
Modification of an oxyhalide solid-solution photocatalyst with an efficient O2-evolving cocatalyst and electron mediator for two-step photoexcitation overall water splitting†
Wenzheng
Sun
 ab,
Ying
Luo
ab,
Jun
Xu
ab,
Qiaoqi
Guo
ab,
Lidan
Deng
c,
Zheng
Wang
*ab and
Hong
He
*ab
ab,
Ying
Luo
ab,
Jun
Xu
ab,
Qiaoqi
Guo
ab,
Lidan
Deng
c,
Zheng
Wang
*ab and
Hong
He
*ab
aState Key Joint Laboratory of Environment Simulation and Pollution Control, Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, Beijing 100085, China. E-mail: zhengwang@rcees.ac.cn; honghe@rcees.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cSchool of Energy and Power Engineering, Huazhong University of Science and Technology, Wuhan 430074, China
First published on 22nd December 2023
Abstract
Two-step photoexcitation overall water splitting based on particulate photocatalysts represents a promising approach for low-cost solar hydrogen production. The performance of an O2-evolution photocatalyst and electron mediator between two photocatalysts crucially influences the construction of an efficient two-step excitation water-splitting system. Bismuth–tantalum oxyhalides are emerging photocatalysts for O2 evolution reactions and can be applied in two-step water-splitting systems. In this study, a highly crystalline Bi4TaO8Cl0.9Br0.1 solid solution with microplatelet morphology was synthesized by the dual flux method. The light absorption intensity and charge transfer efficiency of the Bi4TaO8Cl0.9Br0.1 solid solution were higher than those of Bi4TaO8Cl and Bi4TaO8Br; thus, the sacrificial O2 evolution activity of Bi4TaO8Cl0.9Br0.1 photocatalyst was obviously enhanced. The two-step excitation water splitting with a solid-state electron mediator was successfully constructed using Bi4TaO8Cl0.9Br0.1 as the O2-evolution photocatalyst and Ru/SrTiO3:Rh as the H2-evolution photocatalyst. The CoOx cocatalyst and reduced graphene oxide decorations on the surface of Bi4TaO8Cl0.9Br0.1 promoted the catalytic O2 generation process on Bi4TaO8Cl0.9Br0.1 and electron transfer between CoOx/Bi4TaO8Cl0.9Br0.1 and Ru/SrTiO3:Rh photocatalysts, respectively. As a result, the apparent quantum yield for this overall water-splitting system was 1.26% at 420 nm, which surpassed the present performance of the two-step excitation water-splitting systems consisting of metal oxyhalide photocatalysts. This study demonstrates the validity of high-quality solid-solution photocatalysts with suitable surface modification for efficient solar hydrogen production from water splitting.
 Zheng Wang | Zheng Wang received his PhD under the supervision of Prof. Tsunehiro Tanaka in the Department of Molecular Engineering at Kyoto University in 2015. Then he moved to Prof. Kazunari Domen's group at the University of Tokyo and Shinshu University as the ARPChem project researcher. Since 2020, he joined in Research Center for Eco-Environmental Sciences at Chinese Academy of Sciences as a professor and group leader. He is interested in the utilization and conversion of carbon dioxide and water splitting based on the photocatalysis, photoelectrochemical and photothermocatalysis process. |
Introduction
Solar water splitting using semiconductor particulate photocatalysts for sustainable hydrogen production is one of the promising solutions to tackle the constantly increasing global energy demand and related environmental issues.1–3 However, the large-scale utilization of inexhaustible solar energy faces a series of challenges, the most important being how to attain significant improvement in the efficiency of solar to hydrogen conversion. Under these circumstances, many efficient particulate photocatalysts with narrow bandgaps for water splitting have been extensively explored and achieved immense development of efficient water splitting.4–10 These attempts are mainly accomplished through two approaches, namely, one-step photoexcitation overall water splitting on a single photocatalyst and two-step photoexcitation overall water splitting mimicking photosynthesis in a green plant.11 For one-step photoexcitation water splitting, a lot of metal oxide photocatalysts were active under UV irradiation.11 In order to utilize the solar spectrum over a wide wavelength range, some visible-light-responsive photocatalysts within the (oxy)nitrides,8,12 cation-doped oxides4 and plasmon photocatalysts13 have been reported to function in one-step excitation water splitting. However, the stringent requirements, such as the sufficient band gap and band position and efficient separation and transport of electron–hole pairs, limit the application of various visible-light-driven photocatalysts.11 Thus, two-step photoexcitation water splitting composed of an H2-evolution photocatalyst (HEP) and O2-evolution photocatalyst (OEP) allows the construction of efficient photocatalytic water splitting systems using an increasing number of visible-light-driven photocatalysts.14–16 The O2-evolution reaction involving a four-electron transfer process is generally regarded as a rate-determining step in the water splitting reaction; therefore, it is crucial to develop efficient OEPs to improve the performance of two-step excitation water splitting.17–20Among various visible-light-driven OEPs, Sillén–Aurivillius perovskite-phase oxyhalides, a series of emerging photocatalyst materials, have been studied for Z-scheme water splitting21–24 as well as for H2 or O2 evolution half-reactions using sacrificial reagents.25–28 As a kind of bismuth-based oxyhalides, Bi4TaO8Cl22,29 and Bi4TaO8Br21,30 with the simple layered structures and narrow band gap exhibit outstanding properties in the O2-evolution reaction because of longer charge transfer distance, stability against photocorrosion and potential of extensive band engineering.21,26,31 The hybridization of O 2p orbitals with Bi 6s orbitals in Bi4TaO8Cl and Bi4TaO8Br results in the upward shift of the valence band maximum, which effectively brings the visible light absorption and the photoexcited charge migration promotion.21,25 Moreover, the predominant contribution of O 2p orbitals for valence band maximum allows Bi4TaO8Cl and Bi4TaO8Br to stably produce O2 from water oxidation.21 It is worth noting that Cl 3p orbitals in Bi4TaO8Cl and Br 4p orbitals in Bi4TaO8Br exhibit different energy levels located in the valence band.25 Therefore, the construction of a bismuth-based oxyhalide solid solution using Cl and Br anions will be beneficial for the charge carrier migration towards efficient O2-evolution reaction.
In a two-step photoexcitation water-splitting system, electron transfer from OEPs to HEPs is also an important issue in determining the system's performance.32 Traditionally, ionic redox shuttle couples such as Fe3+/Fe2+, IO3−/I− and [Fe(CN)6]3−/[Fe(CN)6]4− functioning as electron mediators, are necessary building blocks to construct a two-step photoexcitation water splitting system.5,19,21,31,33,34 In the recent work, a Fe3+/Fe2+ redox couple was reported as an active electron mediator in the two-step excitation water splitting with Bi4NbO8Cl or Bi4TaO8X (X = Cl, Br) as OEP.21,31 However, the existence of ionic redox couples in the reaction solution unavoidably leads to backward reactions consuming the photogenerated electrons and holes from HEPs and OEPs, respectively, and a shielding effect for incident light.35 On account of the disadvantages, the solid-state conductive mediators instead of ionic redox couples are applied in the two-step photoexcitation water splitting systems to achieve highly efficient charge transfer between OEPs and HEPs. Au,36 carbon,37 indium tin oxide,38 Ir,39 and reduced graphene oxide (RGO)32,40,41 working as efficient electron mediators have been demonstrated to boost outstanding overall water splitting activity in the two-step excitation photocatalyst sheet systems or suspension systems. Therefore, it is desirable to promote charge transfer efficiency in the two-step photoexcitation water splitting system with bismuth-based oxyhalide OEP by combining the RGO solid-state mediator.
Herein, we present a well-crystallized Bi4TaO8Cl0.9Br0.1 solid solution for photocatalytic O2 evolution and construct a two-step photoexcitation water splitting system by combining it with Ru/SrTiO3:Rh as an HEP and RGO as the solid electron mediator. The incorporation of Br− into the layered Bi4TaO8Cl enhanced the light absorption intensity and facilitated charge separation and migration, thus, the O2-evolution rate of Bi4TaO8Cl0.9Br0.1 using Fe3+ as the electron sacrificial reagent reached 102 μmol h−1 under visible light irradiation. Furthermore, Bi4TaO8Cl0.9Br0.1 solid solution exhibited higher activity for two-step photoexcitation water splitting than Bi4TaO8Cl or Bi4TaO8Br as OEP. Upon the modification of Bi4TaO8Cl0.9Br0.1 with the O2-evolution cocatalyst and solid-state electron mediator, CoOx as O2-evolution cocatalyst and RGO as solid-state conductor dramatically improved the O2 formation process on Bi4TaO8Cl0.9Br0.1 and electron transfer between photocatalysts, resulting in an apparent quantum yield (AQY) of 1.26% at 420 nm for the two-step photoexcitation water splitting system.
Experimental section
Preparation of materials
Bi4TaO8Cl0.9Br0.1 and Bi4TaO8Cl0.5Br0.5 powders were prepared by a flux method. Stoichiometric quantities of Bi2O3, BiOCl, BiOBr, and Ta2O5 were mixed, and NaCl and KCl (molar ratio of NaCl/KCl is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) were used as flux reagents. The fluxes were mixed with Bi2O3, BiOCl, BiOBr, and Ta2O5 at a solute concentration of 10 mol%. The mixture was ground in an agate mortar for 30 min and placed in an alumina crucible and calcined in a muffle furnace at 923 K for 14 h at a heating rate of 10 K min−1. Bi4TaO8Cl0.9Br0.1 and Bi4TaO8Cl0.5Br0.5 obtained in this manner were washed with deionized water several times and collected by filtration to remove residual flux reagents. The yellow powder was then dried at room temperature. For comparison, Bi4TaO8Cl and Bi4TaO8Br samples were prepared using the same procedure.
:1) were used as flux reagents. The fluxes were mixed with Bi2O3, BiOCl, BiOBr, and Ta2O5 at a solute concentration of 10 mol%. The mixture was ground in an agate mortar for 30 min and placed in an alumina crucible and calcined in a muffle furnace at 923 K for 14 h at a heating rate of 10 K min−1. Bi4TaO8Cl0.9Br0.1 and Bi4TaO8Cl0.5Br0.5 obtained in this manner were washed with deionized water several times and collected by filtration to remove residual flux reagents. The yellow powder was then dried at room temperature. For comparison, Bi4TaO8Cl and Bi4TaO8Br samples were prepared using the same procedure.
Rh-doped SrTiO3 (SrTiO3:Rh) HEP was prepared by the solid state reaction method.42 SrCO3, TiO2, and Rh2O3 were mixed in a mortar at the ratio of Sr:Ti:Rh = 1.03:0.99:0.01 and ground for 30 min. The mixture was calcined in a muffle furnace at 1273 K for 10 h in the air using an alumina crucible to obtain SrTiO3:Rh (1%) powder.
Modification of photocatalysts
Firstly, CoOx, IrOx, RuOx, and PtOx were loaded on the obtained metal oxyhalides as cocatalysts for water oxidation by impregnation using aqueous solutions containing Co(NO3)2, Na2IrCl6, RuCl3 or H2PtCl6. Specifically, metal oxyhalide powder (0.22 g) was immersed in an aqueous solution containing the required amount of cocatalyst-precursor solution. The slurry was stirred with sonication for 3 min and then dried by a hot water bath, the resulting mixture was heated at 573–873 K for 2 h in air. Subsequently, metal oxyhalides modified with MOx (M = Co, Ir, Ru, and Pt) cocatalysts were further decorated with graphene oxide by the photodeposition method. Photocatalyst powders and graphene oxide dispersion were suspended in 20 vol% of the aqueous methanol solution. The reaction solution was evacuated to completely remove air and irradiated under visible light (λ ≥ 420 nm) for 2 h to form RGO on photocatalyst powders. Metal oxyhalides loaded with MOx (M = Co, Ir, Ru, and Pt) cocatalyst were also modified with metallic Ir or Au conductive mediator by the impregnation method followed by the H2-reduction treatment. The loading amount of Ir or Au was 0.3 wt%. Pt-modified metal oxyhalide photocatalysts were synthesized by impregnation with H2-reduction treatment, and the Pt content was 0.3 wt%.As for the modification of SrTiO3:Rh, Ru (0.7 wt%) cocatalyst was loaded by the photodeposition method in an aqueous methanol solution (20 vol%) containing the required amount of the Ru precursor under visible light irradiation (λ ≥ 420 nm) for 6 h, denoted as Ru/SrTiO3:Rh.
Characterization of materials
X-ray diffraction (XRD) patterns were obtained using a Bruker D8 Advance X-ray diffractometer with Cu Kα radiation source, operating at 40 kV and 40 mA. Scanning electron microscopy (SEM) images and elemental mapping images were acquired using a Hitachi SU-8020 field-emission scanning electron microscope equipped with an energy dispersive spectrometer (Bruker Nano GmbH). UV-vis diffuse reflectance spectra (DRS) were collected using a spectrophotometer (LAMBDA 650S, PerkinElmer) equipped with an integrating sphere. The binding energies were determined by X-ray photoelectron spectroscopy (XPS) on a Thermo Scientific ESCALAB 250Xi spectrometer with a monochromatic Al Kα source (hν = 1486.6 eV) and normalized to C 1s for each sample.Photoelectrochemical measurements
Photoelectrochemical measurements were performed at room temperature using a CHI 650E electrochemical workstation with a standard three-electrode cell. The prepared electrode, an Ag/AgCl electrode, and Pt wire were used as the working electrode, reference electrode, and counter electrode, respectively. The working electrodes of bare metal oxyhalides and surface-modified metal oxyhalides were prepared by a conventional drop-casting method. The photocurrents of bare metal oxyhalides and surface-modified metal oxyhalides electrodes were measured in an 0.1 M Na2SO4 solution (pH = 7) bubbling with Ar gas under visible light irradiation (λ ≥ 420 nm, 300 W Xe lamp) at 0.8 V vs. RHE. Electrochemical impedance spectroscopy (EIS) was recorded in the frequency range of 105 Hz to 0.1 Hz with the initial potential (0.01 V) in 0.1 M Na2SO4 solution.Photocatalytic activity measurements
The photocatalytic O2 evolution reaction was performed in a gas-closed circulation system with a Pyrex top-illuminated reaction vessel. Pt-modified metal oxyhalide photocatalysts (100 mg) were dispersed in 150 ml of 10 mM aqueous Fe(NO3)3 solution. The reaction temperature was maintained at 285 K by circulating cold water. After completely removing air from the reaction system by evacuation, 10 kPa Ar gas was introduced into the reaction system, and the slurry was irradiated under a 300 W Xenon lamp equipped with a cutoff filter (λ ≥ 420 nm). The gas products were measured by an online thermal conductivity detector-gas chromatography system equipped with a Molecular Sieve 5 Å column using Ar as the carrier gas. Two-step excitation water splitting was carried out under similar experimental conditions except that Ru-modified SrTiO3:Rh (50 mg) as HEP and surface-modified metal oxyhalide as OEP were dispersed in deionized water (150 ml) by adjusting the pH with H2SO4.Apparent quantum yield measurements
The apparent quantum yield (AQY) of two-step photoexcitation water splitting was measured using a 300 W Xenon lamp equipped with a 420 nm band-pass filter. The AQY was calculated using the following equation:| AQY(%) = [4 × n(H2)]/n(photons) × 100 |
Results and discussion
The XRD patterns in Fig. 1a show that Bi4TaO8Cl and Bi4TaO8Br synthesized by the flux method were pure phases of the Sillén–Aurivillius layered perovskite structure and the different diffraction peak positions of Bi4TaO8Cl and Bi4TaO8Br were due to the anionic sizes of Cl− and Br− in the crystal structure. For Bi4TaO8Cl0.9Br0.1 and Bi4TaO8Cl0.5Br0.5, pure phases of the layered perovskite structure were observed and the diffraction peaks were located between those of Bi4TaO8Cl and Bi4TaO8Br, which indicated the formation of the solid solution containing Cl and Br anions. Moreover, the higher XRD peak intensities of the solid solution samples in comparison to Bi4TaO8Cl and Bi4TaO8Br demonstrated that Bi4TaO8Cl0.9Br0.1 and Bi4TaO8Cl0.5Br0.5 were well-crystallized, because the dual fluxes promoted the sufficient diffusion of precursor ions, allowing the rapid growth of single phase at low temperature.43 The morphology of the as-prepared samples is shown in SEM images of Fig. 1b and Fig. S1.† Four samples displayed similar microplatelet morphology with 200–500 nm in size and 50–100 nm in thickness. Bi4TaO8Cl0.9Br0.1 sample exhibited relatively small and thin plates. However, due to the calcination and intrinsic characteristics of the layered crystal structure, the aggregations of particulates were observed for all samples.21 The uniform dispersions of Bi, Ta, O, Cl, and Br atoms shown in Fig. 1c also indicate that Bi4TaO8Cl0.9Br0.1 were homogeneous solid solutions.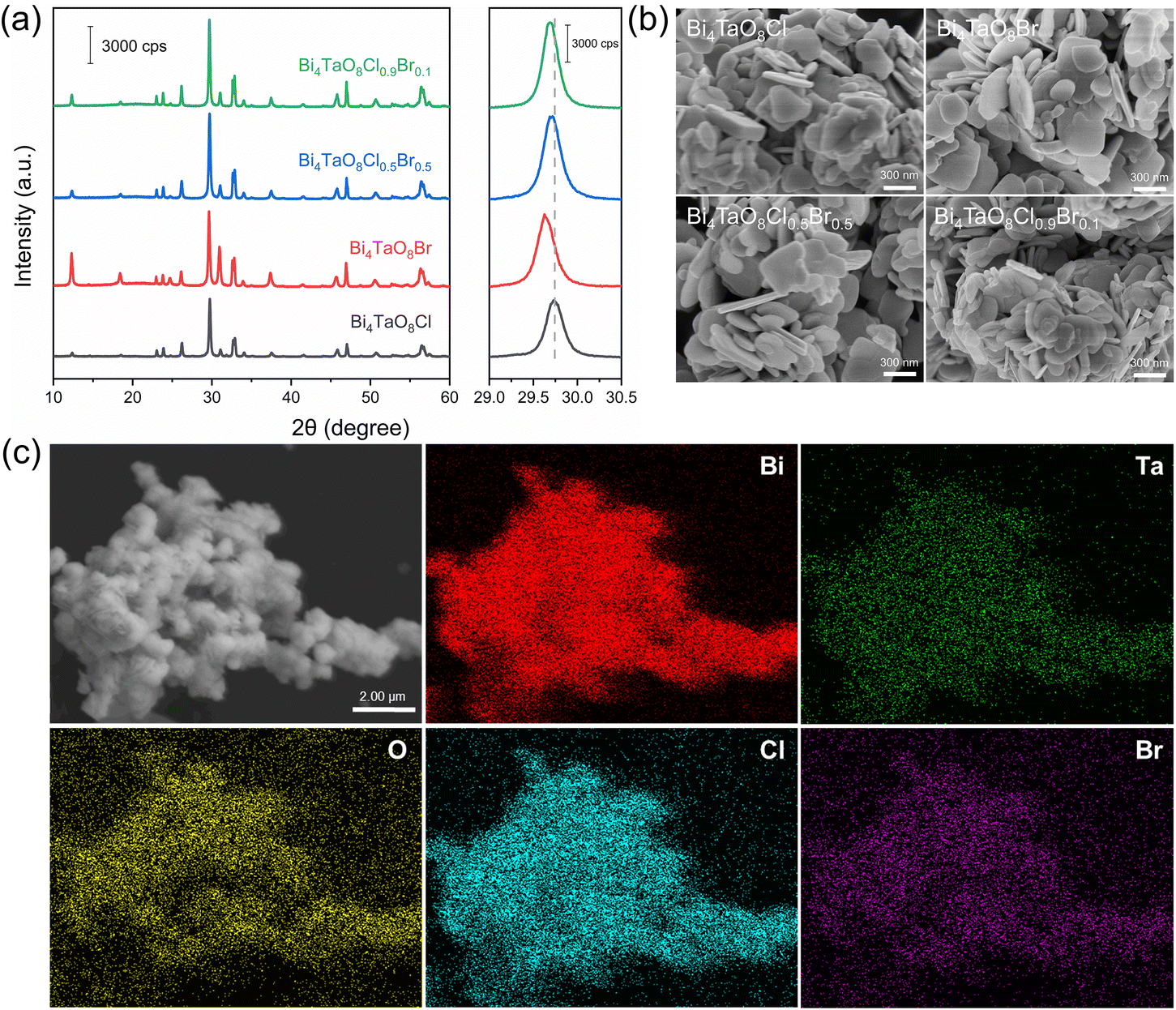 | ||
| Fig. 1 (a) XRD patterns and (b) SEM images of Bi4TaO8Cl, Bi4TaO8Br, Bi4TaO8Cl0.5Br0.5, and Bi4TaO8Cl0.9Br0.1. (c) EDS elemental mapping of Bi4TaO8Cl0.9Br0.1. | ||
The UV-vis DRS of the oxyhalide samples shown in Fig. 2a indicates that the absorption edge of Bi4TaO8Cl, and Bi4TaO8Cl0.9Br0.1 was approximately at 474 nm and the absorption edge of Bi4TaO8Br and Bi4TaO8Cl0.5Br0.5 was approximately at 469 nm. Although the absorption edge and band structures of the four samples are not very different from each other due to the small contribution of the halogen orbitals to the valence band maximum of Bi4TaO8X (X = Cl, Br),21 the light absorption intensity for Bi4TaO8Cl and Bi4TaO8Cl0.9Br0.1 oxyhalides mainly containing Cl− anions was obviously higher than that for Bi4TaO8Br and Bi4TaO8Cl0.5Br0.5, including higher Br− contents. The photocurrent and EIS measurements were carried out and the results are shown in Fig. 2b and c. In comparison to Bi4TaO8Cl, Bi4TaO8Br, and Bi4TaO8Cl0.5Br0.5, Bi4TaO8Cl0.9Br0.1 exhibited a higher photocurrent density for photoelectrochemical water oxidation reactions and lower electrochemical resistance, indicating the formation of Bi4TaO8Cl0.9Br0.1 solid solution with the small Br− content promoting the photogenerated charge separation and diffusion in the bulk. The O2-evolution performance of the Pt-modified oxyhalide photocatalysts was evaluated in the aqueous Fe(NO3)3 solution under the irradiation of visible light, as shown in Fig. 2d and Fig. S2.† The Bi4TaO8Cl0.9Br0.1 sample exhibited a superior O2-evolution activity as compared with the other three oxyhalides, due to high crystallinity, intense light absorption, and rapid charge separation and migration of Bi4TaO8Cl0.9Br0.1. In addition, the O2-evolution performance of these oxyhalides was further investigated in a two-step photoexcitation water-splitting reaction. As shown in Fig. 2e and Fig. S3,† when combined with Ru/SrTiO3:Rh as the HEP, Bi4TaO8Cl0.9Br0.1 showed higher photocatalytic activity for two-step excitation water splitting than the other three oxyhalide photocatalysts. The O2-evolution activities of four oxyhalide photocatalysts are well correlated in the two-step photoexcitation water splitting and half-reactions using the aqueous Fe(NO3)3 solution. Moreover, the XRD patterns (Fig. S4†) and EDS elemental mapping (Fig. S5†) of the photocatalysts after the reaction were collected to analyze the Cl and Br species. The same diffraction peak positions for samples before and after the photocatalytic reaction and the uniform distribution of Bi, Ta, O, Cl, and Br elements after the O2-evolution reaction indicated the stable structure of the Bi4TaO8Cl0.9Br0.1 solid solution containing Cl and Br anions.
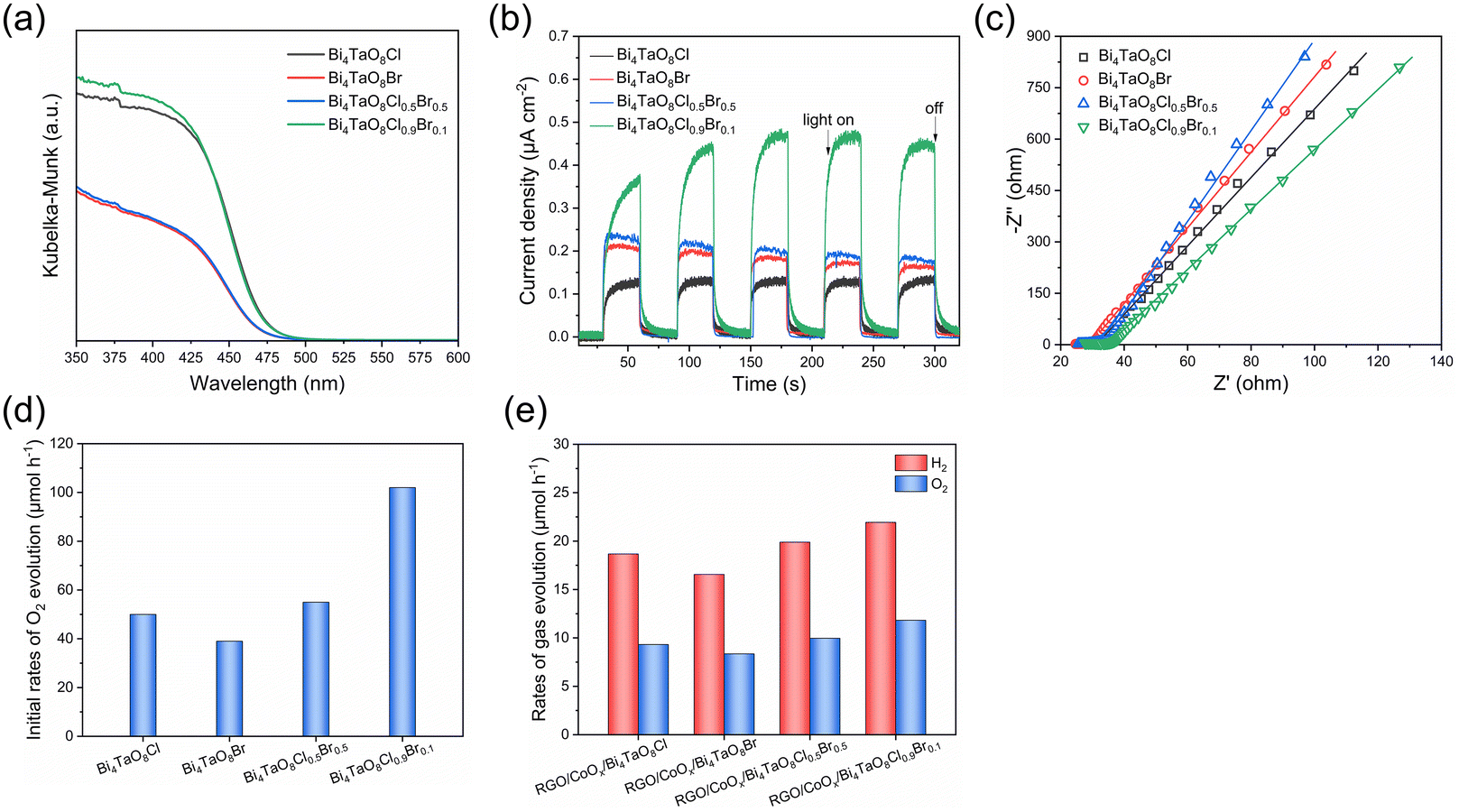 | ||
| Fig. 2 (a) UV-vis DRS of Bi4TaO8Cl, Bi4TaO8Br, Bi4TaO8Cl0.5Br0.5, and Bi4TaO8Cl0.9Br0.1. (b) Photocurrent for Bi4TaO8Cl, Bi4TaO8Br, Bi4TaO8Cl0.5Br0.5, and Bi4TaO8Cl0.9Br0.1 under visible light (λ ≥ 420 nm) at 0.8 V vs. RHE. (c) EIS spectra of Bi4TaO8Cl, Bi4TaO8Br, Bi4TaO8Cl0.5Br0.5, and Bi4TaO8Cl0.9Br0.1. (d) The photocatalytic performance of O2 evolution on Pt-modified oxyhalide photocatalysts from an aqueous Fe(NO3)3 solution. Conditions: photocatalyst, 100 mg; cocatalyst, 0.3 wt% Pt; 10 mM aqueous Fe(NO3)3 solution, 150 mL; light source, 300 W Xenon lamp (λ ≥ 420 nm). (e) H2 and O2 evolution rates of two-step excitation overall water splitting reaction using Ru/SrTiO3:Rh as the HEP and surface-modified oxyhalide photocatalysts as the OEP. Conditions: HEP, 50 mg; OEP (CoOx, 0.5 wt% as Co; RGO, 0.5 wt%), 50 mg; 150 mL H2O, pH = 4; light source, 300 W Xenon lamp (λ ≥ 420 nm). | ||
Considering the essential role of the O2-evolving cocatalyst in the overall water-splitting reaction, the performance of different cocatalysts was examined as shown in Fig. 3 and Fig. S6.† When O2-evolving cocatalysts were loaded on Bi4TaO8Cl0.9Br0.1 by calcining at 573 K in air, CoOx and RuOx cocatalysts gave higher activity for two-step excitation overall water splitting than IrOx and PtOx. Notably, when the calcination temperature of the cocatalyst loading increased to 773 K, the gas evolution rates in the water splitting system using CoOx-modified Bi4TaO8Cl0.9Br0.1 became nearly double that of CoOx-modified Bi4TaO8Cl0.9Br0.1 calcined at 573 K, whereas the performance improvement of the RuOx-modified Bi4TaO8Cl0.9Br0.1 was not so obvious. The results demonstrate that the deposition of the adequate CoOx cocatalyst on Bi4TaO8Cl0.9Br0.1 is significant in preventing adverse electron–hole recombination in Bi4TaO8Cl0.9Br0.1 and boosts the interfacial charge transfer towards the surface O2-evolution reaction, especially in an all-solid-state system.44 Accordingly, the effect of the preparation conditions of CoOx-modified Bi4TaO8Cl0.9Br0.1 on the water-splitting activity was investigated. Table 1 summarizes the photocatalytic activities for the two-step excitation overall water splitting reactions consisting of Bi4TaO8Cl0.9Br0.1 modified with different amounts of CoOx at different calcination temperatures and time courses of water splitting reactions are shown in Fig. S7 and S8.† The activity was improved with increasing calcination temperature from 573 K to 673 K, reaching a maximum of 673 K, and then decreasing at 773 K and 873 K (entries 1–4). Similarly, Bi4TaO8Cl0.9Br0.1 loaded with different CoOx contents exhibited varied activity with volcano-like behavior (entry 2, entries 5–7) and the optimal performance was attained at a Co loading of 0.5 wt%.
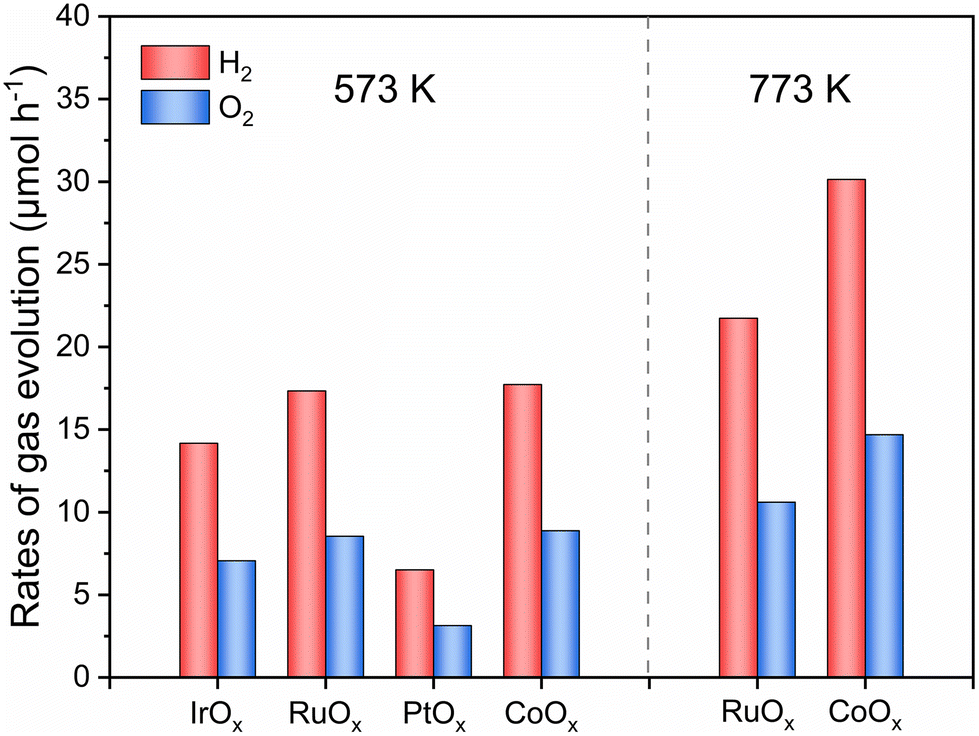 | ||
| Fig. 3 H2 and O2 evolution rates of the two-step excitation overall water splitting reaction using Bi4TaO8Cl0.9Br0.1 as the OEP modified with different O2-evolving cocatalysts calcined at different temperatures. Conditions: Ru/SrTiO3:Rh as HEP, 50 mg; surface-modified Bi4TaO8Cl0.9Br0.1 (IrOx, RuOx, PtOx or CoOx, 0.5 wt% as Ir, Ru, Pt or Co; RGO, 0.5 wt%), 50 mg; 150 mL H2O, pH = 4; light source, 300 W Xenon lamp (λ ≥ 420 nm). | ||
| Entry | Impregnation–calcination temperature of CoOx | Amount of CoOx (wt%) | Evolution rate of H2 (μmol h−1) | Evolution rate of O2 (μmol h−1) |
|---|---|---|---|---|
| a Reaction conditions: Ru/SrTiO3:Rh as HEP, 50 mg; surface-modified Bi4TaO8Cl0.9Br0.1 (RGO, 0.5 wt%), 50 mg; 150 mL H2O, pH = 4; light source, 300 W Xenon lamp (λ ≥ 420 nm). | ||||
| 1 | 573 K | 0.5 | 17.7 | 8.9 |
| 2 | 673 K | 0.5 | 39.8 | 19.7 |
| 3 | 773 K | 0.5 | 32.3 | 15.9 |
| 4 | 873 K | 0.5 | 13.5 | 7.0 |
| 5 | 673 K | 0.3 | 21.8 | 10.5 |
| 6 | 673 K | 0.8 | 27.0 | 13.4 |
| 7 | 673 K | 1.0 | 23.2 | 11.6 |
Besides the importance of the O2-evolution cocatalyst on Bi4TaO8Cl0.9Br0.1, the electron mediator between OEP and HEP also plays a crucial role in the two-step excitation overall water-splitting reaction. Table 2 and Fig. S9, S10† show the influence of different types of electron mediators on the photocatalytic activities of the overall water-splitting system with CoOx-modified Bi4TaO8Cl0.9Br0.1 OEP. All solid-state electron conductors exhibited higher photocatalytic overall water-splitting activities than the Fe3+/Fe2+ ionic shuttle mediator, indicating that the electron transfer between OEP and HEP and the elimination of the reverse reaction benefited from the solid-state conductive mediator.32 As compared with metallic Ir and Au mediators, RGO loaded on Bi4TaO8Cl0.9Br0.1 gave the best photocatalytic performance for two-step excitation water splitting. Furthermore, in comparison to the performance of a two-step excitation water splitting system using RGO as a mediator, the systems without RGO or other conductive mediators and with GO to form physical contact with OEP exhibited much lower activity, which can further demonstrate the significant effect of RGO on the promotion of electron transfer.
| Entry | Mediator | Loading amount (wt%) | Evolution rate of H2 (μmol h−1) | Evolution rate of O2 (μmol h−1) |
|---|---|---|---|---|
| a Reaction conditions: Ru/SrTiO3:Rh as HEP, 50 mg; surface-modified Bi4TaO8Cl0.9Br0.1 (CoOx, 0.5 wt% as Co), 50 mg; 150 mL H2O (150 mL of 2 mM Fe(NO3)3 solution for entry 7), pH = 4 (pH = 2.4 for entry 7); light source, 300 W Xenon lamp (λ ≥ 420 nm). | ||||
| 1 | RGO | 0.1 | 36.1 | 17.8 |
| 2 | RGO | 0.3 | 41.4 | 20.2 |
| 3 | RGO | 0.5 | 39.8 | 19.7 |
| 4 | RGO | 0.7 | 25.9 | 13.1 |
| 5 | Ir | 0.3 | 23.3 | 13.4 |
| 6 | Au | 0.3 | 21.2 | 10.9 |
| 7 | Fe3+/Fe2+ | — | 18.5 | 9.0 |
| 8 | None | — | 17.4 | 8.7 |
| 9 | GO | — | 12.5 | 6.6 |
Fig. 4a and Fig. S11† show the SEM images of the RGO- and CoOx-co-modified Bi4TaO8Cl0.9Br0.1 photocatalyst, in which RGO thin films and CoOx particles were uniformly dispersed on Bi4TaO8Cl0.9Br0.1. Moreover, according to the C 1s XPS spectra for the graphene oxide (GO) sample and RGO-modified Bi4TaO8Cl0.9Br0.1 shown in Fig. 4b, RGO on Bi4TaO8Cl0.9Br0.1 exhibited much weaker C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O peaks than the GO sample. These results demonstrate that oxygen-functional groups in the GO sample were reduced by photogenerated electrons from Bi4TaO8Cl0.9Br0.1 during the photodeposition process45 and RGO adhered to Bi4TaO8Cl0.9Br0.1 intimately to facilitate electron transfer.46 The overall water-splitting activity dependence on the loading amount of RGO was investigated. As shown in Fig. 4c and Fig. S12,† the gas evolution rates increased with the increase of RGO content and reached a maximum at 0.3 wt%, which is because of the efficient electron transfer between Ru/SrTiO3:Rh and CoOx/Bi4TaO8Cl0.9Br0.1 mediated by RGO conductor. The activity began to decrease when RGO content was beyond 0.3 wt%, this is due to incident light shielding and active site blocking effects due to the excessive RGO coverage on CoOx/Bi4TaO8Cl0.9Br0.1.45 Furthermore, the photocurrent densities of RGO- and CoOx-co-modified Bi4TaO8Cl0.9Br0.1 were one or two orders of magnitude higher than those of pristine Bi4TaO8Cl0.9Br0.1 and CoOx-modified Bi4TaO8Cl0.9Br0.1 without RGO, as shown in Fig. 4d, demonstrating the remarkable promotion of charge transfer by the modification of Bi4TaO8Cl0.9Br0.1 with the RGO mediator. In addition, the photoelectrochemical performance for the water oxidation of RGO- and CoOx-co-modified Bi4TaO8Cl0.9Br0.1 with different RGO contents corresponded well to the overall performance of water splitting reactions shown in Fig. 4c.
O peaks than the GO sample. These results demonstrate that oxygen-functional groups in the GO sample were reduced by photogenerated electrons from Bi4TaO8Cl0.9Br0.1 during the photodeposition process45 and RGO adhered to Bi4TaO8Cl0.9Br0.1 intimately to facilitate electron transfer.46 The overall water-splitting activity dependence on the loading amount of RGO was investigated. As shown in Fig. 4c and Fig. S12,† the gas evolution rates increased with the increase of RGO content and reached a maximum at 0.3 wt%, which is because of the efficient electron transfer between Ru/SrTiO3:Rh and CoOx/Bi4TaO8Cl0.9Br0.1 mediated by RGO conductor. The activity began to decrease when RGO content was beyond 0.3 wt%, this is due to incident light shielding and active site blocking effects due to the excessive RGO coverage on CoOx/Bi4TaO8Cl0.9Br0.1.45 Furthermore, the photocurrent densities of RGO- and CoOx-co-modified Bi4TaO8Cl0.9Br0.1 were one or two orders of magnitude higher than those of pristine Bi4TaO8Cl0.9Br0.1 and CoOx-modified Bi4TaO8Cl0.9Br0.1 without RGO, as shown in Fig. 4d, demonstrating the remarkable promotion of charge transfer by the modification of Bi4TaO8Cl0.9Br0.1 with the RGO mediator. In addition, the photoelectrochemical performance for the water oxidation of RGO- and CoOx-co-modified Bi4TaO8Cl0.9Br0.1 with different RGO contents corresponded well to the overall performance of water splitting reactions shown in Fig. 4c.
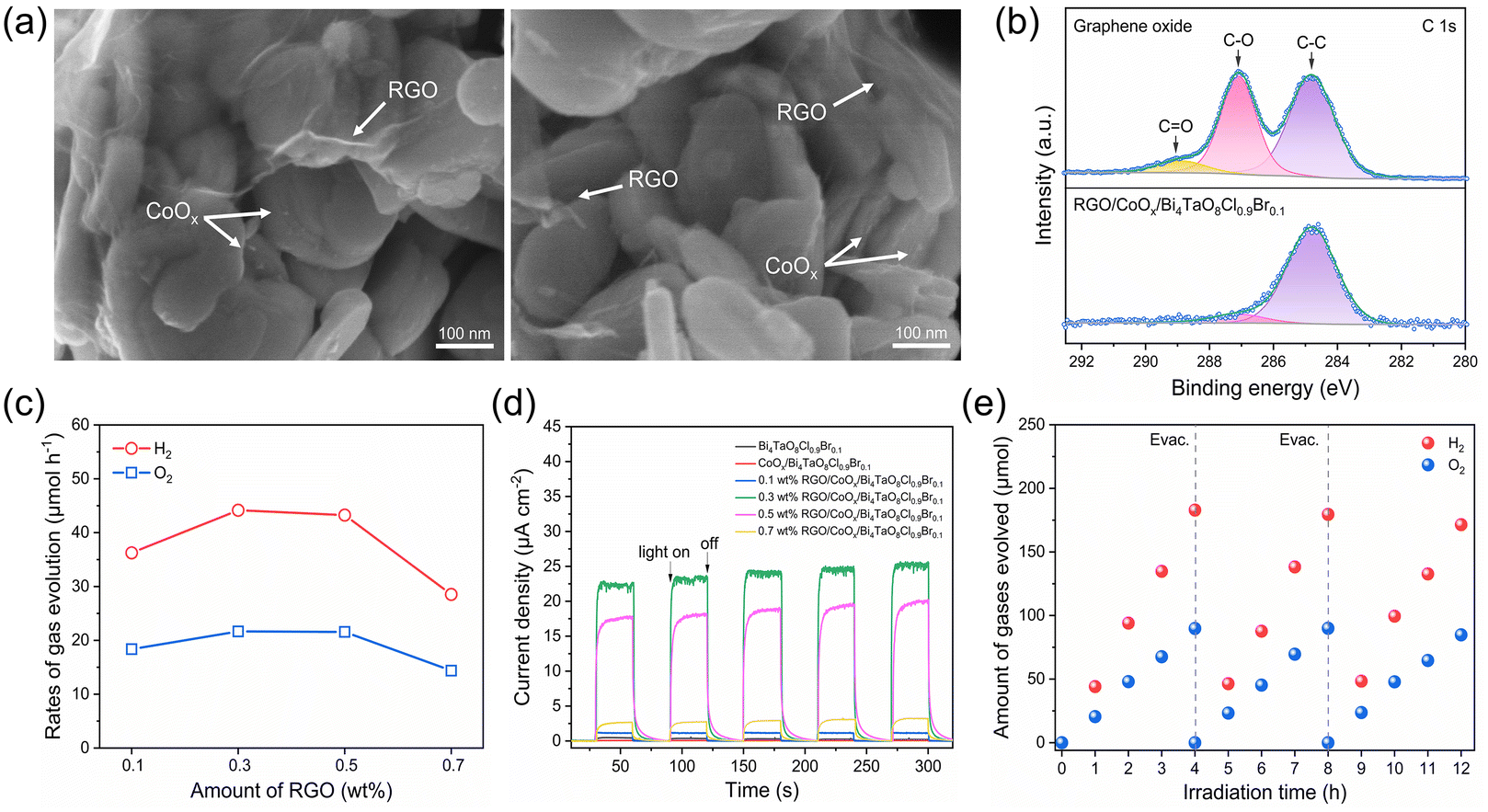 | ||
| Fig. 4 (a) SEM images of RGO- and CoOx-co-modified Bi4TaO8Cl0.9Br0.1. (b) C 1s XPS spectra of graphene oxide (GO) and RGO-modified Bi4TaO8Cl0.9Br0.1. (c) Photocatalytic activity of the two-step excitation water splitting reaction using RGO- and CoOx-modified Bi4TaO8Cl0.9Br0.1 OEP with different RGO contents. Conditions: Ru/SrTiO3:Rh as HEP, 50 mg; surface-modified Bi4TaO8Cl0.9Br0.1 (CoOx, 0.5 wt% as Co), 50 mg; 150 mL H2O, pH = 4; light source, 300 W Xenon lamp (λ ≥ 420 nm). (d) Photocurrent for bare Bi4TaO8Cl0.9Br0.1 and Bi4TaO8Cl0.9Br0.1 modified with RGO and/or CoOx under visible light (λ ≥ 420 nm) at 0.8 V vs. RHE. (e) Time course of gas evolution during two-step photoexcitation water splitting reaction under visible light (λ ≥ 420 nm). Conditions: Ru/SrTiO3:Rh as HEP, 50 mg; surface-modified Bi4TaO8Cl0.9Br0.1 as OEP (CoOx, 0.5 wt% as Co; RGO, 0.3wt%), 50 mg; 150 mL H2O, pH = 4; light source, 300 W Xenon lamp (λ ≥ 420 nm). | ||
Two-step photoexcitation overall water splitting was successfully accomplished with CoOx-modified Bi4TaO8Cl0.9Br0.1 as OEP, Ru/SrTiO3:Rh as the HEP, and RGO as electron mediator. When the RGO conductive mediator and CoOx cocatalyst were decorated on Bi4TaO8Cl0.9Br0.1 under optimal conditions, an efficient overall water-splitting activity was observed under visible light (Fig. 4e) and UV light (Fig. S13†). H2 and O2 were simultaneously evolved at a stoichiometric molar ratio of 2:1. The overall water-splitting reaction was carried out in three cycles with steady evolution of H2 and O2 over 12 h. The AQY value for this two-step photoexcitation water-splitting system was 1.26% at 420 nm, which is superior to previous reports consisting of metal oxyhalides as OEP. Although the photocatalytic performance for solid-state two-step excitation overall water splitting based on oxyhalide photocatalysts was successfully demonstrated, investigations on the Bi4TaO8Cl0.9Br0.1 photocatalyst refinement, RGO decoration methods, and type of solid-state electron mediators are ongoing to further improve the efficiency of overall water splitting.
Conclusions
In summary, an efficient two-step photoexcitation water splitting system using CoOx-modified Bi4TaO8Cl0.9Br0.1 as the OEP and RGO as the solid electron mediator was successfully constructed. The formation of a high-crystallinity Bi4TaO8Cl0.9Br0.1 solid solution synthesized by the dual flux method helped to enhance the light absorption and charge transfer, thus, the O2-evolution activity of Bi4TaO8Cl0.9Br0.1 using Fe3+ as the electron sacrificial reagent was dramatically improved under visible light irradiation. Upon the modification of Bi4TaO8Cl0.9Br0.1 with CoOx cocatalyst and RGO conductive mediator, the charge transfer towards water oxidation reaction and from OEP to HEP was promoted, resulting in the performance enhancement of the two-step excitation water splitting with Bi4TaO8Cl0.9Br0.1 as OEP and Ru/SrTiO3:Rh as HEP with an AQY of 1.26% at 420 nm, which is higher than that of previous systems consisting of metal oxyhalide photocatalysts. This study verifies the significance and effectiveness of solid solution photocatalyst fabrication and adequate surface modification to boost efficient charge transfer towards practical solar-to-hydrogen energy conversion.Author contributions
Funding acquisition: Y. Luo, Z. Wang, and H. He. Experimental design and measurements: W. Sun, Y. Luo, J. Xu, Q. Guo, Z. Wang, and H. He. Manuscript writing, revising, and discussion: W. Sun, Y. Luo, L. Deng, Z. Wang, and H. He. All authors gave their approval to the final version of the manuscript.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was financially supported by the General Program (No. 22376207) and the Excellent Young Scientists Fund Program of The National Natural Science Foundation of China, the General Program (No. 2022M723315) of the China Postdoctoral Science Foundation, and CCUS Strategic Priority Program of Research Center for Eco-Environmental Sciences of the Chinese Academy of Sciences of China.References
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- N. S. Lewis, Science, 2007, 315, 798 CrossRef CAS PubMed.
- T. Hisatomi and K. Domen, Nat. Catal., 2019, 2, 387–399 CrossRef CAS.
- T. Takata, J. Jiang, Y. Sakata, M. Nakabayashi, N. Shibata, V. Nandal, K. Seki, T. Hisatomi and K. Domen, Nature, 2020, 581, 411–414 CrossRef CAS PubMed.
- Y. Qi, Y. Zhao, Y. Y. Gao, D. Li, Z. Li, F. X. Zhang and C. Li, Joule, 2018, 2, 2393–2402 CrossRef CAS.
- Y. Goto, T. Hisatomi, Q. Wang, T. Higashi, K. Ishikiriyama, T. Maeda, Y. Sakata, S. Okunaka, H. Tokudome, M. Katayama, S. Akiyama, H. Nishiyama, Y. Inoue, T. Takewaki, T. Setoyama, T. Minegishi, T. Takata, T. Yamada and K. Domen, Joule, 2018, 2, 509–520 CrossRef CAS.
- Z. Wang, Y. Inoue, T. Hisatomi, R. Ishikawa, Q. Wang, T. Takata, S. Chen, N. Shibata, Y. Ikuhara and K. Domen, Nat. Catal., 2018, 1, 756–763 CrossRef CAS.
- C. Pan, T. Takata, M. Nakabayashi, T. Matsumoto, N. Shibata, Y. Ikuhara and K. Domen, Angew. Chem., Int. Ed., 2015, 54, 2955–2959 CrossRef CAS PubMed.
- F. Hou, F. Liu, H. Wu, M. Qasim, Y. Chen, Y. Duan, Z. Feng and M. Liu, Chin. J. Chem., 2023, 41, 173–180 CrossRef CAS.
- C. Cheng, L. Mao, X. Kang, C.-L. Dong, Y.-C. Huang, S. Shen, J. Shi and L. Guo, Appl. Catal., B, 2023, 331, 122733 CrossRef CAS.
- Z. Wang, C. Li and K. Domen, Chem. Soc. Rev., 2019, 48, 2109–2125 RSC.
- K. Maeda and K. Domen, J. Phys. Chem. C, 2007, 111, 7851–7861 CrossRef CAS.
- A. Tanaka, K. Teramura, S. Hosokawa, H. Kominami and T. Tanaka, Chem. Sci., 2017, 8, 2574–2580 RSC.
- Z. Wang, Y. Luo, T. Hisatomi, J. J. M. Vequizo, S. Suzuki, S. S. Chen, M. Nakabayashi, L. H. Lin, Z. H. Pan, N. Kariya, A. Yamakata, N. Shibata, T. Takata, K. Teshima and K. Domen, Nat. Commun., 2021, 12, 1005 CrossRef CAS PubMed.
- Y. Luo, Q. Q. Guo, J. Xu, H. H. Zhou, Z. Wang and H. He, Energy Mater. Adv., 2022, 2022, 0003 CrossRef.
- J. Xu, Y. Luo, Q. Q. Guo, H. H. Zhou, Z. Wang and H. He, J. Catal., 2022, 415, 19–27 CrossRef CAS.
- S. Lin, H. Huang, T. Ma and Y. Zhang, Adv. Sci., 2021, 8, 2002458 CrossRef CAS PubMed.
- K. Ogawa, O. Tomita, K. Takagi, A. Nakada, M. Higashi and R. Abe, Chem. Lett., 2018, 47, 985–988 CrossRef CAS.
- Y. Qi, J. W. Zhang, Y. Kong, Y. Zhao, S. S. Chen, D. Li, W. Liu, Y. F. Chen, T. F. Xie, J. Y. Cui, C. Li, K. Domen and F. X. Zhang, Nat. Commun., 2022, 13, 484 CrossRef CAS PubMed.
- Z. Wang, J. Seo, T. Hisatomi, M. Nakabayashi, J. Xiao, S. Chen, L. Lin, Z. Pan, M. Krause, N. Yin, G. Smith, N. Shibata, T. Takata and K. Domen, Nano Res., 2022, 16, 4562–4567 CrossRef.
- X. P. Tao, Y. Zhao, L. C. Mu, S. Y. Wang, R. G. Li and C. Li, Adv. Energy Mater., 2018, 8, 1701392 CrossRef.
- A. Nakada, A. Saeki, M. Higashi, H. Kageyama and R. Abe, J. Mater. Chem. A, 2018, 6, 10909–10917 RSC.
- K. Murofushi, K. Ogawa, H. Suzuki, R. Sakamoto, O. Tomita, K. Kato, A. Yamakata, A. Saeki and R. Abe, J. Mater. Chem. A, 2021, 9, 11718–11725 RSC.
- K. Ogawa, H. Suzuki, C. C. Zhong, R. Sakamoto, O. Tomita, A. Saeki, H. Kageyama and R. Abe, J. Am. Chem. Soc., 2021, 143, 8446–8453 CrossRef CAS PubMed.
- H. Kunioku, M. Higashi, O. Tomita, M. Yabuuchi, D. Kato, H. Fujito, H. Kageyama and R. Abe, J. Mater. Chem. A, 2018, 6, 3100–3107 RSC.
- H. Kunioku, A. Nakada, M. Higashi, O. Tomita, H. Kageyama and R. Abe, Sustainable Energy Fuels, 2018, 2, 1474–1480 RSC.
- H. Suzuki, M. Higashi, H. Kunioku, R. Abe and A. Saeki, ACS Energy Lett., 2019, 4, 1572–1578 CrossRef CAS.
- A. Adenle, M. Shi, W. C. Jiang, B. Zeng, C. Li and R. G. Li, J. Mater. Chem. A, 2022, 10, 14293–14299 RSC.
- X. P. Tao, Y. Y. Gao, S. Y. Wang, X. Y. Wang, Y. Liu, Y. Zhao, F. T. Fan, M. Dupuis, R. G. Li and C. Li, Adv. Energy Mater., 2019, 9, 1803951 CrossRef.
- X. P. Tao, W. W. Shi, B. Zeng, Y. Zhao, N. Ta, S. Y. Wang, A. A. Adenle, R. G. Li and C. Li, ACS Catal., 2020, 10, 5941–5948 CrossRef CAS.
- H. Fujito, H. Kunioku, D. Kato, H. Suzuki, M. Higashi, H. Kageyama and R. Abe, J. Am. Chem. Soc., 2016, 138, 2082–2085 CrossRef CAS PubMed.
- A. Iwase, Y. H. Ng, Y. Ishiguro, A. Kudo and R. Amal, J. Am. Chem. Soc., 2011, 133, 11054–11057 CrossRef CAS PubMed.
- K. Maeda, M. Higashi, D. L. Lu, R. Abe and K. Domen, J. Am. Chem. Soc., 2010, 132, 5858–5868 CrossRef CAS PubMed.
- M. X. Hu, S. W. Du, B. B. Dong, Y. Qi, Z. C. Feng and F. X. Zhang, J. Mater. Chem. A, 2022, 10, 16541–16546 RSC.
- Y. Sasaki, H. Nemoto, K. Saito and A. Kudo, J. Phys. Chem. C, 2009, 113, 17536–17542 CrossRef CAS.
- Q. Wang, T. Hisatomi, Q. X. Jia, H. Tokudome, M. Zhong, C. Z. Wang, Z. H. Pan, T. Takata, M. Nakabayashi, N. Shibata, Y. B. Li, I. D. Sharp, A. Kudo, T. Yamada and K. Domen, Nat. Mater., 2016, 15, 611–615 CrossRef CAS PubMed.
- Q. Wang, T. Hisatomi, Y. Suzuk, Z. H. Pan, J. Seo, M. Katayama, T. Minegishi, H. Nishiyama, T. Takata, K. Seki, A. Kudo, T. Yamada and K. Domen, J. Am. Chem. Soc., 2017, 139, 1675–1683 CrossRef CAS PubMed.
- Q. Wang, S. Okunaka, H. Tokudome, T. Hisatomi, M. Nakabayashi, N. Shibata, T. Yamada and K. Domen, Joule, 2018, 2, 2667–2680 CrossRef CAS.
- S. S. K. Ma, K. Maeda, T. Hisatomi, M. Tabata, A. Kudo and K. Domen, Chem. – Eur. J., 2013, 19, 7480–7486 CrossRef CAS PubMed.
- K. Iwashina, A. Iwase, Y. H. Ng, R. Amal and A. Kudo, J. Am. Chem. Soc., 2015, 137, 604–607 CrossRef CAS PubMed.
- Z. Pan, G. Zhang and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 7102–7106 CrossRef CAS PubMed.
- R. Konta, T. Ishii, H. Kato and A. Kudo, J. Phys. Chem. B, 2004, 108, 8992–8995 CrossRef CAS.
- K. Ogawa, A. Nakada, H. Suzuki, O. Tomita, M. Higashi, A. Saeki, H. Kageyama and R. Abe, ACS Appl. Mater. Interfaces, 2019, 11, 5642–5650 CrossRef CAS PubMed.
- X. P. Tao, Y. Zhao, S. Y. Wang, C. Li and R. G. Li, Chem. Soc. Rev., 2022, 51, 3561–3608 RSC.
- Z. H. Pan, T. Hisatomi, Q. Wang, S. S. Chen, A. Iwase, M. Nakabayashi, N. Shibata, T. Takata, M. Katayama, T. Minegishi, A. Kudo and K. Domen, Adv. Funct. Mater., 2016, 26, 7011–7019 CrossRef CAS.
- Y. H. Ng, A. Iwase, A. Kudo and R. Amal, J. Phys. Chem. Lett., 2010, 1, 2607–2612 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr05498e |
| This journal is © The Royal Society of Chemistry 2024 |