
Bifunctional Au–Sn-SiO2 catalysts promote the direct upgrading of glycerol to methyl lactate†
Margot
Van der Verren
 a,
Anna
Corrias
b,
Vit
Vykoukal
c,
Ales
Styskalik
c,
Carmela
Aprile
d and
Damien P.
Debecker
*a
a,
Anna
Corrias
b,
Vit
Vykoukal
c,
Ales
Styskalik
c,
Carmela
Aprile
d and
Damien P.
Debecker
*a
aInstitute of Condensed Matter and Nanoscience (IMCN), UCLouvain, Place Louis Pasteur 1, 1348 Louvain-La-Neuve, Belgium. E-mail: damien.debecker@uclouvain.be
bUniversity of Kent, School of Chemistry and Forensic Science, Ingram Building, Canterbury CT2 NH, UK
cMasaryk University, Department of Chemistry, Kotlarska 2, CZ-61137 Brno, Czech Republic
dUniversité de Namur, Unit of Nanomaterial Chemistry, Department of Chemistry, Namur 5000, Belgium
First published on 27th March 2024
Abstract
Valuable alkyl lactates can be obtained from (waste) glycerol, through a two-step process that entails (i) the oxidation of glycerol to dihydroxyacetone (DHA) catalyzed by support Au nanoparticles and (ii) a rearrangement of DHA with an alcohol effectively catalyzed by Sn-based heterogeneous catalysts. To solve selectivity and processing issues we propose to run the process as a cascade reaction, in one step, and with a single bifunctional catalyst. Tackling the challenge associated with the preparation of such bifunctional catalysts, here, an aerosol-assisted sol–gel route is exploited. The catalysts feature small Au nanoparticles (3–4 nm) embedded at the surface of mesoporous Sn-doped silica microspheres. The preparation successfully leads to insert both active sites in their most active forms, and in close proximity. With the bifunctional catalysts, the yield for the final product of the cascade reaction (methyl lactate) is higher than the DHA yield when only the first reaction is carried out. This highlights a beneficial substrate channeling effect which alleviates side reactions. Interestingly, the bifunctional catalysts also markedly outcompeted mechanical mixtures of the corresponding monofunctional Au- and Sn-based catalysts. Thus, the spatial proximity between the two active sites in bifunctional catalysts is identified as a key to stir the cascade reaction towards high lactate yield.
1. Introduction
Transitioning away from the fossil era will require the sustainable exploitation of abundant (waste) biomass.1,2 In the up-and-coming biorefinery schemes, the upgrading of bio-based compounds into valuable chemicals mostly relies on multistep process, where separate reactions are carried out in different reactors, using different catalysts. In this context, the design of multi-functional nano-catalysts able to perform intensified cascade reactions can pave the way to more sustainable processes.Glycerol is the main by-product (10 wt%) of the biodiesel production via the transesterification of vegetable oils. With the increasing demand for biodiesel, glycerol is being supplied in excess on the market, so that its value has been dropping; it is essential to develop methods to upgrade glycerol into value-added chemicals.3,4 As a platform chemical, glycerol can be converted through redox reactions, esterification, or dehydration to a plethora of industrially attractive compounds.5–7 Among the possible products of glycerol upgrading, lactic acid (LA) and its ester derivatives (alkyl lactates, AL) are standing out. Lactates can be used as green solvents in the cosmetics and as bulk chemicals in various industries. They are also used as building block chemicals for pharmaceutical and healthcare formulations, or as raw materials for the synthesis of bioplastics such as polylactic acids.8 The latter application represents a billion dollars market.8 Lactic acid is currently produced by microbial fermentation. Although it offers eco-friendly synthesis conditions, such biological processes are characterized by low product yield, lack of stability, long processing time, costly purification, and relatively large amount of waste salts produced.1 Thus, robust chemical pathways to synthesize alkyl lactates from (waste) bio-based sources have to be developed, and glycerol is an attractive starting point.
The upgrading of glycerol to lactates is typically achieved in two steps: (1) glycerol is oxidized to dihydroxyacetone (DHA) and (2) DHA is further transformed via a rearrangement either to lactic acid (when working in water) or to alkyl lactate (when working in an alcohol).9 In the first reaction, controlling the level of glycerol oxidation to selectively produce the targeted triose (DHA) represents a challenge, as this compound is prone to further oxidation (Scheme 1).10 Any lack of selectivity in the first part of the process would impose transitional purification steps and/or strongly impair the overall lactate yield in the two-stages process.11
 | ||
| Scheme 1 Transformation of glycerol to methyl lactate via the formation of dihydroxyacetone (DHA), with the possible side products from the overoxidation of DHA in grey. | ||
The oxidation of glycerol was originally performed under basic conditions,12,13 but base-free processes using molecular oxygen as the oxidant and Pt, Pd or Au-based catalysts have also been developed.14,15 The reaction conditions (temperature, reactant concentration, pressure), the nature of the noble metal and of the support can greatly influence the selectivity toward DHA.16 Gold was identified as the best metal to perform the oxidation of glycerol.17 Gold nanoparticles below 10 nm were identified as active.18,19 We recently developed Au-SiO2 catalysts featuring highly dispersed small gold nanoparticles (∼3.7 nm) that perform well in glycerol oxidation in water (59% conversion with a selectivity toward DHA of 30%).20
The catalytic conversion of DHA to alkyl lactates involves acid catalysis. Mechanistic studies were performed and showed that the first step of the reaction requires Brønsted acid sites to catalyze the dehydration of the primary hydroxyl group to form pyruvic aldehyde (PA).21–23 The second step is a combination of two reactions: a nucleophilic addition of the alcohol to the carbonyl carbon atom of the aldehyde and a 1,2-hydride shift via an internal Cannizzarro reaction. This step is catalyzed by Lewis acid sites and leads to the formation of methyl lactate if methanol is used (or lactic acid if working in water).21 Hayashi and Sasaki investigated homogeneous catalysts for the conversion of trioses to alkyl lactates and showed that tin compounds (SnCl2 and SnCl4·5H2O) were the most active,24 for tin exhibits high Lewis acidity compared to other transition metals.25 Based on these results, and aiming at recyclable catalysts, many tin-containing heterogeneous catalysts were developed. Among them, tin-containing zeolites (such as Sn-USY,26 Sn-Beta,27 SnFAU22) showed excellent performance. However, their intricate synthesis and the limited mass transfer in the micropores hamper their effective implementation. It was found that mesoporous materials bearing Lewis acid sites such as Sn-MCM-41 can also be very active and selective in the conversion of DHA to alkyl lactates.28,29 Recently, another type of mesoporous Sn-SiO2 catalyst was disclosed by us.30 Being prepared in one step, using an aerosol-assisted sol–gel process, these tin silicates showed excellent catalytic performance (i.e. yield up to 60%, fully recyclable) in the conversion of dihydroxyacetone to ethyl lactate.
Converting glycerol to lactates via a one-pot cascade reaction is highly attractive, as it may allow boosting the selectivity of the process towards the desired product. Yet only a few research groups have addressed this possibility.9,11,31–33 Pescarmona et al. demonstrated that a mechanical mixture of Au/CuO and Sn-MCM-41-XS could reach a 95% glycerol conversion with a ML yield up to 62% in 10.5 h.11 Similarly, Zhou et al. worked with Au/CuO and Snβ zeolites to obtain similar results – glycerol conversion of 86% and ML yield of 60%.33 Moving further, Tang et al. emphasized that the selectivity of the first step of the cascade reaction should be targeted to increase the overall yields and focused on the improvement of the noble metal catalyst. They reported the use of bimetallic Au–Pd nanoparticles supported carbon nanotubes as catalysts for the oxidation step of glycerol to glyceraldehyde or dihydroxyacetone.9 Combining with Sn-MCM-41-XS, they managed to increase the catalytic performance to a 96% glycerol conversion and 85% ML yield by using a bimetallic catalyst – Au–Pd nanoparticles on carbon nanotubes.
Moving forward, efforts are made to further intensify the processes, by developing multifunctional catalysts, able to run cascade reactions in a unique reactor.34 A strong boost in catalytic performance was sometimes observed with multifunctional catalysts due to the synergistic effect brought by the active site proximity.35–39 Specifically, to perform a cascade reaction such as the transformation of glycerol to ML, effective bifunctional catalysts would be highly desirable because the proximity between the two active sites may favor the rapid channeling of the unstable intermediate (DHA) towards completion of the second step of the cascade reaction thus decreasing the possibility of side reactions. In this way, higher overall yields could be obtained.
However, the synthesis of such materials can be challenging as they usually rely on multistep synthesis processes in which the addition of an active species on a preformed (active) material can modify its morphology or texture, affect the nature of chemical surface species, and hinder the availability of catalytic sites. Indeed, a bifunctional Au/Sn-MCM-41-XS was shown to reach lower ML yields as compared to the corresponding physical mixture Au/CuO and Sn-MCM-41-XS.11 In this example, the process of gold deposition–precipitation onto Sn-MCM-41-XS, and more precisely the basic conditions used to induce the precipitation of gold, resulted in the partial dissolution of the Sn-based catalyst, responsible for a marked decrease in Lewis acidity and activity. In another example, colloidal gold was immobilized on a tin-exchanged montmorillonite but this resulted in relatively large gold nanoparticles and in a decrease in the acidity of the material, leading to modest ML yields.31 On the other hand, an intricate multi-step process was recently reported to prepare an Au/Snβ@mesosilica composite featuring a sandwich-like structure to avoid gold sintering while maintaining the acidity of the zeolites; this material reached high ML yields (but very low specific productivity). As demonstrated through these examples, the synthesis of effective bifunctional catalysts is challenging.
In this work, we propose a one-pot strategy, relying on the aerosol-assisted sol–gel process, for the preparation of a new type of bifunctional heterogeneous catalysts for the upgrading of glycerol to methyl lactate. The aerosol process provides easy access to nanostructured heterogeneous catalysts with tailored properties (texture, composition, dispersion, surface functionality) in a limited number of preparation steps.40,41 Various catalysts including porous oxides, metal and alloys or even hybrid catalysts were synthesized using this technique.42–52 It relies on the rapid thermally induced evaporation of a sprayed solution containing the different precursors (alkoxides or salts) and a templating agent to form dry particles with homogeneous composition and controlled porosity (Scheme S1†). The catalysts developed in this work consist of small Au nanoparticles highly dispersed on a Sn–Si mixed oxide. We demonstrate how combining the two active sites on the same solid particles allows us to boost the overall catalytic performance of the cascade and to lower the formation of over-oxidized by-products.
2. Experimental
2.1 Materials
Tetraethyl orthosilicate (TEOS, >97%), dodecane (>99%), (3-mercaptopropyl)-trimethoxysilane (MPTMS, >96%) and 1,3-dihydroxyacetone dimer (DHA, >96%) were purchased from TCI. Methanol (HPLC Grade, 99.8+%) was purchased from Alfa Aesar. Methyl DL-lactate (ML, >97%), Pluronic® F-127, hydrochloric acid (37%) and tin(IV) chloride pentahydrate (SnCl4·5H2O, 98%) were all purchased from Sigma-Aldrich. Glycerol (Pure, 99%+) and chloroauric acid trihydrate (HAuCl4·3H2O, ACS reagent) were purchased from Acros Organics. Ethanol absolute (ACS reagent) was purchased from VWR Chemicals. n-Hexane (ACS reagent) and phenylethyl mercaptan (PEM, >99%) were purchased from Merck.2.2 Catalysts preparations
For the synthesis of the mesoporous Sn-SiO2 catalyst,30,53 solution A was prepared by mixing 12 g of TEOS with 20 g of acidified water (HCl, 0.01 M). In a second vessel, 3.88 g of Pluronic F127 was dissolved in 45 g of absolute ethanol and 8 g of deionized water (solution B). The two solutions were kept overnight under stirring at room temperature. Then, tin(IV) chloride (SnCl4·5H2O) was added to solution A in order to have a nominal Si/Sn ratio equal to 74 (here obtained with 273 mg of Sn precursor). The resulting solution was stirred for 10 minutes and then added to solution B and stirred for another 30 minutes. The mixture was atomized with a 6-Jet 9306A atomizer from TSI (operating at 30 psi) using compressed air and the droplets formed were dried by passing through a tubular quartz tube heated at 350 °C. The powder was recovered on a cellulose nitrate filter (pore size: 0.45 μm) then dried at 80 °C overnight. The resulting sample was calcined under static air at 550 °C for 6 h (1 °C min−1) and is here denoted SnSiO2.The synthesis of the Au-SiO2 catalyst featuring small gold nanoparticles is based on the stabilization of the gold precursors by MPTMS.20 Solution A was prepared by mixing TEOS and MPTMS in a 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 molar ratio, with 20 g of acidified water (HCl, 0.01 M) while solution B was prepared as explained above. The two solutions were kept overnight under stirring at room temperature. Then, chloroauric acid trihydrate (HAuCl4·3H2O, 69 mg) was added to solution A so as to reach a nominal loading of 1% (w/w) and stirred for 10 minutes. Solutions A and B were mixed and further stirred for another 30 minutes. The mixture was then was atomized as explained above and the recovered powder was calcined in the same conditions. The resulting catalyst is denoted AuSiO2-MPTMS. Note that the addition of MPTMS is the key to control the formation of small gold nanoparticles (in the absence of MPTMS, large Au particles form readily during aerosol processing).20
:5 molar ratio, with 20 g of acidified water (HCl, 0.01 M) while solution B was prepared as explained above. The two solutions were kept overnight under stirring at room temperature. Then, chloroauric acid trihydrate (HAuCl4·3H2O, 69 mg) was added to solution A so as to reach a nominal loading of 1% (w/w) and stirred for 10 minutes. Solutions A and B were mixed and further stirred for another 30 minutes. The mixture was then was atomized as explained above and the recovered powder was calcined in the same conditions. The resulting catalyst is denoted AuSiO2-MPTMS. Note that the addition of MPTMS is the key to control the formation of small gold nanoparticles (in the absence of MPTMS, large Au particles form readily during aerosol processing).20
Bifunctional catalysts were synthesized combining the syntheses described above. Solution B is the same. In solution A, three gold loadings were investigated: 0.5, 1 or 2% (w/w). The gold precursor was added to the pre-hydrolyzed silica precursors (MPTMS and TEOS) (solution A), stirred for 10 minutes. Then, tin precursors (Si/Sn = 74) was added and the solution was stirred for another 10 minutes. Solutions A and B were mixed and further stirred for 30 minutes. Then the mixture is atomized as explained above and the recovered powder is calcined in the same conditions. The catalysts are named Au(x)SnSiO2, where x corresponds to the gold loading.
2.3 Characterization
Nitrogen physisorption analyses were carried out at 77.4 K using a Tristar 3000 (Micromeritics, USA) instrument to determine the textural properties of the different samples. Prior to analysis, the samples were degassed overnight at 200 °C. The specific surface area was determined by the Brunauer–Emmett–Teller (BET) method in the 0.05–0.30 relative pressure range. The total pore volume (Vp) was measured at p/p0 = 0.98. The pore size distribution was obtained from the adsorption part of the isotherm using the Barrett–Joyner–Halenda (BJH) method. The microporous specific surface area was evaluated by the t-plot method in the thickness range of 3.5 to 5.0 Å.The Au and Sn content of the synthesized materials were measured by inductively coupled plasma atomic emission spectroscopy (ICP-OES) on an ICP 6500 instrument (Thermo Scientific Instrument) after dissolution of the samples by peroxide fusion.
The crystallite size of metal nanoparticles was determined by X-ray diffractometry (XRD) using the Scherrer equation. The diffraction patterns were measured at room temperature with a Bruker-D8 Advance diffractometer using Cu Kα radiation (Bragg–Brentano geometry) operated at 40 kV and 30 mA. Diffractograms were taken from 5° to 100° (2θ) with a step size of 0.05° (2θ) and a time step of 1.5 seconds. The resulting data were processed using Winplotr to integrate each peak, determine the full width at half maximum, and to calculate the nanocrystallite size via Scherrer equation.
X-ray photoelectron spectroscopy (XPS) experiments were carried out with an SSX 100/206 photoelectrons spectrometer (Surface Science Instruments, USA) equipped with a monochromatized Al-Kα radiation operated at 10 kV and 20 mA. The calibration of the binding energy scale was performed on the Si 2p peak at 103.5 eV.54 The quantification of Au was based on the 4f7/2 peak (84.3 eV)55 and the one of Sn was based on the 3d5/2 peak (486.5 eV). The data obtained were processed with CasaXPS from Casa Software Ltd, UK.
The TEM images of the sample containing gold (mono- and bimetallic) were obtained with a transmission electron microscope FEI Tecnai F20 (FEI, Eindhoven, The Netherlands) equipped with a field emission gun source, a 4k CCD camera (FEI Eagle) and operated at 200 kV was used for sample visualization (collaboration with the Department of Chemistry, Masaryk University). STEM-EDS measurements were performed on a FEI Titan Themis instrument with a combination of a spherical aberration image (Cs) corrector, a monochromator system, sensitive ChemiSTEM technology, and a high-end GATAN GIF Quantum energy filter for EELS and EFTEM with a new enhanced piezo stage, FEI and GATAN software, and a FEI Ceta 16-megapixel CMOS camera. The TEM images of the tin catalyst (SnSiO2) were obtained using a Philips Tecnai 10 microscope operating at 80 kV.
Diffuse reflectance UV/Vis spectra were measured for the tin-containing powders with a UV-vis spectrophotometer (UV-3600i Plus, Shimadzu) from λ = 200 nm to λ = 500 nm.
The dispersion of gold (defined as the proportion of Au atoms available on the surface) was measured by a thiol titration with a UV-vis spectrophotometer (UV-3600i Plus, Shimadzu) with a protocol adapted from the literature.56 Briefly, the dispersion measurement is based on the difference of absorption of PEM (240–270 nm) in presence of the catalyst. 10 mL of 0.1 mM PEM in hexane is prepared to measure the initial spectra. 50 mg of catalyst is then added and the mixture is stirred for 30 minutes in order to allow the PEM to adsorb on gold sites. The solution is separated from the solid by centrifugation and analyzed by UV-Vis. The absorbance at 273 nm is subtracted from the absorbance at 258 nm to determine the concentration of PEM in solution.56 It is assumed that one mole of PEM adsorbs on one mole of Au. The baseline is measured beforehand with a solution of hexane and the catalyst in the same proportion used during the measurement.
The coordination of Sn atoms was measured by static 119Sn nuclear magnetic resonance (NMR). The spectra were recorded at room temperature on a Varian VNMRS-400 spectrometer operating at 9.4 T using a 5 mm wideline probe. The sample was packed in a 5 mm glass tube and studied in static condition.
Acidity was studied by pyridine adsorption. The FTIR spectra were recorded on a Bruker Equinox 55 spectrometer (transmission mode), performing 256 scans from 4000 to 500 cm−1 with a resolution of 4 cm−1. Catalysts were pelleted and degassed for 2 hours at 350 °C. The pyridine adsorption was carried out at room temperature for 30 minutes. After stabilization, the pyridine was desorbed at 150 °C for 2 hours. The amount of Lewis acid sites was calculated based on the following equation:

X-ray absorption spectra at the Au L3-edge were recorded on the B18 beamline at the DIAMOND synchrotron (Oxfordshire, UK). The Au(1)SnSiO2 sample, in form of powder, was diluted with polyvinylpyrrolidone (PVP) and pressed to form a pellet. Due to the low concentration of absorbing atom the data were collected in fluorescence mode at room temperature, using a Si(311) monochromator. The monochromator energy scale was calibrated using a Au reference foil. The data of the sample were collected simultaneously to those of the foil, placed after the fluorescent detector, measuring the transmitted intensity via an ionization chamber. The data analysis was performed using the ATHENA and ARTEMIS software.58 With ATHENA, the absorption edge, E0, was determined, and the absorption due to the isolated atom was subtracted, by fitting the pre-edge and post-edge regions, to obtain the EXAFS interference functions, χ(k), and the normalized XANES spectra. The Fourier transforms of the EXAFS interference functions were corrected for phase shift, using the phase shifts calculated using ARTEMIS.
2.4 Catalytic studies
The catalytic transformation of glycerol to either DHA (with AuSiO2) or to methyl lactate (with the bifunctional catalyst, AuSnSiO2) were performed in a 160 mL Parr stainless steel autoclave reactor equipped with a Teflon liner. In a typical experiment, a solution of glycerol in methanol (0.125 M, 20 mL) was loaded with 68 mg of dodecane (used as GC internal standard) and a chosen amount of catalysts. The reaction was performed under 15 bar of air (used as oxidant) under vigorous stirring at the desired temperature. After a selected time, the reactor was cooled down in a cold bath and the reaction mixture was centrifuged to separate the catalyst. The solution was analyzed by gas chromatography (Scion Instruments 456-GC) equipped with a Restek Stabilwax column (30-meter length, 0.53 mm ID, 0.25 μm df) and an FID detector. Each component was calibrated with pure chemicals using 7 concentration points. Reusability tests were performed keeping a glycerol/catalyst (w/w ratio) = 1.15 during the different cycles. The reaction was stopped after 2 h. After each cycle, the catalyst was recovered by centrifugation, washed with methanol three times, dried overnight at 40 °C under vacuum and then calcined at 450 °C (1 °C min−1) under flowing air (100 mL min−1). The leaching test was performed under the same reaction conditions. After 1 h, the catalyst was recovered by centrifugation and the supernatant was then allowed to react for 7 h. The glycerol conversion was evaluated by GC after removing the catalyst (1 h) and at the end of the test (8 h).The catalytic tests for the conversion of dihydroxyacetone to methyl lactate were performed in the same reactor. In a typical experiment, 360 mg of dihydroxyacetone was dissolved in 20 ml of methanol with 68 mg of dodecane at 45 °C for 15 minutes. Then the clear solution was loaded in the reactor with 200 mg of catalyst. The reaction was performed in the same conditions described above. After 4 hours of reaction, the reactor was cooled down in a cold bath and the catalyst was separated by centrifugation. The solution was analyzed by GC.
3. Results and discussion
3.1. Mono-functional AuSiO2 and SnSiO2 catalysts
To achieve a good dispersion of Au nanoparticles onto the mesoporous silica material, we relied on the aerosol-assisted sol–gel process and on the strong interactions between gold and the thiol function of a mercapto-silane.20 AuSiO2 catalyst consists of mesoporous microspheres with calibrated mesoporosity (5–7 nm) and homogeneously dispersed Au nanoparticles of about 3–5 nm in size (Fig. 1A–C). The pores result from the templating effect of F127 micelles, which are then removed by calcination. | ||
| Fig. 1 Transmission electron microscopy analysis of (A–C) AuSiO2 and (D) SnSiO2. From left to right: HAADF, STEM-EDX, STEM. | ||
Textural properties were investigated by N2 physisorption. The type IV isotherm with H2 hysteresis loop demonstrated the presence of ink-bottle neck mesopores (Fig. 2A). Overall, the catalyst displayed advantageous textural properties (SSA = 440 m2 g−1; Vp = 0.47 cm3 g−1; Dp = 5 nm) (Table 1). The gold content was verified by inductively couple plasma optical emission spectroscopy (ICP-OES) and was very close to the nominal loading (= 1%) (Table 2). The surface composition was measured by XPS. No sulfur was detected after calcination, proving that the calcination applied to remove the surfactant also leads to the decomposition of mercaptopropyl moieties. XPS analyses showed that the gold present in the sample was in metallic Au0 state. Au 4f7/2 was shifted to relatively low binding energy (∼83 eV) (Fig. S1†). This corresponds to surface Au atoms and is explained by the low coordination number of surface atoms in small nanoparticles.55 This suggests the formation of very small gold nanoparticles in AuSiO2. The crystallite size was estimated by X-ray diffraction, applying the Scherrer equation on the 2θ = 38.1° peak (Fig. 2B). This sample displayed 3.7 nm diameter crystallite (this measure has systematically a standard error of ±0.1 nm), consistent with TEM (Fig. 1) and XPS that pointed to small Au nanoparticles. In good agreement with the N2 physisorption analysis, TEM shows the presence of 5–7 nm interconnected pores. The gold dispersion measured by thiol titration reached a value of 21%, which is fully consistent with ∼3 nm sized Au nanoparticles. It confirms Au is both well dispersed and available – in the pores of the microspheres – for catalysis.
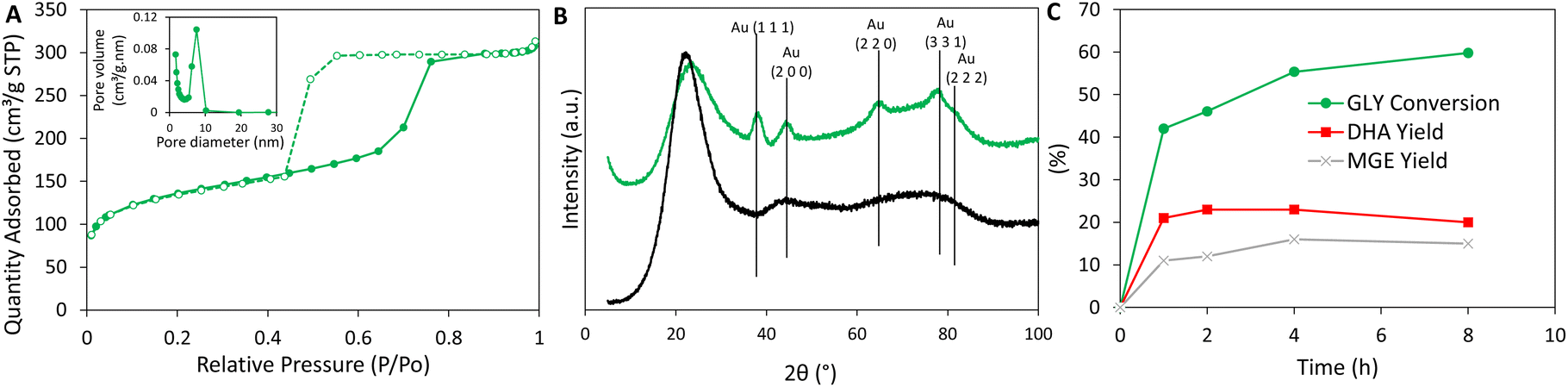 | ||
| Fig. 2 (A) Nitrogen adsorption (plain symbols) and desorption (empty symbols) isotherms of AuSiO2. Insert shows the BJH pore size distribution analysis obtained from the adsorption branch of the isotherm. (B) XRD diffractogram of fresh AuSiO2 (black) and calcined AuSiO2 (green) (the detected metallic Au phase corresponds to JCPDS 65-2870). (C) Kinetic study of the oxidation glycerol (GLY) to dihydroxyacetone (DHA) and methyl glycerate (MGE) with AuSiO2. Reaction conditions: T = 140 °C, pair = 30 bar, 0.25 M glycerol in methanol, and GLY/catalyst ratio = 2.3 (w/w). | ||
| BET surface area (m2 g−1) | BJH pore size (nm) | Pore volume (cm3 g−1) | Micropore volume (cm3 g−1) | |
|---|---|---|---|---|
| AuSiO2 | 440 | 5 | 0.47 | 0.09 |
| SnSiO2 | 330 | 5 | 0.34 | 0.07 |
| Au(0.5)SnSiO2 | 410 | 5 | 0.41 | 0.10 |
| Au(1)SnSiO2 | 440 | 5 | 0.45 | 0.11 |
| Au(2)SnSiO2 | 400 | 5 | 0.38 | 0.10 |
| Surface Au loadinga,c (wt%) | Surface Si/Sn ratioa | Bulk Au loadingb (wt%) | Bulk Si/Sn ratiob | |
|---|---|---|---|---|
| a Determined by XPS analysis. b Determined by ICP-OES analysis. c Calculated based on the surface atomic ratio of O, Si, Sn and Au. | ||||
| AuSiO2 | 0.13 | — | 0.79 | — |
| SnSiO2 | — | 70 | — | 73 |
| Au(0.5)SnSiO2 | 0.13 | 67 | 0.6 | 73 |
| Au(1)SnSiO2 | 0.26 | 82 | 1.1 | 72 |
| Au(2)SnSiO2 | 0.59 | 64 | 2 | 71 |
AuSiO2 was tested in the oxidation of glycerol in conditions that were independently optimized for running the cascade reaction (vide infra): at 140 °C, under 30 bar of air and vigorous stirring. After 4 hours, glycerol conversion reached 55% and the DHA selectivity reached 40% (Fig. 2C). By-products such as methyl glycerate (SMGE = 28%) and traces of dimethyl oxalate (Scheme 1) were also detected during the reaction. During the 4 hours of reaction, glycerol was quickly consumed to form DHA and methyl glycerate (MGE), resulting from the further oxidation of DHA. Increasing the reaction time to 8 hours slightly increased the glycerol conversion to 60% with similar selectivity MGE (= 28%) and to DHA (= 38%). These results are in good agreement with the mechanism already reported by previous studies and with the known relative instability of DHA, prone to overoxidation.16
A mesoporous SnSiO2 catalyst was also prepared by the one-pot aerosol-assisted sol–gel process, using the same templating agent (F127) and targeting a Si/Sn molar ratio of 74. This formulation was already reported as being very active for the conversion of DHA to ethyl lactate.30 TEM shows that the morphology and texture of SnSiO2 is very similar to AuSiO2 (Fig. 1D). The mesopores, clearly visible in TEM, are confirmed by textural analysis (Fig. 3A and Table 1). Type-IV isotherm showed the same H2 hysteresis observed for AuSiO2 and the BJH analysis also highlighted a narrow pore size distribution centered on 5 nm. The BET surface area was slightly lower than for AuSiO2 (330 m2 g−1). Tin was successfully incorporated in the material as evidenced by elemental analysis (ICP) which closely fits with the nominal composition (Table 2).
 | ||
| Fig. 3 (A) Nitrogen adsorption (plain symbols) and desorption (empty symbols) isotherms of SnSiO2. Insert show BJH pore size distribution analysis obtained from the adsorption branch of the isotherm. (B) High resolution XPS spectrum of the Sn 3d region for SnSiO2 with indication (dotted line) of the theoretical binding energy of Sn 3d5/2 in Sn4+ state (487.2 eV).61 (C) Diffuse reflectance UV/Vis spectrum of SnSiO2. | ||
The chemical state of tin in the material was investigated by XPS. The Sn 3d spectra confirmed tin is in Sn4+ state, with a Sn 3d5/2 peak centered at a binding energy of 487.2 eV (Fig. 3B).59–62 The surface atomic fraction Si/Sn reached the value of 70, which is very close to the nominal one (74), suggesting a homogeneous distribution of Sn in the silica matrix. Diffuse reflectance UV/Vis spectroscopy was used to further prove the successful insertion of Sn in the silica framework. The spectra show a broad absorption band at λ = 210–240 nm attesting for the presence of Sn4+ in tetrahedral coordination in the silica framework in this sample, while the minor contribution at λ ∼ 280 nm indicates the presence of a small proportion of hexacoordinated polymeric Sn–O–Sn type species (Fig. 3C).63,64 The insertion of Sn in the silica matrix was also studied by 119Sn NMR spectroscopy under static conditions (Fig. S2†). The signal centered at δ = −695 ppm is characteristic of intra-framework SnIV in tetrahedral coordination, connected with 4 atoms of Si by oxygen bridges.64 Although a minor amount of tin in extra-framework cannot be completely ruled out, significant amounts of SnO2 were not detected. Overall, UV, XPS and NMR results prove the insertion of tin in the silica framework.
The acidity of the sample was assessed by pyridine adsorption and FTIR spectroscopy. The FTIR spectra are displayed in Fig. S3.† The band found at 1455 cm−1 is characteristic of Lewis acid sites and attests of the successful incorporation of Lewis acidity in this sample. The surface density of Lewis acid sites reached 36 μmol g−1. Small amount of Brønsted acidity was observed (band at 1545 cm−1).
The SnSiO2 catalyst was tested for the conversion of DHA to methyl lactate. In the conditions of the reaction (same as above), we verified that DHA was completely transformed in methyl lactate after 4 hours, with a selectivity of >99%. This excellent activity is crucial to the objective of this work, as we target the cascade reaction from glycerol to methyl lactate. Note that SnSiO2 did not shown any activity in the conversion of glycerol.
3.2. Cascade reaction with a physical mixture of AuSiO2 and SnSiO2
The physical mixture of AuSiO2 (1 wt% of Au) and SnSiO2 (Si/Sn = 74) was used to perform the cascade reaction (Fig. 4A). After 8 hours glycerol conversion reached 70%, slightly higher than when using AuSiO2 alone. No DHA was detected, most probably due to its rapid consumption by the tin catalyst. Overall, the yield to the final product of the cascade (YML = 19%) was close to the yield of DHA obtained with AuSiO2 alone. The formation of by-products (formed in abundance via over-oxidation of DHA when AuSiO2 was used alone) could not be avoided. Over time, the conversion of glycerol remained around 70% and the methyl lactate yield plateaued at ∼23%. These results show that – owing to the introduction of the effective SnSiO2 catalyst – the reaction can indeed be pushed from the formed DHA towards the final product (ML). Yet, the ML yield and selectivity remain modest (as dictated by the poorly selective first reaction on the gold catalyst).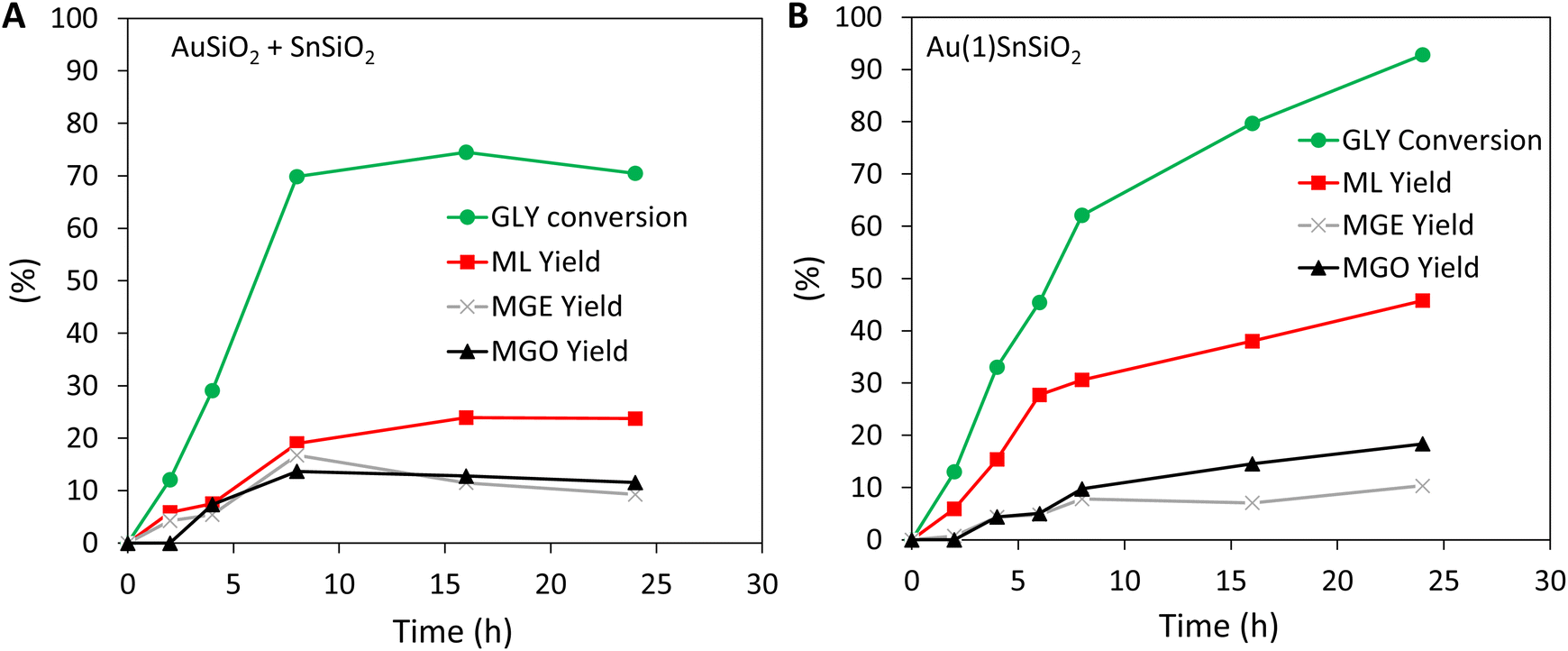 | ||
| Fig. 4 Transformation glycerol (GLY) to methyl lactate (ML), methyl glycerate (MGE) and methyl glycolate (MGO) with (A) the mechanical mixture of AuSiO2 and SnSiO2 (200 mg of each catalyst), (B) the bifunctional catalyst Au(1)SnSiO2 (200 mg). Reaction conditions: T = 140 °C, pair = 30 bar, 0.25 M glycerol in methanol, and GLY/gold-based catalyst ratio = 2.3 (w/w). | ||
3.3. Bifunctional Au(x)SnSiO2 catalysts
We reasoned that bifunctional materials bearing both the tin and the gold active species on the same solid particles would be primed to further boost the ML yield in the cascade reaction. Indeed, a facile substrate channeling between the two active sites could effectively drive the reaction to the desired methyl lactate. The synthesis of such bifunctional catalysts was attempted before (using mostly intricate multi-step preparation procedures),11,31,33,65,66 but the resulting material showed impaired catalytic performance, mostly due to the difficulty to stabilize both the Au and the Sn species in their most active forms. Here, the aerosol-assisted sol–gel synthesis was exploited to synthesize bifunctional catalysts in one step (plus calcination). These samples were denoted Au(x)SnSiO2, where “x” corresponds to the nominal Au loading. The Si/Sn ratio was kept the same in each catalyst (Si/Sn = 74).Similar to the monometallic catalysts described above, the texture of the bifunctional catalysts was characterized by type IV isotherms featuring the typical H2 hysteresis loop (Fig. 5A) corresponding to ink-bottle neck pores. The BJH analysis performed on the adsorption branch of the isotherm showed a narrow pore size distribution centered on 5 nm. All bifunctional catalysts exhibited a high surface area and pore volume, similar to AuSiO2 and SnSiO2 (Table 1).
 | ||
Fig. 5 (A) Nitrogen adsorption (plain symbols) and desorption (empty symbols) isotherms of Au(x)SnSiO2. Inserts show BJH pore size distribution analysis obtained from the adsorption branches of the isotherms. (B) XRD diffractograms of Au(x)SnSiO2 (the detected metallic Au phase corresponds to JCPDS 65-2870). The color of the curves refers to the gold loading: 0.5% ( , orange), 1% ( , orange), 1% ( , red), 2% ( , red), 2% ( , blue). , blue). | ||
Gold and tin contents in the different solids were verified to be very close to the targeted loadings (ICP-OES, Table 2). The Au 4f spectra for each bifunctional catalyst also showed the slight shift toward lower binding energy for the Au 4f7/2 peak (∼83 eV), typically attributed to very small gold nanoparticles (Fig. S4A†). It is not shifted as compared to the Au 4f signal obtained on AuSiO2 (Fig. S1†). Increasing the loading of gold in the catalysts led to a proportionally higher amount of gold found at the surface of the materials (Table 2). The Si/Sn surface ratio remained close to the nominal ratio (Si/Sn = 74) in all bifunctional catalysts, indicating that Sn is homogeneously dispersed in the material and that the presence of gold and MPTMS did not impact tin dispersion. Exactly like in the case of SnSiO2, Sn is inserted in the silica matrix with a Sn 3d5/2 XPS peak centered at ∼487 eV corresponding to Sn4+ state (Fig. S4B†).
In the perspective of a cascade reaction with two different active species, it is important to verify that the two species are not inserted in their appropriate state, and that the presence of one does not affect the chemical state of the other (for example via the formation of alloyed Au–Sn nanoparticles). Thus, Au(1)SnSiO2 was further investigated through XANES and EXAFS analyses, and compared to an Au foil as a reference sample, in order to get selective information about the gold environment. The XANES spectra of the sample and the reference foil are very similar suggesting that gold in the sample is in its metallic form maintaining the same environment to that of pure Au (Fig. 6A). This is confirmed by the comparison of the EXAFS interference functions and of corresponding FTs of the sample and the reference foil, which only show minor differences in the amplitudes of the oscillations and in the height of the peaks, respectively. This indicates that there is a strong similarity between the gold environment in Au(1)SnSiO2 and the gold reference foil (Fig. 6B and C), indicating no strong chemical interaction (e.g. alloying) between gold and tin. The reduced amplitudes of the oscillations and the lower FT peaks intensity of the sample with respect to the reference foil (Fig. 6B and C), are consistent with the small dimensions of the gold nanocrystals.
 | ||
| Fig. 6 (A) Normalized XANES spectra, (B) k2 χ(k) EXAFS interference functions and (C) corresponding FTs moduli (phase shift corrected) of Au(1)SnSiO2 (red curves) and Au foil (black curves). Note that, similarly, the Sn signal was not affected by the presence of Au. | ||
The crystallite size was measured employing X-ray diffraction (Fig. 5B). The addition of tin did not have an impact on the gold crystallites size as it remained very small (3.1 nm) and close to the size in AuSiO2 (Table 3). Similarly, the size remained the same regardless of the gold loading engaged.
The insertion of tin was investigated in the sample Au(1)SnSiO2 (fresh and after calcination) by 119Sn NMR spectroscopy. The spectrum exhibited the same signal as SnSiO2 centered at δ = −695 ppm, indicating the presence of intra-framework SnIV (Fig. S2†). This confirms that the addition of gold in the synthesis does not disrupt the proper insertion of tin in the silica framework. Similarly, the acidity of the sample Au(1)SnSiO2 was not impacted by the presence of gold in the material, with a surface density of Lewis acid sites reaching 38 μmol g−1 (Fig. S3†).
The sample morphology was investigated by transmission electron microscopy (TEM). The Au(x)SnSiO2 samples consist of spherical shaped microspheres with 5–7 nm interconnected pores (Fig. 7). The gold nanoparticle size distribution was estimated through TEM measurements on 200 nanoparticles. Whatever the gold loading, the materials exhibited overall a narrow gold nanoparticles size distribution centered at 3.5 nm, in good agreement with XRD results (Fig. S6†). Gold nanoparticles are homogeneously dispersed in the microspheres. EDX elemental mapping (Fig. 7) allowed highlighting that tin and gold were effectively and homogeneously dispersed in the catalyst microspheres.
 | ||
| Fig. 7 Transmission electron microscope analysis. Respectively from top to bottom: Au(0.5)SnSiO2, Au(1)SnSiO2 and Au(2)SnSiO2; from left to right: HAADF, STEM, STEM-EDX of Au and of Sn. | ||
The dispersion measurements performed on the different samples showed that the dispersion is somewhat lower in the 2%, compared to the 0.5%, 1% bifunctional catalysts and to the 1% monofunctional AuSiO2 catalyst (Table 3). This result shows that the amount of gold actually surface-available in the 2% catalyst is close to the one found in the 1% material. Considering that the gold nanoparticle size remained similar in the two materials, the decreased dispersion suggests that some nanoparticles are partially embed inside the silica walls when the gold loading is higher than 1%.
3.4. Cascade reaction with bifunctional Au(1)SnSiO2 catalysts
The bifunctional catalyst Au(1)SnSiO2 was tested in the one-pot transformation of glycerol to methyl lactate. The kinetic profile of glycerol conversion and ML yield with Au(1)SnSiO2 can be directly compared to the cascade performed with the mechanical mixture (AuSiO2 + SnSiO2) with exactly the same amount of Sn and Au introduced in the reactor (Fig. 4). Overall, the glycerol conversion was higher along the test with the bifunctional catalyst. Close to total conversion was achieved after 24 hours with the bifunctional catalyst while it remained stuck at 70% for the mechanical mixture. More interestingly, the ML selectivity was highly improved when using the bifunctional catalyst, resulting in a markedly higher ML yield. For example, at 4, 8, and 24 h, the ML yield reached 8, 19, and 23% with the mechanical mixture of the two catalysts, but reached 15, 31, and 49% with the bifunctional catalyst.This confirms that Au(1)SnSiO2 is intrinsically more active and selective toward ML. With the characterization and the catalytic results in hands, we reason that the bifunctional catalyst takes advantage of the proximity between tin and gold active sites to rapidly channel the intermediate DHA towards further conversion to ML, resulting in improved selectivity and overall yield. Overall, the formation of MGO and MGE by-products (resulting from the oxidation of DHA on gold nanoparticles) remained low with the bifunctional catalysts.
3.5. Optimization of the reaction conditions
With the high-performance bifunctional catalyst in hand, the reaction conditions were modified with idea to further improve the ML selectivity and yield. By varying the glycerol concentration, the air pressure, and the reaction temperature, new optimized conditions were selected for the rest of this work (vide infra): at 160 °C, under 15 bar of air and vigorous stirring (Table S1†). In these conditions a complete conversion of glycerol was obtained after 16 hours (Fig. 8). More importantly, the ML yield could be improved to around 62% while maintaining the production of by-products (MGO and MGE) very low. | ||
| Fig. 8 Transformation glycerol (GLY) to methyl lactate (ML), methyl glycerate (MGE) and methyl glycolate (MGO) with Au(1)SnSiO2. Reaction conditions: T = 160 °C, pair = 15 bar, 0.125 M glycerol in methanol, and GLY/catalyst ratio = 1.15 (w/w). | ||
The two catalysts Au(0.5)SnSiO2 and Au(2)SnSiO2 were also tested in the new reaction conditions to investigate the effect of the gold loading on the catalytic conditions (Fig. S7†). The catalysts containing 0.5% of gold showed a glycerol conversion limited at 40% after 4 hours while the ML yield reached 33%. The lower conversion is attributed to the lower amount of gold in the material. On the other hand, the Au(2)SnSiO2 sample displayed higher glycerol conversion with a good ML selectivity. Although the dispersion in this material was lower (Table 3), the higher conversion at the beginning of the reaction was ascribed to the higher amount of gold detected at the catalyst surface by XPS. In both cases, methyl glycerate, the first product of the overoxidation of DHA, was detected. This product can actually act as a chelating agent on the gold nanoparticles, hindering the catalytic active sites. The rapid loss of catalytic activity in the two materials was attributed to the poisoning of the gold nanoparticles. Such deactivation was already observed when working with AuSiO2, and we demonstrated that in order to recover its catalytic activity, a thermal treatment was required.20 The adsorption of undesired products on the catalytic species can also be responsible for the carbon loss sometimes observed during the reaction. When comparing the Au(1)SnSiO2 and Au(2)SnSiO2 samples, it appeared that a higher loading of gold led to poorer catalytic performance (lower conversion and ML yield) over time. It is hypothesized that the larger amount of gold located at the surface of the microsphere in the Au 2% would lead to higher amount of methyl glycerate produced and then adsorbed, thus leading to a faster catalyst deactivation. Therefore, Au(1)SnSiO2 was selected as the best for further investigations.
Au(1)SnSiO2 was further studied to shed light on its robustness. A “hot filtration test” confirmed that the activity was solely attributed to the solid catalyst (no leaching of active species acting in the homogeneous phase, see Fig. S8†). The recyclability of Au(1)SnSiO2 was investigated following the same reaction conditions. In a typical recycling experiment, the test was carried out for 2 hours (where conversion is still far from completion) before removing the catalyst by centrifugation. The catalyst was washed with methanol and regeneration was performed at 450 °C under flowing air (100 mL min−1). The catalyst showed excellent recyclability with an overall stable ML yield over 4 cycles (Fig. S9†). The gold nanoparticles remained small (3.8 nm, measured by XRD) after the 4 cycles performed. It is noteworthy to mention that without applying a thermal treatment, the catalyst was no longer active in the cascade reaction, implying that the deactivation was caused by the presence of products adsorbed on the active species.
4. Conclusion
Bifunctional catalysts combining gold nanoparticles dispersed in mesoporous tin silicate microspheres (AuSnSiO2) were prepared in one step by the aerosol-assisted sol–gel process. The materials displayed advantageous mesoporous texture (with large pore volume and high surface area), small surface-accessible gold nanoparticles, and highly dispersed tin species inserted in the silica framework generating abundant Lewis acid sites. Monofunctional AuSiO2 and SnSiO2 catalysts were used as benchmarks. In-depth characterization showed that the intrinsic properties of tin sites and gold sites were not mutually affected in the bifunctional catalyst. While the monofunctional AuSiO2 catalyst showed moderate activity and poor selectivity in the oxidation of glycerol to DHA (producing a.o. large amounts of methyl glycerate), the bifunctional catalyst allowed running the cascade reaction glycerol > DHA > methyl lactate very effectively. In fact, the yield for the final products of the complete cascade reaction (ML) obtained with the bifunctional catalyst markedly surpasses the yield for DHA obtained when running only the first reaction with AuSiO2. Thus, we evidenced that the combination of the two active sites boosts the catalytic performance, as the intermediate product (DHA) is effectively funneled towards the second reaction (rearrangement with methanol) occurring on tin sites. Importantly, higher selectivity and yield are observed for the bifunctional material, if compared with a corresponding mechanical mixture. This confirms the pivotal role of site proximity in the bifunctional catalysts. This work highlights the interest of bringing together in one unique material two catalytic species to run efficiently a cascade reaction. In this context, the aerosol-assisted sol–gel preparation is shown as a key enabling technology that markedly facilitates the preparation of such bifunctional catalysts.Conflicts of interest
The authors declare no competing interests.Acknowledgements
M. Van der Verren is thankful to F.R.S.-FNRS for her PhD fellowship, under the project PDR–T.0058.19. D. P. Debecker thanks the Francqui Foundation for the “Francqui Research Professor” chair. F. Devred, L. Fusaro and C. Charlier are acknowledged for the technical and logistical support. CzechNanoLab project LM2023051 funded by MEYS CR is gratefully acknowledged for the financial support of the measurements/sample fabrication at CEITEC Nano Research Infrastructure. We acknowledge CF CryoEM of CIISB, Instruct-CZ Centre, supported by MEYS CR (LM2023042) and European Regional Development Fund-Project “UP CIISB” (no. CZ.02.1.01/0.0/0.0/18_046/0015974). This research used resources of PC2 (Plateforme Technologique Physico-Chemical Characterization) and MORPH-IM (Morphology & Imaging) technology platforms located at the University of Namur. We thank the Diamond Light Source for the award of beamtime (Rapid Access Proposal SP34013) and the staff beamline for data collection.References
- J. A. Okolie, E. I. Epelle, M. E. Tabat, U. Orivri, A. N. Amenaghawon, P. U. Okoye and B. Gunes, Waste Biomass Valorization for the Production of Biofuels and Value-Added Products: A Comprehensive Review of Thermochemical, Biological and Integrated Processes, Process Saf. Environ. Prot., 2022, 159, 323–344, DOI:10.1016/j.psep.2021.12.049.
- P. Ning, G. Yang, L. Hu, J. Sun, L. Shi, Y. Zhou, Z. Wang and J. Yang, Recent Advances in the Valorization of Plant Biomass, Biotechnol. Biofuels, 2021, 14(1), 102, DOI:10.1186/s13068-021-01949-3.
- Y. Meng, S. Zou, Y. Zhou, W. Yi, Y. Yan, B. Ye, L. Xiao, J. Liu, H. Kobayashi and J. Fan, Activating Molecular Oxygen by Au/ZnO to Selectively Oxidize Glycerol to Dihydroxyacetone, Catal. Sci. Technol., 2018, 8(10), 2524–2528, 10.1039/C8CY00319J.
- G. Bagnato, A. Iulianelli, A. Sanna and A. Basile, Glycerol Production and Transformation: A Critical Review with Particular Emphasis on Glycerol Reforming Reaction for Producing Hydrogen in Conventional and Membrane Reactors, Membranes, 2017, 7(2), 17, DOI:10.3390/membranes7020017.
- C. Carlucci, A Focus on the Transformation Processes for the Valorization of Glycerol Derived from the Production Cycle of Biofuels, Catalysts, 2021, 11(2), 280, DOI:10.3390/catal11020280.
- C. Len and R. Luque, Continuous Flow Transformations of Glycerol to Valuable Products: An Overview, Sustainable Chem. Processes, 2014, 2, 1 CrossRef.
- T. Mizugaki, R. Arundhathi, T. Mitsudome, K. Jitsukawa and K. Kaneda, Highly Efficient and Selective Transformations of Glycerol Using Reusable Heterogeneous Catalysts, ACS Sustainable Chem. Eng., 2014, 2(4), 574–578, DOI:10.1021/sc500006b.
- R. Dziuba, M. Kucharska, L. Madej-Kiełbik, K. Sulak and M. Wiśniewska-Wrona, Biopolymers and Biomaterials for Special Applications within the Context of the Circular Economy, Materials, 2021, 14(24), 7704, DOI:10.3390/ma14247704.
- Z. Tang, D. G. Boer, A. Syariati, M. Enache, P. Rudolf, H. J. Heeres and P. P. Pescarmona, Base-Free Conversion of Glycerol to Methyl Lactate Using a Multifunctional Catalytic System Consisting of Au–Pd Nanoparticles on Carbon Nanotubes and Sn-MCM-41-XS, Green Chem., 2019, 21(15), 4115–4126, 10.1039/C9GC01521C.
- A. Behr, J. Eilting, K. Irawadi, J. Leschinski and F. Lindner, Improved Utilisation of Renewable Resources: New Important Derivatives of Glycerol, Green Chem., 2008, 10(1), 13–30, 10.1039/B710561D.
- Z. Tang, S. L. Fiorilli, H. J. Heeres and P. P. Pescarmona, Multifunctional Heterogeneous Catalysts for the Selective Conversion of Glycerol into Methyl Lactate, ACS Sustainable Chem. Eng., 2018, 6(8), 10923–10933, DOI:10.1021/acssuschemeng.8b02277.
- P. Fordham, R. Garcia, M. Besson and P. Gallezot, Selective Catalytic Oxidation with Air of Glycerol and Oxygenated Derivatives on Platinum Metals, in Studies in Surface Science and Catalysis, Elsevier, 1996, vol. 101, pp. 161–170. DOI:10.1016/S0167-2991(96)80226-6.
- H. Kimura, Selective Oxidation of Glycerol on a Platinum-Bismuth Catalyst by Using a Fixed Bed Reactor, Appl. Catal., A, 1993, 105(2), 147–158, DOI:10.1016/0926-860X(93)80245-L.
- D. Cherni, N. Moussa, M. F. Nsib, C. Evangelisti, L. Prati and A. Villa, Base-Free Glycerol Oxidation over N-TiO2 Supported Au–Pt Catalysts, React. Kinet., Mech. Catal.,, 2019, 128(2), 979–990, DOI:10.1007/s11144-019-01678-7.
- A. El Roz, P. Fongarland, F. Dumeignil and M. Capron, Glycerol to Glyceraldehyde Oxidation Reaction Over Pt-Based Catalysts Under Base-Free Conditions, Front. Chem., 2019, 7, 156, DOI:10.3389/fchem.2019.00156.
- P. M. Walgode, R. P. V. Faria and A. E. Rodrigues, A Review of Aerobic Glycerol Oxidation Processes Using Heterogeneous Catalysts: A Sustainable Pathway for the Production of Dihydroxyacetone, Catal. Rev., 2020, 1–90 Search PubMed.
- T. Mallat and A. Baiker, Oxidation of Alcohols with Molecular Oxygen on Platinum Metal Catalysts in Aqueous Solutions, Catal. Today, 1994, 19(2), 247–283, DOI:10.1016/0920-5861(94)80187-8.
- T. Ishida, T. Murayama, A. Taketoshi and M. Haruta, Importance of Size and Contact Structure of Gold Nanoparticles for the Genesis of Unique Catalytic Processes, Chem. Rev., 2020, 120(2), 464–525, DOI:10.1021/acs.chemrev.9b00551.
- A. Corma and H. Garcia, Supported Gold Nanoparticles as Catalysts for Organic Reactions, Chem. Soc. Rev., 2008, 37(9), 2096, 10.1039/b707314n.
- M. Van der Verren, V. Vykoukal, A. Styskalik, A. S. Malik, C. Aprile and D. P. Debecker, Airborne Preparation of Small Gold Nanoparticles Dispersed on Mesoporous Silica for the Catalytic Oxidation of Glycerol to Dihydroxyacetone, ACS Appl. Nano Mater., 2022, 5(12), 18977–18985, DOI:10.1021/acsanm.2c04786.
- P. P. Pescarmona, K. P. F. Janssen, C. Delaet, C. Stroobants, K. Houthoofd, A. Philippaerts, C. De Jonghe, J. S. Paul, P. A. Jacobs and B. F. Sels, Zeolite-Catalysed Conversion of C3 Sugars to Alkyl Lactates, Green Chem., 2010, 12(6), 1083, 10.1039/b921284a.
- A. Feliczak-Guzik, M. Sprynskyy, I. Nowak and B. Buszewski, Catalytic Isomerization of Dihydroxyacetone to Lactic Acid and Alkyl Lactates over Hierarchical Zeolites Containing Tin, Catalysts, 2018, 8(1), 31, DOI:10.3390/catal8010031.
- L. Yang, X. Yang, E. Tian, V. Vattipalli, W. Fan and H. Lin, Mechanistic Insights into the Production of Methyl Lactate by Catalytic Conversion of Carbohydrates on Mesoporous Zr-SBA-15, J. Catal., 2016, 333, 207–216, DOI:10.1016/j.jcat.2015.10.013.
- Y. Hayashi and Y. Sasaki, Tin-Catalyzed Conversion of Trioses to Alkyl Lactates in Alcohol Solution, Chem. Commun., 2005,(No. 21), 2716 RSC.
- F. R. Abreu, D. G. Lima, E. H. Hamú, S. Einloft, J. C. Rubim and P. A. Z. Suarez, New Metal Catalysts for Soybean Oil Transesterification, J. Am. Oil Chem. Soc., 2003, 80(6), 601–604, DOI:10.1007/s11746-003-0745-6.
- X. Yang, L. Wu, Z. Wang, J. Bian, T. Lu, L. Zhou, C. Chen and J. Xu, Conversion of Dihydroxyacetone to Methyl Lactate Catalyzed by Highly Active Hierarchical Sn-USY at Room Temperature, Catal. Sci. Technol., 2016, 6(6), 1757–1763, 10.1039/C5CY01037C.
- E. Taarning, S. Saravanamurugan, M. S. Holm, J. Xiong, R. M. West and C. H. Christensen, Zeolite-Catalyzed Isomerization of Triose Sugars, ChemSusChem, 2009, 2(7), 625–627, DOI:10.1002/cssc.200900099.
- L. Li, X. Collard, A. Bertrand, B. F. Sels, P. P. Pescarmona and C. Aprile, Extra-Small Porous Sn-Silicate Nanoparticles as Catalysts for the Synthesis of Lactates, J. Catal., 2014, 314, 56–65, DOI:10.1016/j.jcat.2014.03.012.
- L. Li, C. Stroobants, K. Lin, P. A. Jacobs, B. F. Sels and P. P. Pescarmona, Selective Conversion of Trioses to Lactates over Lewis Acid Heterogeneous Catalysts, Green Chem., 2011, 13(5), 1175, 10.1039/c0gc00923g.
- N. Godard, A. Vivian, L. Fusaro, L. Cannavicci, C. Aprile and D. P. Debecker, High-Yield Synthesis of Ethyl Lactate with Mesoporous Tin Silicate Catalysts Prepared by an Aerosol-Assisted Sol–Gel Process, ChemCatChem, 2017, 9(12), 2211–2218, DOI:10.1002/cctc.201601637.
- J. Li, D. Li, C. Liu, C. Xu and W. Dong, The Conversion of Glycerol to Methyl Lactate Catalyzed by Tin–exchanged Montmorillonite–supported Gold Catalysts, J. Chem. Technol. Biotechnol., 2019, 94(6), 1958–1967, DOI:10.1002/jctb.5979.
- R. K. Pazhavelikkakath Purushothaman, J. van Haveren, I. Melián-Cabrera, E. R. H. van Eck and H. J. Heeres, Base-Free, One-Pot Chemocatalytic Conversion of Glycerol to Methyl Lactate Using Supported Gold Catalysts, ChemSusChem, 2014, 7(4), 1140–1147, DOI:10.1002/cssc.201301105.
- L. Zhou, Y. Xu, X. Yang, T. Lu and L. Han, Utilization of Biodiesel Byproduct Glycerol: Production of Methyl Lactate over Au/CuO and Sn-Beta Binary Catalyst under Mild Reaction Conditions, Energy Convers. Manage., 2019, 196, 277–285, DOI:10.1016/j.enconman.2019.06.011.
- S. De, S. Dutta and B. Saha, Critical Design of Heterogeneous Catalysts for Biomass Valorization: Current Thrust and Emerging Prospects, Catal. Sci. Technol., 2016, 6(20), 7364–7385, 10.1039/C6CY01370H.
- D. Y. Murzin, On Spatial Control in Heterogeneous Multifunctional Catalysts, Catal. Lett., 2017, 147(3), 613–621, DOI:10.1007/s10562-017-1970-3.
- Y.-B. Huang, J. Liang, X.-S. Wang and R. Cao, Multifunctional Metal–Organic Framework Catalysts: Synergistic Catalysis and Tandem Reactions, Chem. Soc. Rev., 2017, 46(1), 126–157, 10.1039/C6CS00250A.
- Z. Cai, Y. Wu, Z. Wu, L. Yin, Z. Weng, Y. Zhong, W. Xu, X. Sun and H. Wang, Unlocking Bifunctional Electrocatalytic Activity for CO 2 Reduction Reaction by Win-Win Metal–Oxide Cooperation, ACS Energy Lett., 2018, 3(11), 2816–2822, DOI:10.1021/acsenergylett.8b01767.
- A. Ramli, M. Farooq, A. Naeem, S. Khan, M. Hummayun, A. Iqbal, S. Ahmed and L. A. Shah, Bifunctional Heterogeneous Catalysts for Biodiesel Production Using Low Cost Feedstocks: A Future Perspective, in Frontiers in Bioenergy and Biofuels, ed. E. Jacob-Lopes and L. Q. Zepka, InTech, 2017 Search PubMed.
- D. Ma, B. Li, K. Liu, X. Zhang, W. Zou, Y. Yang, G. Li, Z. Shi and S. Feng, Bifunctional MOF Heterogeneous Catalysts Based on the Synergy of Dual Functional Sites for Efficient Conversion of CO 2 under Mild and Co-Catalyst Free Conditions, J. Mater. Chem. A, 2015, 3(46), 23136–23142, 10.1039/C5TA07026K.
- D. P. Debecker, S. Le Bras, C. Boissière, A. Chaumonnot and C. Sanchez, Aerosol Processing: A Wind of Innovation in the Field of Advanced Heterogeneous Catalysts, Chem. Soc. Rev., 2018, 47(11), 4112–4155, 10.1039/C7CS00697G.
- S. Wintzheimer, L. Luthardt, K. Le Anh Cao, I. Imaz, D. Maspoch, T. Ogi, A. Bück, D. P. Debecker, M. Faustini and K. Mandel, Multifunctional, Hybrid Materials Design via Spray-Drying: Much More than Just Drying, Adv. Mater., 2023, 2306648 CrossRef CAS PubMed.
- A. Vivian, L. Soumoy, L. Fusaro, P. Louette, A. Felten, S. Fiorilli, D. P. Debecker and C. Aprile, The High Activity of Mesoporous Ga-SiO2 Catalysts in the Upgrading of Glycerol to Solketal Explained by in-Depth Characterization, J. Catal., 2021, 400, 83–92, DOI:10.1016/j.jcat.2021.05.017.
- S. Ramesh, K. Indukuri, O. Riant and D. P. Debecker, Synthesis of Carbonate Esters by Carboxymethylation Using NaAlO2 as a Highly Active Heterogeneous Catalyst, Org. Process Res. Dev., 2018, 22(12), 1846–1851, DOI:10.1021/acs.oprd.8b00333.
- S. Maksasithorn, P. Praserthdam, K. Suriye and D. P. Debecker, Preparation of Super-Microporous WO3−SiO2 Olefin Metathesis Catalysts by the Aerosol-Assisted Sol–Gel Process, Microporous Mesoporous Mater., 2015, 213, 125–133, DOI:10.1016/j.micromeso.2015.04.020.
- D. Dochain, A. Van Den Daelen, A. Styskalik, V. Vykoukal and D. P. Debecker, Aerosol-Assisted Sol-Gel Synthesis of Mesoporous Ag-Ta-SiO 2 Catalysts for the Direct Upgrading of Ethanol to Butadiene, RSC Sustainability, 2023, 1, 599–608, 10.1039/D2SU00080F.
- V. Smeets, W. Baaziz, O. Ersen, E. M. Gaigneaux, C. Boissière, C. Sanchez and D. P. Debecker, Hollow Zeolite Microspheres as a Nest for Enzymes: A New Route to Hybrid Heterogeneous Catalysts, Chem. Sci., 2020, 11(4), 954–961, 10.1039/C9SC04615A.
- M. Van der Verren, V. Smeets, A. vander Straeten, C. Dupont-Gillain and D. P. Debecker, Hybrid Chemoenzymatic Heterogeneous Catalyst Prepared in One Step from Zeolite Nanocrystals and Enzyme–Polyelectrolyte Complexes, Nanoscale Adv., 2021, 3(6), 1646–1655, 10.1039/D0NA00834F.
- M. L. De Marco, W. Baaziz, S. Sharna, F. Devred, C. Poleunis, A. Chevillot-Biraud, S. Nowak, R. Haddad, M. Odziomek, C. Boissière, D. P. Debecker, O. Ersen, J. Peron and M. Faustini, High-Entropy-Alloy Nanocrystal Based Macro- and Mesoporous Materials, ACS Nano, 2022, 16(10), 15837–15849, DOI:10.1021/acsnano.2c05465.
- G. Busto, R. Wineh, H. Zamani, E. Shirman, S. Liu, A. V. Shneidman and T. Shirman, Raspberry Colloid Templated Catalysts Fabricated Using Spray Drying Method, Catalysts, 2022, 13(1), 60, DOI:10.3390/catal13010060.
- Y. Mitsuka, N. Ogiwara, M. Mukoyoshi, H. Kitagawa, T. Yamamoto, T. Toriyama, S. Matsumura, M. Haneda, S. Kawaguchi, Y. Kubota and H. Kobayashi, Fabrication of Integrated Copper–Based Nanoparticles/Amorphous Metal–Organic Framework by a Facile Spray–Drying Method: Highly Enhanced CO 2 Hydrogenation Activity for Methanol Synthesis, Angew. Chem., Int. Ed., 2021, 60(41), 22283–22288, DOI:10.1002/anie.202110585.
- C. M. Urruchurto, J. G. Carriazo, C. Osorio, S. Moreno and R. A. Molina, Spray-Drying for the Preparation of Al–Co–Cu Pillared Clays: A Comparison with the Conventional Hot-Drying Method, Powder Technol., 2013, 239, 451–457, DOI:10.1016/j.powtec.2013.02.033.
- N. Wang, Y. Wei, M. Chang, J. Liu and J.-X. Wang, Macro-Meso-Microporous Metal–Organic Frameworks: Template-Assisted Spray Drying Synthesis and Enhanced Catalysis, ACS Appl. Mater. Interfaces, 2022, 14(8), 10712–10720, DOI:10.1021/acsami.1c23297.
- A. Vivian, L. Fusaro, D. P. Debecker and C. Aprile, Mesoporous Methyl-Functionalized Sn-Silicates Generated by the Aerosol Process for the Sustainable Production of Ethyl Lactate, ACS Sustainable Chem. Eng., 2018, 6(11), 14095–14103, DOI:10.1021/acssuschemeng.8b02623.
- M. Jacquemin, M. J. Genet, E. M. Gaigneaux and D. P. Debecker, Calibration of the X-Ray Photoelectron Spectroscopy Binding Energy Scale for the Characterization of Heterogeneous Catalysts: Is Everything Really under Control?, ChemPhysChem, 2013, 14(15), 3618–3626, DOI:10.1002/cphc.201300411.
- J. Radnik, C. Mohr and P. Claus, On the Origin of Binding Energy Shifts of Core Levels of Supported Gold Nanoparticles and Dependence of Pretreatment and Material Synthesis, Phys. Chem. Chem. Phys., 2003, 5(1), 172–177, 10.1039/b207290d.
- B. Panthi, A. Mukhopadhyay, L. Tibbitts, J. Saavedra, C. J. Pursell, R. M. Rioux and B. D. Chandler, Using Thiol Adsorption on Supported Au Nanoparticle Catalysts To Evaluate Au Dispersion and the Number of Active Sites for Benzyl Alcohol Oxidation, ACS Catal., 2015, 5(4), 2232–2241, DOI:10.1021/cs501942t.
- I. S. Pieta, M. Ishaq, P. P. Wells and J. A. Anderson, Quantitative Determination of Acid Sites on Silica-Alumina, Appl. Catal., A, 2010, 390(1–2), 127–134 CrossRef CAS.
- B. Ravel and M. Newville, ATHENA, ARTEMIS, HEPHAESTUS : Data Analysis for X-Ray Absorption Spectroscopy Using IFEFFIT, J. Synchrotron Radiat., 2005, 12(4), 537–541, DOI:10.1107/S0909049505012719.
- M. Kwoka, L. Ottaviano, M. Passacantando, S. Santucci, G. Czempik and J. Szuber, XPS Study of the Surface Chemistry of L-CVD SnO2 Thin Films after Oxidation, Thin Solid Films, 2005, 490(1), 36–42, DOI:10.1016/j.tsf.2005.04.014.
- M. Kwoka, B. Lyson-Sypien, E. Comini, M. Krzywiecki, K. Waczynski and J. Szuber, Surface Properties of SnO 2 Nanolayers Prepared by Spin-Coating and Thermal Oxidation, Nanotechnology, 2020, 31(31), 315714, DOI:10.1088/1361-6528/ab7586.
- E. Peeters, G. Pomalaza, I. Khalil, A. Detaille, D. P. Debecker, A. P. Douvalis, M. Dusselier and B. F. Sels, Highly Dispersed Sn-Beta Zeolites as Active Catalysts for Baeyer–Villiger Oxidation: The Role of Mobile, In Situ Sn(II)O Species in Solid-State Stannation, ACS Catal., 2021, 11(10), 5984–5998, DOI:10.1021/acscatal.1c00435.
- Z. Shen, L. Kong, W. Zhang, M. Gu, M. Xia, X. Zhou and Y. Zhang, Surface Amino-Functionalization of Sn-Beta Zeolite Catalyst for Lactic Acid Production from Glucose, RSC Adv., 2019, 9(33), 18989–18995, 10.1039/C9RA01264H.
- E. A. Alarcón, A. L. Villa and C. M. de Correa, Characterization of Sn- and Zn-Loaded MCM-41 Catalysts for Nopol Synthesis, Microporous Mesoporous Mater., 2009, 122(1–3), 208–215, DOI:10.1016/j.micromeso.2009.03.003.
- K. Chaudhari, T. K. Das, P. R. Rajmohanan, K. Lazar, S. Sivasanker and A. J. Chandwadkar, Synthesis, Characterization, and Catalytic Properties of Mesoporous Tin-Containing Analogs of MCM-41, J. Catal., 1999, 183(2), 281–291 CrossRef CAS.
- B. Tang, D. Wang, A. Li, H.-M. Tang, E.-C. Yang and W. Dai, Constructing Core-Shell Structured Au/Snβ@mesosilica Composite for One-Pot Base-Free Conversion of Glycerol to Methyl Lactate, Microporous Mesoporous Mater., 2023, 347, 112348, DOI:10.1016/j.micromeso.2022.112348.
- T. Lu, X. Fu, L. Zhou, Y. Su, X. Yang, L. Han, J. Wang and C. Song, Promotion Effect of Sn on Au/Sn-USY Catalysts for One-Pot Conversion of Glycerol to Methyl Lactate, ACS Catal., 2017, 7(10), 7274–7284, DOI:10.1021/acscatal.7b02254.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr06518a |
| This journal is © The Royal Society of Chemistry 2024 |