
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBridging colloidal and electrochemical syntheses of metal nanocrystals with seeded electrodeposition for tracking single nanocrystal growth†
Ekta
Verma
a,
Myung-Hoon
Choi‡
b,
Nabojit
Kar
 a,
Lane A.
Baker
b and
Sara E.
Skrabalak
*a
a,
Lane A.
Baker
b and
Sara E.
Skrabalak
*a
aDepartment of Chemistry, Indiana University – Bloomington, Bloomington, Indiana 47405, USA. E-mail: sskrabal@indiana.edu
bDepartment of Chemistry, Texas A&M University, College Station, Texas 77843, USA
First published on 15th March 2024
Abstract
Metal nanocrystals (NCs) produced by colloidal synthesis have a variety of structural features, such as different planes, edges, and defects. Even from the best colloidal syntheses, these characteristics are distributed differently in each NC. This inherent heterogeneity can play a significant role in the properties displayed by NCs. This manuscript reports the use of electrochemistry to synthesize Au NCs in a system evaluated to track individual NC growth trajectories as a first step toward rapid identification of NCs with different structural features. Au nanocubes were prepared colloidally and deposited onto a glassy carbon electrode using either electrospray or an airbrush, resulting in well-spaced Au nanocubes. The Au nanocubes then served as seeds as gold salt was reduced to deposit metal at constant potential. Deposition at constant potential facilitates overgrowth on the Au nanocubes to achieve new NC shapes. The effects of applied potential, deposition time, precursor concentration, and capping agents on NC shape evolution were studied. The outcomes are correlated to results from traditional colloidal syntheses, providing a bridge between the two synthetic strategies. Moreover, scanning electron microscopy was used to image the same NCs before and after deposition, linking individual seed features to differences in deposition. This ability is anticipated to enable tracking of individual growth trajectories of NCs to elucidate sources of heterogeneity in NC syntheses.
1. Introduction
The synthesis of metal nanocrystals (NCs) with defined size and shape is critical to leveraging the properties that arise from their structures, particularly in the fields of plasmonics1 and catalysis.2 Colloidal approaches produce NCs with controlled sizes, shapes, and compositions; however, even the best syntheses produce NCs with some heterogeneity.3 That is, there may be slight variations in size, edge, or corner truncation from one NC to the next in the ensemble or even completely different shapes as minor products arising from different defects (e.g., twin planes). Also, heterogeneity may exist within an individual NC. For example, a metal nanocube may have corners with different degrees of truncation.This heterogeneity arises because the free energy landscape for NC formation offers many low-barrier pathways to closely related metastable structures.4 Yet, mechanistic understanding of these various pathways remains elusive, and such knowledge could inform strategies to achieve higher selectivity in NC syntheses. In this regard, strategies to track growth of individual NCs are required, and in situ liquid-cell transmission electron microscopy (TEM) is the state-of-the-art.5 This approach has been used to study the nucleation, growth, and assembly of metal NCs, revealing complex systems.6–8 For example, growth by both monomer addition and particle coalescence was observed during the synthesis of size-controlled quasi-spherical Pt nanoparticles, which contrasts with the commonly cited LaMer model that accounts for monodisperse nanoparticle formation.9,10 Yet, liquid-cell TEM approaches are limited by beam-induced effects and low throughput.
Here, we evaluate an electrochemical approach for its utility in tracking single NC growth and provide insight broadly into the electrochemical synthesis of NCs with defined structures. Unlike colloidal syntheses which require a molecular reducing agent to nucleate metal NCs from metal salts, electrochemical syntheses use a potentiostat to set the working electrode's potential where metal deposition occurs from metal salts, providing different kinetic pathways to metal NCs compared to colloidal syntheses.11,12 Interest in such syntheses grew when Sun and co-workers demonstrated electrochemical syntheses of Pt and Pd NCs with high-index facets by applying a square-wave potential to a glassy carbon electrode (GCE) with a metal salt growth solution.13–15 As we will show, electrochemical syntheses offer the possibility of tracking NCs as they are created or modified on a GCE exposed to the growth solution. To leverage this capability, however, understanding of different electrochemical parameters on NC shape-evolution must first be attained. Personick and co-workers systematically studied constant current conditions, translating a colloidal synthesis of corrugated Pd nanoparticles to a completely electrochemical synthesis.16 In an important parallel to studies here, their experimental design mimicked the depletion of molecular reducing agents during colloidal syntheses by using constant current conditions.
Nevertheless, constant potential conditions are attractive in electrochemical syntheses because the potential can be selected to match the chosen metal salts, providing a predictive pathway to metal deposition and eliminating the possibility for side reactions.17–19 Precise control of the applied potential also enables targeted deposition on specific facets of the NCs as these surface facets have close but distinct surface energies.20 Moreover, constant potential conditions may be better suited for conditions in which the surface area of the GCE changes, as is the case in metal NC synthesis where NC sizes increase during growth. Here, a systematic study of Au electrodeposition on Au nanocubic seeds was undertaken by considering different constant potential conditions. Au nanocubes were selected as seeds because their overgrowth is well-studied in colloidal syntheses, allowing for comparison to the electrochemical process.21–24 Also, the Au nanocubes orient on GCEs with a (100) facet preferentially parallel to the GCE, making it straightforward to correlate the outcomes from growth to the original seed features in single NC studies as the crystallography is defined.
2. Experimental
2.1. Synthesis of Au nanocube seeds
Synthesis of Au nanocube seeds. The synthesis of Au nanocube seeds proceeds by first preparing Au octahedra to which additional Au is added via a modified literature procedure.25 Briefly, an aqueous solution of 25 μL of HAuCl4·3H2O (100 mM) was added to 8.425 mL of nanopure water and 1.5 mL of CTAB (100 mM) with inversion of the capped reaction vial to facilitate mixing. Then, 50 μL of trisodium citrate (100 mM) was introduced, and the mixture was rapidly inverted once. The vial was heated at 110 °C in an oil bath overnight. Particles were collected by centrifugation (∼11
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g, 10 min), the supernatant decanted, and the pellet redispersed in 3 mL of nanopure water. The Au octahedral particle concentration was calibrated to achieve an absorbance of 0.415 at 400 nm.
000g, 10 min), the supernatant decanted, and the pellet redispersed in 3 mL of nanopure water. The Au octahedral particle concentration was calibrated to achieve an absorbance of 0.415 at 400 nm.
Au nanocubes then were grown from these Au octahedra. Specifically, 21.4 mL of nanopure water, 100 μL of HAuCl4·3H2O (100 mM), and 2 mL of CTAB (200 mM) aqueous solutions were added to a 30 mL reaction vial, and the contents were mixed by inversion. Then, 1.5 mL of freshly made L-ascorbic acid (100 mM) solution was added and the vial inverted for mixing. After, 1.0 mL of Au octahedra solution was added and mixed by inversion. The vial was placed in an oil bath set at 25 °C overnight (this action is taken to prevent crystallization of CTAB in laboratory settings that are slightly cooler). Particles were collected by centrifugation (∼7000g, 15 min), the supernatant was decanted, and the pellet was redispersed in 3 mL of nanopure water. The nanocube seed concentration was calibrated to achieve an absorbance of 0.553 at 400 nm for subsequent experiments.
2.2. Synthesis of sodium gold(I) sulfite
Synthesis of sodium gold(I) sulfite. The method developed by Dietz Jr. et al. was followed to synthesize sodium gold(I) sulfite.26 1 mg of HAuCl4·3H2O was added to 5 ml of nanopure water, and 88.6 mg of finely divided magnesium oxide was added. The pH of the solution was adjusted to 8.3 by adding 1 M HCl dropwise and stirred at 65 °C for 5 min. Further, the temperature was increased to 100 °C for 15 min. The color of the solution became orange, and it was allowed to cool for 2 min. The supernatant was removed after centrifugation of the solution at ∼7000g for 5 min. After centrifugation, the pellet was rinsed with water once, and transferred by spatula to a round-bottom flask containing 187.5 mg of sodium sulfite solution. The sodium sulfite solution with pellet was mixed with stirring and heated on a heating mantle at 74 °C for 10 min. The color of the above solution changed from orange to clear. After that, the solid was removed by centrifugation at ∼7000g for 5 min and the supernatant sodium gold(I) sulfite solution was collected and stored in a vial.
A fraction of the sodium gold(I) sulfite solution was concentrated by quantitative methods and digested in aqua regia to measure the Au content. The solution was analyzed from the liquid-phase on an Agilent 770 ICP-MS at the Indiana University Department of Earth and Atmospheric Sciences. ICP-MS measured an Au concentration of 82.92 μM for the sodium gold(I) sulfite solution.
2.3. Electrospray (ES) deposition of Au nanocubes
ES Deposition. Fig. S2† shows the schematic of the setup used. A procedure from the literature was used for the ES deposition.28 Theta pipette emitters with ∼1 micrometer-diameter tips at each barrel were filled with a solution of Au nanocubes by a microfil needle (World Precision Instruments, Sarasota, FL). The Au nanocube seed concentration was adjusted to achieve an absorbance of 0.165 at 400 nm. One barrel of the theta pipette was filled with the Au nanocube seed solution (500 μL), and an electrolyte solution of 2.5 mM KCl (500 μL) was filled in the second barrel. A platinum wire was introduced into the nanoparticle-filled barrel for an electrical connection. The potential was applied to the Pt wire using a high-voltage power supply from Gamma High Voltage Research (model no. SE30P-5W/DAM). To collect the current generated by the ES, the GCE was placed in electrical contact with a stainless-steel plate, and current was read via a 414A picoammeter (Keithley Instruments). A micromanipulator (Newport ULTRAlign 461-XYZ-M Linear Translation Stage with SM-13 micrometers) was used to accurately position the pipette over the GCE. To monitor position of the pipette tip relative to the GCE, a handheld wireless wi-fi digital microscope from Takmly monitored the live video of the GCE and the pipette emitter. A humidity hygrometer from Govee surrounded the GCE and pipette. During ES a probe–substrate distance of 2 mm, 14–18% relative humidity, and 8 nA of ES current were maintained. ES deposition was carried out for 20 minutes. The ES deposition using the referenced conditions resulted in the well-dispersed Au nanocubes on the GCE.
2.4. Airbrush deposition of nanocubes
Airbrush setup. A schematic of the airbrush used is shown in Fig. S3.† The fluid cup contains the Au nanocube solution with an absorbance of 0.034 at 400 nm (75 μL). The stem of the airbrush is connected to an Ar gas tank to apply pressure. The gas tank passes a stream of Ar gas through it at 10 psi, nebulizing it into a mist. The distance between the airbrush and GC was maintained at 6 mm, holding it in the tilted position of 45° and spraying for ∼3 s. Afterward, the airbrush was cleaned with ethanol between uses. These conditions give well-dispersed Au nanocubes on GCE.
2.5. Seeded electrochemical deposition of Au on Au nanocube seeds
Electrodeposition. Electrochemical measurements were performed using a Pine Instruments WaveDriver 10 Potentiostat (model AFP2). Constant potential electrolysis was performed for the deposition of Au on Au nanocubes. All macroscale measurements were performed in a three-electrode electrochemical cell under an Ar atmosphere and with constant stirring at 350 rpm (Fig. S4a†). The GCE was placed in a custom-made Teflon box to isolate a consistent geometric area of 3 mm for all electrolysis studies; a photograph and schematic of the Teflon box is shown in Fig. S4b and c.† The Au nanocube solutions (0.165 absorbance for ES and 0.034 for airbrush) were used to deposit the Au nanocubes on the GCE. The deposited Au nanocubes were used as seeds without further washing unless designated in the manuscript. For deposition studies, an electrochemical cell with volume of 13 mL was used to accommodate the Pt counter electrode and a Ag/AgCl (3 M NaCl) reference electrode (BASi). All potentials are reported referenced to Ag/AgCl (3 M NaCl). When designated, the Au nanocubes deposited on the GCE were cleaned by soaking the GCE in methanol for 2 min followed by two repetitive cyclic voltammograms carried out in 100 mM HClO4, sweeping from 0 V to −1.0 V vs. Ag/AgCl (3 M NaCl).
2.6. Characterization methods
Electron micrographs were acquired using either an FEI Quanta 600F Environmental SEM operating at an energy of 30 kV and spot size of 3 μm or a Zeiss Auriga focused-ion beam (FIB) in SEM mode with a beam energy of 20 kV and an aperture size of 30 μm. Size analysis was performed manually using ImageJ software. UV-visible spectroscopy was used to characterize the Au nanocube solutions and create solutions of standardized concentrations with a Varian CARY 100 Bio UV-Visible spectrophotometer. The scan rate was set at 600 nm min−1 over a wavelength range of 200–800 nm using disposable plastic cuvettes with a 1 cm path length, and water was used as the blank. TEM samples were prepared by sonicating the GCE in 1.0 mL of methanol for 10 min to release the NCs. The methanol solution was then drop-casted onto carbon-coated Cu TEM grids and the droplet was air-dried for subsequent TEM imaging. Transmission electron microscopy (TEM) images, selected area electron diffraction (SAED) and scanning TEM energy-dispersive X-ray spectroscopy (STEM-EDS) of nanostructures were collected using a JEOL JEM 3200FS microscope operating at 300 keV using a 4k × 4k GatanUltraScan 4000 CCD camera. XPS analysis of the NCs on the GCE was conducted on a PHI 5000 VersaProbe II instrument with a focused monochromatic Al Kα source. An X-ray power of 50 W at 15 keV under ultra-high-vacuum conditions and a beam size of 200 μm was used for all experiments. The C 1s peak from adventitious carbon was calibrated at 284.8 eV and served as an internal standard for the binding energy scale. Fourier transform infrared (FTIR) spectroscopy was taken of Au nanocube samples to characterize the ligands bound on their surfaces and to evaluate the effect of different washing times in removing these ligands. The samples were prepared by drop-casting the Au nanocube solution on a glass slide, which was then dried in air at room temperature. To evaluate the effect of different washing times, the glass slides with deposited Au nanocubes were soaked in methanol for 2 s, 30 s, 1 min or 2 min, followed by drying in air at room temperature prior to FTIR spectroscopy. FTIR spectra was obtained using a Bruker Vertex 70V FTIR spectrometer equipped with a mercury cadmium tellurium (MCT) detector in transmission mode.3. Results and discussion
3.1. ES versus airbrush deposition of Au nanocubes
Drop-casting is the most widely used method for the deposition of colloidal NCs on a substrate. Kumar et al. recently reviewed the effects of drop-casting on nanoparticle dispersions and noted that evaporation of solvent from immobile droplets containing suspended particles under ambient conditions leads to a ring-like pattern known colloquially as the coffee ring; unfortunately, the resulting nanoparticle deposits are irreproducible in terms of aggregation state and nanoparticle placement.29 Here, we observed similar outcomes with drop-casting of the Au nanocubes (Fig. 1a), where many of the Au nanocubes are parts of larger clusters of particles as evident from analyzing 12 representative SEM images (data not included) within the deposition region (Fig. 1b). Such clustering – whether from drop-casting or other deposition methods – is non-ideal as it could give rise to non-uniform overgrowth during electrodeposition due to the proximity of neighboring nanocubes. Also, the ability to track individual NCs in real-time by optical methods would be complicated by such clustering given the diffraction limit of such techniques. To follow the growth trajectories from single NC seeds and to achieve uniform outcomes by electrodeposition, the seeds need to be well-spaced and isolated from one another on the GCE. Thus, we also examined ES and airbrush methods for depositing nanocubes, with both methods resulting in well-spaced and isolated Au nanocubes on the GCE, as also shown in Fig. 1.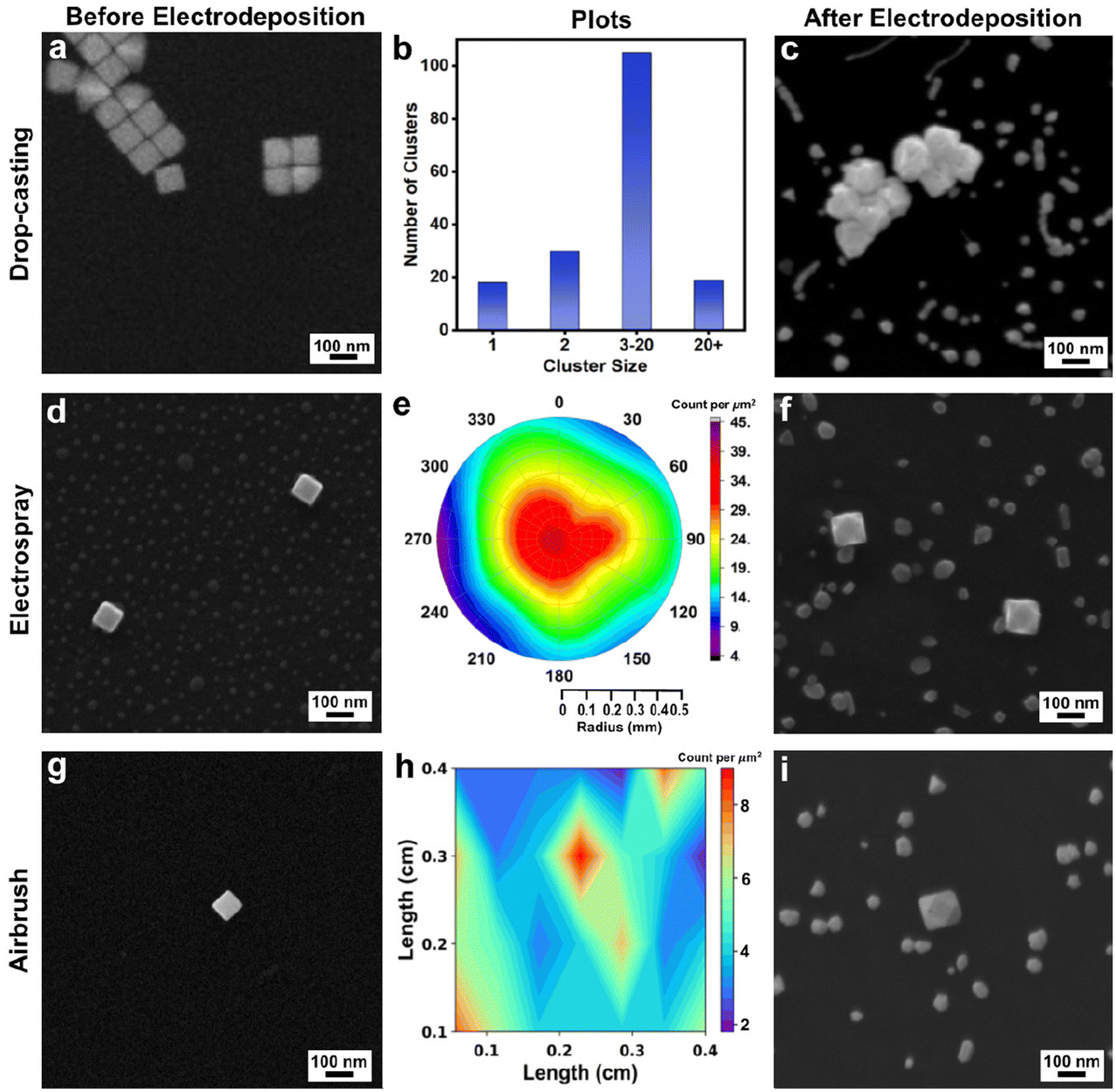 | ||
| Fig. 1 (a) SEM of drop-casted Au nanocubes. (b) Bar graph of cluster size versus number of clusters for Au nanocubes drop-casted on a GCE. (c) SEM of electrodeposited product obtained after constant potential electrolysis for 10 min (−1.175 V vs. Ag/AgCl) with drop-casted Au nanocubes. (d) SEM image of Au nanocubes deposited by ES. (e) Polar plot reporting the Au nanocubes distribution in the ES spot per square micrometer. (f) SEM of product obtained after constant potential electrolysis for 10 min (−1.175 V vs. Ag/AgCl) with electrosprayed Au nanocubes. (g) SEM of Au nanocubes deposited by airbrush. (h) Contour plot reporting the Au nanocube distribution in the airbrushed region per square micrometer. (i) SEM of product obtained after constant potential electrolysis for 10 min (−1.175 V vs. Ag/AgCl) with airbrushed Au nanocubes (all electrodeposition experiment used 3.5 μM Na3Au(SO3)2 with 0.32 M Na2SO3 electrolyte). Note that all SEM images were obtained at the same magnification. | ||
Considering ES deposition, we adapted a method reported by Jagdale et al. in which Au nano-octahedra were deposited on a GCE.28 They noted that the distance between the pipette and the GCE, the ES current, and the pipette diameter were key ES factors to achieve well-spaced, isolated nano-octahedra suitable for single-entity electrochemical studies. Similar optimization was required to achieve the well-spaced and isolated Au nanocubes reported here, which were co-deposited with KCl, the ES electrolyte (Fig. 1d and S5†). Notably, this salt can be removed by soaking the GCE in water after Au nanocube deposition.
Following the analysis protocol from Jagdale et al., the ES-deposited spots from three repeat experiments (n = 3) were investigated to evaluate the dispersion of Au nanocubes within the spray perimeter. SEM images were acquired from each ES-deposited spot at set positions (Fig. S5a;† ∼550 Au nanocubes counted per experiment from the SEM images). Data from one of the ES-deposited spots is shown in Fig. S5b† and the number of Au nanocubes per square micrometer was determined by analyzing the SEM images. These results were then used to create a polar plot (Fig. 1e) given the radius of the ES region and the image positions shown in Fig. S5† while interpolating the non-imaged regions. The number of particles deposited was greatest at the center of the ES spot and decreased gradually when moving away from the center to the perimeter of the ES spot. These results are similar to those previously reported for Au octahedra.28
Considering airbrush deposition of the Au nanocubes, no supporting electrolyte is required, which eliminates the need for GCE washing after deposition of the Au nanocubes. Like ES deposition, well-spaced and isolated single Au nanocubes were observed from airbrush deposition with uniform distribution (Fig. 1g and S6†). The airbrush-deposited spots from three repeat experiments (n = 3) were investigated to evaluate the dispersion of Au nanocubes within the spray areas. SEM images were acquired from each airbrush-deposited spot at set positions, as indicated in Fig. S6a† (∼100 Au nanocubes counted per experiment from the SEM images, as the density of particles is lower from airbrush deposition compared to ES). Data from one of the airbrush-deposited spots is shown in Fig. S6b.† The images were taken from the center of the area, moving outward from the center in all four directions. These particles were counted per square micrometer, and the results were then used to create a contour plot (Fig. 1h) based on image position and interpolating the non-imaged regions. Note that in comparison to ES, the airbrush method results in a deposition area that is more closely approximated as a square or rectangle, thus accounting for the differences in plotting conventions. Notably, the nanocubes appear stochastically distributed over the GCE, without much variation in particle density.
As both ES and airbrush deposition produced well-spaced and isolated Au nanocubes on the GCE, these two methods can be used to deposit the seeds for electrodeposition. To test the feasibility for seeded electrodeposition, Au nanocube seeds were deposited on GCEs by drop-casting, ES, or airbrush. To carry out electrodeposition, a given nanocube-coated GCE was immersed in a solution of 3.5 μM Na3Au(SO3)2 with 0.32 M Na2SO3 supporting electrolyte. For initial experiments, a constant potential of −1.175 V vs. Ag/AgCl was selected (with all potentials herein vs. Ag/AgCl). The selection of these conditions and precursor is discussed in the ESI (see Fig. S7†) and note that the Au nanocubes were used as seeds without any protocol to remove residual CTAB that may remain on their surfaces from synthesis. After electrodeposition for 10 minutes, each sample was investigated with SEM. Structurally varied nanoparticles were observed in the case of drop-casted nanocubes and is consistent with the varied local electrochemical environments that arise from nanoparticle aggregation (Fig. 1c). In contrast, cuboctahedra were observed in both the case of nanocubes deposited by ES and airbrush, with their edge lengths larger than the initial Au nanocubes (Fig. 1f and i). The GCE also has smaller metal deposits of random shape, which will be discussed later.
In both the case of ES and airbrush-deposited Au nanocubes, this electrodeposition experiment was repeated four times to check its reproducibility, as shown in Fig. S8a and S8b.† Cuboctahedra that are larger than the initial Au nanocubes were obtained in all cases, with the increase in average edge length varying between ∼26 nm and ∼48 nm depending on the specific experiment. This variation likely arises from the slight variations in average seed size, a common source of heterogeneity in colloidal NC systems. As similar products were obtained regardless of whether the Au nanocubes were deposited on the GCE by ES or airbrush, the airbrush method is used in subsequent experiments given its compatibility with smaller solution volumes, simpler experimental setup, and the elimination of the GCE washing step.
3.2. Effect of applied potential, deposition time and capping agents on the shape evolution of Au nanocubes during seeded electrodeposition
Heterogeneous nucleation of Au on the GCE, alongside the Au seeds, poses a challenge in distinguishing products. That is, under certain deposition conditions, Au deposits on the GCE may adopt morphologies like the Au seeds or overgrown seeds. Also, in optical studies, distinguishing between Au nanocubes and heterogeneous nucleation on a GCE would become difficult given the increasing particle density on the GCE, yet such distinction would be crucial for in situ studies and correlation of structural information to NC properties. Therefore, heterogeneous nucleation on the GCE should be minimized or eliminated to correlate growth to deposition conditions through single-particle studies.Au deposition on both the GCE and Au nanocubic seeds indicates that the barrier to nucleation is being overcome in both cases with an applied potential of −1.175 V. This observation is surprising as the barrier to nucleation is anticipated to be lower on Au nanocubes than on GCE given the possibility for homoepitaxy.30 Thus, the effects of Au precursor concentration and applied potential on the shape evolution of Au nanocubes during seeded electrodeposition were systematically studied.
These reaction conditions were selected for study as we hypothesized that nucleation on the GCE could be eliminated by decreasing supersaturation. Supersaturation refers to when a solute in a solution exceeds the concentration defined by its solubility at equilibrium and governs the initial stage of crystal formation.31,32 According to classical nucleation theory,33,34 the free energy needed for homogeneous nucleation can be described as a function of local supersaturation as follows:
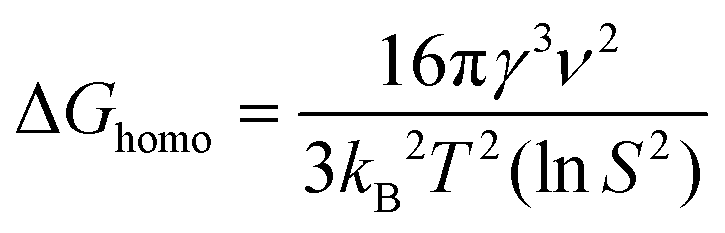 | (1) |
| ΔGhetero = ΔGhomof(θ) | (2) |
Thus, there are two factors from eqn (1) and (2) that can be leveraged to achieve selectivity in nucleation: S and f(θ). When S < 1, the free energy for heterogeneous nucleation will be lower than that for homogeneous nucleation. The wetting factor f(θ) refers to the degree to which a liquid can spread over a solid surface. f(θ) is lower in the case of homoepitaxy (Au–Au bond formation) than the Au-GC interaction, resulting in a lower barrier for heterogeneous nucleation on the Au nanocubes (assuming no residual capping agents on their surfaces) and a higher barrier for heterogeneous nucleation on the GC.36 In this way, the different barriers to nucleation on GCE and the Au nanocubes can be more readily leveraged at low supersaturation.
In colloidal syntheses of NCs, the most common levers to tune supersaturation are reaction temperature, precursor concentration, and strength of the molecular reducing agent. Given reactor differences between colloidal and electrochemical syntheses, we chose not to examine temperature effects. Concentration effects are straightforward to evaluate, where the concentration of the Na3Au(SO3)2 was decreased from 3.5 μM to 0.35 μM, yielding lower supersaturation at an applied potential of −1.175 V. Similar results (i.e., cuboctahedra) were obtained after 10 min of electrodeposition at −1.175 V (Fig. S9†).
Applying a more positive potential during electrodeposition is analogous to switching to a weaker molecular reducing agent; however, we now have the benefit of not changing the solution speciation as is the case with molecular reducing agents. Instead, a range of applied potentials can be readily screened. Therefore, the applied potential was systematically moved more positive, and the products imaged by SEM after 10 min of constant potential electrolysis (in 0.35 μM Na3Au(SO3)2 with 0.32 M Na2SO3 electrolyte; Fig. 2). It is important to note that the electrodeposition solution, which is in contact with the working electrode, is stirred during these experiments, just as traditional colloidal syntheses are stirred. As anticipated, when a potential of −1.075 V was applied, less nucleation on the GCE was observed compared to a reduction potential of −1.175 V (Fig. 2a). Significantly, the Au nanocubes have increased in size (∼120 nm in edge length versus ∼80 nm edge length before deposition; Fig. S10a and b†) and maintain their cubic profile (Fig. 2a). On applying a potential of −0.875 V, heterogeneous nucleation on the GCE is even less; slight deposition at the corners of the Au nanocubes was also observed (Fig. 2b and Fig. S11†). An applied potential of −0.775 V led to Au deposition largely on the corners of the Au nanocubes, and heterogeneous nucleation of Au on the GCE was not observed (Fig. 2c and Fig. S12†). Interestingly, at applied potentials of −0.675 V and −0.575 V, minimal (or possibly no) Au deposition was observed as the Au nanocubes did not increase in size substantially (Fig. 2d, e and Fig. S13a and b†).
 | ||
| Fig. 2 SEM of products from seeded electrodeposition on Au nanocubes deposited by airbrush on a GCE. The horizontal axis is the deposition potential vs. Ag/AgCl. The vertical axis is the deposition time: (a–e) 10 min, (f–j) 15 min, and (k–o) 10 min with removal of CTAB from seeds. All experiments used 0.35 μM Na3Au(SO3)2 with 0.32 M Na2SO3 electrolyte. All scale bars are the same. | ||
The effect of deposition time on final NC shape was also studied by increasing the electrodeposition time from 10 min to 15 min, with all other conditions held constant. At −1.075 V, Au cuboctahedra with edge lengths longer than the initial Au nanocubes were observed along with nucleation on the GCE (Fig. 2f and Fig. S14†). This observation contrasts with the results at 10 min, at which point the NCs had maintained a cubic shape. However, nucleation on the GCE was still observed. At −0.875 V and −0.775 V, 15 min of deposition led to metal deposition on the Au nanocubes (Fig. 2g and h). These results are similar to the same experiments at 10 min (Fig. 2b and c) with more deposition at the corners of the Au nanocubes than the edge. Size analyses for both experiments show an increase in both the edge and the diagonal lengths after electrodeposition compared to both the original seeds as shown in Fig. S15.† At −0.675 V, deposition occurred on Au nanocubes as they increased in size by ∼10 nm (Fig. 2i and S16a†) and −0.575 V, no change in shape and deposition was observed on Au nanocubes (Fig. 2j and S16b†).
Fig. 3a and b include plots of change in average NC edge length and diagonal after 10 and 15 minutes of electrodeposition, respectively, determined from analysis of the size histograms in Fig. S10–S16.† Notably, the largest increases in NC size are observed at an applied potential of −0.775 V for 10 and 15 minutes, aligning with when heterogeneous nucleation on the GCE is suppressed. This finding suggests that heterogeneous nucleation on the GCE is dominating at more negative potentials, depleting growth species from the Au nanocubic seeds. Notably, for both 10 and 15 minutes, the increase in diagonal length is much greater than edge length, consistent with Au deposition at the seed corners dominating; slightly larger NCs produced in the case of 15 minutes on account of the greater electrodeposition time. Growth stops at more positive potentials as there is insufficient driving force for precursor reduction in the presence of Au nanocubes with residual CTAB capping.
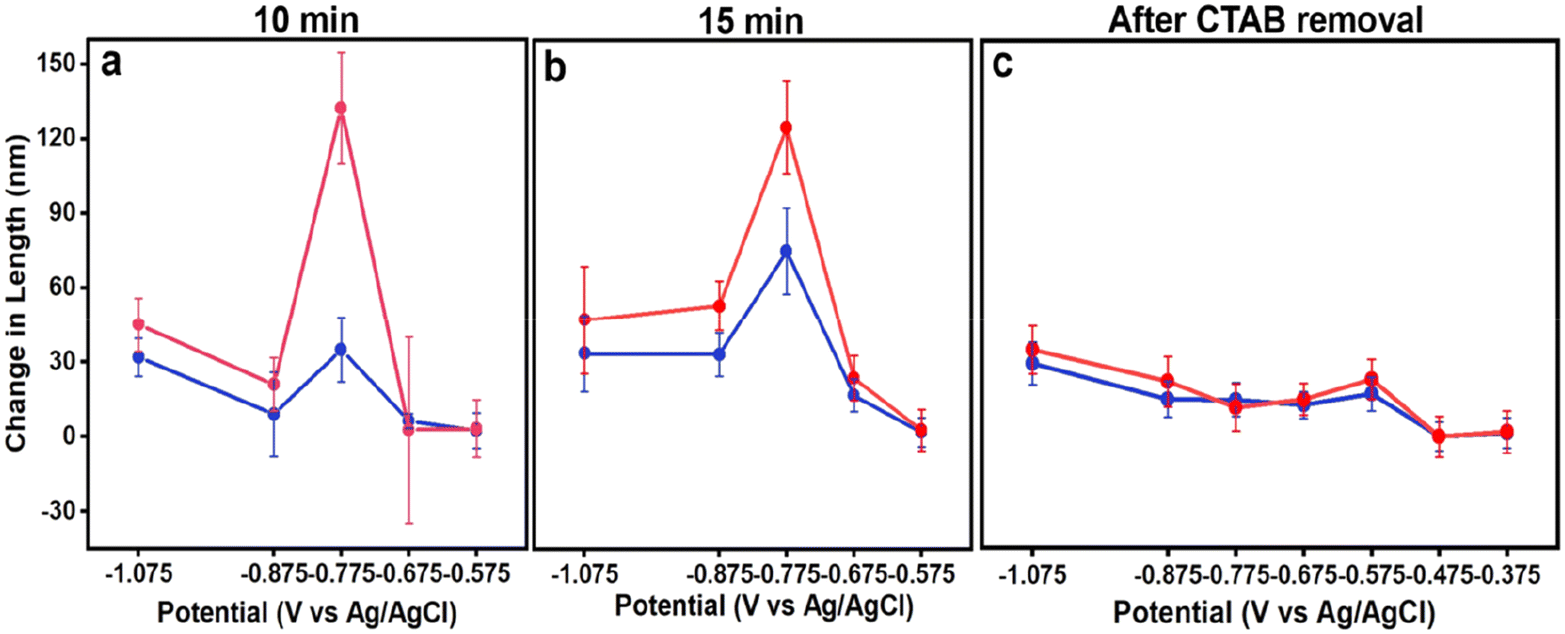 | ||
| Fig. 3 Plots of potential versus change in edge (blue traces) and diagonal (pink traces) length of NCs after electrodeposition (a) for 10 min with CTAB-capped seeds, (b) 15 min with CTAB-capped seeds, and (c) 10 minutes with CTAB removed seeds. All experiments with 0.35 μM Na3Au(SO3)2 with 0.32 M Na2SO3 electrolyte. | ||
Considering the shape evolution observed as a function of applied potential, some parallels between colloidal and electrochemical syntheses can be drawn. First, as supersaturation decreases, heterogeneous nucleation becomes more selective and occurs on preferred surfaces, i.e., the Au seeds rather than GCE; similar effects occur in colloidal syntheses with nucleation occurring on seeds rather than through homogeneous nucleation at lower supersaturation, and even distinguishing between different seed features to induce anisotropy at very low supersaturation.30,37 Second, longer growth periods result in larger NCs when the number of growing NCs is held constant. Third, capping effects of molecules adsorbing to the NC seeds appears to be influencing morphology development.38
This third conclusion arises from considering the Thomson–Gibbs equation, which outlines that the surface energy of crystallites should increase with an increase in supersaturation if no new nuclei form.32 The Au nanocubes with deposition selectively at their corners undoubtedly have higher surface energy than the polyhedral Au forms that form under more reducing/higher supersaturation conditions. This observation is in apparent contradiction unless other factors are contributing to morphological development. In our system, the Au nanocubes were synthesized with CTAB as a capping agent, where bromide stabilizes {100} facets.39 While the Au nanocubes were collected by centrifugation prior to airbrush deposition to remove excess CTAB, FTIR spectroscopy reveals C–H stretching vibrations characteristic of CTAB (Fig. S17†). Selective passivation of the Au nanocube {100} facets, i.e., faces, increases the barrier for nucleation on the faces relative to the unpassivated corners. Thus, as a more positive potential is applied (lower supersaturation), deposition occurs at the sites of highest surface energy (lowest barrier to nucleation), i.e., the corners, to produce the Au nanocubes with selectively overgrown corners (Fig. 2c and h). This capping effect was experimentally tested and is discussed shortly, but this capping effect is augmented by the low precursor concentration which also favors a more regioselective product.36
Capping agents have been shown to influence the size and morphology of products from electrodeposition.40–43 Different adsorbing anions during electrodeposition has facilitated the formation of nanoparticles with precisely controlled sizes. For example, Mastai et al. showed that strongly adsorbing anions such as chloride resulted in smaller nanoparticles than nonadsorbing anions such as perchlorate.40 Additionally, the binding of capping agent to specific facets of NCs can lead to their anisotropic growth. For example, Skibińska et al. found that chloride ions during electrodeposition promoted growth of conically nanostructured nickel layers.43
Considering the role of CTAB in our research, its removal should facilitate greater deposition on the faces of the Au nanocubes and lead to different NC products. To test this hypothesis, the Au nanocubes were further cleaned by a protocol from the literature.27 Specifically, the Au nanocubes deposited on the GCE by the airbrush method were soaked in methanol for 2 min, with excess CTAB dissolving into the methanol. The soaking period was selected such that the majority of the CTAB was removed from the Au nanocubes as revealed by FTIR spectroscopy (Fig. S17†). However, due to the detection limit of FTIR, a trace amount of CTAB may remain on the Au nanocubes. Further, CV cycling was conducted in 100 mM HClO4 to remove additional CTAB from the Au nanocubes as was reported by Choi et al.44
All the potentials applied previously were tested to evaluate the effect of the CTAB cleaning procedure on Au electrodeposition. When a potential of −1.175 V was applied for 10 min (3.5 μM Na3Au(SO3)2 with 0.32 M Na2SO3 electrolyte), a similar electrodeposited product (i.e., larger cuboctahedra along with heterogeneous nucleation of new particles on the GCE) was observed compared to that produced without CTAB removal (Fig. S18†). At an applied potential of −1.075 V for 10 min (in 0.35 μM Na3Au(SO3)2 with 0.32 M Na2SO3 electrolyte), deposition on the {100} facets of the Au nanocubes is evident, along with slight deposition on the {111} facets (Fig. 2k), with the size histogram analysis showing the growth of nanocubes after electrodeposition (Fig. S19a and b†). Similar results were obtained at a potential of −0.875 V and −0.775 V applied for 10 min (Fig. 2l and m), with the size analysis for both indicative of growth (Fig. S19c–f†). Further, slight deposition occurred on the Au nanocubes at applied potentials of −0.675 V and −0.575 V presented in SEM image Fig. 2n and o, and size analysis histograms show that at these potentials Au nanocubes are growing slightly (Fig. S19g–j†). Only by moving to more positive potentials (−0.475 V and −0.375 V) was growth suppressed (Fig. S20 and S21†). The change in morphology observed with removal of CTAB is consistent with lowering the barrier to deposition on the {100} facets of the Au nanocubic seeds. Notably, if the CTAB is only removed partially prior to electrodeposition, then nonuniform deposition is observed that is often characterized by deposition at only a few of the eight vertices of the cubic seeds (Fig. S22†). These findings emphasize the selective capping behavior of CTAB during overgrowth processes, whether they be initiated with molecular reducing agents or electrochemistry.
Fig. 3c includes plots of change in average NC edge length and diagonal after 10 minutes of electrodeposition from Au nanocubes from which CTAB has been removed. The increase in length was greatest at −1.075 V. Growth was still observed until an applied potential of −0.475 V but was less in magnitude, consistent with the decreasing supersaturation with applied potential. While heterogeneous nucleation on the GCE is evident at the most negative potentials and would deplete growth species, its impact on seeded electrodeposition is less as a large increase in NC size is not observed after its suppression. This observation is consistent with an increase in electrochemically active surface area from the Au nanocubes after CTAB removal.
3.3. Mechanistic summary
The morphology development of Au NCs during seeded electrodeposition depends on the applied potential and capping agents. These outcomes correlate with local supersaturation, providing connections to classical nucleation theory and colloidal syntheses of metal NCs. These outcomes also shed insight into the common conditions used in electrochemical syntheses of metal NCs. In this manuscript, constant potential conditions were employed, whereas the overwhelming number of literature reports use either constant current density45–47 or an application of a square wave potential48–50 for size and shape-controlled electrochemical synthesis of NCs. In considering constant current density conditions, the potentiostat changes the cell potential to maintain constant current, which may account for why nucleation on the working electrode is often limited to the initial stages of synthesis. With application of a square wave, often conditions sweep between reducing and oxidizing. During oxidation, the highest energy (likely smallest and most newly formed) metal deposits would be etched at a greater rate, and this quality has likely been leveraged to mitigate heterogeneous nucleation of new NCs on the working electrode.3.4. Single-particle correlation and compositional analysis
With understanding of different experimental parameters on NC morphology during seeded electrodeposition, we then sought to verify the feasibility for single-particle correlation before and after electrodeposition based on pattern matching. That is, the Au nanocubes deposited on the GCE by airbrush create distinct arrangements akin to constellations that can be used to locate the same region for imaging before and after electrodeposition. Shown in Fig. 4 are SEM images from the same region before and after electrodeposition. Fig. 4a shows the SEM image of the Au nanocubes before electrodeposition, and Fig. 4b shows the electrodeposited product obtained after 10 min at −0.775 V in 0.35 μM Na3Au(SO3)2 with 0.32 M Na2SO3 electrolyte. We note that this sample is the same as shown in Fig. 2c and analyzed in Fig. S12.†Fig. 4b and c present high magnification SEM images of the corresponding Au nanocubes shown in Fig. 4a. Similarly, Fig. 4e and f show high magnification SEM images of the electrodeposited product, as depicted in Fig. 4d. Matching the specific Au nanocube seeds to final NCs was facile given the pattern they create on the GCE. | ||
| Fig. 4 SEM images of isolated NCs (a–c) before electrodeposition and (d–f) after electrodeposition, where (b and c) are high magnification images of the Au nanocubes denoted in red and blue in (a) and (e and f) are high magnification images of the product NCs denoted in red and blue in (d). The electrodeposited product was obtained at −0.775 V vs. Ag/AgCl for 10 min in 0.35 μM Na3Au(SO3)2 with 0.32 M Na2SO3 electrolyte. | ||
This workflow is significant as now the slight variations from one NC seed to the next can be identified and potentially correlated to a specific final NC morphology. For example, the top Au nanocube identified in the blue box in Fig. 4a exhibits corner truncation as evident from the higher magnification image in Fig. 4b. The product NC (Fig. 4e) shows spatially diffuse deposition at that corner compared to the other corners. This outcome may be a result of intraparticle heterogeneity; however, a statistically relevant number of NCs would need to be studied to gain such insight.
To confirm that deposition takes place on the bottom edges of the Au nanocubes where they interface with the GCE, SEM imaging of product NCs was conducted at a tilt of 54° as shown for cuboctahedra (−1.175 V for 10 min in 3.5 μM Na3Au(SO3)2 with 0.32 M Na2SO3; Fig. S23a†) and corner-deposited Au nanocubes (−0.775 V for 10 min in 0.35 μM Na3Au(SO3)2 with 0.32 M Na2SO3; Fig. S23b†). For the cuboctahedra, the top and bottom edges of the NC appear similar in length; for the corner-deposited Au nanocubes, arrows are added to the image to guide the viewer to seven regions of deposition, with the eighth corner out of view. Further, the composition of the product obtained at −0.775 V for 10 min (in 0.35 μM Na3Au(SO3)2 with 0.32 M Na2SO3) was characterized, with STEM-EDS elemental mapping and line scan analysis showing largely Au with some amount of S and Na (Fig. 5a–f). SAED supports a face-centered cubic Au crystal structure, with the NC analyzed oriented along the [00![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ] zone axis (Fig. 5g). The XPS survey spectrum is consistent with the elemental information from EDS (Fig. S24†), with high-resolution spectra of the Au 4f, S 2p, and Na 1s regions characteristic of metallic Au, sulfite, and Na+, respectively (Fig. S25; see ESI† for full discussion). The Na:S from XPS is ∼1.5:1, suggesting the presence of Na and S arises from residual electrolyte adsorbed to the Au NC surfaces.
] zone axis (Fig. 5g). The XPS survey spectrum is consistent with the elemental information from EDS (Fig. S24†), with high-resolution spectra of the Au 4f, S 2p, and Na 1s regions characteristic of metallic Au, sulfite, and Na+, respectively (Fig. S25; see ESI† for full discussion). The Na:S from XPS is ∼1.5:1, suggesting the presence of Na and S arises from residual electrolyte adsorbed to the Au NC surfaces.
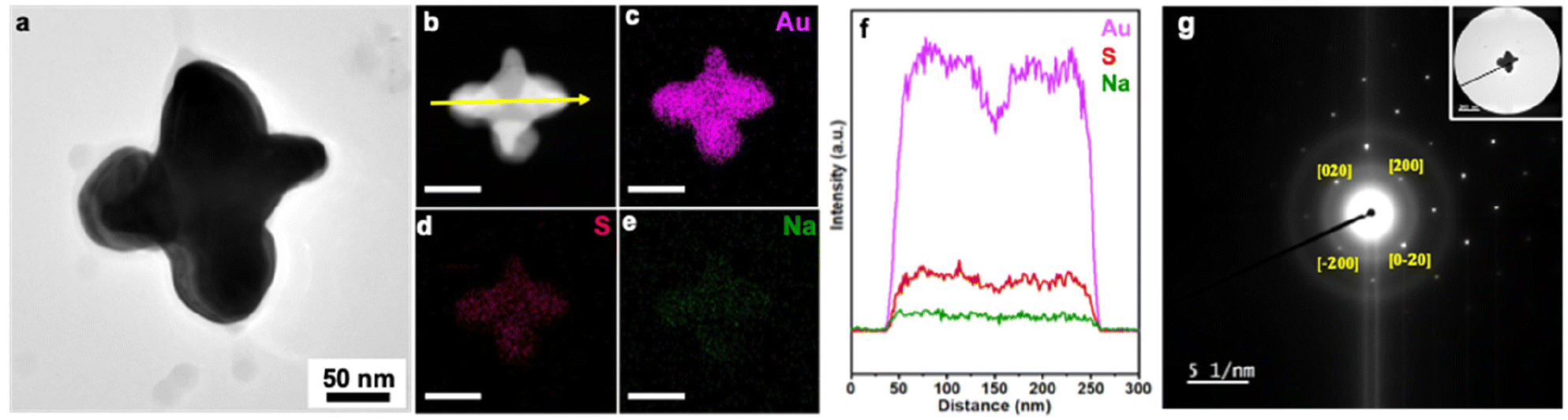 | ||
| Fig. 5 (a) TEM image and (b–e) STEM image and elemental mapping by STEM-EDS of corner deposited Au NCs (scale bar = 100 nm). (f) Line scan analysis by STEM-EDS following the yellow arrow in (b). (g) SAED pattern of single particle along the [00] direction. This sample was obtained from electrodeposition of 0.35 μM Na3Au(SO3)2 with 0.32 M Na2SO3 electrolyte at −0.775 V vs. Ag/AgCl, with Au nanocubes deposited on a GCE by airbrush. | ||
4. Conclusion
Electrochemistry offers a versatile synthetic strategy toward metal NCs with defined shape, with constant potential methods offering a straightforward way to leverage the known reduction potentials of common metal precursors. Moreover, control of potential in electrochemistry enables deposition on specific facets of NCs, where Au NCs with sharp features can be useful platforms for surface-enhanced Raman scattering and plasmon-based sensing applications on account of near-field enhancements.51,52 Notably, when coupling electrodeposition with seeded methods, single-NC tracking becomes possible through colocalization of the initial seeds and final NC products, much in the manner that identical location electron microscopy has advanced understanding of nanoparticle electrocatalysts.53,54 While the current level of development only reveals initial and final states through imaging by electron microscopy, we envision that high-throughput optical characterization methods may be coupled with seeded electrodeposition to provide information on a large number of NCs, including in situ tracking of growth processes. Thus, this demonstration marks a critical step toward elucidating the origins of heterogeneity in NC ensembles.Author contributions
E. V., S. E. S. and L. A. B. were responsible for the project concept and design of experiments. E. V. developed the seeded electrochemical deposition of Au on Au nanocubes, and studied the effect of applied potential, deposition time and capping agent on shape evolution of Au nanocubes and characterized the electrochemical products. M. C. helped in pulling pipettes for ES and N. K. helped with elemental characterization via some STEM-EDS maps. This manuscript was written through the contributions of all authors. All authors have given approval to the final version of this manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
S. E. S., E. V., M. C., N. K. and L. A. B. acknowledge financial support from the National Science Foundation Center for Single-Entity Nanochemsitry and Nanocrystal Design (CHE 2221062) and Indiana University. The authors acknowledge support from Indiana University's Electron Microscopy Center and Nanoscale Characterization Facility for access to instrumentation. They also thanks D. Morgan, Y. Losovyj, A. Dwivedi, M. Knobeloch, and G. Jagdale for their helpful discussions as well as Dr. Amanda J. Haes for inspring the airbrush deposition method.References
- L. Wang, M. H. Kafshgari and M. Meunier, Adv. Funct. Mater., 2020, 30, 2005400 CrossRef CAS.
- H. L. K. S. Stolle, J. J. Kluitmann, A. Csáki, J. M. Köhler and W. Fritzsche, Catalysts, 2021, 11, 1442 CrossRef CAS.
- Q. Chen, Nano Lett., 2022, 22, 3–5 CrossRef CAS PubMed.
- Y. Xia, X. Xia and H.-C. Peng, J. Am. Chem. Soc., 2015, 137, 7947–7966 CrossRef CAS PubMed.
- T. Ngo and H. Yang, J. Phys. Chem. Lett., 2015, 6, 5051–5061 CrossRef CAS PubMed.
- J. Wu, H. Shan, W. Chen, X. Gu, P. Tao, C. Song, W. Shang and T. Deng, Adv. Mater., 2016, 28, 9686–9712 CrossRef CAS PubMed.
- Q. Chen, J. M. Yuk, M. R. Hauwiller, J. Park, K. S. Dae, J. S. Kim and A. P. Alivisatos, MRS Bull., 2020, 45, 713–726 CrossRef.
- B. Yuan and L. Cademartiri, Angew. Chem., Int. Ed., 2021, 60, 6667–6672 CrossRef CAS PubMed.
- H. Zheng, R. K. Smith, Y. Jun, C. Kisielowski, U. Dahmen and A. P. Alivisatos, Science, 2009, 324, 1309–1312 CrossRef CAS PubMed.
- B. Abécassis, M. W. Greenberg, V. Bal, B. M. McMurtry, M. P. Campos, L. Guillemeney, B. Mahler, S. Prevost, L. Sharpnack, M. P. Hendricks, D. DeRosha, E. Bennett, N. Saenz, B. Peters and J. S. Owen, Chem. Sci., 2022, 13, 4977–4983 RSC.
- T. S. Rodrigues, M. Zhao, T.-H. Yang, K. D. Gilroy, A. G. M. da Silva, P. H. C. Camargo and Y. Xia, Chem. – Eur. J., 2018, 24, 16944–16963 CrossRef CAS PubMed.
- S. P. McDarby and M. L. Personick, ChemNanoMat, 2022, 8, e202100472 CrossRef CAS.
- J. Xiao, S. Liu, N. Tian, Z.-Y. Zhou, H.-X. Liu, B.-B. Xu and S.-G. Sun, J. Am. Chem. Soc., 2013, 135, 18754–18757 CrossRef CAS PubMed.
- N. Tian, Z.-Y. Zhou, N.-F. Yu, L.-Y. Wang and S.-G. Sun, J. Am. Chem. Soc., 2010, 132, 7580–7581 CrossRef CAS PubMed.
- N. Tian, Z.-Y. Zhou and S.-G. Sun, J. Phys. Chem. C, 2008, 112, 19801–19817 CrossRef CAS.
- S. P. McDarby, C. J. Wang, M. E. King and M. L. Personick, J. Am. Chem. Soc., 2020, 142, 21322–21335 CrossRef CAS PubMed.
- A. Hickling, Trans. Faraday Soc., 1942, 38, 27–33 RSC.
- J. J. Lingane, C. G. Swain and M. Fields, J. Am. Chem. Soc., 1943, 65, 1348–1353 CrossRef CAS.
- C. Zhu, N. W. J. Ang, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Cent. Sci., 2021, 7, 415–431 CrossRef CAS PubMed.
- N. Mayet, K. Servat, K. B. Kokoh and T. W. Napporn, Surfaces, 2019, 2, 257–276 CrossRef CAS.
- M. R. Langille, M. L. Personick, J. Zhang and C. A. Mirkin, J. Am. Chem. Soc., 2012, 134, 14542–14554 CrossRef CAS PubMed.
- J. Xiao and L. Qi, Nanoscale, 2011, 3, 1383 RSC.
- K. Sohn, F. Kim, K. C. Pradel, J. Wu, Y. Peng, F. Zhou and J. Huang, ACS Nano, 2009, 3, 2191–2198 CrossRef CAS PubMed.
- W. Niu, S. Zheng, D. Wang, X. Liu, H. Li, S. Han, J. Chen, Z. Tang and G. Xu, J. Am. Chem. Soc., 2009, 131, 697–703 CrossRef CAS PubMed.
- C.-C. Chang, H.-L. Wu, C.-H. Kuo and M. H. Huang, Chem. Mater., 2008, 20, 7570–7574 CrossRef CAS.
- G. Dietz Jr., R. M. Skomoroski and R. G. Zobbi, United States, US3966880A, 1976 Search PubMed.
- M. Choi, N. P. Siepser, S. Jeong, Y. Wang, G. Jagdale, X. Ye and L. A. Baker, Nano Lett., 2020, 20, 1233–1239 CrossRef CAS PubMed.
- G. S. Jagdale, M.-H. Choi, N. P. Siepser, S. Jeong, Y. Wang, R. X. Skalla, K. Huang, X. Ye and L. A. Baker, Anal. Methods, 2021, 13, 4105–4113 RSC.
- A. K. S. Kumar, Y. Zhang, D. Li and R. G. Compton, Electrochem. Commun., 2020, 121, 106867 CrossRef.
- Y. Xia, K. D. Gilroy, H.-C. Peng and X. Xia, Angew. Chem., Int. Ed., 2017, 56, 60–95 CrossRef CAS PubMed.
- Y. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS PubMed.
- H. Lin, Z. Lei, Z. Jiang, C. Hou, D. Liu, M. Xu, Z. Tian and Z. Xie, J. Am. Chem. Soc., 2013, 135, 9311–9314 CrossRef CAS PubMed.
- N. T. K. Thanh, N. Maclean and S. Mahiddine, Chem. Rev., 2014, 114, 7610–7630 CrossRef CAS PubMed.
- S. G. Kwon and T. Hyeon, Small, 2011, 7, 2685–2702 CrossRef CAS PubMed.
- S. L. Brock, J. Am. Chem. Soc., 2004, 126, 14679–14679 CrossRef CAS.
- A. N. Chen, M. M. Scanlan and S. E. Skrabalak, ACS Nano, 2017, 11, 12624–12631 CrossRef CAS PubMed.
- X. Y. Liu, J. Chem. Phys., 2000, 112, 9949–9955 CrossRef CAS.
- T.-H. Yang, Y. Shi, A. Janssen and Y. Xia, Angew. Chem., Int. Ed., 2020, 59, 15378–15401 CrossRef CAS PubMed.
- A. Ruditskiy and Y. Xia, J. Am. Chem. Soc., 2016, 138, 3161–3167 CrossRef CAS PubMed.
- Y. Mastai, D. Gal and G. Hodes, J. Electrochem. Soc., 2000, 147, 1435 CrossRef CAS.
- J. M. Lee, K. K. Jung, S. H. Lee and J. S. Ko, Appl. Surf. Sci., 2016, 369, 163–169 CrossRef CAS.
- L. Xu, Y. Guo, Q. Liao, J. Zhang and D. Xu, J. Phys. Chem. B, 2005, 109, 13519–13522 CrossRef CAS PubMed.
- K. Skibińska, M. Huang, G. Mutschke, K. Eckert, G. Włoch, M. Wojnicki and P. Żabiński, J. Electroanal. Chem., 2022, 904, 115935 CrossRef.
- M.-H. Choi, S. Jeong, Y. Wang, S.-J. Cho, S.-I. Park, X. Ye and L. A. Baker, Langmuir, 2021, 37, 7701–7711 CrossRef CAS PubMed.
- F. Pagnanelli, P. Altimari, M. Bellagamba, G. Granata, E. Moscardini, P. G. Schiavi and L. Toro, Electrochim. Acta, 2015, 155, 228–235 CrossRef CAS.
- G.-Q. Yuan, H.-F. Jiang, C. Lin and S.-J. Liao, J. Cryst. Growth, 2007, 303, 400–406 CrossRef CAS.
- N. S. Qu, D. Zhu, K. C. Chan and W. N. Lei, Surf. Coat. Technol., 2003, 168, 123–128 CrossRef CAS.
- X. Liu, K. Wang, L. Zhou, H. Pu, T. Zhang, J. Jia and Y. Deng, ACS Sustainable Chem. Eng., 2020, 8, 6449–6457 CrossRef CAS.
- K. Huang, J. Clausmeyer, L. Luo, K. Jarvis and R. M. Crooks, Faraday Discuss., 2018, 210, 267–280 RSC.
- N. Tian, Z.-Y. Zhou, S.-G. Sun, Y. Ding and Z. L. Wang, Science, 2007, 316, 732–735 CrossRef CAS PubMed.
- Y. Hua, K. Chandra, D. H. M. Dam, G. P. Wiederrecht and T. W. Odom, J. Phys. Chem. Lett., 2015, 6, 4904–4908 CrossRef CAS PubMed.
- I. B. Becerril-Castro, I. Calderon, N. Pazos-Perez, L. Guerrini, F. Schulz, N. Feliu, I. Chakraborty, V. Giannini, W. J. Parak and R. A. Alvarez-Puebla, Analysis Sensing., 2022, 2, e202200005 CrossRef CAS.
- N. Hodnik and S. Cherevko, Curr. Opin. Electrochem., 2019, 15, 73–82 CrossRef CAS.
- M. Zorko, B. Jozinović, M. Bele, N. Hodnik and M. Gaberšček, Ultramicroscopy, 2014, 140, 44–50 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Schematic diagrams, low magnification SEM images, size histograms, FTIR and XPS spectras. See DOI: https://doi.org/10.1039/d4nr00202d |
| ‡ Current Affiliation: Research Application Center, Park Systems Inc., Santa Clara, California, 95054, USA. |
| This journal is © The Royal Society of Chemistry 2024 |