
The role of surface functionalization in quantum dot-based photocatalytic CO2 reduction: balancing efficiency and stability†
Frida
Hernandez
a,
Maggie
Yang
a,
Nejc
Nagelj
 a,
Autumn Y.
Lee
a,
Hasun
Noh
a,
Kyle P.
Hur
a,
Xinyu
Fu
a,
Caleb J.
Savoie
a,
Adam M.
Schwartzberg
b and
Jacob H.
Olshansky
*a
a,
Autumn Y.
Lee
a,
Hasun
Noh
a,
Kyle P.
Hur
a,
Xinyu
Fu
a,
Caleb J.
Savoie
a,
Adam M.
Schwartzberg
b and
Jacob H.
Olshansky
*a
aDepartment of Chemistry, Amherst College, Amherst, MA 01002, USA. E-mail: jolshansky@amherst.edu
bThe Molecular Foundry, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA
First published on 22nd February 2024
Abstract
Photocatalytic CO2 reduction offers a promising strategy to produce hydrocarbons without reliance on fossil fuels. Visible light-absorbing colloidal nanomaterials composed of earth-abundant metals suspended in aqueous media are particularly attractive owing to their low-cost, ease of separation, and highly modifiable surfaces. The current study explores such a system by employing water-soluble ZnSe quantum dots and a Co-based molecular catalyst. Water solubilization of the quantum dots is achieved with either carboxylate (3-mercaptopropionic acid) or ammonium (2-aminoethanethiol) functionalized ligands to produce nanoparticles with either negatively or positively-charged surfaces. Photocatalysis experiments are performed to compare the effectiveness of these two surface functionalization strategies on CO2 reduction and ultrafast spectroscopy is used to reveal the underlying photoexcited charge dynamics. We find that the positively-charged quantum dots can support sub-picosecond electron transfer to the carboxylate-based molecular catalyst and also produce >30% selectivity for CO and >170 mmolCO gZnSe−1. However, aggregation reduces activity in approximately one day. In contrast, the negatively-charged quantum dots exhibit >10 ps electron transfer and substantially lower CO selectivity, but they are colloidally stable for days. These results highlight the importance of the quantum dot–catalyst interaction for CO2 reduction. Furthermore, multi-dentate catalyst molecules create a trade-off between photocatalytic efficiency from strong interactions and deleterious aggregation of quantum dot–catalyst assemblies.
 Jacob H. Olshansky | Jacob Olshansky is an assistant professor in the Department of Chemistry at Amherst College, a small liberal arts college in Western Massachusetts. He received his PhD in 2017 from the University of California Berkeley and performed postdoctoral research at Northwestern University. He has found that he can be promoted to an excited state when he successfully synthesizes highly emissive quantum dots or unravels charge transfer pathways using time-resolved spectroscopy. He has also become fascinated by the various processes that promote (and do not promote) excited state energy transfer from him to his research students. |
Introduction
Fundamental research on the conversion of CO2 to hydrocarbon fuels and value-added carbon compounds is necessary to advance new technologies that reduce reliance on fossil fuels and remove excess atmospheric CO2.1,2 Ideally, such technologies would rely on either renewable electricity or direct sunlight.3,4 Direct photocatalysis is particularly attractive since it can be implemented without intermediate electrical systems, and may be more economically viable than photoelectrochemical systems.5 Semiconducting nanoparticles, or quantum dots (QDs), have recently emerged as promising sensitizers for photocatalytic CO2 reduction.4,6,7 QDs are robust visible light absorbers that offer tunable redox energies via composition8–11 and size control,12–14 and can support significant chemical variety in surface functionalization.15–17 Furthermore, there is extensive literature using QDs for other photocatalytic conversions, including proton reduction to hydrogen,18–23 C–C coupling,24–28 alcohol oxidation,29 and nitrogen fixation.30 A theme of this body of work is the critical role that the surface plays in dictating catalysis,15 thus motivating our exploration of the role of surface functionalization in photocatalytic CO2 reduction using QDs.A variety of metrics must be considered when choosing a specific QD composition for photocatalysis. For example, semiconductors with smaller bandgaps can absorb more of the visible spectrum, but offer less driving force for redox reactions.4 To access photocatalytic CO2 reduction to CO semiconductors must possess a conduction band energy greater than approximately −4.0 vs. vacuum or −0.77 vs. SCE.3 Promising visible light-absorbing semiconducting materials that have been employed for CO2 reduction (bulk conduction band position vs. vacuum) include: CdS (−3.9 V),31–36 ZnSe (−3.4 V),26,31,37–41 CuInS2 (−3.0 V),42–46 and lead halide perovskites (−4.0 to −3.0 V).47,48 Environmental concerns may motivate researchers to use materials that do not contain toxic heavy metals such as Pb and Cd. We have chosen to focus our attention on ZnSe QDs since they are heavy-metal free, provide clear size-dependent excitonic features, and exhibit minimal defect absorption.49,50
A molecular catalyst is typically used in QD-based photocatalytic CO2 reduction schemes to improve selectivity and yield. A large literature exists for molecular and heterogeneous CO2 reduction catalysis,3,51,52 providing a library of potential catalysts that can be combined with the light-absorbing QDs. We are particularly drawn to cobalt tetraphenylporphyrin (Co-TPP) catalysts,53 since they use an earth-abundant metal and are associated with the most productive QD-based CO2 systems for both ZnSe38 and CuInS2.42 Prior work demonstrates the importance of attaching catalysts to the surface of QDs. Kuehnel et al. explored a series of nickel terpyridine catalysts coupled with CdS QDs and found that the anchoring group affected CO selectivity and production rate.34 Huang et al. found a >3× enhancement in CO production when covalently linking a Re catalyst to CuInS2 QDs compared to physically mixing the catalyst and QD.45 We have therefore chosen a Co-TPP catalyst functionalized with carboxylic acids that can bind to QD surfaces.
Surface modification of as-synthesized QDs is nearly always required to prepare photocatalytic systems. Typically, native hydrophobic ligands are exchanged for hydrophilic ligands that impart solubility in common CO2 reduction solvents such as DMF, DMSO, and water. Mercaptopropionic acid (MPA) is a commonly used ligand as its thiol group binds strongly to the surface of metal chalcogenide quantum dots54 and its carboxylate group imparts solubility. MPA has been used in photocatalytic CO2 reduction schemes using CdS,32,33 CuInS2,43,45 and ZnSe26,37 QDs. This work can also be informed by the larger literature of mercaptocarboxylate-QDs used for hydrogen evolution (HER).19,21–23 For example, Wilker et al. explored a series of mercaptocarboxylate ligands with different numbers of carbons (n) on CdS nanorods used for photocatalytic HER. They found that carbon chain length significantly altered hydrogen production rates. Nanorods with mercaptoacetic acid (n = 2) were ∼4× more productive than nanorods with MPA (n = 3), and longer chains were significantly worse.22 Short ligands are therefore critical for enabling efficient charge extraction and subsequent catalysis using QDs; however, trade-offs could exist when aiming to suppress HER relative to CO2 reduction.
Surface functionalization strategies that do not involve carboxylates have also been employed. Work by Reisner and colleagues found that ligand-stripped CdS QDs, stabilized by BF4− ions, produce two orders of magnitude more hydrogen than MPA-functionalized CdS QDs.23 The Reisner group has also used BF4-stabilized ZnSe QDs for photocatalytic CO2 reduction in multiple studies,38–41 producing as much as 79.7 mmolCO gZnSe−1.38 Surface functionalization with ammonium moieties has produced particularly promising results. Feng et al. compared CO production using CdS QDs functionalized with MPA, BF4, and 4-mercaptopyridinium, producing 3 μmol, 0.19 mmol, and 20.3 mmolCO g−1 h−1, respectively.35 The 4-mercaptopyridinium QDs were therefore far superior and, in total, produced ∼450 mmolCO gCdS−1. The Weiss group found similarly high levels of CO production using 2-aminoethanethiol (AET) as a capping ligand on CuInS2 QDs (∼850 mmolCO gQD−1).42 These results were rationalized by the affinity between CO2 and amines.
The prevalence of studies employing carboxylate-functionalized QDs (typically MPA) coupled with the promise of ammonium-functionalized QDs motivates our current work to directly compare these two surface-functionalization strategies for CO2 reduction. Notably, there are no prior reports that make such a comparison using QD–molecular systems that only differ in the terminal functional group of the ligand. The current works directly compares carboxylate (MPA) and ammonium (AET) functionalized QDs. We have performed CO2 reduction experiments with MPA and AET functionalized ZnSe QDs and a Co-TPP catalyst. We show that AET QDs initially produce significantly more CO than MPA QDs with a CO selectivity ∼4× greater than MPA QDs. However, the AET QDs deactivate after approximately one day, while the MPA QDs continue to produce H2 and CO for up to three days. We use both steady state absorption and ultrafast transient absorption spectroscopy to rationalize these observations. In particular, AET QDs facilitate rapid (sub-ps) electron transfer to the catalyst, while electron transfer in the MPA QDs is slower than the intrinsic QD relaxation (tens of ps). However, AET QDs form aggregates, which likely lead to their accelerated deactivation. Both of these observations can be understood as a consequence of stronger catalyst–QD interactions in the AET QDs. This strong interaction and associated sub-picosecond electron transfer allows the AET ZnSe QDs to achieve up to 170 mmolCO gZnSe−1 in water, the highest value reported for photocatalytic CO2 reduction with ZnSe QDs. Furthermore, this work highlights the importance of considering ligand–catalyst interactions.
Results and discussion
Prior to describing the CO2 reduction experiments, we have provided details on the synthesis and relevant energetics of the ZnSe QDs and the Co-TPP catalyst. Stearate-capped ZnSe QDs were synthesized in accordance with prior literature,49 and subsequently ligand-exchanged with either AET or MPA to achieve water solubilization. QD size and concentrations were determined using UV-Vis absorption and standard sizing curves.50 All QDs used for photocatalysis experiments had a diameter of d = 3.6 nm and a bandgap of 3.1 eV. The Co-TPP catalyst was synthesized in accordance with prior literature.55 The relevant energetics for all components of the photocatalytic system are provided in Scheme 1. Oxidation potentials for sacrificial hole scavengers, sodium ascorbate (NaAsc)56 and triethanolamine (TEOA),57 are positioned such that there is a large driving force (>1 V) for hole extraction. The Co-TPP catalyst has been shown to be chemically reduced from Co(III)-TPP to Co(II)-TPP in the presence of hole scavengers such as NaAsc.53 The relevant step for initiating CO2 capture and reduction is therefore photoexcited electron transfer from the QD to Co(II)-TPP, generating Co(I)-TPP with a reduction potential of −0.92 V vs. SCE.53 Reduction potentials for both CO2 to CO and HER are accessible by the reduced catalyst. These two reactions are therefore in competition, and are independently monitored in the present study.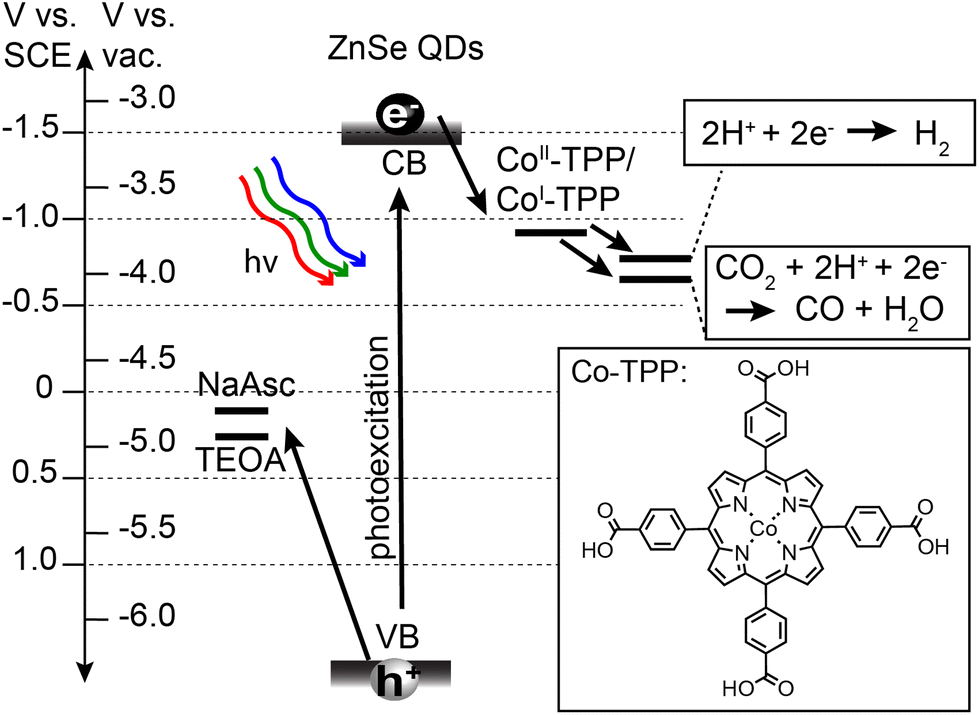 | ||
| Scheme 1 Relevant processes and energetics for photocatalytic CO2 reduction using ZnSe QDs. Oxidation potentials for sacrificial hole scavengers, sodium ascorbate (NaAsc) and triethanolamine (TEOA), are shown as well as reduction potentials for the Co-TPP catalyst and relevant reactions (CO2 reduction and HER). | ||
Photocatalysis experiments were performed on the QD systems using a home-built photoreactor and reaction products were monitored by gas chromatography (GC). In a typical experiment, aqueous samples (2 mL) were prepared containing 0.4 μM ZnSe QD, 0.4 μM Co-TPP, and a hole scavenger (25–400 mM). We used a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of QDs to catalyst based on prior work with the same catalyst.42 Samples were saturated with an atmosphere of CO2 and placed in a six-well photoreactor equipped with 400 nm LEDs. The head space was sampled periodically with an air tight syringe and directly injected into the GC (see Table S1† for complete data). Two metrics are primarily used to assess photocatalytic efficiency: turnover number and selectivity. The total amount of CO (or H2) produced relative to the amount of catalyst (or QD) is the turnover number (TON). The ratio of CO produced relative to total gaseous products (CO and H2) gives the CO selectivity (see ESI† for details of calculations). Control experiments verified that all components were necessary to produce substantial CO (light, CO2, hole scavenger, QD, and catalyst). The QDs and light alone (with or without hole scavenger), however, were sufficient in producing substantial H2 (see Table S2†). Reaction mixture pH was recorded after equilibration to be in the range of 6.4–6.5, which we ascribe to a carbonic acid and bicarbonate buffer created by dissolved CO2.
:1 ratio of QDs to catalyst based on prior work with the same catalyst.42 Samples were saturated with an atmosphere of CO2 and placed in a six-well photoreactor equipped with 400 nm LEDs. The head space was sampled periodically with an air tight syringe and directly injected into the GC (see Table S1† for complete data). Two metrics are primarily used to assess photocatalytic efficiency: turnover number and selectivity. The total amount of CO (or H2) produced relative to the amount of catalyst (or QD) is the turnover number (TON). The ratio of CO produced relative to total gaseous products (CO and H2) gives the CO selectivity (see ESI† for details of calculations). Control experiments verified that all components were necessary to produce substantial CO (light, CO2, hole scavenger, QD, and catalyst). The QDs and light alone (with or without hole scavenger), however, were sufficient in producing substantial H2 (see Table S2†). Reaction mixture pH was recorded after equilibration to be in the range of 6.4–6.5, which we ascribe to a carbonic acid and bicarbonate buffer created by dissolved CO2.
Initial experiments explored the role of the hole scavenger. Fig. 1a shows the two hole scavengers, NaAsc and TEOA, as well as the surface ligands used: MPA and AET. AET QDs were used for comparing the hole scavengers. The results of these experiments are shown in Fig. 1b and c and show a significant suppression of HER by TEOA, which yields a CO selectivity >40%. However, CO production was also significantly lower with TEOA. We therefore decided to use NaAsc as the hole scavenger for all subsequent experiments. Another important trend revealed by this data is that increasing hole scavenger concentration reduces selectivity. A careful look at the specific CO and H2 TONs shows that this trend is driven by enhanced HER at higher hole scavenger concentrations. This suggests that accelerating hole extraction predominantly benefits HER, while CO2 reduction is rate limited by other factors, likely related to the catalyst.
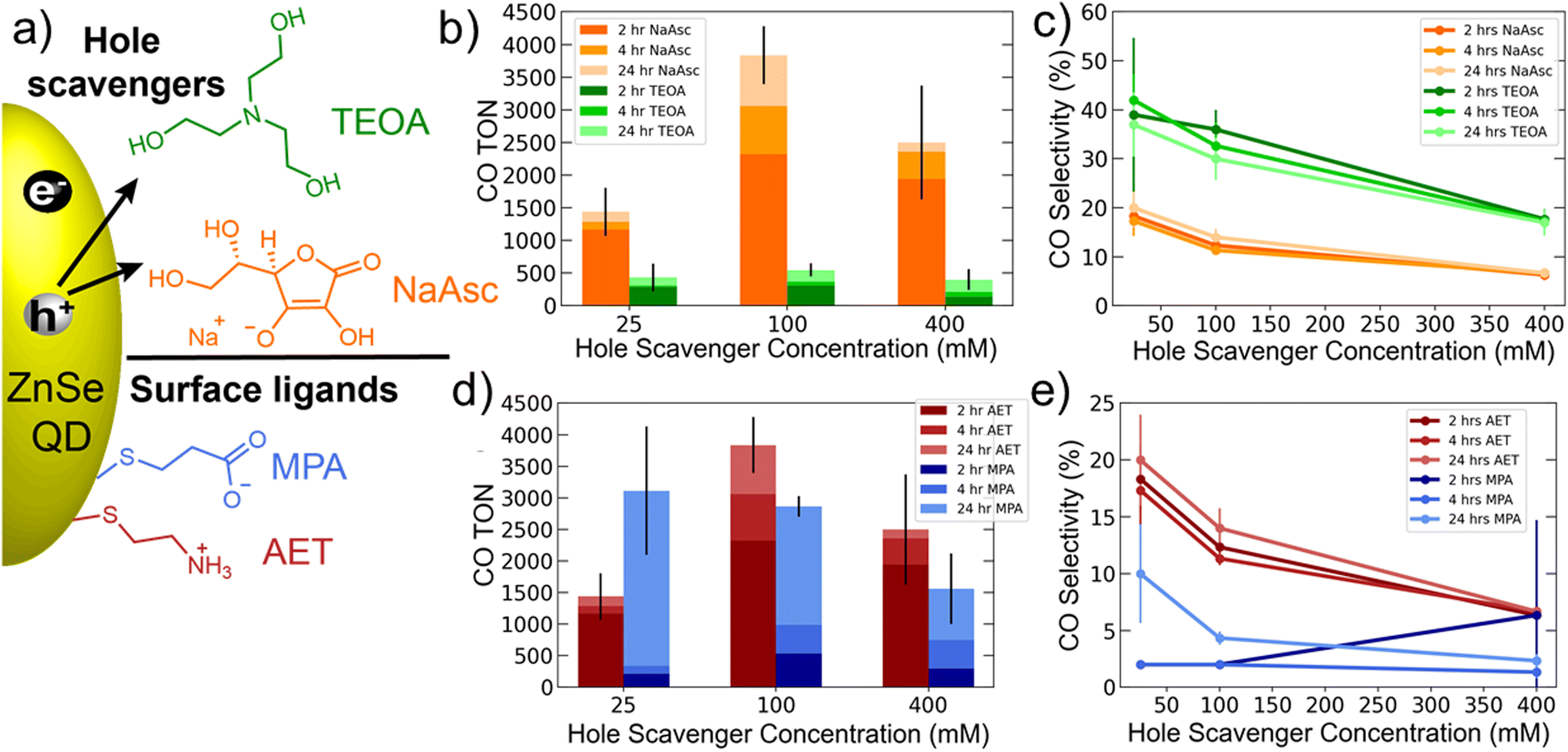 | ||
| Fig. 1 Results of photocatalysis experiments with varying hole scavenger concentrations and identities. (a) Schematic of hole scavengers and surface ligands. (b and c) CO TON and selectivity for AET QDs and both TEOA and NaAsc hole scavengers at different concentrations. (d and e) CO TON and selectivity for both AET and MPA QDs with varying NaAsc concentration. Photocatalysis experiments were performed in triplicate on 2.0 mL aqueous solutions of 0.4 μM ZnSe QDs and 0.4 μM Co-TPP, irradiated with 400 nm LEDs. | ||
A comparison of AET and MPA QDs is presented in Fig. 1d and e. These data show that AET QDs produce CO more selectively and initially in greater quantities than MPA QDs across a range of hole scavenger concentrations. This observation is consistent with prior literature suggesting that ammonium-functionalized QDs are better suited for CO2 reduction owing to their affinity for CO2,42 but the role of catalyst binding must also be considered (see below).
The results comparing AET and MPA QDs (Fig. 1d) suggest that the two systems have differing long-term behavior. Specifically, AET QDs initially produce more CO, but after 24 hours the MPA QDs are able to produce comparable amounts of CO. We therefore performed 4-day experiments (Fig. 2a) that reveal the same trend with greater clarity: the AET QDs produce >2× more CO at four hours, but CO production slows by the second day while MPA QDs continue to produce CO throughout the third day. We hypothesize that the more rapid deactivation of the AET QDs may be a result of aggregation. To verify this hypothesis, UV-Vis absorption spectra of AET and MPA ZnSe QDs in pure water and in the reaction mixture (at identical concentrations) are performed (Fig. 2b). There is a notable increase in light scattering for the AET QDs that is induced either by the addition of NaAsc or Co-TPP.
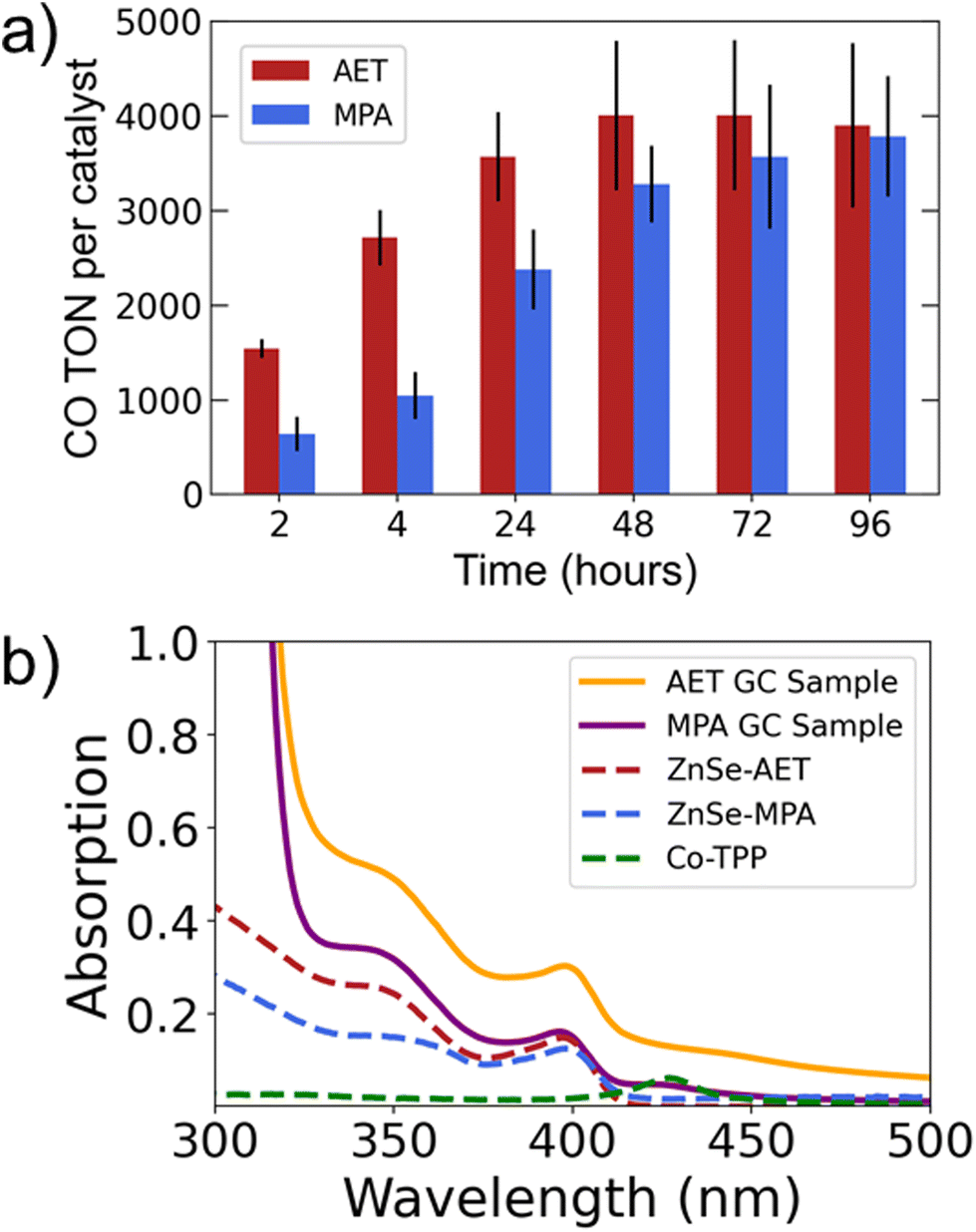 | ||
| Fig. 2 (a) Longer experiments comparing AET and MPA QDs. (b) UV-Vis spectra of reaction mixtures (GC samples) overlain with ligand-functionalized QDs and catalyst at identical concentrations alone in solution (dashed lines). Photocatalysis experiments were performed in triplicate on 2.0 mL aqueous solutions of 0.4 μM ZnSe QDs, 0.4 μM Co-TPP, and 100 mM NaAsc. Samples were irradiated with 400 nm LEDs. | ||
We believe that the aggregation observed in AET QD samples is driven by electrostatic interactions between the positively-charged AET QDs and the negatively charged NaAsc or tetracarboxylate Co-TPP. This hypothesis is supported by work from the Weiss group employing MPA-capped QDs and tetramethylammonium functionalized Fe-TPP catalysts for CO2 reduction.43 In their work, the negatively-charged QDs formed aggregates with the positively-charged catalyst. Furthermore, they found that larger aggregates produced less CO. In our work, with a negatively-charged catalyst, the positively-charged AET QDs aggregate over time while the negatively-charged MPA QDs remain colloidally stable. The aggregated AET QDs reduce available active sites for CO2 reduction leading to deactivation. However, the electrostatic interaction between AET QDs and both hole scavengers and catalysts likely drives their higher initial CO production as compared to MPA QDs.
To better understand and compare the specific interactions and charge dynamics that occur between AET or MPA QDs and the catalyst or hole scavenger, we utilized femtosecond transient absorption spectroscopy. This technique allows us to chart the pathways and timescales for photoexcited charge movement, thus providing insights on chemical interactions and the photophysical processes that underly photocatalysis. Transient absorption experiments were performed on AET and MPA ZnSe QDs with and without Co-TPP and NaAsc (eight samples total). All samples exhibited similar features in the transient spectra. Example spectra for AET QDs with neither catalyst nor NaAsc are shown in Fig. 3a (see Fig. S6 and S7† for all spectra). These spectra contain two notable features: (1) broad photo-induced absorption (450–600 nm) that has previously been assigned to surface-trapped holes,41 but may also be a result of trapped electrons58 and (2) a transient excitonic bleach at 400 nm that is ascribed to electron population in the conduction band.41,58 We cannot distinguish any spectral signatures associated with the Co-TPP, likely because it is substantially broadened in the presence of QDs (Fig. 2b).
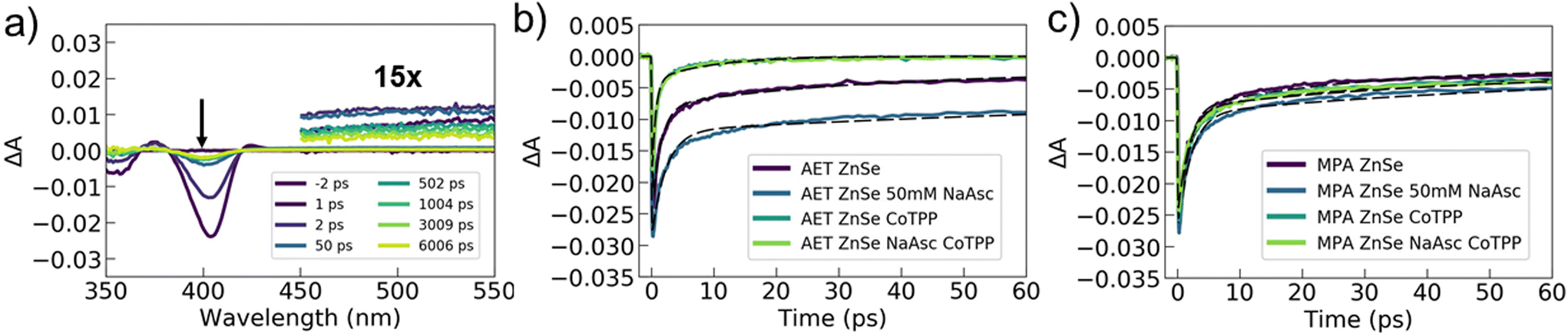 | ||
| Fig. 3 Transient absorption spectroscopy of ZnSe QDs with (and without) the Co-TPP catalyst or NaAsc hole scavenger. (a) Transient spectra of AET ZnSe QDs at varying times after laser excitation. An excitonic bleach feature is observed at 400 nm and broad photo-induced absorption at longer wavelengths (multiplied by 15 for clarity). Kinetic traces of the excitonic bleach feature (400 nm) are shown for (b) AET and (c) MPA QDs. All samples were excited at 350 nm and had 5–6 μM QDs. Co-TPP was included in a 1:1 ratio to the QDs, and NaAsc (50 mM). Bi-exponential fits, convoluted with an instrument response function, are overlaid (dashed black lines). | ||
Transient dynamics were extracted from the data by fitting kinetics at 500 nm (trapped carriers) and 400 nm (conduction band electrons). The 500 nm kinetics for all samples were quite similar, with a ∼500 fs rise-time and a bi-exponential decay with time constants of ∼100–200 ps and 5–15 ns (see Fig. S7 and Table S3†). This indicates that the spectrally accessible trapped carriers are minimally impacted by the addition of catalyst or NaAsc. The excitonic bleach kinetics at 400 nm show clear differences between the AET and MPA QDs (Fig. 3b, c and Table 1). Kinetics of photoexcited electrons in AET QDs are significantly impacted by either the addition of catalyst or hole scavenger, while MPA QDs exhibit similar kinetics under all conditions.
| A 1 | τ 1 (error) | A 2 | τ 2 (error) | A inf | |
|---|---|---|---|---|---|
Data were fit to bi-exponential decay functions of the form 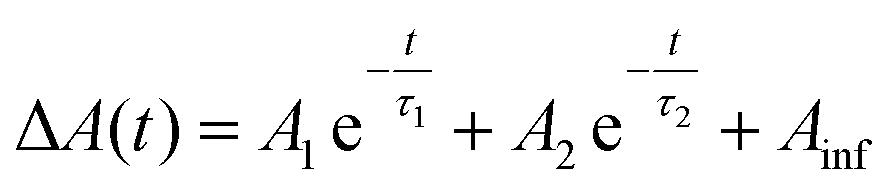 and included convolution of an instrument response function of ∼130 fs.59 and included convolution of an instrument response function of ∼130 fs.59 |
|||||
| AET ZnSe | −0.017 | 1.39 (0.09) | −0.00056 | 33 (4) | −0.0024 |
| AET ZnSe 50 mM NaAsc | −0.017 | 2.29 (0.17) | −0.0063 | 109 (12) | −0.0056 |
| AET ZnSe CoTPP | −0.021 | 0.55 (0.03) | −0.0041 | 7.9 (0.9) | 0 |
| AET ZnSe NaAsc CoTPP | −0.017 | 0.60 (0.04) | −0.0038 | 10 (1) | 0 |
| MPA ZnSe | −0.021 | 1.47 (0.10) | −0.0067 | 26 (3) | −0.0017 |
| MPA ZnSe 50 mM NaAsc | −0.019 | 2.08 (0.15) | −0.0064 | 61 (7) | −0.0026 |
| MPA ZnSe CoTPP | −0.019 | 1.82 (0.13) | −0.0063 | 45 (5) | −0.0018 |
| MPA ZnSe NaAsc CoTPP | −0.017 | 1.81 (0.13) | −0.0058 | 45 (5) | −0.0023 |
The transient data can help us rationalize the differences between the performance of the MPA and AET QDs. The excitonic data for the MPA QDs indicate that electron transfer to Co-TPP proceeds via electron traps, and on a slower time scale than exciton relaxation (1–50 ps). In contrast, photoexcited electrons in AET QDs are rapidly extracted when Co-TPP is included. In these samples, the majority of the excitonic bleach decays with a time constant of ∼500 fs. We ascribe this time constant to electron transfer to Co-TPP, and its rapidity indicates that Co-TPP is bound to the AET QD surface. Furthermore, the addition of NaAsc to AET QDs (without Co-TPP) slows down recovery of the bleach, indicating that electrons are residing in the QD conduction band longer. This is likely a result of efficient hole sequestration by NaAsc, which reduces the availability of holes with which electrons can recombine. It is puzzling that there is no concomitant change in the photoinduced absorption feature ascribed to trapped carriers, but this may be a result of spectrally silent charge carriers playing a role. Overall, these data unambiguously demonstrate that AET QDs support more efficient electron and hole extraction than their MPA QD counterparts. Notably, photoexcited electron transfer from AET QDs to Co-TPP occurs on a sub-picosecond timescale. We posit that the favorable charge extraction kinetics are a result of electrostatic interactions that attract the negatively-charged NaAsc and Co-TPP to the surface of the positively-charged AET QDs.
With a better understanding of the relevant surface interactions and charge dynamics for our system in hand, we aimed to optimize the system for CO production by varying the catalyst number per QD. Since QDs can accommodate multiple molecules on their surface, we explored ratios ranging from 0.25 to 30 Co-TPP per AET QD (Fig. 4). It is important to recall that the work presented in Fig. 1–3 employed a 1:1 ratio. The highest CO TONs per catalyst were achieved at low catalyst concentrations, but increasing catalyst concentration was found to markedly increase selectivity, up to 30%. At high catalyst loadings, we were able to obtain the highest values for mmol CO produced per gram of QD among ZnSe QD-based photocatalytic CO2 reduction systems.
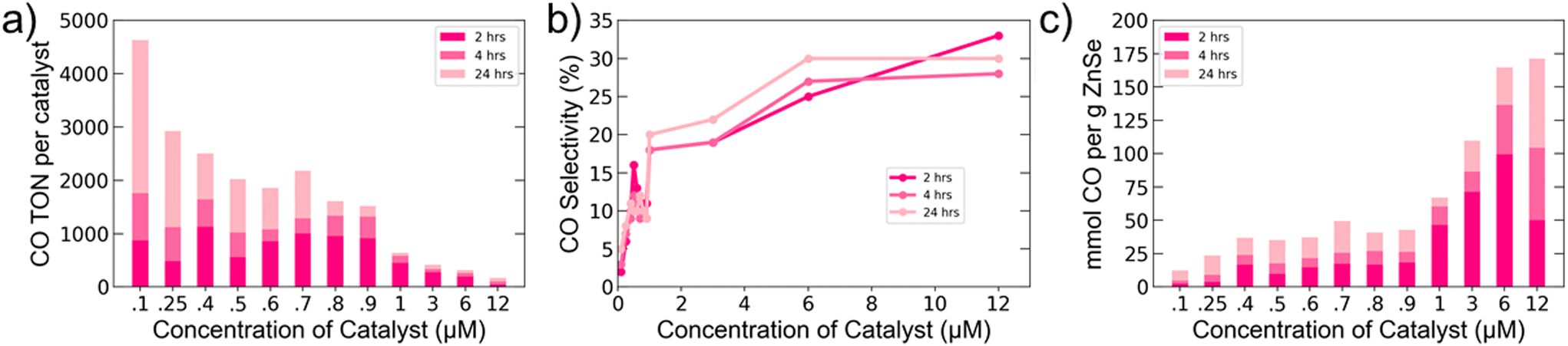 | ||
| Fig. 4 Effect of varying Co-TPP concentration on CO2 reduction. (a) CO TON per catalyst, (b) CO selectivity, and (c) mmol CO produced per gram of ZnSe QDs. Photocatalysis experiments were performed on 2.0 mL aqueous solutions of 0.4 μM ZnSe QDs and 50 mM NaAsc. Samples were irradiated with 400 nm LEDs. | ||
Conclusions
This paper explores the interplay between QD surface functionalization and catalyst chemistry through photocatalysis experiments and ultrafast spectroscopy. Positively-charged QDs were prepared through surface functionalization with 2-aminoethanethiol (AET) while negatively-charged QDs were prepared using 3-mercaptopropionic acid (MPA). These QDs were mixed with a carboxylic acid-functionalized CO2 reduction catalyst, expected to be negatively charged at neutral pH. We found that the positively charged AET QDs support rapid (sub picosecond) photoexcited electron transfer to the catalyst and produced CO in higher quantities and with significantly higher selectivity than the MPA QDs, which supported >10 ps electron transfer. Electrostatic interactions between catalysts’ carboxylate groups and positively-charged AET QDs can explain these observations. Such interactions are not possible for the MPA QDs. Furthermore, after approximately one day, the AET QDs deactivate while the MPA QDs remain active for CO2 reduction. We believe this to be a result of electrostatic-driven QD–catalyst–QD aggregation. Taken as a whole, there is a trade-off between catalytic efficiency and stability when using catalysts with multiple binding groups. A strong catalyst–QD interaction leads to more efficient CO2 reduction, but also deleterious aggregation.The importance of the chemical interaction between the catalyst and the QD surface suggests a variety of potential future experiments. We first observe that the suitability of a specific surface functionalization for a specific photocatalytic reaction is highly dependent on the catalyst chemistry. One strategy to disentangle these effects would be to compare our current system with ammonium-functionalized catalysts and MPA QDs. Furthermore, the potential for QD aggregation must be carefully managed. Altering the ionic strength of the solution or using catalysts functionalized with only one binding group are two strategies that may mitigate the effects of aggregation.
Materials and methods
Synthesis and sample preparation
000 M−1 cm−1).42,53
Instrumentation
Author contributions
F. H., M. Y., N. N., and A. L. contributed equally to this work. F. H. performed data collection, curation, and visualization of photocatalysis data and wrote portions of the materials and methods. M. Y. synthesized ZnSe QDs, developed the methodology for photocatalysis data collection and curation, and collected photocatalysis data. N. N. synthesized the Co-TPP catalyst and developed the methodology for gas chromatography experiments. A. L. performed data curation, developed methodology, and prepared visualizations for transient absorption data and wrote relevant portions of the materials and methods. A. L., K. H., H. N., and X. F. assisted in photocatalysis data collection. C. S. and A. L. prepared ZnSe QDs used in transient absorption experiments. A. S. supervised transient absorption data collection and wrote relevant portions of the materials and methods. J. O. supervised, conceptualized, and obtained funding for this project and wrote the original draft of the manuscript. All authors reviewed and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Acknowledgment is made to the Donors of Petroleum Research Fund, administered by the American Chemical Society, for partial support of this research, under grant number 61874-UNI4. Work at the Molecular Foundry was supported by the Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under contract no. DE-AC02-05CH11231.References
- J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Sustainable Conversion of Carbon Dioxide: An Integrated Review of Catalysis and Life Cycle Assessment, Chem. Rev., 2018, 118(2), 434–504, DOI:10.1021/acs.chemrev.7b00435.
- T. Kong, Y. Jiang and Y. Xiong, Photocatalytic CO2 Conversion: What Can We Learn from Conventional COx Hydrogenation?, Chem. Soc. Rev., 2020, 49(18), 6579–6591, 10.1039/C9CS00920E.
- K. E. Dalle, J. Warnan, J. J. Leung, B. Reuillard, I. S. Karmel and E. Reisner, Electro- and Solar-Driven Fuel Synthesis with First Row Transition Metal Complexes, Chem. Rev., 2019, 119(4), 2752–2875, DOI:10.1021/acs.chemrev.8b00392.
- J. K. Stolarczyk, S. Bhattacharyya, L. Polavarapu and J. Feldmann, Challenges and Prospects in Solar Water Splitting and CO2 Reduction with Inorganic and Hybrid Nanostructures, ACS Catal., 2018, 8(4), 3602–3635, DOI:10.1021/acscatal.8b00791.
- D. M. Fabian, S. Hu, N. Singh, F. A. Houle, T. Hisatomi, K. Domen, F. E. Osterloh and S. Ardo, Particle Suspension Reactors and Materials for Solar-Driven Water Splitting, Energy Environ. Sci., 2015, 8(10), 2825–2850, 10.1039/C5EE01434D.
- H.-L. Wu, X.-B. Li, C.-H. Tung and L.-Z. Wu, Semiconductor Quantum Dots: An Emerging Candidate for CO2 Photoreduction, Adv. Mater., 2019, 31(36), 1900709, DOI:10.1002/adma.201900709.
- Y. Yu, T. Ma and H. Huang, Semiconducting Quantum Dots for Energy Conversion and Storage, Adv. Funct. Mater., 2023, 33(16), 2213770, DOI:10.1002/adfm.202213770.
- N. Nagelj, A. Brumberg, S. Peifer, R. D. Schaller and J. H. Olshansky, Compositionally Tuning Electron Transfer from Photoexcited Core/Shell Quantum Dots via Cation Exchange, J. Phys. Chem. Lett., 2022, 13(14), 3209–3216, DOI:10.1021/acs.jpclett.2c00333.
- D. Prusty, L. Paramanik and K. Parida, Recent Advances on Alloyed Quantum Dots for Photocatalytic Hydrogen Evolution: A Mini-Review, Energy Fuels, 2021, 35(6), 4670–4686, DOI:10.1021/acs.energyfuels.0c04163.
- T. Kameyama, T. Takahashi, T. Machida, Y. Kamiya, T. Yamamoto, S. Kuwabata and T. Torimoto, Controlling the Electronic Energy Structure of ZnS–AgInS2 Solid Solution Nanocrystals for Photoluminescence and Photocatalytic Hydrogen Evolution, J. Phys. Chem. C, 2015, 119(44), 24740–24749, DOI:10.1021/acs.jpcc.5b07994.
- M. D. Regulacio and M.-Y. Han, Multinary I-III-VI2 and I2-II-IV-VI4 Semiconductor Nanostructures for Photocatalytic Applications, Acc. Chem. Res., 2016, 49(3), 511–519 CrossRef CAS PubMed.
- C. B. Murray, D. J. Norris and M. G. Bawendi, Synthesis and Characterization of Nearly Monodisperse CdE (E = Sulfur, Selenium, Tellurium) Semiconductor Nanocrystallites, J. Am. Chem. Soc., 1993, 115(19), 8706–8715, DOI:10.1021/ja00072a025.
- A. Li, T. Wang, C. Li, Z. Huang, Z. Luo and J. Gong, Adjusting the Reduction Potential of Electrons by Quantum Confinement for Selective Photoreduction of CO2 to Methanol, Angew. Chem., Int. Ed., 2019, 58(12), 3804–3808, DOI:10.1002/anie.201812773.
- K. Tvrdy, P. A. Frantsuzov and P. V. Kamat, Photoinduced Electron Transfer from Semiconductor Quantum Dots to Metal Oxide Nanoparticles, Proc. Natl. Acad. Sci. U. S. A., 2011, 108(1), 29–34 CrossRef CAS PubMed.
- E. A. Weiss, Designing the Surfaces of Semiconductor Quantum Dots for Colloidal Photocatalysis, ACS Energy Lett., 2017, 2(5), 1005–1013, DOI:10.1021/acsenergylett.7b00061.
- M. S. Kodaimati, K. P. McClelland, C. He, S. Lian, Y. Jiang, Z. Zhang and E. A. Weiss, Viewpoint: Challenges in Colloidal Photocatalysis and Some Strategies for Addressing Them, Inorg. Chem., 2018, 57(7), 3659–3670, DOI:10.1021/acs.inorgchem.7b03182.
- R. D. Harris, S. Bettis Homan, M. Kodaimati, C. He, A. B. Nepomnyashchii, N. K. Swenson, S. Lian, R. Calzada and E. A. Weiss, Electronic Processes within Quantum Dot-Molecule Complexes, Chem. Rev., 2016, 116(21), 12865–12919, DOI:10.1021/acs.chemrev.6b00102.
- R. Burke, K. L. Bren and T. D. Krauss, Semiconductor Nanocrystal Photocatalysis for the Production of Solar Fuels, J. Chem. Phys., 2021, 154(3), 030901, DOI:10.1063/5.0032172.
- L. Amirav and A. P. Alivisatos, Photocatalytic Hydrogen Production with Tunable Nanorod Heterostructures, J. Phys. Chem. Lett., 2010, 1(7), 1051–1054, DOI:10.1021/jz100075c.
- H. Zhu, N. Song, H. Lv, C. L. Hill and T. Lian, Near Unity Quantum Yield of Light-Driven Redox Mediator Reduction and Efficient H2 Generation Using Colloidal Nanorod Heterostructures, J. Am. Chem. Soc., 2012, 134(28), 11701–11708, DOI:10.1021/ja303698e.
- Z. Han, F. Qiu, R. Eisenberg, P. L. Holland and T. D. Krauss, Robust Photogeneration of H2 in Water Using Semiconductor Nanocrystals and a Nickel Catalyst, Science, 2012, 338(6112), 1321–1324, DOI:10.1126/science.1227775.
- M. B. Wilker, J. K. Utterback, S. Greene, K. A. Brown, D. W. Mulder, P. W. King and G. Dukovic, Role of Surface-Capping Ligands in Photoexcited Electron Transfer between CdS Nanorods and [FeFe] Hydrogenase and the Subsequent H2 Generation, J. Phys. Chem. C, 2018, 122(1), 741–750, DOI:10.1021/acs.jpcc.7b07229.
- C. M. Chang, K. L. Orchard, B. C. M. Martindale and E. Reisner, Ligand Removal from CdS Quantum Dots for Enhanced Photocatalytic H2 Generation in pH Neutral Water, J. Mater. Chem. A, 2016, 4(8), 2856–2862, 10.1039/C5TA04136H.
- Z. Zhang, K. Edme, S. Lian and E. A. Weiss, Enhancing the Rate of Quantum-Dot-Photocatalyzed Carbon-Carbon Coupling by Tuning the Composition of the Dot's Ligand Shell, J. Am. Chem. Soc., 2017, 139(12), 4246–4249 CrossRef CAS PubMed.
- J. A. Caputo, L. C. Frenette, N. Zhao, K. L. Sowers, T. D. Krauss and D. J. Weix, General and Efficient C–C Bond Forming Photoredox Catalysis with Semiconductor Quantum Dots, J. Am. Chem. Soc., 2017, 139(12), 4250–4253, DOI:10.1021/jacs.6b13379.
- Z.-K. Xin, M.-Y. Huang, Y. Wang, Y.-J. Gao, Q. Guo, X.-B. Li, C.-H. Tung and L.-Z. Wu, Reductive Carbon–Carbon Coupling on Metal Sites Regulates Photocatalytic CO2 Reduction in Water Using ZnSe Quantum Dots, Angew. Chem., Int. Ed., 2022, 61(31), e202207222, DOI:10.1002/anie.202207222.
- I. N. Chakraborty, S. Roy, G. Devatha, A. Rao and P. P. Pillai, InP/ZnS Quantum Dots as Efficient Visible-Light Photocatalysts for Redox and Carbon–Carbon Coupling Reactions, Chem. Mater., 2019, 31(7), 2258–2262, DOI:10.1021/acs.chemmater.9b00086.
- Y. Jiang, C. Wang, C. R. Rogers, M. S. Kodaimati and E. A. Weiss, Regio- and Diastereoselective Intermolecular [2+2] Cycloadditions Photocatalysed by Quantum Dots, Nat. Chem., 2019, 11(11), 1034–1040, DOI:10.1038/s41557-019-0344-4.
- Y. Yuan, N. Jin, P. Saghy, L. Dube, H. Zhu and O. Chen, Quantum Dot Photocatalysts for Organic Transformations, J. Phys. Chem. Lett., 2021, 7180–7193, DOI:10.1021/acs.jpclett.1c01717.
- K. A. Brown, D. F. Harris, M. B. Wilker, A. Rasmussen, N. Khadka, H. Hamby, S. Keable, G. Dukovic, J. W. Peters, L. C. Seefeldt and P. W. King, Light-Driven Dinitrogen Reduction Catalyzed by a CdS:Nitrogenase MoFe Protein Biohybrid, Science, 2016, 352(6284), 448–450, DOI:10.1126/science.aaf2091.
- M. Ye, X. Gao, X. Hong, Q. Liu, C. He, X. Liu and C. Lin, Recent Advances in Quantum Dot-Sensitized Solar Cells: Insights into Photoanodes, Sensitizers, Electrolytes and Counter Electrodes, Sustainable Energy Fuels, 2017, 1(6), 1217–1231, 10.1039/C7SE00137A.
- D.-T. Nguyen, A. Chouat and T.-O. Do, Highly Efficient Proton-Assisted Photocatalytic CO2 Reduction over 3-Mercaptopropionic Acid-Capped Quantums Dots, Sustainable Energy Fuels, 2021, 5(16), 4015–4022, 10.1039/D1SE00641J.
- Q.-Q. Bi, J.-W. Wang, J.-X. Lv, J. Wang, W. Zhang and T.-B. Lu, Selective Photocatalytic CO2 Reduction in Water by Electrostatic Assembly of CdS Nanocrystals with a Dinuclear Cobalt Catalyst, ACS Catal., 2018, 8(12), 11815–11821, DOI:10.1021/acscatal.8b03457.
- M. F. Kuehnel, K. L. Orchard, K. E. Dalle and E. Reisner, Selective Photocatalytic CO2 Reduction in Water through Anchoring of a Molecular Ni Catalyst on CdS Nanocrystals, J. Am. Chem. Soc., 2017, 139(21), 7217–7223, DOI:10.1021/jacs.7b00369.
- Y.-X. Feng, H.-J. Wang, J.-W. Wang, W. Zhang, M. Zhang and T.-B. Lu, Stand-Alone CdS Nanocrystals for Photocatalytic CO2 Reduction with High Efficiency and Selectivity, ACS Appl. Mater. Interfaces, 2021, 13(22), 26573–26580, DOI:10.1021/acsami.1c03606.
- Y. S. Chaudhary, T. W. Woolerton, C. S. Allen, J. H. Warner, E. Pierce, S. W. Ragsdale and F. A. Armstrong, Visible Light-Driven CO2 Reduction by Enzyme Coupled CdS Nanocrystals, Chem. Commun., 2011, 48(1), 58–60, 10.1039/C1CC16107E.
- Z.-K. Xin, Y.-J. Gao, Y. Gao, H.-W. Song, J. Zhao, F. Fan, A.-D. Xia, X.-B. Li, C.-H. Tung and L.-Z. Wu, Rational Design of Dot-on-Rod Nano-Heterostructure for Photocatalytic CO2 Reduction: Pivotal Role of Hole Transfer and Utilization, Adv. Mater., 2022, 34(3), 2106662, DOI:10.1002/adma.202106662.
- C. D. Sahm, G. M. Ucoski, S. Roy and E. Reisner, Automated and Continuous-Flow Platform to Analyze Semiconductor–Metal Complex Hybrid Systems for Photocatalytic CO2 Reduction, ACS Catal., 2021, 11(17), 11266–11277, DOI:10.1021/acscatal.1c02921.
- C. D. Sahm, E. Mates-Torres, N. Eliasson, K. Sokołowski, A. Wagner, K. E. Dalle, Z. Huang, O. A. Scherman, L. Hammarström, M. García-Melchor and E. Reisner, Imidazolium-Modification Enhances Photocatalytic CO2 Reduction on ZnSe Quantum Dots, Chem. Sci., 2021, 12(26), 9078–9087, 10.1039/D1SC01310F.
- C. D. Sahm, A. Ciotti, E. Mates-Torres, V. Badiani, K. Sokołowski, G. Neri, A. J. Cowan, M. García-Melchor and E. Reisner, Tuning the Local Chemical Environment of ZnSe Quantum Dots with Dithiols towards Photocatalytic CO2 Reduction, Chem. Sci., 2022, 13(20), 5988–5998, 10.1039/D2SC00890D.
- M. F. Kuehnel, C. D. Sahm, G. Neri, J. R. Lee, K. L. Orchard, A. J. Cowan and E. Reisner, ZnSe Quantum Dots Modified with a Ni(Cyclam) Catalyst for Efficient Visible-Light Driven CO2 Reduction in Water, Chem. Sci., 2018, 9(9), 2501–2509, 10.1039/C7SC04429A.
- F. Arcudi, L. Đorđević, B. Nagasing, S. I. Stupp and E. A. Weiss, Quantum Dot-Sensitized Photoreduction of CO2 in Water with Turnover Number > 80,000, J. Am. Chem. Soc., 2021, 143(43), 18131–18138, DOI:10.1021/jacs.1c06961.
- S. Lian, M. S. Kodaimati and E. A. Weiss, Photocatalytically Active Superstructures of Quantum Dots and Iron Porphyrins for Reduction of CO2 to CO in Water, ACS Nano, 2018, 12(1), 568–575, DOI:10.1021/acsnano.7b07377.
- J. Huang, B. Xu, L. Tian, P. B. Pati, A. S. Etman, J. Sun, L. Hammarström and H. Tian, A Heavy Metal-Free CuInS2 Quantum Dot Sensitized NiO Photocathode with a Re Molecular Catalyst for Photoelectrochemical CO2 Reduction, Chem. Commun., 2019, 55(55), 7918–7921, 10.1039/C9CC04222A.
- J. Huang, M. G. Gatty, B. Xu, P. B. Pati, A. S. Etman, L. Tian, J. Sun, L. Hammarström and H. Tian, Covalently Linking CuInS2 Quantum Dots with a Re Catalyst by Click Reaction for Photocatalytic CO2 Reduction, Dalton Trans., 2018, 47(31), 10775–10783, 10.1039/C8DT01631C.
- J. Zhang, A. Bifulco, P. Amato, C. Imparato and K. Qi, Copper Indium Sulfide Quantum Dots in Photocatalysis, J. Colloid Interface Sci., 2023, 638, 193–219, DOI:10.1016/j.jcis.2023.01.107.
- S. Chen, H. Yin, P. Liu, Y. Wang and H. Zhao, Stabilization and Performance Enhancement Strategies for Halide Perovskite Photocatalysts, Adv. Mater., 2023, 35(6), 2203836, DOI:10.1002/adma.202203836.
- J. Wang, Y. Shi, Y. Wang and Z. Li, Rational Design of Metal Halide Perovskite Nanocrystals for Photocatalytic CO2 Reduction: Recent Advances, Challenges, and Prospects, ACS Energy Lett., 2022, 7(6), 2043–2059, DOI:10.1021/acsenergylett.2c00752.
- M. Banski, M. Afzaal, M. A. Malik, A. Podhorodecki, J. Misiewicz and P. O'Brien, Special Role for Zinc Stearate and Octadecene in the Synthesis of Luminescent ZnSe Nanocrystals, Chem. Mater., 2015, 27(11), 3797–3800, DOI:10.1021/acs.chemmater.5b00347.
- R. Toufanian, X. Zhong, J. C. Kays, A. M. Saeboe and A. M. Dennis, Correlating ZnSe Quantum Dot Absorption with Particle Size and Concentration, Chem. Mater., 2021, 33(18), 7527–7536, DOI:10.1021/acs.chemmater.1c02501.
- A. J. Morris, G. J. Meyer and E. Fujita, Molecular Approaches to the Photocatalytic Reduction of Carbon Dioxide for Solar Fuels, Acc. Chem. Res., 2009, 42(12), 1983–1994, DOI:10.1021/ar9001679.
- H. Takeda, C. Cometto, O. Ishitani and M. Robert, Electrons, Photons, Protons and Earth-Abundant Metal Complexes for Molecular Catalysis of CO2 Reduction, ACS Catal., 2017, 7(1), 70–88, DOI:10.1021/acscatal.6b02181.
- A. Call, M. Cibian, K. Yamamoto, T. Nakazono, K. Yamauchi and K. Sakai, Highly Efficient and Selective Photocatalytic CO2 Reduction to CO in Water by a Cobalt Porphyrin Molecular Catalyst, ACS Catal., 2019, 9(6), 4867–4874, DOI:10.1021/acscatal.8b04975.
- R. R. Knauf, J. C. Lennox and J. L. Dempsey, Quantifying Ligand Exchange Reactions at CdSe Nanocrystal Surfaces, Chem. Mater., 2016, 28(13), 4762–4770, DOI:10.1021/acs.chemmater.6b01827.
- T. Nakazono, A. R. Parent and K. Sakai, Cobalt Porphyrins as Homogeneous Catalysts for Water Oxidation, Chem. Commun., 2013, 49(56), 6325–6327, 10.1039/C3CC43031F.
- T. Matsui, Y. Kitagawa, M. Okumura and Y. Shigeta, Accurate Standard Hydrogen Electrode Potential and Applications to the Redox Potentials of Vitamin C and NAD/NADH, J. Phys. Chem. A, 2015, 119(2), 369–376, DOI:10.1021/jp508308y.
- R. N. Sampaio, D. C. Grills, D. E. Polyansky, D. J. Szalda and E. Fujita, Unexpected Roles of Triethanolamine in the Photochemical Reduction of CO2 to Formate by Ruthenium Complexes, J. Am. Chem. Soc., 2020, 142(5), 2413–2428, DOI:10.1021/jacs.9b11897.
- H. Zhu, Y. Yang, K. Wu and T. Lian, Charge Transfer Dynamics from Photoexcited Semiconductor Quantum Dots, Annu. Rev. Phys. Chem., 2016, 67(1), 259–281, DOI:10.1146/annurev-physchem-040215-112128.
- R. J. Fernández-Terán, E. Sucre-Rosales, L. Echevarria and F. E. Hernández, A Sweet Introduction to the Mathematical Analysis of Time-Resolved Spectra and Complex Kinetic Mechanisms: The Chameleon Reaction Revisited, J. Chem. Educ., 2022, 99(6), 2327–2337, DOI:10.1021/acs.jchemed.2c00104.
- K. M. Watson and J. B. Asbury, Synthesis and Photoinduced Electron Transfer Studies of Ligand Exchanged Mn-Doped ZnSe Nanocrystals in Water, J. Phys. Chem. C, 2022, 126(10), 4995–5003, DOI:10.1021/acs.jpcc.2c00379.
Footnote |
| † Electronic supplementary information (ESI) available: Additional photocatalysis data and details on calculations, additional transient absorption data and analysis. See DOI: https://doi.org/10.1039/d3nr06177a |
| This journal is © The Royal Society of Chemistry 2024 |