, the corresponding Z-cyanated products 29-2 were obtained in good yields. Under two different reaction pathways, a variety of primary, secondary, and tertiary amides were well tolerated. Control experiments revealed that Z-acrylonitriles can be obtained through an isomerization of E-acrylonitriles. The DFT results indicated that the barrier for the Z isomer is lower by 1.9 kcal mol−1 compared to the E isomer, showing that Z-cyanated products were more stable. The stereochemical selectivity is controlled by the coordination mode of the palladium catalyst with two different ligands. The bidentate ligand L20 allows the catalyst to accommodate the substrate, forming a five-membered cyclopalladium ring, thereby achieving the E to Z isomerization through cyclopalladation/isomerization. However, when using the monodentate ligand DPPF, the corresponding cyclopalladium ring is not formed, thereby inhibiting isomerization and resulting in the formation of the E-isomer.
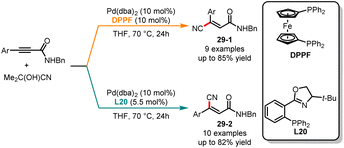 |
| | Scheme 29 Pd-catalyzed diastereoselective hydrocyanation of propiolamides. | |
4.2 Ligand-controlled stereodivergent cycloaddition reactions
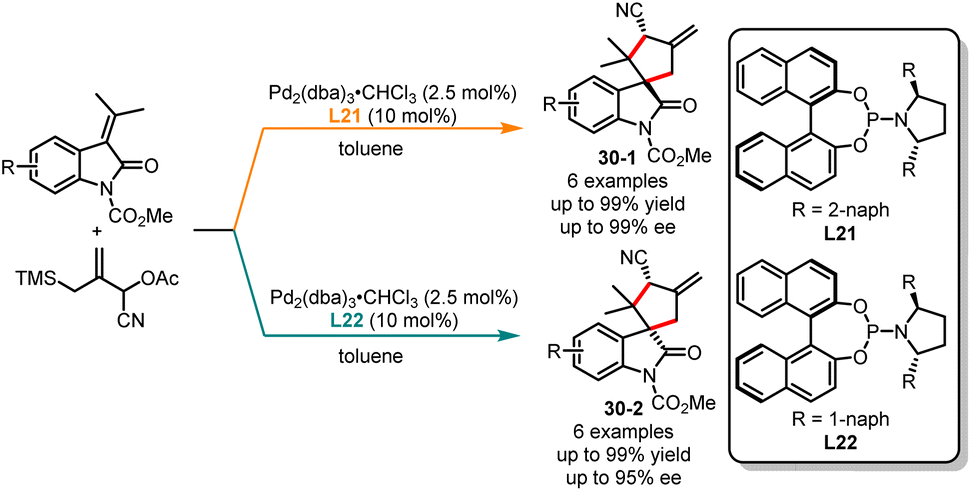
Although the palladium-catalyzed asymmetric [3 + 2] trimethylenemethane (TMM) cycloaddition of the TMM precursor is well-known, the diastereodivergent synthesis of multi-chiral center compounds, controlled by ligands, remains highly challenging.78 In 2007, Trost and co-workers reported an elegant ligand-controlled highly diastereo- and stereoselective [3 + 2] cycloaddition of cyano-substituted TMM precursor with oxindoles, yielding a range of spirocyclic oxindolic cyclopentanes with good yields and enantiomeric excesses (Scheme 30).79 In the presence of bulky 1-naphthyl substituents of L21 in toluene, the highly trans-cyclopentanes 30-1 were achieved in good yields with excellent enantioselectivities. Conversely, the cis-cyclopentanes 30-2 were obtained as a major isomer when 2-naphthyl substituents of L22 were used. Later, in 2020, Ruijter and co-workers published a novel ligand-controlled stereodivergent synthesis of β-lactams through palladium-catalyzed intramolecular Tsuji–Trost allylation.80
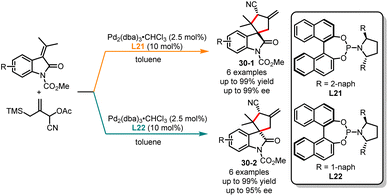 |
| | Scheme 30 Pd-catalyzed diastereoselective [3 + 2] cycloaddition of TMM precursor with oxindoles. | |
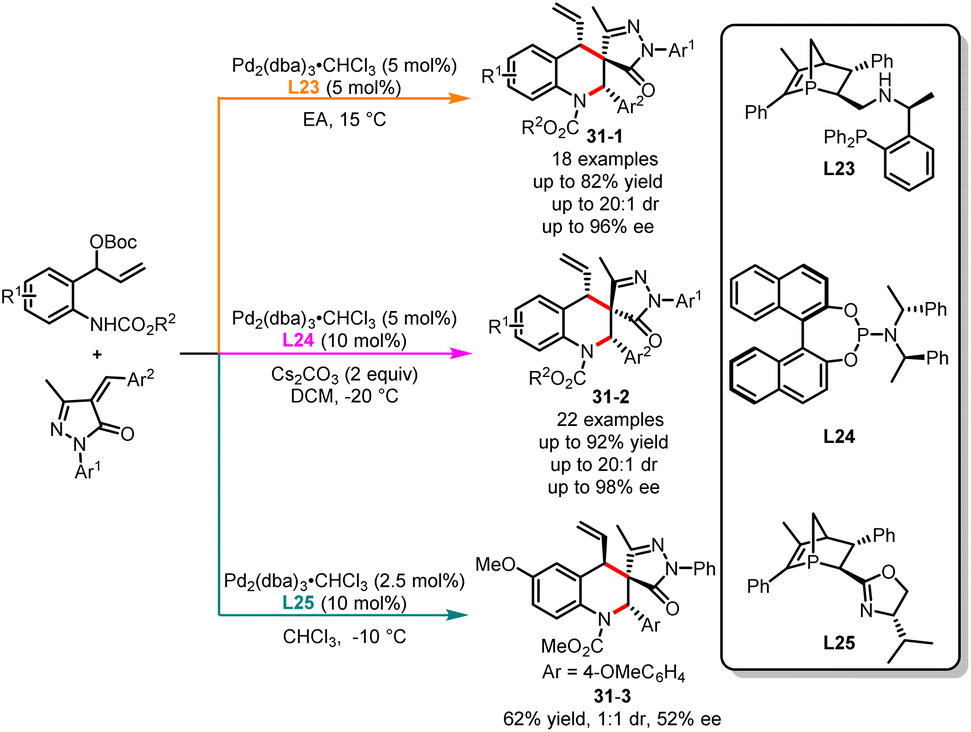
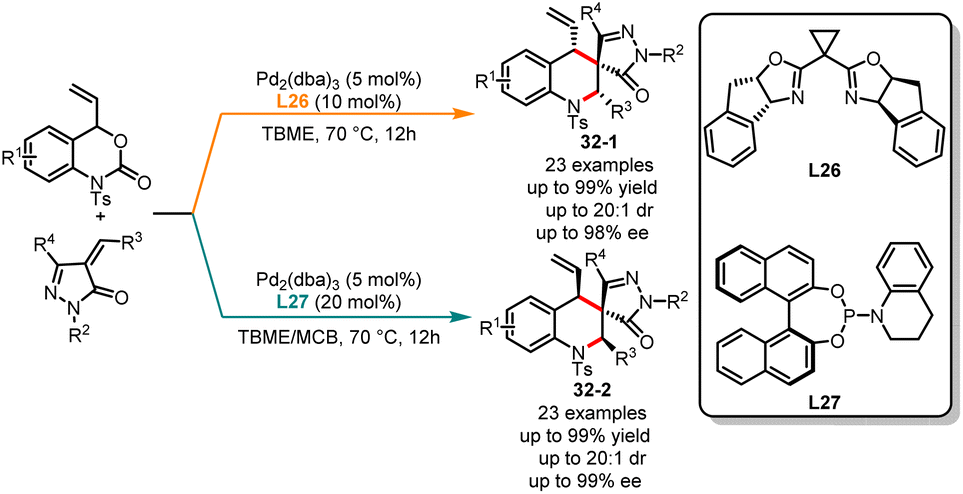
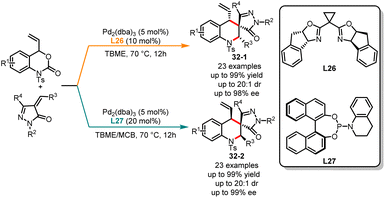
The physiological properties of a biologically active substance can be altered by its chirality.81 Obtaining each stereoisomer accurately for compounds with two or more chiral centers is of great significance for biological systems, medicinal development and material properties.82 In the past decade, significant progress has been made in the stereodivergent synthesis of C–C bond coupling compounds.83 However, developing a general strategy to selectively obtain any diastereomer of reaction products with multiple stereocenters by switching different chiral ligands remains a challenge. In the past few years, Li, Duan and co-workers have been actively engaged in the development of novel phosphorus chiral center ligands.84 These ligands, based on the 1-phosphanorbornene framework, have been successfully applied in transition metal-catalyzed asymmetric cyclization reactions. In 2022, an interesting example of ligand-controlled palladium-catalyzed asymmetric [4 + 2] cycloaddition was exemplified by Li, Duan and co-workers in the stereodivergent synthesis of tetrahydroquinolines (Scheme 31).85 Six stereoisomers were easily generated in excellent yields with high diastereo- and enantioselectivities from the same starting substrates. It was observed that a novel bisphosphine ligand L23 used in ethyl acetate selectively delivered (S,R,S)-tetrahydroquinolines 31-1. However, the use of phosphoramidite ligand L24 along with Cs2CO3 as the base in DCM affords alternative (S,S,S)-tetrahydroquinolines 31-2. Through additional ligand screening, it was discovered that a new P-chiral ligand L25, recently developed by us, can also facilitate the construction of tetrahydroquinolines, yielding the corresponding product 31-3 with moderate yield and stereoselectivity. Very recently, Huang and co-workers have also achieved the stereodivergent synthesis of tetrahydroquinolines (Scheme 32).86 Starting from vinyl benzoxazinanones, by employing either a phosphinoamide ligand L26 or a nitrogen-based ligand L27, the corresponding products 32-1 and 32-2 were obtained with excellent yields and enantioselectivities.
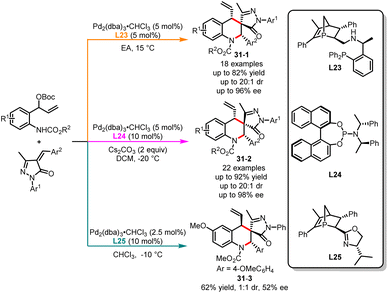 |
| | Scheme 31 Pd-catalyzed diastereoselective [4 + 2] cycloaddition of carbamate-functionalized carbonates with α-phenylidene succinimides. | |
 |
| | Scheme 32 Pd-catalyzed diastereoselective [4 + 2] cycloaddition of vinyl benzoxazinanones with α-phenylidene succinimides. | |
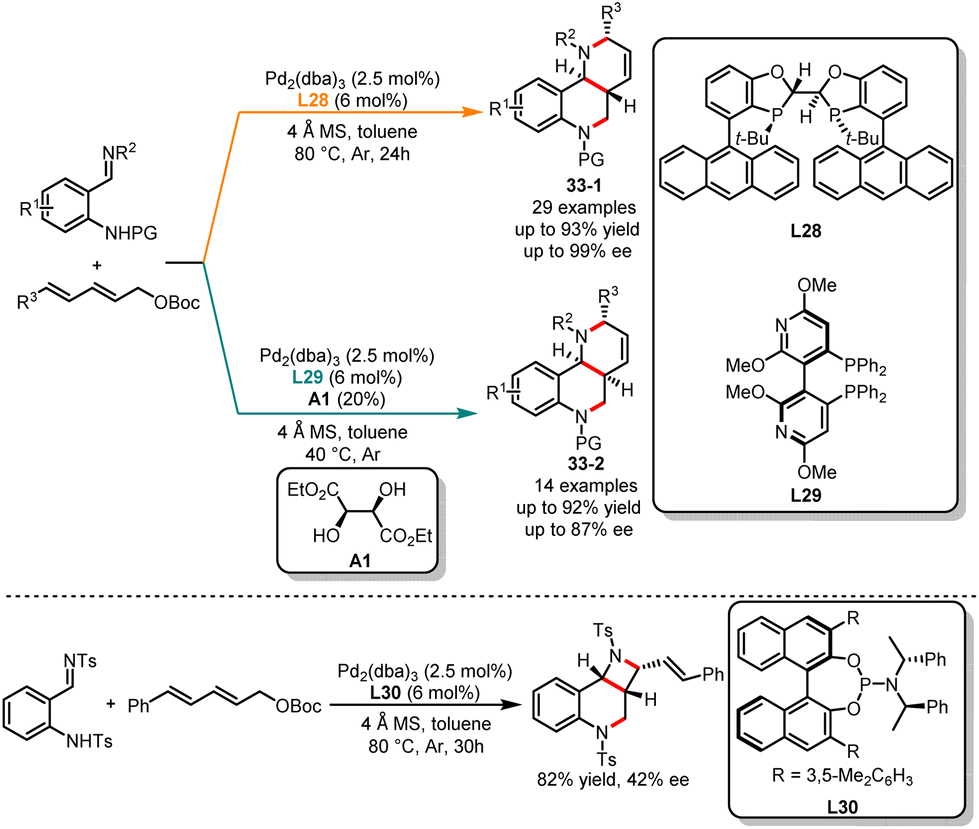
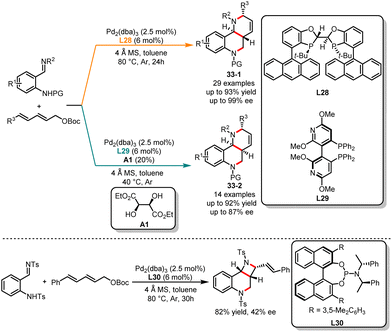
Auto-tandem catalysis has gained significant attention in the field of synthetic chemistry in recent years.87 This single catalytic process in which multiple reactions occur sequentially within a single catalytic system, without the need for intermediate isolation or purification steps, enables a more efficient and streamlined synthesis of complex molecules. While the field of transition metal and organo-catalyzed auto-tandem reactions has rapidly progressed, there have been limited reports on auto-tandem reactions with a divergent synthesis pattern. In 2022, Chen and co-workers reported an unprecedented ligand-controlled palladium-catalyzed cascade reaction of 2,4-dienyl carbonates with o-TsNH arylimines (Scheme 33).88 The diastereodivergent synthesis of tetrahydroquinoline derivatives can be achieved by using different chiral bisphosphine ligands. When 2,4-dienyl carbonates were treated with a combination of Pd2dba3 catalyst and chiral bisphosphine ligand L28, the selective formation of multiple-substituted tetrahydroquinoline derivatives 33-1 was observed. In contrast, when employing L20 as a ligand and (2R,3R)-diethyl-tartrate as an additive in toluene, the diastereomers 33-2 were obtained. In addition, the authors discovered through ligand screening that the combination of phosphinoamide ligand L30 and Pd2dba3 enabled the regioselective synthesis of a unique vinyl azetidine derivative in high yield and low enantioselectivity.
 |
| | Scheme 33 Pd-catalyzed diastereoselective cascade reaction of o-TsNH arylimines with 2,4-dienyl carbonates. | |
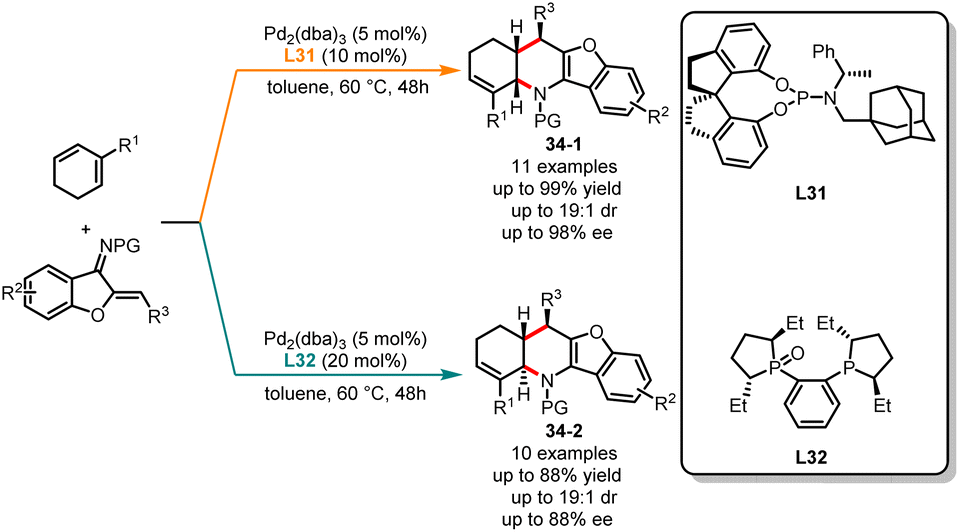
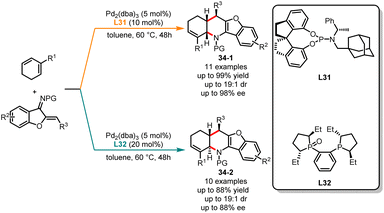
Subsequently, the same group explored an example of ligand-controlled diastereodivergent [4 + 2] annulations of cyclic 1,3-dienes and 1-azadienes, leading to both cis- and trans-fused tetrahydropyridine derivatives with high yields and acceptable enantioselectivities (Scheme 34).89 The treatment of substrates with Pd2(dba)3 and bulky monophosphorous ligand L31 in toluene provided cis-fused tetrahydropyridine 34-1 in good yields. Contrarily, by switching to the (R,R)-Me-DuPhos monoxide L32, trans-fused tetrahydropyridines 34-2 were produced as the sole products in excellent yields. In this catalytic system, various linear dienes and 1-heterodienes were also tolerated, generating a range of structurally diverse cyclization products.
 |
| | Scheme 34 Pd-catalyzed diastereoselective [4 + 2] annulations of 1-azadienes and cyclic 1,3-dienes. | |
5. Conclusions
In conclusion, we have reviewed the recent achievements in the field of ligand-enabled palladium-catalyzed divergent synthesis. The cutting-edge catalytic divergent strategies are truly captivating to derive distinct molecular frameworks from the same starting materials while employing the different ligands under nearly identical reaction conditions. These methods pave the way for the design and discovery of novel catalytic divergent approaches for enabling the realization of challenging organic transformations. Moreover, palladium-catalyzed divergent synthesis exhibits remarkable vitality and offers a valuable perspective within the realm of synthetic chemistry. It's noted that the control of the selectivities remains a notable challenge. Typically, the formation of one specific isomer is inherently favoured, while the other isomers cannot be readily accessed with high efficiency. Additionally, the ligand-directed divergent synthesis is serendipitous. To overcome these challenges, it is imperative to develop more catalytic transformations with broader synthetic utility, for instance the continuous development of widely applicable catalytic divergent strategies, the elucidation of the principles governing catalytic divergent synthesis, and the exploration of reaction mechanisms through modern techniques such as DFT calculations and AI-driven approaches. We expect that breakthroughs in palladium-catalyzed divergent synthesis will open avenues for the development of innovative reactions in the future.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful to the National Natural Science Foundation of China (21702189), the Key Scientific Science and Technology Research and Development Plan joint fund (cultivation of superior disciplines) project (222301420042) and the Start-up Grant of Henan University of Technology (No. 31401507).
References
-
(a) J. A. Frearson and I. T. Collie, Drug Discovery Today, 2009, 14, 1150 CrossRef;
(b) R. Macarron, M. N. Banks, D. Bojanic, D. J. Burns, D. A. Cirovic, T. Garyantes, D. V. S. Green, R. P. Hertzberg, W. P. Janzen, J. W. Paslay, U. Schopfer and G. S. Sittampalam, Nat. Rev. Drug Discovery, 2011, 10, 188 CrossRef CAS.
- S. Krautwald, D. Sarlah, M. A. Schafroth and E. M. Carreira, Science, 2013, 340, 1065 CrossRef CAS PubMed.
- Selected examples:
(a) S. Singha, E. Serrano, S. Mondal, C. G. Daniliuc and F. Glorius, Nat. Catal., 2020, 3, 48 CrossRef CAS;
(b) D. Kaldre, I. Klose and N. Maulide, Science, 2018, 361, 664 CrossRef CAS;
(c) L. Wei, Q. Zhu, S.-M. Xu, X. Chang and C.-J. Wang, J. Am. Chem. Soc., 2018, 140, 1508 CrossRef CAS PubMed;
(d) H. Wang, R. Zhang, Q. Zhang and W. Zi, J. Am. Chem. Soc., 2021, 143, 10948 CrossRef CAS PubMed;
(e) Y. Meng, L. Chen and E.-Q. Li, Chem. Rec., 2022, 22, e202100276 CrossRef CAS;
(f) H.-F. Tu, P. Yang, Z.-H. Lin, C. Zheng and S.-L. You, Nat. Chem., 2020, 12, 838 CrossRef CAS.
-
(a) Y. Liu, W. Li and J. Zhang, Natl. Sci. Rev., 2017, 4, 326 CrossRef CAS;
(b) S. S. Ichake, B. K. Villuri, S. R. Reddy, V. Kavala and C.-F. Yao, Org. Lett., 2019, 21, 2256 CrossRef CAS;
(c) S. R. Reddy and J. Jayakumar, Phys. Sci. Rev., 2023, 8, 3859 Search PubMed.
-
(a) I. P. Beletskaya, C. Nájera and M. Yus, Chem. Rev., 2018, 118, 5080 CrossRef CAS PubMed;
(b) G. Zhan, W. Du and Y.-C. Chen, Chem. Soc. Rev., 2017, 46, 1675 RSC;
(c) L. Lin and X. Feng, Chem. – Eur. J., 2017, 23, 6464 CrossRef CAS;
(d) C. Nájera, F. Foubelo, J. M. Sansano and M. Yus, Org. Biomol. Chem., 2020, 18, 1232 RSC;
(e) G. G. Victoriaab and S. R. Reddy, New J. Chem., 2021, 45, 8386 RSC;
(f) P. Sridhar, M. Alagumuthu, B. Ram, S. Arumugam and S. R. Reddy, ChemistrySelect, 2017, 2, 842 CrossRef;
(g) M. Dai, Z. Sun and L.-A. Chen, Angew. Chem., Int. Ed., 2022, 61, e202203835 CrossRef CAS;
(h) C. Wu, F. Zhao, Y. Du, L. Zhao, L. Chen, J. Wang and H. Liu, RSC Adv., 2016, 6, 70682 RSC;
(i) F.-P. Wu, J.-B. Peng, L.-S. Meng, X. Qi and X.-F. Wu, ChemCatChem, 2017, 9, 3121 CrossRef CAS;
(j) J. Huo, K. Zhong, Y. Xue, M. Lyu, Y. Ping, W. Ouyang, Z. Liu, Y. Lan and J. Wang, Chem. – Eur. J., 2022, 28, e202200191 CrossRef CAS;
(k) Y.-Q. Zhu, R. Zhang, W. Sang, H.-J. Wang, Y. Wu, B.-Y. Yu, J.-C. Zhang, H. Cheng and C. Chen, Org. Chem. Front., 2020, 7, 1981 RSC;
(l) Y. Ogiwara, Y. Sakurai, H. Hattori and N. Sakai, Org. Lett., 2018, 20, 4204 CrossRef CAS.
- Y. Ke, W. Li, W. Liu and W. Kong, Sci. China: Chem., 2023, 66, 2951 CrossRef CAS.
-
(a) V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471 CrossRef CAS;
(b) C. Locatelli, F. B. Filippin-Monteiro and T. B. Creczynski-Pasa, Eur. J. Med. Chem., 2013, 60, 233 CrossRef CAS PubMed.
-
(a) S. D. Friis, T. Skrydstrup and S. L. Buchwald, Org. Lett., 2014, 16, 4296 CrossRef CAS PubMed;
(b) J. Chen, K. Natte, A. Spannenberg, H. Neumann, P. Langer, M. Beller and X.-F. Wu, Angew. Chem., Int. Ed., 2014, 53, 7579 CrossRef CAS.
- T. Xu and H. Alper, J. Am. Chem. Soc., 2014, 136, 16970 CrossRef CAS.
-
(a) J. Tsuji, M. Morikawa and N. Iwamoto, J. Am. Chem. Soc., 1964, 86, 2095 CrossRef CAS;
(b) A. Schoenberg and R. F. Heck, J. Am. Chem. Soc., 1974, 96, 7761 CrossRef CAS;
(c) X.-F. Wu, H. Neumann and M. Beller, Chem. Rev., 2013, 113, 1 CrossRef CAS PubMed.
- C. Shen, Z. Wei, H. Jiao and X.-F. Wu, Chem. – Eur. J., 2017, 23, 13369 CrossRef CAS.
- J.-B. Peng, H.-Q. Geng, F.-P. Wu, D. Li and X.-F. Wu, J. Catal., 2019, 375, 519 CrossRef CAS.
-
(a) S. Das, D. Addis, L. R. Knöpke, U. Bentrup, K. Junge, A. Brückner and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 9180 CrossRef CAS;
(b) K. Orito, A. Horibata, T. Nakamura, H. Ushito, H. Nagasaki, M. Yuguchi, S. Yamashita and M. Tokuda, J. Am. Chem. Soc., 2004, 126, 14342 CrossRef CAS;
(c) L.-B. Zhang, Z.-C. Wang, S.-Z. Sun, S.-F. Ni, L.-R. Wen and M. Li, Chin. J. Chem., 2021, 39, 903 CrossRef CAS.
- C. S. Cho, L. H. Jiang, D. Y. Lee, S. C. Shim, H. S. Lee and S.-D. Cho, J. Heterocycl. Chem., 1997, 34, 1371 CrossRef CAS.
- J. Takaya, K. Sangu and N. Iwasawa, Angew. Chem., Int. Ed., 2009, 48, 7090 CrossRef CAS PubMed.
- J. Yan, Y. Ding and H. Huang, J. Org. Chem., 2023, 88, 5194 CrossRef CAS.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS.
-
(a) X.-B. Chen, L. Li, W.-C. Yang, K.-L. Song, B. Wu, W.-E. Gan, J. Cao and L.-W. Xu, Chin. J. Chem., 2021, 39, 1611 CrossRef CAS;
(b) X. Li, H. Yang, Y. Teng, Y. Wang, D. Yin and Y. Tian, Chin. Chem. Lett., 2022, 33, 4223 CrossRef CAS.
- A. F. Littke, C. Dai and G. C. Fu, J. Am. Chem. Soc., 2000, 122, 4020 CrossRef CAS.
- R. Sakakibara, K. Itoh and H. Fujii, J.
Org. Chem., 2019, 84, 11474 CrossRef CAS PubMed.
- E. K. Reeves, J. N. Humke and S. R. Neufeldt, J. Org. Chem., 2019, 84, 11799 CrossRef CAS PubMed.
-
(a) Y. J. Hu, L.-X. Li, J.-C. Han, L. Min and C.-C. Li, Chem. Rev., 2020, 120, 5910 CrossRef CAS PubMed;
(b) B. M. Trost and G. Mata, Acc. Chem. Res., 2020, 53, 1293 CrossRef CAS PubMed;
(c) J. Wang, S. A. Blaszczyk, X. Li and W. Tang, Chem. Rev., 2021, 121, 110 CrossRef CAS PubMed.
- P. Li, R. Pan, M. Wang, H. Yao and A. Lin, Org. Lett., 2021, 23, 2292 CrossRef PubMed.
- F. Xie, Y. Sun, H. Song, S. Dong, Q. Zhao, J. Liu and Y. Miao, Org. Lett., 2022, 24, 268 CrossRef CAS PubMed.
- P. Nava, H. Clavier, Y. Clavier, L. Giordano, G. Buono and S. Humbel, ChemCatChem, 2015, 7, 3848 CrossRef CAS.
-
(a) H. Zheng, Y. Zhu and Y. Shi, Angew. Chem., Int. Ed., 2014, 53, 11280 CrossRef CAS PubMed;
(b) T. Yao and D. He, Org. Lett., 2017, 19, 842 CrossRef CAS PubMed;
(c) T. Yao, T. Liu and C. Zhang, Chem. Commun., 2017, 53, 2386 RSC;
(d) S. Fang, H. Jiang and W. Wu, Chin. J. Chem., 2023, 41, 181 CrossRef CAS.
- T. Yao, B. Wang, D. He, X. Zhang, X. Li and R. Fang, Org. Lett., 2020, 22, 6784 CrossRef CAS PubMed.
- X.-L. Liu, Y.-Y. Zhang, L. Li, L.-Q. Tan, Y.-A. Huang, A.-J. Ma and J.-B. Peng, Org. Lett., 2022, 24, 6692 CrossRef CAS PubMed.
-
(a) X.-F. Wu, X. Fang, L. Wu, R. Jackstell, H. Neumann and M. Beller, Acc. Chem. Res., 2014, 47, 1041 CrossRef CAS PubMed;
(b) J.-B. Peng and X.-F. Wu, Angew. Chem., Int. Ed., 2018, 57, 1152 CrossRef CAS PubMed.
-
(a) T. Satoh, M. Ikeda, Y. Kushino, M. Miura and M. Nomura, J. Org. Chem., 1997, 62, 2662 CrossRef CAS PubMed;
(b) M. Chen, Y. Li and Z.-H. Guan, Org. Lett., 2023, 25, 2571 CrossRef CAS PubMed.
- J. Liu, Q. Liu, R. Franke, R. Jackstell and M. Beller, J. Am. Chem. Soc., 2015, 137, 8556 CrossRef CAS PubMed.
-
(a) G. Kiss, Chem. Rev., 2001, 101, 3435 CrossRef CAS PubMed;
(b) A. Brennführer, H. Neumann and M. Beller, ChemCatChem, 2009, 1, 28 CrossRef.
-
(a) L. Wu, Q. Liu, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2014, 53, 6310 CrossRef CAS;
(b) N. Armanino, M. Lafrance and E. M. Carreira, Org. Lett., 2014, 16, 572 CrossRef CAS PubMed.
- W. Ren, W. Chang, Y. Wang, J. Li and Y. Shi, Org. Lett., 2015, 17, 3544 CrossRef CAS PubMed.
- W. Liu, W. Ren, J. Li, Y. Shi, W. Chang and Y. Shi, Org. Lett., 2017, 19, 1748 CrossRef CAS PubMed.
- M. Feng, B. Feng, N. Wang, H.-X. Xu and X. Jiang, Angew. Chem., Int. Ed., 2015, 54, 14960 CrossRef CAS PubMed.
- T. Xu, F. Sha and H. Alper, J. Am. Chem. Soc., 2016, 138, 6629 CrossRef CAS PubMed.
- F. Sha and H. Alper, ACS Catal., 2017, 7, 2220 CrossRef CAS.
- H.-J. Ai, W. Lu and X.-F. Wu, Angew. Chem., Int. Ed., 2021, 60, 17178 CrossRef CAS PubMed.
- D. Ding, G. Zhu and X. Jiang, Angew. Chem., Int. Ed., 2018, 57, 9028 CrossRef CAS PubMed.
- J.-B. Liu, X. Zhang, Y.-Y. Tian, X. Wang, X.-K. Zhu and D.-Z. Chen, Dalton Trans., 2019, 48, 15059 RSC.
- N. Miyaura and A. Suzuki, J. Chem. Soc., Chem. Commun., 1979, 866 RSC.
-
(a) S. A. Ohnmacht, P. Mamone, A. J. Culshaw and M. F. Greaney, Chem. Commun., 2008, 1241 RSC;
(b) H. Hichiya, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2009, 11, 1737 CrossRef PubMed.
- N. A. Strotman, H. R. Chobanian, J. He, Y. Guo, P. G. Dormer, C. M. Jones and J. E. Steve, J. Org. Chem., 2010, 75, 1733 CrossRef CAS PubMed.
- Y. Yang and S. L. Buchwald, J. Am. Chem. Soc., 2013, 135, 10642 CrossRef CAS PubMed.
- X. Dai, Y. Chen, S. Garrell, H. Liu, L.-K. Zhang, A. Palani, G. Hughes and R. Nargund, J. Org. Chem., 2013, 78, 7758 CrossRef CAS PubMed.
- M. Yamaguchi, S. Fujiwara and K. Manabe, Org. Lett., 2019, 21, 6972 CrossRef CAS PubMed.
-
(a) J. Xuan and A. Studer, Chem. Soc. Rev., 2017, 46, 4329 RSC;
(b) Y. Hu, M. Bai, Y. Yang and Q. Zhou, Org. Chem. Front., 2017, 4, 2256 RSC.
-
(a) B. M. Trost and J. T. Masters, Chem. Soc. Rev., 2016, 45, 2212 RSC;
(b) Y. Makida, Y. Takayama, H. Ohmiya and M. Sawamura, Angew. Chem., Int. Ed., 2013, 52, 5350 CrossRef CAS PubMed.
- T. R. Pradhan, H. W. Kim and J. K. Park, Angew. Chem., Int. Ed., 2018, 57, 9930 CrossRef CAS PubMed.
- M. H. Shayegan, Z.-Y. Li and X. Cui, Chem. – Eur. J., 2022, 28, e202200191 CrossRef PubMed.
- C. Cordovilla, C. Bartolomé, J. M. Martínez-Ilarduya and P. Espinet, ACS Catal., 2015, 5, 304 Search PubMed.
- S. Minière and J.-C. Cintrat, Synthesis, 2001, 705 CrossRef.
- K. de la Vega-Hernández, E. Romain, A. Coffinet, K. Bijouard, G. Gontard, F. Chemla, F. Ferreira, O. Jackowski and A. Perez-Luna, J. Am. Chem. Soc., 2018, 140, 17632 CrossRef PubMed.
- V. Debrauwer, A. Turlik, L. Rummler, A. Prescimone, A. Blanchard, K. N. Houk and V. Bizet, J. Am. Chem. Soc., 2020, 142, 11153 CrossRef CAS PubMed.
-
(a) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS PubMed;
(b) W. Wu, Y. You and Z. Weng, Chin. Chem. Lett., 2022, 33, 4517 CrossRef CAS.
- T. W. Butcher, J. L. Yang, W. M. Amberg, N. B. Watkins, N. D. Wilkinson and J. F. Hartwig, Nature, 2020, 583, 548 CrossRef CAS PubMed.
- Z.-T. Jiang, J. Huang, Y. Zeng, F. Hu and Y. Xia, Angew. Chem., Int. Ed., 2021, 60, 10626 CrossRef CAS PubMed.
- L. Lv and C. Li, Angew. Chem., Int. Ed., 2021, 60, 13098 CrossRef CAS PubMed.
- L. Wu, M. Wang, Y. Liang and Z. Shi, Chin. J. Chem., 2022, 40, 2345 CrossRef CAS.
- O. Pàmies, J. Margalef, S. Cañellas, J. James, E. Judge, P. J. Guiry, C. Moberg, J.-E. Bäckvall, A. Pfaltz, M. A. Pericàs and M. Diéguez, Chem. Rev., 2021, 121, 4373 CrossRef PubMed.
- X. Liu, C. Hou, Y. Peng, P. Chen and G. Liu, Org. Lett., 2020, 22, 9371 CrossRef CAS PubMed.
- X. Wang, H.-Z. Miao, G.-Q. Lin and Z.-T. He, Angew. Chem., Int. Ed., 2023, 62, e202301556 CrossRef CAS PubMed.
-
(a) B. M. Trost and S. Schneider, J. Am. Chem. Soc., 1989, 111, 4430 CrossRef CAS;
(b) D. C. Witkowski, M. S. McVeigh, G. M. Scherer, S. M. Anthony and N. K. Garg, J. Am. Chem. Soc., 2023, 145, 10491 CrossRef CAS PubMed;
(c) X. Zhang, C. Zhang, B. Jiang, Y. Gao, X. Xu and Z. Miao, Org. Lett., 2022, 24, 3097 CrossRef CAS PubMed;
(d) Z. Zhao, X.-X. Yang, G.-Y. Ran, Q. Ouyang, W. Du and Y.-C. Chen, Org. Lett., 2021, 23, 4791 CrossRef CAS PubMed.
- B. M. Trost, Z. Huang and G. M. Murhade, Science, 2018, 362, 564 CrossRef CAS PubMed.
- Y. Zou, S. Chen and K. N. Houk, J. Am. Chem. Soc., 2019, 141, 12382 CrossRef CAS PubMed.
- Y. Xia, Q.-F. Bao, Y. Li, L.-J. Wang, B.-S. Zhang, H.-C. Liua and Y.-M. Liang, Chem. Commun., 2019, 55, 4675 RSC.
- W. Chai, Q. Zhou, W. Ai, Y. Zheng, T. Qin, X. Xu and W. Zi, J. Am. Chem. Soc., 2021, 143, 3595 CrossRef CAS PubMed.
- Y. Li, D. Wu, H.-G. Cheng and G. Yin, Angew. Chem., Int. Ed., 2020, 59, 7990 CrossRef CAS PubMed.
- T. Tsuda, Y. Kawakami, S.-M. Choi and R. Shintani, Angew. Chem., Int. Ed., 2020, 59, 8057 CrossRef CAS PubMed.
- T. Tsuda, S.-M. Choi and R. Shintani, J. Am. Chem. Soc., 2021, 143, 1641 CrossRef CAS PubMed.
- X. Yan, M. Yang, Y.-B. She and Y.-F. Yang, Dalton Trans., 2023, 52, 737 RSC.
- B. M. Trost and D. L. VanVranken, Chem. Rev., 1996, 96, 395 CrossRef CAS PubMed.
- M. Luparia, M. T. Luparia, D. Luparia, F. Frébault, R. Goddard and N. Maulide, Angew. Chem., Int. Ed., 2011, 50, 12631 CrossRef CAS PubMed.
- S.-Z. Tan, P. Chen, L. Zhu, M.-Q. Gan, Q. Ouyang, W. Du and Y.-C. Chen, J. Am. Chem. Soc., 2022, 144, 22689 CrossRef CAS PubMed.
- J. Han, R. Liu, Z. Lin and W. Zi, Angew. Chem., Int. Ed., 2023, 62, e202215714 CrossRef CAS PubMed.
- J. Long, R. Long, G.-J. Long and X. Fang, Angew. Chem., Int. Ed., 2023, 62, e202304543 CrossRef CAS PubMed.
- M. Lautens, W. Klute and W. Tam, Chem. Rev., 1996, 96, 49 CrossRef CAS PubMed.
- B. M. Trost, N. Cramer and S. M. Silverman, J. Am. Chem. Soc., 2007, 129, 12396 CrossRef CAS PubMed.
- M. Faltracco, V. Sukowski, M. Druenen, T. A. Hamlin, F. M. Bickelhaupt and E. Ruijter, J. Org. Chem., 2020, 85, 9566 CrossRef CAS PubMed.
- E. Geist, A. Kirschning and T. Schmidt, Nat. Prod. Rep., 2014, 31, 441 RSC.
-
(a) S. Singha, E. Serrano, S. Mondal, C. G. Daniliuc and F. Glorius, Nat. Catal., 2020, 3, 48 CrossRef CAS;
(b) I. P. Beletskaya, C. Nájera and M. Yus, Chem. Rev., 2018, 118, 5080 CrossRef CAS PubMed.
- C. C. Chintawar, A. K. Yadav, A. Kumar, S. P. Sancheti and N. T. Patil, Chem. Rev., 2021, 121, 8478 CrossRef CAS PubMed.
-
(a) S. Jia, M. Ma, E.-Q. Li, Z. Duan and F. Mathey, Org. Lett., 2021, 23, 3337 CrossRef CAS PubMed;
(b) H. Cui, K. Li, Y. Wang, M. Song, C. Wang, D. Wei, E.-Q. Li, Z. Duan and F. Mathey, Org. Biomol. Chem., 2020, 18, 3740 RSC;
(c) M. Zhi, Z. Gan, R. Ma, H. Cui, E.-Q. Li, Z. Duan and F. Mathey, Org. Lett., 2019, 21, 3210 CrossRef CAS PubMed;
(d) Z. Gan, M. Zhi, R. Han, E.-Q. Li, Z. Duan and F. Mathey, Org. Lett., 2019, 21, 2782 CrossRef CAS PubMed;
(e) Z. Gan, K. Li, H. Zhang and E.-Q. Li, Synthesis, 2021, 53, 1331 CrossRef CAS;
(f) Q. Wang, Y. Meng, L. Wu and E.-Q. Li, Chin. Chem. Lett., 2023, 34, 108544 CrossRef CAS;
(g) M. Gou, P. Zhang and E.-Q. Li, Top. Curr. Chem. (Z), 2023, 381, 33 CrossRef PubMed.
- Y. Wang, E.-Q. Li and Z. Duan, Chem. Sci., 2022, 13, 8131 RSC.
- Z.-C. Ma, L.-W. Wei, S. Liu and Y. Huang, Chem Catal., 2023, 3, 100669 CrossRef CAS.
-
(a) Y. Hayashi, Chem. Sci., 2016, 7, 866 RSC;
(b) H. Pellissier, Chem. Rev., 2013, 113, 442 CrossRef CAS PubMed.
- J.-X. Zhu, Z.-C. Chen, W. Du and Y.-C. Chen, Angew. Chem., Int. Ed., 2022, 61, e202200880 CrossRef CAS PubMed.
- Y. Hu, J.-Y. Huang, R.-J. Yan, Z.-C. Chen, Q. Ouyang, W. Du and Y.-C. Chen, Chem. Sci., 2023, 14, 1896 RSC.
Footnote |
| † All authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *b,
Zhenhua
Jia†
*b,
Zhenhua
Jia†