
Cu(II) salts as terminal oxidants in visible-light photochemical oxidation reactions
Grace A.
Lutovsky
 and
Tehshik P.
Yoon
*
and
Tehshik P.
Yoon
*
Department of Chemistry, University of Wisconsin–Madison, 1101 University Avenue, Madison, WI 53706, USA. E-mail: tyoon@chem.wisc.edu
First published on 22nd November 2023
Abstract
Photochemistry provides an important platform for the discovery of synthetically useful transformations. The development of new oxidative photoreactions, however, has proven to be relatively challenging. The importance of the identity of the terminal oxidant has been an underappreciated consideration in the design of these reactions. Many of the most common terminal oxidants used in ground-state catalytic methods are poorly compatible with the one-electron oxidation state changes characteristic of photoredox reactions and result in hard-to-control deleterious side reactions. As an alternative, Cu(II) salts have emerged as versatile terminal oxidants in photochemical oxidation reactions that are terrestrially abundant, cost-effective, and readily compatible with one-electron oxidation state changes. This review highlights recent reaction methods that leverage Cu(II) oxidation in combination with the photochemical activation of substrates or that use Cu(II) salts as both the active chromophore and terminal oxidant.
 Grace A. Lutovsky, Tehshik P. Yoon | Grace Lutovsky received her B.Sc. in Chemistry from Iowa State University. In 2018, she joined the research group of Prof. Tehshik Yoon at the University of Wisconsin–Madison where she was a 3M Science & Technology Fellow developing photochemical oxidation reactions. Grace is currently a synthetic chemist at Corteva Agriscience. Tehshik Yoon received his M.S. and Ph.D. from Caltech studying with Prof. Erick Carreira and Prof. David MacMillan, respectively. He then completed his postdoctoral studies with Prof. Eric Jacobson at Harvard University. Tehshik is currently a Professor in the Department of Chemistry at the University of Wisconsin–Madison. His research focuses on designing novel photochemical reactions to use in organic synthesis. |
1. Introduction
Over the past decade, the generation of highly reactive open-shelled intermediates under mild photochemical conditions has increasingly become recognized as a powerful strategy for reaction development. Among the remarkable range of new synthetic methods that have emerged from this recent effort, however, the majority have been redox-neutral transformations.1 This can be rationalized as a consequence of the balanced electron transfer steps required to initiate and terminate a photocatalytic cycle (Scheme 1). In a generic oxidatively triggered photoredox reaction, for example, visible light irradiation enables the excited-state photocatalyst to oxidize a substrate by a single electron transfer (SET) event to afford a radical cation intermediate. Regardless of the number or nature of the steps through which this photogenerated intermediate evolves, the regeneration of the photocatalyst requires a complementary single electron transfer event in which a radical intermediate accepts an electron from the reduced photocatalyst. This description details a reductive quenching cycle of the photocatalyst; an oxidative quenching cycle can also occur involving electron transfer steps with opposite directionality. Both cycles are redox-neutral, as two complementary single electron transfer events occur to turn over the photocatalyst. For a net-oxidative photochemical transformation to occur, the overall mechanism must involve two additional single-electron transfer steps to a suitable terminal oxidant from an intermediate in the organoradical process and from the photocatalyst. The identity of the oxidant is a crucial consideration in the design of effective net-oxidative photoreactions. | ||
| Scheme 1 Photoredox catalysis is well-suited for redox-neutral reactions. | ||
The identification of suitable terminal oxidants for photoredox reactions has proven to be a nontrivial problem.2 Molecular oxygen is often considered an ideal terminal oxidant for many conventional transition metal-catalyzed oxidative processes because of its low cost and because its reduction results in relatively benign byproducts.3 The application of molecular oxygen in photochemistry however, can often be problematic.4 Oxygen can deactivate the excited states of photocatalysts at rates that outcompete productive photocatalytic processes; photocatalyst quenching by molecular oxygen can produce singlet oxygen or superoxide, each of which are highly reactive oxidants that engage in many deleterious decomposition pathways (Scheme 2A). Other oxidants have been used in oxidative photoredox reactions, including peroxides,5 halocarbons,6 and persulfates (Scheme 2B).7 One-electron reduction of these oxidants, however, generates heteroatom-centered radicals that are highly reactive towards organic substrates, which can also result in uncontrollable and often unpredictable decomposition pathways.
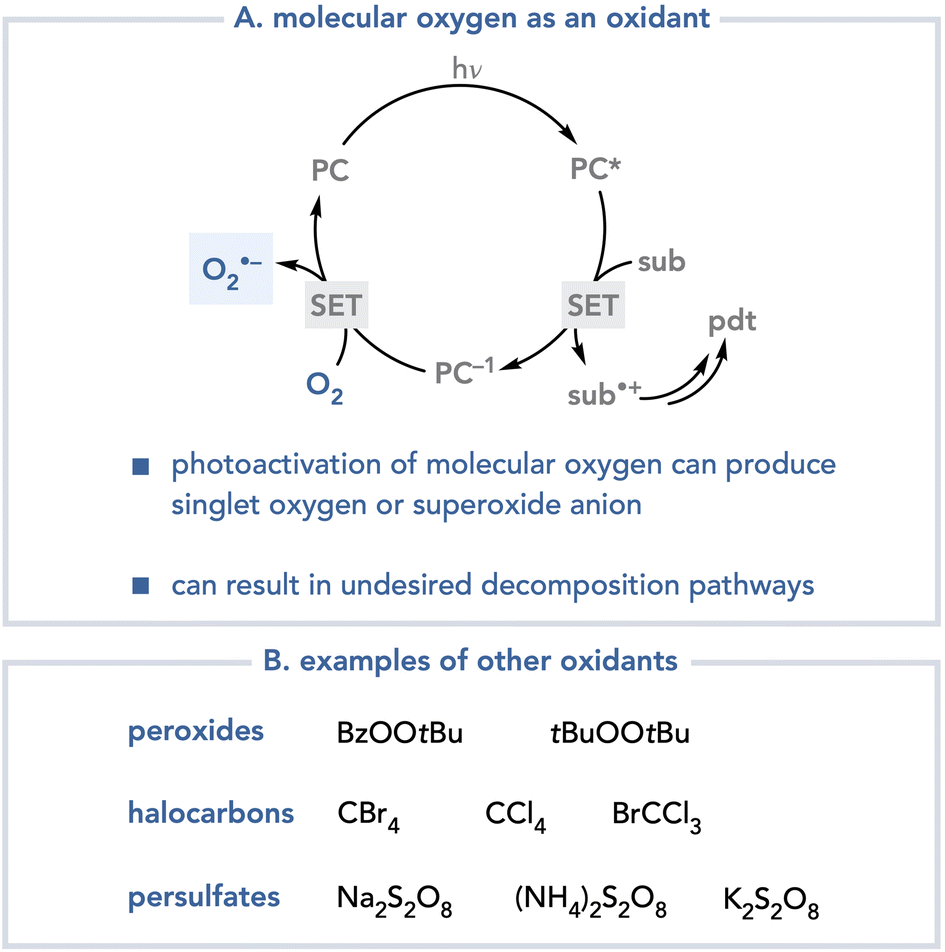 | ||
| Scheme 2 Selected classes of terminal oxidants in photoredox catalysis. | ||
A recent development in this field has been the recognition that Cu(II) salts are excellent, practical terminal oxidants that can be used in a range of net-oxidative photochemical reactions (Scheme 3). Cu(II) salts are terrestrially abundant, and they are classified as metals of low safety concern in drug substances: current international guidelines permit a daily exposure limit of 2500 μg day−1 (compared to <250 μg day−1 for Ni or Pd).8 The commercial cost of Cu(II) salts varies, but in general their per mole prices are comparable to or lower than those of the most common oxidants used in thermal oxidation methods (Scheme 3A). Additionally, the byproducts arising from the one- and two-electron reduction of Cu(II) salts are relatively benign and can be easily removed from a reaction mixture by an aqueous workup or by filtration through silica. Mechanistically, Cu(II) salts have been extensively studied for their reactions with radicals. Cu(II) salts oxidize organoradicals at diffusion-controlled rates to generate polar, cationic intermediates that can be exploited in a variety of useful transformations (Scheme 3B).9 Consequently, many examples of thermally initiated radical reactions using Cu(II) terminal oxidants are known.10 Additionally, Cu is often used as a co-catalyst in photochemical reactions.11 As one example, Sanford and coworkers demonstrated that the trifluoromethylation of aryl boronic acids can be accomplished using a combination of photoredox and copper catalysis (Scheme 3C).12 In this reaction, the copper undergoes a series of one-electron oxidation state changes spanning Cu(I), Cu(II), and Cu(III). If the one-electron oxidation state changes of Cu salts are readily tolerated in catalytic metallaphotoredox reactions, it stands to reason that they should also be compatible with photocatalytic reactions when Cu(II) is used as a stoichiometric terminal oxidant.
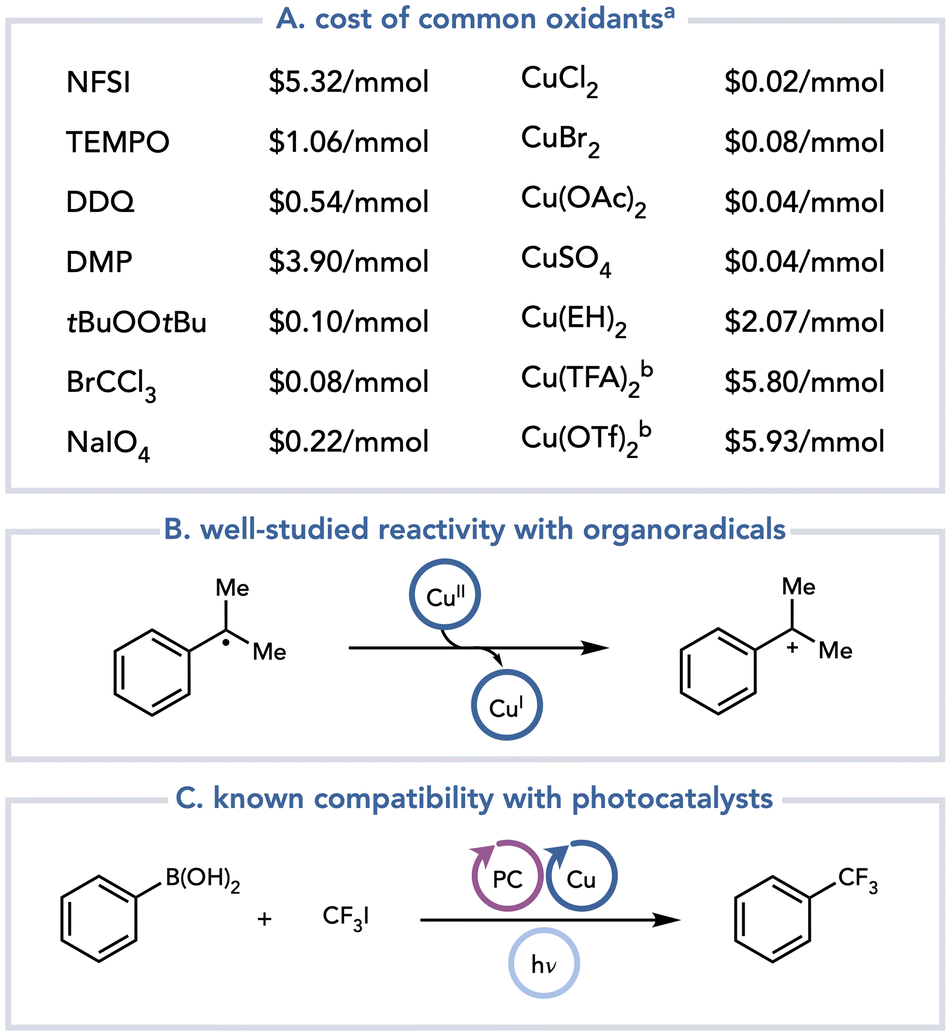 | ||
Scheme 3 Selected classes of terminal oxidants in photoredox catalysis. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Prices were found on the Sigma-Aldrich website on July 27, 2023. Prices will vary by vendor. bCan be synthesized from CuSO4 or CuCl2. Prices were found on the Sigma-Aldrich website on July 27, 2023. Prices will vary by vendor. bCan be synthesized from CuSO4 or CuCl2. | ||
This review summarizes photochemical oxidative reaction methods that have been published using Cu(II) as a terminal oxidant. First, we discuss methods that use photoredox catalysis in combination with oxidation by Cu(II), and second, we describe methods that harness Cu(II) as the active chromophore in addition to its role as a terminal oxidant.
2. Cu(II) salts as terminal oxidants in photoredox reactions
2.1 Oxidative substitution reactions of photochemically generated organoradicals
In 2018, Yoon and coworkers reported an alkene oxyamination reaction enabled by photoredox catalysis that utilized Cu(II) salts as terminal oxidants.13 In this proof-of-concept study, these authors proposed that a redox-neutral hydroamination reaction developed by Nicewicz14 could be diverted towards net-oxidative reactivity by introducing a Cu(II) salt as a terminal oxidant (Scheme 4A). After the oxidation of styrenyl olefin 1 and nucleophilic attack on radical cation 2, it was proposed that organoradical 3 could be oxidized by Cu(II) to afford carbocation 5, which could be trapped by the pendant oxygen nucleophile to afford oxyamination product 6. After optimization, 1.2 equiv. of Cu(TFA)2 was determined to deliver the oxyamination products in good yield. The identity of the Cu(II) salt proved to be critical, as Cu(TFA)2 exhibits superior reactivity over other Cu(II) salts (Scheme 4B, entries 1–4). However, Cu(II) salts appear to be unique in their suitability for this transformation, as other common terminal oxidants result in no production of 8 (entries 6–11). This method is applicable to several oxyaminations, diaminations, and dioxygenations of styrenyl olefins (Scheme 4C, 9–14).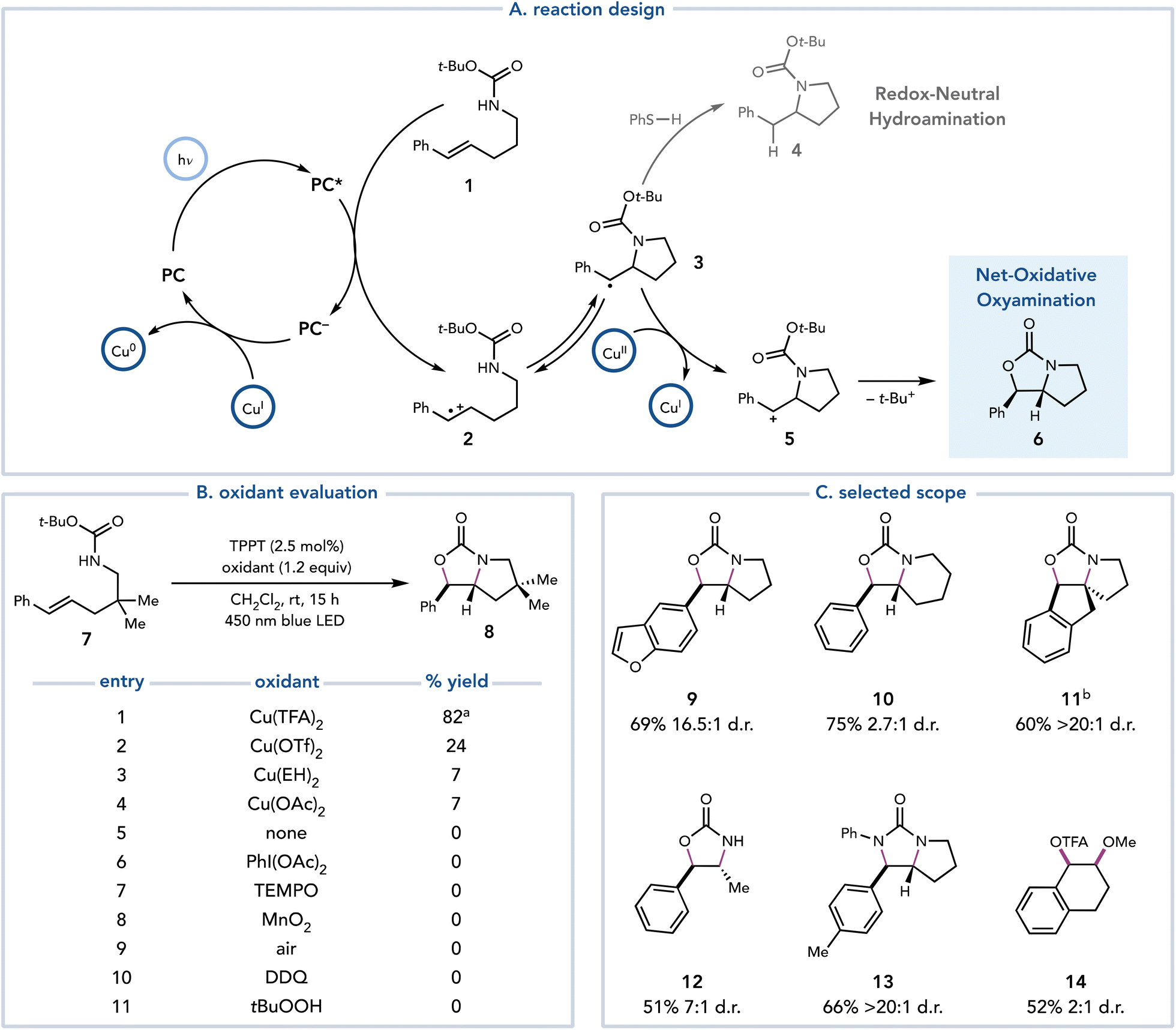 | ||
| Scheme 4 Oxidative difunctionalization of alkenes enabled by photoredox catalysis and Cu(II)-mediated oxidation. aIsolated in 72% yield, >20:1 dr. bReaction time: 72 h. TPPT, triphenylpyrylium tetrafluoroborate; EH, 2-ethylhexanoate; DDQ, 2,3-dichloro-5,6-dicyano-p-benzoquinone. | ||
The mechanism by which Cu(II) salts act as terminal oxidants is agnostic towards the mechanism by which the organoradical intermediate is generated; therefore, oxidation by Cu(II) should be compatible with any method of radical generation. Recognizing this feature, Tunge and coworkers developed a photocatalytic method for decarboxylative acetoxylation of alkyl carboxylic acids (Scheme 5).15 Single-electron photooxidation by an excited-state acridinium photocatalyst initiates a decarboxylation event to afford a carbon-centered radical, which is oxidatively functionalized by Cu(OAc)2. The acetate ligand of the Cu(II) salt serves as the nucleophile to achieve the decarboxylative acetoxylation reaction. While this reaction is conducted with stoichiometric Cu(II), the authors demonstrated that this reaction also proceeded in high yields with catalytic Cu(OAc)2 when NaOAc is added as an external acetate source and MnO2 is added as a terminal oxidant. However, a cost comparison between the two methods revealed that it is less expensive to conduct the reaction with stoichiometric Cu(II).
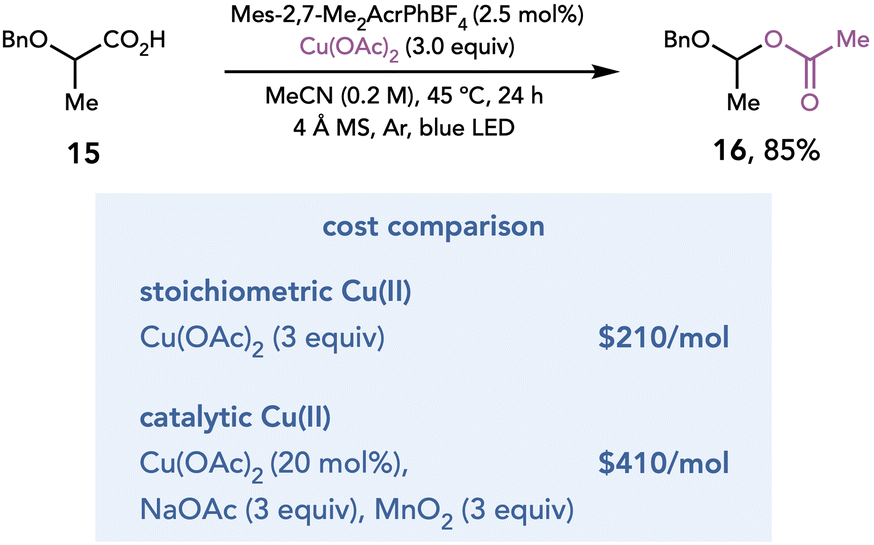 | ||
| Scheme 5 Decarboxylative acetoxylation. | ||
Yoon and coworkers later developed a photocatalytic strategy for the oxidative etherification of benzylic C–H bonds (Scheme 6).16 Upon irradiation with visible light, an excited state iridium photocatalyst can oxidize an electron-rich arene. The acidified benzylic C–H bond of the resulting radical cation can be readily deprotonated by a weak base to afford a benzylic radical, which the authors proposed could be oxidized by Cu(II) and functionalized by alcohol nucleophiles. The mild nature of photoredox catalysis in combination with Cu(II) oxidation in this method enabled the C–H benzylic alkoxylation to be applied to complex natural product scaffolds and structurally diverse alcohols containing acid- and base-sensitive functional groups (17–21).
 | ||
| Scheme 6 Benzylic C–H alkoxylation. [Ir]: [Ir(dF(CF3)ppy)2(5,5′-dCF3bpy)]PF6. a8 equiv. MeOH. bReaction conducted at 0.05 M. cReaction conducted at 0.10 M. | ||
Subsequently, Zhu and coworkers exploited Cu(II) oxidants in a photocatalytic synthesis of lignans, a class of natural products derived from coniferyl alcohol.17 Aryltetralin cyclic ethers could be synthesized from dimeric or psuedodimeric-cinnamyl ethers in the presence of a highly oxidizing photocatalyst, Cu(TFA)2 as a terminal oxidant, and MeOH as a nucleophile (Scheme 7). The authors proposed that the reaction cascade is initiated by the oxidation of styrene 22 by the excited-state photocatalyst, which undergoes cyclization to afford distonic 1,4 radical cation 24. Nucleophilic attack by the alcohol on radical cation 24, oxidation by Cu(II), and Friedel–Crafts cyclization of the electron-rich arene affords lignan 26. Employing the alcohol in large excess results in dialkoxylation (27) becoming favored over Friedel–Crafts cyclization.
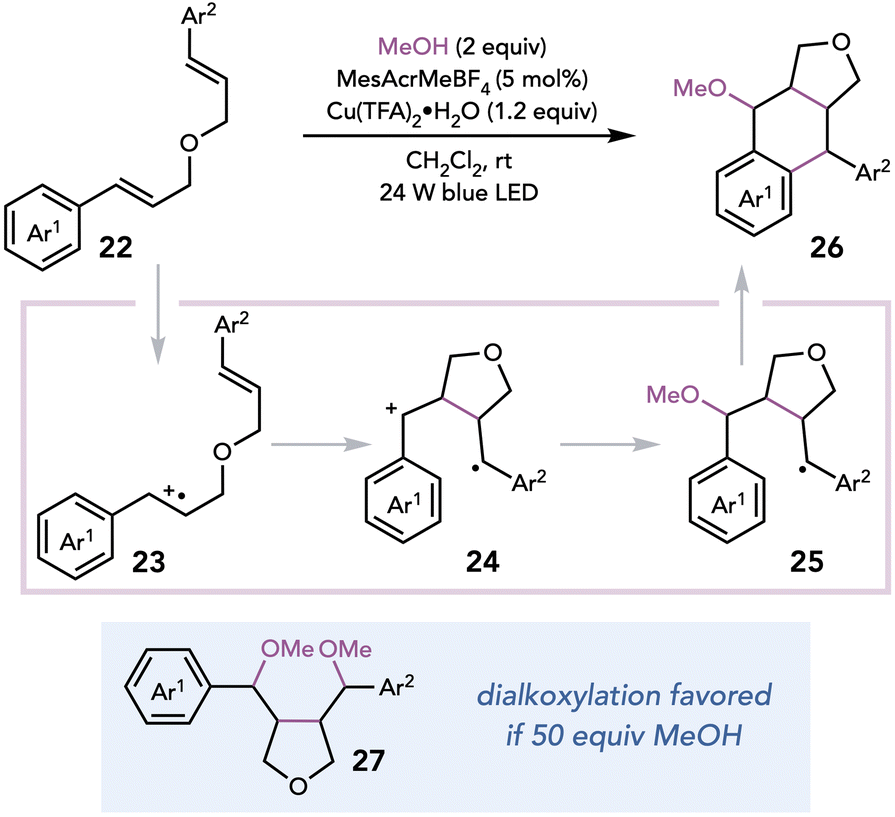 | ||
| Scheme 7 Synthesis of lignans. | ||
The merger of photoredox catalysis and Cu(II)-mediated oxidation has been further expanded to the oxidative ring-opening of arylcyclopropanes (Scheme 8).18 Feng and coworkers demonstrated that photooxidation of the electron-rich arene triggers ring-opening of the cyclopropane to afford both 1,3- and 1,5-dioxygenated products. Additionally, the facile synthesis of acetals was achieved when a diol was employed as a nucleophile.
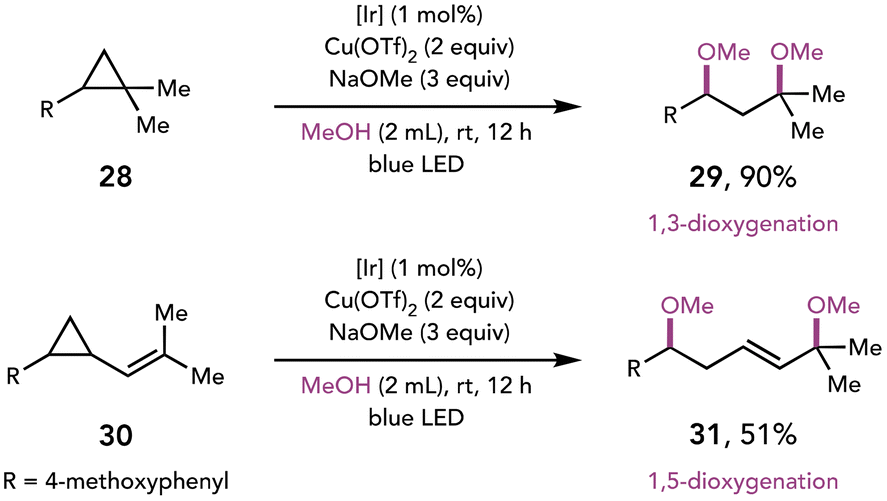 | ||
| Scheme 8 Oxidative cyclopropane ring opening affords dioxygenated products. [Ir]: [Ir(dF(CF3)ppy)2(5,5′-dCF3bpy)]PF6. | ||
2.2 Oxidative elimination reactions of photochemically generated organoradicals
While each of the previous examples highlighted nucleophilic substitution reactions of polar intermediates accessed with Cu(II) salts, these intermediates have also been shown to participate in elimination reactions. Kochi first demonstrated that the outcome of organoradical oxidation can be diverted towards substitution or elimination reactions depending on the identity of the Cu(II) salt and the reaction conditions.9a,19 Many years later, Tunge and coworkers applied this insight to an elimination reaction starting from the photochemical decarboxylation of carboxylic acids (Scheme 9).20 Interestingly, while many Cu(II) salts generated the olefin, the identity of the Cu(II) salt influenced the E:Z ratio of the enamide product.
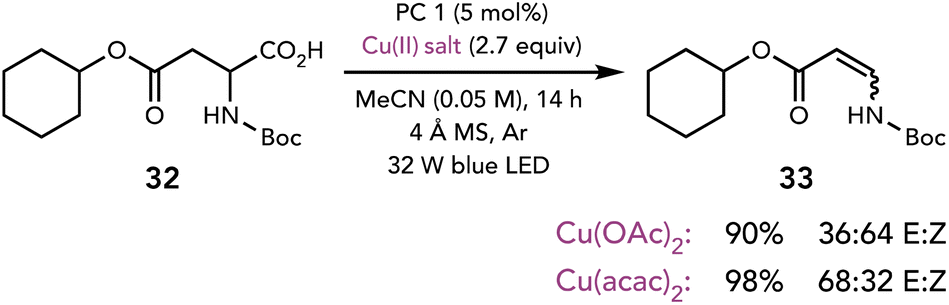 | ||
| Scheme 9 Decarboxylative elimination. PC 1: Mes-2,7-Me2AcrPhBF4. | ||
Nicewicz later utilized this mode of reactivity for the formal β-functionalization of piperidines and other saturated heterocycles (Scheme 10).21 This two-step, C–H functionalization reaction proceeds by desaturation of the nitrogen heterocycle, followed by an anti-Markovnikov hydrofunctionalization of the resulting enamine. The desaturation of piperidine 34 was proposed to occur by a photoinduced single-electron oxidation of nitrate, which reacts by hydrogen atom transfer with the heterocycle to afford an α-amino radical. Oxidation, deprotonation, and elimination furnishes the desaturated piperidine. The authors found the reaction provides optimal yields with substoichiometric amounts of Cu(OAc)2 (0.5 equiv.) which they attributed to the disproportionation of Cu(I) to Cu(II) and Cu(0). Additionally, LiNO3 (1.25 equiv.) was present in the reaction, which they proposed acts as a hydrogen atom transfer reagent, although LiNO3 could also function as an oxidant.
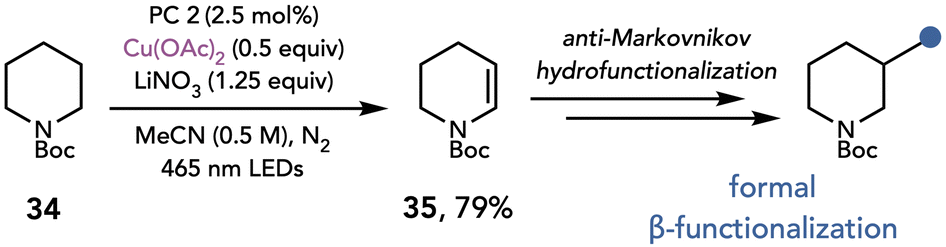 | ||
| Scheme 10 Desaturation of saturated heterocycles. PC 2: Mes-3,6-tBu2AcrPhBF4. | ||
Yoon and coworkers expanded Cu(II)-mediated elimination reactions to include oxidative alkene heterofunctionalization reactions (Scheme 11).22 Intramolecular alkene heterofunctionalization reactions are an efficient means to synthesize saturated heterocycles while still maintaining the alkene functionality to serve as a linchpin for future synthetic manipulations. The authors proposed that an excited-state photocatalyst could oxidize a tri- or tetrasubstituted alkene to the corresponding alkene radical cation. Cyclization by a pendent heteroatomic nucleophile would afford a carbon-centered radical that could undergo Cu(II)-mediated oxidative elimination. Copper(II) ethylhexanoate (Cu(EH)2) was empirically found to be the optimal Cu(II) source. This strategy proved general to synthesize a variety of saturated heterocycles, including pyrrolidines, piperidines, morpholines, tetrahydrofurans, and lactones (37–41).
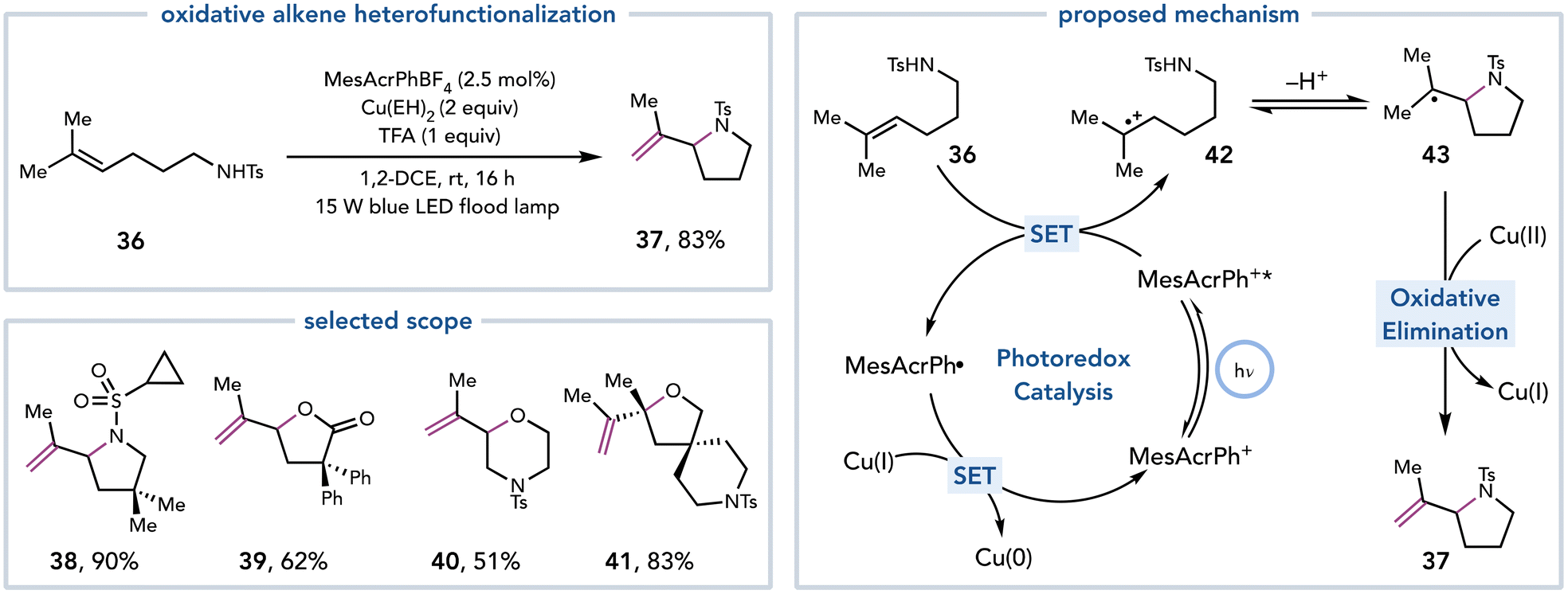 | ||
| Scheme 11 Synthesis of saturated heterocycles. | ||
Yoon next applied this strategy to an intermolecular allylic amination reaction between highly substituted alkenes and heteroatomic nucleophiles (Scheme 12).23 When trisubstituted alkene 44 and sulfonamide 45 are irradiated in the presence of a pyrylium photocatalyst and Cu(OAc)2, nucleophilic substitution was observed at C3 instead of C2, affording an unusual regioisomer for allylic amination (46) in 81% yield. This regioselectivity was proposed to arise from bimolecular nucleophilic attack on the alkene radical cation occurring at a slower rate than deprotonation to access the allylic radical; the observed product is consistent with formation of the more stable allylic radical regioisomer.
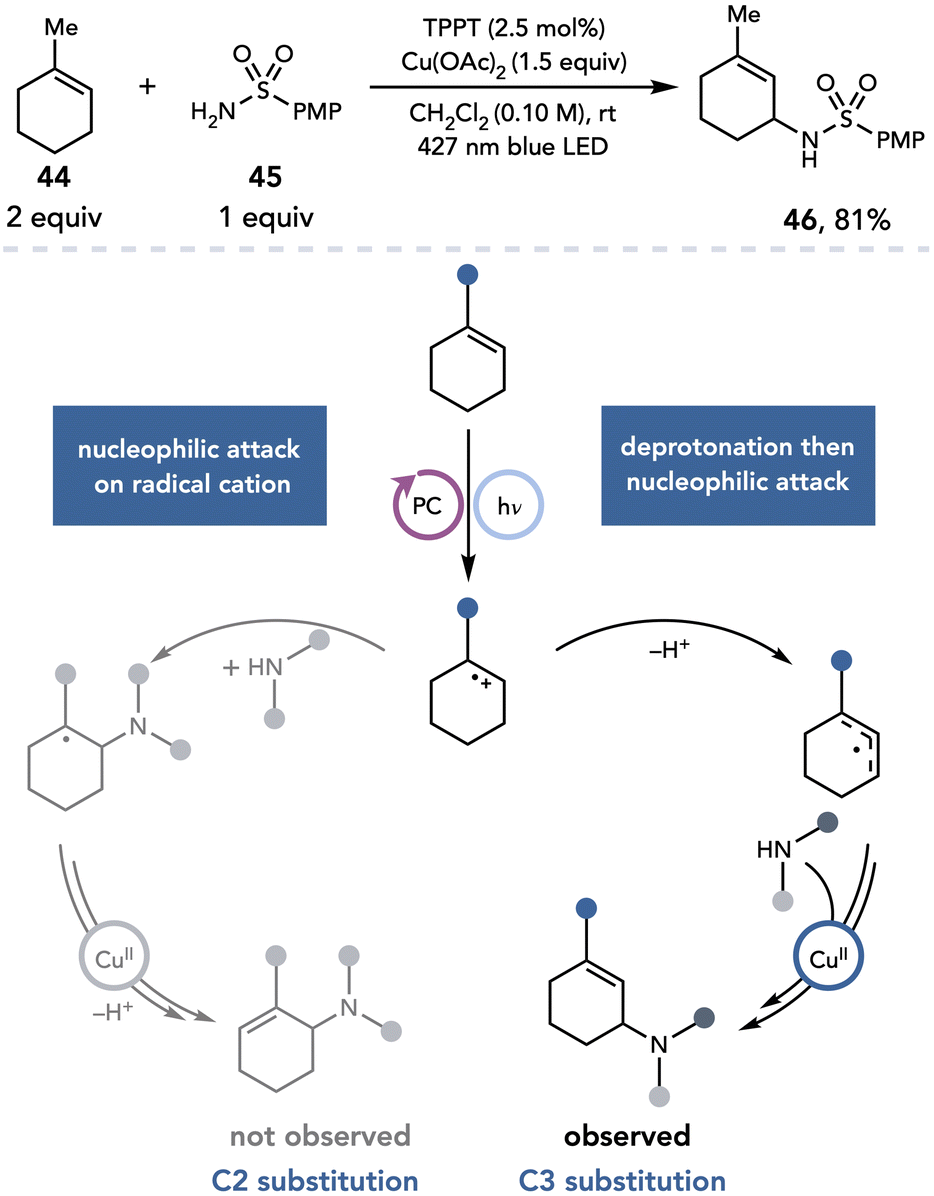 | ||
| Scheme 12 Intermolecular allylic amination of highly substituted alkenes. PMP, p-methoxyphenyl. | ||
3. Cu(II) salts as photoactive species and terminal oxidants
The direct irradiation of copper complexes to generate radical intermediates has emerged as an alternate strategy for initiating oxidative photochemical transformations.24 When an appropriately substituted copper complex is irradiated, it can undergo a ligand-to-metal charge transfer (LMCT) transition in which an electron is formally promoted from a filled orbital centered on the nucleophilic ligand to an empty orbital on copper. This event results in a single-electron reduction of the copper center and generates a radical on the ligand, which can be involved in a variety of transformations (Scheme 13). This section will discuss the methods that have leveraged LMCT reactivity of copper complexes in combination with Cu(II) as an oxidant to enable net-oxidative photochemical transformations.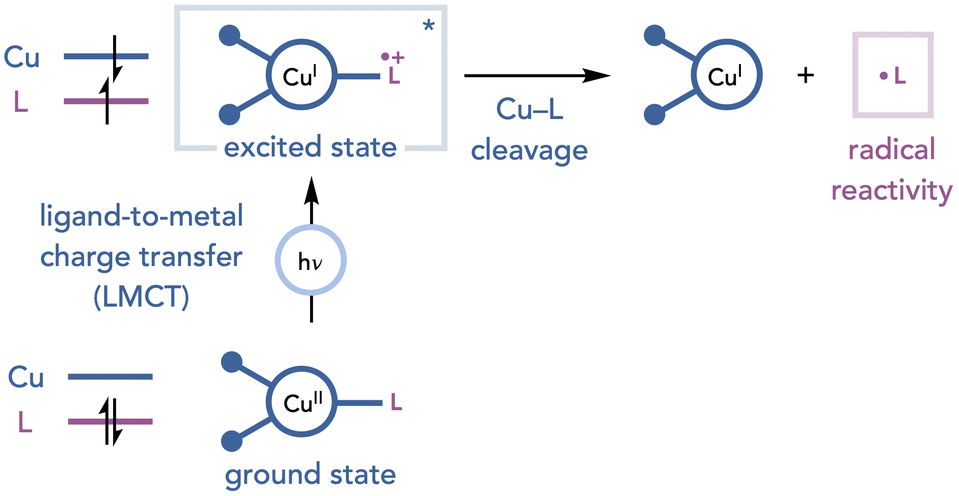 | ||
| Scheme 13 General depiction of LMCT reactivity of Cu(II) complexes. | ||
Kochi reported one of the earliest examples of LMCT reactivity of Cu complexes in which chlorine radicals were generated upon irradiation of a copper chloride complex with an unfiltered mercury lamp (Scheme 14).25 It was proposed that the active complex is an anionic halocuprate complex formed upon the addition of LiCl to the reaction media. After the homolysis of the copper–chloride bond, the chlorine radical reacts with many organic substrates to enable a range of reactions including the oxidation of alcohols to ketones (line 1), chlorination of weak C–H bonds (line 2), and dichlorination of alkenes (line 3).
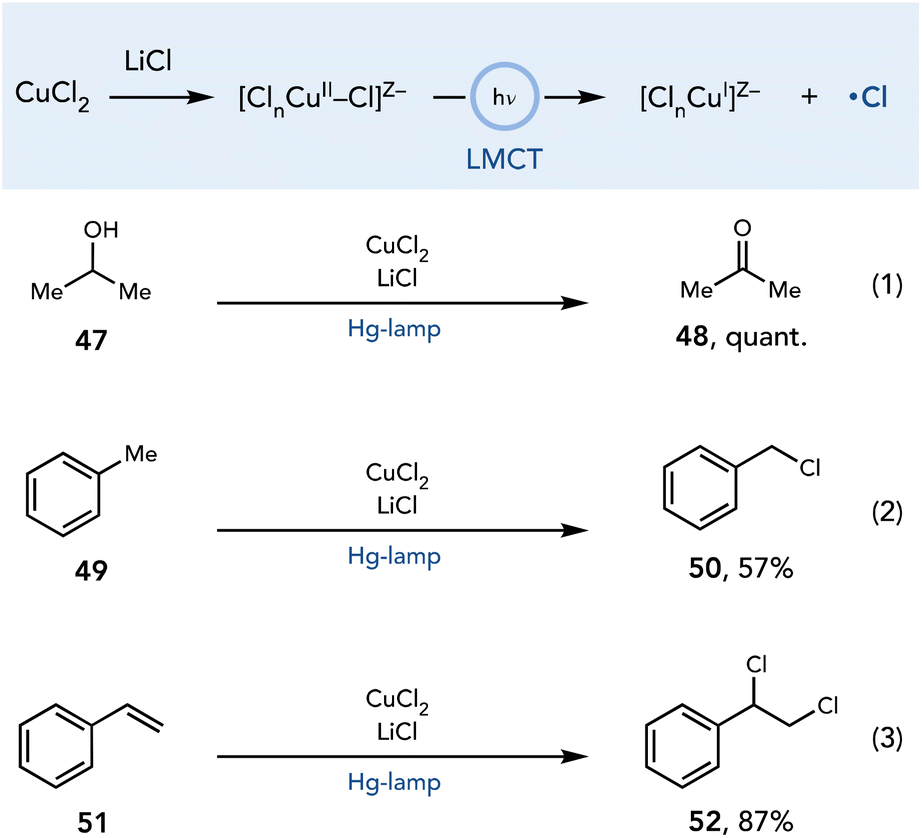 | ||
| Scheme 14 Initial observations of LMCT reactivity with CuCl2. | ||
Wan and coworkers improved upon Kochi's seminal studies in 2020, achieving copper–chloride bond homolysis of CuCl2 with visible light irradiation.26 Dichlorination of activated alkenes employed 4 equivalents of CuCl2 with white LEDs (Scheme 15). Dichlorination was proposed to occur sequentially, first by the addition of the chlorine radical into the olefin, followed by a halogen-atom-transfer from CuCl2. For unactivated alkenes, the authors used catalytic CuCl2, HCl as the external chloride source, and air as the oxidant.
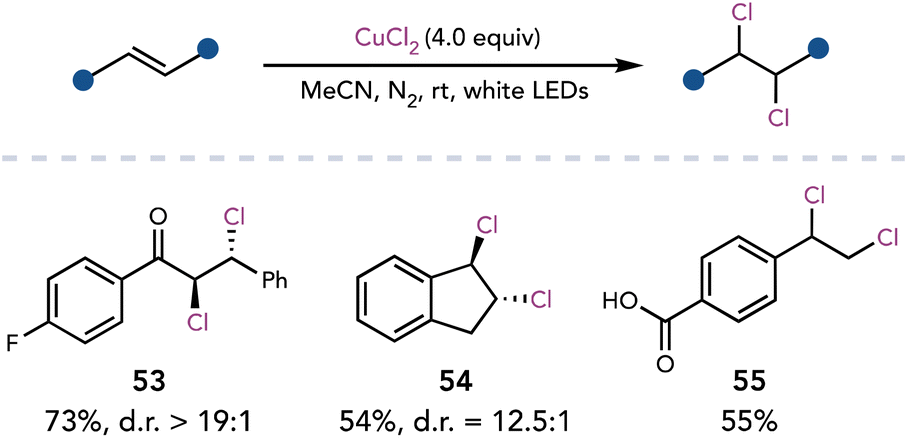 | ||
| Scheme 15 Dichlorination of olefins with CuCl2. | ||
Recently, the photoexcitation of CuCl2 complexes has been exploited to facilitate transformations beyond chlorinations. Dai and coworkers employed CuCl2 as a radical initiator and terminal oxidant to achieve the C–H borylation of alkyl C–H bonds (Scheme 16).27 The authors proposed chlorine radical is generated from LMCT when irradiated with 365 nm light and subsequently abstracts a hydrogen atom from an alkyl substrate. Trapping of the intermediate alkyl radical by B2cat2 affords the desired C–H borylation product. The C–H borylation was demonstrated on a variety of linear and cyclic aliphatic alkanes (56–59); the authors note the selectivity of this transformation is dependent on both the HAT and the C–B bond forming steps specific to B2cat2, consistent with observations made previously by Aggarwal.28 It is possible that another species may be acting as the terminal oxidant in this transformation; Aggarwal published a C–H borylation reaction under similar reaction conditions and observed the copper-catalyzed in situ formation of O(Bcat)2 with residual water.29 This oxidation of B2cat2 is balanced by the reduction of the water to molecular hydrogen, allowing the C–H borylation to proceed with catalytic CuCl2.
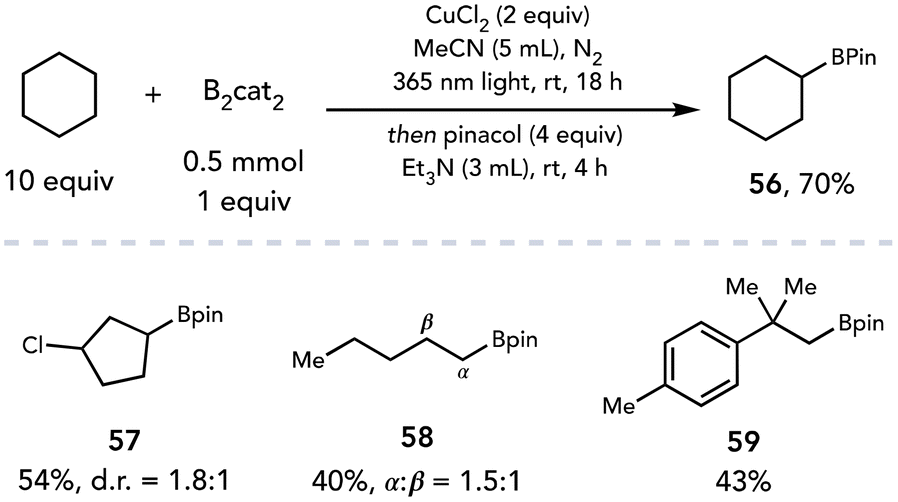 | ||
| Scheme 16 C–H borylation of aliphatic C–H bonds. | ||
The ability to generate radical intermediates from photoactive Cu complexes has been extended beyond the excitation of complexes formed from CuCl2. DeGraff and Faust demonstrated that irradiation of Cu(II)–malonate complexes with UV light results in decarboxylation and nucleophilic substitution by water to afford glycolic acid.30 In 2021, Ritter and coworkers expanded these seminal reports to the decarboxylative fluorination of benzoic acids upon visible light irradiation (Scheme 17).31 The authors anticipated that decarboxylative fluorination could be achieved by first photoexciting the in situ generated Cu(II)–carboxylate complex to induce an LMCT event that results in homolysis of the Cu–O bond. Subsequent decarboxylation of carboxy radical 62 would release CO2 and aryl radical 63, which could be rapidly captured by Cu(II). Facile reductive elimination from a Cu(III) arylfluoride complex would yield desired aryl C–F product 66. Indeed, irradiation with purple light (390 nm) in the presence of a benzoic acid, TBAF·(tBuOH)4 (2.5 equiv.), Cu(OTf)2 (2.5 equiv.), Cu(MeCN)4BF4 (2.5 equiv.) in MeCN affords high yields of the fluorinated products (67–70).
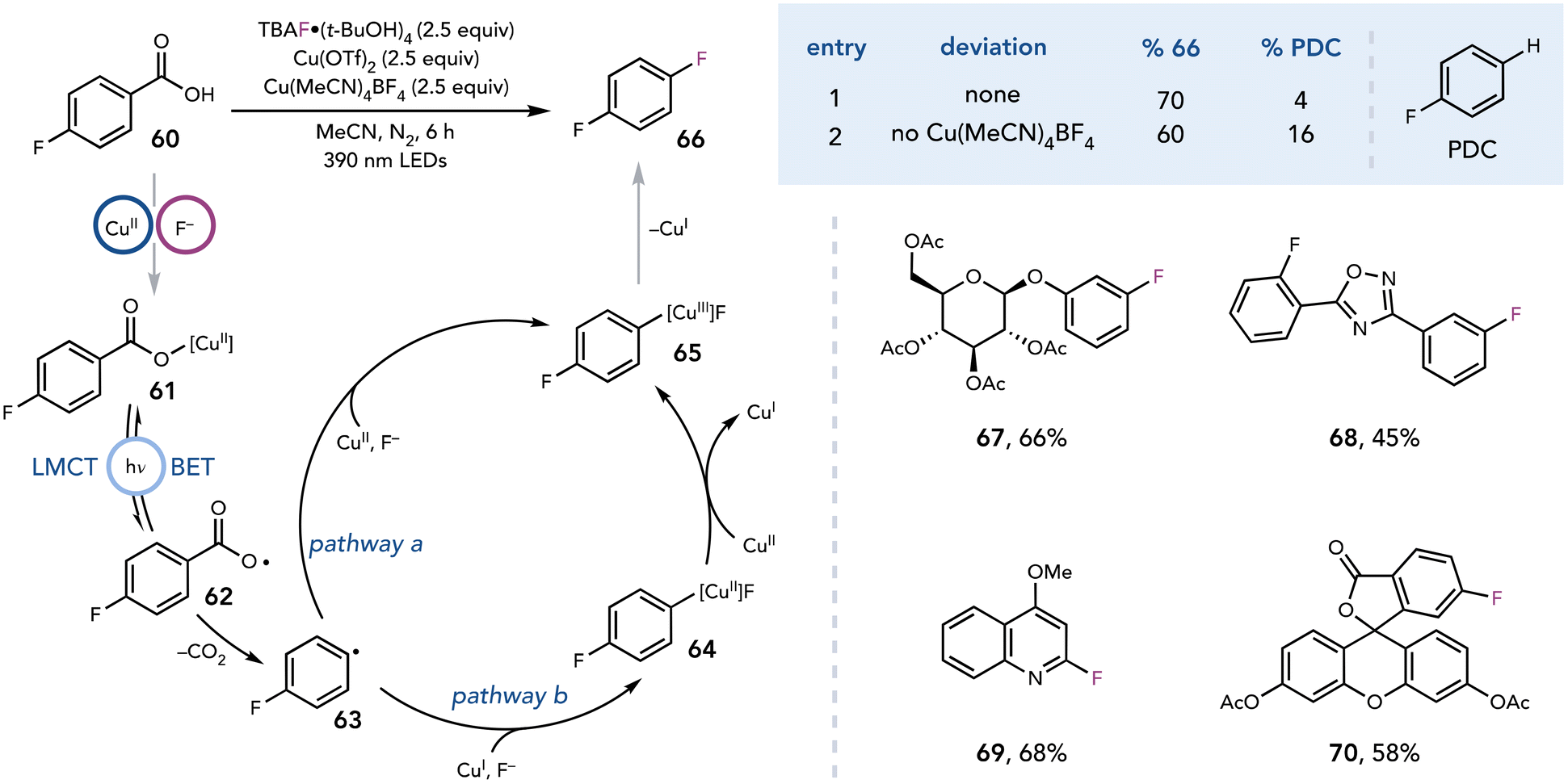 | ||
| Scheme 17 Decarboxylative fluorination. PDC, protodecarboxylation; TBAF, tetrabutylammonium fluoride. | ||
During the investigation of the decarboxylative fluorination reaction, Ritter observed high yields of a C–O dimer when the fluorine source was omitted and proposed that this dimer is formed when another equivalent of the carboxylic acid acts as the nucleophile (Scheme 18A).32 This process has a theoretical yield of 50% as two equivalents of the benzoic acid combine to afford one equivalent of the product. Alternatively, the authors speculated that if two different benzoic acids were introduced to the reaction system to achieve a 100% yield of C–O bond formation, the selectivity of the cross-dimerization to heterodimerization would be difficult to control. One strategy to achieve crossed selectivity could be to control the relative rates of decarboxylation. Presumably, both aryl carboxylates would bind to Cu and form carboxyl radicals upon irradiation with purple light. If one carboxyl radical underwent back electron transfer (BET) or hydrogen atom transfer (HAT) preferentially over decarboxylation to the aryl radical, it could selectively act as the nucleophile for the C–O coupling. Gratifyingly, they identified thiophene-2-carboxylate (TC) as a carboxylic acid that undergoes decarboxylation at a slower rate than an electron-neutral benzoic acid. The C–COO˙ is in conjugation with the sulfur atom, stabilizing and strengthening the C–C bond and slowing the rate of decarboxylation (Scheme 18B). After optimization, the reaction was found to proceed with reasonable selectivity when irradiating a lithium aryl carboxylate in the presence of Cu(TC) (1.5 equiv.) and Cu(OTf)2 (2.5 equiv.) with purple light. Upon hydrolysis with aqueous 1 M LiOH, high yields of the corresponding phenols could be isolated (Scheme 18C). This method provides a mild platform for the decarboxylative hydroxylation of benzoic acids.
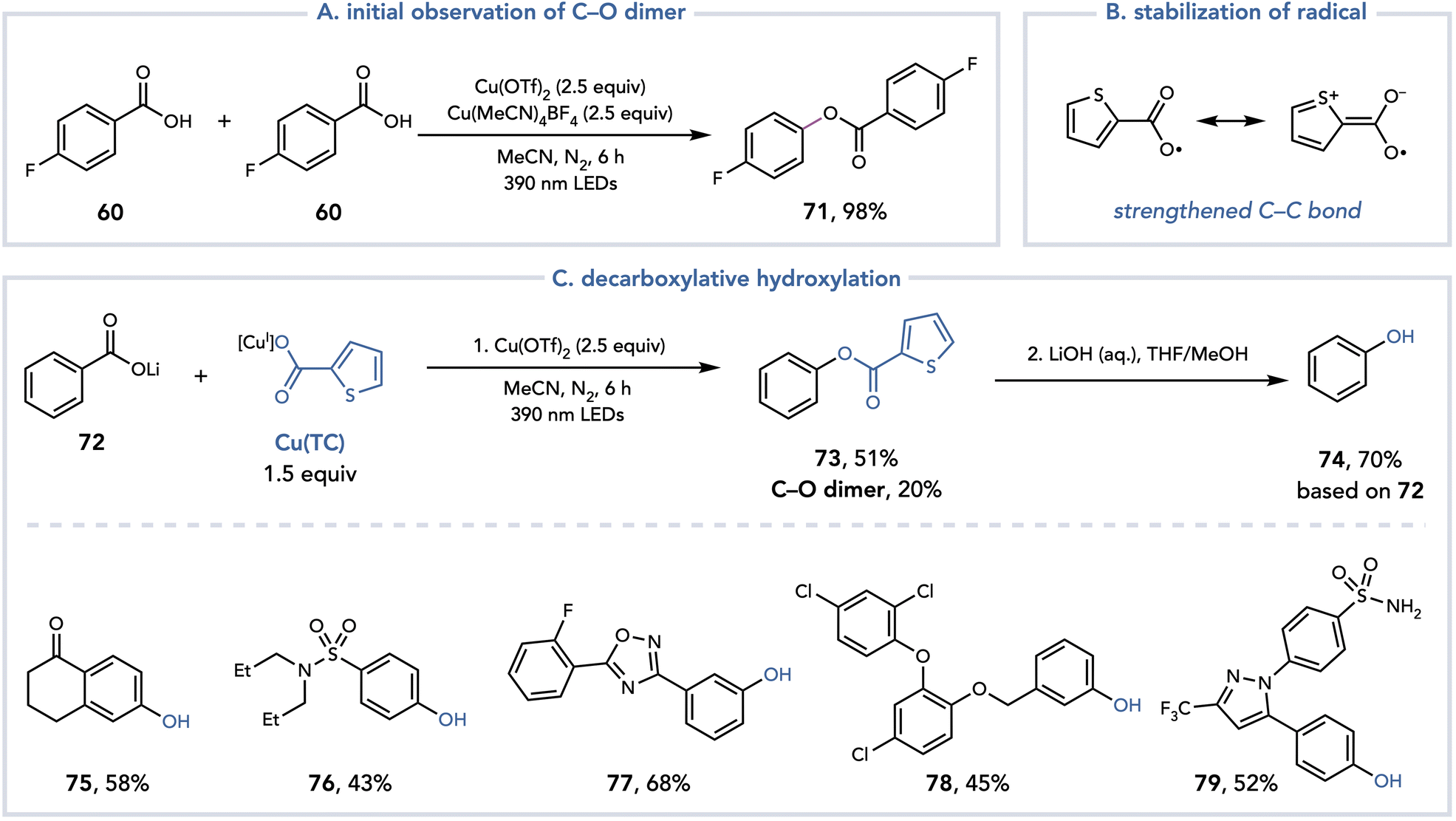 | ||
| Scheme 18 Decarboxylative hydroxylation to afford phenols. | ||
Ritter next applied this strategy to enable decarboxylative sulfoximination (Scheme 19).33 Sulfoximines can be challenging nucleophiles as their deprotonated form can coordinate to Cu. This coordination may compete with coordination of carboxylates required for decarboxylative LMCT photochemistry, and therefore may inhibit decarboxylation if the desired Cu(II)–carboxylate is not formed. The authors found the addition of 2,6-di-tert-butylpyridine (DTBP) and LiOMe in MeCN enables formation of the desired C–N coupling product. The addition of sterically bulky and weakly coordinating DTBP was hypothesized to favor the active Cu(II)–carboxylate complex or to assist in the reductive elimination from the Cu(III) complex. The authors are more uncertain of why LiOMe is significant; they proposed that it may help to form sulfoximine lithium salts, which are poorly soluble and therefore decrease the coordination of the sulfoximines to the Cu complex.
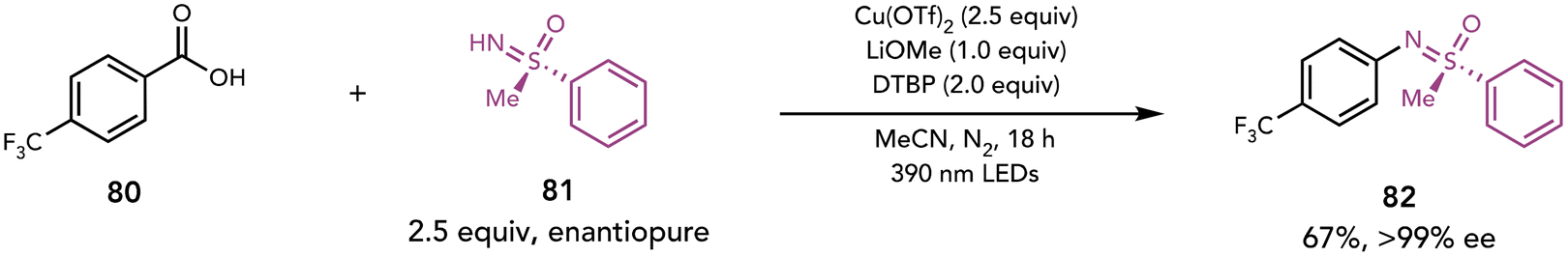 | ||
| Scheme 19 Decarboxylative sulfoximination. DTBP, 2,6-di-tert-butylpyridine. | ||
Around the same time, MacMillan and coworkers reported a similar strategy for the halogenation of benzoic acids (Scheme 20).34 A photoinduced LMCT event followed by decarboxylation afforded an aryl radical that could either be trapped by Cu or undergo halogen atom transfer to afford the halogenated product. Each halogenating coupling partner required slightly different conditions, most notably differing in the amount of copper necessary for each reaction. The iodination proceeded with catalytic Cu, N-iodosuccinimide (NIS) as an iodine source, and 1-fluoro-2,4,6-trimethylpyridinium tetrafluoroborate (NFTPT) as an oxidant (Scheme 20A). The bromination was achieved under similar conditions with 1,3-dibromo-5,5-dimethylhydantoin or bromotrichloromethane as brominating agents (Scheme 20B). The authors also found that 1 equiv. Cu could be employed to achieve similar yields without the use of NFTPT as an oxidant. Decarboxylative chlorination was achieved with [Cu(MeCN)4]BF4, NFTPT (1.5 equiv.) as an oxidant and ZnCl2 (1 equiv.) as the Cl source (Scheme 20C). [Cu(MeCN)4]BF4 was most often employed at stoichiometric loading to ensure rapid capture of the aryl radical and to outcompete production of C–O dimer, which is formed when another equivalent of the carboxylic acid acts as the coupling partner. The authors proposed that this reaction proceeds through an aryl Cu(III)–Cl species, which reductively eliminates to form the desired C–Cl bond. When CuCl2, CuCl, or FeCl2 are utilized as the chlorinating reagents in the reaction, however, significantly reduced reactivity is observed. Finally, a decarboxylative fluorination reaction was developed using [Cu(MeCN)4]BF4 (3.0 equiv.), NFTPT (2.0 equiv.), and CsF (1.0 equiv.) (Scheme 20D).
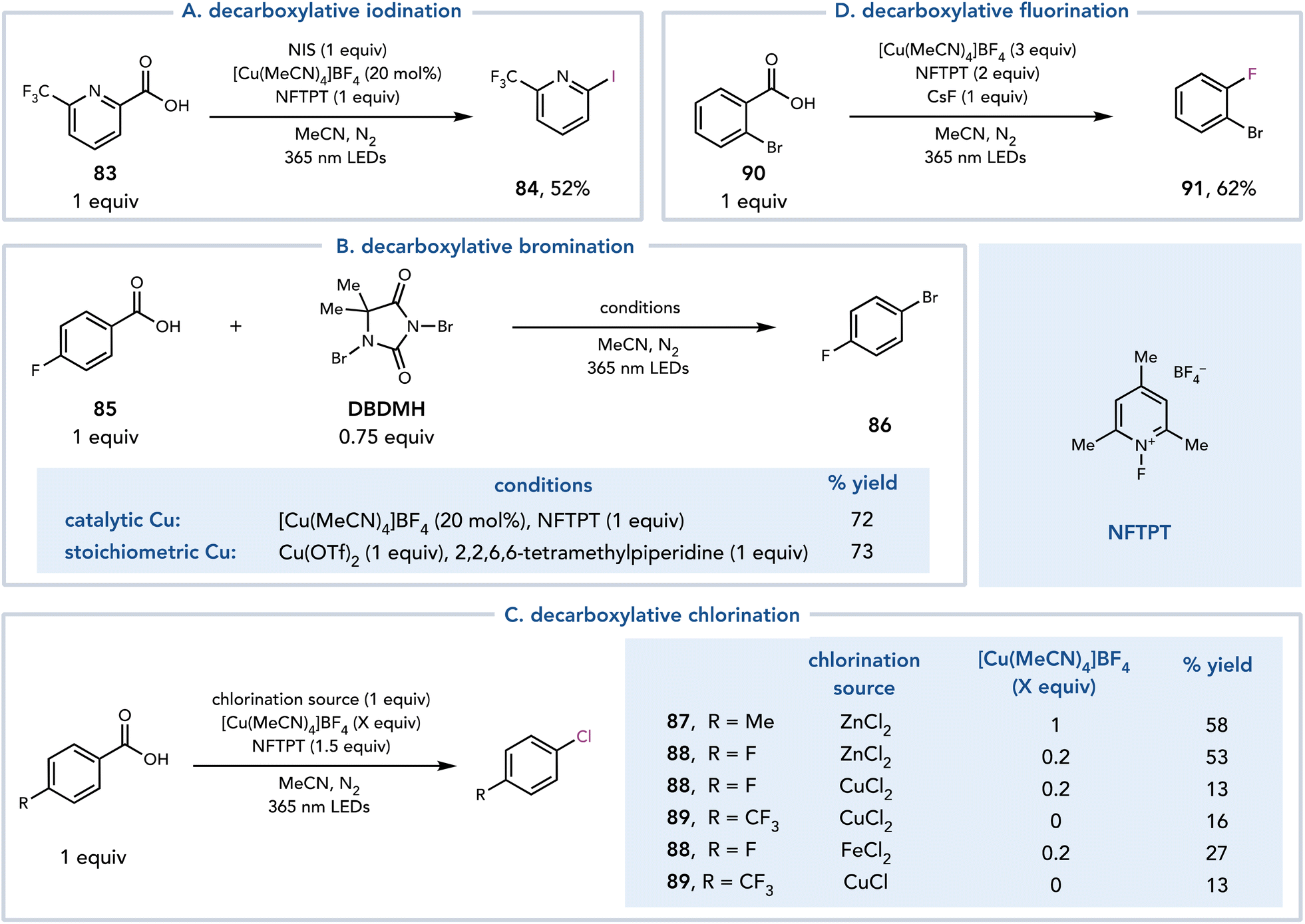 | ||
| Scheme 20 Decarboxylative halogenation. | ||
A conceptually analogous approach was also identified for the decarboxylative functionalization of C(sp3) carboxylic acids with nucleophiles (Scheme 21).35 Yoon and coworkers leveraged Cu(OTf)2 to access an alkyl radical through a visible light-promoted decarboxylation, followed by a Kochi-type oxidation36 of the resulting organoradical to access a polar intermediate that can trap a variety of nucleophiles. This method proved general with respect to the nucleophile, as sulfonamides (92, 93), alcohols (94, 95) carbamates (96), amides (97, 98), and electron-rich (hetero)arenes (98) were readily tolerated. Yoon investigated the nature of the photoactive copper carboxylate complex that was formed in situ. A series of UV-vis titration experiments revealed that at low concentrations of carboxylate, an absorption band with λmax = 304 nm dominates the UV-vis spectrum, consistent with the LMCT bands of a monomeric Cu(II) carboxylate. As the concentration of the carboxylic acid was gradually increased to 2:1 excess relative to Cu(OTf)2, an absorption feature with λmax = 260 nm began to emerge, consistent with the LMCT bands of dimeric Cu(II) carboxylate. These data support the empirical observation that 2.5 equiv. of Cu(OTf)2 was optimal for high yields of the cross-coupling products, presumably to minimize the formation of Cu(II) carboxylate dimers, which are inactive under visible light irradiation.
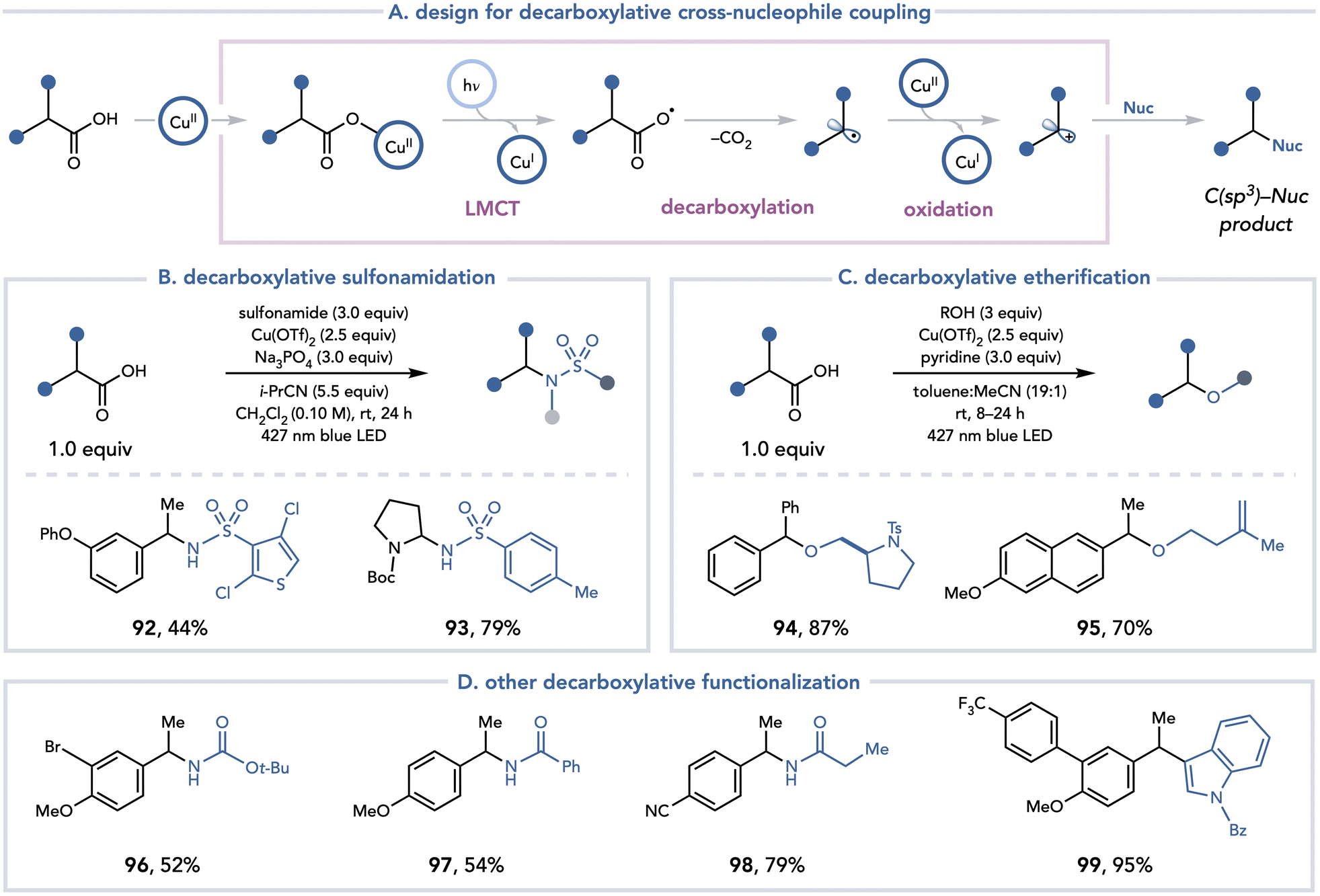 | ||
| Scheme 21 Decarboxylative cross-nucleophile coupling. | ||
Radical generation by a photoactive copper complex was recently expanded to the activation of sulfoximines.37 Ritter and coworkers reported a method for the C–H sulfoximination of arenes, which proceeds in the presence of Cu(OTf)2 (2 equiv.), LiOMe (1 equiv.), DTBP (2 equiv.), and NFTPT (0.75 equiv.) in MeCN (Scheme 22). The authors propose a sulfoximine-to-copper charge transfer upon irradiation with violet light to afford a sulfoximinyl radical. This radical could add into an unactivated arene. Subsequent oxidation with Cu(II) affords a Wheland intermediate, and upon deprotonation affords the aryl sulfoximine product.
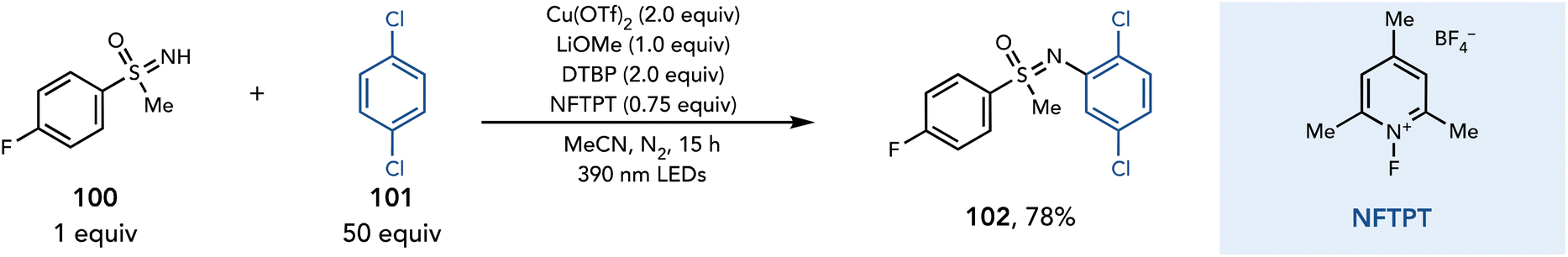 | ||
| Scheme 22 C–H sulfoximination of arenes. DTBP, 2,6-di-tert-butylpyridine. | ||
4. Conclusions
The use of Cu(II) salts as terminal oxidants has enabled the recent development of a diverse range of net-oxidative photochemical transformations of significant potential synthetic utility. The variety of reactions enabled by this platform to date spans a range of oxidative functionalizations, which opens the door for chemists to creatively apply these insights to the discovery of other new, synthetically important photochemical oxidation reactions. These reactions benefit from the ability of Cu(II) to support the one-electron oxidation state changes characteristic of photoredox reactions without generating highly reactive heteroatom-centered radical intermediates that can be incompatible with densely functionalized complex molecules. They also benefit from the terrestrial abundance of copper, the commercial availability of many Cu(II) salts at modest cost, and the relatively low environmental and toxicity concerns associated with the use of copper reagents in chemical synthesis. Collectively, these studies support the view that because the mechanisms of photochemical reactions often differ substantially from those of their ground-state counterparts, the reagents that are unconventional in one context can be ideal in another. In the case of photochemical oxidation reactions, Cu(II) salts appear to be uniquely suitable as terminal oxidants, and recent developments in this field justify further investigation into their use.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding for our laboratory's research in oxidative photoredox catalysis is provided by NIH (R35 GM144129), an ACS GCI Pharmaceutical Roundtable Research Grant, and Pfizer. G. A. L. is the recipient of a 3M Science & Technology Fellowship.References
- For selected reviews, see: (a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS; (b) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed; (c) J. M. R. Naarayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- N. L. Reed and T. P. Yoon, Chem. Soc. Rev., 2021, 50, 2954–2967 RSC.
- S. S. Stahl, Science, 2005, 309, 1824–1826 CrossRef CAS PubMed.
- For selected examples, see: (a) M. Haayyan, M. A. Haashim and I. M. AlNashef, Chem. Rev., 2016, 116, 3029–3085 CrossRef PubMed; (b) J. S. Winterle, D. S. Kliger and G. S. Hammond, J. Am. Chem. Soc., 1976, 98, 3719–3721 CrossRef CAS; (c) B. Maillard, K. U. Ingold and J. C. Scaiano, J. Am. Chem. Soc., 1983, 105, 5095–5099 CrossRef CAS.
- H. G. Yayla, F. Peng, I. K. Mangion, M. McLaughlin, L.-C. Campeau, I. W. Davies, D. A. DiRocco and R. R. Knowles, Chem. Sci., 2016, 7, 2066–2073 RSC.
- D. B. Freeman, L. Furst, A. G. Condie and C. R. J. Stephenson, Org. Lett., 2012, 14, 94–97 CrossRef CAS PubMed.
- For selected examples, see: (a) T. R. Blum, Y. Zhu, S. A. Nordeen and T. P. Yoon, Angew. Chem., 2014, 126, 11236–11239 CrossRef; (b) J. Jin and D. W. C. MacMillan, Angew. Chem., 2015, 127, 1585–1589 CrossRef.
- D. R. Abernethy, A. J. DeStafano, T. L. Cecil, K. Zaidi, R. L. Williams and USP Metal Impurities Advisory Panel, Pharm. Res., 2010, 27, 750–755 CrossRef CAS.
- For selected examples, see: (a) J. K. Kochi, A. Bemis and C. L. Jenkins, J. Am. Chem. Soc., 1968, 90, 4616–4625 CrossRef CAS; (b) J. K. Kochi and A. Bemis, J. Am. Chem. Soc., 1968, 90, 4038–4051 CrossRef CAS; (c) C. L. Jenkins and J. K. Kochi, J. Am. Chem. Soc., 1972, 94, 856–865 CrossRef CAS.
- For selected examples, see: (a) B. B. Snider and T. Kwon, J. Org. Chem., 1990, 55, 1965–1968 CrossRef CAS; (b) B. B. Snider and B. A. McCarthy, J. Org. Chem., 1993, 58, 6217–6223 CrossRef CAS; (c) L. H. Powell, P. H. Docherty, D. G. Hulcoop, P. D. Kemmitt and J. W. Burton, Chem. Commun., 2008, 2559–2561 RSC.
- For selected examples, see: (a) Y. Liang, X. Zhang and D. W. C. MacMillan, Nature, 2018, 559, 83–88 CrossRef CAS PubMed; (b) C. Le, T. Q. Chen, T. Liang, P. Zhang and D. W. C. MacMillan, Science, 2018, 360, 1010–1014 CrossRef CAS PubMed; (c) R. Mao, A. Frey, J. Balon and X. Hu, Nat. Catal., 2018, 1, 120–126 CrossRef CAS; (d) W.-J. Yoo, T. Tsukamoto and S. Kobayashi, Angew. Chem., Int. Ed., 2015, 54, 6587–6590 CrossRef CAS.
- Y. Ye and M. S. Sanford, J. Am. Chem. Soc., 2012, 134, 9034–9037 CrossRef CAS.
- N. L. Reed, M. I. Herman, V. P. Miltchev and T. P. Yoon, Org. Lett., 2018, 20, 7345–7350 CrossRef CAS.
- (a) T. M. Nguyen and D. A. Nicewicz, J. Am. Chem. Soc., 2013, 135, 9588–9591 CrossRef CAS; (b) T. M. Nguyen, N. Manohar and D. A. Nicewicz, Angew. Chem., Int. Ed., 2014, 53, 6198–6201 CrossRef CAS.
- S. Senaweera, K. C. Cartwright and J. A. Tunge, J. Org. Chem., 2019, 84, 12553–12561 CrossRef CAS PubMed.
- B. J. Lee, K. S. DeGlopper and T. P. Yoon, Angew. Chem., Int. Ed., 2020, 59, 197–202 CrossRef CAS.
- J.-C. Xiang, C. Fung, Q. Wang and J. Zhu, Nat. Commun., 2022, 13, 3481 CrossRef CAS PubMed.
- C. Pan, Y. Xu, B. Zhang, L. Ge, C. Zhang and C. Feng, Cell Rep. Phys. Sci., 2023, 4, 101233 CrossRef CAS.
- (a) J. K. Kochi, Science, 1967, 155, 415–424 CrossRef CAS PubMed; (b) J. K. Kochi, J. Am. Chem. Soc., 1963, 85, 1958–1968 CrossRef CAS.
- K. C. Cartwright, D. B. Lang and J. A. Tunge, J. Org. Chem., 2019, 84, 2933–2940 CrossRef CAS PubMed.
- N. Holmberg-Douglas, Y. Choi, B. Aquila, H. Huynh and D. A. Nicewicz, ACS Catal., 2021, 11, 3153–3158 CrossRef CAS PubMed.
- N. L. Reed, G. A. Lutovsky and T. P. Yoon, J. Am. Chem. Soc., 2021, 143, 6065–6070 CrossRef CAS PubMed.
- G. A. Lutovsky, E. F. Plachinski, N. L. Reed and T. P. Yoon, Org. Lett., 2023, 25, 4750–4754 CrossRef CAS PubMed.
- For selected reviews, see: (a) Y. Abderrazak, A. Bhattacharyya and O. Reiser, Angew. Chem., Int. Ed., 2021, 60, 21100–21115 CrossRef CAS; (b) F. Juliá, ChemCatChem, 2022, 14, e202200916 CrossRef; (c) J. Beaudelot, S. Oger, S. Peruško, T.-A. Phan, T. Teunens, C. Moucheron and G. Evano, Chem. Rev., 2022, 122, 16365–16609 CrossRef CAS PubMed; (d) S. Gavelle, M. Innocent, T. Aubineau and A. Guérinot, Adv. Synth. Catal., 2022, 364, 4189–4230 CrossRef CAS; (e) A. Reichle and O. Reiser, Chem. Sci., 2023, 14, 4449 RSC.
- J. K. Kochi, J. Am. Chem. Soc., 1962, 84(11), 2121–2127 CrossRef CAS.
- P. Lian, W. Long, J. Li, Y. Zheng and X. Wan, Angew. Chem., Int. Ed., 2020, 59, 23603–23608 CrossRef CAS PubMed.
- W. Fang, H.-Q. Wang, W. Zhou, Z.-W. Luo and J. J. Dai, Chem. Commun., 2023, 59, 7108–7111 RSC.
- C. Shu, A. Noble and V. K. Aggarwal, Nature, 2020, 586, 714–719 CrossRef CAS PubMed.
- R. Sang, W. Han, H. Zhang, C. M. Saunders, A. Noble and V. K. Aggarwal, J. Am. Chem. Soc., 2023, 145, 15207–15217 CrossRef CAS PubMed.
- (a) J. Y. Morimoto and B. A. DeGraff, J. Phys. Chem., 1975, 79, 326–331 CrossRef CAS; (b) J. Y. Morimoto and B. A. DeGraff, J. Phys. Chem., 1972, 76, 1387–1388 CrossRef CAS; (c) L. Sun, C.-H. Wu and B. C. Faust, J. Phys. Chem. A, 1998, 102, 8664–8672 CrossRef CAS; (d) L. Sun, C.-H. Wu and B. C. Faust, J. Phys. Chem. A, 2000, 104, 4989–4996 CrossRef.
- P. Xu, P. López-Rojas and T. Ritter, J. Am. Chem. Soc., 2021, 143, 5349–5354 CrossRef CAS PubMed.
- W. Su, P. Xu and T. Ritter, Angew. Chem., Int. Ed., 2021, 60, 24012–24017 CrossRef CAS PubMed.
- P. Xu, W. Su and T. Ritter, Chem. Sci., 2022, 13, 13611 RSC.
- T. Q. Chen, P. S. Pedersen, N. W. Dow, R. Fayad, C. E. Hauke, M. C. Rosko, E. O. Danilov, D. C. Blakemore, A.-M. Dechert-Schmitt, T. Knauber, F. N. Castellano and D. W. C. MacMillan, J. Am. Chem. Soc., 2022, 144, 8296–8305 CrossRef CAS PubMed.
- Q. Y. Li, S. N. Gockel, G. A. Lutovsky, K. S. DeGlopper, N. J. Baldwin, M. W. Bundesmann, J. W. Tucker, S. W. Bagley and T. P. Yoon, Nat. Chem., 2022, 14, 94–99 CrossRef CAS PubMed.
- J. K. Kochi, A. Bemis and C. L. Jenkins, J. Am. Chem. Soc., 1968, 90, 4616–4625 CrossRef CAS.
- W. Su, P. Xu, R. Retzold, J. Yan and T. Ritter, Org. Lett., 2023, 25, 1025–1029 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |