
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControl of metal–support interaction for tunable CO hydrogenation performance over Ru/TiO2 nanocatalysts†
Heyun
Lin
abc,
Wenzhe
Zhang
c,
Huachen
Shen
 ab,
Hailing
Yu
ab,
Yunlei
An
a,
Tiejun
Lin
*ab and
Liangshu
Zhong
*abc
ab,
Hailing
Yu
ab,
Yunlei
An
a,
Tiejun
Lin
*ab and
Liangshu
Zhong
*abc
aKey Laboratory of Low-Carbon Conversion Science and Engineering, Shanghai Advanced Research Institute, Chinese Academy of Sciences, Shanghai, 201210, China. E-mail: lintj@sari.ac.cn; zhongls@sari.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
cSchool of Physical Science and Technology, ShanghaiTech University, Shanghai, 201210, China
First published on 20th February 2024
Abstract
The catalytic behavior of CO hydrogenation can be modulated by metal–support interactions, while the role of the support remains elusive. Herein, we demonstrate that the presence of strong metal–support interactions (SMSI) depends strongly on the crystal phase of TiO2 (rutile or anatase) and the treatment conditions for the TiO2 support, which could critically control the activity and selectivity of Ru-based nanocatalysts for CO hydrogenation. High CO conversion and olefin selectivity were observed for Ru/rutile-TiO2 (Ru/r-TiO2), while catalysts supported by anatase (a-TiO2) showed almost no activity. Characterization confirmed that the SMSI effect could be neglected for Ru/r-TiO2, while it is dominant on Ru/a-TiO2 after reduction at 300 °C, resulting in the coverage of Ru nanoparticles by TiOx overlayers. Such SMSI could be suppressed by H2 treatment of the a-TiO2 support and the catalytic activity of the as-obtained Ru/a-TiO2(H2) can be greatly elevated from almost inactive to >50% CO conversion with >60% olefin selectivity. Further results indicated that the surface reducibility of the TiO2 support determines the SMSI state and catalytic performance of Ru/TiO2 in the CO hydrogenation reaction. This work offers an effective strategy to design efficient catalysts for the FTO reaction by regulating the crystal phase of the support.
 Tiejun Lin | Tiejun Lin earned his BS degree from Zhejiang University of Technology and received his PhD degree in 2015 from East China University of Science and Technology. He then joined in Shanghai Advanced Research Institute (SARI), Chinese Academy of Science (CAS), where he was promoted to full professor in 2023. His research interests focus on heterogeneous catalysis for C1 chemistry-related reactions, including Fischer–Tropsch synthesis to olefins, higher alcohols synthesis, CO2 hydrogenation and industrial catalysis. |
1. Introduction
Olefins, one of the most important organic chemical raw materials, can be employed to produce cosmetics, lubricants, rubber, detergents and polymers and a wide range of commodities. The steam cracking and catalytic cracking of naphtha are commonly used to produce light olefins industrially.1 Due to the depletion of oil resources, the demand for olefin production from alternative feedstocks is attracting great interest.2 Fischer–Tropsch synthesis (FTS) technology allows carbon sources such as natural gas, coal and biomass to replace petroleum by producing a variety of liquid fuels and chemicals through syngas (H2 + CO).3–5 Traditional FT metals contain Fe, Co and Ru. Among these metals, Ru-based catalysts exhibit intrinsically higher catalytic activity and long-chain hydrocarbon selectivity, along with much lower selectivity for CH4 and CO2, and are considered to be ideal FT catalysts.6 Very recently, Yu et al. reported that alkali-modified supported Ru metal could catalyze CO hydrogenation to produce olefins, instead of traditional saturated hydrocarbons, with 80.1% selectivity for olefins with ultralow total selectivity for CH4 and CO2 (<5%) at CO conversion of 45.8%.7 Subsequent work also suggests that the types of promoter and support could greatly affect the performance of Fischer–Tropsch synthesis to olefins (FTO) for supported Ru-based catalysts.8,9For these supported catalysts, the metal–support interaction (MSI) plays a crucial role in tuning activity, selectivity and stability. In particular, the strong metal-support interaction (SMSI), typically occurring between a transition metal and a partially reducible oxide support (i.e., TiO2, CeO2, and MoO), has emerged as an effective strategy to modulate the catalytic behavior of a supported catalyst, which refers to the migration of support-derived species to the surface of the metal nanoparticles (NPs) to form an encapsulation overlayer under high-temperature reduction conditions.10–13 The application of the SMSI effect in enhancing catalytic reactivity and product selectivity of a metal catalyst has been commonly reported for CO hydrogenation toward long-chain hydrocarbons.14–18 Zhang et al. reported that the activity in FTS shows a volcano-like trend with increasing reduction temperature from 200 to 600 °C.14 Such a variation in activity is characterized as being related to the as-formed metal–support interface, in which the TiOx overlayer at Ru/TiO2 interfaces promotes CO dissociation. Xu et al. fabricated an SMSI-type interfacial TiO2−x/Ni catalyst by varying the reduction temperature, and the TiO2−x overlayers around the Ni nanoparticles could facilitate C–C chain propagation to produce a C2+ hydrocarbon, which is quite different from a pure metallic Ni surface with CH4 as the dominant product.17 However, some cases indicate that the encapsulation structure will also hinder contact between active site and reactant, resulting in a reduction in catalytic activity. Lyu et al. employed a DA sacrificial coating strategy to prepare Ru/TiO2-T-H catalysts with well-distributed Ru NPs (∼3 nm).19 The DA-derived carbon shells could form a complete coating on the catalysts, which could efficiently isolate Ru NPs and TiO2 during the annealing process and hence suppress the encapsulation of metal NPs by the TiO2 support in the H2 reduction procedure. Besides, TiO2 has different crystal structures, which also influence catalytic activity. Katsuya Shimura et al. suggest that the crystal phase of the TiO2 support would greatly affect the degree of Co metal reduction and the exposed surface area of Co metal, and the highest activity was obtained for a rutile-TiO2-supported catalyst (CoCa/r-TiO2).20 Clearly, the properties of the support significantly influence the MSI effect and FTS performance. However, the role of the crystal phase of the TiO2 support on a supported Ru-based catalyst for the FTO reaction has rarely been reported, and it remains of interest to explore the crystal-phase-dependent MSI effect in CO hydrogenation.
Herein, the correlation between the SMSI effect and the fine structure of TiO2 crystal phases (rutile, anatase) for an Ru/TiO2 catalyst was investigated. It is revealed that the interfacial structure of the Ru/TiO2 catalyst was strongly dependent on the crystal phases and treatment conditions of the support. The SMSI dominates on Ru/a-TiO2 after H2 treatment at 300 °C, resulting in Ru NPs covered by a TiOx overlayer and reduced catalytic activity. Further experiments confirmed that the H2-treated a-TiO2 support could inhibit the SMSI effect and the catalytic performance of Ru/a-TiO2 could be completely changed from inactive to active. Various characterization techniques were used to elucidate the evolution of the structure and the structure-performance relationship was also explored in detail.
2. Experimental section
2.1 Materials
Ruthenium nitrosyl nitrate (14.14 wt%, aqueous solution, AR) was purchased from Heraeus Precious Metal Technology Co., Ltd. Sodium nitrate (NaNO3, AR) was purchased from Sinopharm Chemical Reagent Co., Ltd. All materials were used as received without further purification. Titanium oxide, rutile (99.8 wt%, metal basis, 40 nm) was purchased from Shanghai Aladdin Biochemical Technology Co., Ltd. Titanium oxide, anatase (99.8 wt%, metal basis, 40 nm) was purchased from Shanghai Aladdin Biochemical Technology Co., Ltd.2.2 Catalyst preparation
The Ru/TiO2 catalysts were prepared by an impregnation method using rutile TiO2 and anatase TiO2 as the support. In a typical synthesis, 5.43 g of aqueous Ru(NO3)3 solution (9.2 wt%, 0.0356 g Ru per gram of solution, AR) and 0.184 g of NaNO3 was diluted to 50 mL with deionized water. 9.27 g of TiO2 was then added to the solution, and the resulting suspension was stirred in a water bath at 80 °C to evaporate and dry. The resulting solid was dried at 80 °C overnight, followed by calcination in air at 400 °C for 3 h. The as-obtained sample was labeled xNayRu/r-TiO2 or xNayRu/a-TiO2, whereas x and y denote the theoretical weight percent of Na and Ru elements, respectively. Generally, the weight ratio of Na/Ru for all samples is fixed at 0.1, and the samples are also abbreviated as yRu/r-TiO2 or yRu/a-TiO2. In addition, samples with an x value of 0.5% and a y value of 5% are mainly used for discussion in this work unless otherwise specified, and these samples are also further abbreviated to Ru/r-TiO2 or Ru/a-TiO2. For example, Ru/r-TiO2 denotes the sample with 5 wt% Ru loading and 0.5 wt% Na loading; 2Ru/a-TiO2 denotes the sample with 2 wt% Ru loading and 0.2 wt% Na loading.For the Ru/a-TiO2(H2) catalysts, a-TiO2 was firstly thermally treated under H2 flow at 600 °C for 4 h, and the as-obtained TiO2 was denoted a-TiO2(H2). The preparation of the Ru/a-TiO2(H2) catalyst was similar to Ru/a-TiO2 except for the use of a-TiO2(H2) as support.
2.3 Catalyst characterization
Power X-ray diffraction (XRD) data were acquired using a Rigaku Ultima IV X-ray diffractometer (40 kV, 40 mA) equipped with Cu Kα radiation (λ = 1.54056 Å) with scanning angle from 5 to 90° at a scanning speed of 2° min−1. Structural phases were identified from the JCPDS standard card.Transmission electron microscopy (TEM) and high-solution transmission electron microscopy (HRTEM) images were obtained on FEI Tecnai G2 F20 S-TWIN equipment with 200 kV accelerating voltage. Typically, the samples were dispersed into ethanol. After ultrasonication for 10 min, the suspension was deposited on copper grids for measurement. The average particle size was calculated from more than 150 particles. High-angle annular dark-field scanning transmission electron microscopy and energy dispersive X-ray spectroscopy (EDS) elemental mapping measurements were performed on an FEI-TALOS-F200X instrument.
Electron paramagnetic resonance (EPR) spectra at room temperature or low temperature (110 K) were collected on a Bruker A300 EPR spectrometer operated at the X-band frequency.
The elemental content was measured by inductively coupled plasma optical emission spectrometry (ICP-OES, PerkinElmer).
Hydrogen temperature-programmed reduction (H2-TPR) was tested on a Micromeritics Autochem-II 2920 instrument. 50 mg samples were loaded into a U-shaped quartz tube and then purged in He flow (30 mL min−1) at 200 °C for 1 h. The temperature was then cooled to 50 °C and switched to 5%H2/95%Ar (30 mL min−1). After the baseline was stable, the temperature was raised from 50 to 800 °C at a heating rate of 10 °C·min−1. The reduction curve was recorded continuously.
X-ray photoelectron spectroscopy (XPS) was recorded on a Thermo Fisher Scientific K-Alpha spectrometer with an Al Kα source (12 kV, 4 mA, hν = 1486.6 eV). The results were calibrated by setting the C 1s peak at 284.8 eV.
The CO temperature-programmed surface reaction (CO-TPSR) was performed on a Micromeritics Autochem-II 2920 instrument. Approximately 20 mg of reduced catalyst was loaded into a U-tube reactor; then the reactor was purged by Ar flow (30 mL min−1) at 200 °C for 1 h. After that, the reactor was cooled to 50 °C and the Ar flow was replaced with 30 mL min−1 CO for saturated adsorption. Subsequently, the CO flow was replaced with Ar (30 mL min−1) to purge the reactor for 30 min, and then H2 was introduced into the reactor. As the temperature increased to 800 °C at a rate of 10 °C min−1, the effluent gas was monitored using MS.
H2–D2 exchange experiments were carried out in a homemade quartz U-tube reactor system at atmospheric pressure. A fresh sample (0.2 g) was added to the U-tube reactor and then heated at 300 °C for 2 h under a 30 mL min−1 H2 atmosphere. After cooling to 50 °C, the reactor inlet flow was switched from H2 to D2 (30 mL min−1) via a 4-way valve. After the switch, D atoms react with H atoms on the surface of the sample, resulting in the appearance of an HD signal peak (m/z = 3). The off-gas was continuously monitored by a mass spectrometer (INFICON, Transpector CPM).
2.4 Catalytic evaluation
The catalytic performance was evaluated in a continuous-flow fixed-bed reactor. 1.0 g of catalyst (40–60 mesh) was diluted with quartz sand (2 g, 40–60 mesh) and then loaded into the constant-temperature zone of the reactor. Prior to the catalytic reaction, the catalysts were reduced with pure H2 (100 mL min−1) at a specified temperature for 4 h. The treated sample is denoted as Ru/r-TiO2-XR or Ru/a-TiO2-XR, or Ru/a-TiO2(H2)-XR, where X represents the reduction temperature. After the reactor was cooled to 180 °C, syngas (H2/CO/N2 = 64.7/32.3/3) was introduced into the reactor and the pressure was increased to 1.0 MPa, where N2 was used as internal standard. The gaseous product flow after passing through a hot trap (120 °C) and a cold trap (1 °C) was analyzed online by an Agilent chromatograph (GC) equipped with a flame ionization detector (FID) and a thermal conductivity detector (TCD). H2, N2, CO, CH4 and CO2 were detected by the TCD, and the hydrocarbons with carbon number in the range of 1–7 (C1–C7) were detected by the FID. The liquid or solid organic products were collected by the hot trap and cold trap, and were analyzed by an off-line Shimadzu GC.7 The carbon balance, mass balance and oxygen balance were calculated and maintained at 100 ± 5%. All experiments were repeated more than twice to keep the results convincing.CO conversion (XCO) and product selectivity (Si) were calculated with the following equation:
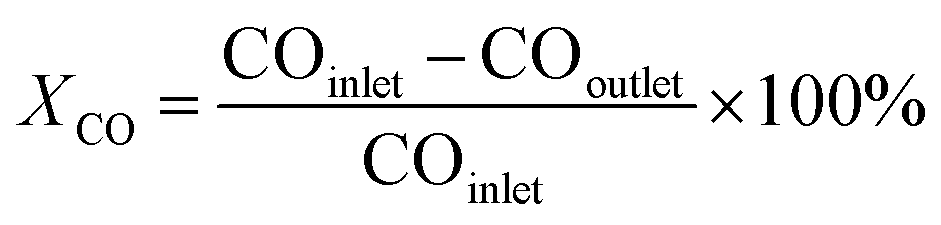 | (1) |
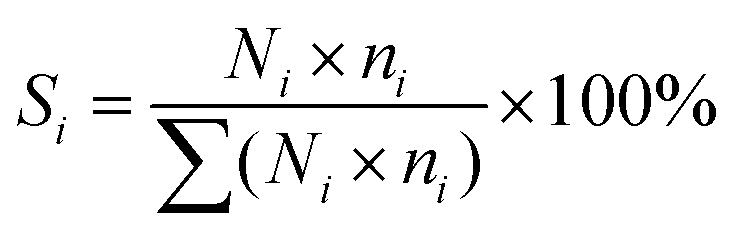 | (2) |
3. Results and discussion
3.1 Catalytic performance
The catalytic performance of the Ru/TiO2 catalyst with two crystal phases of TiO2 support was evaluated at 1 MPa, 3000 mL g−1 h−1, H2/CO = 2 and 260 °C. Before reaction, the catalysts were reduced with pure H2 at 300 °C for 4 h. Since prior work suggested that Na plays a crucial role in shifting the product selectivity of a metallic Ru-based catalyst from paraffins to olefins during CO hydrogenation,7,21 most catalysts described here contained an Na promoter with a fixed weight ratio Na/Ru of 0.1. As shown in Fig. 1a and Table S1† (entry 1), the rutile-TiO2-supported Ru-based catalysts showed typical FTO performance. For example, the olefin selectivity was as high as 69.4% at 26.4% CO conversion over the Ru/r-TiO2-300R catalyst, which also exhibited excellent catalytic stability (Fig. S1a†). However, once the support was changed to anatase-TiO2(r-TiO2), the obtained Ru/a-TiO2-300R catalyst showed almost no reactivity under the same reaction conditions regardless of the amount of Ru loading (entries 2 and 3). To exclude the possible influence of the Na promoter, an Ru/a-TiO2-300R(0Na) catalyst without adding an Na promoter (entry 4) was prepared and evaluated. No obvious CO conversion could be detected in this case. Such significant differences in reactivity are surprising since the Ru loading amount, chemical composition of the support and the preparation and treatment procedures were the same for both catalysts. Obviously, a strong crystal-phase-dependent effect of Ru/TiO2 is observed for CO hydrogenation.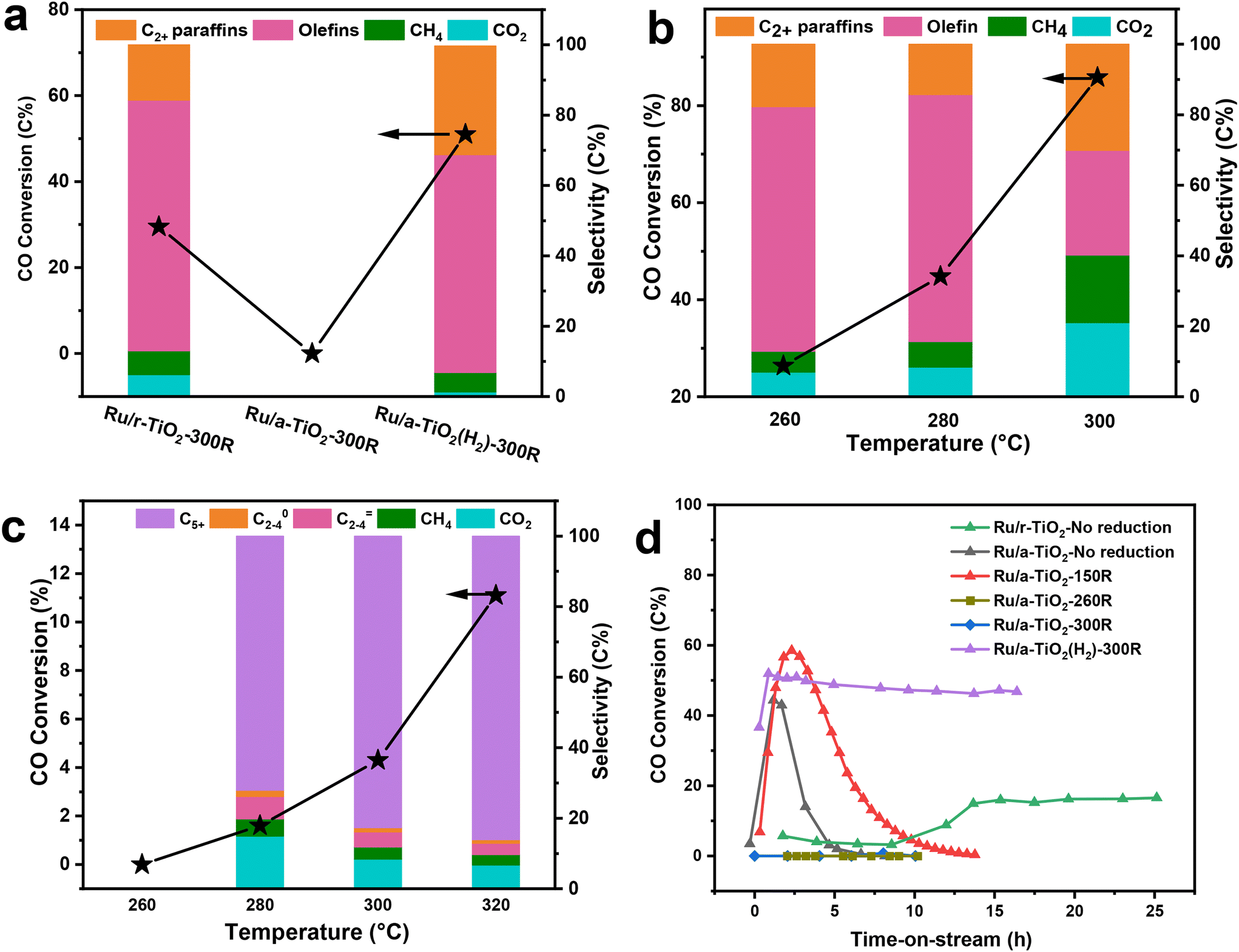 | ||
| Fig. 1 Detailed catalytic results over various supported catalysts. (a) Comparison of CO conversion and product selectivity over various Ru/TiO2 catalysts. Reaction conditions: 1 MPa, 3000 mL g−1 h−1, H2/CO = 2, 260 °C. (b and c) Effect of reaction temperature on catalytic performance of Ru/r-TiO2 (b) and Ru/a-TiO2 (c). (d) Evolution of CO conversion over Ru/TiO2 catalysts treated under different reduction conditions. Reaction conditions: 1 MPa, 3000 mL g−1 h−1, H2/CO = 2, 260 °C. | ||
We further investigated the effect of reaction temperature. As shown in Fig. 1b and c, the CO conversion surged to 85.8% for Ru/r-TiO2 when the reaction temperature increased to 300 °C, while that of Ru/a-TiO2 was only 4.3%, confirming that the catalytic activity of the a-TiO2-supported catalyst is still greatly suppressed.
It was previously reported that TiO2 as a type of reducible support would lead to surface coverage of metal NPs by TiOx suboxide under high-temperature reduction and H2 atmosphere, thus decreasing the catalytic activity.14 Such a phenomenon is known as the strong metal–support interaction (SMSI) effect. To explore the possible SMSI effect on the catalytic performance of Ru/TiO2 catalysts, the influence of reduction temperature was studied (Fig. S2†). For the case of Ru/r-TiO2, the CO conversion showed a decreasing trend with increasing reduction temperature, which further dropped to 8.9% for Ru/r-TiO2 reduced at 600 °C. As for Ru/a-TiO2, the reduction temperature of 600 °C still led to undetectable reactivity (Ru/a-TiO2-600R). By comparison, it can be reasonably inferred that thermal treatment of Ru/r-TiO2 at high temperature under H2 flow would lead to a similar phenomenon observed over Ru/a-TiO2. In other words, the active site structure of Ru/r-TiO2-600R may be similar to that of Ru/a-TiO2-300R. Prior study demonstrated that the Ru/r-TiO2 catalyst reduced at a temperature >300 °C can cause the coverage of Ru NPs by a layer of TiOx suboxide.14 Therefore, the SMSI effect might dominate Ru/a-TiO2-300R even at a reduction temperature as low as 300 °C. But, what really surprised us is that the reactivity of Ru/a-TiO2 could be restored when the a-TiO2 support was pre-treated with H2 at 600 °C. The as-obtained Ru/a-TiO2(H2)-300R catalyst exhibited 55.5% CO conversion with 61.9% olefin selectivity (Fig. 1a and Table S1,† entry 5), whose activity is far higher than that of Ru/a-TiO2-300R and can be comparable to that of Ru/r-TiO2-300R. Ru/a-TiO2(H2)-300R also shows excellent catalytic stability for the FTO reaction (Fig. S1b†). This result indicates that the H2 treatment strategy could change the active structure of Ru/a-TiO2, causing it to exhibit similar catalytic behavior to Ru/r-TiO2 in the FTO reaction.
The evolution of CO conversion with reaction time over Ru/TiO2 catalysts treated under different reduction conditions was further investigated in detail and compared. As shown in Fig. 1d, the Ru/r-TiO2 catalyst without any reduction showed an obvious activation period, which might due to the gradual reduction of Ru2O to Ru metal as active sites. After ∼14 h of time-on-stream, CO conversion gradually increased to ∼17% and remained almost unchanged at a reaction temperature of 260 °C. However, for the Ru/a-TiO2 catalyst without any reduction (Ru/a-TiO2-No reduction) or reduction at 150 °C under H2 flow (Ru/a-TiO2-150R), CO conversion first increased to a certain value and then was deactivated quickly until no CO conversion could be detected. When the Ru/a-TiO2 catalyst was directly pretreated by H2 at 260 °C or 300 °C, there was almost no CO conversion. Since the reaction temperature of the FTO reaction was fixed at 260 °C, it can be inferred that the Ru/a-TiO2 reduced at low temperature (i.e. <200 °C) might undergo similar structural evolution to that directly H2-reduced at high temperature (i.e. ≥260 °C) after exposure to syngas at 260 °C. However, Ru/a-TiO2(H2)-300R shows constant high CO conversion even at the initial reaction stage. Evidently, the reduction at a low temperature of 260 °C for Ru/a-TiO2 could cause significant structural evolution, while that of Ru/r-TiO2 and Ru/a-TiO2(H2) remained almost unchanged.
3.2 Structural identification of Ru/TiO2 catalysts
ICP analysis suggested that Ru/r-TiO2 and Ru/a-TiO2 show similar Ru loading (4.44 wt% and 4.02 wt%) and Na content (0.40 wt% and 0.43 wt%). XRD results indicated that there are no changes in the crystal phase of a-TiO2 after H2 treatment at 600 °C (Fig. 2a). Fig. 2b–d shows that no obvious characteristic peaks ascribed to Ru species could be detected for any of the catalysts at different stages (fresh, reduced or spent), indicating a high degree of Ru dispersion on the support.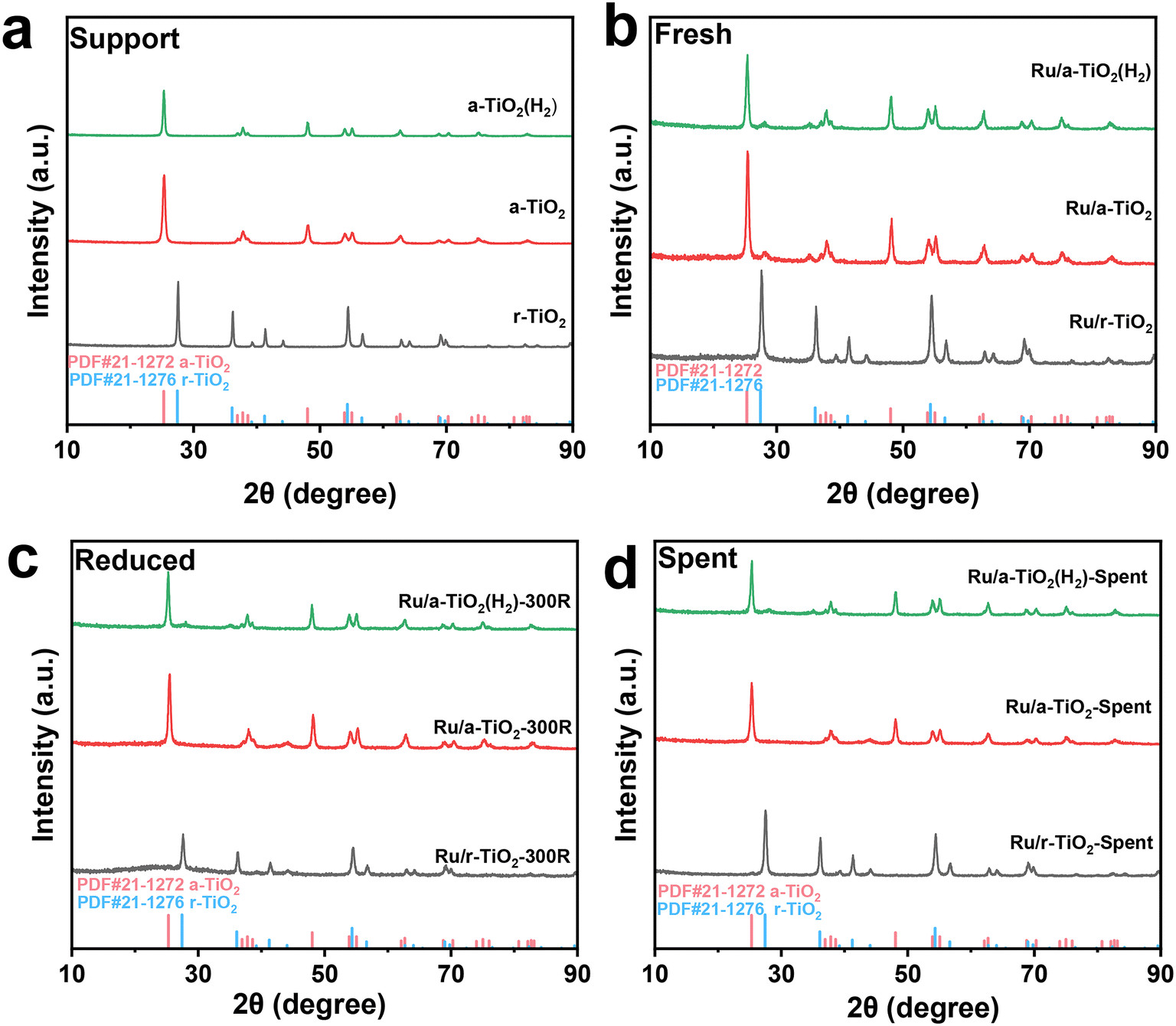 | ||
| Fig. 2 XRD patterns of support and various catalysts: (a) support, (b) fresh catalysts, (c) reduced catalysts, (d) spent catalysts. | ||
H2-TPR was used to determinate the reducibility of Ru species and the possible metal–support interaction. As shown in Fig. 3a, no obvious reduction peak for r-TiO2 was detected, while a large broad peak centered at 544 °C was observed for a-TiO2. This comparison suggests that anatase-type TiO2 exhibits much higher reducibility of surface-oxygen atoms. As for the Ru/r-TiO2 catalyst, there are three main peaks centered at 100 °C (peak I), 156 °C (peak II), and around 180 °C (peak III), attributed to reduction of surface adsorbed O, surface RuO2, and interfacial RuOx species, respectively (Fig. 3b).22 The decrease in reduction temperature of a-TiO2 from 544 °C to 332 °C suggested that the presence of Ru species greatly promotes the reduction of a-TiO2 due to the H-spillover effect. In addition, the reduction temperature of Ru species in r-TiO2 is significantly higher than that of a-TiO2, suggesting the existence of stronger interfacial adhesion between Ru NPs and the r-TiO2 support due to the same lattice structure between RuO and rutile.22 However, Ru/a-TiO2(H2) mainly shows the reduction peaks of Ru species, and the reduction process of the TiO2 support disappeared. Note that the a-TiO2 support has been pretreated by H2 at 600 °C, so the reduction of the a-TiO2 support and the possible migration of TiOx suboxides might be greatly suppressed, leading to the observed differences in catalytic performance of various Ru/TiO2 catalysts.
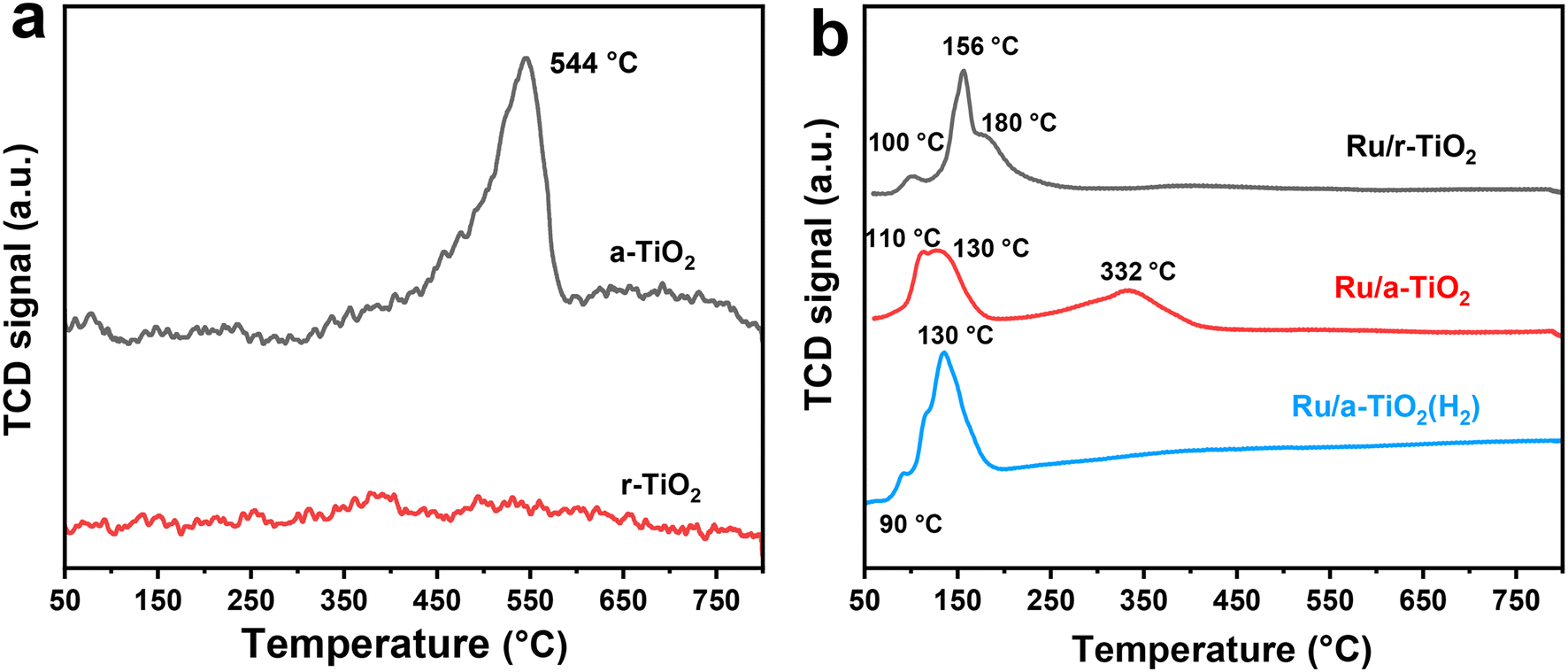 | ||
| Fig. 3 H2-TPR profiles of support (a) and various Ru/TiO2 catalysts (b). | ||
The representative TEM images and the corresponding Ru particle size distribution for various reduced Ru/TiO2 catalysts are shown in Fig. 4 and Fig. S3.† It is evident that the Ru/r-TiO2 catalysts (Fig. 4a and b) show a much narrower distribution than that of the Ru/a-TiO2 catalysts (Fig. 4c and d), indicating stronger interfacial interaction between Ru NPs and rutile TiO2 due to the high degree of interfacial compatibility.22 The similar Ru size for Ru/r-TiO2 (1.4 nm) and Ru/a-TiO2 (2.0 nm) reduced at 300 °C suggested that the quite different catalytic behaviors that were observed may not originate from the Ru size. The particle size of Ru increased to 3.8 nm for the Ru/r-TiO2 catalyst after reduction at 600 °C (Fig. S3†). The H2 pretreatment of a-TiO2 at 600 °C would lead to the as-obtained Ru/r-TiO2(H2) showing a slightly increased Ru size (Fig. 4e and f).
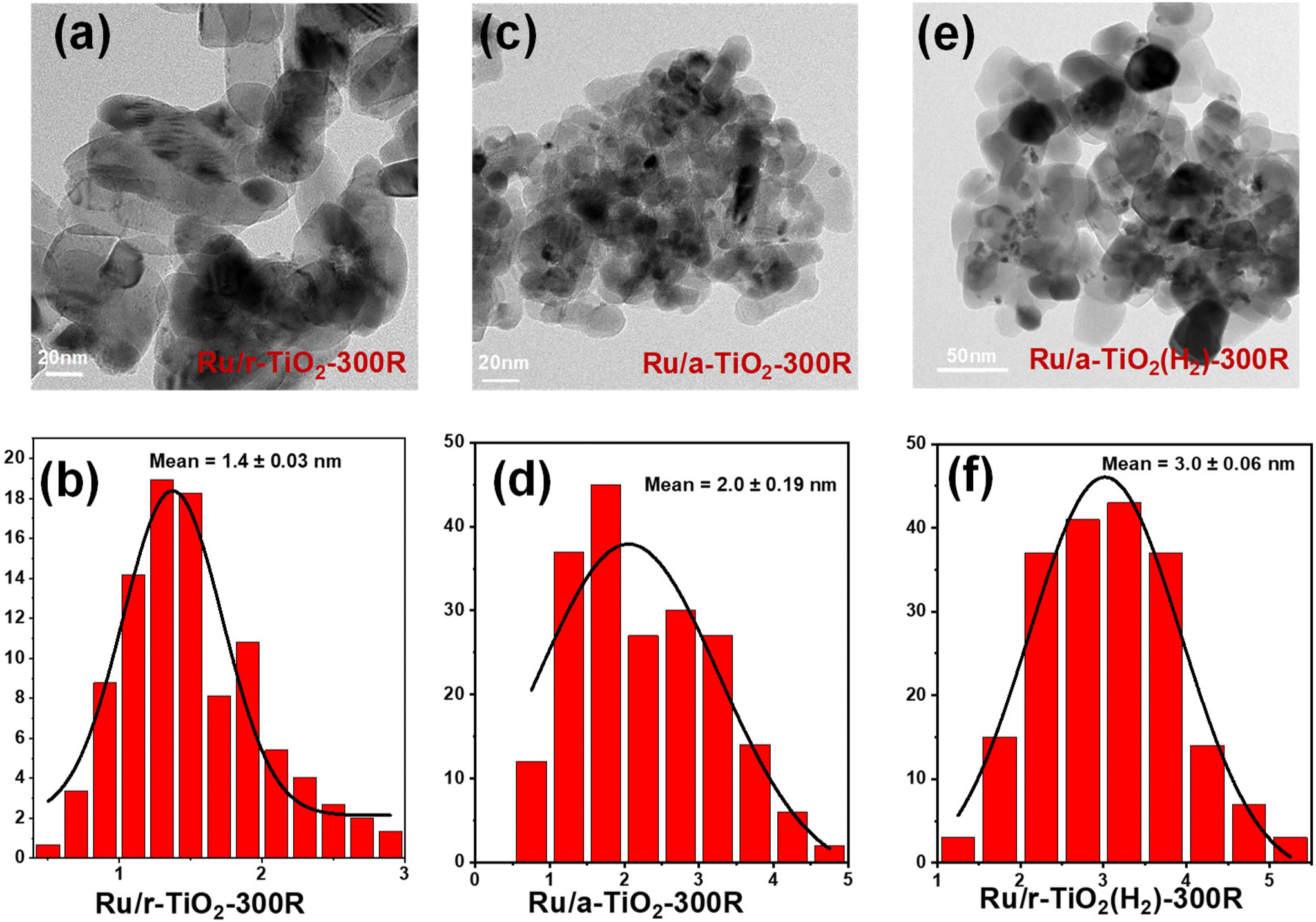 | ||
| Fig. 4 TEM images and the corresponding particle size distribution of (a and b) Ru/r-TiO2-300R, (c and d) Ru/a-TiO2-300R, (e and f) Ru/a-TiO2(H2)-300R. | ||
HRTEM observations were performed to determinate the chemical state and surface morphology of Ru/TiO2 catalysts with different TiO2 crystal phases. Schematic illustrations of the structural evolution of Ru/TiO2 at different stages of reduction are also inserted in the corresponding figures. As shown in Fig. 5a and Fig. S4,† bare Ru NPs with a lattice spacing of 0.205 nm corresponding to Ru (101) are anchored on the surface of rutile TiO2 (101) with a lattice fringe of 0.249 nm for the Ru/r-TiO2-300R catalyst. Due to the same lattice structure for RuO2 and r-TiO2, these Ru species tend to form epitaxial overlayers on R-TiO2 with extremely low contact angles;22 thus flat Ru NPs on Ru/r-TiO2-300R were commonly observed. The HAADF-STEM-mapping images in Fig. S5† suggest that the Ru NPs are well dispersed on the r-TiO2 support, and the lattice-matching properties can help to resist the aggregation of metal NPs under high-temperature treatment. However, for Ru/a-TiO2-300R, a low-contrast coating or a very thin overlayer around Ru NPs could be recognized (Fig. 5b). Considering the wide investigation of the SMSI effect for TiO2-based catalytic systems in the references15,22–24 as well as the chemical composition in all Ru/TiO2 catalysts, the overlayer could be reasonably ascribed to TiOx suboxides. The HAADF-STEM-mapping images suggested that a slight aggregation of Ru NPs for the Ru/a-TiO2 sample due to the lattice misfit interfacial structure and the signal for Ti can also be simultaneously detected around Ru species (Fig. S6†). This feature suggests that the encapsulation of Ru NPs by TiOx at a low reduction temperature of 300 °C occurred for anatase TiO2, but is not observed for rutile TiO2. The following characterizations, such as XPS, surface adsorption and reaction could further confirm the existence of surface encapsulation. In fact, for r-TiO2-supported Ru-based catalysts, the SMSI behavior shows a temperature-dependent effect, and it has been reported that SMSI would not occur at such a low temperature as 300 °C.23 While the crystal-dependent SMSI effect was clearly observed for Ru/r-TiO2 and Ru/a-TiO2 at a reduction temperature of 300 °C, which can be attributed to the facile reduction of a-TiO2 compared to that of r-TiO2, as determined by the H2-TPR results. In addition, the different lattice structures of RuO2 and a-TiO2 would lead to the formation of an Ru-TiO2 interface with a high contact angle. Therefore, the reduced TiOx suboxides would easily migrate to the surface of Ru NPs to form an encapsulation structure for the Ru/a-TiO2 case, causing a decrease in the number of exposed Ru surface sites. Another interesting phenomenon is that there is no observable TiOx overlayer around Ru NPs on Ru/a-TiO2(H2)-300R, as shown in Fig. 5c and Fig. S7,† indicating that the SMSI effect for Ru on the H2-treated anatase support is greatly suppressed. Since the high-temperature H2 reduction has not changed the crystal phase of anatase TiO2, a possible reason for the different performance between Ru/a-TiO2(H2) and Ru/a-TiO2 might be related to the surface or bulk properties of a-TiO2, which is also confirmed by the different H2-TPR behaviors.
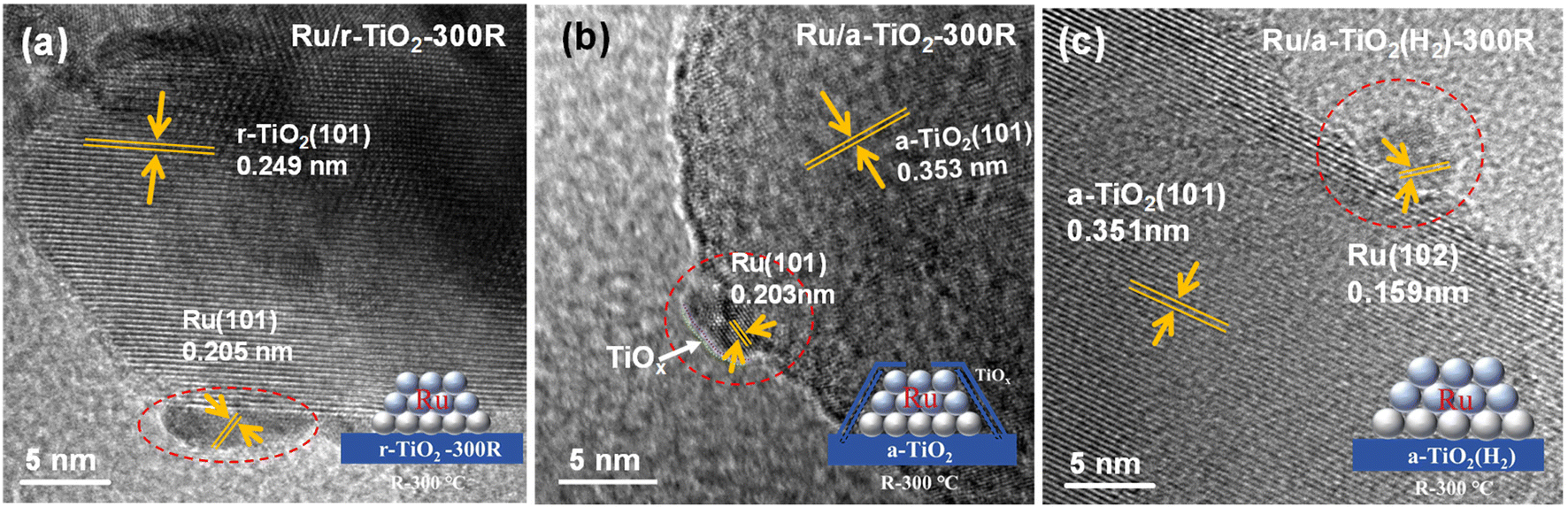 | ||
| Fig. 5 HRTEM images of (a) Ru/r-TiO2-300R, (b) Ru/a-TiO2-300R, (c) Ru/a-TiO2(H2)-300R. | ||
Electron paramagnetic resonance (EPR) characterization was performed to investigate the properties of various supports and Ru/TiO2 catalysts. As shown in Fig. 6a, the room-temperature EPR signal corresponding to a g-value of 2.004 could be observed for all supports. The EPR signal intensity of these supports follows the order: a-TiO2(H2) > a-TiO2 > r-TiO2. In particular, the H2 treatment greatly increases the EPR signal of a-TiO2. It is widely considered that the EPR signal intensity shows a positive correlation with support defects, such as Ti3+ and oxygen vacancies.25–29 The H2 reduction treatment would lead to the formation of a high concentration of bulk Ti3+ species and oxygen vacancies in a-TiO2. To detect the Ti3+ species more accurately, low-temperature EPR characterization was performed. The EPR signal corresponding to a g-value of ∼1.997 can be ascribed to the Ti3+ species.25,27,28 As shown in Fig. S8,† no EPR signals could be observed for a-TiO2 or r-TiO2. However, upon H2 treatment at 300 °C, the characteristic peak at a g-value of 1.997 appeared. This signal intensity becomes stronger as the reduction temperature increases to 600 °C. Based on previous studies,25 the EPR signal at g = 1.997 can reflect the relative concentration of bulk Ti3+ species. XPS spectra of TiO2 were also performed and the content of Ti3+ was estimated (Fig. S9†). As expected, the H2 treatment greatly increased the content of Ti3+, which is in line with the result of low-temperature EPR. The existence of bulk Ti3+ species in the TiO2(H2) support may greatly inhibit the migration of surface-reduced TiOx suboxides to the surface of Ru NPs over Ru/a-TiO2(H2). Such a phenomenon was also observed in the Ni/TiO2 catalytic system.25Fig. 6b shows the room-temperature EPR signal of various reduced catalysts. Clearly, the EPR signal intensity of these catalysts follows the same order as that of the individual supports. The high concentration of oxygen vacancies may also help the activation of the C–O bond.30
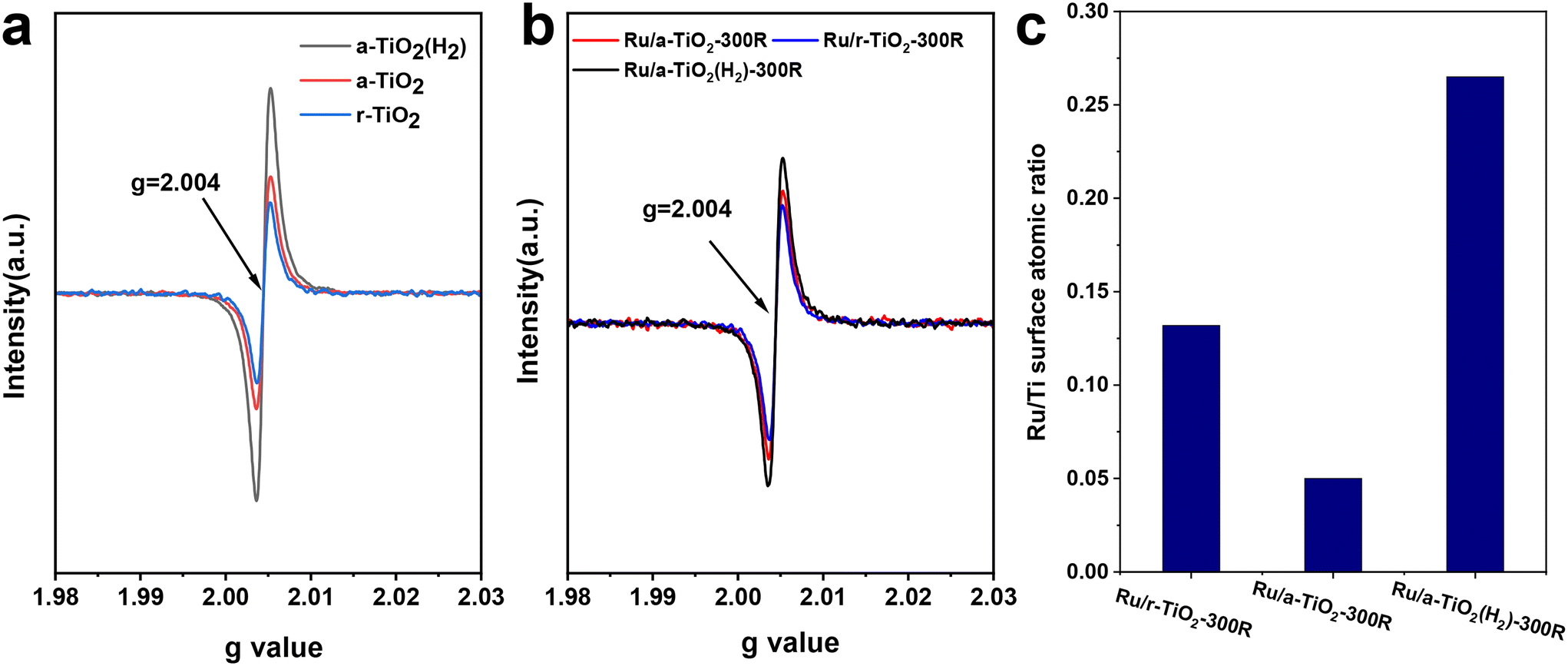 | ||
| Fig. 6 Room-temperature EPR spectra recorded for supports (a) and reduced catalysts (b). (c) Atomic ratio of surface Ru/Ti estimated from the XPS spectra over various Ru/TiO2 catalysts. | ||
Encapsulation of metal NPs (i.e., Ru,14,15,24 Ni,17,31 Co,16 and Ir32) by a reducible TiO2 support has been reported previously. Another effective approach to verify the migration of TiOx suboxides to Ru NPs is to detect the atomic ratio of surface Ru/Ti for various Ru/TiO2 catalysts. The XPS spectra of various reduced catalysts are shown in Fig. S10.† After fitting, the surface Ru/Ti atomic ratio was estimated. As shown in Fig. 6c, the surface Ru/Ti atomic ratio for Ru/r-TiO2-300R was 0.13, which decreased to 0.05 for Ru/a-TiO2-300R. Since the same Ru loading, the same chemical composition of TiO2, and the same preparation and pretreatment methods were applied to the two cases, the rather low surface Ru/Ti atomic ratio for Ru/a-TiO2-300R indicated that a more pronounced SMSI effect was observed for Ru supported on an a-TiO2 support. However, this value inversely increased to 0.26 for Ru/a-TiO2(H2)-300R, suggesting that the surface Ru NPs were less covered by a TiOx overlayer. Obviously, the H2 treatment of a-TiO2 can weaken the SMSI effect, agreeing well with the results of the HRTEM characterization.
3.3 Structure-performance relationship
The above structural characterization results powerfully confirm that the SMSI state of Ru/TiO2 catalysts can be controlled by changing the crystal phase of TiO2. The anatase-TiO2-supported-Ru catalysts tend to form an SMSI-induced TiOx overlayer, which would block the catalytic sites and result in a decreased number of exposed Ru sites under an H2-containing environmental atmosphere at a temperature ≤300 °C. The formation of a TiOx coating by SMSI critically affects the adsorption and activation of reactants. To obtain a qualitative comparison of the exposed Ru sites after encapsulation by the TiOx overlayer, the chemisorption of CO was measured. As shown in Fig. 7a, Ru/r-TiO2-300R shows 2.6-fold higher CO uptake than Ru/a-TiO2-300R. However, after H2 treatment of r-TiO2, the CO uptake of Ru/a-TiO2(H2)-300R greatly increased to 553.6 μmol g−1, which is ∼7.9-fold higher than for Ru/a-TiO2-300R and 3-times higher than for Ru/r-TiO2-300R. Therefore, it is reasonable to infer that the Ru sites are severely covered on Ru/a-TiO2-300R, while more Ru sites are exposed on Ru/a-TiO2(H2)-300R.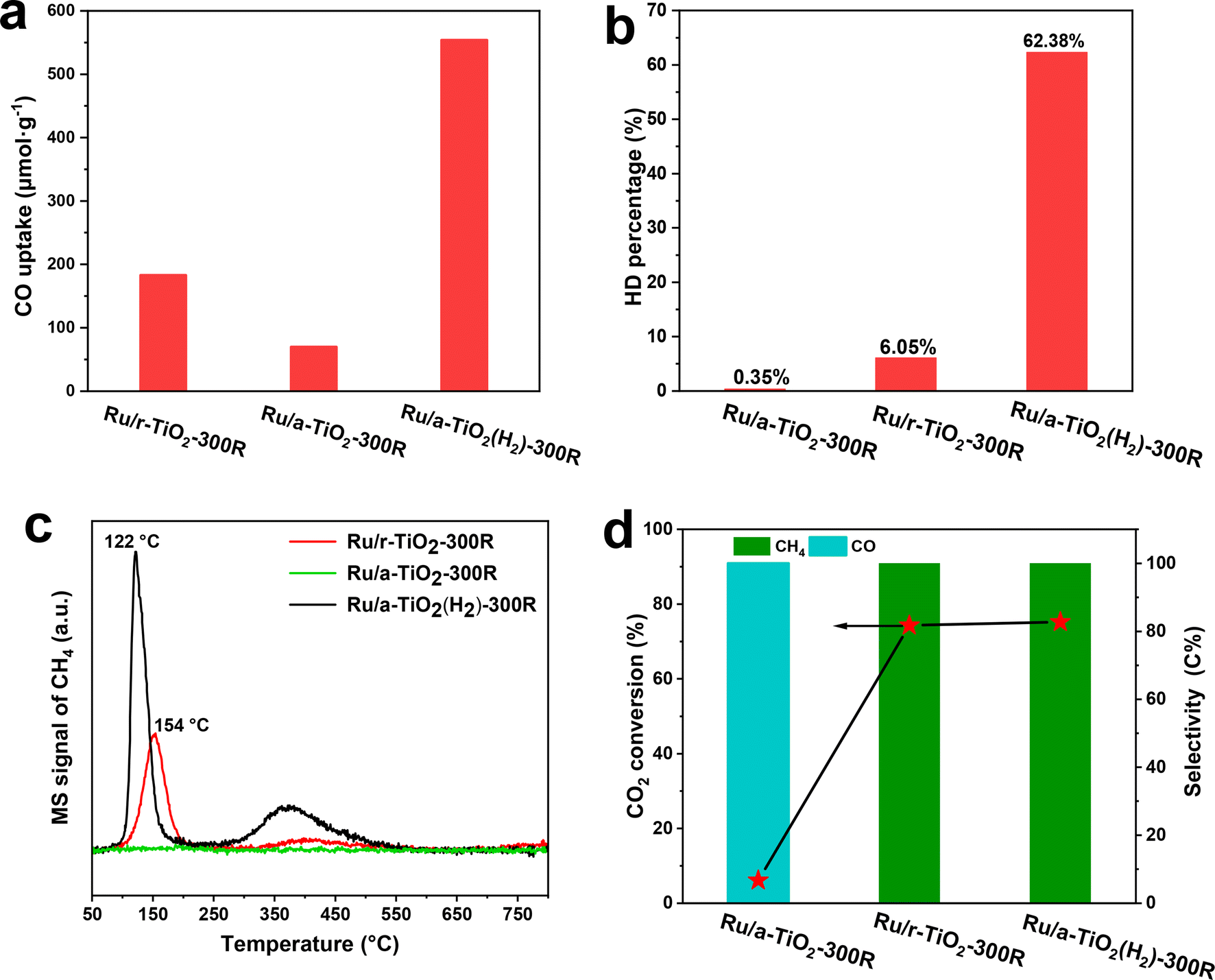 | ||
| Fig. 7 Surface adsorption and reaction for various reduced Ru/TiO2 catalysts: (a) CO uptake; (b) H–D exchange experiments measured at room temperature, (c) CO-TPSR profile, (d) catalytic performance for CO2 hydrogenation. Reaction conditions: 1.0 g, 1 MPa, CO2/H2/Ar = 24/73/3, 3000 mL gcat−1 h−1. | ||
H–D exchange is another useful tool with which to identify surface hydrogenation ability and to probe the exposed Ru sites.12,33 The adsorption and dissociation of H2 on Ru sites will form H* species, which can exchange with D* species derived from D2 dissociation to form HD species. As shown in Fig. 7b, the HD percentage of the Ru/a-TiO2-300R catalyst was as low as 0.35%, while that of the Ru/r-TiO2-300R catalyst reached 6.05%, which is about 17-fold higher than the former. While for the Ru/a-TiO2(H2) catalyst, the HD percentage surged to 62.3%, demonstrating considerably more surface Ru sites than for the other two. By combining this with the structural characterization, we can conclude that the coating structure strongly inhibits the activation of H2 on the Ru surface, and the pretreatment of a-TiO2 with H2 significantly promotes the exposure of surface Ru sites.
To further correlate the structural evolution with the corresponding catalytic performance, CO-TPSR experiments were performed (Fig. 7c). The adsorbed CO species can dissociate to surface carbon species, which are then hydrogenated to form CH4 during a temperature-programmed process under H2 flow.7 The peak area and intensity of the CH4 signal are used to determine the strength of CO adsorption on Ru sites and the dissociation rate. It was observed that Ru/r-TiO2-300R exhibits a major CH4 signal peak at 154 °C, while no peak could be detected over Ru/a-TiO2-300R, suggesting that there are no CO molecules adsorbed or activated on the Ru/a-TiO2-300R sample. This result corresponds to the observed difference in performance between Ru/r-TiO2-300R and Ru/a-TiO2-300R, where the latter shows undetectable CO conversion under 260 °C. Also note that the Ru/a-TiO2(H2)-300R catalyst displays CH4 peaks at temperature as low as 122 °C, suggesting that surface carbon species are facilely hydrogenated to form CH4. In particular, the higher peak area and strong intensity of the CH4 signal for Ru/a-TiO2(H2)-300R confirmed that more CO can be adsorbed and activated. By calculation, the peak area of CH4 for Ru/a-TiO2(H2)-300R is 1.9- and 2.1-times higher than that of Ru/r-TiO2-300R and Ru/r-TiO2-600R, respectively. It should also be noted that Ru/a-TiO2(H2)-300R exhibited 2.1-fold higher CO conversion than Ru/r-TiO2-300R at the same reaction temperature of 260 °C. These results match well with each other, and the H2 treatment of the a-TiO2 support would be an effective strategy to improve the catalytic performance of Ru/a-TiO2 for CO hydrogenation.
CO2 hydrogenation is a structure-sensitive reaction that can be used to explore the evolution of the surface structure of Ru/TiO2. Typically, the exposed metal sites can catalyze CO2 hydrogenation to CH4via a methanation reaction, while the encapsulation of metal NPs benefits the reverse-water–gas-shift (RWGS) reaction with CO as the dominant product.12,32,34 As demonstrated in Fig. 7d, Ru/a-TiO2-300R shows nearly 100% CO product selectivity at a rather low CO2 conversion (∼6%). However, the product selectivity completely shifted to CH4 at a CO conversion as high as ∼75% for Ru/r-TiO2-300R and Ru/a-TiO2(H2)-300R. Evidently, it can be inferred that the surface Ru sites of Ru/a-TiO2-300R were covered, while the other two cases possess many more exposed Ru sites, in line with the above experimental and characterization results.
3.4 Discussion
Fischer–Tropsch synthesis is a structure-sensitive reaction and the changes in the surface structure of metal sites would greatly influence the catalytic behavior.35–38 A suitable metal–support interaction could effectively disperse metal species and prevent their aggregation and sintering, providing a sufficiently exposed surface metallic area.3,39 This work demonstrates that the SMSI state could be controlled for tunable CO hydrogenation performance by selecting the appropriate crystal phase of TiO2 and support-treatment strategy.According to the above characterization results, the reducibility of TiO2 and the metal–support interfacial configuration play an essential role for the crystal-phase-dependent metal–support interaction. The SMSI-induced encapsulation of Ru NPs by TiOx typically involves the reduction of TiO2 as well as the migration of TiOx suboxides from the bulk to the surface of Ru sites. Previous study has indicated that the anatase form is more readily reduced in the bulk than the rutile form under the same conditions due to the lower formation energies of the oxygen vacancies of anatase (101) than of rutile (110) with a value of 4.95 eV vs. 5.70 eV.40,41 The H2-TPR and EPR results also confirm the same conclusions. At a low temperature of 300 °C, r-TiO2 is hard to reduce, while the surface oxygen-atoms of a-TiO2 could easily be removed. The formed oxygen vacancies can induce the occurrence of an SMSI effect with the coating of Ru NPs by TiOx suboxides,40 leading to decreased CO uptake and diminished activity. Actually, SMSI started to play a major role for Ru/a-TiO2 at a reaction temperature of 260 °C, but to a lesser extent than for Ru/r-TiO2. The pre-reduction of a-TiO2 at 600 °C would not change its crystal phase while producing a high concentration of bulk Ti3+ species and oxygen vacancies in a-TiO2(H2), which would suppress the formation of TiOx and its migration to Ru NPs for Ru/a-TiO2(H2).25 Moreover, the same lattice parameters for RuO2 and r-TiO2 determine the higher matching degree of atomic configuration on the metal–support contacting interface. Therefore, these Ru species tend to form epitaxial overlayers on r-TiO2, and the migration of TiOx suboxides to Ru NPs would be greatly inhibited. For Ru/a-TiO2, the misfit lattice structure easily causes different surface energies between metal and support, so the migration of TiOx suboxides would be promoted. When comparing Ru/a-TiO2(H2) and Ru/r-TiO2 without any surface coating, the strong interfacial coupling of Ru/r-TiO2 would cause the production of small-sized Ru (i.e., <2 nm), while the metal–support interaction of Ru/a-TiO2(H2) would be to some extent weakened by the formation of Ru NPs of ∼3.0 nm in size. It is reported that the Ru-based FTS showed a size-dependent effect,37,42 in the which activity and TOF increased with Ru size within a critical level of Ru size (approximately 6–7 nm). Therefore, Ru/a-TiO2(H2) can show a much higher CO activation rate based on the CO-TPSR result and catalytic activity in the FTO reaction.
4. Conclusions
In summary, the metal–support interaction could be controlled to achieve tunable CO hydrogenation performance over Ru/TiO2 nanocatalysts. The Ru/r-TiO2 catalyst with a negligible SMSI effect at a reduction temperature of 300 °C shows high CO conversion, high olefin selectivity and high stability. However, SMSI plays a major role for Ru/a-TiO2 under identical reduction and reaction conditions, resulting in Ru NPs being covered by a TiOx overlayer and showing undetectable catalytic activity. Whereas, the H2-pretreated a-TiO2 support (a-TiO2(H2)) led to a great improvement in CO conversion (>50%) and olefin selectivity (>60%) for Ru/a-TiO2(H2), which are far higher than those of Ru/r-TiO2 and Ru/a-TiO2(H2). Structural characterizations suggest that the facile reduction of surface-oxygen atoms for an a-TiO2 support and its mismatched lattice structure with RuO2 readily lead to the SMSI phenomenon and the decreased number of Ru sites, causing the reduced CO uptake, hydrogenation capacity and degraded FTO performance. The pretreatment of a-TiO2 by H2 yielded more bulk Ti3+ species that could inhibit the formation of an SMSI-induced encapsulated structure. Moreover, Ru/a-TiO2(H2) provides a suitable size of Ru for enhanced activity. These results indicate that Ru/TiO2 catalysts could be modified by tailoring the SMSI state through changing the crystal phase of the TiO2 support and a corresponding thermal treatment strategy to enhance FTO performance.Author contributions
H. L.: validation, investigation, data curation, formal analysis, materials characterization, writing – original draft. W. Z.: formal analysis, materials characterization. H. S.: investigation, formal analysis. H. Y.: investigation, formal analysis. Y. A.: formal analysis, validation. T. L.: conceptualization, supervision, methodology, validation, writing – original draft, writing – review & editing, project administration, funding acquisition. L. Z.: project administration, supervision, writing – review& editing, conceptualization, funding acquisition.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We appreciate the financial support from the National Key Research and Development Program of China (2023YFB4103104), Natural Science Foundation of China (U22B20136, 22293023, 22072177, and 22202230), the Natural Science Foundation of Shanghai (22JC1404200 and 21ZR1471700), and the Youth Innovation Promotion Association of Chinese Academy of Science.References
- H. M. Torres Galvis, J. H. Bitter, C. B. Khare, M. Ruitenbeek, A. I. Dugulan and K. P. de Jong, Science, 2012, 335(6070), 835–838 CrossRef CAS PubMed.
- T. Lin, F. Yu, Y. An, T. Qin, L. Li, K. Gong, L. Zhong and Y. Sun, Acc. Chem. Res., 2021, 54(8), 1961–1971 CrossRef CAS PubMed.
- K. Cheng, J. C. Kang, D. L. King, V. Subramanian, C. Zhou, Q. H. Zhang and Y. Wang, Adv. Catal., 2017, 60, 125–208 CAS.
- J. Zhao, J. Liu, Z. Li, K. Wang, R. Shi, P. Wang, Q. Wang, G. I. N. Waterhouse, X. Wen and T. Zhang, Nat. Commun., 2023, 14, 1909 CrossRef CAS PubMed.
- J. Zhao, Z. Li, P. Wang, P. Miao, R. Shi, G. I. N. Waterhouse and T. Zhang, Nano Energy, 2023, 110, 108350 CrossRef CAS.
- A. Y. Khodakov, W. Chu and P. Fongarland, Chem. Rev., 2007, 107, 1692–1744 CrossRef CAS PubMed.
- H. Yu, C. Wang, T. Lin, Y. An, Y. Wang, Q. Chang, F. Yu, Y. Wei, F. Sun, Z. Jiang, S. Li, Y. Sun and L. Zhong, Nat. Commun., 2022, 13(1), 5987 CrossRef PubMed.
- H. Yu, Y. Wei, T. Lin, C. Wang, Y. An, F. Yu, F. Sun, Z. Jiang, Y. Sun and L. Zhong, ACS Catal., 2023, 13, 3949–3959 CrossRef CAS.
- Y. An, X. Wang, H. Yu, X. Qi, D. Lv, T. Lin and L. Zhong, ACS Catal., 2023, 13(20), 13245–13256 CrossRef CAS.
- Z. Luo, G. Zhao, H. Pan and W. Sun, Adv. Energy Mater., 2022, 12, 2201395 CrossRef CAS.
- T. Pu, W. Zhang and M. Zhu, Angew. Chem., Int. Ed., 2023, 62, e202212278 CrossRef CAS PubMed.
- H. Xin, L. Lin, R. Li, D. Li, T. Song, R. Mu, Q. Fu and X. Bao, J. Am. Chem. Soc., 2022, 144(11), 4874–4882 CrossRef CAS PubMed.
- T. W. van Deelen, C. H. Mejia and K. P. de Jong, Nat. Catal., 2019, 2(11), 955–970 CrossRef CAS.
- Y. Zhang, X. Yang, X. Yang, H. Duan, H. Qi, Y. Su, B. Liang, H. Tao, B. Liu, D. Chen, X. Su, Y. Huang and T. Zhang, Nat. Commun., 2020, 11(1), 3185 CrossRef CAS PubMed.
- Y. Zhang, X. Su, L. Li, H. Qi, C. Yang, W. Liu, X. Pan, X. Liu, X. Yang, Y. Huang and T. Zhang, ACS Catal., 2020, 10(21), 12967–12975 CrossRef CAS.
- C. Hernández Mejía, T. W. van Deelen and K. P. de Jong, Nat. Commun., 2018, 9, 4459 CrossRef PubMed.
- M. Xu, X. Qin, Y. Xu, X. Zhang, L. Zheng, J.-X. Liu, M. Wang, X. Liu and D. Ma, Nat. Commun., 2022, 13(1), 6720 CrossRef CAS PubMed.
- X. Qi, T. Lin, Y. An, X. Wang, D. Lv, Z. Tang and L. Zhong, ACS Catal., 2023, 13, 11566–11579 CrossRef CAS.
- S. Lyu, Q. Cheng, Y. Liu, Y. Tian, T. Ding, Z. Jiang, J. Zhang, F. Gao, L. Dong, J. Bao, Q. Ma, Q.-H. Yang and X. Li, Appl. Catal., B, 2020, 278, 119261 CrossRef CAS.
- K. Shimura, T. Miyazawa, T. Hanaoka and S. Hirata, Catal. Today, 2014, 232, 2–10 CrossRef CAS.
- H. Yu, Y. Wei, T. Lin, C. Wang, Y. An, F. Yu, F. Sun, Z. Jiang, Y. Sun and L. Zhong, ACS Catal., 2023, 13(6), 3949–3959 CrossRef CAS.
- J. Zhou, Z. Gao, G. Xiang, T. Zhai, Z. Liu, W. Zhao, X. Liang and L. Wang, Nat. Commun., 2022, 13(1), 327 CrossRef CAS PubMed.
- Y. Zhang, X. Yang, X. Yang, H. Duan, H. Qi, Y. Su, B. Liang, H. Tao, B. Liu, D. Chen, X. Su, Y. Huang and T. Zhang, Nat. Commun., 2020, 11(1), 3185 CrossRef CAS PubMed.
- Y. Zhang, W. Yan, H. Qi, X. Su, Y. Su, X. Liu, L. Li, X. Yang, Y. Huang and T. Zhang, ACS Catal., 2022, 12(3), 1697–1705 CrossRef CAS.
- J. Li, Y. Lin, X. Pan, D. Miao, D. Ding, Y. Cui, J. Dong and X. Bao, ACS Catal., 2019, 9, 6342–6348 CrossRef CAS.
- A. Teleki and S. E. Pratsinis, Phys. Chem. Chem. Phys., 2009, 11(19), 3742–3747 RSC.
- S. Hoang, S. P. Berglund, N. T. Hahn, A. J. Bard and C. B. Mullins, J. Am. Chem. Soc., 2012, 134(8), 3659–3662 CrossRef CAS PubMed.
- H. Tang, Y. Su, B. Zhang, A. F. Lee, M. A. Isaacs, K. Wilson, L. Li, Y. Ren, J. Huang, M. Haruta, B. Qiao, X. Liu, C. Jin, D. Su, J. Wang and T. Zhang, Sci. Adv., 2017, 3, e1700 Search PubMed.
- H. Khan and I. K. Swati, Ind. Eng. Chem. Res., 2016, 55(23), 6619–6633 CrossRef CAS.
- K. Gong, T. Lin, F. Yu, Y. An, X. Wang, L. Zhong and Y. Sun, Catal. Sci. Technol., 2021, 11(15), 5232–5241 RSC.
- M. Monai, K. Jenkinson, A. E. M. Melcherts, J. N. Louwen, E. A. Irmak, S. Van Aert, T. Altantzis, C. Vogt, W. van der Stam, T. Duchon, B. Smid, E. Groeneveld, P. Berben, S. Bals and B. M. Weckhuysen, Science, 2023, 380(6645), 644–651 CrossRef CAS PubMed.
- Y. Zhang, Z. Zhang, X. Yang, R. Wang, H. Duan, Z. Shen, L. Li, Y. Su, R. Yang, Y. Zhang, X. Su, Y. Huang and T. Zhang, Green Chem., 2020, 22(20), 6855–6861 RSC.
- F. Jiao, B. Bai, G. Li, X. Pan, Y. Ye, S. Qu, C. Xu, J. Xiao, Z. Jia, W. Liu, T. Peng, Y. Ding, C. Liu, J. Li and X. Bao, Science, 2023, 380(6646), 727–730 CrossRef CAS PubMed.
- X. Yuan, T. Pu, M. Gu, M. Zhu and J. Xu, ACS Catal., 2021, 11(19), 11966–11972 CrossRef CAS.
- F. Bertella, P. Concepción and A. Martínez, Catal. Today, 2017, 296, 170–180 CrossRef CAS.
- J. Hong, B. Wang, G. Xiao, N. Wang, Y. Zhang, A. Y. Khodakov and J. Li, ACS Catal., 2020, 10(10), 5554–5566 CrossRef CAS.
- Q. Zhang, K. Cheng, J. Kang, W. Deng and Y. Wang, ChemSusChem, 2014, 7(5), 1251–1264 CrossRef CAS PubMed.
- P. Zhai, Y. Li, M. Wang, J. Liu, Z. Cao, J. Zhang, Y. Xu, X. Liu, Y.-W. Li, Q. Zhu, D. Xiao, X.-D. Wen and D. Ma, Chem, 2021, 7(11), 3027–3051 CAS.
- M. Ahmadi, H. Mistry and B. Roldan Cuenya, J. Phys. Chem. Lett., 2016, 7(17), 3519–3533 CrossRef CAS PubMed.
- H. Li, Y. Guo and J. Robertson, J. Phys. Chem. C, 2015, 119(32), 18160–18166 CrossRef CAS.
- B. Cui, H. Wang, J. Han, Q. Ge and X. Zhu, J. Catal., 2022, 413, 880–890 CrossRef CAS.
- W. Chen, T. Lin, Y. Dai, Y. An, F. Yu, L. Zhong, S. Li and Y. Sun, Catal. Today, 2018, 311, 8–22 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr06208b |
| This journal is © The Royal Society of Chemistry 2024 |