
Novel ternary Zintl phosphide halides Ba3P5X (X = Cl, Br) with 1D helical phosphorus chains: synthesis, crystal and electronic structure†
Mohd
Ishtiyak
,
S. M. Gayomi K.
Samarakoon
,
Thimira
Kandabadage Don
,
Spencer R.
Watts
and
Sviatoslav
Baranets
 *
*
Department of Chemistry, Louisiana State University, Baton Rouge, Louisiana 70803, USA. E-mail: sbaranets@lsu.edu
First published on 6th March 2024
Abstract
Black single crystals of two novel ternary phosphide halides, Ba3P5Cl and Ba3P5Br, were grown using molten metal Pb-flux high-temperature reactions. These compounds were structurally characterized with the aid of the single-crystal X-ray diffraction (SCXRD) method at 100(2) K. The SCXRD shows that both compounds are isostructural and adopt a new structure type (space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c, No. 167, Z = 6) with unit cell parameters a = 14.9481(16) Å, c = 7.3954(11) Å and a = 15.045(4) Å, c = 7.537(3) Å for Ba3P5Cl and Ba3P5Br, respectively. Cl− and Br− anions are octahedrally coordinated by Ba2+ cations, thus composing a face-sharing 1D infinite chain 1∞[XBa3]5+ running along the [001] direction. Moreover, the crystal structures feature peculiar one-dimensional disordered infinite helical chains of 1∞P−, composed of partially occupied phosphorous atoms, each being a superposition of three symmetrical copies of the ordered phosphorus chain, with continuity along the c-axis. Ba3P5X (X = Cl, Br) compounds are charge-balanced heteroanionic Zintl phases according to the charge-partitioning scheme (Ba2+)3[P−]5X−. The presumed semiconducting behavior of both compounds corroborates well with the results of the electronic structure calculations performed with the aid of the TB-LMTO-ASA code.
c, No. 167, Z = 6) with unit cell parameters a = 14.9481(16) Å, c = 7.3954(11) Å and a = 15.045(4) Å, c = 7.537(3) Å for Ba3P5Cl and Ba3P5Br, respectively. Cl− and Br− anions are octahedrally coordinated by Ba2+ cations, thus composing a face-sharing 1D infinite chain 1∞[XBa3]5+ running along the [001] direction. Moreover, the crystal structures feature peculiar one-dimensional disordered infinite helical chains of 1∞P−, composed of partially occupied phosphorous atoms, each being a superposition of three symmetrical copies of the ordered phosphorus chain, with continuity along the c-axis. Ba3P5X (X = Cl, Br) compounds are charge-balanced heteroanionic Zintl phases according to the charge-partitioning scheme (Ba2+)3[P−]5X−. The presumed semiconducting behavior of both compounds corroborates well with the results of the electronic structure calculations performed with the aid of the TB-LMTO-ASA code.
 Sviatoslav Baranets | Dr. Sviatoslav Baranets is an Assistant Professor of Chemistry at Louisiana State University. He obtained his Ph.D. in Inorganic Chemistry from the Institute of General and Inorganic Chemistry of the National Academy of Sciences of Ukraine in 2015. Later, he was a DAAD scholarship holder and worked as a visiting researcher at the Technische Universität Dresden, Germany. He was also a postdoctoral researcher at the University of Delaware. His research area primarily focuses on the design, synthesis, and characterization of emerging heteroanionic inorganic functional materials for energy applications. |
Introduction
Heteroanionic quantum materials have recently received increased attention due to unusual structural characteristics and exceptional properties.1,2 These compositionally and structurally complex phases that are marked by an anionic sublattice comprising of multiple anion types remain relatively underdeveloped compared to the families of homoanionic compounds.1 However, they have already demonstrated a broad range of physical properties and have recently emerged as promising platforms for nanotechnology,3,4 energy-related applications,5–11 catalysis,12,13 and developing topological quantum materials.14A particularly intriguing subset of heteroanionic compounds is composed of pnictogens (i.e., electronegative elements of group 15, Pn = P, As, Sb, Bi) combined with halogens (i.e., nonmetallic elements of group 17, X = F, Cl, Br, I). These heteroanionic ternary and multinary solid-state phases contain negatively charged pnictogen and halogen anions and are termed pnictide halides (e.g. [Ba2+]2[P7]3−[Cl−]). They are distinct from perovskite-like pnictohalides materials, where pnictogens exhibit positive oxidation states (e.g. [Ag+][Bi3+]2[I−]7),15 These compounds are known for their rich structural chemistry due to their potential to stabilize various types of homoatomic finite and infinite pnictogen-based units or metal cluster compounds.16–20 Recently discovered pnictide halides and pnictohalides, such as A2As7X (A = Ba, Sr, Eu; X = Cl, Br, I) or W6PCl17, are considered unique low-dimensional semiconductors with promising tunability that may lead to the discovery of promising electronic and spintronic materials.20,21
Our research group is interested in advancing novel heteroanionic compounds that are characterized by distinct charge transfer, aiming to develop narrow-gap semiconducting materials suitable for energy applications. Within this domain, polar intermetallic Zintl phases are known for their abundant chemical compositions, variability of crystal structures, and wide range of physical properties.22 The bonding in these charge-balanced compounds is a blend of ionic and covalent interactions. Typically, the electropositive elements from groups 1 and 2 donate electrons to the more electronegative elements, which then use these electrons to form covalent bonds and create complex anionic substructures.23
Heteroanionic pnictide halides, such as A2Pn7X,16,20,24 A5Pn3X,25 or A2PnX (A = Ca, Sr, Ba, Eu; Pn = P–Bi; X = Cl–I)26–28 exemplify charge-balanced double Zintl compounds, showing a range of band gaps and an excellent tunability of crystal structures and electronic properties. Several A2Pn7X phases crystallize in the chiral space group P213 – a desirable prerequisite for the materials requiring chiral control and enantioselectivity.20 As these phosphide and arsenide halides do not have inversion symmetry, they can be used for application in second harmonic generation and ferroelectricity.29
Among pnictogens, phosphorous exhibits remarkable homoatomic bonding formation capability, which results in the constant discovery of novel phosphorus allotropes, like recently reported P11,30 structurally diverse polyanions, such as infinite 1∞P− chains in BaP2,31 oligomeric [P9] units in Ba5P9, or 0D [P7]3− clusters in A2Pn7X.16,32 Being motivated by the rich structural chemistry of Zintl phosphides and developing frontiers for the synthesis of heteroanionic materials that crystallize in the non-centrosymmetric and chiral space groups, we discovered two novel isostructural phosphide halides, Ba3P5Cl and Ba3P5Br. These compounds adopt a novel structure type featuring a peculiar infinite helical phosphorus chain. We report their synthesis, structural characterization, and chemical bonding study.
Results and discussion
Synthesis
The Ba–P–X (X = Cl, Br) ternary compositional diagrams are relatively underexplored, with a limited number of compounds currently known. These include Ba2P7X of Ba2P7Cl structure type,16 Ba2PCl of α-NaFeO2 structure type,28 and the solid solution Ba16P10.14Br1.59 of Th3P4 structure,28 which is rather believed to be a defect binary Ba4P2.67 phase (Fig. S1†).31The initial discovery of Ba3P5Cl was made during an experiment targeting a quaternary Ba–In–Cl–P composition. This process, mirroring the synthetic procedure described in the Experimental section, included InCl3 in the reaction mixture instead of BaCl2. However, the resulting product contained no known In-bearing compounds, yielding only crystals of the new Ba3P5Cl phase. EDS studies also confirmed the absence of foreign elements in the obtained crystals. Subsequently, we attempted the synthesis of the title phase without InCl3 to elucidate its role. Initially, it was hypothesized that the partial decomposition of InCl3 at high temperatures might provide a source of chlorine to the reaction mixture. However, our findings revealed that indium halides are not essential for synthesizing Ba3P5Cl or Ba3P5Br. We found that the presence of anhydrous barium halides alone is sufficient, serving as an equally excellent source of halogens and yielding nearly high-purity samples of the title phases.
We also attempted to explore periodic trends and synthesize the isostructural Ba3P5I phase, but several synthetic attempts were unsuccessful, and the crystals of Ba2P7I were mainly found as a major phase.16 Experimental efforts to synthesize this hitherto unknown iodide using InI3 also yielded the same ternary 2–7–1 phase.
The single crystals of Ba3P5Cl and Ba3P5Br synthesized in our study exhibit extreme sensitivity to air and moisture, posing significant challenges in the study of their properties. These crystals rapidly decompose upon brief exposure to ambient conditions and undergo slow deterioration even with the presence of trace amounts of moisture.
Crystal structure description of Ba3P5X (X = Cl, Br)
The detailed crystallographic analysis of the Ba3P5X phases uncovers a great degree of complexity, as depicted in Fig. 1. Both phosphide halides are isostructural and crystallize in the rhombohedral centrosymmetric space group Rc (No. 167) within the trigonal crystal system with six formula units per unit cell. The crystalline arrangement of both compounds, being unprecedented in existing literature, represents a novel structure type. As can be observed from Table 1, the unit cell parameters for the bromide analog are substantially larger, aligning with the larger atomic radius of bromine compared to chlorine.
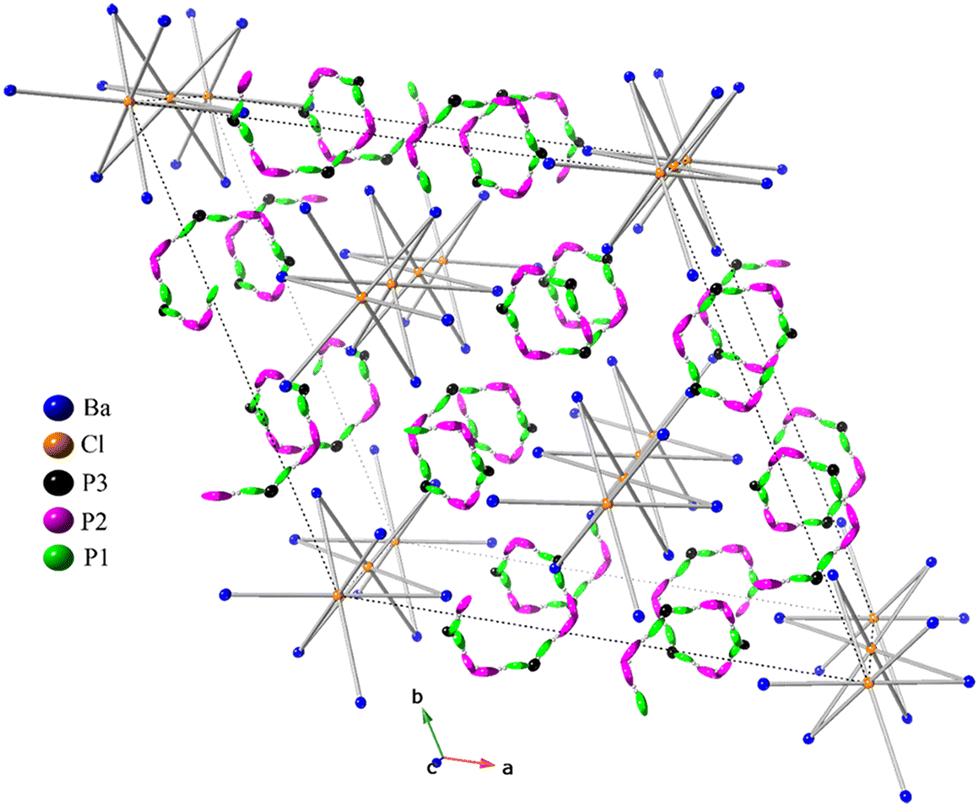 | ||
| Fig. 1 A unit cell view of the crystal structure of Ba3P5X (X = Cl and Br) approximately along the [001] direction. Barium atoms are blue, halogen atoms are orange, whereas three different phosphorus atoms are depicted as green, pink, and black. Ellipsoids for phosphorus atoms are plotted with the 50% probability level. The unit cell is dashed. | ||
| Refined formula | Ba3P5Cl | Ba3P5Br |
|---|---|---|
| a R 1 = ∑||Fo| − |Fc||/∑|Fo|. wR2 = {∑[w(Fo2 − Fc2)2]/∑wFo4}1/2, w = 1/[σ2(Fo2) + (AP)2 + (BP)], where P = (Fo2 + 2Fc2)/3; A and B are weight coefficients. | ||
| fw/g mol−1 | 602.32 | 646.78 |
| Space group |
Rc |
|
| a/(Å) | 14.9481(16) | 15.045(4) |
| c/(Å) | 7.3954(11) | 7.537(3) |
| V (Å3) | 1431.1(4) | 1477.6(10) |
| Z | 6 | |
| ρ cal./g cm−3 | 4.193 | 4.361 |
| μ(Mo Kα)/cm−1 | 132.8 | 166.5 |
| Collected/independent reflections | 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 569/493 569/493 |
5390/471 |
| R 1 (I > 2σ(I))a | 0.025 | 0.048 |
| wR2 (I > 2σ(I))a | 0.040 | 0.106 |
| R 1 (all data)a | 0.030 | 0.066 |
| wR2 (all data)a | 0.041 | 0.111 |
| Δρmax,min/e− Å−3 | 0.83, −2.09 | 1.82, −2.45 |
| CCDC | 2313247 | 2313248 |
The asymmetric unit of Ba3P5X (X = Cl, Br) comprises a total of 5 crystallographically independent sites with respective site symmetries: Ba1 (.2), X1 (.), P1 (1), P2 (1), and P3 (.2). The atomic arrangement in these 3–5–1 phases demonstrates that the structures of both compounds can be considered as pseudo-one-dimensional (Fig. 1). The halogen atoms are octahedrally coordinated by six equidistant Ba atoms, with Ba–X distances of ca. 3.32 Å and 3.41 Å for the chloride and the bromide phases, respectively. These values are slightly longer than the sum of Ba2+ and X− ionic radii33 or those observed in the Ba2P7Cl phase,16 indicating a high degree of ionicity for these contacts, vide infra. [XBa6] octahedra are interconnected by sharing two opposite faces, forming a 1D chain 1∞[XBa3]5+ along the [001] direction. Compared to the edge- and vertex-sharing polyhedral arrangements, the face-sharing of polyhedral units is often regarded as the least stable arrangement,34 primarily because of the closest positioning of two ions (X−) to each other.
However, the most interesting component of the reported crystal structures is the arrangement of phosphorus atoms. Their connectivity forms an anionic substructure identified as 1D infinite disordered chains also oriented along [001] direction. These anionic 1∞P− helical chains are a hallmark of the Ba3P5X crystal structure and exemplify the structural abundance of phosphorus Zintl chemistry.
To better understand the observed structural disorder in these anionic helical chains, we provide the reader with the details of the structure determination flow for Ba3P5Cl. The initial structural model, derived from the first iteration of the structure solution, indicated four crystallographically independent sites. Based on the geometrical parameters and coordination environments, these atomic positions were assigned as one Ba, one Cl, and two P atoms. In this model, each Ba atom can be viewed as 6-coordinated by two chlorine atoms and four P–P pairs. However, we observed immediate problems with this disorder-free structure with the refined composition of Ba3P6Cl. Among them were (i) anomalously large atomic displacement parameters for both P atoms, (ii) a residual electron density peak of 4.5 e− Å−3 and a hole of −4.9 e− Å−3 near one phosphorus atom, and (iii) a relatively short interatomic P–P distance of ca. 1.84 Å. These observations clearly pointed to the disorder of the P atoms. Allowing free occupancies for these atoms indicated a clear underoccupancy of one phosphorus atom (P1) and a refined formula approaching the Ba3P5Cl composition. However, the second phosphorus atom (P2) still exhibited large ADP alongside a significant residual electron density peak, located ca. 0.9 Å away. By assigning this peak as another partially occupied P atom (P3) and splitting the P1 atom, we achieved a nearly featureless Fourier difference map with reasonable ADP values without introducing additional constraints. This structural model was successfully applied to the bromide analog, yielding an identical structural solution with heavy disorder on phosphorus sites.
Refined occupancies of phosphorus atomic sites for both chloride and bromide phases were found to be close to 1/3, yielding nearly stoichiometric refined compositions of Ba3P4.8(3)Cl and Ba3P4.9(5)Br, respectively. Given the large calculated ESD values, we considered the “off-stoichiometry” as an artifact arising from differences in Ueq values for all three phosphorus atomic sites. Constraining their occupancies to 1/3 did not affect the quality of the structural refinement and resulted in minor changes in ADP values. On the contrary, such a structural model represents a stoichiometric Ba3P5X (X = Cl, Br) composition, which corroborates with the idea of charge balance, as discussed further below.
The rationalization of the disorder within the 1∞P chain aligns well with the model containing constrained occupancies. The observed minimal P–P interatomic distances are too short to be classified as covalent bonds, so the simultaneous existence of all phosphorus atomic sites can only be achieved through their partial SOFs. At first glance, the chain looks like a continuous flow of phosphorus atoms within the helix with almost no breaches, leading to nearly uniformly distributed or “smeared” electron density, as can be seen from the alignment of refined ellipsoids in Fig. 2a. However, the partial occupancies of phosphorus atoms allows for various structural scenarios with specific atomic sequence. Given the covalent radius of 1.07 Å for P,35 one should expect an interatomic P–P distance of ca. 2.15 Å. In the presented structural model, we observe a wide range of similar distances for P1–P1 (2.20–2.22 Å), P1–P2 (2.18–2.22 Å), and P2–P3 (2.15–2.19 Å) contacts so that each disordered helix can be viewed as a superposition of three symmetry copies of the same ordered chain sequenced to the {P1–P1–P2–P3–P2} fragment of partially occupied phosphorus atoms (Fig. S6†), each with SOF of 1/3. The random shift of adjacent chains in the trigonal ab plane with respect to each other justifies the observed disorder. Lastly, the described sequence of five phosphorus atoms exemplifies one complete turn of the helix, with a pitch equal to the c lattice parameter.
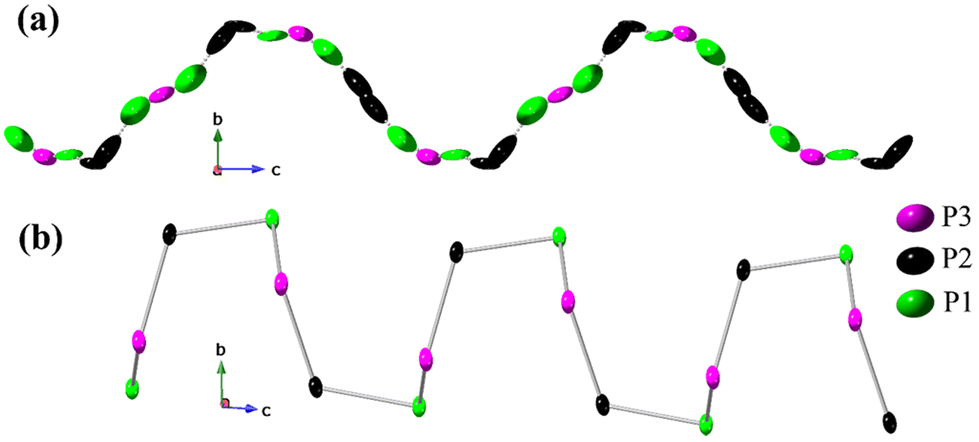 | ||
| Fig. 2 Propagation of 1D 1∞P1− helical chains found in (a) the crystal structures of Ba3P5X and (b) AP2 (A = Sr, Ba). The P–P bonding interactions have been represented with dotted lines in Ba3P5X, while solid-bond has been used in the case of AP2. Ellipsoids are plotted with a 50% probability level. | ||
Described charged helical phosphorus chains are stabilized by the 1∞[ClBa3]5+ matrix, yet their fundamental existence arises from the ability of pnictogen elements to form homoatomic Pn–Pn contacts (Pn = P, As, Sb, Bi) and to construct electron-rich polyanionic networks.36 Such structural fragments are commonly observed in the realm of Zintl pnictides or heteroanionic Zintl-like salts.23,37 In addition, phosphorus is known for forming a variety of allotropes of different dimensionalities,30,38–42 including some exotic predicted forms like helical coil phosphorus allotropes.43
The disordered helical phosphorus chains described in this work (Fig. 2a) have never been previously reported, although disorder-free phosphorus helices are known for binary phosphides AP2 (A = Sr, Ba) (Fig. 2b)31 and ternary heteroanionic compounds like SnPX (Fig. S6†)44,45 and Cd2P3X (X = Cl, Br, and I).46 The crystal structure of the SnPI compound reported by Pfister et al. in 201644 can be described as a double helical SnIP strand composed of inner 1∞[P−] and outer 1∞[SnI]+ chains oriented along the [100] direction. Reported 1∞P chains in SnIP (Fig. S6†) are structurally similar, yet not identical to those presented in this work. Bond angles within the {P1–P1–P2–P3–P2} fragment in Ba3P5Cl range from 127.2° to 130.2°, whereas observed angles within the phosphorus chain in SnPI are much smaller, being within 95.5–96.7° range. The zigzag phosphorus chains were also found running along the c-axis in the structures of valence-precise compound Cd2P3X (X = Cl, Br, and I) and monoclinic-NiP2.47 1D helical chains of 1∞P− were also reported in α-CdP2 and β-CdP2 phases.48 These compounds exhibit P–P interatomic distances within the range of 2.10 to 2.35 Å, similar to those in the title Ba3P5X phases close to the sum of their covalent radii.35 It would be worth noting that one-dimensional heteroanionic semiconducting material, SnPI, exhibited anisotropic mechanical properties like steel, which strongly motivate the further exploration of novel helix-containing compounds.44
Although heavy disorder significantly complicates the bonding analysis, we observe that the shortest and longest Ba–P bond distances in Ba3P5X range from ca. 3.13(1) to 3.29(1) Å, comparable with those in BaP2.31 Similar Ba–P distances are also observed in Ba2P7X (X = Br and I).16 As Table 2 shows, Ba–X bond distances are longer in Ba3P5Br compared to Ba3P5Cl, which aligns with the larger ionic radius of Br, while Ba–P distances do not show significant variation in Ba3P5X.
| Ba3P5Cl | Distances (Å) | Ba3P5Br | Distances (Å) |
|---|---|---|---|
| Ba1–Cl1 | 3.3207(4) | Ba1–Br1 | 3.4110(11) |
| Ba1⋯P1 | 3.229(6) | Ba1⋯P1 | 3.254(13) |
| Ba1⋯P2 | 3.161(9) | Ba1⋯P2 | 3.176(14) |
| Ba1⋯P3 | 3.236(4) | Ba1⋯P3 | 3.257(6) |
| P1⋯P1 | 2.204(12) | P1⋯P1 | 2.22(3) |
| P1⋯P2 | 2.178(10) | P1⋯P2 | 2.219(19) |
| P2⋯P3 | 2.144(7) | P2⋯P3 | 2.194(10) |
The described structural model of Ba3P5X (X = Cl, Br) with constrained occupancies of phosphorus atoms, as detailed in Table 3, leads to the charge-balanced composition, where disordered phosphorus helix fully accounts for a −5 charge necessary for electron balancing the space-fillers between the 1D chains of 1∞[ClBa3]5+. A charge-balanced composition can be represented as (Ba2+)3(P−)5X− (X = Cl, Br), satisfying the Zintl count and pointing to the potential semiconducting behavior.
| Atoms | Site | x | y | z | U eq |
|---|---|---|---|---|---|
| a U eq is the one-third value of the trace of orthogonalized Uij tensor. Atom site occupancies of P1, P2, and P3 were constrained as 1/3. | |||||
| Ba3P5Cl | |||||
| Ba1 | 18e | 0.8154(1) | 0 | ¼ | 0.014(1) |
| P1 | 36f | 0.0254(5) | 0.2674(5) | 0.1776(8) | 0.027(2) |
| P2 | 36f | 0.0861(4) | 0.3150(7) | 0.1047(18) | 0.032(2) |
| P3 | 18e | 0.2383(8) | 0 | ¼ | 0.021(2) |
| Cl1 | 6b | 0 | 0 | 0 | 0.012(1) |
| Ba3P5Br | |||||
| Ba1 | 18e | 0.8110(1) | 0 | ¼ | 0.015(1) |
| P1 | 36f | 0.0256(11) | 0.2664(12) | 0.1746(15) | 0.023(3) |
| P2 | 36f | 0.0857(9) | 0.3142(10) | 0.105(2) | 0.023(3) |
| P3 | 18e | 0.2362(11) | 0 | ¼ | 0.016(3) |
| Br1 | 6b | 0 | 0 | 0 | 0.013(1) |
XPS studies
To further elucidate the validity of the proposed charge-partitioning and understand the chemical states in Ba3P5X, we conducted X-ray Photoelectron Spectroscopy (XPS) analysis on the polycrystalline samples. The results of these studies are presented in the ESI (Fig. S4 and S5).† The survey spectra (Fig. S4a and S5a†) of Ba3P5X indicated the presence of peaks corresponding to the assigned elements, corroborating well with the SEM-EDS analysis, vide supra. The characteristic binding energy peaks of Ba 3d5/2 (780.26 eV for Ba3P5Cl and 780.41 eV for Ba3P5Br) confirmed that Ba is in the +2 oxidation state, in agreement with previous reports on Ba2+ ions.49 The high-resolution spectrum of Cl exhibited 2p3/2 and 2p1/2 peaks at 198.66 eV and 200.43 eV, respectively, while the Br 3d5/2 and 3d3/2 peaks were observed at 69.11 eV and 70.17 eV, corresponding to the binding energies of Cl− and Br−, respectively.49The high-resolution P spectra for both compounds reveal two broad asymmetric peaks. The deconvolution of these peaks suggested the presence of binding energies indicative of several phosphorus states, categorized into higher (BE > 132.5 eV), mid-range (BE ∼ 130 eV), and lower binding (BE ∼ 129 eV) energy regions. Such spectral behavior has already been previously reported for transition metal phosphides, indicating the presence of oxidized, unreacted, and reduced P chemical states.50
As previously mentioned, both title compounds were found to be air-sensitive, hence, a certain degree of surface oxidation was anticipated. This was further evidenced by the presence of an O 1s peak (∼531 eV) and the higher binding energy (P 2p) peaks of 132.40 eV and 132.57 eV found in the XPS spectra of Ba3P5Cl and Ba3P5Br, respectively. These bands are characteristic of oxidized species, such as phosphates, although this phenomenon was also observed during the XPS studies of metal phosphides, such as Ba2Si3P6 and NiP2, indicating similar surface oxidation.47,49–51
The phosphorus peaks observed in the lower binding energy regions of the recorded XPS spectra are particularly noteworthy, as they indicate reduced chemical states. This feature is commonly seen in the XPS spectra of metal phosphides, such as Ni2+(P1−)2, which typically exhibit characteristic P 2p3/2 peaks in this region. For instance, the bands at 128.7 eV and 129.3 eV correspond to P1− in NiP2.47 Similarly, the XPS spectra of Ba3P5Cl and Ba3P5Br displayed major P 2p3/2 peaks at 128.98 eV and 129.71 eV, respectively. These binding energies suggest that phosphorous predominantly exists in a −1 oxidation state in Ba3P5X, aligning well with the proposed charge-partitioning scheme. Distinct peaks corresponding to the phosphorous 2p3/2 and 2p1/2 were not observed, likely due to the high sensitivity of the compounds to air and moisture. Additionally, the use of high pass energy during the XPS measurement and the presence of oxidized species impurities may have contributed to the broadening of the P 2p peaks for both compounds.50
Electronic structure and chemical bonding
While the Zintl–Klemm formalism, considerations of electronegativity differences, and experimental studies of oxidation states have provided insights into an empirical understanding of the bonding situation in the title Ba3P5X (X = Cl, Br) phases, a more comprehensive understanding of the electronic structure and stability of these compounds was gained from our electronic structure calculations. Due to extensive structural disorder in both title phases, calculations using experimentally obtained atomic coordinates were not feasible. Consequently, we considered a structurally similar model that keeps structural features and chemical composition of the title phases.As we discussed above, the structural disorder likely originates from the different packing of the phosphorus chains in the trigonal ab plane. With the assistance of the ISODISTORT tool,52,53 we transformed the trigonal structure with the space group Rc to a monoclinic one with the space group C2/c, in which the original three phosphorus sites split into eight with lower multiplicities.
We can remove some of the P sites, leaving three that are symmetrically equivalent to those in the starting structure. The resulted structural model (Fig S7a and Tables S1, S2†) retained the helical 1∞P− chains while maintaining full occupancy of all atoms, refined 3–5–1 composition, and physically meaningful P–P distances. The periodicity of the helixes remained identical to those in the refined structures. We could reduce the symmetry even further by transforming the structure to the non-centrosymmetric space group Cc while yielding a reasonable structural model (Fig. S7b and Tables S1, S4†). The results of the electronic structure calculations for Ba3P5Cl and Ba3P5Br compositions using the C2/c structural model are presented in Fig. 3 and 4, respectively, whereas the Total/PDOS calculations for the Cc structural model are included and displayed in Fig. S8 of the ESI.† Crystal Orbital Hamilton Population curves are presented in Fig. 5.
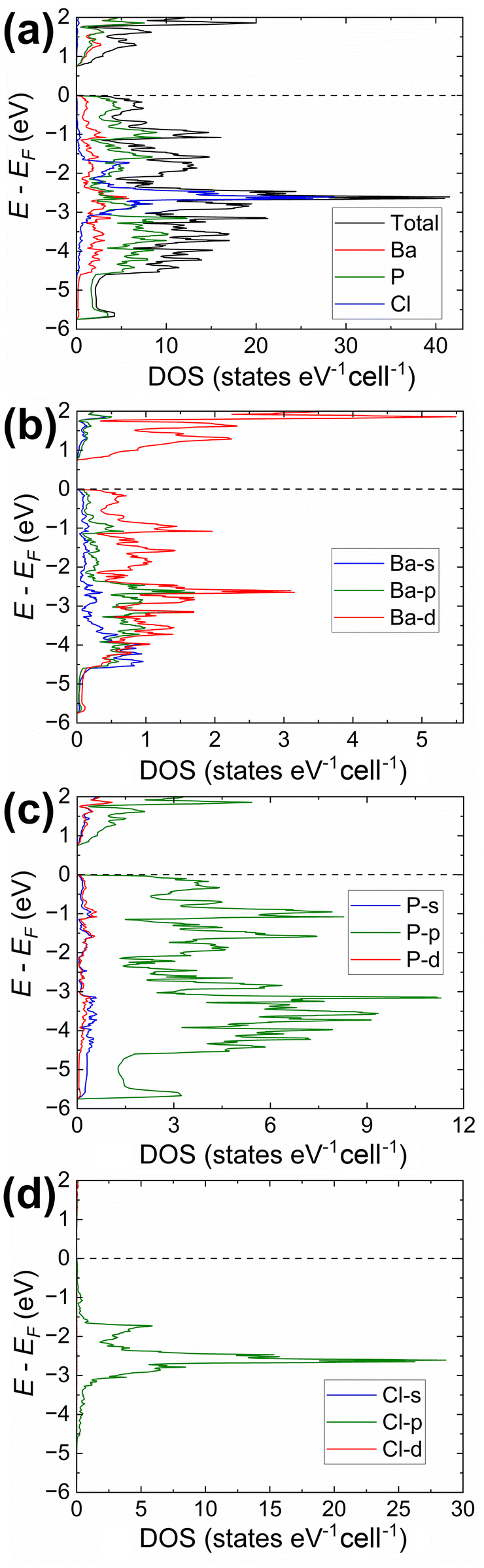 | ||
| Fig. 3 Electronic structure in Ba3P5Cl (C2/c structural model; Fig. S7a and Tables S1, S2†): (a) total DOS together with projected densities of states for constituting elements; (b)–(d) partial DOS calculated for the different orbitals of Ba, P, and Cl, respectively. | ||
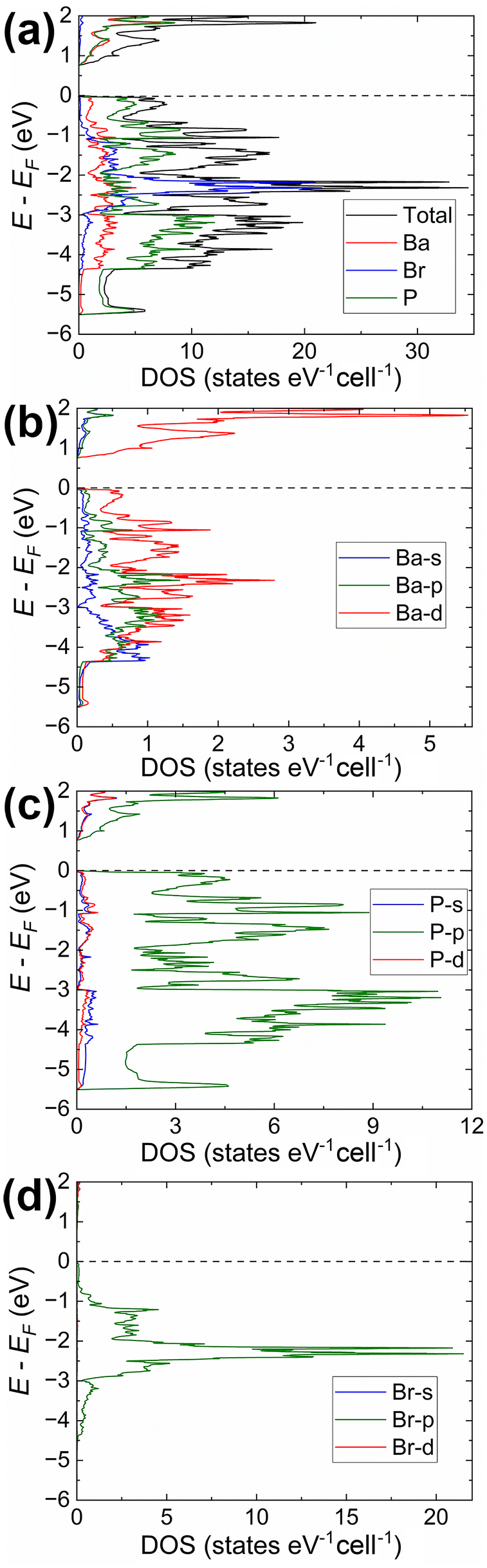 | ||
| Fig. 4 Electronic structure in Ba3P5Br (C2/c structural model; Fig. S7a and Tables S1, S3†): (a) total DOS together with projected densities of states for constituting elements; (b)–(d) partial DOS calculated for the different orbitals of Ba, P, and Br, respectively. | ||
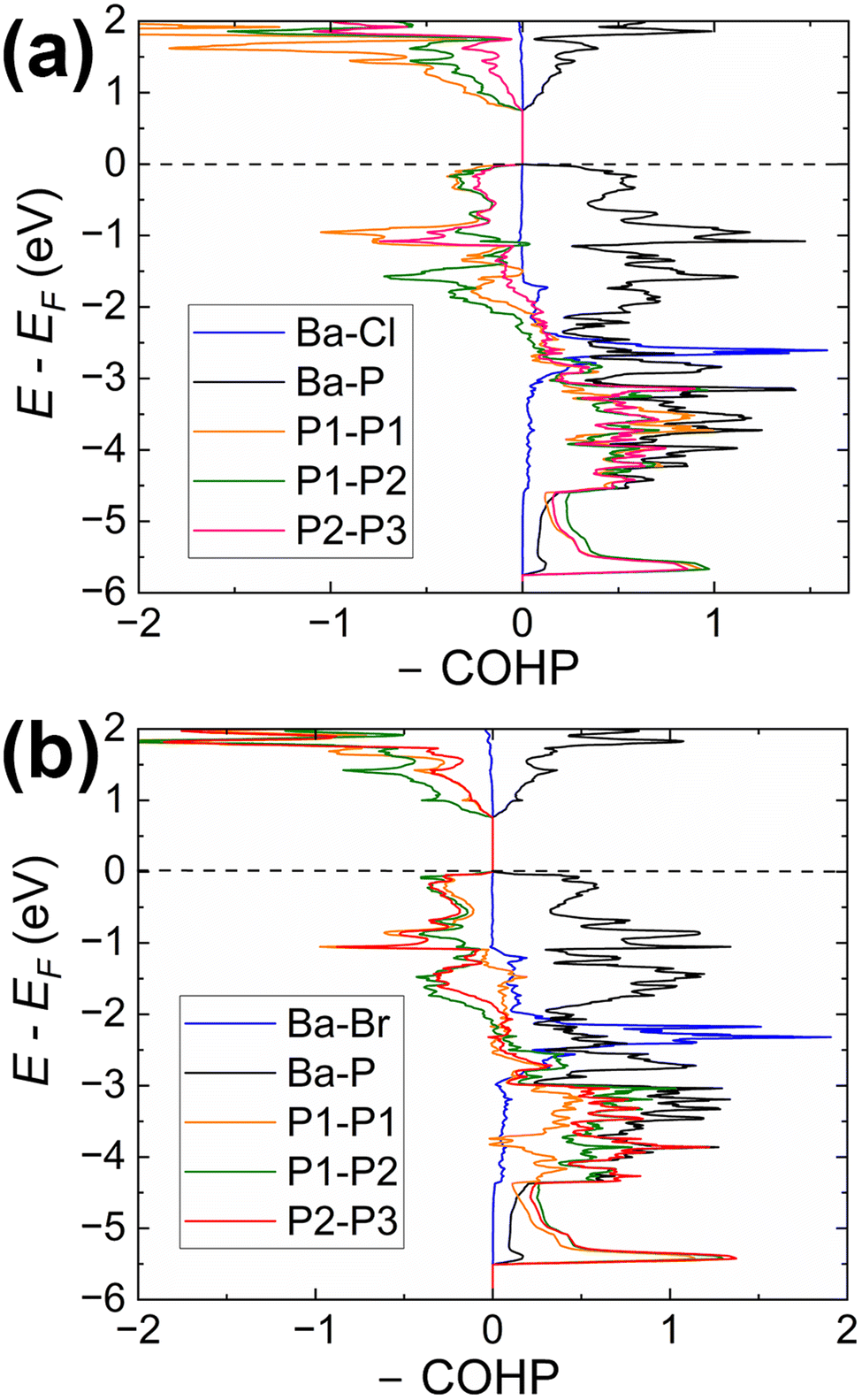 | ||
| Fig. 5 (a) COHP curves for selected averaged Ba–X and Ba–P interactions and three specific interactions (P1–P1, P1–P2, and P2–P3) within the helical 1∞P chain in Ba3P5Cl (a) and Ba3P5Br (b) calculated for the C2/c structural model. | ||
The analysis of the electronic structure indicates an almost identical band gap opening of ca. 0.73 eV for both compounds, even though it is expected to observe smaller band gaps for compounds with heavier elements within the same family of isostructural phases.54 On the contrary, the calculated band gap for the Ba3P5Cl model in space group Cc is significantly larger (ca. 1.2 eV, Fig. S8†). Here we should note, that none of the discussed ordered models were geometrically optimized, which is believed to be a major contributing factor for the observed inconsistency. However, details of the electronic structure can provide insightful results for the understanding of the bonding of the title compounds, primarily due to the similar crystal structures of Ba3P5X in C2/c and Rc models.
Our calculations indicate that the electronic structures of the chloride and bromide compounds are nearly identical; therefore, we will focus our discussion primarily on the characterization of the chloride phase. The total (TDOS) and partial (PDOS) density of states diagrams demonstrate that the conduction band is predominantly populated by the Ba-d and P-p orbitals, with minor contributions from chlorine. Conversely, the states in the valence band are contributed mainly by P-p and Cl-p orbitals. Cl-p states are located in a relatively narrow energy window between −0.5 eV and −4.5 eV, with Ba-s, Ba-p, and Ba-d orbitals also contributing within this range. Such orbital mixing may be indicative of Ba–Cl bonding interactions, as this region exhibits domains of bonding character in the computed COHP curves (Fig. 5).
States above −5.7 eV are significantly contributed by P-p orbitals, while in the lower energy regions (not shown in Fig. 3), P-s orbital contributions emerge, indicating a small degree of hybridization in the phosphorus atoms. Empty Ba-d orbitals also contribute between the VBM and −5.7 eV, where the significant contributions from P-p orbitals are observed, pointing to the orbital mixing between atoms and, therefore, Ba–P interactions. The involvement and mixing of Ba-d orbitals with P-p and Cl-p orbitals clearly illustrates the departure from the fully ionic approximation of the Zintl–Klemm concept, which posits the role of metal cations as electron donors and space-filling entities.
The Crystal Orbital Hamilton Population curves (Fig. 5) provide insights into the bonding situation in the title compounds. COHP analysis indicates optimized Ba–P and Ba–X contacts, while P–P contacts are underoptimized, as evidenced by domains of antibonding character in both the valence and conduction bands. This pattern is characteristic for homoatomic electron-rich pnictogen–pnictogen bonds and was previously observed in several Zintl pnictides, such as Ba2Cd2P3 and Ba3Cd2P4 with homoatomic P–P contacts.55
The average integrated COHP (–ICOHP) values for Ba–X and Ba–P contacts are almost identical, falling within the ranges of 0.20–0.23 eV per bond and 0.28–0.38 eV per bond, respectively. These values indicate a significant charge transfer from electron-donating Ba to electron-accepting P and halogen atoms. –ICOHP values for P–P bonds in Ba3P5Cl range between 2.96–3.58 eV per bond, pointing to the large degree of covalency for these interactions.
Experimental
Synthesis
The synthesis of barium phosphide halides, Ba3P5Cl and Ba3P5Br, was accomplished utilizing the following supplied chemicals: Ba rod (99.9%, Thermo Scientific), BaCl2 powder (99.9%, J.T. Baker), BaBr2 powder (99.9%, Thermo Scientific), P powder (99.9%, Beantown Chemical) and Pb powder (99.99%, Strem Chemicals). All synthetic and post-synthetic procedures were performed inside an argon-filled glovebox with oxygen and moisture levels below 1 ppm. An oxide layer was removed from the barium rod, whereas other reagents were used as received. Black-colored, irregular-shaped single crystals of the phosphide halide phases can be readily synthesized utilizing a metal-flux reaction in alumina crucibles. Ba3P5Cl can be produced in a reaction with stoichiometric amounts of barium chloride (86.4 mg, 0.415 mmol), phosphorous (128.5 mg, 4.149 mmol), barium (285.0 mg, 2.075 mmol), and lead flux (2500 mg, 12.065 mmol). The reaction mixture was loaded into an alumina crucible in the argon-filled glove box. Subsequently, the crucible was placed inside a fused silica tube, which was then sealed under vacuum. Quartz wool was used to cover the crucible, serving as a filter during the centrifugation step. After that, the tube was placed inside a muffle furnace for the heat treatment using the following temperature profile. The furnace temperature was initially ramped up from 373 K to 1273 K at a rate of 100 K h−1, followed by a sustained hold at 1273 K for 20 h. Subsequently, the temperature was gradually decreased to 923 K at a cooling rate of 5 K h−1. Upon reaching the target temperature, the tube was removed from the furnace, and the sample was centrifugated, enabling the separation of the lead flux from the reaction product. Once cooled, the ampoule was transferred to the glovebox and crack-opened. The crystalline reaction product was retrieved from the alumina crucible and stored under the protective atmosphere of the argon-filled glovebox.After the successful synthesis of single crystals of Ba3P5Cl, we succeeded in producing the isostructural bromide analog by loading stoichiometric amounts of barium bromide (114.9 mg, 0.529 mmol), phosphorous (119.7 mg, 3.865 mmol), barium (264.4 mg, 1.925 mmol), and the excess of lead (2500 mg, 12.065 mmol) using the same reaction procedure and temperature profile that were used for the synthesis of single crystals of Ba3P5Cl. It was noted that the crystals of the bromine-containing compound were found to be more stable than the crystals of the chlorine-containing compound. However, neither phase's crystals are sufficiently stable in the presence of air to allow for the comprehensive analysis of physical properties.
X-ray diffraction methods
Powder X-ray diffraction (PXRD) patterns were recorded in the reflection mode at room temperature using a Rigaku Miniflex 600 diffractometer (Cu Kα radiation, λ = 1.5406 Å, Ge monochromator). The PXRD data were acquired using a θ–θ step-scan mode with 0.05° increments and a 2 s collection period per step. Due to the air sensitivity of the samples, they were covered by a thin amorphous film that prevented immediate oxidation and allowed for the data collection. Collected PXRD patterns indicate the presence of the title phases, yet we cannot assert the phase purity due to the high degree of phase amorphization (Fig. S3†).Structural studies were performed with the aid of the single-crystal X-ray diffraction (SCXRD) method. Low-temperature data were collected at 100(2) K on a Bruker APEX II CCD diffractometer equipped with a graphite-monochromized Mo Kα radiation (λ = 0.71073 Å). Suitable-sized single crystals of Ba3P5Cl and Ba3P5Br were selected, cut to the desired dimensions under the Paratone-N oil, and mounted on a low-background MiTeGen plastic loop attached to the goniometer head. A constant flow of chilled nitrogen gas stream was applied throughout the data collection process to solidify the oil and protect the crystals.
Single crystal data for both compounds were acquired with a scan width of 0.5° and a scan time of 20 s per frame, maintaining a crystal-to-detector distance of 50 mm. The intensity data acquisition, data reduction, cell refinement processes, and absorption correction were conducted using the APEX3 software package.56,57 The single-crystal data of both compounds were integrated with a Rhombohedral R setting. The centrosymmetric Rc space group was used to solve the crystal structures. The determination of atomic positions was successfully located by the direct method utilizing the SHELXT program, whereas the structure refinement was performed with full-matrix least-squares methods on F2 with SHELXL.58,59 Olex2 program was used as a graphical interface.60 Atomic coordinates were standardized using the STRUCTURE TIDY Program.61 The crystal structures were visualized with the help of CrystalMaker software.62
Selected details of the data collection and relevant crystallographic parameters are given in Tables 1–3. CSD 2313247 and 2313248 contain the complete supplementary crystallographic data for the compounds discussed in this paper.†
Elemental microanalysis
Irregularly shaped black crystals of Ba3P5Cl and Ba3P5Br were carefully selected and mounted on carbon tape affixed to an aluminum stub. All sample preparation procedures were carried out inside the glovebox due to the extreme air sensitivity of the title compounds.Energy-dispersive X-ray spectroscopy (EDS) analysis was performed using a Thermo Scientific (TFS Helios G5 PFIB CXe) scanning electron microscope equipped with an OXFORD Instruments Ultim Max Detector spectrometer. For both sets of crystals, EDX data were acquired at an operating acceleration voltage of 20 kV. Semiquantitative analyses were executed on various crystals at multiple sites to verify the uniformity of elemental compositions throughout the crystals. The EDS studies detected the presence of elements barium, phosphorous, and chlorine/bromine in the crystals, with approximate elemental ratios of Ba:P:X ≈ 3:5:1, as shown in Fig. S2.† These compositions are consistent with the ratios obtained from the SCXRD structure refinements.
X-ray photoelectron spectroscopy (XPS) studies
The XPS measurements were performed to investigate the formal oxidation states of elements in both compounds (Ba3P5Cl and Ba3P5Br). The polycrystalline samples were first ground into fine powder and then pressed into pellets using a hydraulic press. These pellets were placed on the surface of conductive adhesive carbon tape, which was affixed to an aluminum sheet. All these procedures were performed inside the argon-filled glove box to minimize the chances of oxidation, as both compounds are susceptible to air and moisture. The prepared samples were kept inside an argon-filled sealed container for transportation to the XPS instrument and loaded instantly inside the analysis chamber. The pressure in the analysis chamber was maintained between 2.7 × 10−9 and 6 × 10−9 mbar during the measurements. The data were collected on Scienta Omicron ESCA 2SR X-ray Photoelectron Spectroscope instrument equipped with a high-performance monochromatic Al-Kα X-ray radiation source. The operating working voltage and current were set to 15 kV and 20 mA throughout the data measurement. Due to the semiconducting nature of samples, no charge correction was performed. Before plotting the spectra, the observed binding energies for both samples were calibrated with respect to carbon (C1s = 284.8 eV). The spectra of polycrystalline samples were analyzed using CasaXPS software.63 Survey spectra of both compounds were collected in the binding energy range of 0 to 1200 eV.Electronic structure calculations
Electronic structure calculations were performed for the Ba3P5Cl and Ba3P5Br compositions within the framework of the TB-LMTO-ASA code.64 Two disorder-fee structural models with lowered symmetries in space groups C2/c and Cc were considered for calculations. Unit cell parameters and fractional atomic coordinates used for calculations are presented in Tables S1–S4.† The visualization of the crystal structures used for calculations is presented in Fig. S7.† The von-Barth-Hedin exchange–correlation functional was utilized, and empty spheres were introduced to satisfy the atomic sphere approximation (ASA) requirements.65 Integration over the k-space was achieved using the tetrahedron method.66 After confirming convergence, a 10 × 10 × 10 k-point grid was employed to sample the Brillouin zone. The Fermi level, denoted as EF = 0 eV, was set as the reference energy level. We carried out density of states (DOS) and atom-projected density of states (PDOS) calculations to elucidate the electronic structure of these novel phosphide halide compounds. The calculations employed a basic set of orbitals including Ba [5s, (6p), 5d], P [3s, 3p, (3d)], Cl [(4s) 3p, (3d)], Br [(5s) 4p, (4d)]; the downfolded orbitals are in parentheses. Furthermore, Crystal Orbital Hamilton population (COHP) calculations were performed to provide deeper insights into the chemical bonding within these compounds.67 All calculations were facilitated with the assistance of the modules available in the LMTO program.Conclusions
Two new ternary phosphide halides, Ba3P5Cl and Ba3P5Br, have been synthesized using the Pb flux technique. Both compounds represent a novel structure type and crystallize in the rhombohedral space group Rc. These double Zintl salts are charge-balanced and are presumed to function as narrow band gap semiconductors. Ba3P5Cl and Ba3P5Br phases exhibit peculiar one-dimensional infinite helical 1∞P− chains of partially occupied phosphorus atoms. The discovery of these previously unknown compounds with unique structural unit opens a possibility for further exploration and development of novel functional inorganic materials and nanomaterials. The Ba3P5X compounds may act as precursors for synthesizing hitherto unknown non-centrosymmetric solids by utilizing preexisting helical phosphorus chains.68 Further exploratory work involving different synthetic approaches to sequester these phosphorus chains will be worthwhile pursuing, as it could lead to the stabilization of this novel phosphorus form at the nanoscale or within a more suitable matrix than 1∞[XBa3]5+, potentially enhancing the ambient stability of compounds featuring this unique building block.
Author contributions
Mohd Ishtiyak: writing – original draft, methodology, formal analysis, investigation, data curation, and validation. S. M. Gayomi K. Samarakoon, Thimira Kandabadage Don, Spencer R. Watts: formal analysis, validation. Sviatoslav Baranets: conceptualization, methodology, supervision, validation, project administration, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the College of Science and Department of Chemistry at Louisiana State University (start-up funding). SB also acknowledges the LSU Provost's Fund for Innovation in Research – Council on Research Summer Stipend Program for the summer support. We thank Dr Dongmei Cao for helping with XPS measurements and data processing. The XPS was performed at the Shared Instrumentation Facility (SIF) at Louisiana State University.References
- H. Kageyama, K. Hayashi, K. Maeda, J. P. Attfield, Z. Hiroi, J. M. Rondinelli and K. R. Poeppelmeier, Nat. Commun., 2018, 9, 772 CrossRef PubMed.
- J. K. Harada, N. Charles, K. R. Poeppelmeier and J. M. Rondinelli, Adv. Mater., 2019, 31, 1805295 CrossRef.
- Y. Nalawade, J. Pepper, A. Harvey, A. Griffin, D. Caffrey, A. G. Kelly and J. N. Coleman, ACS Appl. Electron. Mater., 2020, 2, 3233–3241 CrossRef CAS.
- B. Chitara, T. B. Limbu, J. D. Orlando, Y. Tang and F. Yan, Nanoscale, 2020, 12, 16285–16291 RSC.
- K. Xhaxhiu, C. Kvarnström, P. Damlin and K. Bente, Mater. Res. Bull., 2014, 60, 88–96 CrossRef CAS.
- L.-D. Zhao, J. He, D. Berardan, Y. Lin, J.-F. Li, C.-W. Nan and N. Dragoe, Energy Environ. Sci., 2014, 7, 2900–2924 RSC.
- P. Vaqueiro, G. Guélou, M. Stec, E. Guilmeau and A. V. Powell, J. Mater. Chem. A, 2012, 1, 520–523 RSC.
- Y. Kamihara, T. Watanabe, M. Hirano and H. Hosono, J. Am. Chem. Soc., 2008, 130, 3296–3297 CrossRef CAS.
- Y.-F. Shi, W.-B. Wei, X.-T. Wu, H. Lin and Q.-L. Zhu, Dalton Trans., 2021, 50, 4112–4118 RSC.
- B. M. Oxley, J. B. Cho, A. K. Iyer, M. J. Waters, J. He, N. C. Smith, C. Wolverton, V. Gopalan, J. M. Rondinelli, J. I. Jang and M. G. Kanatzidis, J. Am. Chem. Soc., 2022, 144, 13903–13912 CrossRef CAS.
- M. Yang, W. Liu and S.-P. Guo, Inorg. Chem., 2022, 61, 14517–14522 CrossRef CAS PubMed.
- Y. Subramanian, A. Dhanasekaran, L. A. Omeiza, M. R. Somalu and A. K. Azad, Catalysts, 2023, 13, 173 CrossRef CAS.
- K. Chatterjee and S. E. Skrabalak, ACS Appl. Mater. Interfaces, 2021, 13, 36670–36678 CrossRef CAS PubMed.
- B. Khamari and B. R. K. Nanda, Mater. Res. Express, 2019, 6, 066309 CrossRef CAS.
- A. Chakraborty, N. Pai, J. Zhao, B. R. Tuttle, A. N. Simonov and V. Pecunia, Adv. Funct. Mater., 2022, 32, 2203300 CrossRef CAS.
- J.-A. Dolyniuk and K. Kovnir, Crystals, 2013, 3, 431–442 CrossRef CAS.
- M. Ströbele, O. Oeckler, M. Thelen, R. F. Fink, A. Krishnamurthy, S. Kroeker and H.-J. Meyer, Inorg. Chem., 2022, 61, 17599–17608 CrossRef PubMed.
- M. Ströbele, K. Eichele and H.-J. Meyer, Eur. J. Inorg. Chem., 2011, 2011, 4063–4068 CrossRef.
- J.-A. Dolyniuk, N. Tran, K. Lee and K. Kovnir, Z. Anorg. Allg. Chem., 2015, 641, 1422–1427 CrossRef CAS.
- J.-A. Dolyniuk, S. Lee, N. Tran, J. Wang, L.-L. Wang and K. Kovnir, J. Solid State Chem., 2018, 263, 195–202 CrossRef CAS.
- Y. Qiao and H. Yin, Nanoscale, 2023, 15, 9835–9842 RSC.
- R. Nesper, Z. Anorg. Allg. Chem., 2014, 640, 2639–2648 CrossRef CAS.
- S. Baranets, G. M. Darone and S. Bobev, Z. Kristallogr. - Cryst. Mater., 2022, 237, 1–26 CrossRef CAS.
- E. R. Franke and H. Schäfer, Z. Naturforsch., B: Anorg. Chem., Org. Chem., Biochem., Biophys., Biol., 1972, 27, 1308–1315 CrossRef CAS.
- W. M. Hurng and J. D. Corbett, Chem. Mater., 1989, 1, 311–319 CrossRef CAS.
- C. Hadenfeldt and W. Held, J. Less-Common Met., 1986, 123, 25–35 CrossRef CAS.
- C. Hadenfeldt and H. Herdejürgen, Z. Anorg. Allg. Chem., 1988, 558, 35–40 CrossRef CAS.
- C. Hadenfeldt, Z. Anorg. Allg. Chem., 1977, 436, 113–121 CrossRef CAS.
- M. D. Donakowski, R. Gautier, J. Yeon, D. T. Moore, J. C. Nino, P. S. Halasyamani and K. R. Poeppelmeier, J. Am. Chem. Soc., 2012, 134, 7679–7689 CrossRef CAS PubMed.
- G. Cicirello, M. Wang, Q. P. Sam, J. L. Hart, N. L. Williams, H. Yin, J. J. Cha and J. Wang, J. Am. Chem. Soc., 2023, 145, 8218–8230 CrossRef CAS PubMed.
- J.-A. Dolyniuk, H. He, A. S. Ivanov, A. I. Boldyrev, S. Bobev and K. Kovnir, Inorg. Chem., 2015, 54, 8608–8616 CrossRef CAS PubMed.
- B. Eisenmann and U. Rößler, Z. Anorg. Allg. Chem., 2003, 629, 459–462 CrossRef.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- M. Ishtiyak, G. Panigrahi, S. Jana, J. Prakash, A. Mesbah, C. D. Malliakas, S. Lebègue and J. A. Ibers, Inorg. Chem., 2020, 59, 2434–2442 CrossRef CAS PubMed.
- B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008, 2832–2838 RSC.
- G. A. Papoian and R. Hoffmann, Angew. Chem., Int. Ed., 2000, 39, 2408–2448 CrossRef.
- S. Baranets, A. Ovchinnikov and S. Bobev, in Handbook on the Physics and Chemistry of Rare Earths, eds. J.-C. G. Bünzli and V. K. Pecharsky, Elsevier, 2021, vol. 60, pp. 227–324 Search PubMed.
- M. Aykol, J. W. Doak and C. Wolverton, Phys. Rev. B, 2017, 95, 214115 CrossRef.
- J. Liu, Y. Guo, S. Zhang, Q. Wang, Y. Kawazoe and P. Jena, J. Phys. Chem. C, 2015, 119, 24674–24680 CrossRef CAS.
- A. Pfitzner, Angew. Chem., Int. Ed., 2006, 45, 699–700 CrossRef CAS PubMed.
- A. Pfitzner, M. F. Bräu, J. Zweck, G. Brunklaus and H. Eckert, Angew. Chem., Int. Ed., 2004, 43, 4228–4231 CrossRef CAS.
- Z. Sun, B. Zhang, Y. Zhao, M. Khurram and Q. Yan, Chem. Mater., 2021, 33, 6240–6248 CrossRef CAS.
- D. Liu, J. Guan, J. Jiang and D. Tománek, Nano Lett., 2016, 16, 7865–7869 CrossRef CAS.
- D. Pfister, K. Schäfer, C. Ott, B. Gerke, R. Pöttgen, O. Janka, M. Baumgartner, A. Efimova, A. Hohmann, P. Schmidt, S. Venkatachalam, L. van Wüllen, U. Schürmann, L. Kienle, V. Duppel, E. Parzinger, B. Miller, J. Becker, A. Holleitner, R. Weihrich and T. Nilges, Adv. Mater., 2016, 28, 9783–9791 CrossRef CAS.
- F. Reiter, M. Pielmeier, A. Vogel, C. Jandl, M. Plodinec, C. Rohner, T. Lunkenbein, K. Nisi, A. Holleitner and T. Nilges, Z. Anorg. Allg. Chem., 2022, 648, e202100347 CrossRef CAS.
- P. C. Donohue, J. Solid State Chem., 1972, 5, 71–74 CrossRef CAS.
- B. Owens-Baird, J. Xu, D. Y. Petrovykh, O. Bondarchuk, Y. Ziouani, N. González-Ballesteros, P. Yox, F. M. Sapountzi, H. Niemantsverdriet, Y. V. Kolen'ko and K. Kovnir, Chem. Mater., 2019, 31, 3407–3418 CrossRef CAS.
- J. Chen, C. Lin, F. Xu, S. Yang, Y. Sun, X. Zhao, X. Jiang, B. Li, T. Yan and N. Ye, Chem. Mater., 2020, 32, 10246–10253 CrossRef CAS.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-ray Photoelectron Spectroscopy; Physical Electronics, Inc.: Eden Prairie, MN, 1995 Search PubMed.
- P. E. R. Blanchard, A. P. Grosvenor, R. G. Cavell and A. Mar, Chem. Mater., 2008, 20, 7081–7088 CrossRef CAS.
- J. Mark, J. Wang, K. Wu, J. G. Lo, S. Lee and K. Kovnir, J. Am. Chem. Soc., 2019, 141, 11976–11983 CrossRef CAS PubMed.
- H. T. Stokes, D. M. Hatch and B. J. Campbell, ISODISTORT, ISOTROPY Software Suite, https://iso.byu.edu Search PubMed.
- B. J. Campbell, H. T. Stokes, D. E. Tanner and D. M. Hatch, J. Appl. Crystallogr., 2006, 39, 607–614 CrossRef CAS.
- S. Baranets, A. Balvanz, G. M. Darone and S. Bobev, Chem. Mater., 2022, 34, 4172–4185 CrossRef CAS.
- A. Balvanz, S. Baranets and S. Bobev, J. Solid State Chem., 2020, 289, 121476 CrossRef CAS.
- SAINT, BrukerAXS Inc., 2014 Search PubMed.
- SADABS, BrukerAXS Inc., 2014 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- L. M. Gelato and E. Parth'e, STRUCTURE TIDY - a computer program to standardize and crystal structure data, J. Appl. Crystallogr., 1987, 20, 139–143 CrossRef.
- D. C. Palmer, CrystalMaker, CrystalMaker Software Ltd, Begbroke, Oxfordshire, England, 2014. https://journals.iucr.org/m//services/stdswrefs.htm Search PubMed.
- N. Fairley, CasaXPS, version 2.2.19, Casa Software, Ltd, Teignmouth, Devon, U.K., 2003, https://www.casaxps.com Search PubMed.
- O. Jepsen and O. K. Andersen, The Stuttgart TB-LMTO program, 1999 Search PubMed.
- U. von Barth and L. Hedin, J. Phys. C: Solid State Phys., 1972, 5, 1629 CrossRef CAS.
- P. E. Blöchl, O. Jepsen and O. K. Andersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 16223–16233 CrossRef.
- S. Steinberg and R. Dronskowski, Crystals, 2018, 8, 225 CrossRef.
- P. A. Maggard, C. L. Stern and K. R. Poeppelmeier, J. Am. Chem. Soc., 2001, 123, 7742–7743 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Ba–P–X compositional diagrams, EDS and XPS spectra, PXRD patterns, structural and computational details of the electronic structure calculations. CCDC 2313247-2313248. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3nr06492a |
| This journal is © The Royal Society of Chemistry 2024 |