
Recent advances in ring-opening of cyclobutanone oximes for capturing SO2, CO or O2via a radical process
Long-Jin
Zhong†
 ,
Jian-Hong
Fan†
,
Pu
Chen
,
Peng-Fei
Huang
*,
Bi-Quan
Xiong
,
Ke-Wen
Tang
and
Yu
Liu
*
,
Jian-Hong
Fan†
,
Pu
Chen
,
Peng-Fei
Huang
*,
Bi-Quan
Xiong
,
Ke-Wen
Tang
and
Yu
Liu
*
Department of Chemistry and Chemical Engineering, Hunan Institute of Science and Technology, Yueyang 414006, China. E-mail: pengfeihuang@whu.edu.cn; lyxtmj_613@163.com
First published on 23rd November 2023
Abstract
Cyclobutanone oximes and their derivatives are pivotal core structural motifs in organic chemistry. Iminyl-radical-triggered C–C bond cleavage of cyclobutanone oximes delivers an efficient strategy to produce stable distal cyano-substituted alkyl radicals, which can capture SO2, CO or O2 to form cyanoalkylsulfonyl radicals, cyanoalkylcarbonyl radicals or cyanoalkoxyl radicals under mild conditions. In the past several years, cyanoalkylsulfonylation/cyanoalkylcarbonyaltion/cyanoalkoxylation has attracted a lot of interest. In this updated report, the strategies for trapping SO2, CO or O2via iminyl-radical-triggered ring-opening of cyclobutanone oximes are summarized.
 Long-Jin Zhong | Long-Jin Zhong was born in Ganzhou, Jiangxi, China. He received his BSc from Jinggangshan University in 2015, and his PhD from Hunan University (under the supervision of Prof. Jin-Heng Li and Prof. De-Lie An) in 2022. At present, he is working as an assistant professor at the Hunan Institute of Science and Technology. His current research interest is the application of selective remote inert C–H/C–C bond functionalization via a radical relay strategy and novel methods in organic photochemistry synthesis. |
 Jian-Hong Fan | Jian-Hong Fan was born in Shaoyang, Hunan, China. He received his BSc from Hunan Normal University in 2012, and obtained his MS degree from Hunan University in 2015 under the direction of Professor Jin-Heng Li. He completed his doctorate under the direction of Professor Chuang-Chuang Li at Southern University of Science and Technology in 2020. He joined the Hunan Institute of Science and Technology in 2021. His current research interests include the development of new synthetic methods and total synthesis of biologically active natural products. |
 Peng-Fei Huang | Peng-Fei Huang was born in Shaoyang, Hunan, China. He received his BSc from Hunan University in 2016, and his PhD from Wuhan University (under the supervision of Prof. Aiwen Lei) in 2021. He joined the Hunan Institute of Science and Technology in 2021 as a lecturer. His current research interests include photochemical and electrochemical radical reactions. |
 Bi-Quan Xiong | Bi-Quan Xiong was born in Hunan province, China, in 1987. He obtained his B.Sc. degree from Jishou University, and his Ph.D. degree from Hunan University in 2015. In 2015, he joined Prof. Tang's group at the Hunan Institute of Science and Technology. He was promoted to associate professor in 2020. His current research interests focus on the transition-metal-catalyzed activation of inert chemical bonds and P–H/P–OH/P–OR bonds and developing green synthetic protocols for the construction of P–C and P–Z bonds in organophosphorus chemistry. |
 Ke-Wen Tang | Ke-Wen Tang was born in Hunan, China, in 1970. He received his Ph.D. degree from Central South University in 2003 under the supervision of Prof. Chun-Shan Zhou in 1999. After postdoctoral associate work at Central South University from 2004 to 2009 with Prof. Yuan-Jian Li, he joined the Hunan Institute of Science and Technology in 1999 and was promoted to professor in 2005. His interests include modern chemical separation technology, catalysts for the petrochemical industry and new separation materials. |
 Yu Liu | Yu Liu was born in Hunan, China, in 1985. He received his BSc from Hunan Normal University in 2008, and received his Ph.D. degree in organic chemistry from Hunan University in 2015 under the supervision of Prof. Jin-Heng Li. In July 2015, he joined the Hunan Institute of Science and Technology. His current research is focused on radical ring-opening/cyclization, insertion of sulfur dioxide through a radical strategy and visible-light-catalyzed reactions. |
1. Introduction
Alkylnitriles, a pivotal class of structural motifs, are widely distributed in organic synthesis, medicines and pharmaceutical compounds with a wide range of biological properties (Scheme 1).1–3 Additionally, the cyano group can be easily and efficiently converted into other functional groups, such as amidines, amides, carboxylic acids, esters, tetrazoles, aldehydes, and heterocycles.4,5 Various alkylnitriles are constructed through dehydrogenation of amines, dehydration of aldoximes or amides, and cyanation of alkyl halides.4–8 As a result, efficient methods for introducing alkylnitrile skeletons into organic molecules are still in high demand.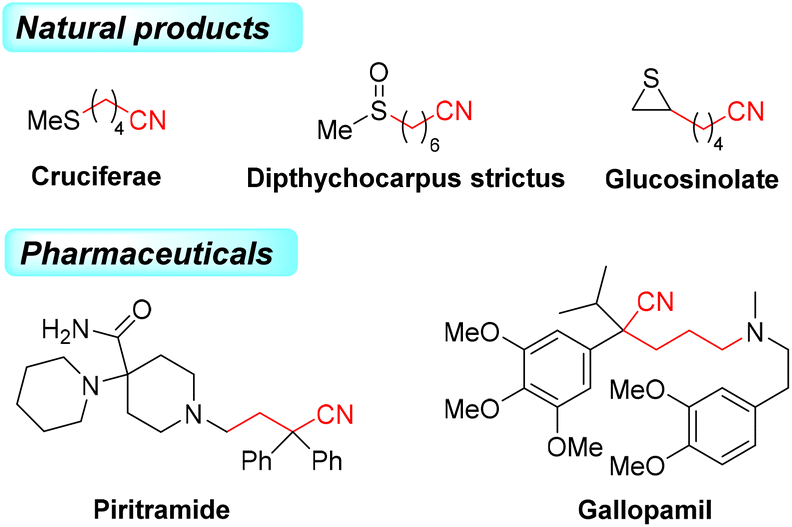 | ||
| Scheme 1 Natural products and pharmaceuticals containing alkylnitriles. | ||
In the past several years, cyclobutanone oximes have been employed as mild and efficient reagents for cyanoalkylation, and they can introduce distal cyano-substituted alkyl moieties into different chemical compounds.9–12 Recently, significant processes for the cleavage of cyclobutanone oximes via transition-metal-catalyzed C–C bond cleavage13,14 or radical-mediated oxidative ring-opening15–18 have attracted considerable attention. The transition-metal-catalyzed cyclobutanone oxime is induced via β-carbon elimination with the assistance of Pd0 (Scheme 2), and the radical-mediated oxidative ring opening of cyclobutanone oxime is achieved by selecting various oxidants to initiate a process of single electron transfer (SET) (Scheme 2). The first example of using cyclobutanone sulfenylimines and carboxymethyl oximes to obtain various alkyl nitriles via radical C–C bond cleavage was proposed by Zard and co-workers.19
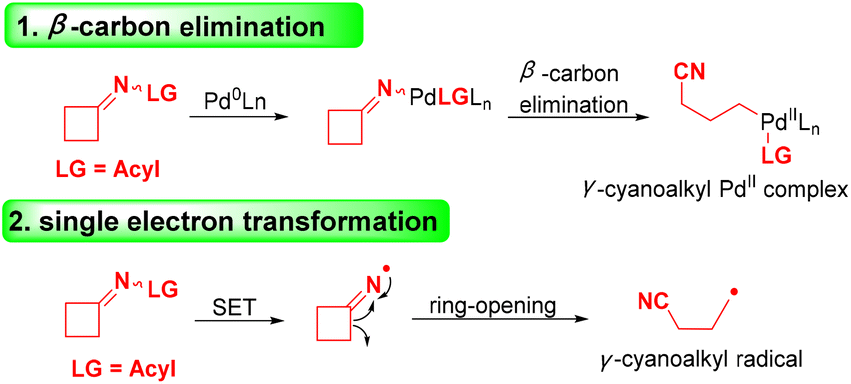 | ||
| Scheme 2 Ring-opening of cyclobutanone oximes. | ||
Cyclobutanone oximes go through iminyl-radical-triggered ring-opening to form the stable cyanoalkyl radical, which can capture SO2, CO, and O2. These transformations have captured the attention of organic chemists in the past several years. All of the recently reported approaches contain a radical process, which is shown in Scheme 3. The cyclobutanone oximes provide the iminyl radicals Avia SET reduction. Then, the radicals B, generated from cleavage of the C–C σ-bond in intermediates A, can trap SO2 to form the sulfonyl radicals C. The intermediates C react with radical acceptors to furnish the desired products D (Scheme 3, path I). The cyanoalkyl radicals B can capture CO to form acyl radicals E, which react with radical acceptors to deliver the target products F (Scheme 3, path II). The cyanoalkyl radicals B can also trap O2 to generate alkoxy radicals G and react with similar radical acceptors to construct the corresponding products H (Scheme 3, path III). In this updated report, we conclude recent main themes in trapping SO2, CO or O2 by cyanoalkyl radicals, which come from iminyl-radical-triggered C–C bond cleavage of cyclobutanone oxime derivatives.
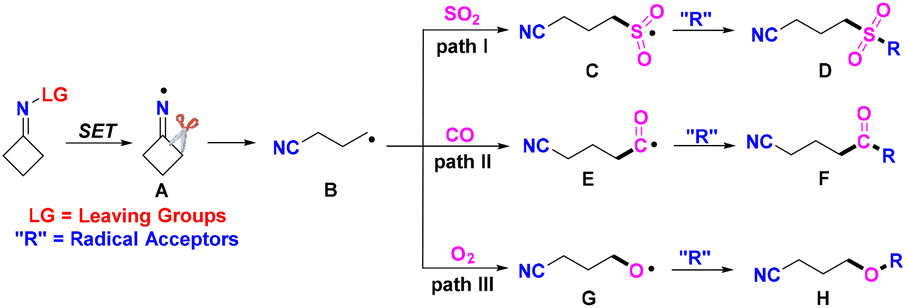 | ||
| Scheme 3 Iminyl-radical-triggered ring-opening of cyclobutanone oximes for the insertion of SO2, CO or O2. | ||
2. Cyanoalkylsulfonylation via the insertion of SO2
Due to their diversified biological properties, sulfones are highly significant structural motifs in organic synthesis and the pharmaceutical industry.20–22 As a simple and convenient strategy for the construction of sulfones, sulfonylation has received continuous attention.23–30 Recently, the insertion of SO2 has become a kind of powerful and efficient sulfonylation method.31,32 Compared with the toxic, gaseous and irritant SO2, DABCO·(SO2)2, thiourea dioxide and inorganic sulfites were employed as the source of sulfur dioxide in the past several years. Several cases of sulfonylation of cyanoalkyl radicals via the insertion of sulfur dioxide are summarized.In 2019, Wu and co-workers proposed a photoredox-catalyzed four component oxyalkylation/cyanoalkylsulfonylation of alkenes 2 with cyclobutanone oximes 1, K2S2O5 and nucleophiles to prepare the β-alkoxy or β-hydroxyl sulfones 3 with broad functional group tolerance (Scheme 4).33 In this transformation, commercially and easily available potassium metabisulfite was employed as the source of sulfur dioxide. As expected, a series of cyclobutanone oximes and alkenes bearing various substituents were suitable for the optimal conditions. Surprisingly, both alcohols and water were good nucleophiles in the sulfonylation. Notably, the secondary and tertiary alcohols were less effective than primary alcohols according to the yields. The reaction in methan-d3-ol instead of methanol afforded the target product in moderate yield. A radical-trapping experiment was conducted by utilizing TEMPO as the radical inhibitor under the standard conditions. This transformation was suppressed by TEMPO, which proved that a radical process was involved in the sulfonylation. The plausible mechanism was proposed based on the results and the literature. First, iminyl radicals 4 formed from the reduction of cyclobutanone oximes in the presence of a photocatalyst under visible light irradiation.34–37 Further cleavage of C–C σ-bonds occurred to provide cyanoalkyl radicals 5. Then, the radical 5 could capture SO2, which was generated from K2S2O5 to afford sulfonyl radicals 6. The carbon radical 7 was furnished from the addition of cyanoalkylsulfonyl radicals 6 to the carbon–carbon double bonds of alkenes 2. The radical 7 could undergo SET oxidation via the oxidized form of the photocatalyst to provide the intermediate 8. Employing MeOH or H2O 4 as a nucleophile to react with the intermediate 8 offered the desired β-alkoxy or β-hydroxyl sulfones in the presence of a base.
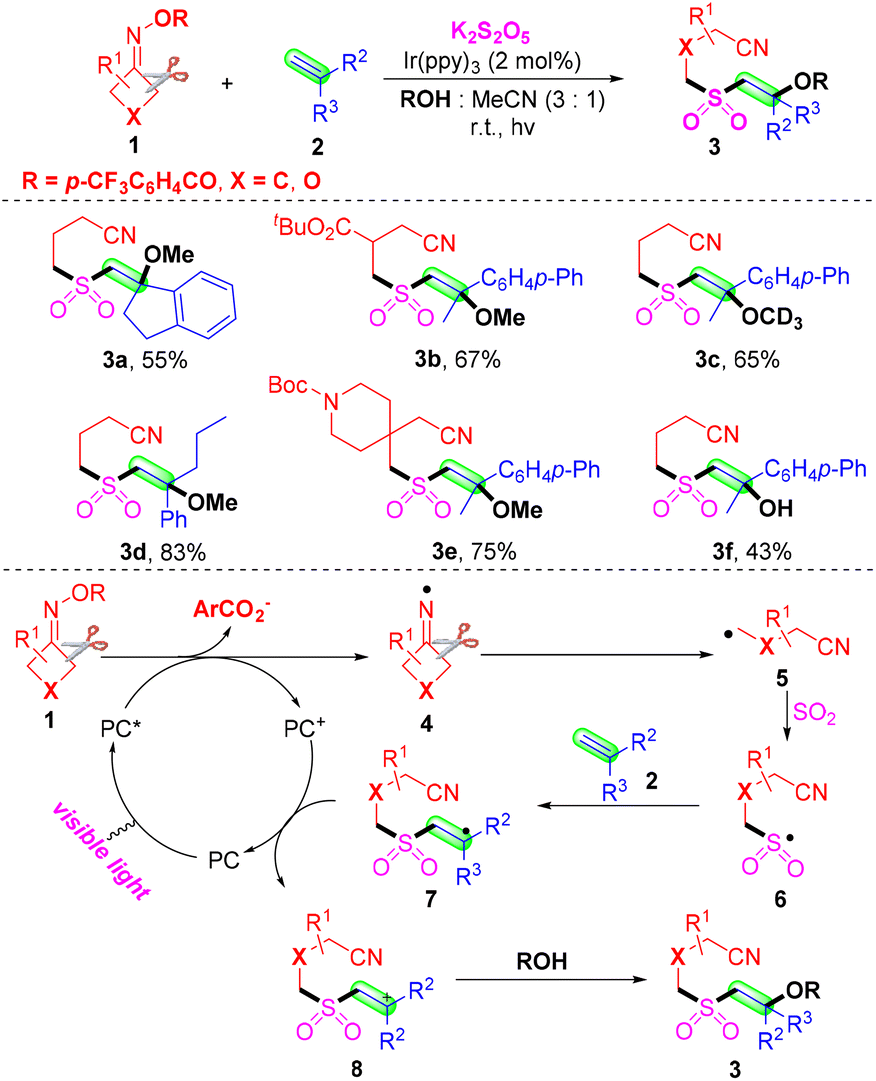 | ||
| Scheme 4 Photoinduced oxyalkylation/cyanoalkylsulfonylation of alkenes with cyclobutanone oximes, K2S2O5 and nucleophiles. | ||
Subsequently, Zhou's group developed a copper-catalyzed multicomponent oxysulfonylation of alkenes 2 with cyclobutanone oxime esters 1 and hydroxamic acids 9, using K2S2O5 as a sulfur dioxide surrogate and the in situ insertion of sulfur dioxide. A series of desired β-amidoxy sulfone products and late-stage functionalized bioactive molecules were obtained in good yields (Scheme 5).38 The control experiment results suggested that a radical–radical coupling reaction of the alkyl radical and amidoxyl radical might be involved, and cyanoalkyl, cyanoalkylsulfonyl and amidoxyl radicals were generated in this transformation. Based on the above-mentioned experiments and previous reports, a proposed mechanism is shown at the end of Scheme 5. Initially, TBHP reacts with the Cu(II) catalyst under heating to form tBuOO*, which can abstract a hydrogen atom from hydroxamic acid 9 to produce TBHP and amidoxyl radical 11. A single-electron transfer process between cyclobutanone oxime esters 1 and Cu(I) species would lead to the formation of iminyl radical intermediate 4 and Cu(II) species. Then, the iminyl radical 4 undergoes ring opening to deliver the cyanoalkyl radical 5, which is converted into the radical species 6 by capturing sulfur dioxide from K2S2O5. Subsequently, radical species 6 attacks alkene 2 to afford carbon radical intermediate 7. Finally, radical 7 is captured by amidoxyl radical 11, affording the oxysulfonylation product 10.
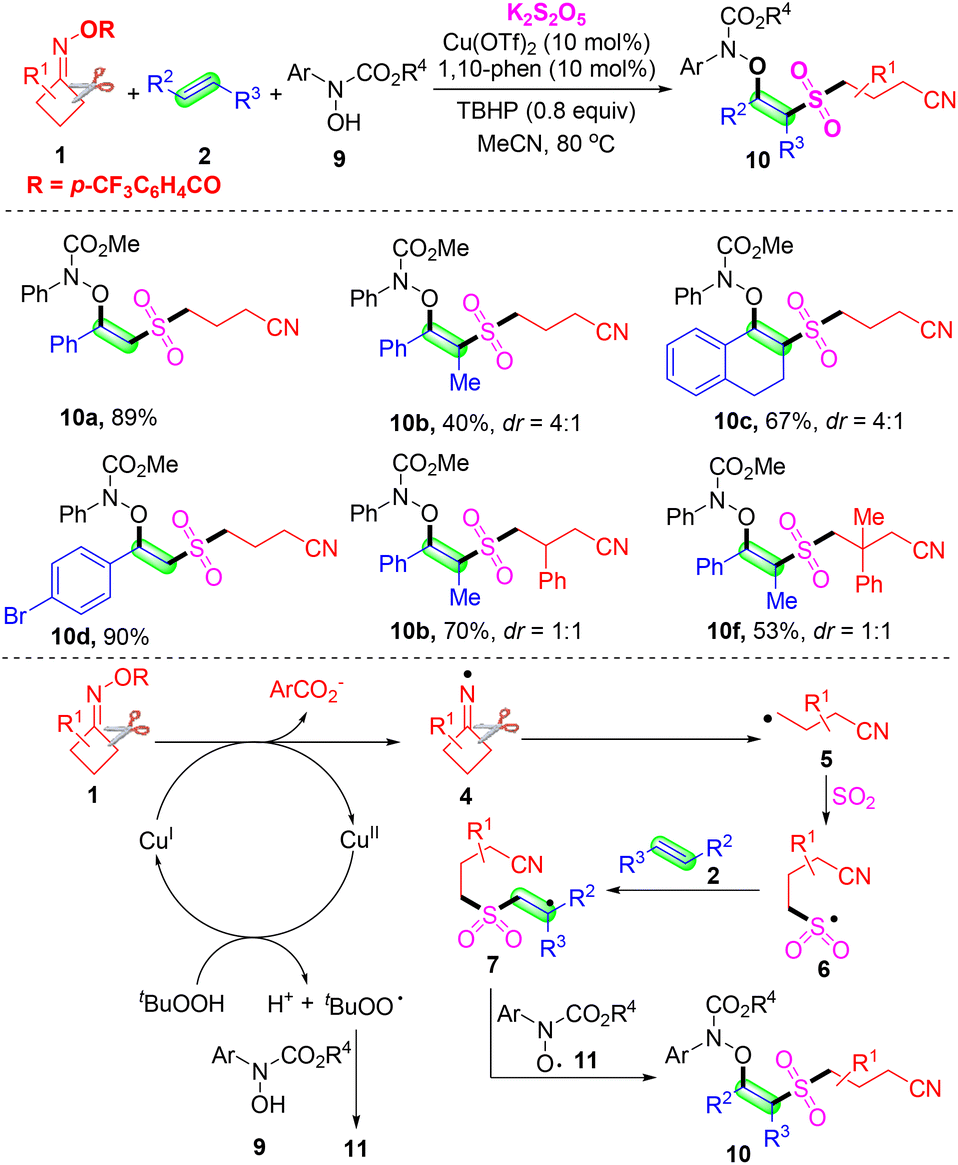 | ||
| Scheme 5 Copper-catalyzed oxysulfonylation of alkenes with cyclobutanone oximes, K2S2O5 and hydroxamic acids. | ||
Cyanoalkylsulfonyl alkenes are particularly attractive and versatile building blocks in organic synthetic chemistry, due to the presence of the useful cyano group, which can be readily transformed into other valuable functional groups.39,40 Recently, Wu and co-workers developed a copper-catalyzed three-component reaction of alkenes 2 with cyclobutanone oxime esters 1, employing DABCO·(SO2)2 as a sulfur dioxide surrogate for the synthesis of diverse (E)-cyanoalkylsulfonyl alkenes (Scheme 6).41 This method exhibited excellent regio- and stereoselectivity and functional group tolerance. A plausible radical pathway is proposed, which involves C–C bond cleavage of cyclobutanone oxime esters, insertion of sulfur dioxide and deprotonation of the cation intermediate. As shown below in Scheme 6, the cycloketone oxime ester 1 through a single-electron transfer process is reduced by Cu(I) to afford iminyl radical species 4 and a Cu(II) complex. And then, the iminyl radical intermediate 4 undergoes β-C–C bond cleavage giving rise to a new cyanoalkyl radical intermediate 5, which is transformed into cyanoalkylsulfonyl radical species 6 through capturing sulfur dioxide from DABCO·(SO2)2. Next, this cyanoalkylsulfonyl radical 6 would react with alkene 2 further forming a more stable benzyl radical intermediate 7. Finally, an oxidative single electron transfer between benzyl radical 7 and Cu(II) species would provide the Cu(I) species to complete the catalytic cycle. Meanwhile, cation intermediate 13 would be formed and further deprotonation of cation intermediate 13 would result in the formation of target products (E)-cyanoalkylsulfonyl alkenes 12.
 | ||
| Scheme 6 Copper-catalyzed cyanoalkylsulfonylation of alkenes with cyclobutanone oximes and DABCO·(SO2)2. | ||
Meanwhile, Akondi's group used enamides 14 instead of alkenes 2 and accomplished cyanoalkylsulfonylation of enamides 14 with cyclobutanone oxime esters 1, using K2S2O5 as a sulfur dioxide surrogate (Scheme 7).42 This radical tandem reaction proceeds with excellent regio- and stereoselectivity to generate diverse (E)-β-(sulfonyl)enamines 15 under organophotoredox catalytic rose bengal conditions. Based on the result of control experiments, a similar process of photoredox-catalyzed cyanoalkylsulfonylation of enamides involves C–C bond cleavage of cyclobutanone oxime esters, insertion of sulfur dioxide and deprotonation of the cation intermediate.
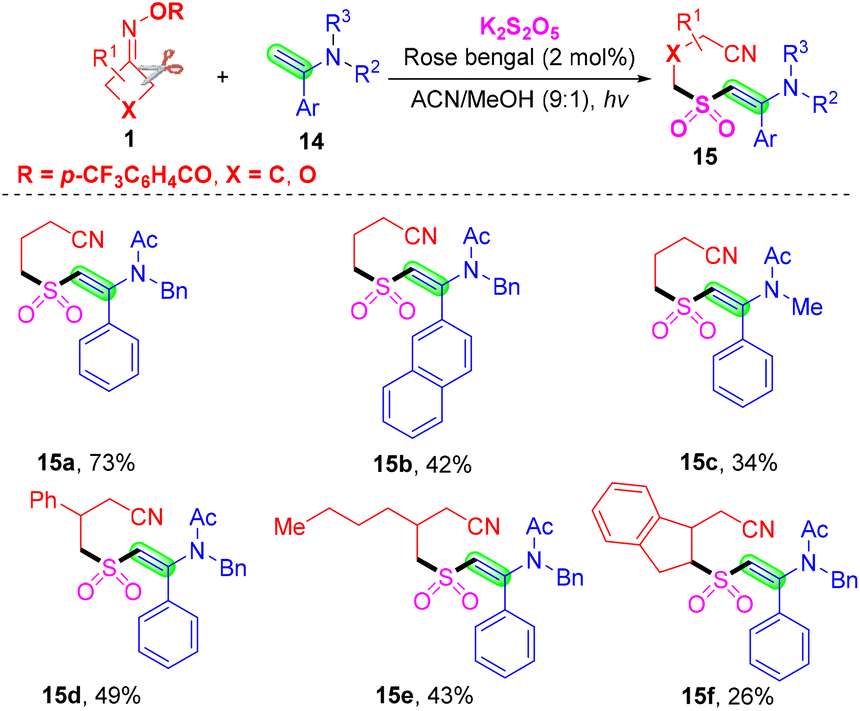 | ||
| Scheme 7 Visible-light-catalyzed cyanoalkylsulfonylation of enamides with cyclobutanone oximes and K2S2O5. | ||
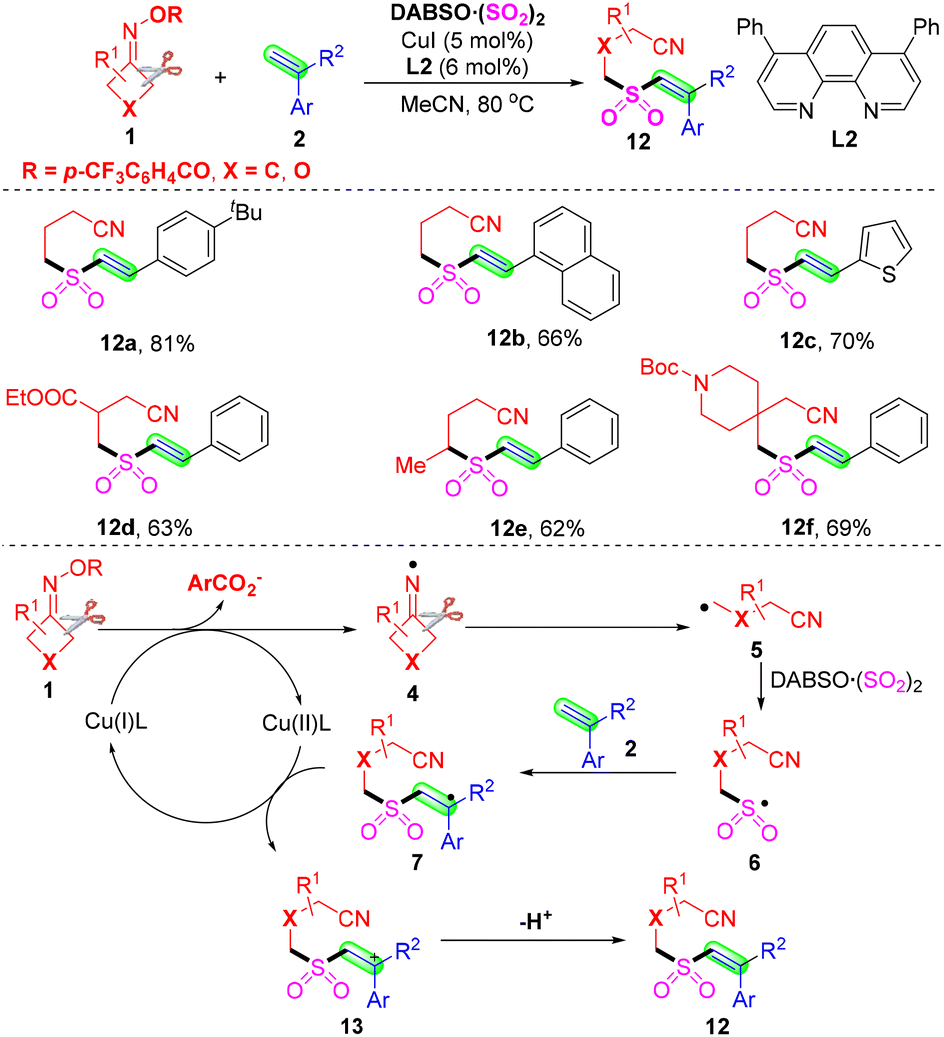
It is well known that the tetrasubstituted alkene core scaffolds are broadly encountered in pharmaceuticals and bioactive molecules.43,44 Wu's group disclosed a copper-catalyzed three-component cyanoalkylsulfonylation of 2H-azirines 16 with cyclobutanone oximes 1 and DABCO·(SO2)2 for the preparation tetrasubstituted β-sulfonyl N-unprotected enamines 17 (Scheme 8).45 It was the first example where 2H-azirines were employed as efficient and novel synthons for the generation of β-functionalized N-unprotected enamines. The alkyl radicals, which are formed from the cleavage of C–C σ-bonds in cyclobutanone oximes, would prefer to react with sulfur dioxide to offer sulfonyl radicals. The plausible mechanism revealed that the iminyl radicals 4, generated from single-electron reduction of cyclobutanone oximes with the aid of Cu(I), could initiate the reaction. Then, the radicals 4 could undergo C–C σ-bond cleavage to afford the alkyl radicals 5. The sulfonyl radicals 6 could be produced from intermediates 5 by capture of sulfur dioxide. Meanwhile, the ring-opening of 2H-azirines 18 and protonation occurred to form α-carbon radicals 19 with the aid of a copper catalyst. The sulfonyl radicals 6 and α-carbon radicals 19 underwent selective cross-coupling to deliver intermediates 20. Finally, isomerization of the intermediates 20 occurred to construct the corresponding products 17.
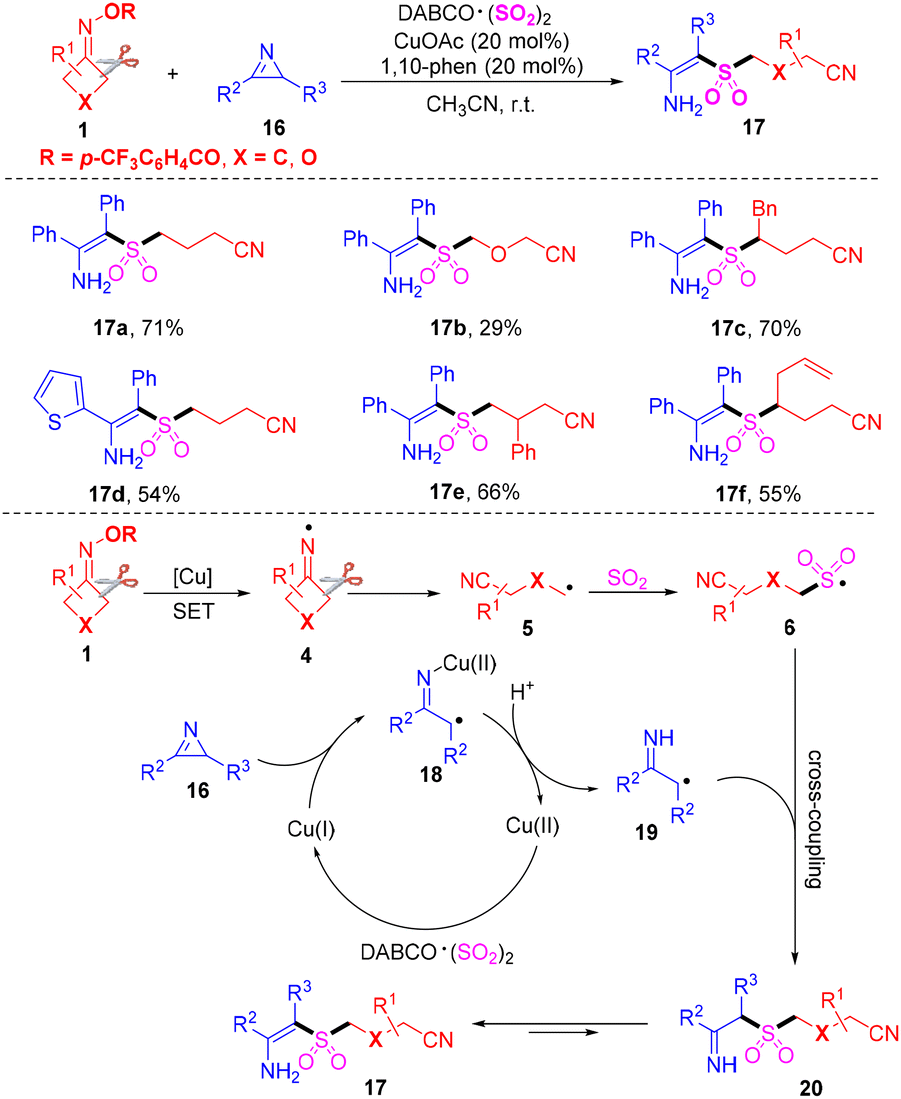 | ||
| Scheme 8 Copper-catalyzed cyanoalkylsulfonylation of 2H-azirines with cyclobutanone oximes and DABCO·(SO2)2. | ||
Different from cyanoalkylsulfonylation of alkenes with cyclobutanone oximes and a sulfur dioxide source, Wu's group described a facile copper-catalyzed four-component selenosulfonylation of alkynes 21, cycloketone oxime esters 1, DABCO·(SO2)2 and diselenides 22, and the rapid assembly of β-cyanoalkylsulfonylated vinyl selenides in one-pot (Scheme 9).46 Advantages of this method include broad substrate scope, good functional group tolerance and late-stage functionalization of complex molecules, meanwhile, the products β-cyanoalkylsulfonylated vinyl selenides allow accessing synthetically important intermediate alkynyl sulfones with hydrogen peroxide. On the basis of the control experiment results and literature reports, the author postulated that initially the Cu(I)-mediated single-electron reduction of cyclobutanone oxime esters 1 would afford iminyl radical 4, which would produce a highly reactive cyanoalkyl radical 5 by selective C–C σ-bond cleavage. Subsequently, radical 5via capturing sulfur dioxide from DABCO·(SO2)2 would lead to the generation of cyanoalkylsulfonyl radical 6. The subsequent addition of alkyne 21 would afford vinyl radical 24, which might react with diselenide 22 to provide the desired product 23.
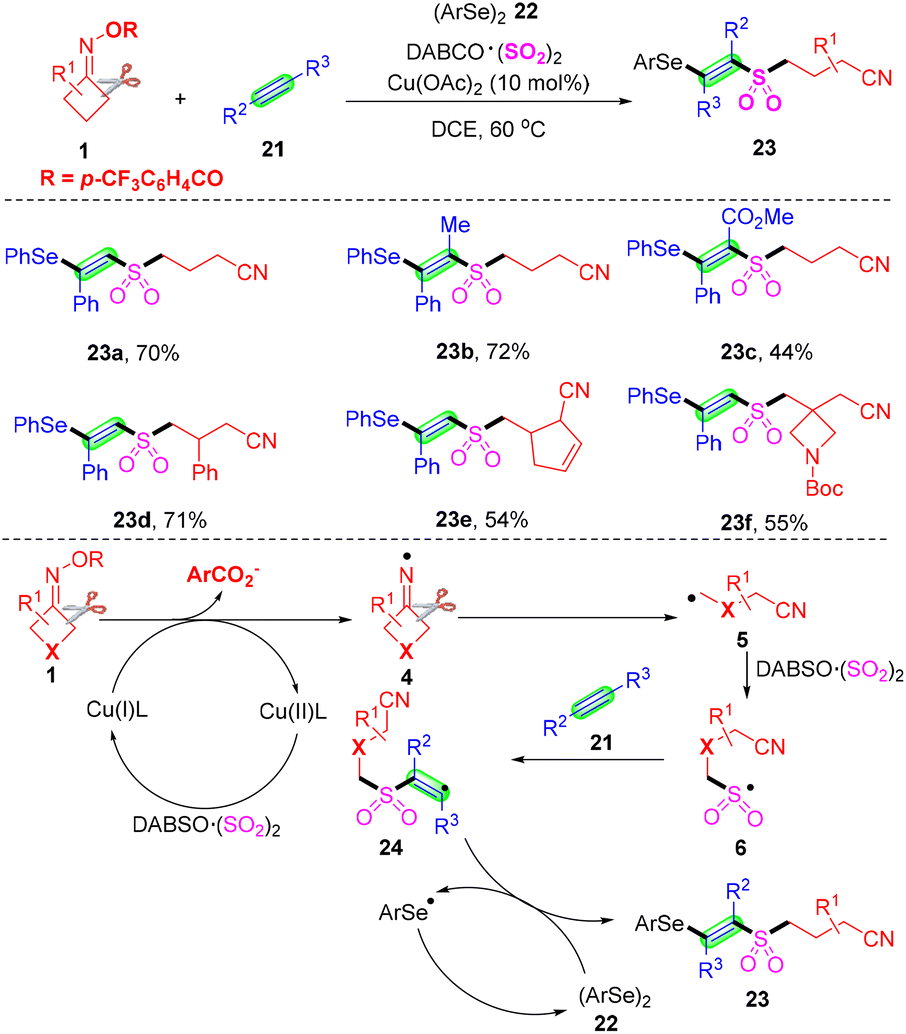 | ||
| Scheme 9 Copper-catalyzed four-component selenosulfonylation of alkynes with cyclobutanone oximes, diselenides and DABCO·(SO2)2. | ||
β-Keto sulfones are valuable structural moieties widely found in natural products and functional materials, and as important synthetic intermediates in the synthesis of bioactive molecules and pharmaceutical molecules.47–49 Very recently, Wei's group reported an iron-catalyzed multi-component reaction employing terminal alkynes 25, cyclobutanone oximes 1 and eco-friendly sulfur dioxide source Na2S2O5 for the synthesis of β-keto sulfones under mild reaction conditions (Scheme 10).50 In this method, naturally abundant and readily available FeCl2 was used as the catalyst and additional oxidants and ligands were not required. Moreover, this synthesis method revealed good compatibility toward functional groups, a wide range of substrates, and high chemoselectivity. This is a new, efficient and rapid method for the preparation of β-keto sulfones via oxosulfonylation of alkynes.
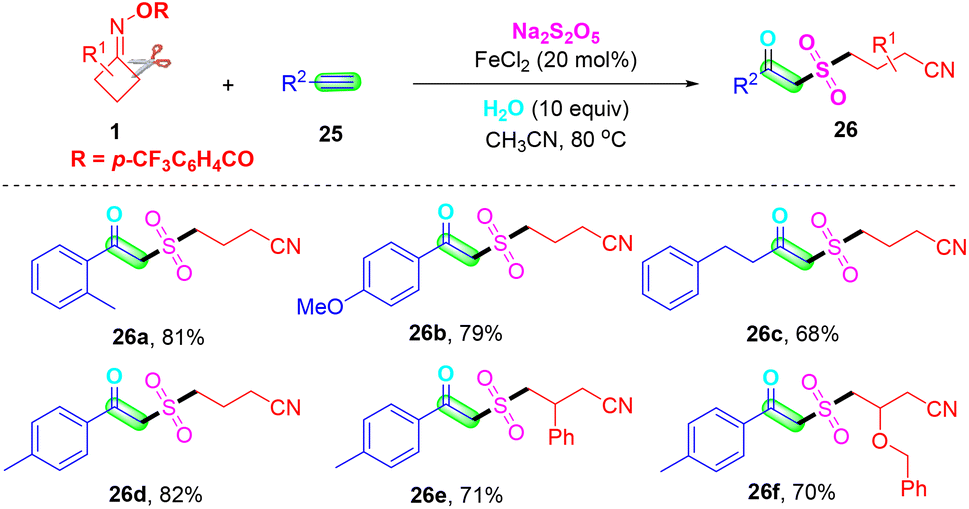 | ||
| Scheme 10 Iron-catalyzed multi-component oxosulfonylation of alkynes with cyclobutanone oximes and Na2S2O5. | ||
In 2023, Ge, Wei and co-workers further enriched the methodology for the preparation of cyanoalkyl-substituted β-ketone sulfones via a simple and efficient strategy under mild reaction conditions. They described a copper-catalyzed radical cascade reaction of vinyl azides 27, oxime esters 1 and Na2S2O5 for the synthesis of polyfunctionalized β-ketone sulfones (Scheme 11).51 Based on the control experiment results, a series of processes were involved in this transformation such as radical relay, radical addition, hydrogen abstraction, and hydrolysis or oxidation. More importantly, a one-pot four-component radical cascade reaction, which directly utilizes the terminal alkyne and TMSN3 instead of the vinyl azide reaction with oxime esters and Na2S2O5, can also generate the corresponding desired β-ketone sulfone products in moderate yields.
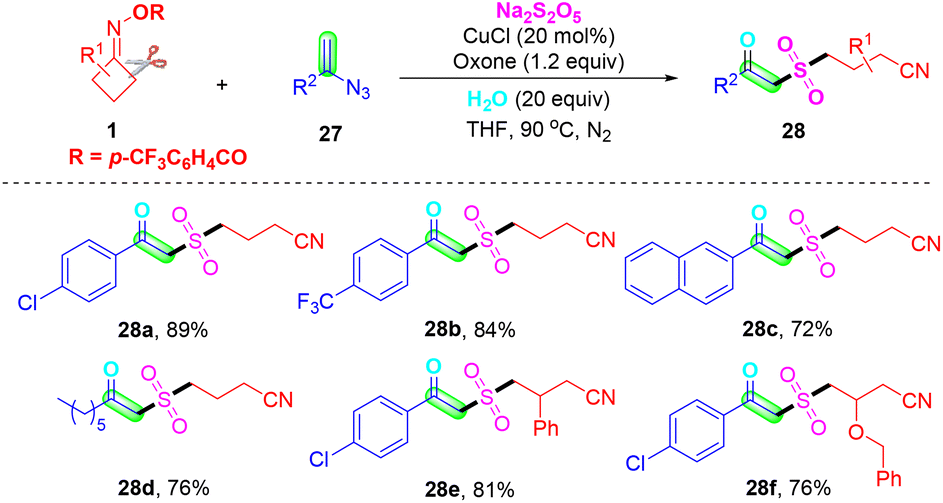 | ||
| Scheme 11 Copper-catalyzed three-component radical cascade reaction of vinyl azides with cyclobutanone oximes and Na2S2O5. | ||
As important and useful synthetic building blocks and fragments, β-ketosulfones and allylsulfones have been broadly applied in the fields of pharmaceuticals and organic synthesis.52–54 Lu and co-workers developed the first Cu-catalyzed or visible-light-catalyzed cyanoalkylsulfonylation of vinyl triflates or allyl trifluoromethylsulfones with cyclobutanone oximes via the in situ capture of SO2 by cyanoalkyl radicals, obtained from cyclobutanone oximes (Scheme 12).55 It was found that various functional groups such as halogen, nitro, cyano, ester or methoxy groups were all suitable for the optimal catalytic system. The Ar atmosphere played a vital role in this transformation, because the in situ generated SO2 could easily diffuse into the atmosphere when the reaction was conducted under ambient open-air conditions, and the yields of the target products decreased obviously. Mechanistic studies proved that the trifluoromethyl group was indispensable in this transformation, and the solvent had a positive effect in the aspect of minimized side reactions by capturing active trifluoromethyl radicals.
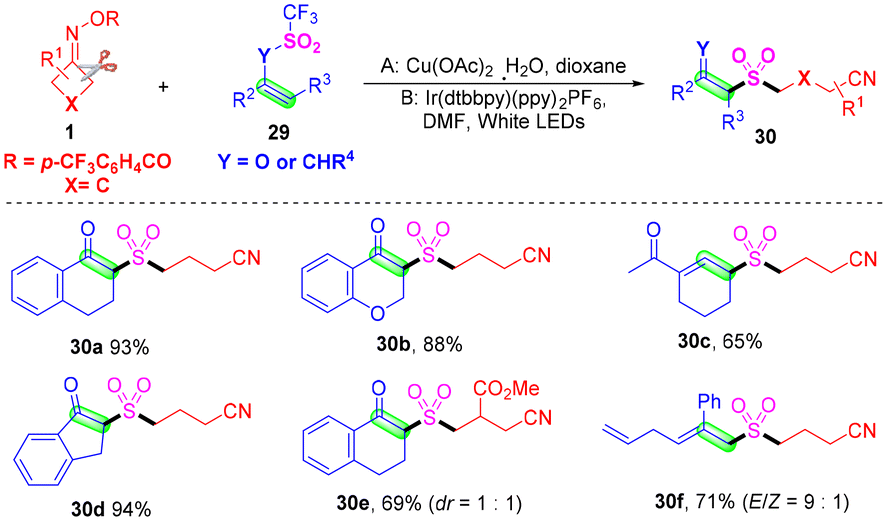 | ||
| Scheme 12 Cu- or visible-light-catalyzed cyanoalkylsulfonylation of vinyl triflates or allyl trifluoromethylsulfones with cyclobutanone oximes. | ||
The photoinduced cyanoalkylsulfonylation of methylenecyclopropanes (MCPs) with cyclobutanone oximes and potassium metabisulfite for the synthesis of 2-cyanoalkylsulfonated 3,4-dihydronaphthalenes with broad functional group tolerance was proposed by Liu, Tang et al. (Scheme 13).56 This cyanoalkylsulfonylation process proceeded via radical-mediated dual cleavage of C–C σ-bonds in methylenecyclopropanes (MCPs) and cyclobutanone oximes, in which the hindrance of the substituents and the electronic effect showed obvious influence on the reaction yields. The mechanism is outlined, and indicates that iminyl radical 4 was produced from cyclobutanone oximes in the presence of [Ru(bpy)3]2+* under visible light irradiation via a SET. Then, the iminyl radical 4 would undergo intermolecular ring-opening to furnish the alkyl radical 5, which would capture sulfur dioxide offering cyanoalkylsulfonyl radical 6. Then it would attack the double bond of methylenecyclopropanes affording alkyl radical 33. A ring-opening would occur to produce the terminal alkyl radical 34, which would undergo intermolecular cyclization to afford intermediate 35. Finally, oxidative SET and deprotonation were achieved in the presence of a photocatalyst and a base, and the 2-cyanoalkylsulfonated 3,4-dihydronaphthalenes 32 would be constructed in moderate to excellent yields.
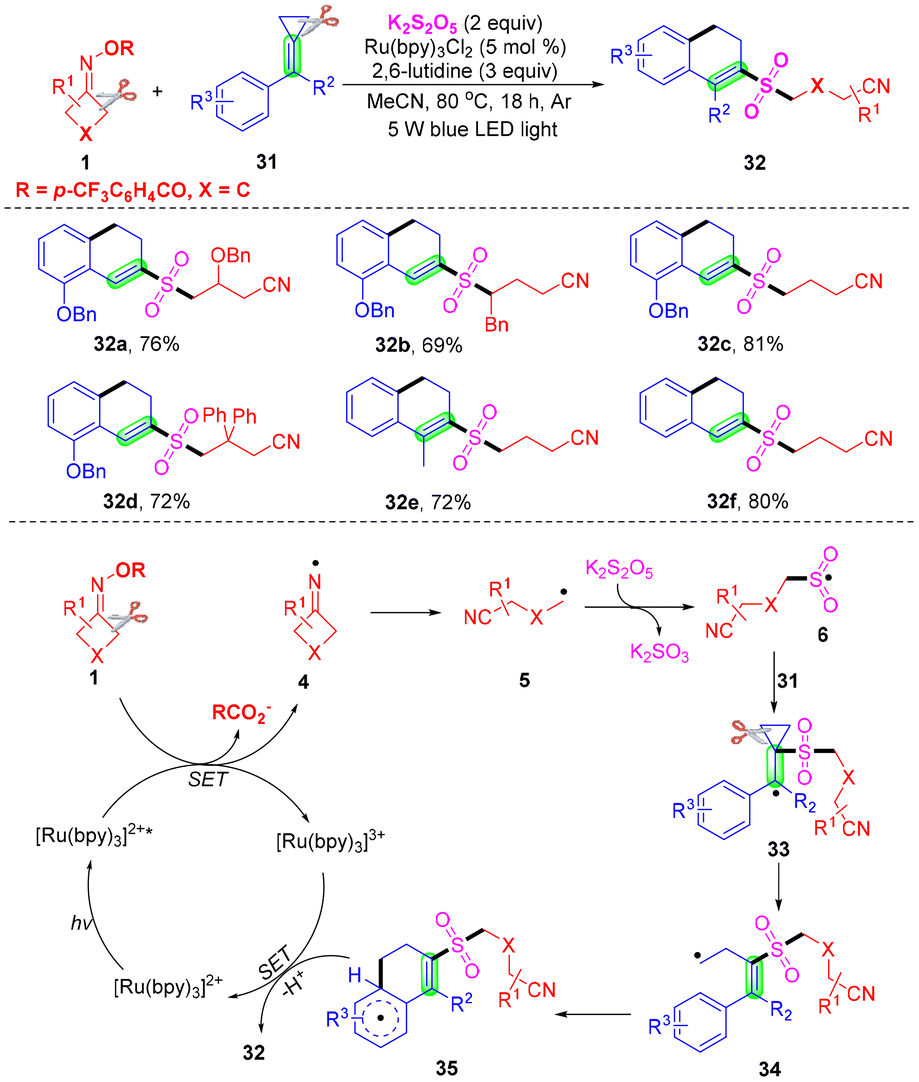 | ||
| Scheme 13 Photoinduced cyanoalkylsulfonylation of MCPs, cyclobutanone oximes and K2S2O5. | ||
Construction of cyanoalkylsulfonylated oxindoles was accomplished through an iron-mediated reaction of active alkenes, potassium metabisulfite and cyclobutanone oximes with good functional group tolerance and without the addition of any external base, oxidant and ligand (Scheme 14).57 This cascade radical cyanoalkylsulfonylation/arylation with insertion of sulfur dioxide was conducted by Liu and co-workers. In this transformation, potassium metabisulfites can afford sulfur dioxide. On the basis of control experiments and previous reports, it was shown that cyclobutanone oximes 1 would undergo SET reduction and intermolecular ring-opening to generate the alkyl radical 5 in the presence of an iron catalyst, which would subsequently react with SO2 to produce intermediate 6. Radical 38 was furnished from the addition of intermediate 6 to the C–C double bond of acrylamide 36. Finally, intermolecular cyclization occurred, with the following SET oxidation to construct cyanoalkylsulfonylated oxindoles 37 in the presence of a catalyst.
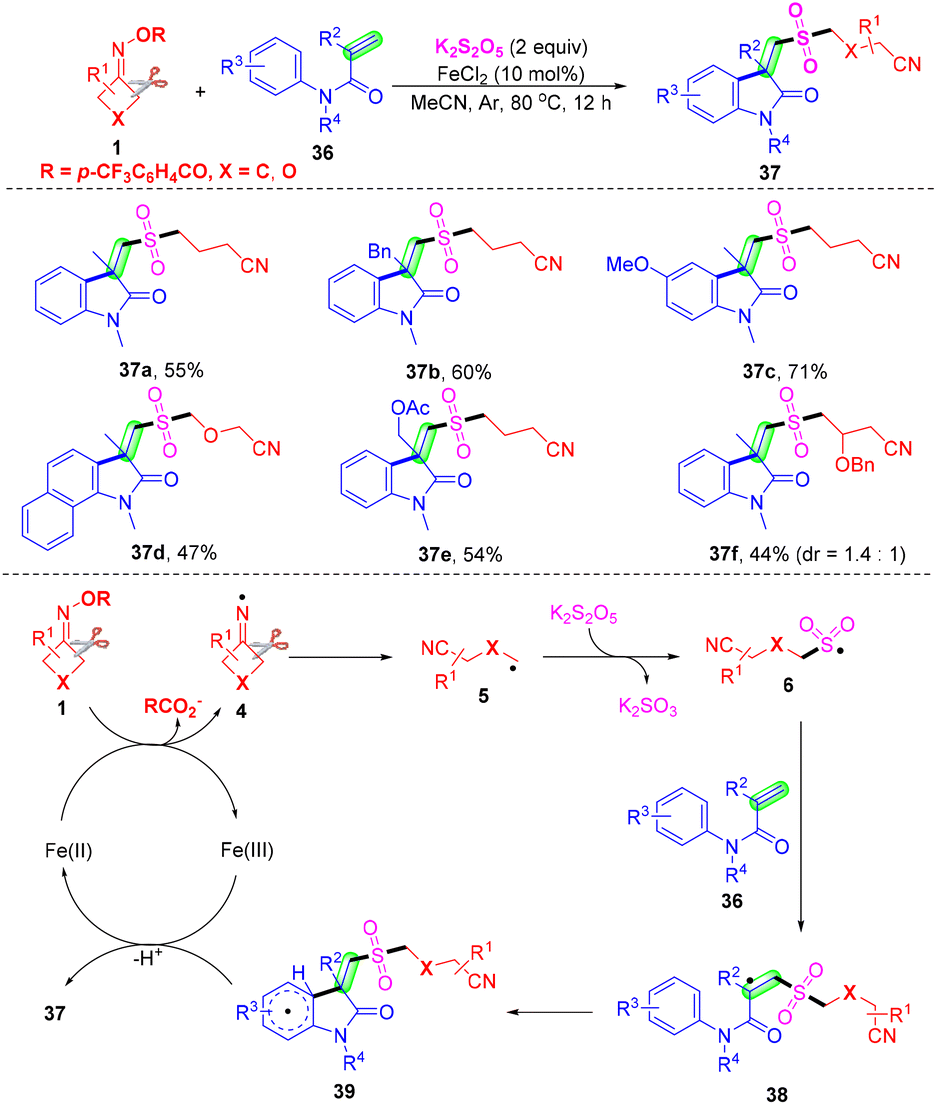 | ||
| Scheme 14 Iron-catalyzed three-component reaction of active alkenes, cyclobutanone oximes and K2S2O5. | ||
Even though the insertion of SO2 into the alkyl nitrile radical has been established, the use of transition metals or additive photosensitizers was unavoidable. In 2021, a visible-light-induced radical tandem cyanoalkyl sulfonylation/arylation reaction of active alkenes 36 with cycloketone oxime esters 1 using K2S2O5 as the sulfur dioxide surrogate was established by Gui and co-workers (Scheme 15).58 This method provided a convenient pathway for the preparation of cyanoalkylsulfonylated oxindoles, and just required 10 W irradiation of a 390 nm LED source with the assistance of transition metals, photosensitizers, bases, or oxidants. The mechanism of this radical tandem reaction has two different points from the works of Liu's group (Scheme 14). On the one hand, oxime ester 1 undergoes SET reduction under visible light to generate the iminyl radical 4 without a reductive metal catalyst. On the other hand, radical 39 undergoes SET oxidation under visible light and deprotonation by the 4-(trifluoromethyl)benzoate ion to deliver product 37 without an oxidation state metal catalyst.
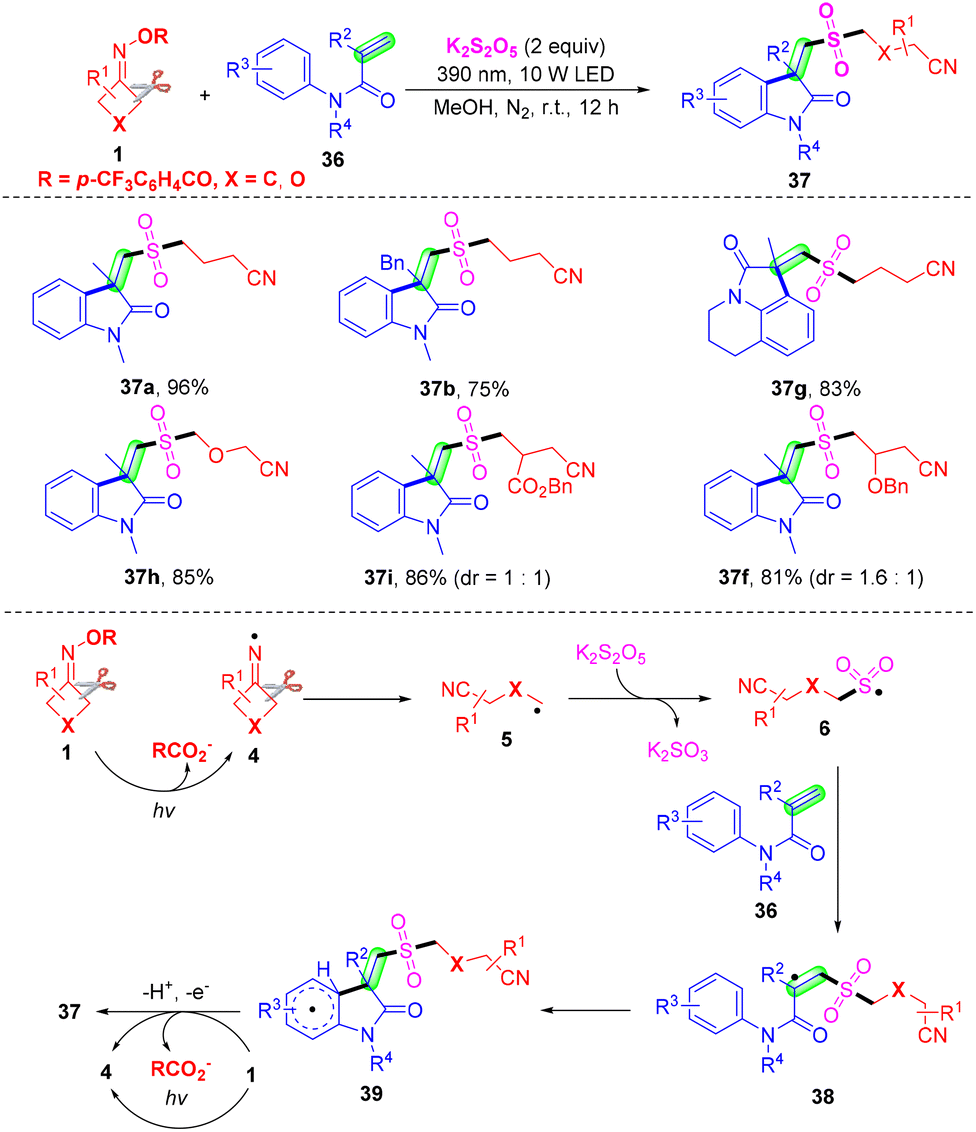 | ||
| Scheme 15 Photoinduced cyanoalkylsulfonylation of active alkenes, cyclobutanone oxime and K2S2O5. | ||
It is generally reported that the cyanoalkyl group is used to capture SO2 in cyanoalkylsulfonylation reactions, and the carbon–carbon double bond is used as a cyanoalkyl acceptor, while the report of the cyanoalkylsulfonylation strategy with a carbon–carbon triple bond is relatively scarce. Liu et al. proposed a series of cyanoalkylsulfonylation/cyclization reactions of alkynes mediated by visible light without a transition metal. 2-Cyanoalkylsulfonyl-9H-pyrrolo[1,2-a]indoles were prepared from N-propargylindoles and cyclobutanone oxime derivatives by SO2 insertion (Scheme 16).59 The mechanism shows that eosin Y2− is converted into eosin Y2−* by light excitation. Then, iminos group 4 formed by reduction of eosin Y2−* in cyclobutanone oxime 1 is further ring-opened to afford the cyanoalkyl group 5. Then, intermediate 5 reacts with potassium pyrosulfite to afford cyanoalkylsulfonyl 6. Intermediate 6 attacks the C–C triple bond in alkyne 40 to generate free radical 42, which is intramolecularly cyclized to generate free radical 43, which is then converted into carbocation 44 by eosin Y−, and eosin Y− is reduced to eosin Y2−. Finally, intermediate 44 was deprotonated and isomerized to obtain the target product 41 in excellent yield.
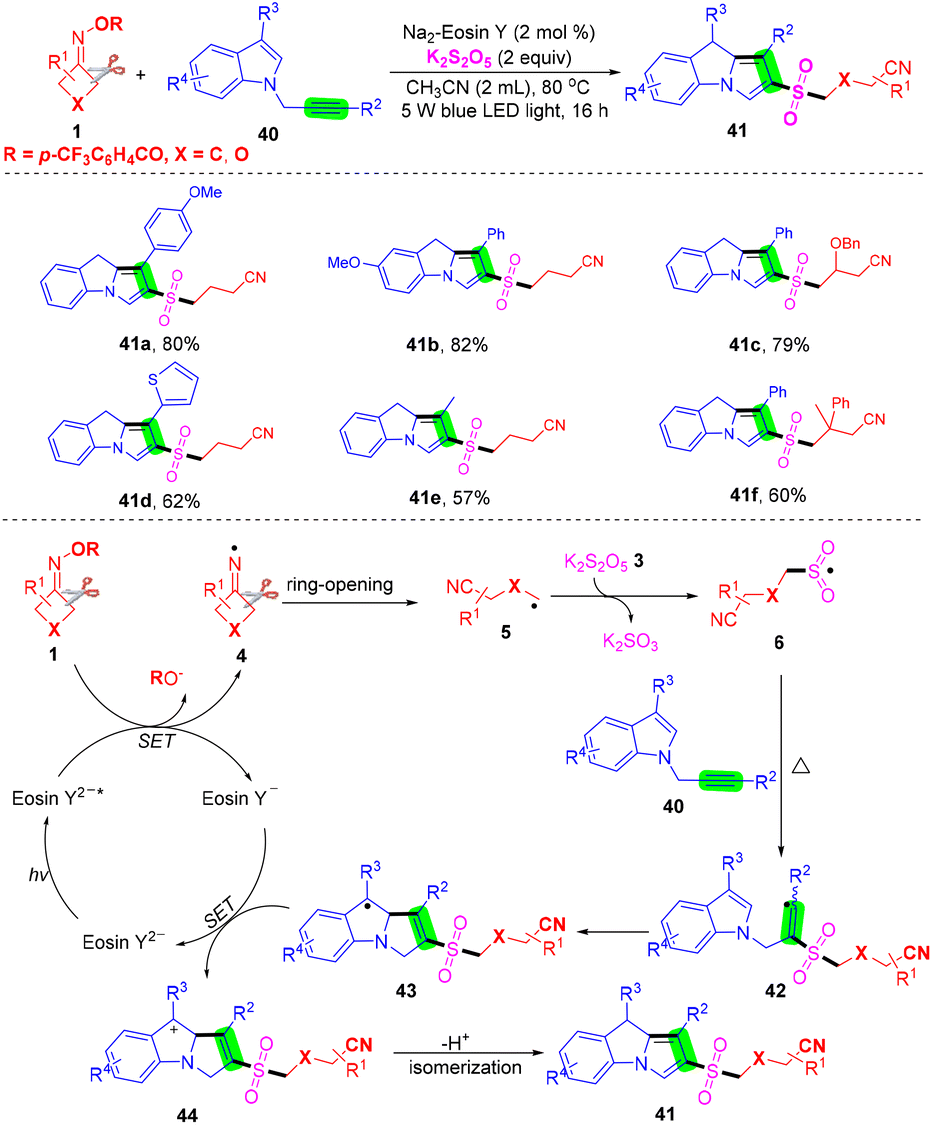 | ||
| Scheme 16 Visible-light-mediated three-component reaction of N-propargylindoles, cyclobutanone oxime and potassium metabisulfite. | ||
In recent years, a strategy has been established for the preparation of 3-arylsulfonylquinolines, and methods for the synthesis of 3-alkylsulfonylquinolines are still retained. Zhou's group reported a visible-light-induced multicomponent radical cascade cyclization of N-propargyl aromatic amines 45 with cyclobutanone oxime esters 1 and K2S2O5 to access 3-cyanoalkylsulfonylated quinolines via the insertion of sulfur dioxide (Scheme 17).60 This cascade reaction features a broad substrate scope and excellent functional group compatibility, providing a convenient pathway for the synthesis of 3-cyanoalkylsulfonylated quinolines by the selective C–C σ-bond cleavage and the formation of one C–C bond and two C–S bonds under mild reaction conditions. A plausible reaction mechanism is outlined in Scheme 17. Initially, eosin Y was excited by visible-light irradiation to produce its excited species eosin Y*. Then, the cycloketone oxime ester 1 through a SET process was reduced by eosin Y* to afford iminyl radical species 4 and eosin Y+˙ species. Next, the radical intermediate 4 further underwent ring-opening to deliver the cyanoalkyl radical 5, which would capture sulfur dioxide in the presence of K2S2O5 to obtain cyanoalkylsulfonyl radical 6. The cyanoalkylsulfonyl radical 6 attacked the C–C triple bond of 45 to afford the radical intermediate 47, which then underwent intramolecular cyclization, a single electron oxidation process by eosin Y+˙ species and further deprotonation to acquire intermediate 50. Finally, intermediate 50 was ultimately aromatized to produce the target product 46.
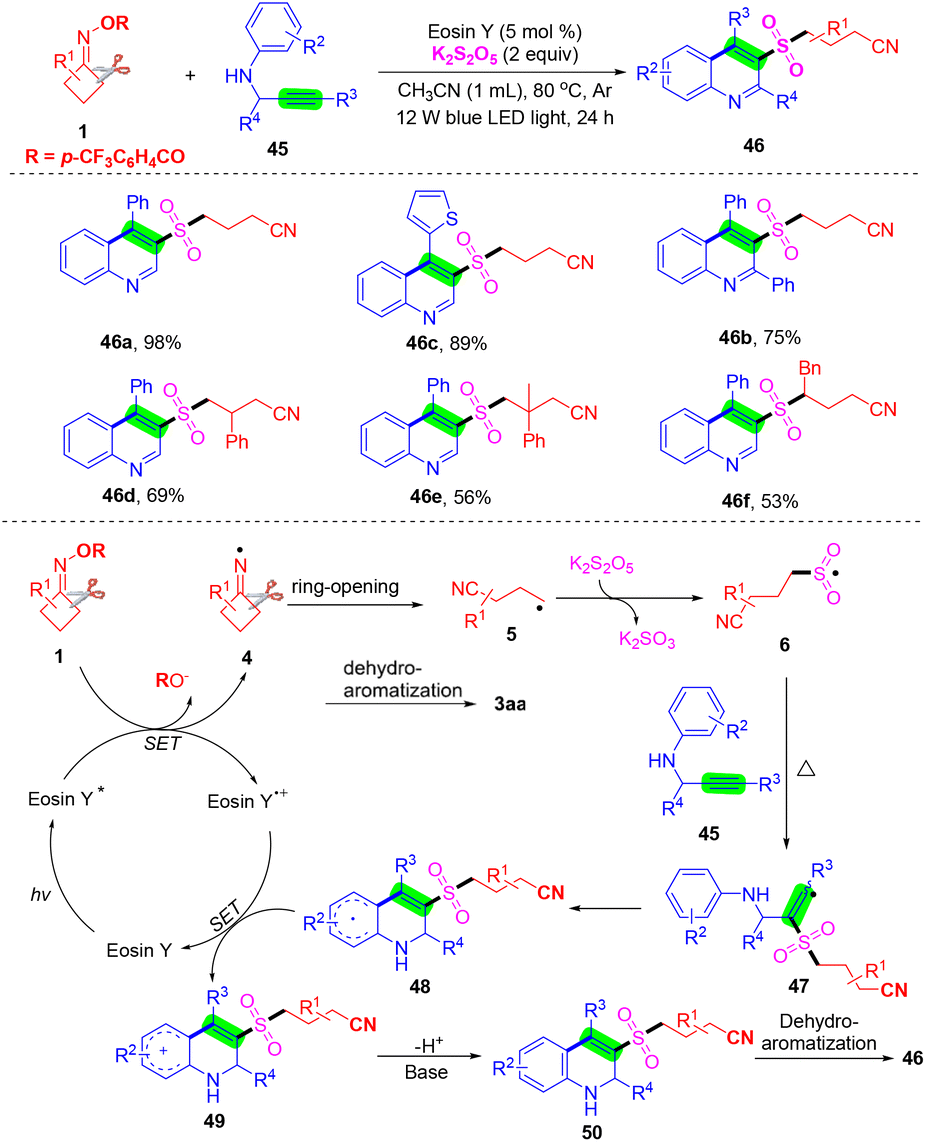 | ||
| Scheme 17 Visible-light-induced cyanoalkylsulfonylated quinolones via cascade cycloaddition of N-propargyl aromatic amines, cyclobutanone oxime esters, and K2S2O5. | ||
Liu et al. discovered a cyanoalkylsulfonylation/cyclization reaction method of alkynate and cyclobutanone oxime compounds mediated by visible light, and prepared 3-cyanoalkylsulfonylcoumarin by SO2 insertion (Scheme 18).61 This method provides a convenient way for the preparation of cyanoalkylsulfonylated coumarins. Mechanism study shows that the NCR process is involved in the transformation. Initially, IrIII was converted into IrIII* and excited by light irradiation. Then, under the reduction of IrIII* ((E1/2 IrIV/IrIII*) = −1.73 versus SCE), the imino group generated by cyclobutanone oximes 4 (E6 = −1.64 versus SCE) underwent ring-opening to afford intermediate 5. Then cyanoalkyl group 5 reacts with K2S2O5 to afford intermediate 6. Cyanoalkylsulfonyl group 6 attacks the C–C triple bond in alkynate 51 (E49 = −1.88 versus SCE) to furnish the radical 53, which underwent ipso-cyclization to give the radical 54. Next, the group provides a spirocyclic intermediate 55 through ion exchange of IrIV, and IrIV is reduced to IrIII. Finally, the intermediate 55 undergoes 1,2-ester migration and deprotonation to form the target product 52.
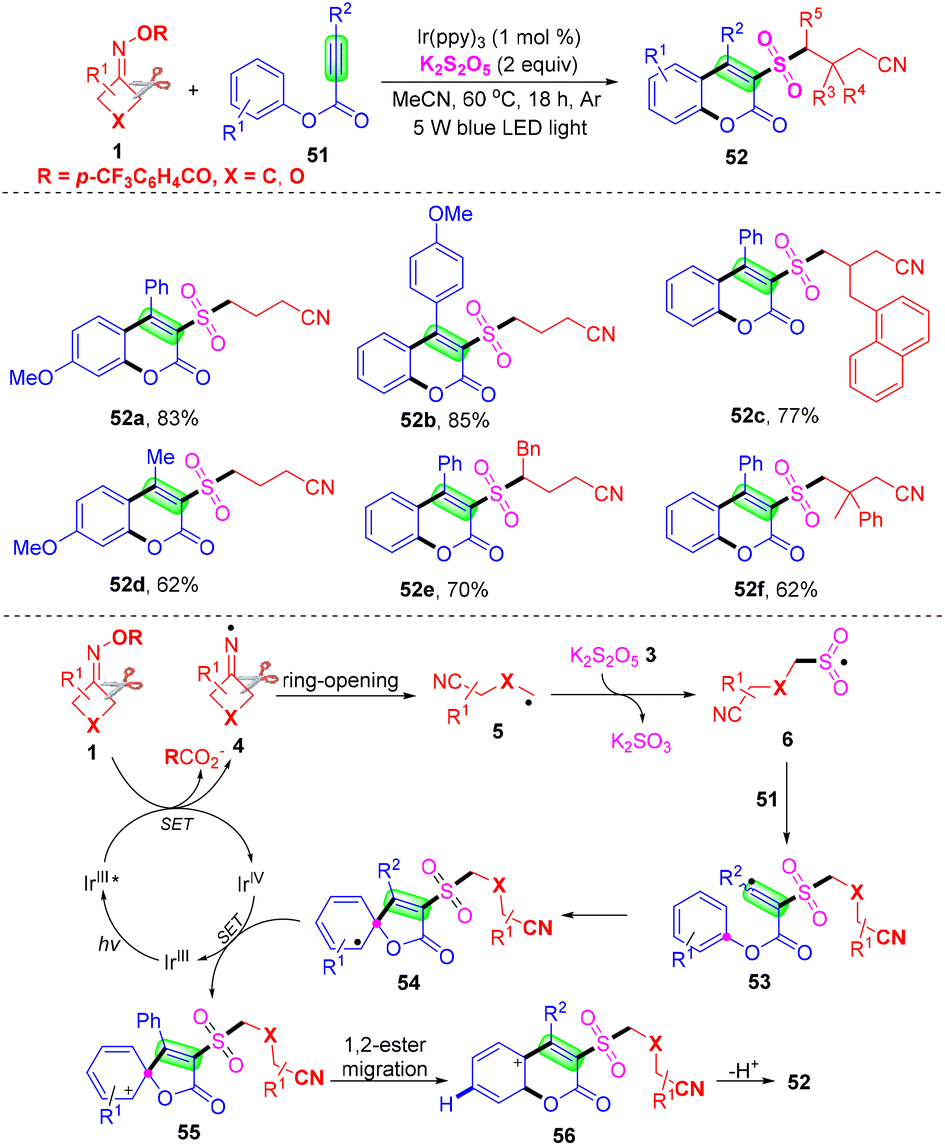 | ||
| Scheme 18 Visible-light-mediated three-component reaction of alkynoates, cyclobutanone oxime and K2S2O5. | ||
Furan-2(5H)-ones represent a prominent class of structural scaffolds widely found in natural products and biologically active compounds.62 Owing to allenoic acids having high reactivity and easy preparation methods, they are intensively employed as synthons for acquiring furan-2(5H)-one derivatives. In 2021, Yu's group described an iron-catalyzed three-component cyanoalkylsulfonylation of 2,3-allenoic acids 55, cyclobutanone oxime esters 1, and K2S2O5 for the preparation of a variety of cyanoalkylsulfonylated butenolides under mild conditions (Scheme 19).63 Moreover, further transformations of the cyanoalkylsulfonylated butenolides including lactone ring opening/butanolysis were achieved and the cyano group can easily be transformed through a Ritter reaction, which provided the corresponding derivatives. Based on the results of control experiments discussed above and previous studies, a possible free radical pathway for the present cascade cyclization was proposed. Initially, SET reduction of the cycloketone oxime ester 1 by the Fe(II) catalyst generated iminyl radical species 4 and Fe(III) species. Subsequently, the C–C bond cleavage of iminyl radical 4 yields the alkyl radical intermediate 5, which reacts with K2S2O5 to give sulfonyl radical 6, followed by addition to 2,3-allenoic acid 57 to form the intermediate 59. The allylic cation 60 was generated from the single electron oxidation process of Fe(III) species, furnishing the Fe(II) species to complete the catalytic cycle. Finally, the intermediate 60 underwent intramolecular nucleophilic attack and produced the cyclization product furan-2(5H)-one derivative 58.
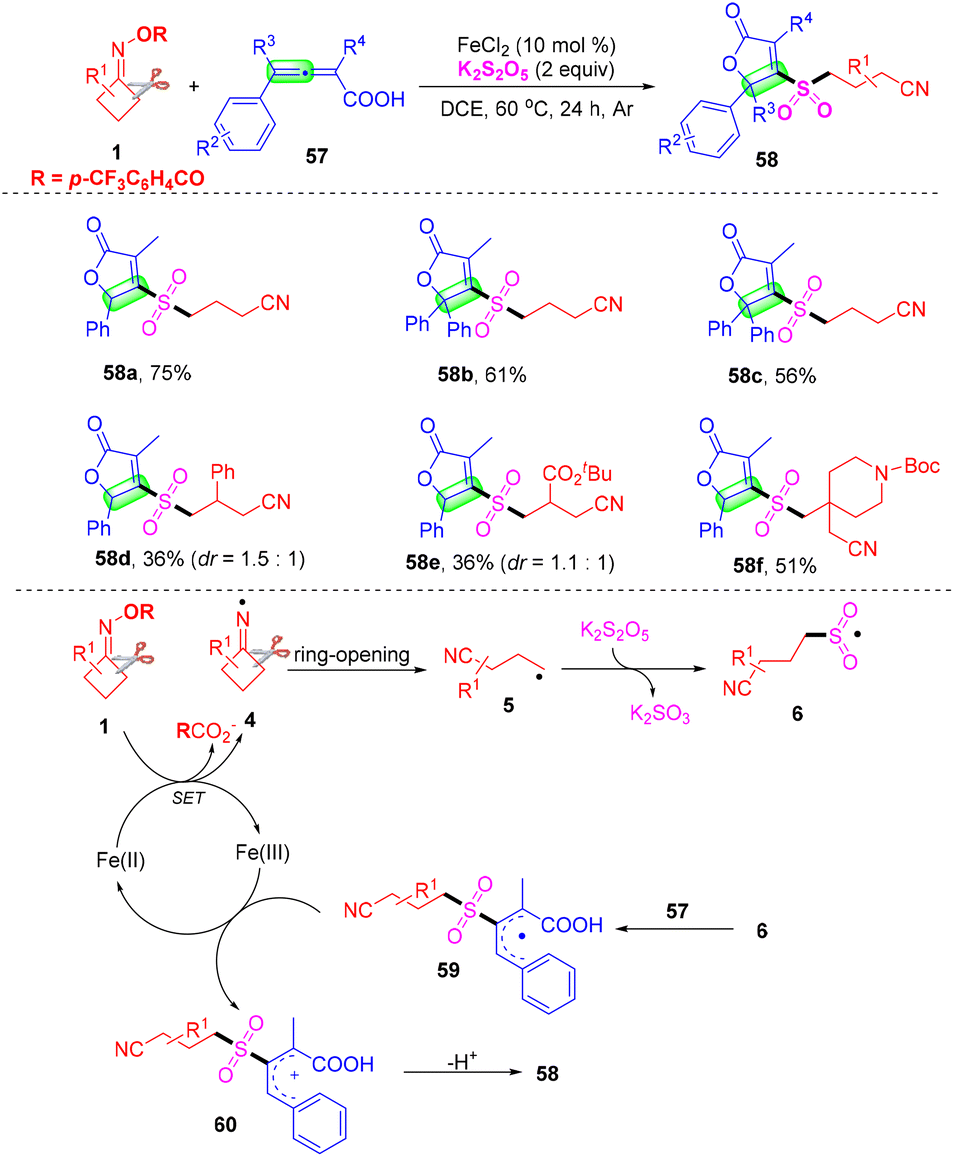 | ||
| Scheme 19 Iron-catalyzed three-component cyanoalkylsulfonylation of 2,3-allenoic acids, cycloketone oxime esters and K2S2O5. | ||
At the same time, Zhou's group developed a photoredox- or iron-catalyzed radical cyanoalkylsulfonylation/cyclization reaction of unactivated olefins 61, cyclobutanone oxime esters 1, and K2S2O5 for constructing cyanoalkylsulfonylated benzoxepines 62 (Scheme 20).64 This SO2-trapping strategy provides a direct method for constructing the cyanoalkylsulfonylated substituted benzoxepine scaffold, exhibiting a wide substrate scope and excellent functional group tolerance. This reaction proceeded efficiently with a cascade of C–C σ-bond cleavage and C–S/C–S/C–C bond formation to provide the corresponding cyanoalkylsulfonylated benzoxepine derivatives.
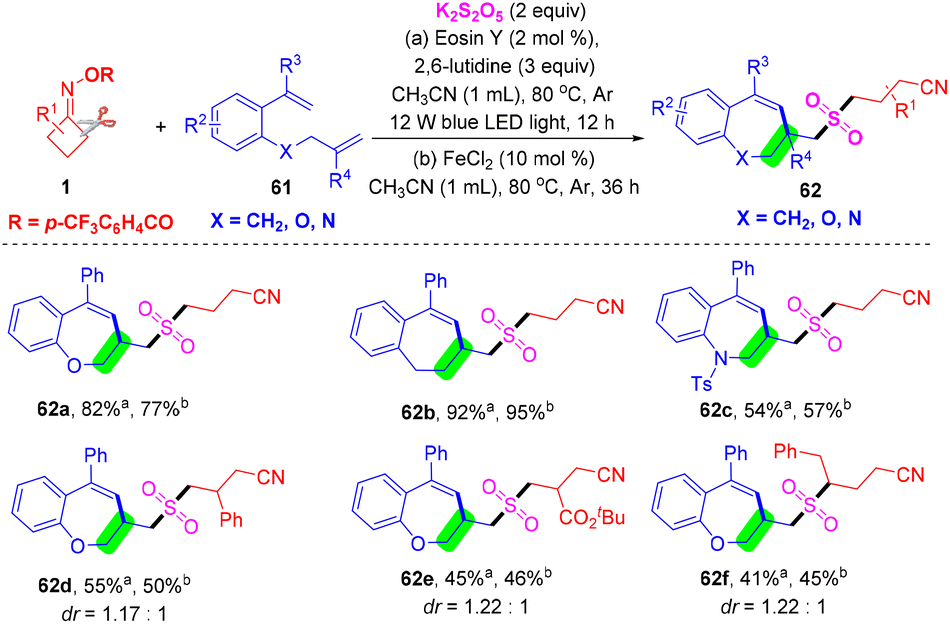 | ||
| Scheme 20 A photoredox- or metal-catalyzed three-component cyanoalkylsulfonylation reaction of 1-(allyloxy)-2-(1-arylvinyl)benzenes, cycloketone oxime esters and K2S2O5. | ||
Over the past decade, Nevado65,66 and other groups employed different kinds of radical addition to the double bond of the tosyl acrylamide system for the synthesis of oxindole and amide derivatives, which have been successfully achieved through the intramolecular 1,4-aryl migration/5-ipso cyclization and desulfonylation process. On the ground, Liang's group have established a copper-catalyzed radical cross-coupling of oxime esters 1 and tosyl acrylamides 63 to prepare various cyanoalkylsulfonylated oxindoles, and sulfone addition/desulfonylation/cyclization processes were involved in this transformation (Scheme 21).67 Significantly, in this method in situ desulfonylation inserts sulfur dioxide, avoiding the addition of extra sulfur dioxide (such as K2S2O5 and DABSO). The mechanism research indicates that a series of processes including β-C–C bond scission, in situ sulfur dioxide insertion, intramolecular 1,4-aryl migration/5-ipso cyclization/desulfonylation and cyclization were involved in this transformation.
 | ||
| Scheme 21 Copper-catalyzed radical aryl migration approach for the preparation of cyanoalkylsulfonylated oxindoles. | ||
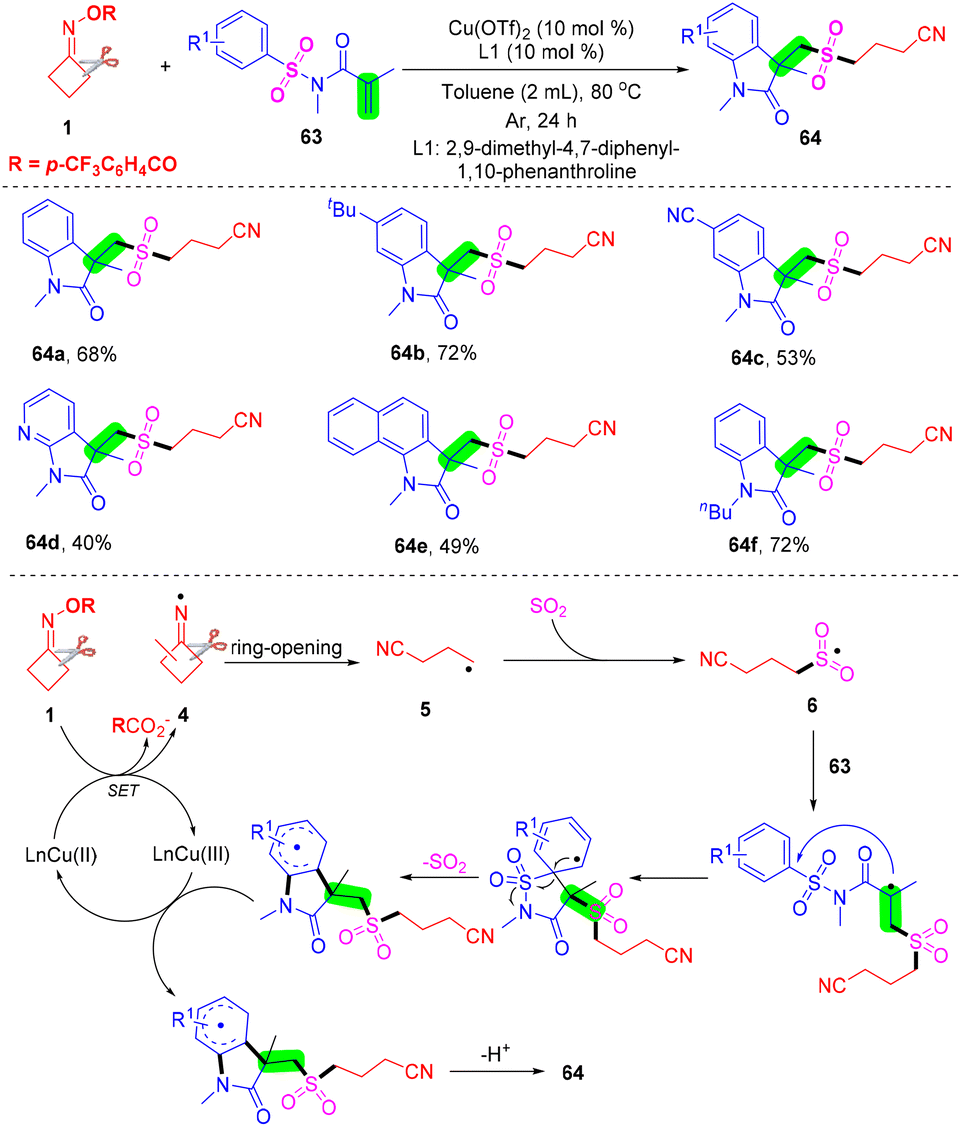
With Zhou's group continuing studies on the construction of organosulfone compounds by radical cyclization and annulation, they recently developed a palladium-catalyzed radical cascade cyanoalkylsulfonylation/cyclization of 3-arylethynyl-[1,10-biphenyl]-2-carbonitriles 65 with cyclobutanone oxime esters 1 and DABCO·(SO2)2 for the synthesis of cyanoalkylsulfonylation cyclopenta[gh]phenanthridines in moderate to good yields (Scheme 22).68 A series of cyanoalkylsulfone substituted cyclopenta[gh]phenanthridines were obtained via cleavage of a C–C single bond and in situ capture of SO2 under mild reaction conditions. This strategy not only represents a novel and efficient mode for sulfur dioxide insertion but also realizes straightforward methods for directly obtaining structurally important cyanoalkylsulfone-containing cyclopenta[gh]phenanthridines.
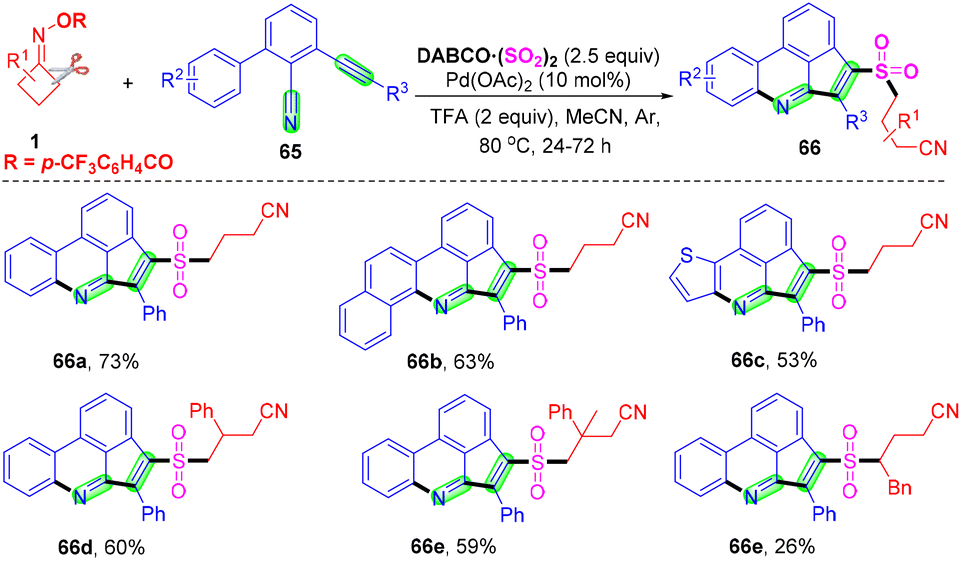 | ||
| Scheme 22 Palladium-catalyzed radical cascade cyanoalkylsulfonylation/cyclization of 3-arylethynyl-[1,1′-biphenyl]-2-carbonitriles with cyclobutanone oxime esters and DABCO·(SO2)2. | ||
3. Cyanoalkylcarbonylation via the insertion of CO
Carbonyl compounds broadly exist in many complex pharmaceuticals, materials and organic compounds.69,70 They are valuable building blocks in organic synthesis.71–73 For these reasons, the development of carbonylation reactions via novel and efficient methods has received considerable attention. Different kinds of organic reagents,74–77 such as CO, aldehyde, ketone, acyl chlorides, α-ketoacids and so on, as the source of carbonyl have been successfully used in the carbonylation reactions. In recent years, developing a radical strategy for carbonylation has attracted high attention. Compared with the transition metal-catalyzed carbonylation reaction, the direct use of a radical intermediate led to in situ capture of CO due to the lack of stabilization with transition metals resulting in low reaction efficiency and serious side reactions. Generally, the use of high-pressure CO in many processes avoids these problems. Herein, several cases of carbonylation of cyanoalkyl radicals via the capture of CO have been summarized.α,β-Unsaturated ketones are versatile and important building blocks with broad application in organic synthesis.78–80 Wu and co-workers reported a novel and efficient vinylboron self-promoted carbonylative coupling with cyclobutanone oxime to develop α,β-unsaturated ketones with broad functional group tolerance, without the use of any external radical initiators, oxidants and catalysts (Scheme 23).80 Additionally, CO was used as the source of carbonyl in this radical coupling reaction. Extensive substrate scope was proved, and a series of functional groups such as alkyl, methoxy, halogen or ester groups, were all compatible. Additionally, the reaction was not suitable for cyclopentanone oxime esters or cyclohexanone oxime esters to react with styrylboronic acid and CO under the optimal conditions. The control experiments proved that an iminyl radical 4 was obtained from cyclobutanone oxime esters via cleavage of the N–O bond and it would initiate the reaction. After intermolecular C–C cleavage of radical 4, the alkyl radical 5 would be produced, which would react with CO to afford carbonyl radical intermediate 69. This intermediate 69 would attack the double bond of trans-styrylboronic acid 67 leading to a benzyl radical 70. Lastly, the benzyl radical reacted with 1 leading to the desired α,β-unsaturated ketone 68.
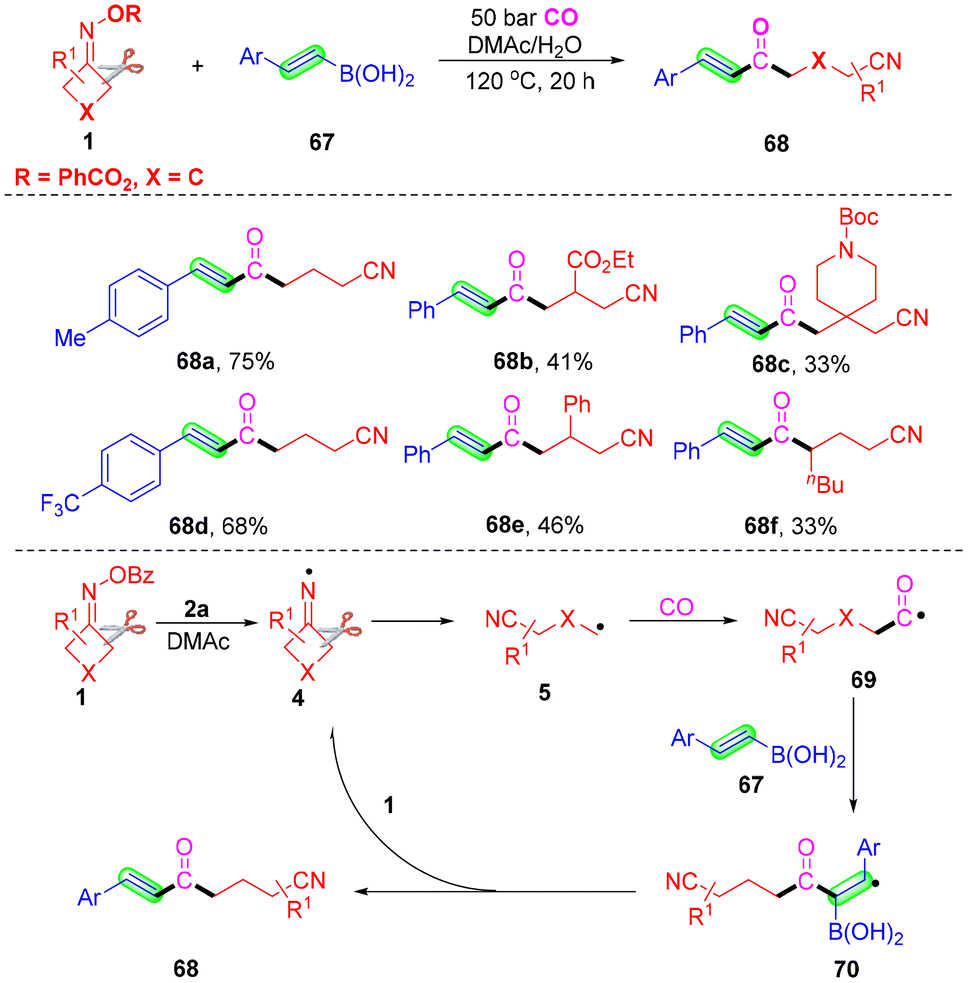 | ||
| Scheme 23 Generation of α,β-unsaturated ketones from cyclobutanone oxime, vinylboron and CO. | ||
Xiao and co-workers proposed a carbonylation to generate cyanoalkylated amides between cyclobutanone oxime, amines and CO gas under visible light-induced and exogenous photosensitizer-free, copper-catalyzed conditions (Scheme 24).81 A series of cyclobutanone oximes, aryl amines or alkyl amines were all suitable under the standard conditions. However, the reaction did not occur when cyclopentanone oxime esters or cyclohexanone oxime esters were used to react with amines and CO gas. Control experiments proved that visible-light irradiation and a copper catalyst were essential in this carbonylation. The mechanism study indicated that the iminyl radical 4 was obtained from a SET-mediated reduction of cyclobutanone oxime in the presence of photoexcited LnCu(I)-NHPh complex 75* (path a) or the ground state LnCu(I)-NHPh species 76 (path b), which would initiate the reaction. Next, a C–C bond selective cleavage occurred to furnish radical 5, which could be captured by complex 76 to generate high-valence Cu(III) complex 77. The complex 77 would react with CO gas to afford complex 78 or 79. Finally, the target product 72 would be furnished with the reductive elimination of complex 78 or 79.
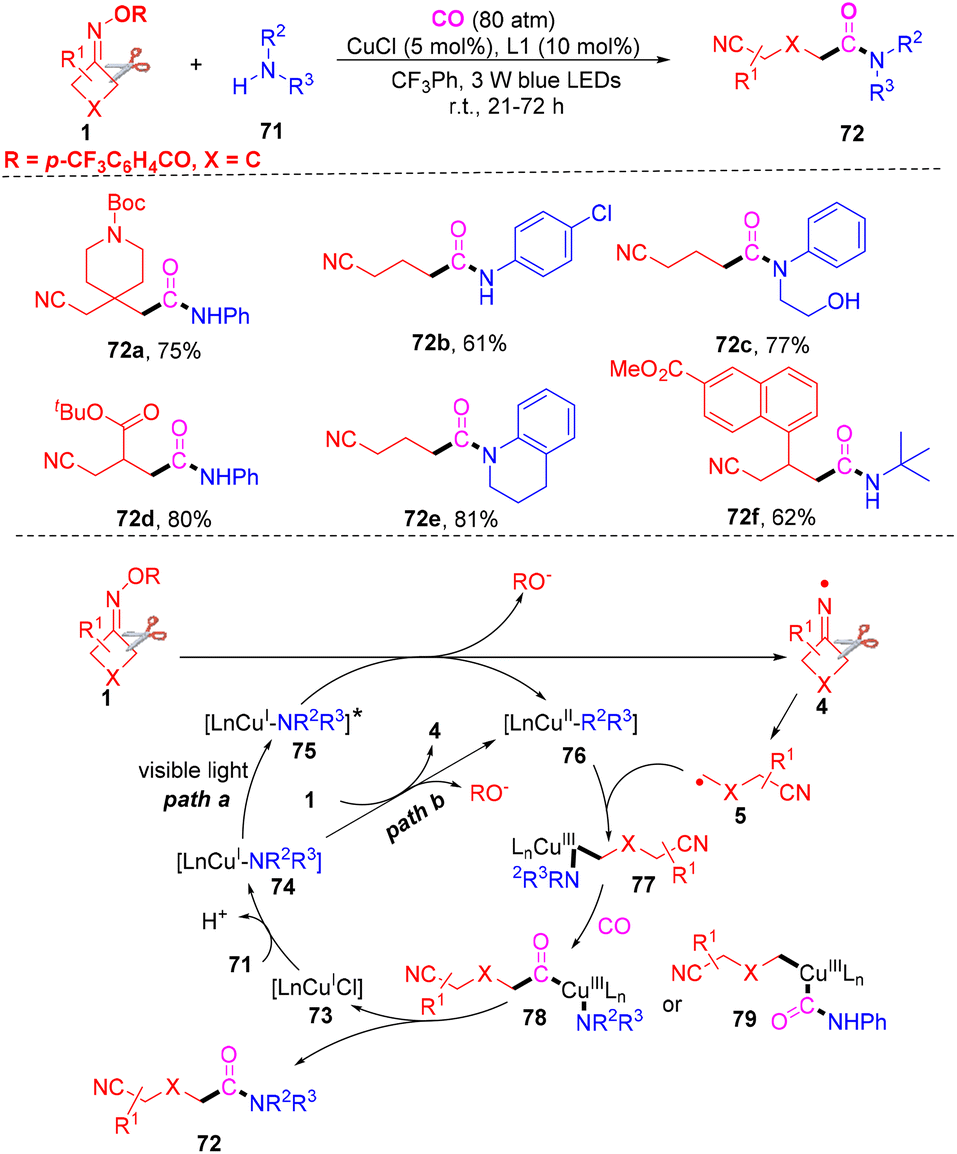 | ||
| Scheme 24 Copper-catalyzed reaction of cyclobutanone oximes, amines and CO. | ||
A novel and first gallic acid-catalyzed carbonylation of cyclobutanone oximes and olefins was developed by Wu and co-workers (Scheme 25).82 This carbonylation selected green and sustainable gallic acid as the catalyst, without the need for any transition metals. The EPR investigation and control experiments definitely proved that the carbonylation began from cyclobutanone oximes and gallic acid. The mechanism showed that iminyl radical 4 was constructed from gallic-catalyzed SET reduction of cyclobutanone oximes. The cyanoalkyl 5 was generated via a homolytic C–C single bond cleavage, and it would capture CO before the reaction with 1,1-diphenyl ethylene 80 to form radical 83. Finally, intermediate 83 would produce the target carbonylation products 82 with the aid of gallic acid 81.
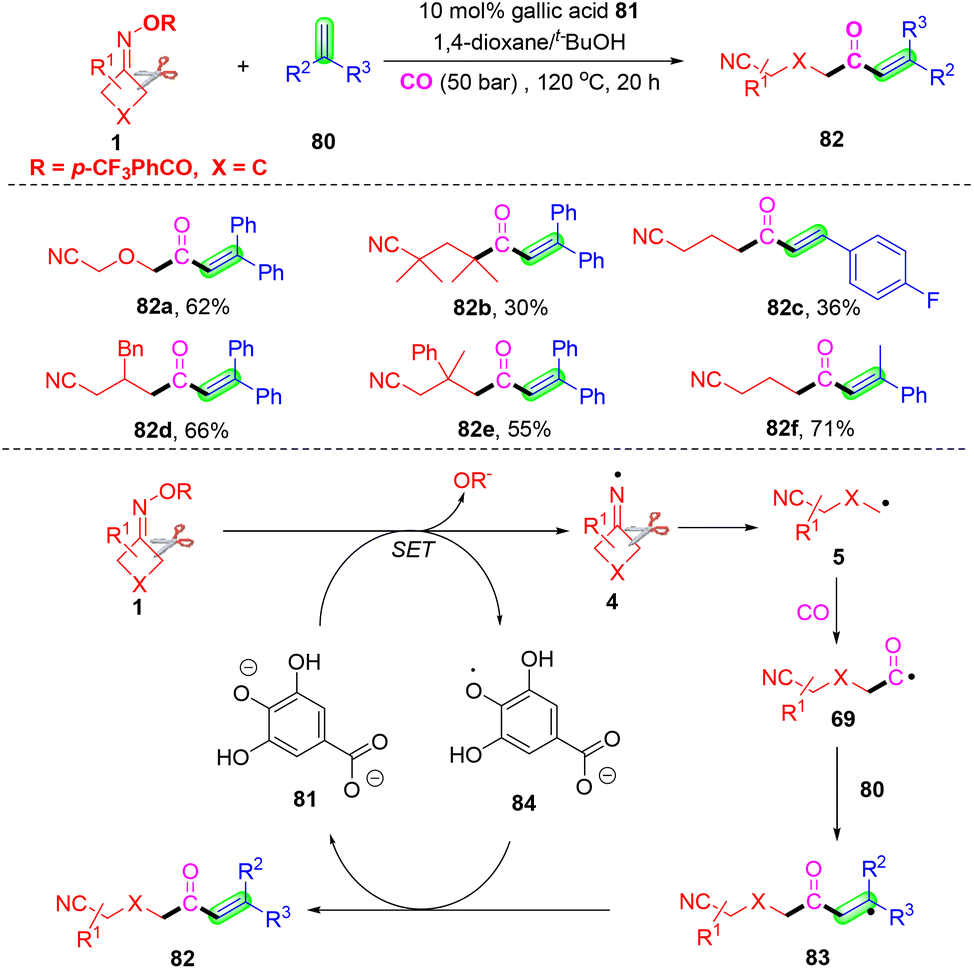 | ||
| Scheme 25 Gallic acid-catalyzed reaction of cyclobutanone oximes, olefins and CO. | ||
Thiazole and oxazole derivatives are found in materials, organic molecules and natural products.83,84 As yet, the carbonylation of thiazole or oxazole has been extensively explored in organic chemistry. Wu and co-workers found that thiazole or oxazole could be used as a coupling partner with CO and cyclobutanone oximes (Scheme 26).85 Alkyl heteroaryl ketones were obtained with excellent functional group tolerance, good chemoselectivity and satisfactory yields. In this transformation, thiazoles or oxazoles were used as the substrates for the introduction of CO into organic motifs. Control experiments proved that a radical pathway was excluded. A similar mechanism was developed; first, cyanoalkyl 5 was generated from cyclobutanone oximes with the assistance of an iron catalyst. Subsequent carbonylation and addition to thiazole or oxazole 85 would form the nitrogen radical 87. Finally, single electron oxidation and deprotonation followed to provide the final products 86.
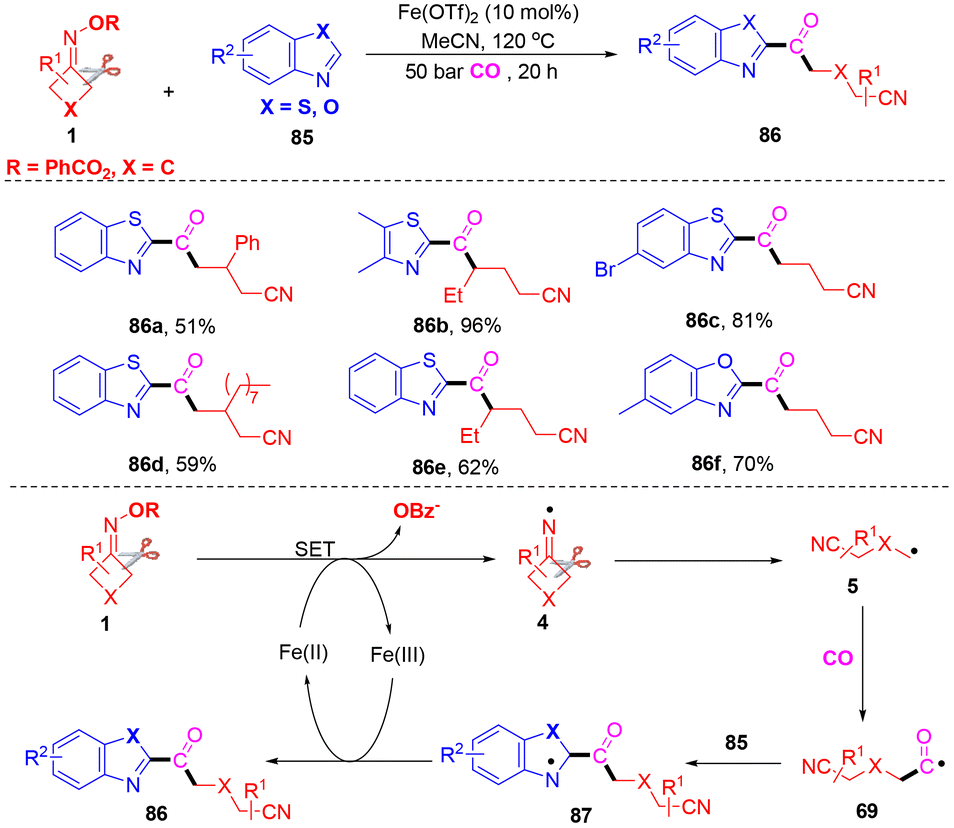 | ||
| Scheme 26 Reaction of thiazole or oxazole, cyclobutanone oximes and CO under iron catalysis. | ||
4. Cyanoalkoxylation via the insertion of O2
Synthesis of cyanoalkoxylated tetrasubstituted alkenes through a reaction of cycloketone oxime esters and alkenes under a dioxygen atmosphere was accomplished in the presence of CuCl2 (Scheme 27).86 This transition-metal-catalyzed alkyl-Heck-type cross-coupling of olefinic C–H bond was first disclosed by Yu and co-workers. Control experiments proved that the cleavage of the internal olefinic C–H bond in 88 was not a rate-determining step in this reaction. It was postulated that iminyl radical 4 was developed initially from cyclobutanone oximes with the assistance of a catalyst, followed by ring-opening of radical 4 to develop radical 5, which could easily be trapped with the O2 molecule to generate peroxy radical 90 under an oxygen atmosphere. Then, the reduction of radical 90 occurred to deliver alkoxy radical 91 with the assistance of Cun species through the Fenton-type mechanism.87,88 The species 91 would attack the internal alkene 88 to give radical 92, which could be further oxidized in the presence of Cun+1 to generate cationic intermediate 93. Finally, the cationic intermediate 93 would undergo hydrogen abstraction to produce Heck-like cyanoalkoxylation products 89.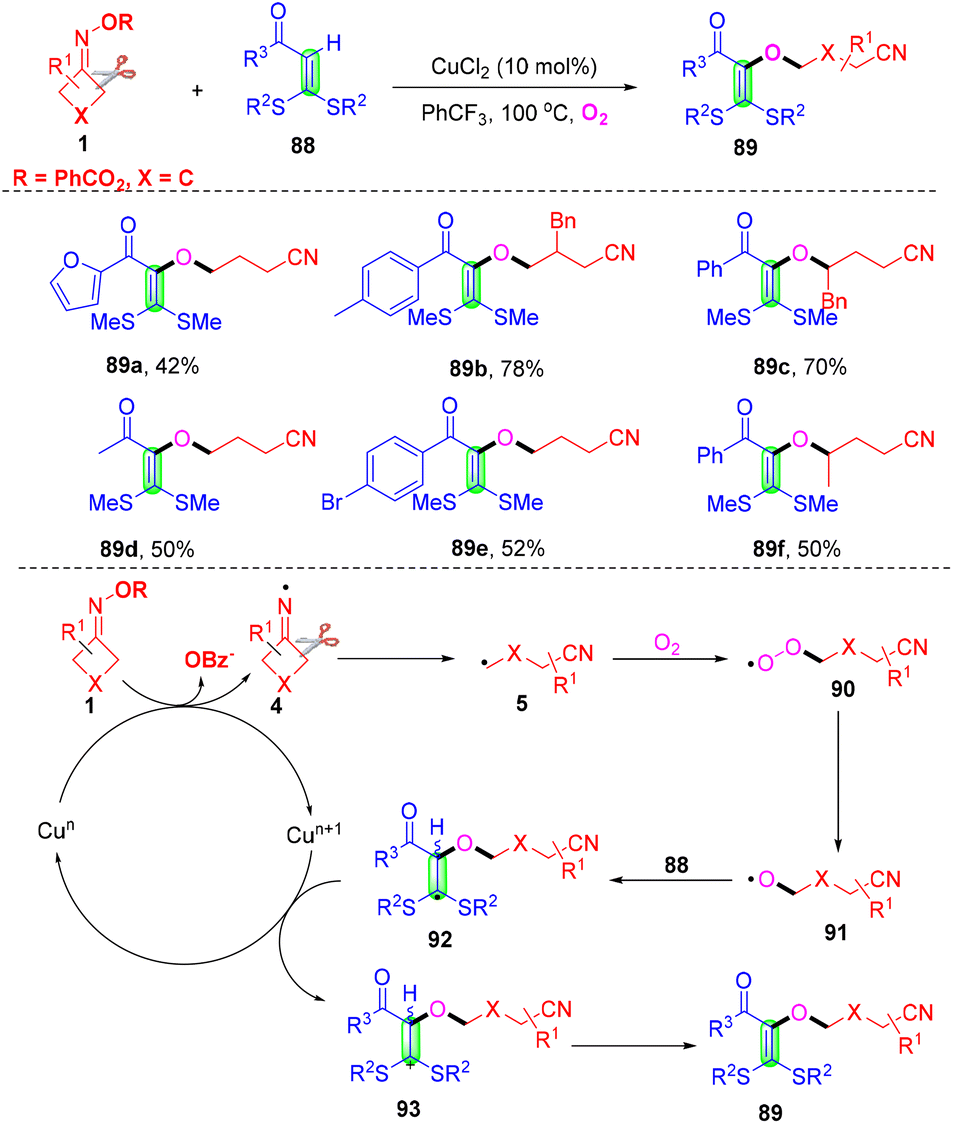 | ||
| Scheme 27 Construction of cyanoalkoxylation tetrasubstituted alkene from cycloketone oxime esters, alkenes and O2. | ||
5. Conclusion
In this updated report, the sulfonylation/carbonylation/oxidation reactions of the cyanoalkyl radical via the insertion of SO2, CO or O2 are summarized, and the cyanoalkyl radical is produced from iminyl-radical-triggered C–C bond cleavage of cyclobutanone oxime derivatives. In this field, these transformation reactions have attracted considerable attention in recent years. As mentioned above, these methods are fascinating and attractive, since cyclobutanone oximes can provide stable distal cyano-substituted alkyl radicals without the use of toxic cyanide reagents. Diverse cyanoalkylsulfonylated molecules, cyanoalkylcarbonylated molecules, and cyanoalkyloxylated molecules can be constructed synthetically and conveniently. In all of the reactions, the radical process is involved.However, despite these advances, some challenges still have to be explored. Most of the present research studies concentrate on distal cyano-substituted alkyl radical capture of SO2, CO or O2, and then reaction with radical acceptors. However, studies on the significance of the distal cyano-substituted alkyl radical with insertion of SO2, CO or O2 and the further exploration of target products are still absent. In these cases, where radical reactions involve well-established radical acceptors, novel and efficient radical acceptors still need to be explored and discovered. In addition, the concept of green chemistry has concentrated the attention of chemists in the past several years. Thus, more green and economical approaches, such as without a catalyst and non-toxic solvent and so on, are highly desired.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of Hunan Provincial Education Department (no. 21B0589, 22B0674 and 22B0678), the Natural Science Foundation of Hunan Province (no. 2023JJ30273, 2023JJ40301 and 2023JJ40304) and the National Natural Science Foundation of China (no. 22078084, 22201070 and 51874132) for financial support.References
- F. F. Fleming, L. Yao, P. C. Ravikumar, L. Funk and B. C. Shook, J. Med. Chem., 2010, 53, 7902 CrossRef CAS.
- E. L. May, A. E. Jacobson, M. V. Mattson, J. R. Traynor, J. H. Woods, L. S. Harris, E. R. Bowman and M. D. Aceto, J. Med. Chem., 2000, 43, 5030 CrossRef CAS PubMed.
- R. Turnaturi, A. Marrazzo, C. Parenti and L. Pasquinucci, Eur. J. Med. Chem., 2018, 148, 410 CrossRef CAS PubMed.
- F. F. Fleming and Q. Wang, Chem. Rev., 2003, 103, 2035 CrossRef CAS PubMed.
- R. López and C. Palomo, Angew. Chem., Int. Ed., 2015, 54, 13170 CrossRef.
- I. Dutta, S. Yadav, A. Sarbajna, S. De, M. Hölscher, W. Leitner and J. K. Bera, J. Am. Chem. Soc., 2018, 140, 8662 CrossRef CAS.
- Y. Ueda, N. Tsujimoto, T. Yurino, H. Tsurugi and K. Mashima, Chem. Sci., 2019, 10, 994 RSC.
- L. Yang, Y. T. Liu, Y. Park, S. W. Park and S. Chang, ACS Catal., 2019, 9, 3360 CrossRef CAS.
- T. Nishimura and S. Uemura, J. Am. Chem. Soc., 2000, 122, 12049 CrossRef CAS.
- X.-Y. Yu, J.-R. Chen, P.-Z. Wang, M.-N. Yang, D. Liang and W.-J. Xiao, Angew. Chem., Int. Ed., 2018, 57, 738 CrossRef CAS.
- X.-Y. Yu, Q.-Q. Zhao, J. Chen, J.-P. Chen and W.-J. Xiao, Angew. Chem., Int. Ed., 2018, 57, 15505 CrossRef CAS PubMed.
- J.-J. Zhang, X.-H. Duan, Y. Wu, J.-C. Yang and L.-N. Guo, Chem. Sci., 2019, 10, 161 RSC.
- T. Nishimura, Y. Nishiguchi, Y. Maeda and S. Uemura, J. Org. Chem., 2004, 69, 5342 CrossRef CAS.
- J.-X. Yu, F. Teng, J.-N. Xiang, W. Deng and J.-H. Li, Org. Lett., 2019, 21, 9434 CrossRef CAS PubMed.
- W.-Y. Ai, Y.-Q. Liu, Q. Wang, Z.-L. Lu and Q. Liu, Org. Lett., 2018, 20, 409 CrossRef CAS.
- Y.-Q. Tang, J.-C. Yang, L. Wang, M. Fan and L.-N. Guo, Org. Lett., 2019, 21, 5178 CrossRef CAS.
- W. Zhang, Y.-L. Pan, C. Yang, X. Li and B. Wang, Org. Chem. Front., 2019, 6, 2765 RSC.
- M. Murakami and N. Ishida, Chem. Rev., 2021, 121, 264 CrossRef CAS PubMed.
- J. Boivin, E. Fouquet and S. Z. Zard, J. Am. Chem. Soc., 1991, 113, 1055 CrossRef CAS.
- A.-N. R. Alba, X. Companyó and R. Rios, Chem. Soc. Rev., 2010, 39, 2018 RSC.
- D. Kaiser, I. Klose, R. Oost, J. Neuhaus and N. Maulide, Chem. Rev., 2019, 119, 8701 CrossRef CAS PubMed.
- D. Joseph, M. A. Idris, J. Chen and S. Lee, ACS Catal., 2021, 11, 4169 CrossRef CAS.
- A. S. Deeming, C. J. Russell and M. C. Willis, Angew. Chem., Int. Ed., 2016, 55, 747 CrossRef CAS PubMed.
- H. P. Wang, S. Sun and J. Cheng, Org. Lett., 2017, 19, 5844 CrossRef CAS PubMed.
- M. Wang, S. Chen and X. Jiang, Org. Lett., 2017, 19, 4916 CrossRef CAS.
- X. Gong, J. Chen, L. Lai, J. Cheng, J. Sun and J. Wu, Chem. Commun., 2018, 54, 11172 RSC.
- T. H. Zhu, X. C. Zhang, K. Zhao and T. P. Loh, Org. Chem. Front., 2019, 6, 94 RSC.
- Y. Liu, Q.-L. Wang, Z. Chen, P. Chen, K.-W. Tang, Q. Zhou and J. Xie, Org. Biomol. Chem., 2019, 17, 10020 RSC.
- Y. Li, T. Liu, G. Qiu and J. Wu, Adv. Synth. Catal., 2019, 361, 1154 CrossRef CAS.
- Y.-H. Lu, C. Wu, J.-C. Hou, Z.-L. Wu, M.-H. Zhou, X.-J. Huang and W.-M. He, ACS Catal., 2023, 13, 13071 CrossRef CAS.
- K. Sun, D. Zhao, Q. Li, S. Ni, G. Zheng and Q. Zhang, Sci. China: Chem., 2023, 66, 2309 CrossRef CAS.
- J. Zhang, H. Ma, C. Yang, J. Dang, J. Xia and J. Wu, Org. Lett., 2023, 25, 8043 CrossRef CAS.
- X. Gong, M. Wang, S. Ye and J. Wu, Org. Lett., 2019, 21, 1156 CrossRef CAS.
- B. Zhao and Z. Shi, Angew. Chem., Int. Ed., 2017, 56, 12727 CrossRef CAS.
- P.-Z. Wang, X.-Y. Yu, C.-Y. Li, B.-Q. He, J.-R. Chen and W.-J. Xiao, Chem. Commun., 2018, 54, 9925 RSC.
- J. Zhang, X. Li, W. Xie, S. Ye and J. Wu, Org. Lett., 2019, 21, 4950 CrossRef CAS PubMed.
- D. Anand, Y. He, L. Li and L. Zhou, Org. Biomol. Chem., 2019, 17, 533 RSC.
- N. Zhou, Q. Xu, Z. Xia, F. Zhao, L. Wang and M. Zhang, Adv. Synth. Catal., 2022, 364, 4020 CrossRef CAS.
- M.-X. Wang, Acc. Chem. Res., 2015, 48, 602 CrossRef CAS PubMed.
- X. Yang and F. F. Fleming, Acc. Chem. Res., 2017, 50, 2556 CrossRef CAS PubMed.
- Y. Liu, L. Wang, L.-H. Zeng, Y. Zhao, T. Zhu and J. Wu, Chin. Chem. Lett., 2022, 33, 2383 CrossRef CAS.
- D. Golagani, A. M. Ghouse, S. Ajmeeraa and S. M. Akondi, Org. Biomol. Chem., 2022, 20, 8599 RSC.
- A. B. Flynn and W. W. Ogilvie, Chem. Rev., 2007, 107, 4698 CrossRef CAS PubMed.
- E. Negishi, Z. Huang, G. Wang, S. Mohan, C. Wang and H. Hattori, Acc. Chem. Res., 2008, 41, 1474 CrossRef CAS.
- J. Zhang, M. Yang, J.-B. Liu, F.-S. He and J. Wu, Chem. Commun., 2020, 56, 3225 RSC.
- F.-S. He, Y. Yao, Z. Tang, Y. Qiu, W. Xie and J. Wu, Chem. Commun., 2021, 57, 12603 RSC.
- R. J. Reddy, A. H. Kumari and J. J. Kumar, Org. Biomol. Chem., 2021, 19, 3087 RSC.
- B. Dam, A. K. Sahoo and B. K. Patel, Green Chem., 2022, 24, 7122 RSC.
- C. Liu, Z. Liang, A. Jialingbieke, J. Gao and D. Du, Org. Lett., 2023, 25, 2657 CrossRef CAS PubMed.
- L.-B. Li, H. Qiu, M.-H. Li, N.-N. Dai, L. Mei, Y. He, L. Yu, T. Li and W.-T. Wei, Org. Chem. Front., 2023, 10, 5190 RSC.
- C.-M. Luo, Y.-B. Sun, L.-T. Wang, N.-N. Dai, J.-Z. Li, J. Zhang, J.-Y. Chen, W.-T. Wei and G.-P. Ge, Org. Chem. Front., 2023, 10, 4023 RSC.
- M.-Y. Chang and Y.-C. Cheng, Org. Lett., 2016, 18, 1682 CrossRef CAS PubMed.
- T. Markovic, P. R. D. Murray, B. N. Rocke, A. Shavnya, D. C. Blakemore and M. C. Willis, J. Am. Chem. Soc., 2018, 140, 15916 CrossRef CAS PubMed.
- B. Lin, J.-Q. Kuang, J.-J. Chen, Z.-G. Hua, V. Khakyzadeh and Y.-Z. Xia, Org. Chem. Front., 2019, 6, 2647 RSC.
- M. Zheng, G. Li and H. Lu, Org. Lett., 2019, 21, 1216 CrossRef CAS PubMed.
- Y. Liu, Q.-L. Wang, Z. Chen, H. Li, B.-Q. Xiong, P.-L. Zhang and K.-W. Tang, Chem. Commun., 2020, 56, 3011 RSC.
- Z. Chen, Q. Zhou, Q.-L. Wang, P. Chen, B.-Q. Xiong, Y. Liang, K.-W. Tang and Y. Liu, Adv. Synth. Catal., 2020, 362, 3004 CrossRef CAS.
- F. Teng, J. Du, C. Xun, M. Zhu, Z. Lu, H. Jiang, Y. Chen, Y. Li and Q.-W. Gui, Org. Biomol. Chem., 2021, 19, 8929 RSC.
- Y. Liu, Z. Chen, P. Chen, B.-Q. Xiong, J. Xie, A. Liu, Y. Liang and K.-W. Tang, Chin. J. Org. Chem., 2021, 41, 2290 CrossRef CAS.
- N. Zhou, Z. Xia, S. Wu, K. Kuang, Q. Xu and M. Zhang, J. Org. Chem., 2021, 86, 15253 CrossRef CAS PubMed.
- P. Chen, Z. Chen, B.-Q. Xiong, Y. Liang, K.-W. Tang, J. Xie and Y. Liu, Org. Biomol. Chem., 2021, 19, 3181 RSC.
- E. Adelin, M. T. Martin, M. F. Bricot, S. Cortial, P. Retailleau and J. Ouazzani, Phytochemistry, 2012, 84, 135 CrossRef CAS PubMed.
- X. Zheng, T. Zhong, X. Yi, Q. Shen, C. Yin, L. Zhang, J. Zhou, J. Chen and C. Yu, Adv. Synth. Catal., 2021, 363, 3359 CrossRef CAS.
- N. Zhou, S. Wu, K. Kuang, Z. Xia, Q. Xu and M. Zhang, Org. Chem. Front., 2021, 8, 6032 RSC.
- W. Kong, M. Casimiro, E. Merino and C. Nevado, J. Am. Chem. Soc., 2013, 135, 14480 CrossRef CAS PubMed.
- N. Fuentes, W. Kong, L. Fernández-Sánchez, E. Merino and C. Nevado, J. Am. Chem. Soc., 2015, 137, 964 CrossRef CAS PubMed.
- M. Li, C.-T. Wang, Q.-F. Bao, Y.-F. Qiu, W.-X. Wei, X.-S. Li, Y.-Z. Wang, Z. Zhang, J.-L. Wang and Y.-M. Liang, Org. Lett., 2021, 23, 751 CrossRef CAS PubMed.
- N. Zhou, Q. Xu, Z. Xia, K. Kuang, S. Wu, W. Li and M. Zhang, Chem. Commun., 2022, 58, 2335 RSC.
- C. Chatgilialoglu, D. Crich, M. Komatsu, I. Ryu, X.-F. Wu, H. Neumann and M. Beller, Chem. Rev., 2013, 113, 1 CrossRef PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075 CrossRef CAS PubMed.
- X.-F. Wu, X. Fang, L. Wu, R. Jackstell, H. Neumann and M. Beller, Acc. Chem. Res., 2014, 47, 1041 CrossRef CAS PubMed.
- Y. Chen, L.-Q. Lu, D.-G. Yu, C.-J. Zhu and W.-J. Xiao, Sci. China: Chem., 2019, 62, 24 CrossRef CAS.
- C.-C. Zhang, H.-L. Wu, X.-C. Yu, L.-T. Wang, Y. Zhou, Y.-B. Sun and W.-T. Wei, Org. Lett., 2023, 25, 5862 CrossRef CAS PubMed.
- J.-B. Peng, H.-Q. Geng and X.-F. Wu, Chem, 2019, 5, 526 CAS.
- R. Cheng, H.-Y. Zhao, S. Zhang and X.-G. Zhang, ACS Catal., 2020, 10, 36 CrossRef CAS.
- Y. Liu, Q.-L. Wang, C.-S. Zhou, B.-Q. Xiong, P.-L. Zhang, C.-A. Yang and K.-W. Tang, J. Org. Chem., 2018, 83, 4657 CrossRef CAS PubMed.
- Y. Liu, Z. Chen, Q.-L. Wang, P. Chen, J. Xie, B.-Q. Xiong, P.-L. Zhang and K.-W. Tang, J. Org. Chem., 2020, 85, 2385 CrossRef CAS PubMed.
- Y. K. Jang, M. Magre and M. Rueping, Org. Lett., 2019, 21, 8349 CrossRef CAS PubMed.
- S. Zhang and M. Beller, Chem. Commun., 2019, 55, 5938 RSC.
- Z. Yin, J. Rabeah, A. Brückner and X.-F. Wu, Org. Lett., 2019, 21, 1766 CrossRef CAS PubMed.
- B. Lu, Y. Cheng, L.-Y. Chen, J.-R. Chen and W.-J. Xiao, ACS Catal., 2019, 9, 8159 CrossRef CAS.
- Z. Yin, J. Rabeah, A. Brückner and X.-F. Wu, ACS Catal., 2018, 8, 10926 CrossRef CAS.
- J. Senger, J. Melesina, M. Marek, C. Romier, I. Oehme, O. Witt, W. Sippl and M. Jung, J. Med. Chem., 2016, 59, 1545 CrossRef CAS PubMed.
- D. Verga, C. H. N'Guyen, M. Dakir, J. L. Coll, M. P. Teulade-Fichou and A. Molla, J. Med. Chem., 2018, 61, 10502 CrossRef CAS PubMed.
- Z. Yin, J. F. Soulé, P. H. Dixneuf and X.-F. Wu, J. Catal., 2019, 372, 272 CrossRef CAS.
- J. Lou, Y. He, Y.-L. Li and Z.-K. Yu, Adv. Synth. Catal., 2019, 361, 3787 CrossRef CAS.
- Y.-F. Liang and N. Jiao, Acc. Chem. Res., 2017, 50, 1640 CrossRef CAS PubMed.
- Y. L. Tnay, G. Y. Ang and S. Chiba, Beilstein J. Org. Chem., 2015, 11, 1933 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |