
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multi-stimuli-responsive polymer degradation by polyoxometalate photocatalysis and chloride ions†
Chen
Gu
a,
Chifeng
Li
a,
Noriyuki
Minezawa
 b,
Susumu
Okazaki
*b,
Kazuya
Yamaguchi
a and
Kosuke
Suzuki
*a
b,
Susumu
Okazaki
*b,
Kazuya
Yamaguchi
a and
Kosuke
Suzuki
*a
aDepartment of Applied Chemistry, School of Engineering, The University of Tokyo, Tokyo, Japan. E-mail: ksuzuki@appchem.t.u-tokyo.ac.jp
bDepartment of Applied Materials Science, Graduate School of Frontier Sciences, The University of Tokyo, Chiba, Japan. E-mail: okazaki@edu.k.u-tokyo.ac.jp
First published on 18th March 2024
Abstract
Photocatalytic polymer degradation based on harnessing the abundant light energy present in the environment is one of the promising approaches to address the issue of plastic waste. In this study, we developed a multi-stimuli-responsive photocatalytic polymer degradation system facilitated by the photocatalysis of a polyoxometalate [γ-PV2W10O40]5− in conjunction with chloride ions (Cl−) as harmless and abundant stimuli. The degradation of various polymers was significantly accelerated in the presence of Cl−, which was attributed to the oxidation of Cl− by the polyoxometalate photocatalysis into a highly reactive chlorine radical that can efficiently generate a carbon-centered radical for subsequent polymer degradation. Although organic and organometallic photocatalysts decomposed under the conditions for photocatalytic polymer degradation in the presence of Cl−, [γ-PV2W10O40]5− retained its structure even under these highly oxidative conditions.
Introduction
Plastic waste has become a pressing environmental concern, demanding the development of effective technologies for polymer degradation.1 Given that most plastics demonstrate stability and resist natural degradation processes in the environment, their improper disposal results in severe environmental harm. To address this issue, diverse waste management methods have been developed at both the fundamental research and industrial levels.2 Among these, photocatalytic polymer degradation stands out as one of the promising techniques, as it is based on harnessing the readily available and abundant light energy from the environment without the need for specific polymer structures, excess amounts of reactants, and high reaction temperature.3 Photocatalytic reactions truncate polymer chains and induce oxygenation, facilitating subsequent degradation by microorganisms. However, most photocatalytic systems rely solely on light as a stimulus and have difficulty in controlling the polymer degradation (Fig. 1a).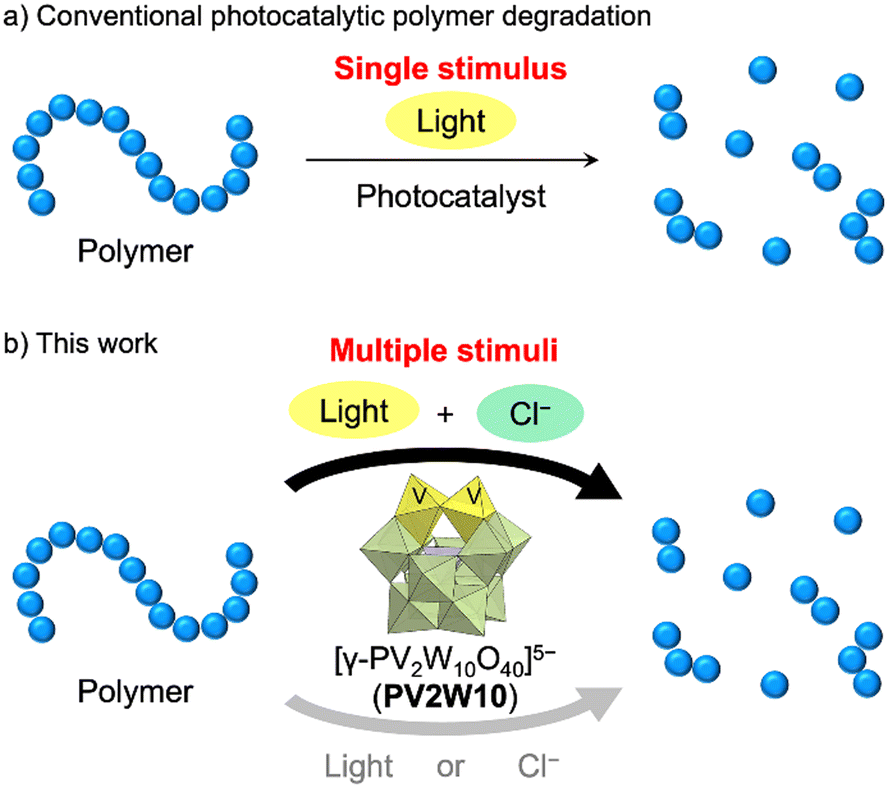 | ||
| Fig. 1 Schematic representations of (a) a conventional photocatalytic polymer degradation system utilizing a single stimulus, light, and (b) this work: multi-stimuli-responsive polymer degradation by polyoxometalate photocatalysis utilizing both light and chloride ions. | ||
To achieve more controlled photocatalytic polymer degradation, we aimed to develop a photocatalytic system that requires multiple stimuli for polymer degradation beyond just light. The stimulus we focused on was chloride ions (Cl−), which are innocuous and abundant in various salt forms in the environment and, more importantly, can be converted to highly reactive chlorine radicals (Cl˙) via electrochemical or photochemical processes.4,5 Photocleavage of the M–Cl bonds of chloride salts of various metals, such as Cu2+, Fe3+, Ni3+, and Ti4+, via ligand-to-metal charge transfer has been employed to generate Cl˙.6 In addition, since the one-electron oxidation potential of Cl− is ca. +1.5 V vs. the normal hydrogen electrode,7 photocatalytic oxidation processes with photoredox catalysts can also be harnessed to generate Cl˙ from Cl−.4,8,9 Importantly, the generated Cl˙ exhibits a remarkably high hydrogen atom transfer (HAT) ability, enabling it to abstract hydrogen atoms from various organic molecules, consequently generating carbon radicals. This propensity arises from the higher bond dissociation energy of H–Cl (103 kcal mol−1) compared to that of typical H–C(sp3) bonds (e.g., H–CH2CH3, 101 kcal mol−1; H–CH(CH3)2, 98.6 kcal mol−1).10 The carbon radicals generated via Cl˙-mediated HAT can react with O2, making this approach suitable for polymer degradation. While organic dye photocatalysts, like 9-mesityl-10-methylacridinium ion (Acr+-Mes), have been reported to generate Cl˙ from Cl− by photo-irradiation,8 they are susceptible to decomposition by Cl˙ and are therefore unsuitable for polymer degradation.
Polyoxometalates (POMs) are anionic metal oxide clusters considered as emerging materials in the field of photocatalysts.11,12 Their redox potentials and reactivities can be finely controlled by selecting their structures and constituent elements. In addition, POMs offer a distinct advantage as they have a significantly greater oxidative durability compared to organic dye and organometallic photocatalysts. We have recently reported a highly efficient polymer degradation system based on the photocatalysis of decatungstate [W10O32]4−.13 However, owing to its pronounced HAT ability, decatungstate photocatalysis for polymer degradation relies exclusively on photo-irradiation, without requiring additional stimuli. We have previously reported the visible-light-responsive photocatalysis of a vanadium-containing polyoxotungstate [γ-PV2W10O40]5− (PV2W10, Fig. S1†) for the aerobic oxygenation of organic substrates via single electron transfer (SET) initiated by the photoactive catalyst.14 In this study, by harnessing the photocatalytic SET property of PV2W10, we developed a system for multiple-stimuli-responsive polymer degradation that allows efficient degradation of various polymers when multiple stimuli, specifically, light and Cl−, are present (Fig. 1b).
Results and discussion
We investigated the multi-stimuli-responsive photocatalytic degradation of polycaprolactone (PCL), our selected model polymer, using various photocatalysts, including POMs, organic dyes, organometallic complexes, and inorganic semiconductor photocatalysts. The PCL degradation experiments were conducted under photo-irradiation by a xenon lamp (λ > 350 nm) in acetonitrile with an O2 atmosphere (1 atm) (Table 1, Fig. S2†). After 4 h of the photocatalytic reaction, the number-average and weight-average molecular weights of PCL (Mn and Mw) were determined via gel permeation chromatography. Degradation efficiency was evaluated based on the degradation rate, which was defined as (Mw0 − Mw)/Mw0 (%) (where Mw0 is Mw before reaction, Table 1, entry 1). PCL degradation by the tetra-n-butylammonium (TBA) salt of PV2W10 (TBA4H[γ-PV2W10O40], TBAPV2W10) hardly proceeded, resulting in a degradation rate of only 7% (Table 1, entry 2). In stark contrast, the addition of TBACl (tetra-n-butylammonium chloride) as a Cl− source to the reaction solution significantly enhanced the degradation, achieving a degradation rate of 51% (Table 1, entry 3 and Fig. S3†). The degradation also proceeded when using visible light (λ > 400 nm, Table 1, entry 4). It is important to highlight that degradation did not proceed without photo-irradiation or under an argon (Ar) atmosphere (Table 1, entries 5 and 6). Furthermore, without TBAPV2W10, degradation did not proceed even when light, Cl−, and O2 were all present (Table 1, entry 7). These results firmly established that this multi-stimuli-responsive polymer degradation was realized by the photocatalysis of MgCl TBAPV2W10. Even when the amount of TBAPV2W10 was reduced, the acceleration effect of Cl− was still observed (Table S1†). Remarkably, this system was efficient also under sunlight. After 10 h of sunlight irradiation (Fig. S4†), the PCL degradation rates in the absence and presence of TBAPV2W10 were only <1% and 11%, respectively (Table 1, entries 8 and 9). On the other hand, the coexistence of TBAPV2W10 and Cl− significantly accelerated PCL degradation, leading to a degradation rate of 30% (Table 1, entry 10).|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | TBACl | M w (kg mol−1) | (Mw0 − Mw)/Mw0 (%) | M w/Mn |
| Reaction conditions: PCL (40 mg), catalyst (0.0011 mmol, equivalent to 4 mg of TBAPV2W10), TBACl (4 mg), O2 (1 atm), acetonitrile (4 mL), photo-irradiation (xenon lamp, λ > 350 nm), 4 h. After the reaction, Mw and Mn were determined by gel permeation chromatography.a Photo-irradiation (xenon lamp, λ > 400 nm).b Sunlight 10 h.c Sunlight 10 h, TBAPV2W10 (9 mg).d TBACl (8 mg).e TiO2 (4 mg). | |||||
| 1 | (Before reaction) | — | 20.8 (Mw0) | — | 1.72 |
| 2 | TBAPV2W10 | — | 19.4 | 7 | 1.94 |
| 3 | TBAPV2W10 | Yes | 10.2 | 51 | 1.89 |
| 4a | TBAPV2W10 | Yes | 10.8 | 48 | 1.95 |
| 5 | TBAPV2W10 (dark) | Yes | 20.7 | <1 | 1.78 |
| 6 | TBAPV2W10 (Ar) | Yes | 22.0 | <1 | 1.67 |
| 7 | — | Yes | 20.9 | <1 | 1.76 |
| 8b | — (sunlight) | — | 21.0 | <1 | 1.72 |
| 9c | TBAPV2W10 (sunlight) | — | 18.5 | 11 | 1.80 |
| 10c,d | TBAPV2W10 (sunlight) | Yes | 14.6 | 30 | 1.81 |
| 11 | TBAW10 | — | 3.98 | 81 | 1.53 |
| 12 | TBAW10 | Yes | 7.40 | 64 | 2.17 |
| 13 | TBA4H2[γ-SiV2W10O40] | — | 20.5 | 1 | 1.71 |
| 14 | TBA4H2[γ-SiV2W10O40] | Yes | 17.8 | 15 | 1.88 |
| 15 | TBA3[α-PW12O40] | — | 19.6 | 6 | 1.75 |
| 16 | TBA3[α-PW12O40] | Yes | 19.4 | 7 | 1.75 |
| 17 | TBA4[α-PVW11O40] | — | 19.9 | 4 | 1.73 |
| 18 | TBA4[α-PVW11O40] | Yes | 19.8 | 5 | 1.75 |
| 19 | TBA6[α-PV3W9O40] | — | 20.2 | 3 | 1.70 |
| 20 | TBA6[α-PV3W9O40] | Yes | 20.0 | 4 | 1.74 |
| 21 | Eosin Y | — | 17.6 | 15 | 1.83 |
| 22 | Eosin Y | Yes | 14.9 | 29 | 1.92 |
| 23 | Acr+-Mes ClO4− | — | 19.6 | 6 | 1.80 |
| 24 | Acr+-Mes ClO4− | Yes | 16.3 | 22 | 1.94 |
| 25 | Ru(bpy)3Cl2 | — | 19.8 | 5 | 1.78 |
| 26 | Ru(bpy)3Cl2 | Yes | 19.6 | 6 | 1.83 |
| 27e | TiO2 (ST-01) | — | 18.1 | 13 | 2.35 |
| 28e | TiO2 (ST-01) | Yes | 18.4 | 11 | 1.87 |
We also explored the utility of other photocatalysts. As we recently reported, TBAW10, the TBA salt of decatungstate (TBA4[W10O32]) is an exceptionally reactive photocatalyst for polymer degradation.13 With TBAW10, PCL degradation proceeded both in the absence and presence of Cl−, and multi-stimuli-responsive polymer degradation could not be achieved (Table 1, entries 11 and 12). TBA4H2[γ-SiV2W10O40], which has the same structure as TBAPV2W10 but contains different heteroatoms (Si and P), also exhibited enhanced reactivity for PCL degradation when Cl− was present (Table 1, entries 13 and 14). However, the enhancement was much lower than that obtained with TBAPV2W10, resulting in a degradation rate of only 15% even in the presence of Cl− (Table 1, entry 14). In contrast, TBA3[α-PW12O40], TBA4[α-PVW11O40], and TBA6[α-PV3W9O40] hardly showed any activity for PCL degradation under photo-irradiation (Table 1, entries 15–20).
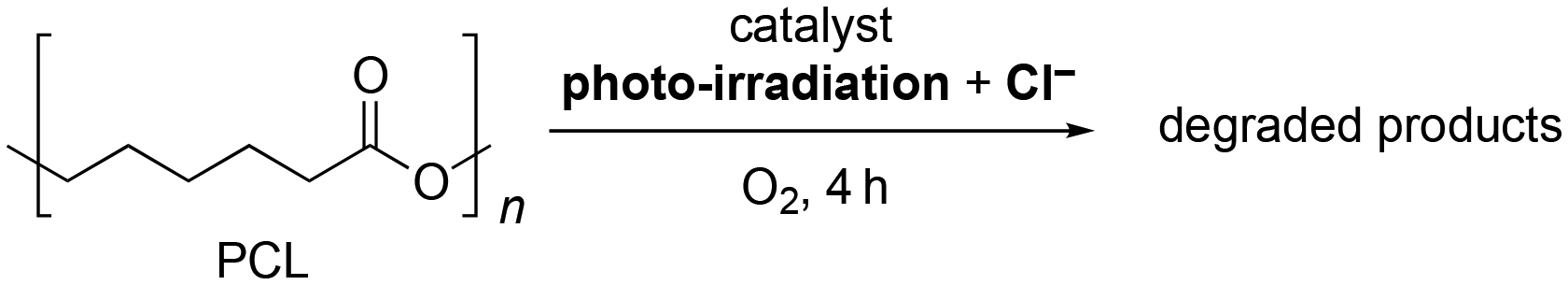
Organic dye photocatalysts, such as Eosin Y and Acr+-Mes ClO4−, proved ineffective in this system (Table 1, entries 21–24). Although Eosin Y is known to promote various reactions via both SET and HAT processes,15 the UV-Vis spectra revealed its decomposition during PCL degradation under photo-irradiation (Fig. 2a and b).16 In addition, previous reports have shown that Acr+-Mes ClO4− can generate Cl˙ from Cl−via photo-irradiation;8 however, reactivity for PCL degradation remained modest, achieving a degradation rate of only 22% in the presence of Cl− (Table 1, entries 23 and 24). Notably, although Acr+-Mes ClO4− is stable under photo-irradiation in the absence of Cl− (Fig. 2c), the absorbance of Acr+-Mes ClO4− in the UV-Vis spectrum significantly decreased under photo-irradiation in the presence of Cl−, revealing an issue with its durability (Fig. 2d). This was likely caused by the decomposition of Acr-Mes+ ClO4− by the highly reactive Cl˙ generated from Cl−. In contrast, TBAPV2W10 can maintain its structure even under oxidative conditions. The UV-Vis and 31P and 51V NMR spectra proved that the structure of TBAPV2W10 was maintained even after polymer degradation in the presence of Cl− (Fig. 2e–h). Slight changes in the absorbance of the UV-Vis spectrum and 31P NMR chemical shifts were likely attributed to TBAPV2W10 protonation during the reaction rather than decomposition of the catalyst (Fig. S5–S7†). The durable TBAPV2W10 provided superior catalytic activity compared to these organic dyes. The organometallic photocatalyst Ru(bpy)3Cl2 was not effective for polymer degradation, either in the absence or presence of Cl− (Table 1, entries 25 and 26). This was likely due to its structural change, as observed in the UV-Vis spectra (Fig. S8†). These results revealed that organic and organometallic photocatalysts have durability issues under the conditions of polymer degradation where various reactive radical species, including Cl˙, are generated. TiO2, a well-studied photocatalyst for polymer degradation,3 was also assessed. Although TiO2 showed low efficiency in polymer degradation (degradation rate, 13%) without Cl− by photo-irradiation, it did not exhibit multi-stimuli-responsive polymer degradation in the presence of Cl− (Table 1, entries 27 and 28).
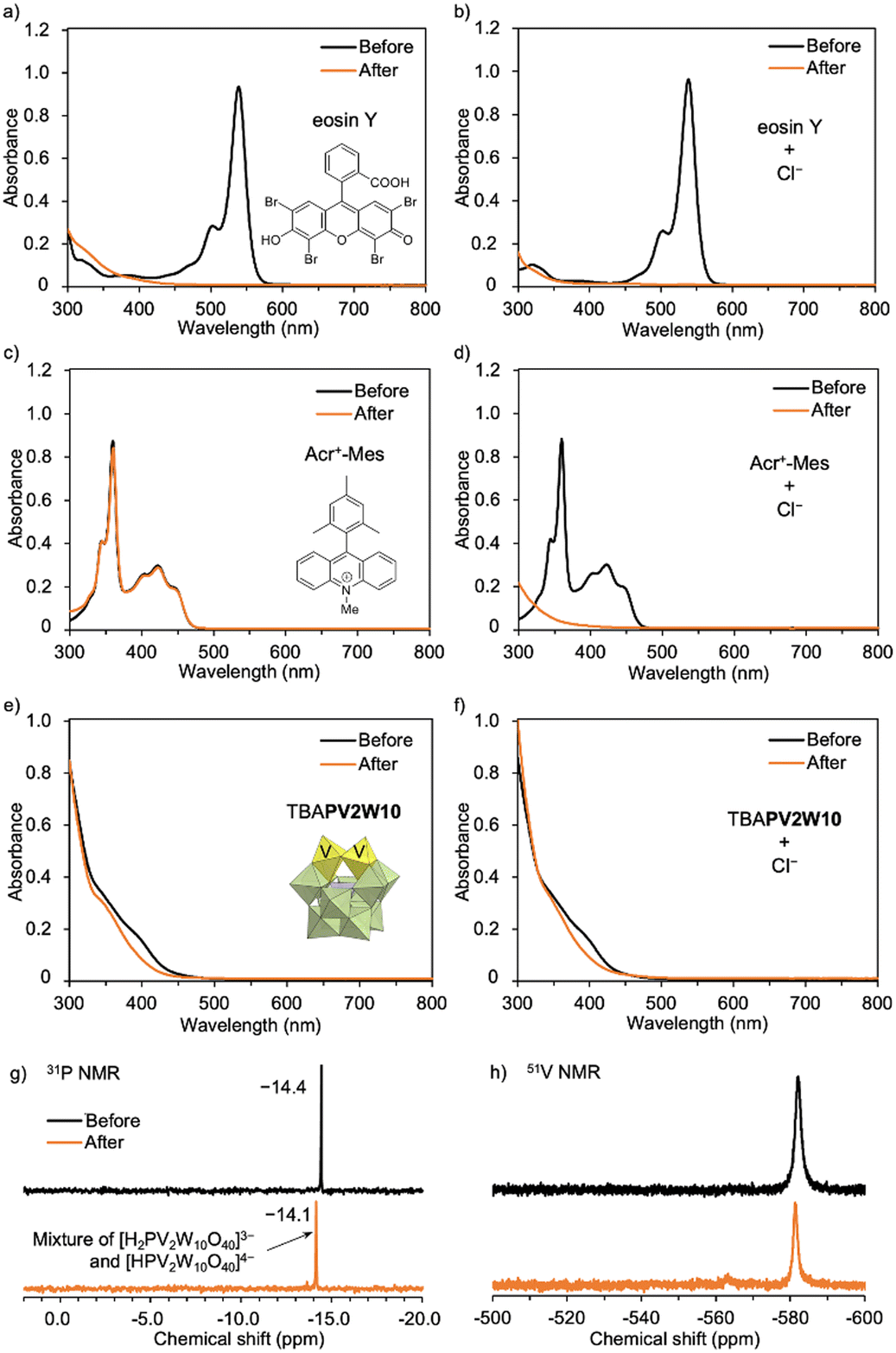 | ||
| Fig. 2 Stability test for various photocatalysts. UV-vis spectra of (a and b) Eosin Y, (c and d) Acr+-Mes ClO4−, (e and f) TBAPV2W10 before and after PCL degradation in the absence and presence of Cl− by photo-irradiation (λ > 350 nm) for 4 h. (g) 31P and (h) 51V NMR spectra of TBAPV2W10 before and after PCL degradation in the presence of Cl− by photo-irradiation (λ > 350 nm) for 4 h. | ||
Next, we investigated the effect of Cl− by employing various counter cations of Cl− and other anions as additives while using TBAPV2W10 as a photocatalyst (Table 2). When tetramethylammonium (TMA), tetrabutylphosphonium (TBP), and tetraphenylphosphonium (TPP) were employed as counter cations instead of TBA, they exhibited a comparable PCL degradation activity (in terms of degradation rate) to that under the system employing TBACl (Table 2, entries 1–6). This indicated that the choice of counter cation did not have any noticeable effect on this system. Furthermore, the inorganic salts LiCl and NaCl also accelerated PCL degradation under TBAPV2W10 photocatalysis, despite their incomplete dissolution in the solution (Table 2, entries 7 and 8). In contrast, when bromide (Br−), iodide (I−), and hydrogen sulfate (HSO4−) ions were employed instead of Cl−, the degradation rates significantly changed depending on the type of anion used (Table 2, entries 9–13 vs. entries 3–6). While Br− and HSO4− showed PCL degradation capabilities via TBAPV2W10 photocatalysis, the degradation rates were significantly lower than when Cl− was employed. When TBAI was employed, the degradation hardly proceeded (Table 2, entry 12). Interestingly, the PCL degradation rates decreased in the order of TBACl > TBABr > TBAI, aligning with the order of the electron detachment energies of halogen anions (X−),17 the order of the bond dissociation energies of H–X (X = Cl, Br, and I), and the HAT ability of the corresponding halogen radicals (X˙).10 These results indicated that this photocatalytic degradation system proceeded via oxidation of X− to the corresponding radicals by TBAPV2W10 photocatalysis, followed by halogen-radical-mediated HAT, resulting in the generation of carbon radicals on the polymer.
|
|
||||
|---|---|---|---|---|
| Entry | Additive | M w (kg mol−1) | (Mw0 − Mw)/Mw0 (%) | M w/Mn |
| Reaction conditions: PCL (40 mg), TBAPV2W10 (4 mg), additive (0.014 mmol), O2 (1 atm), acetonitrile (4 mL), photo-irradiation (λ > 350 nm), 4 h. | ||||
| 1 | (Before reaction) | 20.8 (Mw0) | — | 1.72 |
| 2 | — | 19.4 | 7 | 1.94 |
| 3 | TBACl | 10.2 | 51 | 1.89 |
| 4 | TMACl | 8.65 | 58 | 1.92 |
| 5 | TBPCl | 8.82 | 58 | 1.97 |
| 6 | TPPCl | 9.16 | 56 | 1.90 |
| 7 | LiCl | 11.4 | 45 | 2.06 |
| 8 | NaCl | 16.4 | 21 | 2.00 |
| 9 | TBABr | 16.5 | 21 | 1.86 |
| 10 | TBPBr | 15.1 | 27 | 1.80 |
| 11 | TPPBr | 15.0 | 28 | 1.82 |
| 12 | TBAI | 20.0 | 4 | 1.73 |
| 13 | TBAHSO4 | 16.7 | 20 | 2.04 |

To investigate this hypothesis, we conducted the reaction in the presence of 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO) or 2,6-di-tert-butyl-p-cresol (BHT), which are commonly used radical scavengers. When TEMPO was added to the reaction, the degradation rate decreased from 51% to 24% (Table S2,† entries 2 and 3). Similarly, the addition of BHT also led to a decrease in the degradation rate (Table S2,† entries 2 and 4). PCL degradation was inhibited by radical scavengers, suggesting that the process is regulated by a radical-mediated mechanism. In particular, TEMPO showed a greater suppression than BHT, suggesting that degradation proceed with the generation of carbon radicals followed by oxygen insertion.18,19 The 1H NMR spectrum of PCL after photocatalytic degradation by TBAPV2W10 in the presence of Cl− showed the formation of formate esters and aldehydes, which derived from oxidative chain cleavage (Fig. S9†).
To confirm the generation of Cl˙ during the photocatalytic reaction, we applied photo-irradiation to an acetonitrile solution containing TBAPV2W10, Cl−, and styrene in an O2 atmosphere. The subsequent GC-mass analysis of the solution confirmed the production of phenacyl chloride along with other oxidation products (Fig. S10 and S11†). Phenacyl chloride likely formed through the addition of Cl˙ to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of styrene, followed by the introduction of O2 at the benzyl position.20 These results supported the conversion of Cl− to Cl˙ by the TBAPV2W10 photocatalysis.
C bond of styrene, followed by the introduction of O2 at the benzyl position.20 These results supported the conversion of Cl− to Cl˙ by the TBAPV2W10 photocatalysis.
Density functional theory calculations were employed to delve deeper into the mechanism of this system. First, the reactivity of the photo-excited PV2W10 with Cl− was evaluated. The spin density distribution of the lowest triplet state of the PV2W10–Cl− complex showed that one of the unpaired electrons was located at the Cl atom, indicating that SET occurred from the photocatalyst to the Cl atom, resulting in PV2W10˙−–Cl˙ (Fig. S12†). Thermodynamic calculations also confirmed the experimental results. As shown in Table S3,† the one-electron oxidation of Cl− by the lowest triplet state of PV2W10 was exergonic. The Gibbs free energy of the reaction for PV2W10 (T1) + Cl− → PV2W10− + Cl˙ was negative (−18.3 kcal mol−1), revealing that this electron transfer was thermodynamically favorable. Importantly, the one-electron oxidation by the ground state PV2W10 was 42.3 kcal mol−1 weaker than in the excited state, and the electron abstraction from Cl− was thermodynamically unfavorable (+23.9 kcal mol−1). These results indicated that Cl˙ may have formed through the oxidation of Cl− by the photo-excited PV2W10 rather than the ground-state one. Furthermore, we confirmed that Cl˙ can abstract a hydrogen atom from PCL to produce a carbon-centered radical. The Gibbs free energy of the reaction for abstraction of a hydrogen atom from model PCL using Cl˙ ranged from −11 to −17 kcal mol−1 (Table S4†). Moreover, the calculations corresponded to the order of the PCL degradation rates: Cl− > Br− > I− (Table 2). Both the electron detachment energy17 and the Gibbs free energy of the reaction (Table S3†) indicated that the heavier halide ion was more easily oxidized, but the reactivity of the formed radicals reversed (Table S4†). In particular, the iodine-mediated HAT reactions were highly endergonic in all cases, which is consistent with the experimental results showing essentially no degradation reaction.
Based on these findings, we propose a plausible mechanism regulating this multi-stimuli-responsive polymer degradation (Fig. 3). Cl− was oxidized by photo-exited PV2W10 to form Cl˙, which performed HAT on the polymer to generate a carbon radical, followed by oxidative chain cleavage.
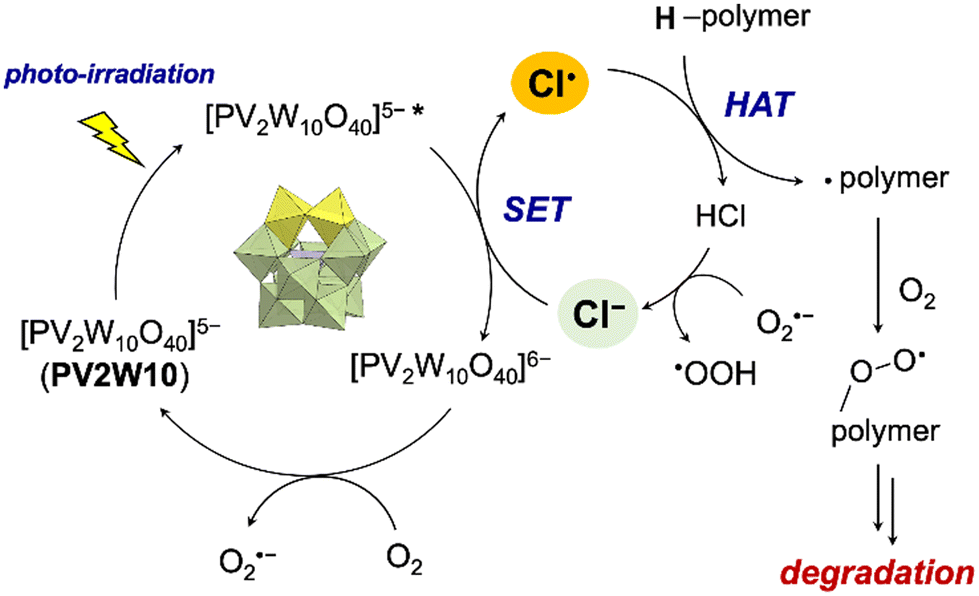 | ||
| Fig. 3 Proposed mechanism regulating multi-stimuli-responsive polymer degradation by TBAPV2W10 and Cl−. | ||
Finally, we investigated the applicability of the multi-stimuli-responsive polymer degradation system to various polymers (Fig. 4 and Table S4†). In addition to degrading PCL, this system can efficiently degrade various other polymers, such as poly(1,4-butylene adipate) (PBA), polyvinyl acetate (PVAC), and cellulose acetate (CA), by photo-irradiation in the presence of both TBAPV2W10 and Cl−. In particular, the degradation rate of these polymers was significantly enhanced in the presence of Cl−. In contrast, polyethers with low bond dissociation energies and low redox potentials, such as poly(propylene glycol) (PPG), underwent degradation via TBAPV2W10 photocatalysis, with moderate enhancement of the degradation rate in the presence of Cl−. The system was also shown to degrade polymers in water by using a water-soluble Cs salt of PV2W10 (Cs5[γ-PV2W10O40], CsPV2W10). By employing the photocatalysis of CsPV2W10, polyethylene glycol (PEG) degradation was facilitated in the presence of NaCl in water.
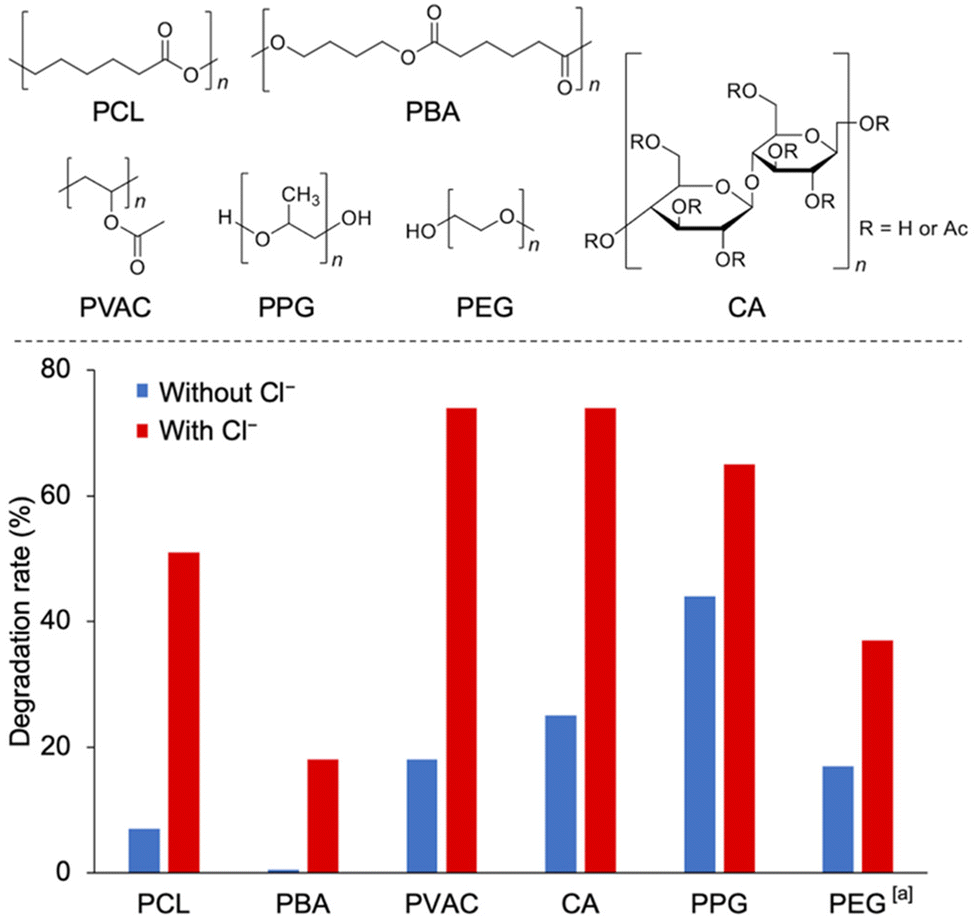 | ||
| Fig. 4 Photocatalytic degradation of various polymers by TBAPV2W10 in the absence and presence of Cl−. Reaction conditions: polymer (40 mg), TBAPV2W10 (4 mg), TBACl (4 mg), O2 (1 atm), acetonitrile (4 mL), photo-irradiation (λ > 350 nm), 4 h. a CsPV2W10 (1.8 mg), NaCl (0.42 mg), water (4 mL), 2 h. | ||
The results revealed a good correlation between the degradation rate of the polymers and their redox properties. Table S3† shows that the Gibbs free energy of the reaction for polyethers (PPG and PEG) was comparable to that of Cl−. Thus, a direct electron transfer from the polymer to the photocatalyst is a plausible mechanism that works similarly in the absence of Cl− ions, and such a mechanism is consistent with the experiment (Fig. 4), showing that polyethers can degrade quickly even without Cl−. The carbon radical cations generated by the transfer of electrons to the photocatalyst may also cause fragmentation. The enhancement of polymer degradation by Cl− may be due to an increase in ion-derived radical initiators. In contrast, the SET reaction of polyesters (PCL and PBA) and PVAC was slightly endergonic, and the SET mechanism leading to the formation of carbon radical cations was less favorable (Table S3†). Furthermore, the HAT by Cl˙ provided an exergonic reaction pathway to the polymer's radical species (Table S4†).
Conclusions
In conclusion, we developed a photocatalytic polymer degradation system that requires multiple stimuli. This system harnesses the photoredox property of a polyoxometalate, TBA4H[γ-PV2W10O40] (TBAPV2W10), requiring both light and chloride ions (Cl−) as stimuli. In the presence of Cl−, the degradation rate of PCL, defined as (Mw0 − Mw)/Mw0 (%) (where Mw0 is Mw before reaction), using TBAPV2W10 under photo-irradiation was significantly enhanced (degradation rate 7% without Cl−, 51% with Cl−). This enhancement was attributed to the oxidation of Cl− to highly reactive chlorine radical (Cl˙) by the photo-activated TBAPV2W10. The Cl˙ species exhibited a notable ability to transfer hydrogen atoms, allowing their abstraction from polymers to generate carbon-centered radicals for subsequent polymer degradation. Although organic and organometallic photocatalysts significantly decomposed under the conditions of photocatalytic polymer degradation in the presence of Cl−, TBAPV2W10 retained its structure even under these highly oxidative conditions. Moreover, this multi-stimuli-responsive polymer degradation system demonstrated its effectiveness across various types of polymer and was capable of operating even under sunlight and in water. We believe that this system will open new possibilities for the development of polymer degradation methods in environments where Cl− and light can be harnessed, such as the ocean.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We gratefully acknowledge financial support from the New Energy and Industrial Technology Development Organization (NEDO), JST FOREST (JPMJFR213M), JSPS KAKENHI (22H04971), and the JSPS Core-to-Core program. We thank Prof. K. Ito and Dr S. Ando (The University of Tokyo) for their help in polymer analysis and fruitful discussion.References
- (a) D. K. A. Barnes, F. Galgani, R. C. Thompson and M. Barlaz, Philos. Trans. R. Soc. London, Ser. B, 2009, 364, 1985–1998 CrossRef CAS PubMed; (b) J. R. Jambeck, R. Geyer, C. Wilcox, T. R. Siegler, M. Perryman, A. Andrady, R. Narayan and K. L. Law, Science, 2015, 347, 768–771 CrossRef CAS PubMed; (c) R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed; (d) M. MacLeod, H. P. H. Arp, M. B. Tekman and A. Jahnke, Science, 2021, 373, 61–65 CrossRef CAS PubMed; (e) A. Chamas, H. Moon, J. Zheng, Y. Qiu, T. Tabassum, J. H. Jang, M. Abu-Omar, S. L. Scott and S. Suh, ACS Sustainable Chem. Eng., 2020, 8, 3494–3511 CrossRef CAS; (f) A. Cózar, F. Echevarria, J. I. González-Gordillo, X. Irigoien, B. Úbeda, S. Hernández-León, A. T. Palma, S. Navarro, J. García-de-Lomas, A. Ruiz, M. L. Fernández-de-Puelles and C. M. Duarte, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 10239–10244 CrossRef PubMed.
- (a) J. Hopewell, R. Dvorak and E. Kosior, Philos. Trans. R. Soc. London, Ser. B, 2009, 364, 2115–2126 CrossRef CAS PubMed; (b) S. C. Kosloski-Oh, Z. A. Wood, Y. Manjarrez, J. P. de los Rios and M. E. Fieser, Mater. Horiz., 2021, 8, 1084–1129 RSC; (c) I. Vollmer, M. J. F. Jenks, M. C. P. Roelands, R. J. White, R. J. White, T. van Harmelen, P. de Wild, G. P. Van der Laan, F. Meirer, J. T. F. Keurentjes and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2020, 59, 15402–15423 CrossRef CAS PubMed; (d) T. P. Haider, C. Völker, J. Kramm, K. Landfester and F. R. Wurm, Angew. Chem., Int. Ed., 2019, 58, 50–62 CrossRef CAS PubMed; (e) M. S. Qureshi, A. Oasmaa, H. Pihkola, I. Deviatkin, A. Tenhunen, J. Mannila, H. Minkkinen, M. Pohjakallio and J. Laine-Ylijoki, J. Anal. Appl. Pyrolysis, 2020, 152, 104804 CrossRef CAS; (f) A. Rahimi and J. M. García, Nat. Rev. Chem., 2017, 1, 0046 CrossRef; (g) J. Zhou, T. G. Hsu and J. Wang, Angew. Chem., Int. Ed., 2023, 62, e202300768 CrossRef CAS; (h) R. Miandad, M. A. Barakat, A. S. Aburiazaiza, M. Rehan and A. S. Nizami, Process Saf. Environ. Prot., 2016, 102, 822–838 CrossRef CAS.
- (a) W. Li, W. Zhao, H. Zhu, Z. J. Li and W. Wang, J. Mater. Chem. A, 2023, 11, 2503–2527 RSC; (b) Z. Ouyang, Y. Yang, C. Zhang, S. Zhu, L. Qin, W. Wang, D. He, Y. Zhou, H. Luo and F. Qin, J. Mater. Chem. A, 2021, 9, 13402–13441 RSC; (c) I. Nabi, A. U. R. Bacha, F. Ahmad and L. Zhang, J. Environ. Chem. Eng., 2021, 9, 105964 CrossRef CAS; (d) P. Ebrahimbabaie, K. Yousefi and J. Pichtel, Sci. Total Environ., 2022, 806, 150603 CrossRef CAS PubMed; (e) S. Chu, B. Zhang, X. Zhao, H. S. Soo, F. Wang, R. Xiao and H. Zhang, Adv. Energy Mater., 2022, 12, 2200435 CrossRef CAS.
- N. Fu, G. S. Sauer and S. Lin, J. Am. Chem. Soc., 2017, 139, 15548–15553 CrossRef CAS PubMed.
- (a) Y. Itabashi, H. Asahara and K. Ohkubo, Chem. Commun., 2023, 59, 7506–7517 RSC; (b) S. Bonciolini, T. Noël and L. Capaldo, Eur. J. Org. Chem., 2022, e202200417 CrossRef CAS.
- (a) J. K. Kochi, J. Am. Chem. Soc., 1962, 84, 2121–2127 CrossRef CAS; (b) Y. Abderrazak, A. Bhattacharyya and O. Reiser, Angew. Chem., Int. Ed., 2021, 60, 21100–21115 CrossRef CAS; (c) F. Juliá, ChemCatChem, 2022, 14, e202200916 CrossRef; (d) B. J. Shields and A. G. Doyle, J. Am. Chem. Soc., 2016, 138, 12719–12722 CrossRef CAS; (e) S. M. Treacy and T. Rovis, J. Am. Chem. Soc., 2021, 143, 2729–2735 CrossRef CAS PubMed; (f) Y. C. Kang, S. M. Treacy and T. Rovis, ACS Catal., 2021, 11, 7442–7449 CrossRef CAS PubMed; (g) S. Oh and E. E. Stache, J. Am. Chem. Soc., 2022, 144, 5745–5749 CrossRef CAS PubMed.
- (a) R. Bevernaegie, S. A. M. Wehlin, E. J. Piechota, M. Abraham, C. Philouze, G. J. Meyer, B. Elias and L. Troian-Gautier, J. Am. Chem. Soc., 2020, 142, 2732 CrossRef CAS PubMed; (b) A. M. Deetz, L. Troian-Gautier, S. A. M. Wehlin, E. J. Piechota and G. J. Meyer, J. Phys. Chem. A, 2021, 125, 9355 CrossRef CAS PubMed.
- (a) K. Ohkubo, A. Fujimoto and S. Fukuzumi, Chem. Commun., 2011, 47, 8515–8517 RSC; (b) H. P. Deng, Q. Zhou and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 12661–11284 CrossRef CAS.
- (a) S. Rohe, A. O. Morris, T. McCallum and L. Barriault, Angew. Chem., Int. Ed., 2018, 57, 15664–15669 CrossRef CAS; (b) M. Zidan, A. O. Morris, T. McCallum and L. Barriault, Eur. J. Org. Chem., 2020, 1453–1458 CrossRef CAS; (c) C. Y. Huang, J. Li and C. J. Li, Nat. Commun., 2021, 12, 4010 CrossRef CAS PubMed.
- S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255–263 CrossRef CAS PubMed.
- (a) M. T. Pope, Heteropoly and Isopoly Oxometalates, Springer, Berlin, 1983 CrossRef; (b) C. L. Hill and C. M. Prosser-McCartha, Coord. Chem. Rev., 1995, 143, 407–455 CrossRef CAS; (c) N. Mizuno and M. Misono, Chem. Rev., 1998, 98, 199–218 CrossRef CAS PubMed; (d) R. Neumann, Prog. Inorg. Chem., 1998, 47, 317 CAS; (e) D. L. Long, R. Tsunashima and L. Cronin, Angew. Chem., Int. Ed., 2010, 49, 1736–1758 CrossRef CAS PubMed; (f) S. S. Wang and G. Y. Yang, Chem. Rev., 2015, 115, 4893–4962 CrossRef CAS; (g) A. Misra, K. Kozma, C. Streb and M. Nyman, Angew. Chem., Int. Ed., 2020, 59, 596–612 CrossRef CAS; (h) J. M. Cameron, G. Guillemot, T. Galambos, S. S. Amin, E. Hampson, K. M. Haidaraly, G. N. Newton and G. Izzet, Chem. Soc. Rev., 2022, 51, 293–328 RSC; (i) K. Yonesato, D. Yanai, S. Yamazoe, D. Yokogawa, T. Kikuchi, K. Yamaguchi and K. Suzuki, Nat. Chem., 2023, 15, 940–947 CrossRef CAS; (j) N. Ogiwara and S. Uchida, Chem Catal., 2023, 3, 100607 CrossRef CAS.
- (a) K. Suzuki, N. Mizuno and K. Yamaguchi, ACS Catal., 2018, 8, 10809–10825 CrossRef CAS; (b) C. Streb, Dalton Trans., 2012, 41, 1651–1659 RSC; (c) H. Lv, Y. V. Geletii, C. Zhao, J. W. Vickers, G. Zhu, Z. Luo, J. Song, T. Lian, D. G. Musaev and C. L. Hill, Chem. Soc. Rev., 2012, 41, 7572–7589 RSC; (d) J. Lan, Y. Wang, B. Huang, Z. Xiao and P. Wu, Nanoscale Adv., 2021, 3, 4646–4658 RSC; (e) X. Chen, H. Wu, X. Shi and L. Wu, Nanoscale, 2023, 15, 9242–9255 RSC.
- (a) C. Li, C. Gu, K. Yamaguchi and K. Suzuki, Nanoscale, 2023, 15, 15038–15042 RSC; (b) N. Minezawa, K. Suzuki and S. Okazaki, Phys. Chem. Chem. Phys. 10.1039/D4CP00362D.
- C. Li, K. Suzuki, N. Mizuno and K. Yamaguchi, Chem. Commun., 2018, 54, 7127–7130 RSC.
- (a) D. P. Hari and B. König, Chem. Commun., 2014, 50, 6688–6699 RSC; (b) X. Z. Fan, J. W. Rong, H. Wu, Q. Zhou, H. P. Deng, J. D. Tan, C. W. Xue, L. Z. Wu, H. Tao and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 8514–8518 CrossRef CAS PubMed; (c) D. M. Yan, J. R. Chen and W. J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 378–380 CrossRef CAS PubMed.
- A. Alvarez-Martin, S. Trashin, M. Cuykx, A. Covaci, K. De. Wael and K. Janssens, Dyes Pigm., 2017, 145, 376–384 CrossRef CAS.
- B. Winter, R. Weber, I. V. Hertel, M. Faubel, P. Jungwirth, E. C. Brown and S. E. Bradforth, J. Am. Chem. Soc., 2005, 127, 7203–7214 CrossRef CAS PubMed.
- T. Vogler and A. Studer, Synthesis, 2008, 1979–1993 CAS.
- W. A. Yehye, N. A. Rahman, A. Ariffin, S. B. A. Hamid, A. A. Alhadi, F. A. Kadir and M. Yaeghoobi, Eur. J. Med. Chem., 2015, 101, 295–312 CrossRef CAS PubMed.
- S. Tian, X. Jia, L. Wang, B. Li, S. Liu, L. Ma, W. Gao, Y. Wei and J. Chen, Chem. Commun., 2019, 55, 12104–12107 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr00394b |
| This journal is © The Royal Society of Chemistry 2024 |