
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Investigating the mechanism of phosphorene nanoribbon synthesis by discharging black phosphorus intercalation compounds†
Rebecca R. C.
Shutt
 a,
Eva S. Y.
Aw
a,
Qili
Liu
b,
Jasper
Berry-Gair
b,
Hector J.
Lancaster
a,
Samia
Said
c,
Thomas S.
Miller
c,
Furio
Corà
b,
Christopher A.
Howard
*a and
Adam J.
Clancy
*b
a,
Eva S. Y.
Aw
a,
Qili
Liu
b,
Jasper
Berry-Gair
b,
Hector J.
Lancaster
a,
Samia
Said
c,
Thomas S.
Miller
c,
Furio
Corà
b,
Christopher A.
Howard
*a and
Adam J.
Clancy
*b
aDepartment of Physics and Astronomy, University College London, London, WC1E 6BT, UK. E-mail: c.howard@ucl.ac.uk
bDepartment of Chemistry, University College London, London, WC1E 0AJ, UK. E-mail: a.clancy@ucl.ac.uk
cElectrochemical Innovation Laboratory, Department of Chemical Engineering, University College London, Gower Street, London, WC1E 6BT, UK
First published on 27th December 2023
Abstract
Phosphorene nanoribbons (PNRs) can be synthesised in intrinsically scalable methods from intercalation of black phosphorus (BP), however, the mechanism of ribbonisation remains unclear. Herein, to investigate the point at which nanoribbons form, we decouple the two key synthesis steps: first, the formation of the BP intercalation compound, and second, the dissolution into a polar aprotic solvent. We find that both the lithium intercalant and the negative charge on the phosphorus host framework can be effectively removed by addition of phenyl cyanide to return BP and investigate whether fracturing to ribbons occurred after the first step. Further efforts to exfoliate mechanically with or without solvent reveal that the intercalation step does not form ribbons, indicating that an interaction between the amidic solvent and the intercalated phosphorus compound plays an important role in the formation of nanoribbons.
 Adam J. Clancy | Dr Adam J. Clancy is a Royal Society URF and Lecturer of Inorganic Chemistry at UCL Chemistry. His work revolves around the redox chemistry and spontaneous dissolution of high aspect ratio nanomaterials which are both used in practical energy applications and studied intrinsically to provide fundamental insight into dissolution and nanoscale charged interfaces. Prior to his current position, he undertook his PhD at Imperial College London under Prof Shaffer on carbon nanotube structural composites (and still holds the record strength for PVC composites), having initially studied his MChem at Cardiff University. |
Introduction
Predictions of phosphorene nanoribbons (PNRs) as a promising functional material for energy harvesting and storage devices preceded their experimental bulk synthesis in 2019.1–3 PNRs retain 2D phosphorene's high specific capacity4–6 (433 mA h g−1 for Li(1/2)P), and high carrier mobilities,7 which are on the order of 105 cm2 V−1 s−1. Additional to their 2D phosphorene counterpart, PNRs’ lateral electronic confinement, edge states,8–10 and quasi-1D dimensionality could enable greater tunability in properties and emergent exotic phenomena have been predicted.11 For this reason, the width of nanoribbons is an important characteristic to distinguish between properties exclusive to nanoribbon-like phosphorene instead of sheet-like 2D phosphorene, with the most compelling PNR properties thought to require significant lateral confinement (i.e., narrow widths of <100 nm) and well understood edges (e.g. crystallographically straight).The additional routes to modify the intrinsic optoelectronic properties open applications beyond traditional phosphorene devices, such as water splitting photocatalysts, where edge-controlled chemistries provide a route to devices with a high theoretical maximum efficiency7 of 20%, as well as charge transport layers in perovskite solar cells.10 Experimentally, PNRs have already shown promise, including as hole carrier layers in MAPbI3-based inverted perovskite solar cells, with improved efficiencies12 from ∼19.6% to >21%, and in lithium-metal batteries, for which thin PNR coatings on Li anodes were found to suppress lithium dendrite formation more efficiently than 2D phosphorene.13 Phosphorene-based materials are particularly promising for Li-ion batteries, due to the moderate Li atom binding energy (2.0 eV) and low anisotropic barrier to Li diffusion (0.10 eV) in the zigzag (ZZ) direction (i.e. along the a axis, Fig. 1a).2
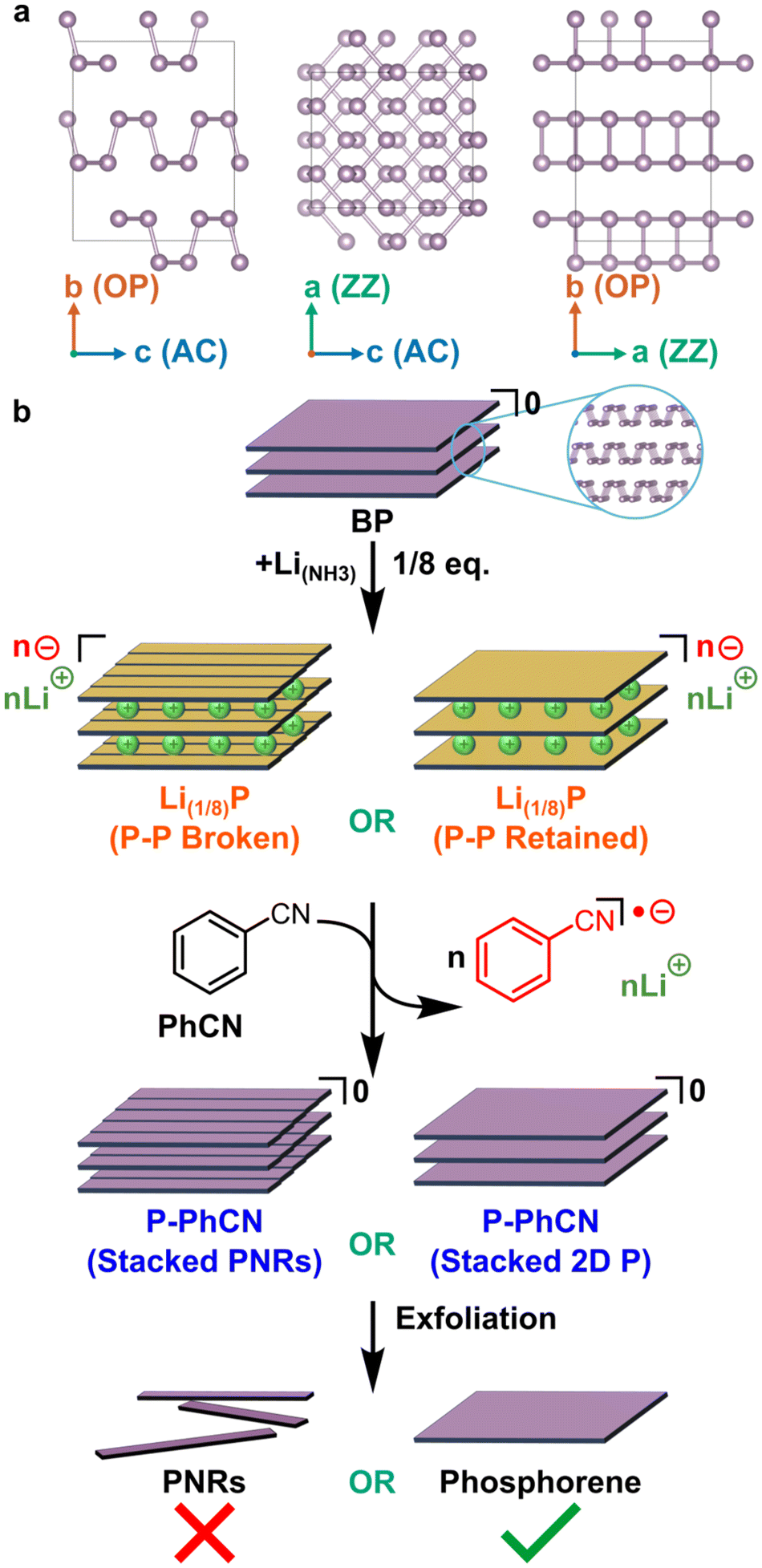 | ||
| Fig. 1 (a) Structure of BP along crystallographic axes, from left to right ‘a’ (zig-zag, ZZ), ‘b’ (out of plane, OP), ‘c’ (armchair, AC). (b) Schematic of reaction pathway of BP intercalation to Li(1/8)P (forming either stacked PNRs from cutting during intercalation, or returning stacked phosphorene sheets) followed by discharging to P-PhCN and exfoliation to PNRs/phosphorene respectively, providing insight into P-framework structure after intercalation. | ||
For use in lab-scale microdevices, tape exfoliation of black phosphorus (BP, Fig. 1a) followed by electron beam lithography has been a popular route to PNRs of well-defined length and a high degree of width uniformity along the length of the ribbons.2,14–16 More recently, tape exfoliation has been shown to directly yield PNRs by partially ripping the surface layer of few-layer BP with widths down to 14 nm via partial tape exfoliation, which can be subsequently extracted with polydimethylsiloxane.17 However, routes that use tape-exfoliation and electron beam lithography are difficult to scale, with the latter requiring challenging synthesis for each individual PNR.
Chemical vapour transport to grow narrow, high aspect ratio BP columns out of BP crystals (later exfoliated through sonication in amidic solvent) could be a promising route to scalable PNR synthesis.18 However, at present the thinnest reported species grown in this manner are multiple layers thick and have widths “around 1 μm”15 (referred to as ‘nanobelts’), which is insufficient for the lateral confinement of PNRs and are thus expected to have similar properties to few-layer 2D phosphorene.
Alternatively, intrinsically scalable approaches have unzipped zigzag-edged nanoribbons (ZZ-PNRs) from intercalated BP, either via electrochemical charging or intercalation of group 1 metals with highly reducing metal electride solutions followed by exfoliation in amidic solvents.13,19,20 These reduction-based techniques rely on the relative stability of orthorhombic phosphorus, which can form stable anionic frameworks.21 The exclusive formation of ZZ-PNRs as opposed to armchair (AC) nanoribbons through these intercalation-based syntheses indicates breaking of the longer, partially out-of-plane P–P bonds. All methods involving intercalation of BP followed by sonication produce nanoribbons with high aspect ratio (>20), however the distributions of nanoribbon width, number of layers, and the morphology at the edges of the nanoribbons vary between syntheses. The solvent chosen also appears to impact the yield and layer number of produced PNRs, e.g., dimethylformamide (DMF) provides high yield, while N-methyl-2-pyrrolidone (NMP) produces fewer, but mostly monolayer, species.20 The Watts et al. method involves a two-step process, with separate steps for formation of the BP intercalation compound and subsequent spontaneous dissolution in amidic solvent, as well as being technically challenging as it involves manipulation of ammonia over the substrate inside a sealed gas manifold. However, significant proportion of the formed nanoribbons have narrow widths, are predominately monolayer species, and, in contrast to other approaches, have an extremely high degree of width uniformity. Even narrower nanoribbons (∼10 nm) are accessible through electrochemical Na intercalation of BP, although these nanoribbons lack uniformity of width along the length of the ribbon, attributed to etching of phosphorene to amorphous phosphides preferentially (but not exclusively) down the zig-zag axis either side of residual PNRs.22 Although different characteristics of nanoribbons are apparent from different reported syntheses, the causal link between synthesis method and nanoribbon properties remains unclear.
Insights into the mechanism of nanoribbon formation is pivotal for improving PNR synthesis to increase yield, as well as to diversify and control nanoribbon structure (width, length, edge chemistry, chirality, etc.) and thus control their properties.
Here, we decouple the two steps of the lithium electride PNR synthesis by reducing BP, and subsequently chemically discharging the Li(1/8)P material by addition of phenyl cyanide (PhCN, Fig. 1b), in a reaction analogous to that performed by Hirsch for graphite intercalation compounds.23 We show through X-ray photoelectron spectroscopy (XPS) and Raman spectroscopy analysis that the negative charge introduced to the phosphorus framework on formation of Li(1/8)P via lithium electride solutions can be removed by addition of PhCN. For clarity, the resulting deintercalated material is termed P-PhCN in the subsequent discussion of the paper. Density functional theory (DFT) calculations, transmission electron microscopy (TEM), and atomic force microscopy (AFM) of mechanically exfoliated P-PhCN crystals are used to postulate the role of solvent interaction with the phosphorus intercalation compound in the formation of nanoribbons.
Results
The potential formation of discrete ZZ-PNRs during lithium intercalation is predicated on the weakening of the out-of-plane P–P bonds. These bonds have previously been shown to elongate upon intercalation of Na or K by Abellán et al.,24 and have been computationally predicted to eventually rupture upon further sodiation to Na(∼1/2)P.25 However, while the electrochemical intercalation route (involving lateral etching to leave less-reacted residual PNRs) is most effective with sodium intercalation,22 the electride synthesis of Watts et al.20 involves lithium (1/8 equivalent) with no notable difference found using sodium. However, the structure of BP intercalated with lithium at the required stoichiometry has not been explored computationally to date.To study the behaviour of lithium intercalation in comparison to the heavier group 1 metals, we investigated the lithium doping of BP with first principles calculations (B3LYP functional with D3 correction). A BP supercell of 32 phosphorus atoms consisting of two phosphorene sheets/galleries was modelled with 0, 1, 2, 4, and 8 lithium atoms present, for LixP stoichiometries of x = 0, 1/32, 1/16, 1/8, and 1/4. The 1/16 system was modelled with the two Li atoms initially placed in both the same and in different interlayer galleries (Fig. 2f and g).
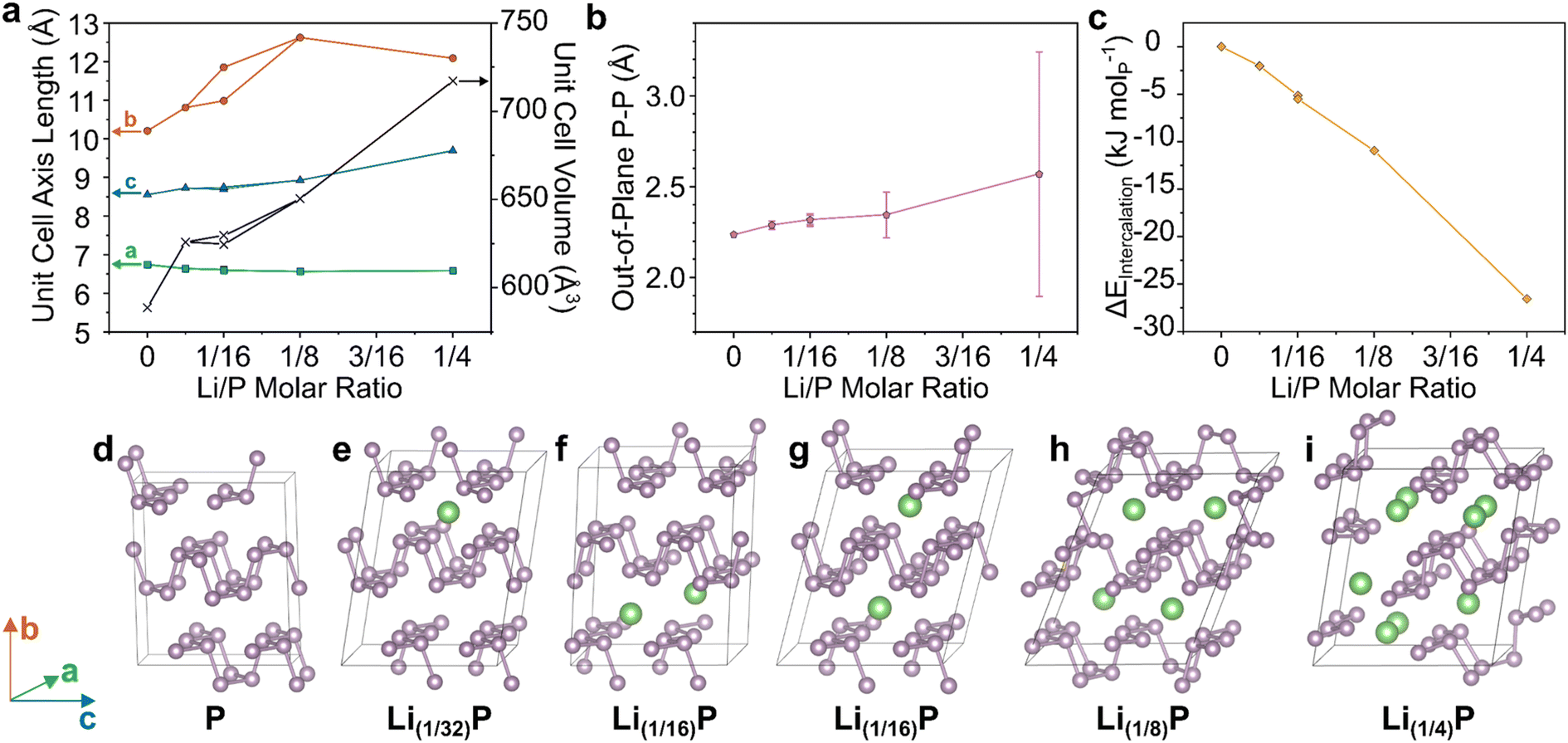 | ||
| Fig. 2 DFT modelling of BP upon intercalation of Li at different stoichiometries. (a) Unit cell axes lengths and unit cell volume of modelled supercells. (b) Lengths of out-of-plane P–P bonds, with error bars representing one standard deviation from the mean. (c) Enthalpy of formation of Li–BP intercalation compound normalised per P atom. Full data tabulated in ESI.† (d–i) supercell structures after relaxation with increasing Li content. Schematic axes assignments provided on the left. Visualizations along crystallographic axes provided in ESI.† | ||
With increasing Li content up to Li(1/8)P, the interlayer b-axis dramatically increased (Fig. 2a), predominantly to accommodate the lithium ions, but also due to lengthening of the out-of-plane P–P bonds (Fig. 2b), as seen for Na/K intercalation.24 Concurrently, the in-plane bonds shorten, decreasing the zig-zag parallel a-axis, indicative of bond strengthening. The initial AA stacking of BP (Fig. 2d) changes upon Li intercalation with layers appearing to shear, giving an AB structure, leading to the corrugations of adjacent sheets forming pockets where Li ions reside (Fig. 2e and g–i). The exception is the Li(1/16) model with both ions in the same layer (Fig. 2f) which better retains the AA stacking. However, as the difference in energy of both Li(1/16)P models is very small (<1 kJ molP−1), it appears that forming Li pockets is comparable in energy to retaining AA stacking of neutral sheets. The lack of a driving force for evenly distributing Li across layers suggests that ordered staging is unlikely and Li will be intercalated stochastically. Multiple initial configurations are also possible for Li(1/8)P which also show negligible energy differences, further supporting this conclusion (ESI discussion & Fig. S16†). When increasing the lithium fraction further to Li(1/4)P, there is an increase in the mean interatomic length alongside a very dramatic increase in the standard deviation, due to a notable fraction of out-of-plane P–P distances increasing significantly, indicating the breaking of bonds (Fig. 2i). This behaviour implies formation of molecular lithium phosphides at the expense of discrete phosphorene sheets, as seen previously24,26 experimentally for K(≤1/4)P and the related Li(1/4.5)(As0.45P0.55). Notably, intercalation becomes increasingly thermodynamically favourable for increasing Li content, akin to the behaviour of the sodiation of BP.25 Taken together, these data support the possible formation of PNRs during the intercalation step of the synthesis, prior to contact with, and dissolution in, solvent. Although, while the P–P bonds are lengthened in the Li(1/8)P model, which is known to form PNRs (and is the targeted stoichiometry for the experimental part of this study), total bond cleavage to nanoribbons was not seen in the computation at the degrees of lithiation explored.
To investigate whether ribbons are present in significant concentration in the phosphorous intercalation compound after reaction with 1/8 molar equivalent of lithium electride and prior to any amidic solvent interaction, we characterised tape-exfoliated material with AFM. Attempts to apply tape exfoliation on the intermediate lithium intercalation compound was unsuccessful, attributed to the replacement of BP's relatively weak interlayer van der Waals bonding with strong ionic bonds between the anionic phosphorus layers and the intercalated lithium cations.
Instead, the Li(1/8)P was discharged to return a phosphorus framework and remove the interlayer ionic bonding. If intercalation alone had fractured the constituent crystalline phosphorene sheets to PNRs, then subsequent exfoliation through traditional methods would be expected to lead to the presence of ribbons and/or poorly crystalline sheets if the ribbons reformed P–P bonds upon discharging. For the discharging, an oxidant is necessary with redox potential comparable with the neutral Fermi level,27,28 must involve species unreactive with the neutral species,29 and ideally is simple to separate after reaction. Fitting these criteria, we selected PhCN which has previously been shown to be capable of discharging graphite intercalation compounds to return neutral graphene by extraction of molar equivalent electrons (as PhCN radical anions) and Li counter-ions.23
Here, intercalated Li(1/8)P was mildly deintercalated through introduction of PhCNto form a delithiated compound termed hereafter P-PhCN (Fig. 1b). The reaction formed a solution of PhCN−/Li+ with a characteristic visible red colour that could be seen to diffuse from the Li(1/8)P crystal on addition of PhCN. The effect appears to match that seen by Vecera23 for PhCN with KC8, and is consistent with an increased absorption in the blue region (450–495 nm) of the UV-Vis absorption spectrum compared to unreacted PhCN, and is indicative of formation of a stabilised radical anion from charge extraction from the anionic phosphorene (ESI Fig. S1†).
Analysis of the chemistry at the surface of the pristine BP, intercalated Li(1/8)P, and P-PhCN, as analysed by XPS further supports the chemical changes inferred from the UV-vis data. The most intense P 2p3/2 component observed in the P 2p high resolution XPS spectrum of BP is centred at 130.1 eV (Fig. 3). This component is observed to shift by ∼1 eV to lower binding energy (129.2 eV) for the intercalated Li(1/8)P sample, attributed to the donation of electrons to the phosphorus framework upon intercalation of lithium. After reaction between Li(1/8)P and PhCN, the neutral P 2p3/2 component is restored (130.2 eV). The comparable binding energies observed for both BP and P-PhCN, and the lower Li content (ESI Table S2†) is attributed to successful removal of electrons and lithium from the phosphorus framework.
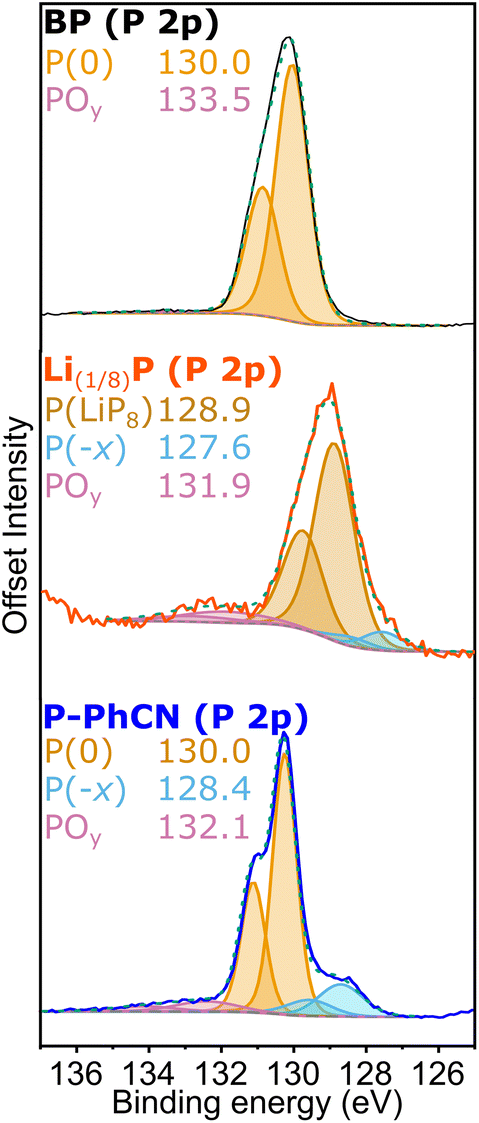 | ||
| Fig. 3 Analysis of surface phosphorus chemical states for BP, Li(1/8)P, and P-PhCN. High-resolution X-ray photoelectron spectroscopy (XPS) phosphorus 2p spectra fitted with several P-bonding environments. The sample identity is annotated with the fitted centres of the P 2p3/2 component of each assigned bonding environment (data, black/red/blue solid; fitted P environments, orange/blue/red solid; total fit, green dotted; background, grey dashed). | ||
An additional low energy shoulder peak (P(−x)) observable for Li(1/8)P between 128–128.5 eV is assigned to P with higher fractional negative charge and increased Li content; this shoulder remains in P-PhCN, suggesting a fraction of the phosphide with higher fractional negative charge (and associated lithium) remains.
The Raman spectrum of pristine BP is typified by three characteristic peaks arising from the A1g, B2g, and A2g phonon modes at 361, 438, and 467 cm−1, respectively (Fig. 4a).20,24,30 On intercalation to Li(1/8)P, the air-free Raman spectrum shows the characteristic three peaks of BP are convoluted by multiple contributions (Fig. 4b), which have been attributed to the introduction of new “intercalation modes” in previous work.20,24 One of these intercalation modes is observed in the spectrum of the Li(1/8)P sample at ∼390 cm−1, which has been previously experimentally observed in potassium and sodium intercalation of black phosphorous.24 On addition of the PhCN, the three characteristic peaks of BP are again deconvoluted, suggesting successful deintercalation. Consistent with previous experimental observations of Li(1/8)P material discharged in air,20 the intercalation mode at 390 cm−1 is not present after treatment with PhCN.
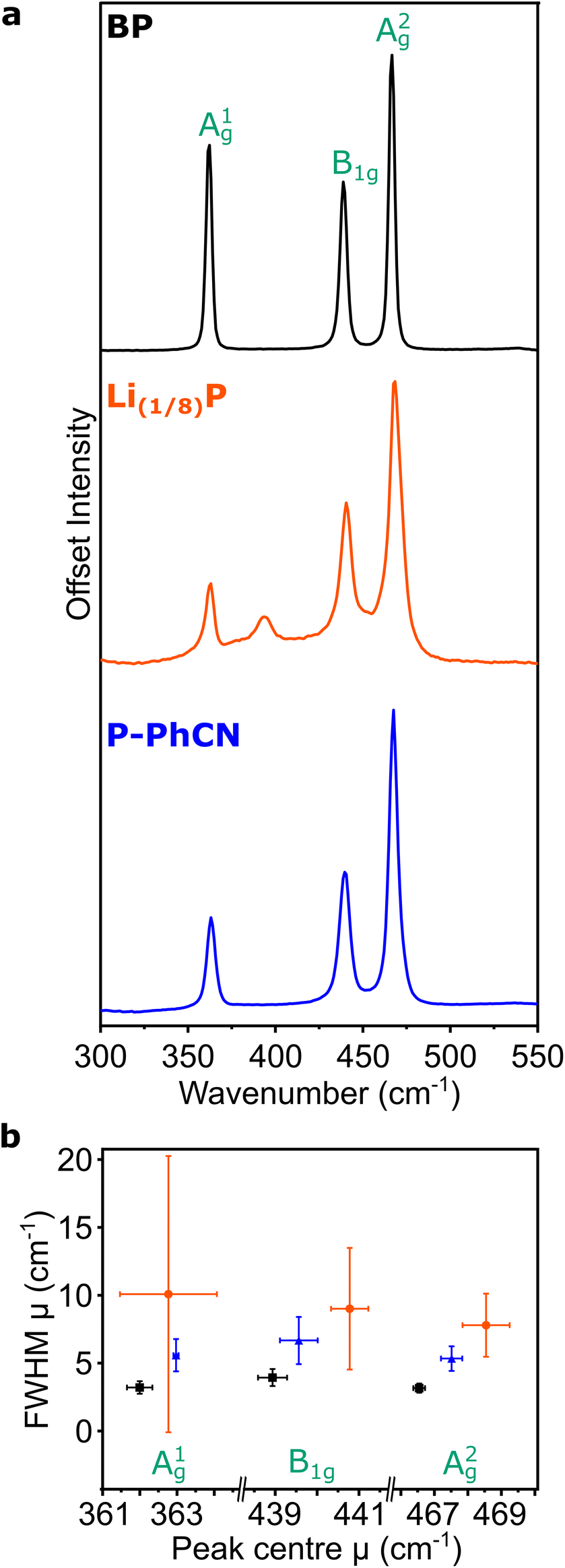 | ||
| Fig. 4 Raman spectroscopy of BP, Li(1/8)P, and P-PhCN samples. (a) Averaged 100-point Raman spectra. (b) Plots the mean fitted (μ) FWHM versus the mean fitted peak centres for A1g, B2g, A2g of BP (black), Li(1/8)P (orange) and P-PhCN (blue) from 100-point spectra taken of each sample over an area of 2500 μm2. The error bars represent one standard deviation from the mean value of peak centre and FWHM extracted from the distribution of fitted peak positions and FWHMs of the 100-point spectra (sampling area = 2500 μm2). The instrument resolution has a FWHM of approximately 1.5 cm−1. | ||
The integrated intensity ratio of A1g to A2g (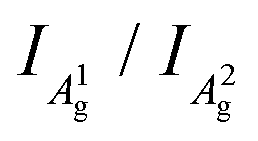 ) of P-PhCN is 0.3 (compared to BP = 0.7, Li(1/8)P = 0.3), consistent with numbers reported for few-layer phosphorene and PNRs in other syntheses.20,31 It is noted that the
) of P-PhCN is 0.3 (compared to BP = 0.7, Li(1/8)P = 0.3), consistent with numbers reported for few-layer phosphorene and PNRs in other syntheses.20,31 It is noted that the 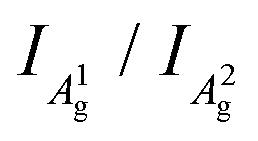 is dependent on multiple factors, including the number of BP layers, degradation from exposure in air, the orientation of the BP crystal,30 and other affects that increase the disorder of the material or otherwise break the long P–P bonds in the BP layers.32 The observation that the integrated intensity ratio of A1g/A2g of the de-intercalated material doesn't return to that observed in the BP could be contributed to by an increased degree of disorder in the material after the chemical intercalation/deintercalation process, potentially linked to the formation of a fraction of amorphous material. In addition, we postulate that shearing of the P-atom planes as indicated by DFT calculations could also be a contributing factor, which is not reversed upon deintercalation.
is dependent on multiple factors, including the number of BP layers, degradation from exposure in air, the orientation of the BP crystal,30 and other affects that increase the disorder of the material or otherwise break the long P–P bonds in the BP layers.32 The observation that the integrated intensity ratio of A1g/A2g of the de-intercalated material doesn't return to that observed in the BP could be contributed to by an increased degree of disorder in the material after the chemical intercalation/deintercalation process, potentially linked to the formation of a fraction of amorphous material. In addition, we postulate that shearing of the P-atom planes as indicated by DFT calculations could also be a contributing factor, which is not reversed upon deintercalation.
The FWHM is related to the lifetime of the phonon, where a broader FWHM indicates a shorter lifetime, which can be a result of scattering events, such as interaction with an electron or with defects in the crystal. The Raman modes of Li(1/8)P show a significant broadening of the FWHM versus BP which is partially recovered on discharging by addition of PhCN. Due to the lack of broad Raman active modes indicative of amorphous phosphorus in P-PhCN, it is more likely that the residual negative charge indicated by XPS is due to a fraction of residual Li intercalant, as opposed to amorphous lithium phosphides.24,33 Given the expansion/layer shearing of the BP framework upon charging, it is assumed that some of the strain effects on the BP crystal from intercalation of Li remain after the crystal is discharged with PhCN, which would be concurrent with some P–P bond weakening, consistent with work by Karki et al.,32 and shown recently from ex situ Raman spectroscopy of BP electrodes electrochemically intercalated and de-intercalated with Na.34
It is noted that there is a large standard deviation in the Li(1/8)P data (Fig. 4b). Our values are extracted from the distribution of fitted modes of 100 spectra taken at different locations on the sample (5 μm spacings across a 50 μm × 50 μm area), and the large standard deviations are indicative of varying local signals across a heterogeneous sample. Based on our DFT calculations, we attribute this wide variation of fitted modes to local variation in the degree of intercalation across the Li(1/8)P sample (ESI, Fig. S2†).17 We note that the heterogeneity is lost upon deintercalation with PhCN.
These trends were further supported by powder XRD of the initial, intercalated, and discharged material (ESI, Fig. S5†). The intercalation of lithium leads to a slight broadening of all peaks, an upshift of the in-plane peaks (e.g. 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ(002) = 41.2° from initial 40.7°), although the significant downshift of the inter-plane reflections expected from DFT were not seen; similar to the XRD of K(1/8)P measured by Abellán.24 In agreement with this prior work, we also saw the emergence of a new peak at ∼32° and further low-angle peaks centred at 6.0° and 12.5°. The discharging reaction led to a removal of these new peaks and a restoration of the in-plane modes (2θ(002) = 40.5°), however, all peaks with an out of plane b component were significantly upshifted implying a layer separation not present in the intercalation compound. We tentatively assign this to a consequence of the layer shearing upon intercalation seen by DFT, whereby the P lone pairs in the AB configuration point towards the lithium pockets in Li(1/8)P, but point towards each other upon the removal of Li+ ions, leading to significant repulsion.
θ(002) = 41.2° from initial 40.7°), although the significant downshift of the inter-plane reflections expected from DFT were not seen; similar to the XRD of K(1/8)P measured by Abellán.24 In agreement with this prior work, we also saw the emergence of a new peak at ∼32° and further low-angle peaks centred at 6.0° and 12.5°. The discharging reaction led to a removal of these new peaks and a restoration of the in-plane modes (2θ(002) = 40.5°), however, all peaks with an out of plane b component were significantly upshifted implying a layer separation not present in the intercalation compound. We tentatively assign this to a consequence of the layer shearing upon intercalation seen by DFT, whereby the P lone pairs in the AB configuration point towards the lithium pockets in Li(1/8)P, but point towards each other upon the removal of Li+ ions, leading to significant repulsion.
To investigate whether the BP had been cleaved into PNRs after undertaking a Li-charging/PhCN discharging cycle (i.e., ribbonised during intercalation, before addition of solvent), exfoliation attempts were undertaken. Samples were sonicated in NMP and deposited by drop-casting for study with transmission electron microscopy (TEM), and mechanically exfoliated with nitto tape for atomic force microscopy (AFM). The same exfoliation method and subsequent characterisation by AFM and TEM was also applied to pristine BP for comparison (ESI, Fig. S4 and S5†). We note that unlike Li(1/8)P which spontaneously forms solutions of PNRs upon addition to tertiary amide solvents, driven by the charged nature of the phosphorus framework, here the P-PhCN did not dissolve in NMP, further supporting successful discharging.
TEM data of sonicated P-PhCN clearly demonstrates the formation of 2D phosphorene sheets and small fragments lacking the high aspect ratio of PNRs (Fig. 5). The presence of large (>1 μm) crystalline phosphorene flakes, as confirmed in selected area electron diffraction, implies that P–P bonds are unlikely to have been first broken during intercalation and then reformed during PhCN treatment, as a notable degree of disorder would be expected during reformation. AFM of tape exfoliated P-PhCN also primarily showed small 2D species, which were mostly <10 phosphorene layers thick (Fig. 6).35 A few high aspect ratio species were also observed (Fig. 6c and d), however, we note that recent work has shown that tape exfoliation may in some cases intrinsically form low quality PNRs from BP, similar to what is observed here.14 The behaviours observed in this work are notably distinct from PNR synthesis from dissolution of Li(1/8)P, which shows near-exclusive formation of >1 μm PNRs in both AFM and TEM.
 | ||
| Fig. 5 TEM characterisation of exfoliated P-PhCN. (a–e) TEM micrographs; (f) Selected area electron diffraction of Fig. 5e. Scalebars: (a–c) = 500 nm, (d and e) = 100 nm, (f) = 10 nm−1, (f)-inset = 2 nm−1. | ||
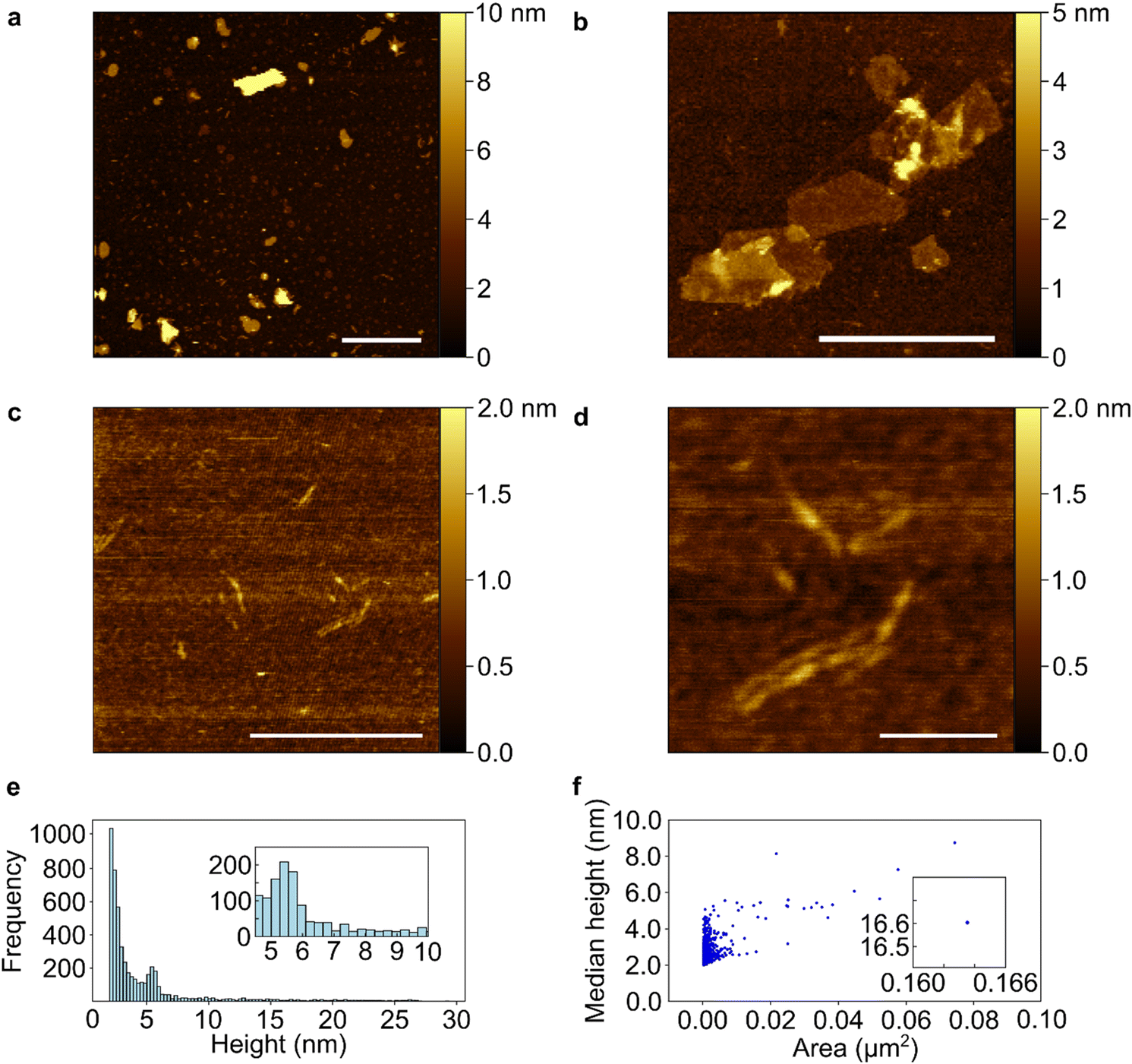 | ||
| Fig. 6 AFM characterisation of tape exfoliated P-PhCN on HOPG substrate. Images with scalebars representing (a) 1 μm, (b and c) 500 nm, and (d) 100 nm. (e) Height distribution of 5184 pixels above a 2 nm threshold from (a) (bin size = 100), with inset zoomed in on 4.5–10 nm. (f) Median height against individual flake area showing a positive correlation, with inset displaying anomaly corresponding to the largest flake in (a). | ||
Conclusion
In conclusion, this study indicates that the interaction of the phosphorus intercalation compound with the amidic solvent is important for the formation of PNRs via the lithium electride synthesis, and that they are not formed purely from bond cleavage during intercalation, nor from anisotropic etching to amorphous phosphides between formed PNRs, as reported for Na electrochemical intercalation.22 This conclusion goes some way to rationalising the previously observed solvent-dependent yield of PNRs and their AsP alloy counterparts.20,26Application of PhCN can be used to de-intercalate lithium from intercalated black phosphorus (Li(1/8)P), and the extent of deintercalation and discharging can be monitored using XPS and Raman spectroscopy. The deintercalated crystal appears to recover long range order, but retain some of the strain induced by occupation of lithium in the interlayer galleries, analogous to that seen by Wen et al. when expanding graphite with Hummers’ method.36 It is concluded therefore that P–P bonds are weakened, but not broken, during intercalation with lithium at this stoichiometry.
The specific role of the solvent in the formation of nanoribbons is yet to be elucidated, but may be related to providing edge passivation, e.g., as an H-atom source for hydrogenation, or stabilising anionic edge sites via solvation. It could also be that on contact of the Li(1/8)P crystal with amidic solvent, some solvent is pulled into the BP framework to co-intercalate with the Li, and seeds the breakage of the out-of-plane P-P bonds and subsequent unzipping of the nanoribbons. Future studies could seek to model the solvation of BP intercalation compounds with various amidic solvents to gain further insights to the mechanism.
Experimental
All sample handling was conducted inside an argon filled glovebox (O2, H2O, less than 0.1 ppm), unless otherwise stated.Intercalation of black phosphorus
Intercalation of BP was performed using lithium electride solutions adapted from a method reported previously.20 Macroscopic BP crystals (99.998% purity; Smart Elements) were outgassed at 100 °C under dynamic vacuum until the pressure dropped to less than 10−6 mbar. Without exposure to air, the outgassed crystals were loaded along with lithium metal (99.9% purity; Sigma Aldrich) at a stoichiometric ratio of Li/P = 1/8 into a fused silica reaction tube and attached via a glass–metal transition to a leak-tight, custom, gas-handling manifold. The argon headspace above the sample is first evacuated to a pressure less than 10−6 mbar and cooled to less than −45 °C using an isopropanol bath and Julabo FT902. High-purity ammonia gas (greater than 99.99% purity; Sigma Aldrich) was then condensed onto the black phosphorus and lithium, dissolving the alkali metal, until the black phosphorus is fully submerged in liquid ammonia and left submerged for at least 12 h, during which time intercalation occurred. Following this process, the ammonia was slowly removed by cryo-pumping, and the intercalated black phosphorus crystal was then further dried by vacuum treatment (<10−6 mbar for 1 h).Deintercalation of Li(1/8)P by addition of PhCN
Neat PhCN (anhydrous, ≥99%, Sigma Aldrich) was added in excess to intercalated black phosphorus crystals (Li(1/8)P) in a glass vial. Small electron acceptors like PhCN can form stable negatively charged radicals, which can be represented through resonance structures. These react via single electron transfer (SET) reactions to form negatively charged radicals, and have been utilized to remove charge from 2D materials.37 Red colour can be seen to diffuse from the crystal as the PhCN reacts to form a stabilized negatively charged radical. After at least 1 h, the excess PhCN was decanted off the crystal using a micropipette and retained for UV-vis analysis. A hotplate at 65 °C was used to evaporate off the remaining PhCN to leave a dry, deintercalated Li(1/8)P + PhCN crystal.AFM characterisation
Atomic force microscopy (AFM) was used to measure the height and lateral size of the flakes. AFM was performed using Bruker Dimension Icon in “PeakForce” tapping mode. ScanAsyst-Air mode was used at a scan rate of 2 Hz, 256 samples per line, with Bruker probes of k = 0.4 N m−1 and tip radius of 2 nm. Nitto tape was used to exfoliate the solid samples by peeling several times and transferred onto freshly cleaved highly ordered pyrolytic graphite (HOPG). All samples were prepared and maintained inside a high-purity argon glovebox environment. Samples were exposed to air only immediately before imaging under AFM. Image and data processing were executed in NanoScope Analysis (2.0) software and Gwyddion (2.56). Further AFM analysis provided in ESI, Fig. S8 and S9.†XPS characterisation
X-ray photoelectron spectroscopy (XPS) was used to determine the surface elemental composition using K-ALPHA Surface Analysis spectrometer (Thermo Scientific) with monochromatic Al Kα radiation of 1486.6 eV as an excitation source, using a spot size of 400 μm. Small pieces of the samples were adhered to the sample holder using indium foil (99.999%, Sigma Aldrich). The sample holder was introduced into a load-lock chamber using a transfer vessel (Thermo Scientific 831-57-100-2) without air exposure. Further XPS Spectra (Li 1s, C 1s, O1s, Survey) provided in ESI, Fig. S4 & Table S2.†TEM characterisation
Transmission electron microscopy were performed using a JEOL 2100F microscope with a W source filament and operated at 200 kV. The samples were prepared first by sonication of the crystal in NMP (1 mg mL−1 concentration) for 30 minutes, and subsequent centrifugation (∼100g, 15 minutes, Hettich EBA 20). The supernatant was dropcast (20 μL) onto lacey carbon copper TEM grids (Agar Scientific) supported on Whatman cellulose filter paper in an Ar glovebox and left to dry at room temperature overnight. Further TEM analysis provided in ESI, Fig. S6 and S7.†Raman spectroscopy characterisation
Samples were loaded in a custom-made leak-tight glass cell in an argon environment. Raman spectroscopy was collected using a Renishaw inVia Raman microscope fitted with a 488 nm laser through a ×20 objective. Laser power at the sample was kept <150 mW. Raman spectra were acquired over a 50 μm × 50 μm sample area over 100 grid points. Spectra were fitted with a linear background and Lorentzian functions. For BP and PhCN, three Lorentzian peaks for the A1g, B2g, and A2g modes were fitted. The convolution of peaks for Li(1/8)P was fitted with the minimum number of Lorentzian functions (five are used) to avoid over-fitting, and the peaks with fitted centres closest to those A1g, B2g, and A2g modes detected in the spectrum of BP were likewise assigned to these modes in Li(1/8)P. Further Raman analysis including xy-maps of peak centres, optical microscopy of measured regions, and full fitted population details provided in ESI, Fig. S2, S3 and Table S1.†Powder X-ray diffraction
Samples were loaded in 0.5 mm glass capillaries sealed with wax in an argon glovebox. Measurements were performed on a STOE XRD using a Cu kα X-ray source, on a rotating sample (∼5 Hz) in 2θ 0.2° steps for 10s at each step. Peak assignment was performed using VESTA to predict peaks from structures derived from DFT calculations.Computational DFT
Density functional theory (DFT) calculations were performed with CRYSTAL17 using the B3LYP hybrid exchange functional with D3 dispersion correction and a triple zeta valence plus polarisation basis set for all atoms,38 assuming a closed shell configuration. A [2 × 1 × 2] supercell of black phosphorus consisting of 32 phosphorus atoms in two atomic layers was used, with 1, 2, 4, and 8 lithium inserted into the interlayer gallery(s). For Li2P32, two configurations were tested to allow for lithium atoms to reside in different/the same gallery, as a stage-1/stage-2 intercalation compounds respectively. Input scripts are provided in the ESI.† Intercalation energies were calculated as the difference in energy between the relaxed structure, and the sum of the constituent separate initial BP cell and lithium metal. Due to the established errors associated with calculating lithium metal with hybrid exchange functionals, its energy was calculated from the B3LYP energy of the Li+ cation using both the ionisation energy (−1.963 × 104 kJ mol−1) and cohesive energy (−1.982 × 104 kJ mol−1). DFT structures down all crystallographic axes for all models, extracted energy values, input scripts, and further models provided in ESI, Fig. S10–S16 and Table S3.†Conflicts of interest
There are no conflicts to declareAcknowledgements
A. J. C. would like to thank The Royal Society for funding through the University Research Fellowship Scheme (URF\R1\221476 and RF\ERE\221017). R. R. C. S., E. A. and S. S. would like to thank the Engineering and Physical Sciences Research Council (EPSRC UK) for studentship funding from the Centre for Doctoral Training in Advanced Materials Characterisation (EP/S023259/1). The authors acknowledge the use of the UCL Kathleen High Performance Computing Facility (Kathleen@UCL), and associated support services, in the completion of this work.References
- T. J. Macdonald, A. J. Clancy, R. R. Shutt and C. A. Howard, Joule, 2022, 6, 2441–2446 CrossRef.
- Q. Yao, C. Huang, Y. Yuan, Y. Liu, S. Liu, K. Deng and E. Kan, J. Phys. Chem. C, 2015, 119, 6923–6928 CrossRef CAS.
- W. Hu, L. Lin, R. Zhang, C. Yang and J. Yang, J. Am. Chem. Soc., 2017, 139, 15429–15436 CrossRef CAS PubMed.
- J. Sun, H.-W. Lee, M. Pasta, H. Yuan, G. Zheng, Y. Sun, Y. Li and Y. Cui, Nat. Nanotechnol., 2015, 10, 980–985 CrossRef CAS PubMed.
- S. Zhao, W. Kang and J. Xue, J. Mater. Chem. A, 2014, 2, 19046–19052 RSC.
- C. Zhang, M. Yu, G. Anderson, R. R. Dharmasena and G. Sumanasekera, Nanotechnology, 2017, 28, 075401 CrossRef PubMed.
- L. Li, Y. Yu, G. J. Ye, Q. Ge, X. Ou, H. Wu, D. Feng, X. H. Chen and Y. Zhang, Nat. Nanotechnol., 2014, 9, 372–377 CrossRef CAS PubMed.
- Z. Yi, M. Wu and R. Jia, J. Mater. Sci., 2022, 57, 5482–5496 CrossRef CAS.
- J. Ou and Q. Zhang, Nanomaterials, 2022, 12, 2350 CrossRef CAS PubMed.
- J.-J. Yang, F. Li, X.-L. Zhang, X.-Y. Liu and L. Li, Appl. Surf. Sci., 2023, 158946 Search PubMed.
- A. Ashoka, A. J. Clancy, N. A. Panjwani, N. J. Popiel, A. Eaton, T. G. Parton, L. Picco, S. Feldmann, R. R. Shutt and R. Carey, arXiv, 2022, preprint arXiv:2211.11374, https://doi.org/10.48550/arXiv.2211.11374.
- T. J. Macdonald, A. J. Clancy, W. Xu, Z. Jiang, C.-T. Lin, L. Mohan, T. Du, D. D. Tune, L. Lanzetta and G. Min, J. Am. Chem. Soc., 2021, 143, 21549–21559 CrossRef CAS PubMed.
- W. Yu, J. Yang, J. Li, K. Zhang, H. Xu, X. Zhou, W. Chen and K. P. Loh, Adv. Mater., 2021, 33, 2102083 CrossRef CAS PubMed.
- X. Feng, X. Huang, L. Chen, W. C. Tan, L. Wang and K. W. Ang, Adv. Funct. Mater., 2018, 28, 1801524 CrossRef.
- S. Lee, F. Yang, J. Suh, S. Yang, Y. Lee, G. Li, H. S. Choe, A. Suslu, Y. Chen and C. Ko, Nat. Commun., 2015, 6, 1–7 Search PubMed.
- P. Masih Das, G. Danda, A. Cupo, W. M. Parkin, L. Liang, N. Kharche, X. Ling, S. Huang, M. S. Dresselhaus and V. Meunier, ACS Nano, 2016, 10, 5687–5695 CrossRef CAS PubMed.
- B. Hu, T. Zhang, K. Wang, L. Wang, Y. Zhang, S. Gao, X. Ye, Q. Zhou, S. Jiang and X. Li, Small, 2023, 19, 2207538 CrossRef CAS PubMed.
- L. Macewicz, K. Pyrchla, R. Bogdanowicz, G. Sumanasekera and J. B. Jasinski, J. Phys. Chem. Lett., 2021, 12, 8347–8354 CrossRef CAS PubMed.
- Z. Liu, Y. Sun, H. Cao, D. Xie, W. Li, J. Wang and A. K. Cheetham, Nat. Commun., 2020, 11, 3917 CrossRef CAS PubMed.
- M. C. Watts, L. Picco, F. S. Russell-Pavier, P. L. Cullen, T. S. Miller, S. P. Bartuś, O. D. Payton, N. T. Skipper, V. Tileli and C. A. Howard, Nature, 2019, 568, 216–220 CrossRef CAS PubMed.
- R. R. Shutt, T. Ramireddy, E. Stylianidis, C. Di Mino, R. A. Ingle, G. Ing, A. A. Wibowo, H. T. Nguyen, C. A. Howard and A. M. Glushenkov, Chem. – Eur. J., 2023, 29, e202301232 CrossRef CAS PubMed.
- U. O. Abu, S. Akter, B. Nepal, K. A. Pitton, B. S. Guiton, D. R. Strachan, G. Sumanasekera, H. Wang and J. B. Jasinski, Adv. Sci., 2022, 9, 2203148 CrossRef CAS PubMed.
- P. Vecera, J. Holzwarth, K. F. Edelthalhammer, U. Mundloch, H. Peterlik, F. Hauke and A. Hirsch, Nat. Commun., 2016, 7, 12411 CrossRef CAS PubMed.
- G. Abellán, C. Neiss, V. Lloret, S. Wild, J. C. Chacón-Torres, K. Werbach, F. Fedi, H. Shiozawa, A. Görling and H. Peterlik, Angew. Chem., Int. Ed., 2017, 56, 15267–15273 CrossRef PubMed.
- K. Hembram, H. Jung, B. C. Yeo, S. J. Pai, S. Kim, K.-R. Lee and S. S. Han, J. Phys. Chem. C, 2015, 119, 15041–15046 CrossRef CAS.
- F. F. Zhang, E. Aw, A. G. Eaton, R. R. C. Shutt, J. Lim, J. H. Kim, T. J. Macdonald, C. I. D. L. Reyes, A. Ashoka, R. Pandya, O. D. Payton, L. Picco, C. E. Knapp, F. Corà, A. Rao, C. A. Howard and A. J. Clancy, J. Am. Chem. Soc., 2023, 145, 18286–18295 CrossRef CAS PubMed.
- S. A. Hodge, D. J. Buckley, H. C. Yau, N. T. Skipper, C. A. Howard and M. S. Shaffer, Nanoscale, 2017, 9, 3150–3158 RSC.
- S. A. Hodge, H. H. Tay, D. B. Anthony, R. Menzel, D. J. Buckley, P. L. Cullen, N. T. Skipper, C. A. Howard and M. S. Shaffer, Faraday Discuss., 2014, 172, 311–325 RSC.
- A. J. Clancy, M. K. Bayazit, S. A. Hodge, N. T. Skipper, C. A. Howard and M. S. Shaffer, Chem. Rev., 2018, 118, 7363–7408 CrossRef CAS PubMed.
- H. Ribeiro, C. Villegas, D. Bahamon, D. Muraca, A. Castro Neto, E. De Souza, A. Rocha, M. Pimenta and C. De Matos, Nat. Commun., 2016, 7, 12191 CrossRef CAS PubMed.
- A. Favron, E. Gaufrès, F. Fossard, A.-L. Phaneuf-L'Heureux, N. Y. Tang, P. L. Lévesque, A. Loiseau, R. Leonelli, S. Francoeur and R. Martel, Nat. Mater., 2015, 14, 826–832 CrossRef CAS PubMed.
- B. Karki, B. Freelon, M. Rajapakse, R. Musa, S. S. Riyadh, B. Morris, U. Abu, M. Yu, G. Sumanasekera and J. B. Jasinski, Nanotechnology, 2020, 31, 425707 CrossRef CAS PubMed.
- C. Ferrara, E. Vigo, B. Albini, P. Galinetto, C. Milanese, C. Tealdi, E. Quartarone, S. Passerini and P. Mustarelli, ACS Appl. Energy Mater., 2019, 2, 2794–2802 CrossRef CAS.
- S. Said, Z. Zhang, R. R. Shutt, H. J. Lancaster, D. J. Brett, C. A. Howard and T. S. Miller, ACS Nano, 2023, 17, 6220–6233 CrossRef CAS PubMed.
- N. Nitta, D. Lei, H.-R. Jung, D. Gordon, E. Zhao, G. Gresham, J. Cai, I. Luzinov and G. Yushin, ACS Appl. Mater. Interfaces, 2016, 8, 25991–26001 CrossRef CAS PubMed.
- Y. Wen, K. He, Y. Zhu, F. Han, Y. Xu, I. Matsuda, Y. Ishii, J. Cumings and C. Wang, Nat. Commun., 2014, 5, 4033 CrossRef CAS PubMed.
- T. Wei, M. Kohring, H. B. Weber, F. Hauke and A. Hirsch, Nat. Commun., 2021, 12, 552 CrossRef CAS PubMed.
- M. F. Peintinger, D. V. Oliveira and T. Bredow, J. Comput. Chem., 2013, 34, 451–459 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr05416k |
| This journal is © The Royal Society of Chemistry 2024 |