
The enhanced electrocatalytic performance of nanoscopic Cu6Pd12Fe12 heterometallic molecular box encaged cytochrome c†
Shazia
Nabi
a,
Feroz Ahmad
Sofi
a,
Qounsar
Jan
 a,
Aamir Y.
Bhat
b,
Pravin P.
Ingole
b,
Maryam
Bayati
c and
Mohsin Ahmad
Bhat
*a
a,
Aamir Y.
Bhat
b,
Pravin P.
Ingole
b,
Maryam
Bayati
c and
Mohsin Ahmad
Bhat
*a
aDepartment of Chemistry, University of Kashmir, Srinagar-190006, J & K, India. E-mail: mohsin@kashmiruniversity.ac.in; Tel: +91 9419033125
bDepartment of Chemistry, Indian Institute of Technology (IIT) Delhi, New Delhi 110016, India
cDepartment of Mechanical & Construction Engineering, Faculty of Engineering and Environment, Northumbria University, Newcastle upon Tyne, NE1 8ST, UK
First published on 16th November 2023
Abstract
Designing molecular cages for atomic/molecular scale guests is a special art used by material chemists to harvest the virtues of the otherwise vile idea known as “the cage”. In recent years, there has been a notable surge in research investigations focused on the exploration and utilization of the distinct advantages offered by this art in the advancement of efficient and stable bio-electrocatalysts. This usually is achieved through encapsulation of biologically accessible redox proteins within specifically designed molecular cages and matrices. Herein, we present the first successful method for encaging cytochrome c (Cyt-c), a clinically significant enzyme system, inside coordination-driven self-assembled Cu6Pd12Fe12 heterometallic hexagonal molecular boxes (Cu-HMHMB), in order to create a Cyt-c@Cu-HMHMB composite. 1H NMR, FTIR, and UV-Vis spectroscopy, ICP-MS, TGA and voltammetric investigations carried out on the so-crafted Cyt-c@Cu-HMHMB bio-inorganic composite imply that the presented strategy ensures encaging of Cyt-c in a catalytically active, electrochemically stable and redox-accessible state inside the Cu-HMHMB. Cyt-c@Cu-HMHMB is demonstrated to exhibit excellent stability and electrocatalytic activity toward very selective, sensitive electrochemical sensing of nitrite exhibiting a limit of detection as low as 32 nanomolar and a sensitivity of 7.28 μA μM−1 cm−2. Importantly, Cyt-c@Cu-HMHMB is demonstrated to exhibit an excellent electrocatalytic performance toward the 4ē pathway oxygen reduction reaction (ORR) with an onset potential of 0.322 V (vs. RHE) and a Tafel slope of 266 mV dec−1. Our findings demonstrate that Cu-HMHMB is an excellent matrix for Cyt-c encapsulation. We anticipate that the entrapment-based technique described here will be applicable to other enzyme systems and Cyt-c for various electrochemical and other applications.
1. Introduction
The exceptional catalytic activity, specificity and selectivity of enzymes (biocompatible, bio-renewable catalysts) have sparked intense research efforts to translate biochemical pathways into various electrochemical applications, such as the design and development of bio-fuel cells and bio-electrosynthesis/electrosensing setups.1,2 These applications demand the routing of appropriately energized and kinetically facile heterogeneous electron-transfer (ET) reactions mediated by the electrochemically stable and redox-accessible native conformations of metalloproteins.3,4 Therefore, the practical utility of such applications demands the design of appropriate strategies that ensure the reusability, long-term stability and kinetically facile redox accessibility of enzyme active sites in electrochemical setups. Since enzymes can mediate the passage of electrons across electrode/electrolyte interfaces only in specific orientations, which are usually extremely sensitive to the polarity and other features of the surrounding milieu,5–8 the design of such strategies is always a tough endeavor. These promises and concerns associated with metalloprotein/enzyme mediated heterogenous electron transfer processes demand the development of novel strategies for immobilization of enzymes and metalloproteins for the fabrication of efficient and reliable bio-electrocatalysts.7,8 The practical utility of any such newly developed strategy is decided by its capacity to assure electrochemical stability, redox accessibility, and bio-physical, biochemical, and structural stability of the immobilized enzyme during electrochemical applications/investigations.In nature, the activity and stability concerns associated with enzymes are addressed through their compartmentalization into very specialized compartments formed via the cooperative self-assembly of some very special amphiphilic molecules.9 Recently, it has been shown that immobilizing or encasing enzyme molecules on or inside strong solid matrices opens up exciting opportunities for electrochemical and electrocatalytic uses of enzymes.10,11The use of mesoporous silica,12 mesoporous titania,13 sol–gel matrices,14 fullerenes,15 carbon nanotubes,16 macro-porous polymeric beads such as Eupergit C17 and metal–organic frameworks (MOFs)18–20 as solid supports for enzyme encapsulation has been successfully reported. Recently, metal–organic cage-type porous materials known as metal–organic cages (MOCs), which are highly adaptable and dynamic, have drawn a lot of attention for enzyme immobilization. Their design, which incorporates a thoughtful selection of organic linkers, offers several benefits, including variable pore size, wide surface areas, and greater void volumes, which are useful for the intended target applications. Moreover, MOCs provide adequate structural and chemical flexibility for the construction of appropriate support matrices for targeted enzyme encapsulation.21 All these specialties have established the entrapment of enzymes inside correctly constructed supramolecular assemblies as a potential method for utilizing enzymes as bio-renewable electrocatalysts for diverse electrochemical applications. It is anticipated that coordination-driven self-assembled nanoscopic supramolecular complexes containing molecular motifs with desired functionalities and morphologies hold considerable potential in this context. Given the increased interest in this new subject, we successfully developed a straightforward method for encasing cytochrome c (Cyt-c) inside nanoscale Cu6Pd12Fe12 heterometallic hexagonal molecular boxes (Cu-HMHMB). The choice of Cyt-c was made considering its well-established biological functions, stability, compact structure, small molecular mass (ca.12 kDa), and especially its size (3.2 nm × 2.7 nm × 3.3 nm), which closely fits the dimensions of the inner cavity of Cu-HMHMB.22–24 It is important to note that Cyt-c is one of the most thoroughly studied metalloenzymes and is highly recommended as a model system for research targeted at exploring and using biological electron transfer reactions in electrochemistry.22 Furthermore, it has been noted that Cyt-c possesses high electro-oxidation activity for nitrite and the ORR.25,26 For the study described here, a Cu-HMHMB entrapped Cyt-c composite – referred to as Cyt-c@Cu-HMHMB in the text – was crafted. After its chemical and morphological characterization, the so-crafted Cyt-c@Cu-HMHMB composite was tested for its electrocatalytic performance toward nitrite-ion oxidation and the oxygen reduction reaction (ORR). The voltammetric (conventional and hydrodynamic), electrochemical impedance spectroscopy (EIS), and chronoamperometric investigations presented herein establish Cyt-c@Cu-HMHMB as an electrode material with great potential for electro-oxidation/electrochemical sensing of nitrite and the ORR. The results further imply that the Cyt-c@Cu-HMHMB composite has outstanding electrochemical activity, stability, and remarkable selectivity for electrochemical sensing of nitrite and the ORR. We anticipate that the strategy described here will be adaptable for use with various enzyme systems for electrocatalytic applications and will not be restricted to Cyt-c.
2. Results and discussion
2.1. Physicochemical and electrochemical characterization
To confirm the insertion of Cyt-c within the Cu-HMHMB units, the as-synthesized samples of Cu-HMHMB and the Cyt-c@Cu-HMHMB nanocomposite were subjected to spectroscopic and electroanalytical investigations. Solid-state UV-Vis spectroscopy was employed to elucidate the structural changes and the interactions exhibited by Cu-HMHMB encapsulated Cyt-c. Fig. 1A (traces b and d) depicts the UV-Vis diffuse reflectance (DR) spectra of Cu-HMHMB and the Cyt-c@Cu-HMHMB composite, respectively. For a better comparison of the structures, the UV-Vis spectra of HMHMB without Cu(II) metal and free Cyt-c were also recorded and are depicted in Fig. 1A as traces a and c respectively. As displayed in Fig. 1A (trace a), the spectrum of HMHMB without Cu(II) exhibits a sharp and intense Soret band at 429 nm. Additionally, four weak absorption peaks in the visible region at 522 nm, 556 nm, 595 nm and 650 nm are the characteristic Q-bands that arise from the π–π* transition in a conjugated porphyrin ring system.24As is clear from trace b in Fig. 1A, the inclusion of Cu(II) into the HMHMB units results in significant changes in their UV-Vis characteristics. In contrast to the UV-Vis spectrum of HMHMB, the UV-Vis spectrum of Cu-HMHMB units shows a drop in intensity and a modest blue shift in the Soret band to 425 nm and only two Q-bands at 545 nm and 580 nm instead of four. These changes can be attributed to the complete coordination of Cu2+ ions with the N-atoms of the porphyrinic cores at the six faces of the HMHMB units, thereby confirming the successful synthesis of Cu6Pd12Fe12 heterometallic hexagonal molecular boxes.27The comparative features in the UV-Vis spectra recorded for Cyt-c and Cyt-c@Cu-HMHMB composite offered the first evidence establishing the insertion of the former into Cu-HMHMB. As evident from Fig. 1A (trace c), the UV-Vis spectrum of Cyt-c shows the presence of two broad bands at 410 nm and 525 nm (attributed to π–π* transition of the porphyrin ring) corresponding to its characteristic Soret band and Q-band respectively.28 The UV-Vis spectrum of Cyt-c@Cu-HMHMB depicts broadening in the Soret band compared to that in Cu-HMHMB only, along with a red shift in the Cyt-c characteristic Soret band to 423 nm and the presence of two Q bands that appear at 546 nm and 583 nm.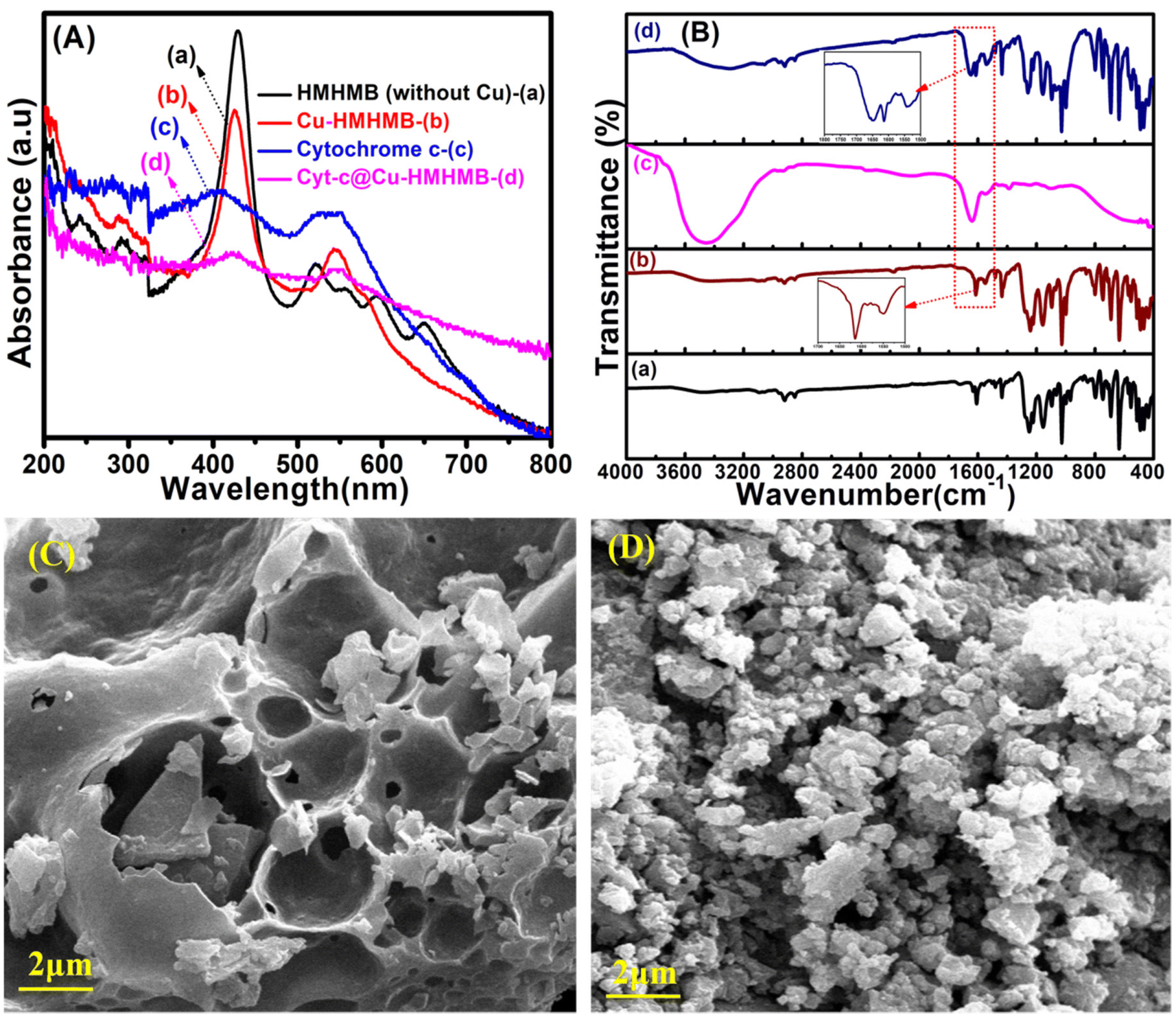 | ||
| Fig. 1 Solid state diffuse reflectance UV-Vis spectrum of HMHMB (without Cu2+) (a), Cu-HMHMB (b), free cytochrome c (c) and Cyt-c@Cu-HMHMB (d) [panel A]; FTIR spectrum of HMHMB (a), Cu-HMHMB (b), free cytochrome c (c) and Cyt-c@Cu-HMHMB (d) [panel B]; SEM images of Cu-HMHMB and Cyt-c@Cu-HMHMB [panel C and D] respectively. | ||
The UV-Vis spectra of Cu-HMHMB similarly exhibit two Q bands, but the Cyt-c@Cu-HMHMB composite exhibits a modest red shift and a corresponding decrease in the intensity of these two bands [Fig. 1A (trace d)]. These conclusions from the UV-Vis studies of these materials provided the initial proof that Cyt-c was successfully loaded into the Cu-HMHMB units. The red shift noted in the absorption characteristics of encapsulated Cyt-c in the nanocomposite compared to its free form most likely refers to its involvement in a novel type of hydrophilic/hydrophobic interactions within the Cu-HMHMB units. The FTIR analysis of the crafted units further confirmed this. Fig. 1B (traces a, b, c and d) depicts typical FTIR spectra recorded for the HMHMB, Cu-HMHMB, Cyt-c and Cyt-c@Cu-HMHMB samples, respectively. The FTIR spectrum for HMHMB [Fig. 1B (trace a)] shows peaks characteristic of TPyP units. The bands at 3314 cm−1 and 969 cm−1 are related to the N–H and C–N bond stretching frequency of the pyrrole ring, respectively. The zones of 3100–2800 cm−1 and 1500–1600 cm−1 are assigned to the C–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibrations of the porphyrin ring respectively. The intense vibration at 799 cm−1 is attributed to the vibration of C–H from pyrrole. The peak at 885.2 cm−1 is assigned to the C–C bond of the pyrrole ring.29 Linking of the TPyP unit to cis-[Pd(dppf)]2+as a vertex in the HMHMB is confirmed by the splitting noticed in the strong band at 1625 cm−1 attributed to CN stretching of the meso-attached pyridyl substituent in TPyP. The appearance of two split peaks positioned at 1609 cm−1and 1631 cm−1, along with the formation of the Pd–N bond as reflected by the peak at 466 cm−1 (ref. 30 and 31) confirms the linking of TPyP units to cis[Pd(dppf)]2+. Compared to HMHMB, similar features are observed in the FTIR spectrum of Cu-HMHMB [Fig. 1B (trace b)]. The slight shift noted in the absorption bands of the TPyP unit is attributed to the metalation of porphyrin molecules due to their binding with Cu(II). This is further confirmed by the presence of a peak at 359 cm−1 in the FTIR spectrum of Cu-HMHMB, which is a typical characteristic of the Cu–N vibration.32 Compared to the FTIR spectrum of HMHMB, a slight shift from 466 cm−1 to 468 cm−1 is noticed for the vibrational frequency of Pd–N bond for Cu-HMHMB. Moreover, the absorption peak assigned to the CN bond of the pyridyl substituent in the Cu(TPyP) unit of Cu-HMHMB coordinated to Pd of cis-[Pd(dppf)]2+ is observed to shift to 1614 cm−1 and 1634 cm−1 respectively. In the case of Cyt-c@Cu-HMHMB, the FTIR spectrum shows the presence of all the peaks of Cu-HMHMB albeit with slightly reduced peak intensities. This suggests that the Cu-HMHMB structure is effectively preserved in the Cyt-c@Cu-HMHMB nanocomposite, and the decreased intensities may result from Cyt-c's interaction with Cu-HMHMB. Fig. 1B (trace c) depicts the FTIR spectrum recorded for a typical pristine sample of Cyt-c. It clearly shows the characteristic peaks at 1642 cm−1 and 1547 cm−1 corresponding to absorptions resulting from amide-I, H2N-CO-R and amide-II, -NH-CO-R, respectively.33The FTIR spectra of Cyt-c, Cu-HMHMB, and Cyt-c@Cu-HMHMB in the wavelength range of 1500–1650 cm−1 show peaks at 1649 cm−1 and 1567 cm−1 corresponding to Cyt-c, which are absent in Cu-HMHMB, proving the successful integration of cytochrome c in the Cu-HMHMB structure. Following its encapsulation inside Cu-HMHMB units, no change is noticed for the amide I and amide II bands of Cyt-c. In view of these FTIR signatures, it is safe to presume that the biologically active secondary structure of Cyt-c is intact in the Cyt-c@Cu-HMHMB composite. The composite is therefore anticipated to display all the biological characteristics that are unique to Cyt-c. These conclusions are further supported by the 1H NMR spectra recorded for the HMHMB, Cu-HMHMB and Cyt-c@Cu-HMHMB samples. As depicted in Fig. S1,† the 1H NMR spectrum of the HMHMB sample presents all the signals expected from a non-metalated TPyP moiety. The pronounced shift of these peaks towards a low δ value is attributed to the constrictions developed from the symmetrical arrangement of TPyP molecules in this self-assembled cage structure. The 1H NMR of the HMHMB is in good accordance with that reported in the literature.27 The appearance of the signals at δ = 8.4 ppm and δ = 8.01 ppm corresponds to the α and β protons of the pyridyl moieties while the two sets of signals at δ = 8.21 and δ = 8.26 ppm correspond to the pyrrolyl protons of the TPyP unit in the HMHMB structure. A significant shift of the α and β pyridyl protons (from δ = 8.8 and δ = 8.15 ppm in TPyP precursor) is in support of the coordination of the pyridyl nitrogen atoms to the cis-[Pd(dppf)]2+ unit and moreover the presence of two sets of CH pyrrolyl protons (contrary to a single set in the case of TPyP) supports the formation of a hexagonal open barrel type structure.34 In the Cu-HMHMB, a significant up-field shift in the proton signals of porphyrin corresponding to the region of δ = 8.0–8.8 ppm and those of phenyl protons present at δ = 7.35–7.8 ppm in the HMHMB is evident suggesting the presence of Cu2+ ions in the TPyP moiety. In comparison with the host Cu-HMHMB, the striking finding in the 1H NMR of Cyt-c@Cu-HMHMB system is that the characteristic proton peaks of the former are split in the region of δ = 7.38–7.8 ppm while some new peak signals characteristic of amide protons of cytochrome c appear at δ = 7.5–7.8 ppm.35 These observations from the 1H NMR spectra further validate the incorporation of Cyt-c into the Cu-HMHMB units and the existence of specific interactions between the Cyt-c guest with its host cage in the Cyt-c@Cu-HMHMB composite. Additionally, we used inductively coupled plasma mass spectrometry (ICP-MS) to conduct elemental analyses in both Cu-HMHMB and Cyt-c@Cu-HMHMB to validate the existence of cytochrome c in the composite. The ICP-MS investigation was predicated on the discovery of Fe in Cyt-c@Cu-HMHMB because Fe is the element that makes up the active site in cytochrome c. As reflected from the entries of Table S1,† the amount of Fe in Cyt-c@Cu-HMHMB determined by ICP-MS is twice as much as the amount of Fe present in Cu-HMHMB alone. This excess Fe in the Cyt-c@Cu-HMHMB composite is thought to originate from cytochrome c. The elemental composition of the Cyt-c@Cu-HMHMB composite was further explored via energy dispersive X-ray (EDX) spectroscopy. Fig. S2† depicts a typical EDX spectrum recorded over the Cyt-c@Cu-HMHMB sample. The spectrum clearly establishes the presence of elements nitrogen (N), copper (Cu), palladium (Pd) and iron (Fe). It is pertinent to mention here that the EDX spectrum was recorded on a copper grid, and the high intensity of the copper peak in the spectrum results in the suppression of peaks characteristic of N, Cu, Pd and Fe. Nevertheless, the elemental composition of Cyt-c@Cu-HMHMB estimated from the EDX records clearly establishes the presence of these elements with % composition that matches well with that observed in the ICP-MS investigations. Thermal gravimetric analysis (TGA) further supports the embedding of cytochrome c in the Cu-HMHMB structure. The TGA curves shown in Fig. S3† indicate that the thermally induced mass changes in Cu-HMHMB and Cyt-c@Cu-HMHMB almost stop and the mass vs. temperature plots reach stable plateaus at temperatures of 490 °C and 520 °C, respectively. The thermal breakdown of the Cyt-c loaded in the latter is responsible for the weight difference in the plateau region of the TGA curves of Cu-HMHMB and Cyt-c@Cu-HMHMB. The observed mass difference points to a Cyt-c loading of approximately 31.43% (wt%) in the Cyt-c@Cu-HMHMB composite. SEM imaging was used to characterize the surface morphology of the Cu-HMHMB and Cyt-c@Cu-HMHMB samples, and the resulting sample images are shown in Fig. 1C and D, respectively. From Fig. 1C, it is clear that although being continuous, the Cu-HMHMB surface exhibits a number of grooves throughout. In contrast, granular features can be seen all over the sample surface in the SEM picture of the Cyt-c@Cu-HMHMB composite [Fig. 1D]. In its surface-specific physicochemical applications, this morphology is anticipated to greatly improve the performance of this composite. In order to substantiate the encapsulation of the cytochrome c within the Cu-HMHMB nanocage, TEM images of the Cu-HMHMB and
cytochrome c loaded within the Cu-HMHMB nanostructure were recorded. In the TEM micrograph of Cyt-c@Cu-HMHMB, dark spots of Cyt-c were clearly observed within the nanocage structure [Fig. S4(B)†] which were absent in the TEM image of Cu-HMHMB heterostructures [Fig. S4(A)†]. This observation further validates the successful encapsulation of cytochrome c within the Cu-HMHMB heterometallic cage.
C stretching vibrations of the porphyrin ring respectively. The intense vibration at 799 cm−1 is attributed to the vibration of C–H from pyrrole. The peak at 885.2 cm−1 is assigned to the C–C bond of the pyrrole ring.29 Linking of the TPyP unit to cis-[Pd(dppf)]2+as a vertex in the HMHMB is confirmed by the splitting noticed in the strong band at 1625 cm−1 attributed to CN stretching of the meso-attached pyridyl substituent in TPyP. The appearance of two split peaks positioned at 1609 cm−1and 1631 cm−1, along with the formation of the Pd–N bond as reflected by the peak at 466 cm−1 (ref. 30 and 31) confirms the linking of TPyP units to cis[Pd(dppf)]2+. Compared to HMHMB, similar features are observed in the FTIR spectrum of Cu-HMHMB [Fig. 1B (trace b)]. The slight shift noted in the absorption bands of the TPyP unit is attributed to the metalation of porphyrin molecules due to their binding with Cu(II). This is further confirmed by the presence of a peak at 359 cm−1 in the FTIR spectrum of Cu-HMHMB, which is a typical characteristic of the Cu–N vibration.32 Compared to the FTIR spectrum of HMHMB, a slight shift from 466 cm−1 to 468 cm−1 is noticed for the vibrational frequency of Pd–N bond for Cu-HMHMB. Moreover, the absorption peak assigned to the CN bond of the pyridyl substituent in the Cu(TPyP) unit of Cu-HMHMB coordinated to Pd of cis-[Pd(dppf)]2+ is observed to shift to 1614 cm−1 and 1634 cm−1 respectively. In the case of Cyt-c@Cu-HMHMB, the FTIR spectrum shows the presence of all the peaks of Cu-HMHMB albeit with slightly reduced peak intensities. This suggests that the Cu-HMHMB structure is effectively preserved in the Cyt-c@Cu-HMHMB nanocomposite, and the decreased intensities may result from Cyt-c's interaction with Cu-HMHMB. Fig. 1B (trace c) depicts the FTIR spectrum recorded for a typical pristine sample of Cyt-c. It clearly shows the characteristic peaks at 1642 cm−1 and 1547 cm−1 corresponding to absorptions resulting from amide-I, H2N-CO-R and amide-II, -NH-CO-R, respectively.33The FTIR spectra of Cyt-c, Cu-HMHMB, and Cyt-c@Cu-HMHMB in the wavelength range of 1500–1650 cm−1 show peaks at 1649 cm−1 and 1567 cm−1 corresponding to Cyt-c, which are absent in Cu-HMHMB, proving the successful integration of cytochrome c in the Cu-HMHMB structure. Following its encapsulation inside Cu-HMHMB units, no change is noticed for the amide I and amide II bands of Cyt-c. In view of these FTIR signatures, it is safe to presume that the biologically active secondary structure of Cyt-c is intact in the Cyt-c@Cu-HMHMB composite. The composite is therefore anticipated to display all the biological characteristics that are unique to Cyt-c. These conclusions are further supported by the 1H NMR spectra recorded for the HMHMB, Cu-HMHMB and Cyt-c@Cu-HMHMB samples. As depicted in Fig. S1,† the 1H NMR spectrum of the HMHMB sample presents all the signals expected from a non-metalated TPyP moiety. The pronounced shift of these peaks towards a low δ value is attributed to the constrictions developed from the symmetrical arrangement of TPyP molecules in this self-assembled cage structure. The 1H NMR of the HMHMB is in good accordance with that reported in the literature.27 The appearance of the signals at δ = 8.4 ppm and δ = 8.01 ppm corresponds to the α and β protons of the pyridyl moieties while the two sets of signals at δ = 8.21 and δ = 8.26 ppm correspond to the pyrrolyl protons of the TPyP unit in the HMHMB structure. A significant shift of the α and β pyridyl protons (from δ = 8.8 and δ = 8.15 ppm in TPyP precursor) is in support of the coordination of the pyridyl nitrogen atoms to the cis-[Pd(dppf)]2+ unit and moreover the presence of two sets of CH pyrrolyl protons (contrary to a single set in the case of TPyP) supports the formation of a hexagonal open barrel type structure.34 In the Cu-HMHMB, a significant up-field shift in the proton signals of porphyrin corresponding to the region of δ = 8.0–8.8 ppm and those of phenyl protons present at δ = 7.35–7.8 ppm in the HMHMB is evident suggesting the presence of Cu2+ ions in the TPyP moiety. In comparison with the host Cu-HMHMB, the striking finding in the 1H NMR of Cyt-c@Cu-HMHMB system is that the characteristic proton peaks of the former are split in the region of δ = 7.38–7.8 ppm while some new peak signals characteristic of amide protons of cytochrome c appear at δ = 7.5–7.8 ppm.35 These observations from the 1H NMR spectra further validate the incorporation of Cyt-c into the Cu-HMHMB units and the existence of specific interactions between the Cyt-c guest with its host cage in the Cyt-c@Cu-HMHMB composite. Additionally, we used inductively coupled plasma mass spectrometry (ICP-MS) to conduct elemental analyses in both Cu-HMHMB and Cyt-c@Cu-HMHMB to validate the existence of cytochrome c in the composite. The ICP-MS investigation was predicated on the discovery of Fe in Cyt-c@Cu-HMHMB because Fe is the element that makes up the active site in cytochrome c. As reflected from the entries of Table S1,† the amount of Fe in Cyt-c@Cu-HMHMB determined by ICP-MS is twice as much as the amount of Fe present in Cu-HMHMB alone. This excess Fe in the Cyt-c@Cu-HMHMB composite is thought to originate from cytochrome c. The elemental composition of the Cyt-c@Cu-HMHMB composite was further explored via energy dispersive X-ray (EDX) spectroscopy. Fig. S2† depicts a typical EDX spectrum recorded over the Cyt-c@Cu-HMHMB sample. The spectrum clearly establishes the presence of elements nitrogen (N), copper (Cu), palladium (Pd) and iron (Fe). It is pertinent to mention here that the EDX spectrum was recorded on a copper grid, and the high intensity of the copper peak in the spectrum results in the suppression of peaks characteristic of N, Cu, Pd and Fe. Nevertheless, the elemental composition of Cyt-c@Cu-HMHMB estimated from the EDX records clearly establishes the presence of these elements with % composition that matches well with that observed in the ICP-MS investigations. Thermal gravimetric analysis (TGA) further supports the embedding of cytochrome c in the Cu-HMHMB structure. The TGA curves shown in Fig. S3† indicate that the thermally induced mass changes in Cu-HMHMB and Cyt-c@Cu-HMHMB almost stop and the mass vs. temperature plots reach stable plateaus at temperatures of 490 °C and 520 °C, respectively. The thermal breakdown of the Cyt-c loaded in the latter is responsible for the weight difference in the plateau region of the TGA curves of Cu-HMHMB and Cyt-c@Cu-HMHMB. The observed mass difference points to a Cyt-c loading of approximately 31.43% (wt%) in the Cyt-c@Cu-HMHMB composite. SEM imaging was used to characterize the surface morphology of the Cu-HMHMB and Cyt-c@Cu-HMHMB samples, and the resulting sample images are shown in Fig. 1C and D, respectively. From Fig. 1C, it is clear that although being continuous, the Cu-HMHMB surface exhibits a number of grooves throughout. In contrast, granular features can be seen all over the sample surface in the SEM picture of the Cyt-c@Cu-HMHMB composite [Fig. 1D]. In its surface-specific physicochemical applications, this morphology is anticipated to greatly improve the performance of this composite. In order to substantiate the encapsulation of the cytochrome c within the Cu-HMHMB nanocage, TEM images of the Cu-HMHMB and
cytochrome c loaded within the Cu-HMHMB nanostructure were recorded. In the TEM micrograph of Cyt-c@Cu-HMHMB, dark spots of Cyt-c were clearly observed within the nanocage structure [Fig. S4(B)†] which were absent in the TEM image of Cu-HMHMB heterostructures [Fig. S4(A)†]. This observation further validates the successful encapsulation of cytochrome c within the Cu-HMHMB heterometallic cage.
In order to follow the structural/conformational changes if any in the Cyt-c following its encapsulation inside Cu-HMHMB, circular dichroism (CD) spectroscopic investigations were carried out over the Cyt-c@Cu-HMHMB composites. Fig. S5 in the ESI† depicts the typical CD spectra recorded over Cyt-c@Cu-HMHMB composites in the far-UV and near-UV region. The far-UV CD spectrum provides information regarding the secondary structure (orientation of peptide backbone) of proteins [Fig. S5(A)†]. The far-UV CD spectra of both native Cyt-c and Cyt-c@Cu-HMHMB exhibit two negative minima at 210 nm and 226 nm which result- from the chirality of the α-helical structure of the peptide bonds. A slight shift observed in the spectral peak at 226 nm due to the Cyt-c entrapped within the Cu-HMHMB heterostructures indicates some sort of hydrophilic/hydrophobic interactions which agrees with the UV-Vis absorption studies. The negative minimum below 240 nm as observed in the CD spectrum of Cyt-c@Cu-HMHMB is in conformity with that reported for Cyt-c in the literature and it implies that its entrapment inside Cu-HMHMB results in no change in its secondary structure.36 The near-UV CD spectra recorded to establish the tertiary structure of cytochrome c in its native state and within Cu-HMHMB are presented in Fig. S5(B).† It shows two negative minima in the region 280–286 nm and 290–306 nm corresponding to the amino acids Tyr and Trp respectively. These results are in agreement with the previously published reports for Cyt-c.47 In light of these observations, it is safe to conclude that the synthetic strategy used in the present work ensures encaging of Cyt-c within the Cu-HMHMB framework without any significant conformational change in the structure of the former. Therefore, it is safe to assume that the Cyt-c in Cyt-c@Cu-HMHMB, as crafted for the present work, exists in its biologically active state.
Using dynamic light scattering experiments, the particle diameter of Cu-HMHMB was determined to be 944.78 nm with a polydispersity index (PDI) of 2.96. The value was found to decrease to 240.70 nm (one-fourth of its original size) post encaging of cytochrome c with a reduction in the polydispersity index to 0.44 [Fig. S5(C)†]. This significant change in the particle size of Cu-HMHMB post the formation of the Cyt-c@Cu-HMHMB composite clearly indicates that Cu-HMHMB units are engaged in some sort of interaction with each other and hence exist as aggregates. However, post the treatment with cytochrome c, the Cu-HMHMB heterostructures are aligned to more favorable interactions with the cytochrome c molecules resulting in the breaking of aggregates to discrete Cyt-c@Cu-HMHMB composite material. This validates the successful encaging of cytochrome c within the Cu-HMHMB heterostructures and is quite consistent with the TEM reports of the samples.
To evaluate the redox activity, electrochemical stability, and electrocatalytic performance of the heterometallic structures viz. Cu-HMHMB and Cyt-c@Cu-HMHMB, detailed voltammetric investigations were carried out with these crafted materials. Fig. 2A depicts a typical set of CVs recorded with a scan rate of 50 mV s−1 in 0.1 M phosphate buffer (pH = 7.2) over GCE, and Cu-HMHMB or Cyt-c@Cu-HMHMB modified GCE. While CVs recorded over the bare GCE exhibit no significant faradaic response in the potential range from +1.0 V to −1.0 V [Fig. 2A (trace a)], a pair of clearly distinct redox peaks can be noted in the CVs recorded over Cu-HMHMB modified GCE [Fig. 2A (trace b)]. In the cathodic scan, a reduction peak positioned (Epc) at −0.328 V with a peak current (Ipc) of 18.89 μA and a sharp oxidation peak (Epa) centered at −0.028 V with a peak current (Ipa) of 12.54 μA in the anodic scan is noted. The peak-to-peak separation (ΔEp = Epa − Epc) between the two peaks was estimated to be ca. 300 mV. These faradaic responses noted in the CVs recorded with the Cu-HMHMB modified GCE are attributed to the two-electron redox process involving the Cu2+/Cu0 couple of Cu-HMHMB. This reflects the redox accessibility of Cu(II) sites in these heterometallic molecular boxes. The concentration of the Cu(II) sites present in the Cu-HMHMB modified GCE as calculated from the peak area of the CVs recorded in 0.1 M phosphate buffer (pH = 7.2) was found to be equal to 3.78 × 10−8 moles per cm2. We also attempted to perform voltammetric investigations on cytochrome c loaded over bare GCE under similar electrochemical conditions. However, due to the instability of the Cyt-c-film over GCE, the redox response of cytochrome c was observed to be highly unstable, with redox signals exhibiting a fast decay with an increase in the scan number [Fig. S6†]. The very first scan recorded over the Cyt-c/GCE film, implies a redox response from an electrochemically irreversible redox event (Fe(III)/Fe(II) redox process) with ΔEp equal to 278 mV. Similar observations have also been reported by other groups for Cyt-c modified electrodes.37
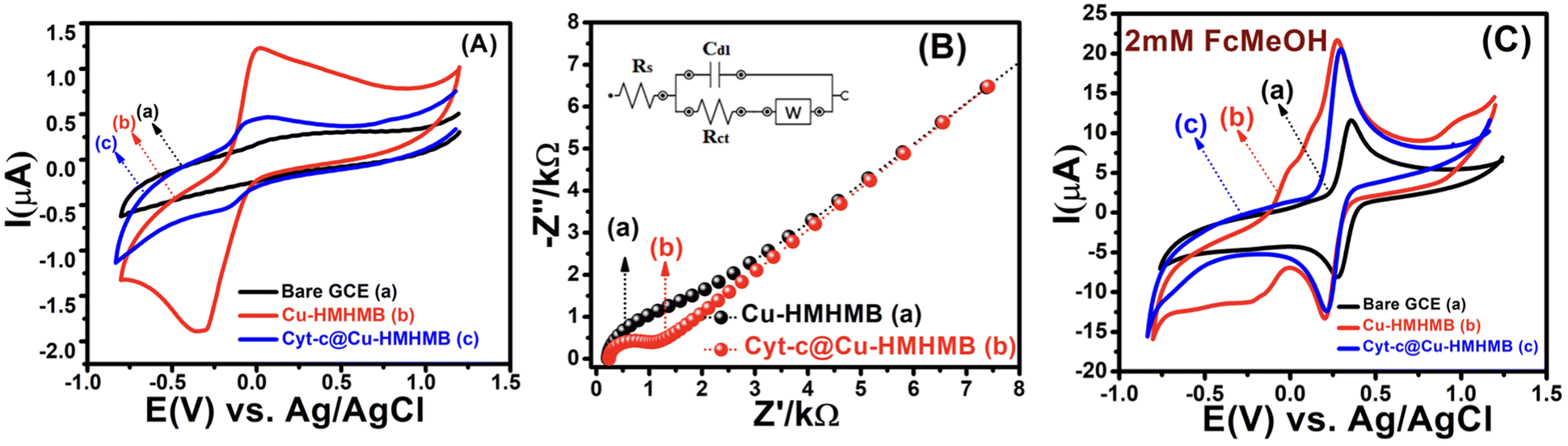 | ||
| Fig. 2 CV curves recorded for the bare GCE (a), Cu-HMHMB/GCE (b) and Cyt-c@Cu-HMHMB/GCE (c) in 0.1 M phosphate buffer solution (pH = 7.2) at a scan rate of 50 mV s−1 [panel A]; electrochemical impedance spectra of Cu-HMHMB (a) and Cyt-c@Cu-HMHMB (b) in the absence of any redox probe-using the Randles equivalent circuit model [panel B]. CVs recorded for 2 mM Fc(CH2OH) in 0.1 M KNO3 over the bare GCE (a), Cu-HMHMB/GCE (b) and Cyt-c@Cu-HMHMB/GCE (c) in 0.1 M phosphate buffer solution (pH = 7.2) at a scan rate of 50 mV s−1 [panel C]. | ||
To further assess the nature of the redox response observed for Cu-HMHMB, the CV investigations were carried out by changing scan rate (ν). The anodic and cathodic peak currents were observed to exhibit a linear dependence over ν1/2 with an R2 value of 0.99. Moreover, as depicted in Fig. S7(A),† in the CVs recorded over Cu-HMHMB, an increase in ν was observed to increase the peak current associated with the anodic as well as cathodic peak and a slight shift in their peak positions and an increase in the potential gap (ΔEp) between their peak potentials. The peak current was found to vary linearly with ν1/2 (inset of Fig. S7A†), implying a diffusion-controlled process. These voltammetric characteristics represent a faradaic response typical to a diffusion-controlled, electrochemically irreversible heterogeneous electron transfer mechanism. The CVs recorded over the Cyt-c@Cu-HMHMB composite [Fig. 2A (trace c)] were significantly different from those recorded over Cu-HMHMB. Though two redox peaks like those observed over the Cu-HMHMB were also noticed in the CVs recorded with the Cyt-c@Cu-HMHMB composite [Fig. 2A (trace c)], these were differently positioned over the potential axes with significantly reduced peak currents and peak-to-peak separation. The peak potentials of these redox events in the Cyt-c@Cu-HMHMB composite were observed to shift to Epc = −0.177 and Epa = 0.017 V with a peak separation of just 194 mV. This reflects more facile electron transfer kinetics, almost quasi-reversible redox behavior of Cyt-c@Cu-HMHMB compared to the electrochemically irreversible redox response of Cu-HMHMB. It is pertinent to mention here that compared to Cu-HMHMB, the peak current values are significantly smaller for Cyt-c@Cu-HMHMB, with Ipc = 5.39 μA and Ipa = 4.51 μA with an Ipc/Ipa ratio nearly equal to 1. The decrease in the peak current due to cytochrome c loading is in good accordance with the earlier reports wherein protein loading has been found to significantly decrease the faradaic response of the substrates on account of the surface coverage of their electrochemically active redox sites by the redox inactive protein matrix.38 We further recorded the CVs with changing scan rates over Cyt-c@Cu-HMHMB in 0.1 M phosphate buffer at pH 7.2, and a sample set of these CVs is presented in Fig. S7(B).† The peak currents showed a significant increase with an increase in the ν. Furthermore, following repeated potential scanning for the CV measurements, the CV responses remained unaltered. As displayed in the inset of Fig. S7(B),† both the anodic and cathodic peak currents were found to increase linearly with ν within the scan rate range of 10–200 mV s−1. The Ipvs. ν data fit well to linear regression with R2 of 0.99. These observations imply a surface-controlled redox response being responsible for the faradaic events noticed in the CV records of Cyt-c@Cu-HMHMB. The linear fits of Ipvs. ν data were employed for the estimation of the surface concentration (Γ /mol cm−2) of redox active sites in Cyt-c@Cu-HMHMB following the Brown–Anson model [eqn (1)]:
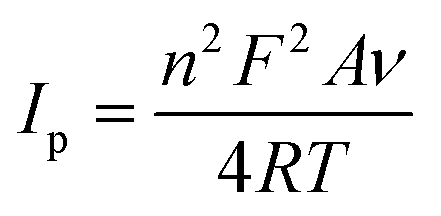 | (1) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1), R is the gas constant and T is the temperature. Following eqn (1), Γ was estimated to be ca. 1.612 × 10−8 mol cm−2, which is much higher than the theoretically expected surface coverage of 1.14 × 10−12 mol cm−2 (ref. 39) and the reported surface coverage values for Cyt-c modified electrodes in the literature; for example, Cyt-c immobilized on poly-3-methylthiophene/MWCNT (Γ = 1.6 × 10−11 mol cm−2, Eguílaz et al., 2010);40 DNA incorporated MWCNT (Γ = 2.34 × 10−11 mol cm−2, Shie et al., 2008);41 binary SAMs (Γ = 9.2 × 10−12 mol cm−2, Ji et al., 2007);42 MWCNT/ciprofloxacin films (Γ = 5.35 × 10−10 mol cm−2, Kumar et al., 2010).43 The higher value of Γ is probably due to the multilayered structure of the Cyt-c@Cu-HMHMB film. Interestingly, the surface concentration of the redox-active sites in the Cyt-c@Cu-HMHMB composite is almost half the value of the concentration of redox-active centers estimated for Cu-HMHMB (as mentioned above). Following the encapsulation of Cyt-c by these molecular boxes to create the Cyt-c@Cu-HMHMB composite, the surface concentration of redox-active sites in Cu-HMHMB appears to decrease significantly (as shown by the peak currents). In contrast, the reduced ΔEp values suggest that the encapsulation of Cyt-c inside Cu-HMHMB significantly improves the kinetic feasibility of heterogeneous ET to the redox active sites of the Cyt-c@Cu-HMHMB composite. The Cyt-c specific redox response as observed for Cyt-c@Cu-HMHMB herein seems kinetically more facile than that reported for it post its immobilization over electronically conducting substrates like Au nanoparticles.44 The effect of the scan rate over Ep (as depicted in the inset to Fig. S7(B)†) was employed for the estimation of the charge transfer coefficient (α) and, thereby, the heterogeneous ET rate constant (ks) following Laviron formalism45 [eqn (2)]:
485 C mol−1), R is the gas constant and T is the temperature. Following eqn (1), Γ was estimated to be ca. 1.612 × 10−8 mol cm−2, which is much higher than the theoretically expected surface coverage of 1.14 × 10−12 mol cm−2 (ref. 39) and the reported surface coverage values for Cyt-c modified electrodes in the literature; for example, Cyt-c immobilized on poly-3-methylthiophene/MWCNT (Γ = 1.6 × 10−11 mol cm−2, Eguílaz et al., 2010);40 DNA incorporated MWCNT (Γ = 2.34 × 10−11 mol cm−2, Shie et al., 2008);41 binary SAMs (Γ = 9.2 × 10−12 mol cm−2, Ji et al., 2007);42 MWCNT/ciprofloxacin films (Γ = 5.35 × 10−10 mol cm−2, Kumar et al., 2010).43 The higher value of Γ is probably due to the multilayered structure of the Cyt-c@Cu-HMHMB film. Interestingly, the surface concentration of the redox-active sites in the Cyt-c@Cu-HMHMB composite is almost half the value of the concentration of redox-active centers estimated for Cu-HMHMB (as mentioned above). Following the encapsulation of Cyt-c by these molecular boxes to create the Cyt-c@Cu-HMHMB composite, the surface concentration of redox-active sites in Cu-HMHMB appears to decrease significantly (as shown by the peak currents). In contrast, the reduced ΔEp values suggest that the encapsulation of Cyt-c inside Cu-HMHMB significantly improves the kinetic feasibility of heterogeneous ET to the redox active sites of the Cyt-c@Cu-HMHMB composite. The Cyt-c specific redox response as observed for Cyt-c@Cu-HMHMB herein seems kinetically more facile than that reported for it post its immobilization over electronically conducting substrates like Au nanoparticles.44 The effect of the scan rate over Ep (as depicted in the inset to Fig. S7(B)†) was employed for the estimation of the charge transfer coefficient (α) and, thereby, the heterogeneous ET rate constant (ks) following Laviron formalism45 [eqn (2)]: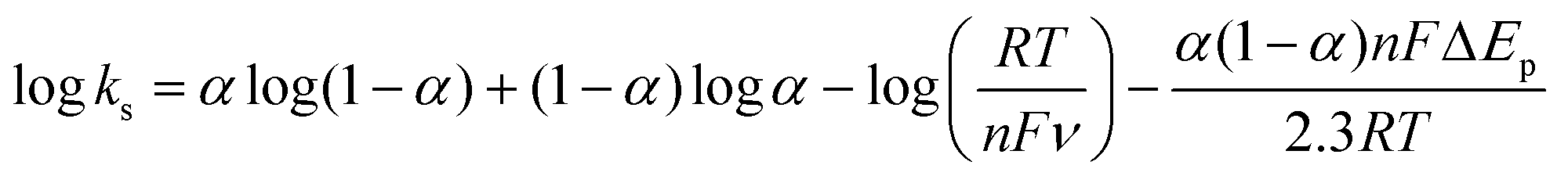 | (2) |
Electrochemical impedance spectroscopy (EIS) was employed to estimate the kinetic parameters associated with the heterogeneous electron transfer responsible for the faradaic responses observed in the CV records [Fig. 2A] for the Cu-HMHMB/GCE and Cyt-c@Cu-HMHMB/GCE interfaces. The EIS measurements were carried out in the frequency range from 0.1 Hz to 100 kHz in 0.1 M phosphate buffer solution (pH = 7.2). The impedance data were fitted using the Randles equivalent circuit, and the typical impedance spectra are presented as Nyquist plots in Fig. 2B. The smaller diameter of the semicircle portion for Cyt-c@Cu-HMHMB/GCE as compared to Cu-HMHMB/GCE indicates high conductivity and low charge transfer resistance (Rct). The values of Rct were estimated as 0.727 kΩ and 1.27 kΩ for Cyt-c@Cu-HMHMB/GCE and Cu-HMHMB/GCE respectively. These estimates suggest that the presence of Cyt-c significantly facilitates the heterogeneous electron transfer process across the Cyt-c@Cu-HMHMB/GCE interface. We also tested these interfaces for their ability to mediate heterogeneous electron transfer to solution phase redox species. In this regard, we explored the CV response of these interfaces for heterogeneous electron transfer to ferrocene methanol (FcMeOH) as a model redox probe. Fig. 2C shows typical CV curves recorded with a scan rate of 50 mV s−1 for 2 mM FcMeOH in 0.1 M KNO3 over bare GCE (a), Cu-HMHMB/GCE (b) and Cyt-c@Cu-HMHMB/GCE (c). The voltammetric characteristics of CVs recorded for FcMeOH over these electrodes are typical to a reversible one electron redox process. Compared to the CVs recorded over the bare GCE, the CVs recorded over modified GCE exhibited narrower peaks associated with higher peak currents. Importantly compared to the CVs recorded over Cu-HMHMB/GCE, the CVs recorded over Cyt-c@Cu-HMHMB/GCE exhibited slightly less peak currents but with smaller half-peak widths. These features of the CVs depicted in Fig. 2C suggest that the electrochemically active surface area (ECSA) for Cu-HMHMB/GCE and Cyt-c@Cu-HMHMB/GCE is almost identical but significantly higher than that of the bare GCE. Interestingly the CVs depicted in Fig. 2C suggest that the concentration of surface-confined redox active sites in Cu-HMHMB/GCE is much higher than that in Cyt-c@Cu-HMHMB/GCE. However, both Fig. 2A and B suggest that heterogeneous electron transfer over Cyt-c@Cu-HMHMB is kinetically more facile than that over Cu-HMHMB. Importantly, these voltammetric characteristics imply that while the ECSA of GCE is greatly improved by the casting of Cu-HMHMB and Cyt-c@Cu-HMHMB films over it, the concentration of redox-active sites (thought to give the GCE specificity) appears to be lower in the case of Cyt-c@Cu-HMHMB films. This again confirms the presence of redox-active Cyt-c in the Cyt-c@Cu-HMHMB composite. CVs were also recorded at changing scan rates for 2 mM FcMeOH over Cyt-c@Cu-HMHMB. In the CVs so recorded, as depicted in Fig. S8(A),† an increase in ν resulted in a slight shift in peak potentials and an increase in the peak current. In the scan rate range of 10–500 mV s−1, the FcMeOH-specific peak currents exhibited a linear dependence over ν1/2. The Ipvs. ν1/2 data recorded over Cyt-c@Cu-HMHMB fitted well to a linear equation with R2 = 0.99. This implies an outer sphere, diffusion-controlled redox response of FcMeOH over Cyt-c@Cu-HMHMB. Large amplitude potential step chrono coulometry with FcMeOH as the standard redox probe was employed for the more reliable estimation of ECSA of Cyt-c@Cu-HMHMB/GCE and Cu-HMHMB/GCE. The ECSA was estimated from the slopes of Cottrell plots by assuming the diffusion coefficient of FcMeOH in 0.1 M KNO3 as 7.5 × 10−6 cm2 s−1.53 The Cottrell plots corresponding to the chronocoulometric responses recorded over various electrode systems for 2 mM FcMeOH solutions are displayed in Fig. S8(B).† The electroactive surface area estimated from these plots turns out as 8.68 × 10−2 cm2 for Cu-HMHMB/GCE and 9.71 × 10−2 cm2 for Cyt-c@Cu-HMHMB/GCE. It is, therefore, clear that with the loading of Cyt-c, the roughness factor of the nanocomposite-modified electrode increases by a factor of 1.2 (as expected from the SEM image). All these observations vis-à-vis the electrochemical properties of Cyt-c@Cu-HMHMB along with the charge transfer studies of solution phase redox probe, thereby validate the successful formation of the Cyt-c@Cu-HMHMB composite. To establish the nature of redox-accessible sites in Cyt-c@Cu-HMHMB, detailed square wave voltammetric investigations were carried out over Cyt-c@Cu-HMHMB/GCE in phosphate buffer solutions of varying pH [Fig. S9(A)†]. The details of these investigations are presented in the ESI.† As depicted in Fig. S9,† the change in pH was observed to result in changes (shift in position and the broadening of the voltammetric peaks) in the square wave voltammograms in the forward and reverse scans. These changes reveal strong pH sensitivity of the voltammetric response from the Cyt-c@Cu-HMHMB nanocomposite. To estimate the number of electrons involved in the electron transfer, the square wave voltammetric data were analyzed for the effect of pH on the formal peak potential (E′0). A plot of E′0vs. pH depicted the linear variation of E′0 with the slope of −40.4 mV per pH [Fig. S9(B)†], which is close to that expected for the reversible, one-proton-coupled single electron transfer.54 Based on these observations, it is safe to assume that the voltammetric response of the Cyt-c@Cu-HMHMB nanocomposite is due to a one-electron-one proton-transfer characteristic of the iron centre of Cyt-c:
| Cyt c FeIII + e− + H+ ↔ Cyt c FeII |
This conclusion from our square wave voltammetric studies over the Cyt-c@Cu-HMHMB/GCE interface shows that the redox response shown for this composite (Fig. 2A) is most likely caused by the redox activity of the Fe(III)/Fe(II) couple of Cyt-c.
2.2. Electrocatalytic investigations
Cytochrome c is a well-known redox active hemeprotein that mediates the electron transfer between membrane-bound Cyt-c reductase and Cyt-c oxidase complexes in biological systems. In addition to being thoroughly examined as a model redox protein for comprehending biological electron transfer reactions, Cyt-c has been shown to possess high electrocatalytic activity for a select few electro-oxidations and electro-reductions. While there are several publications of Cyt-c-mediated electro-reductions in the literature, there are very few reports about electrocatalytic oxidations by Cyt-c.39,55,56 Lei and coworkers have demonstrated the iron porphyrin (electroactive center of Cyt-c) complex as an amazingly effective electrocatalyst for the electro-oxidation of NO− and NO2−.57 Recently, Cyt-c catalyzed electro-oxidations have been employed for selective electrochemical sensing of nitric oxide,58 superoxide radical anion,59 halogen oxyanions, ascorbic acid and L-cysteine.41 Cyt-c has also been proved to effectively catalyze the reductive reaction of nitrite, and the same has been employed for the electrochemical sensing of nitrite.60 Cyt-c electro catalyzed ORR is one of the several electro-reductions that has been investigated so far, and it is being thoroughly investigated with an eye toward potential commercial-scale applications. This is due to the possibility of its application in creating and developing metal–air batteries and biofuel cells, the newest technologies for producing and storing renewable energy. Currently, a number of platinum group metals (PGM) and their composites are used to address the problem of slow ORR kinetics in these devices.61,62 But the practical utility of these catalysts is constrained by their high cost and poor durability.63,64 This necessitates the discovery and design of suitable alternatives to PGGM-based electrocatalysts for the ORR, and metalloproteins have emerged as a viable option in this area. In addition to having outstanding activity and selectivity that facilitate the design simplification of biofuel and metal–air batteries, metalloproteins are considered bio-renewable and can electro catalyze processes under ambient pH, temperature, and pressure settings.65,66 These facts and voltammetric investigations (as presented in the preceding section) motivated us to explore the Cyt-c@Cu-HMHMB composite for its electrocatalytic performance toward the electro-oxidation of nitrite and the ORR in neutral pH conditions.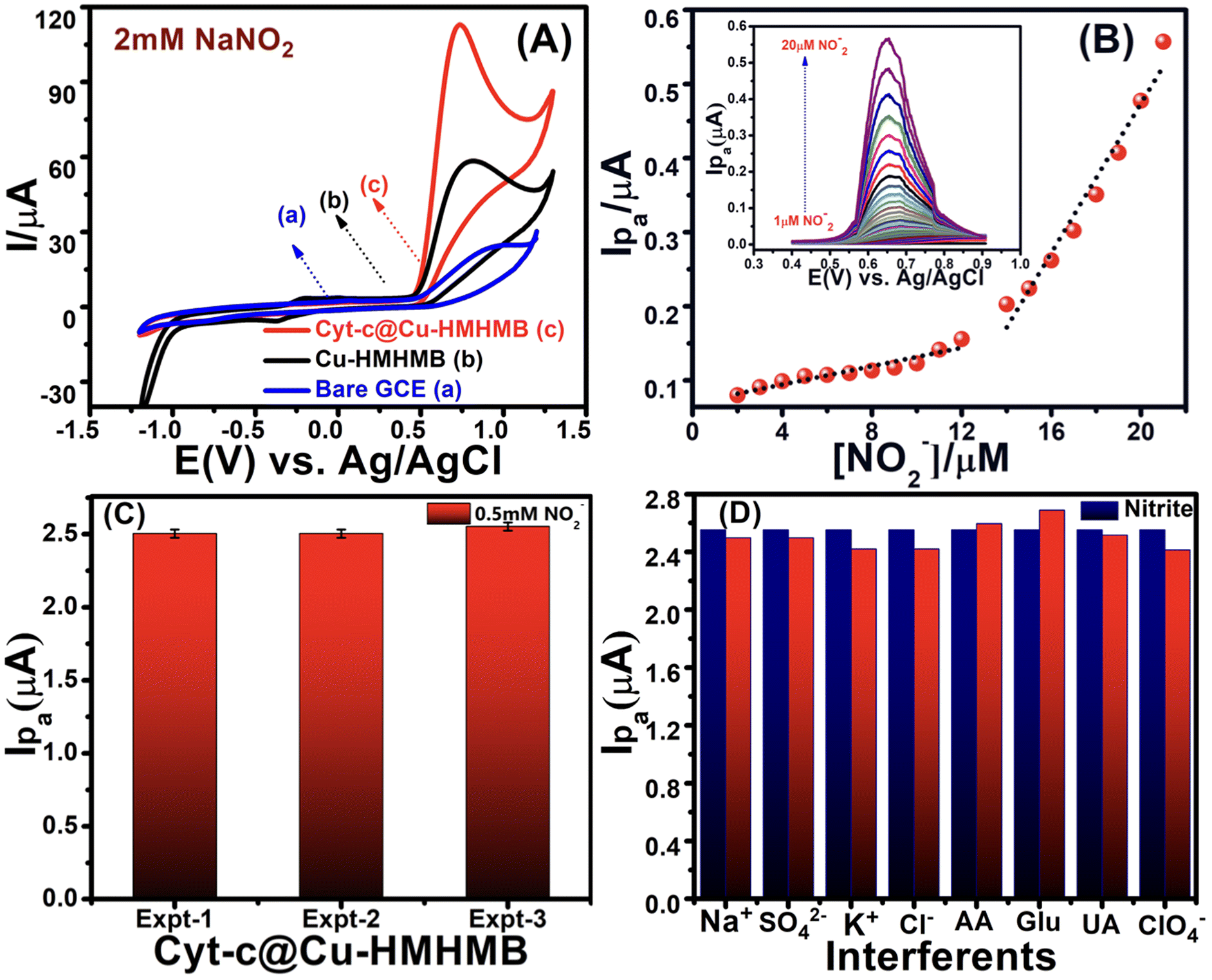 | ||
| Fig. 3 CVs recorded for 2 mM NaNO2 over the bare GCE (a), Cu-HMHMB/GCE (b) and Cyt-c@Cu-HMHMB/GCE (c) in 0.1 M phosphate buffer solution (pH = 7.2) at a scan rate of 50 mV s−1 [panel A]. DPVs recorded for changing concentrations of nitrite (NO2−) in 0.1 M PBS (pH 7.2) over Cyt-c@Cu-HMHMB/GCE [inset, panel B]. Calibration plots between [Ip] vs. [NO2−] with two linear ranges from 1 μM to 12 μM, Ip = 3.112 + 0.7072 [NO2−], (R2 = 0.98) and from 13 μM to 20 μM, Ip = −26.69 + 5.0393 [NO2−], (R2 = 0.97) [panel B]; reproducibility of the Cyt-c@Cu-HMHMB/GCE electrode system for the electro-detection of the same concentration (0.5 mM) of nitrite ions in 0.1 M PBS (pH 7.2) recorded via DPV measurements over three prepared electrode systems [panel C]; interference study of inorganic compounds and biomolecules on the current response of 0.5 mM nitrite ions in 0.1 M phosphate buffer solution (pH = 7.2) over the Cyt-c@Cu-HMHMB/GCE bio-composite [panel D]. | ||
A distinct oxidative peak attributed to the electro-oxidation of NO2− is noticed in the CVs recorded with both the modified electrodes. The significantly better peak current and slightly less positive onset and peak potentials noticed for the electro-oxidation of nitrite over Cyt-c@Cu-HMHMB/GCE imply its significantly better electrocatalytic performance. The Epa value for NO2− oxidation was observed at ca. +0.933 V for the bare GCE and +0.82 V over Cu-HMHMB/GCE, whereas it was shifted to +0.72 V (shift of 100 mV) over Cyt-c@Cu-HMHMB/GCE. The oxidation peak current in the CV records recorded over Cyt-c@Cu-HMHMB/GCE (113.53 μA) is about two times that observed for the CVs recorded over Cu-HMHMB/GCE (58.76 μA) under similar conditions. The smaller value of Ep – Ep/2 noticed for the anodic peak in the CVs recorded over Cyt-c@Cu-HMHMB/GCE compared to that noticed for the CV records of bare GCE and Cu-HMHMB/GCE implies a comparatively more facile kinetics for the electro-oxidation of NO2− over Cyt-c@Cu-HMHMB. Similar observations reported for the MWCNTs-TiN/Cyt-c/GCE interfaces have been attributed to the electrocatalytic performance of Cyt-c.50 Considering our abovementioned observations and these reports,50 it is safe to infer that encaging of Cyt-c, besides enhancing the stability, offers a promising means to exploit the bio-electrocatalytic performance of this hemeprotein. Fig. S10(A)† depicts a typical set of CVs, recorded with changing potential scan rates (10 mV s−1–500 mV s−1) over the Cyt-c@Cu-HMHMB/GCE interface in a 2 mM solution of nitrite ions. These CVs suggested a linear dependence of the anodic peak current over ν1/2 with R2 = 0.99 (as depicted in the inset of Fig. S10(A)).† The NO2− specific electro-oxidation peak was also seen to shift to a more negative potential with an increase in ν. These features imply that the electro-oxidation of NO2− over Cyt-c@Cu-HMHMB is an electrochemically irreversible, diffusion-controlled heterogeneous ET.
The electro-oxidation of nitrite ions over Cyt-c@Cu-HMHMB/GCE can proceed via a simple irreversible electrode process (Ei mechanism) or an electron transfer coupled chemical reaction consuming the oxidized species, namely EC- or catalytic-type mechanisms.67,68 To establish the exact mechanism of ET, chronoamperometric measurements were carried out for the electro-oxidation of nitrite over Cyt-c@Cu-HMHMB/GCE composite material. The sample chronoamperometric records were analyzed in light of Cottrell behavior typical to simple ET processes and electrocatalytic mechanism (EC′) of nitrite ion electro-oxidation [eqn (3)]:
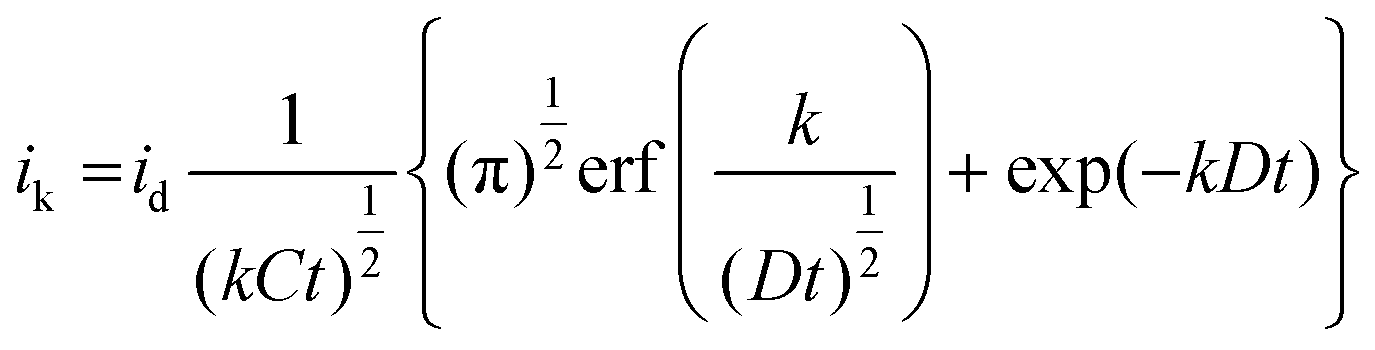 | (3) |
485 C mol−1); A = area of electrode (0.07 cm2); D = diffusion coefficient (cm2 s−1); t = time (seconds) and; erf(x) = error function.
Considering the diffusion coefficient (D) and nitrite concentration (C) = 1.8 × 10−5 cm2 s−1 and 2 × 10−6 mM cm−3 respectively, theoretical fits of the experimental chronoamperograms were generated following simple Cottrell and EC′ mechanisms. As depicted in Fig. S11,† the experimental data fit better to the EC′ mechanism of irreversible nitrite ion electrooxidation. This endorses that the electro-oxidation of nitrite in phosphate buffer of pH 7.2 over the Cyt-c@Cu-HMHMB/GCE composite follows the electrocatalytic mechanism (EC′). Compton and co-workers have reported similar observations for the electro-oxidation of nitrite over GCE.67
CVs were also recorded at changing concentrations of nitrite ions. As clearly reflected by the CVs depicted in Fig. S10(B),† an increase of NO2− concentration was noticed to increase the peak current of the anodic peak corresponding to its electro-oxidation. As is clear from the inset of Fig. S10(B),† the peak current of the anodic peak in these CVs exhibited a linear dependence over the nitrite ion concentration with R2 of more than 0.99. Motivated by the CV measurements reflecting the potential utility of Cyt-c@Cu-HMHMB toward electrochemical sensing of nitrite ions, we explored the same via differential pulse voltammetric (DPV) investigations. A typical set of DPVs, recorded with optimized parameters of scan rate = 50 mV s−1; deposition/accumulation time = 5 s and modulation amplitude = 50 mV for changing nitrite ion concentration in 0.1 M phosphate buffer solution (pH 7.2), is presented in the inset of Fig. 3B. As is clear from these DPV records, an increase in [NO2−] increases the peak current in these voltammograms. A graphic representation of the dependence of peak current over [NO2−] as seen in the DPV investigations is shown in Fig. 3B. As is clear from the depicted data, the plot of Ipvs. [NO2−] exhibits two regions of linear dependence. The peak current vs. concentration data in the low concentration range (1–12 μM) follows the linear equation Ip = 3.112 + 0.7072 [NO2−] (with R2 = 0.96). In the tested concentration range of 13 μM–20 μM, the data followed the linear equation Ip = −26.69 + 5.0393 [NO2−] with R2 = 0.97. These dependencies observed for Ip (μA) vs. [NO2−] in the DPV records were employed for the estimation of the limit of detection (LOD) based on triple signal-to-noise ratio (S/N = 3) using the equation, LOD = 3σ/S (where σ is the standard deviation of error of the intercept and S is the slope of the calibration plot) and sensitivity of Cyt-c@Cu-HMHMB toward electrochemical sensing of nitrite ions. The estimates yielded a limit of detection as low as 32 nanomolar and a sensitivity of 7.28 μA μM−1 cm−2 for the electrochemical sensing of nitrite ions over Cyt-c@Cu-HMHMB.
To appreciate the significance of these numbers, we present a comparison of similar estimates as reported for electrochemical sensing of nitrite ions over the various so-called state-of-the-art electrochemical sensing materials, in Table 1. The entries in Table 1 show the excellent capabilities of the Cyt-c@Cu-HMHMB composite for the electrochemical sensing of nitrite.40,69–76 The practical value of an electrochemical sensor is significantly influenced by the reproducibility and repeatability of its estimates. The reproducibility of the Cyt-c@Cu-HMHMB nanocomposite-based electrode towards the detection of nitrite was assessed via DPV analysis. The same concentration of nitrite (0.5 mM in 0.1 M PBS (pH 7.2)) was electrochemically sensed using three different but similarly prepared Cyt-c@Cu-HMHMB/GCE interfaces. Fig. 3C reflects a typical set of our observations in these investigations. Our overall estimates of reproducibility and repeatability reflected an RSD of ca. 2.3%, which is very impressive for such kinds of sensors.
| Nitrite (NO2−) | ||||
|---|---|---|---|---|
| Modified electrode | Linear range | LOD (μM) | Sensitivity (μA μM−1 cm−2) | Ref. |
| Cyt-c/L-Cys/P3MT/MWCNT/GCE | 10–100 | 0.5 | — | 40 |
| Cyt-c/PANI-g-ND/GNPs/GCE | 0.5–3000 | 0.16 | 0.88 | 69 |
| Cyt-c/Pt-NPs/ITO | 5–800 | 0.4 | — | 70 |
| Cyt-c/TiO2(G) NWs@PANI(SH)-Au MCNC/3DNE/GCE | 10–720000 |
0.05 | — | 71 |
| NPCF-GNs/GCE | 0.1–100 | 0.08 | 3.1 | 72 |
| AuNPs/CS/MXene/GCE | 0.5–335.5, 335.5–3300 | 0.069 | 0.517, 0.403 | 73 |
| MWCNTs/PPy-C/GCE | 5–9500 | 3.06 | 0.12 | 74 |
| PEDOT/PEDOT-SH/Au/GCE | 0.15–1000 | 0.051 | 0.301 | 75 |
| CeO2-SnO2/Pd/GCE | 0.36–2200 | 0.1 | 0.652 | 76 |
| Cyt-c@Cu-HMHMB/GCE | 1–20 | 0.032 | 7.28 | Present work |
We also investigated the impact of various potential interferents on the electrochemical sensing ability of Cyt-c@Cu-HMHMB for nitrite ions. For this, DPVs with optimized parameters were recorded for 0.5 mM NO2− in the presence of 1 mM each of K+, Na+, Cl−, SO42−, ClO4−, glucose (Glu), ascorbic acid (AA) and uric acid in 0.1 M phosphate buffer (pH 7.2) solution. The impact of the presence of these interferents on the current signal of the nitrite, as observed in these measurements, is presented in the form of a bar graph in Fig. 3D. It is apparent that even at such high concentrations, these potential interferents result in an insignificant change with a maximum of ±0.05% in the NO2− specific current signal. These observations establish the high selectivity and excellent anti-interference ability of the Cyt-c@Cu-HMHMB nanocomposite toward electrochemical sensing of nitrite ions. To assess the potential use of Cyt-c@Cu-HMHMB for the electrochemical sensing of nitrite ions in real-life samples, recovery experiments in tap water samples using the standard addition method were carried out. A typical set of overall averaged results from these investigations are summarized and presented in Table S2.† The recovery results obtained are in the range of 98.5%–99.8% with the relative standard deviation ranging between 1.0% and 1.02%, revealing the excellent reliability of the designed Cyt-c@Cu-HMHMB sensor in the effective electrochemical sensing of nitrite in real samples.
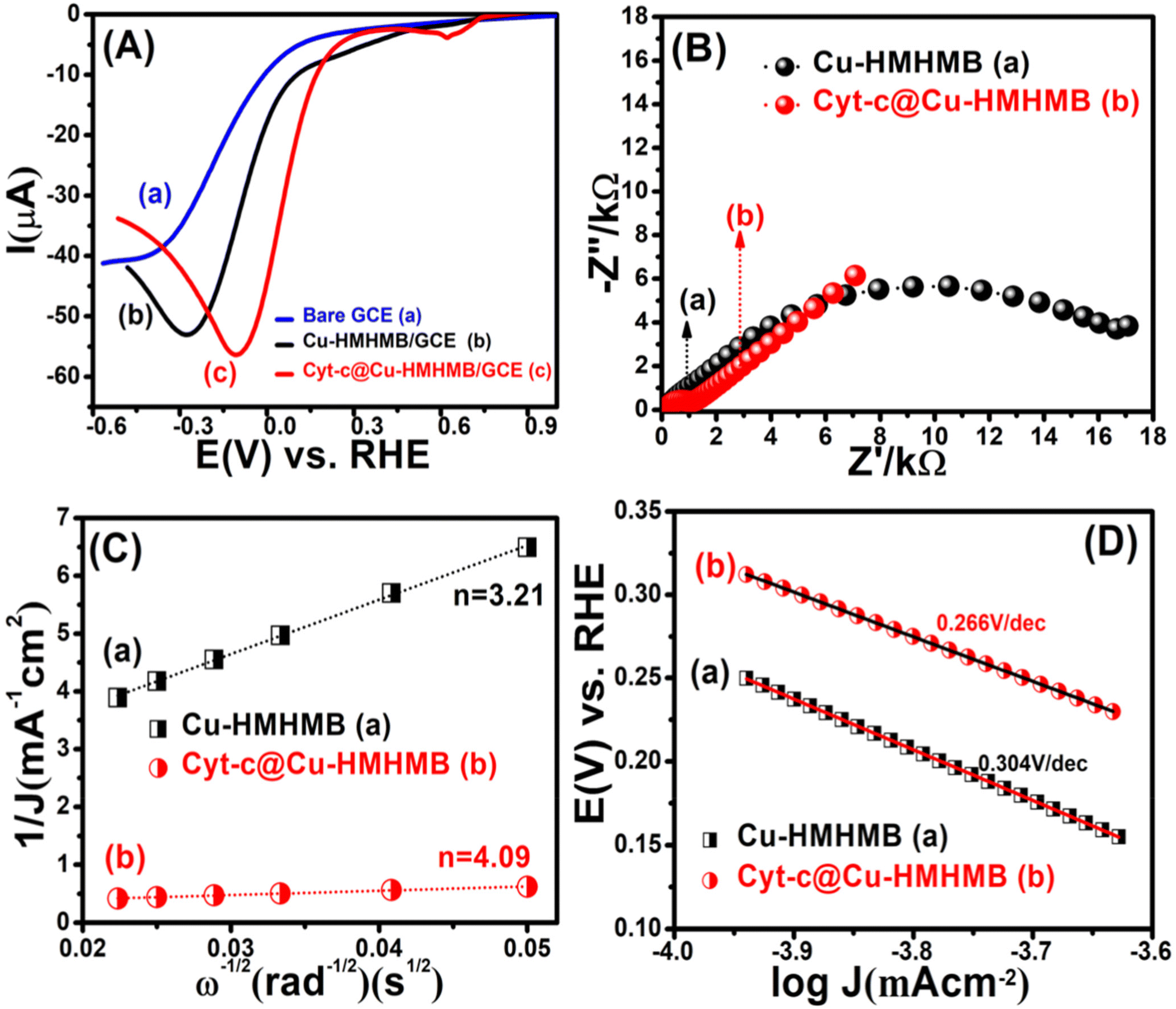 | ||
| Fig. 4 LSVs recorded for the oxygen reduction reaction over the bare GCE [panel A], Cu-HMHMB/GCE [panel B] and Cyt-c @Cu-HMHMB/GCE [panel C] in 0.1 M phosphate buffer solution (pH = 7.2) at scan rate of 50 mV s−1. EIS studies showing Nyquist plots of the oxygen reduction reaction catalyzed by Cu-HMHMB/GCE (a) and Cyt-c@Cu-HMHMB/GCE (b) [panel-B]; Koutecky–Levich (K–L) plots (J−1vs. ω−1/2) corresponding to the RDE voltammetry data recorded over Cu-HMHMB/GCE and Cyt-c@Cu-HMHMB/GCE in the diffusion controlled region of the ORR [panel C]; Tafel plots corresponding to the LSV recorded in O2 saturated solution of 0.1 M PBS (pH = 7.2)over Cu-HMHMB/GCE (a) and Cyt-c@Cu-HMHMB (b) [panel-D]. | ||
Depicted LSV traces recorded in O2-saturated PBS solutions clearly reflect ORR-specific faradaic transients with the onset and peak potentials following the order bare GCE < Cu-HMHMB/GCE < Cyt-c@Cu-HMHMB/GCE. The redox parameters related to the ORR characteristic LSVs recorded over bare GCE, Cu-HMHMB/GCE and Cyt-c@Cu-HMHMB/GCE in O2 saturated 0.1 M PBS (pH = 7.2) are presented as Table S3 in the ESI.† As evident from the polarization curves depicted in Fig. 4(A), while both Cu-HMHMB and Cyt-c@Cu-HMHMB show electrocatalytic performance toward the ORR, the electrocatalytic performance of the latter is significantly much better than that observed for the former. This is evident from the onset values and potentials corresponding to the peak currents corresponding to ORR over these materials (Table S3 in the ESI†). Theoretical and experimental investigations reported in the recent past have amply established the electrocatalytic potential of Cyt-c and its synthetic mimics toward the ORR.77–79 However, the fast denaturation of Cyt-c under harsh electrochemical conditions continues to be a major reason to limit its practical utility as an ORR electrocatalyst.78,79 Mechanistically, it is now well established that the electrocatalytic activity of Cyt-c is mediated by its Fe(II)-centre, which ensures selective binding of O2, proton-coupled electron transfer and O–O lysis via a complex mechanism involving a series of intermediates.77,79 Our results suggest that entrapment inside the Cu-HMHMB not only improves the electrocatalytic performance but also improves the electrochemical stability of Cyt-c. This was further confirmed by our voltammetric investigations carried out with RDE and EIS investigations. Fig. S12 (A and B)† depicts typical hydrodynamic polarization curves recorded with different rotation speeds (100–2000 rpm) for the ORR over Cu-HMHMB /GCE and Cyt-c@Cu-HMHMB/GCE. As is clear from these depicted curves (Fig. S12†), the current density increased with an increase in the rotation speeds, implying a diffusion-controlled redox activity. The current potential data from the diffusion limiting zone of the RDE-LSV records were sampled for the construction of J−1vs. ω−1/2 (ω is the angular velocity, ω = 2πN and N is the linear rotation speed) plots also regarded as Koutecky–Levich (K–L) plots. Fig. 4C depicts K–L plots for potentials −0.41 V, −0.27 V vs. RHE as recorded with Cu-HMHMB/GCE and Cyt-c@Cu-HMHMB/GCE for the ORR in 0.1 M phosphate buffer of pH 7.2. The linearity of the K–L plots implies that the ORR over these electrode systems follows first-order kinetics with respect to the concentration of dissolved O2. The K–L plots were employed for the estimation of various kinetic parameters employing the Koutecky–Levich equation:
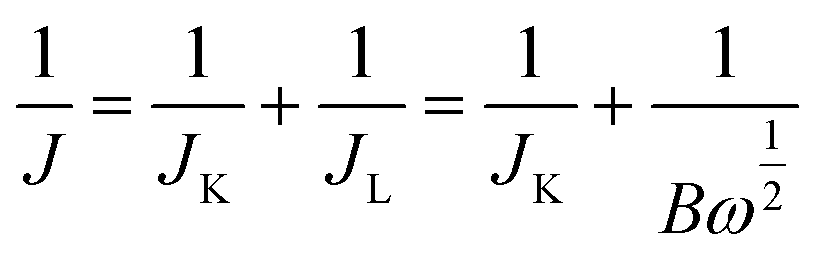 | (4) |
| B = 0.62nFC0(D0)2/3ν−1/6 | (5) |
| JK = nFkC0 | (6) |
485 C mol−1), C0 is the bulk concentration of O2, ν is the kinematic viscosity of the electrolyte, D0 is the diffusion coefficient of O2 and k is the electron transfer rate constant. The value of parameters: C0 = 0.26 mM, D0 = 1.8 × 10−5 cm2 s−1 and ν = 0.009 cm2 s−1 at 25 °C in 0.1 M PBS (pH = 7.2)77 was assumed for the quantification. From the slope of the K–L plot, the number of electrons (n) transferred per O2 molecule in the kinetically controlled region for Cu-HMHMB and Cyt-c@Cu-HMHMB was estimated to be 3.21 and 4.09 respectively. From these estimates, it is safe to assume that Cyt-c@Cu-HMHMB favors a direct 4-electron reduction pathway for the ORR as desired for the bio-fuel cell and air battery designs. Besides n, the rate constant ‘k’ was also determined from the intercept value of the K–L plots, and the values thus obtained were 0.048 × 102 and 0.27 × 102 cm s−1 for Cu-HMHMB/GCE and Cyt-c@Cu-HMHMB/GCE respectively. These n and k estimations unequivocally demonstrate that Cyt-c@Cu-HMHMB exhibits considerably better electrocatalytic activity toward the ORR than Cu-HMHMB. This was further confirmed by the Tafel plots corresponding to ORR kinetics over Cu-HMHMB and Cyt-c@Cu-HMHMB and the same are presented in Fig. 4D. Thus, while the voltammetric data recorded over Cu-HMHMB suggest a Tafel slope of 304 mV dec−1 for the ORR, the Tafel slope was found to be appreciably lower (266 mV dec−1) for the ORR over Cyt-c@Cu-HMHMB, implying its excellent electrocatalytic performance. Our EIS investigations further confirmed this conclusion.
Fig. S13(A and B)† shows the Nyquist plots corresponding to EIS data recorded at varied potentials (chosen from LSV plots depicted in Fig. 4) for the ORR over Cu-HMHMB and Cyt-c@Cu-HMHMB in O2 saturated 0.1 M PBS (pH = 7.2). As is clear from the figure, the Rct for the ORR over Cyt-c@Cu-HMHMB is appreciably smaller than that observed for Cu-HMHMB. Typical Nyquist plots corresponding to these records are presented in Fig. 4B. These plots clearly suggest that the electrocatalytic performance of Cyt-c@Cu-HMHMB toward the ORR is significantly much better than that of Cu-HMHMB. These observations and inferences from the EIS investigations confirm the observations and inferences observed in and arrived at from the voltammetric investigations as presented in the above section.
A comparison of various electrocatalytic parameters regarding the oxygen reduction reaction over Cyt-c@Cu-HMHMB in the present study and those reported over the various state-of-the-art electrode materials in the literature80–87 are listed in Table 2. The entries of Table 2 show that the electrocatalytic performance of Cyt-c@Cu-HMHMB toward the ORR in the neutral medium is either equivalent to or superior to most similar cathodic electrode materials that have been described thus far in the literature.
| Electrocatalyst | Medium | Onset potential (vs. RHE) | N | Ref. |
|---|---|---|---|---|
| Fe3O4@N/Co-C | 0.1 M PBS (pH = 7.0) | 0.23 | 3.5–3.7 | 80 |
| N-Fe/Fe3C@C | 0.1 M PBS (pH = 7.0) | 0.21 | 3.98 | 81 |
| FePc | 0.05 M PBM (pH = 7.0) | 0.429 | 4.0 | 82 |
| G65YCuBMb | 100 mM PBS (pH 7.0) | −0.263 | 4.0 | 83 |
| FeN5 SAs | 0.1 M PBS (pH 7.0) | 0.93 | 3.96 | 84 |
| MWNT-FeP(9) | 0.1 M PBS (pH 7.0) | 0.65 | 3.96 | 85 |
| Co(II)Porp/Cu(TACNA) | 0.1 M NaClO4 + 0.025 M PBS (pH 7.3) | 0.21 | 4.0 | 86 |
| Fe(III)TMPyP/Ru(NH4)6Cl3 | 0.1 M Triflic acid (pH 1.0) | 0.40 | 3.2 | 87 |
| Cyt-c@Cu-HMHMB | 0.1 M PBS (pH 7.2) | 0.322 | 4.09 | Present work |
In addition to electrocatalytic performance, electrochemical stability is crucial in deciding the practical utility of potential electrode materials. The electrochemical stability of Cyt-c@Cu-HMHMB in ORR setups was tested via chronoamperometry. Fig. S13(C)† depicts a typical chronoamperogram recorded over Cyt-c@Cu-HMHMB with a three-electrode setup in O2 saturated 0.1 M PBS (pH = 7.2) at a potential near to E1/2 value of 0.1 V vs. RHE. As is clear from the depicted chronoamperogram, the current corresponding to the ORR does not exhibit any notable change even after 30000 seconds (about eight and a half hours). Moreover, in order to confirm the structural integrity post-electrolysis for about 30000 seconds, TEM images were also recorded for the Cyt-c@Cu-HMHMB samples pre and post their use for bulk scale electrolysis and the same are depicted in Fig. S14.† The TEM images, suggest almost insignificant changes in the structure and morphology of the Cyt-c@Cu-HMHMB nanocomposite and thereby clearly manifest its excellent structural stability and electrochemical durability towards the ORR in neutral pH conditions. Based on our observations and projections from voltammetric, EIS, and stability tests, it is safe to say that the Cyt-c@Cu-HMHMB composite is a promising bio-electrocatalyst for nitrite ion sensing and the ORR. Our results suggest that the Cyt-c@Cu-HMHMB composite is a potentially promising material that can be employed in the design of electrochemical sensors for nitrite ion sensing and the ORR in bio-fuel cells and metal–air batteries.
3. Conclusion
In summary, we demonstrate the successful encaging of cytochrome c within Cu-HMHMB. Analysis through various spectroscopic and electrochemical techniques establishes that in the Cyt-c@Cu-HMHMB bio-composite, Cyt-c exists in a biologically active and redox-accessible state. The Cyt-c@Cu-HMHMB bio-composite is demonstrated to exhibit excellent electrocatalytic performance toward the electro-oxidation of nitrite ions and the ORR in neutral medium (pH = 7.2). To the best of our knowledge, the presented work is the first of its kind to report the use of heterometallic molecular boxes for the encapsulation of metalloproteins to design and fabricate electrochemically stable, electrocatalytically active bio-composites. The current work presents a novel approach toward designing and fabricating novel bio-composites for electrochemical sensing and bio-fuel cell applications.Conflicts of interest
There are no conflicts to declare.References
- J. C. Ruth and A. M. Spormann, Enzyme electrochemistry for industrial energy applications: A perspective on future area of focus, ACS Catal., 2021, 11, 5951–5967 CrossRef CAS.
- C. Cadoux and R. D. Milton, Recent enzymatic electrochemistry for reductive reactions, ChemElectroChem, 2020, 7, 1974–1986 CrossRef CAS.
- U. T. Bornscheuer, G. W. Huisman, R. J. Kazlauskas, S. Lutz, J. C. Moore and K. Robins, Engineering the third wave of biocatalysis, Nature, 2012, 485, 185 CrossRef CAS.
- D. Feng, T. F. Liu, J. Su, M. Bosch, Z. Wei, W. Wan, D. Yuan, Y. P. Chen, X. Wang and K. Wang, Stable metal-organic frameworks containing single-molecule traps for enzyme encapsulation, Nat. Commun., 2015, 6, 5979 CrossRef PubMed.
- X. Gao, S. Yang, C. Zhao, Y. Ren and D. Wei, Artificial Multienzyme Supramolecular Device: Highly Ordered Self-Assembly of Oligomeric Enzymes In Vitro and In Vivo, Angew. Chem., Int. Ed., 2014, 53, 14027–14030 CrossRef CAS.
- W. Zhang and G. Li, Third-generation biosensors based on the direct electron transfer of proteins, Anal. Sci., 2004, 20, 603 CrossRef CAS.
- J. Zhang, Q. Chi, A. M. Kuznetsov, A. G. Hansen, H. Wackerbarth, H. E. M. Christensen, J. E. T. Andersen and J. Ulstrup, Electronic Properties of Functional Biomolecules at Metal/Aqueous Solution Interfaces, J. Phys. Chem. B, 2002, 106, 1131–1152 CrossRef CAS.
- B. Bonanni, L. Andolfi, A. R. Bizzarri and S. Cannistraro, Functional Metalloproteins Integrated with Conductive Substrates: Detecting Single Molecules and Sensing Individual Recognition Events, J. Phys. Chem. B, 2007, 111, 5062–5075 CrossRef CAS PubMed.
- C. A. Kerfield, S. Heinhorst and G. C. Cannon, Bacterial microcompartments, Annu. Rev. Microbiol., 2010, 64, 391–408 CrossRef.
- L. S. Wong, F. Khan and J. Micklefield, Selective covalent protein immobilization: Strategies and applications, Chem. Rev., 2009, 109, 4025–4053 CrossRef CAS.
- P. N. Catalano, A. Wolosiuk, G. J. A. A. Soler-Illia and M. G. Bellino, Wired enzymes in mesoporous materials: A benchmark for fabricating biofuel cells, Bioelectrochemistry, 2015, 106, 14–21 CrossRef CAS.
- Y. Zhang, Q. Yue, M. M. Zagho, J. Zhang, A. A. Elzatahry, Y. Jiang and Y. Deng, Core–Shell Magnetic Mesoporous Silica Microspheres with Large Mesopores for Enzyme Immobilization in Biocatalysis, ACS Appl. Mater. Interfaces, 2019, 11, 10356–10363 CrossRef CAS.
- N. Frančič, M. G. Bellino, G. J. A. A. Soler-Illiab and A. Lobnik, Mesoporous titania thin films as efficient enzyme carriers for paraoxon determination/detoxification: effects of enzyme binding and pore hierarchy on the biocatalyst activity and reusability, Analyst, 2014, 139, 3127–3136 RSC.
- J. Yu and H. Ju, Preparation of Porous Titania Sol-Gel Matrix for Immobilization of Horseradish Peroxidase by a Vapor Deposition Method, Anal. Chem., 2002, 74, 3579–3583 CrossRef CAS PubMed.
- K. Guo, N. Li, L. Bao and X. Lu, Fullerenes and derivatives as electrocatalysts: Promises and challenges, Green Energy Environ., 2024, 9, 7–27 CrossRef CAS.
- B. Kowalewska, M. Skunik, K. Karnicka, K. Miecznikowski, M. Chojak, G. Ginalska, A. Belcarz and P. J. Kulesza, Enhancement of bio-electrocatalytic oxygen reduction at the composite film of cobalt porphyrin immobilized within the carbon nanotube-supported peroxidase enzyme, Electrochim. Acta, 2008, 53, 2408–2415 CrossRef CAS.
- T. Boller, C. Meier and S. Menzler, Eupergit Oxirane Acrylic Beads: How to Make Enzymes Fit for Biocatalysis, Org. Process Res. Dev., 2002, 6, 509–519 CrossRef CAS.
- M. K. Lee, M. Shokouhimehr, S. Y. Kim and H. W. Jang, Two-Dimensional Metal–Organic Frameworks and Covalent–Organic Frameworks for Electrocatalysis: Distinct Merits by the Reduced Dimension, Adv. Energy Mater., 2022, 12, 2003990 CrossRef CAS.
- Y. S. Wei, M. Zhang, R. Zou and Q. Xu, Metal–Organic Framework-Based Catalysts with Single Metal Sites, Chem. Rev., 2020, 120, 12089–12174 CrossRef CAS.
- X. Wang, P. C. Lan and S. Ma, Metal–Organic Frameworks for Enzyme Immobilization: Beyond Host Matrix Materials, ACS Cent. Sci., 2020, 6, 1497–1506 CrossRef CAS PubMed.
- H. Xia, N. Li, X. Zhong and Y. Jiang, Metal-Organic Frameworks: A Potential Platform for Enzyme Immobilization and Related Applications, Front. Bioeng. Biotechnol., 2020, 8, 695 CrossRef PubMed.
- R. A. Scott and A. G. Mauk, Cytochrome c: A multidisciplinary approach, University Science Books, Sausalito, CA, 1996, eISBN: 0-935702-33-4 Search PubMed.
- Y. Chen, F. Jimenez-Angeles, B. Qiao, M. D. Krzyaniak, F. Sha, S. Kato, X. Gong, C. T. Buru, Z. Chen, X. Zhang, N. C. Gianneschi, M. R. Wasielewski, M. Olvera de la Cruz and O. K. Farha, Insights into the Enhanced Catalytic Activity of Cytochrome c When Encapsulated in a Metal−Organic Framework, J. Am. Chem. Soc., 2020, 142, 18576–18582 CrossRef CAS.
- A. K. Bar, R. Chakrabarty, G. Mostafa and P. S. Mukherjee, Self-Assembly of a Nanoscopic Pt12Fe12 Heterometallic Open Molecular Box Containing Six Porphyrin Walls, Angew. Chem., Int. Ed., 2008, 47, 8455–8459 CrossRef CAS.
- S. Chatterjee, K. Sengupta, S. Hematian, K. D. Karlin and A. Dey, A. Electrocatalytic oxygen reduction by synthetic Cyt-c Oxidase mimics: Identification of a “bridging peroxo” intermediate involved in facile 4e−/4H+ Oxygen reduction, J. Am. Chem. Soc., 2015, 137, 12897–12905 CrossRef CAS PubMed.
- M. Mukherjee and A. Dey, Rejigging electron and proton transfer to transition between Dioxygenase, Monooxygenase, Peroxygenase and oxygen reduction activity: Insights from bioinspired constructs of heme enzymes, JACS Au, 2021, 1, 1296–1311 CrossRef CAS PubMed.
- H. P. Oliveira, C. F. O. Graeff and J. M. Rosolen, Synthesis and structural characterization of tetrakis(N-methyl-4-pyridyl) porphyrin copper into V2O5 xerogel, Mater. Res. Bull., 1999, 34, 1891–1903 CrossRef CAS.
- M. Tong, S. Dong, K. Wang and G. Suo, A porphyrin MOF and Ionic Liquid biocompatible matrix for the direct electrochemistry and electrocatalysis of Cytochrome c, J. Electrochem. Soc., 2017, 164, 200–204 CrossRef.
- E. Fagadar-Cosma, C. Enache, D. Dascalu, G. H. Fagadar-Cosma and R. Gavrila, FT-IR, fluorescence and electronic spectra for monitoring the aggregation process of tetra-pyridylporphyrine entrapped in silica matrices, Optoelectron. Adv. Mater., Rapid Commun., 2008, 2, 437–441 CAS.
- W. Sun, H. Wang, D. Qi, L. Wang, K. Wang, J. Kan, W. Li, Y. Chen and J. Jiang, 5,10,15,210-tetra(4-pyridyl)porphyrinato Zinc coordination polymeric particles with different shapes and luminescent properties, CrystEngComm, 2012, 14, 7780–7786 RSC.
- J. R. Durig, R. Layton, D. W. Sink and B. R. Mitchell, Far infrared spectra of palladium compounds—I. The influence of ligands upon the palladium chloride stretching frequency, Spectrochim. Acta, 1965, 21, 1367–1378 CrossRef CAS.
- Sajila and H. Mohabey, IR Spectra, Magnetic and Thermal Studies of Copper(II) Complex of N-Hydroxy –N-(4-Chloro) Phenyl N′(4-Fluoro) Phenyl Benzamidine Hydrochloride, Mater. Sci. Res. India, 2014, 11, 63–65 Search PubMed.
- A. S. Kumar, N. Vishnu and B. Dinesh, Studies on Controlled Protein Folding versus Direct Electron-Transfer Reaction of Cytochrome c on MWCNT/Nafion Modified Electrode Surface and Its Selective Bioelectrocatalytic H2O2 Reduction and Sensing Function, ACS Symp. Ser., 2020, 1342, 185–205 CrossRef CAS.
- E. Fagadar-Cosma, C. Enache, I. Armeanu and G. Fagadar-Cosma, Comparative investigations of the absorption and fluorescence spectra of the tetrapyridylporphyrine and Zn(II) tetrapyridyl porphyrin, Dig. J. Nanomater. Biostructures, 2007, 2, 175–183 Search PubMed.
- R. M. Keller and K. Wuthrich, 1H-NMR studies of structural homologies between the heme environments in horse Cytochrome-c and in Cytochrome c-552 from Euglena Graciliis, Biochim.Biophys Acta, 1981, 668, 307–320 CrossRef CAS PubMed.
- L. Santiago-Rodríguez, J. Méndez, G. M. Flores-Fernandez, M. Pagán, J. A. Rodríguez-Martínez, C. R. Cabrera and K. Griebenow, Enhanced stability of a nanostructured cytochrome c biosensor by PEGylation, J. Electroanal. Chem., 2011, 663, 1–7 CrossRef.
- H.-Z. Zhao, Q. Du, Z.-S. Li and Q.-Z. Yang, Mechanisms for the Direct Electron Transfer of Cytochrome c Induced by Multi-Walled Carbon Nanotubes, Sensors, 2012, 12, 10450–10462 CrossRef CAS.
- J. Juan He, H. Yang, Y. Zhang, J. Yu, L. Miao, Y. Song and L. Wang, Smart nanocomposites of Cu-Hemin metal organic framework for electrochemical glucose biosensing, Sci. Rep., 2016, 6, 36637 CrossRef PubMed.
- R. E. Dickerson, T. Takano, D. Eisenberg, O. B. Kallai, L. Samson and A. Cooper, et al. Ferricytochrome c: I. General features of the horse and bonito proteins at 2.8 Å resolution, J. Biol. Chem., 1971, 246, 1511–1535 CrossRef CAS.
- M. Eguílaz, L. Agüí, P. Yáñez-Sedeño and J. M. Pingarrón, A biosensor based on cytochrome c immobilization on a poly-3-methylthiophene/multi-walled carbon nanotubes hybrid-modified electrode. Application to the electrochemical determination of nitrite, J. Electroanal. Chem., 2010, 644, 30–35 CrossRef.
- J. W. Shie, U. Yogeswaran and S. M. Chen, Electroanalytical properties of cytochrome c by direct electrochemistry on multi-walled carbon nanotubes incorporated with DNA bio-composite film, Talanta, 2008, 74, 1659–1669 CrossRef CAS.
- X. Ji, J. Ren, J. Jin and T. Nakamura, A sensor for superoxide in aqueous and organic/aqueous media based on immobilized Cytochrome c on binary self-assembled maonolayers, Biosens. Bioelectron., 2007, 23, 241–247 CrossRef CAS.
- S. A. Kumar, S. F. Wang, C. T. Yeh, H. C. Lu, J. C. Yang and Y. T. Chang, Direct electron transfer of Cytochrome c and its electrocatalytic properties on multiwalled carbon nanotubes/ciprofloxacin films, J. Solid State Electrochem., 2010, 14, 2129–2135 CrossRef CAS.
- H. Liu, Y. Tian and D. Zifeng, Morphology dependent electrochemistry and electrocatalytic activity of Cytochrome c, Langmuir, 2007, 23, 9487–9494 CrossRef CAS PubMed.
- A. Fatahi, R. Malakooti and M. Shahlaei, Electrocatalytic oxidation and determination of dexamethasone at an Fe3O4/PANI–Cu(II) microsphere modified carbon ionic liquid electrode, RSC Adv., 2017, 7, 11322 RSC.
- H. X. Ju, Q. S. Liu, X. B. Ge, F. Lisdat and W. F. Scheller, Electrochemistry of cytochrome c immobilized on colloidal gold modified carbon paste electrodes and its electrocatalytic activity, Electroanalysis, 2002, 14, 141–147 CrossRef CAS.
- Y. C. Liu, S. Q. Cui, J. Zhao and Z. S. Yang, Direct electrochemistry behaviour of cytochrome c/L-Cysteine modified electrode and its electrocatalytic oxidation to nitric oxide, Bioelectrochemistry, 2007, 7, 416–420 CrossRef PubMed.
- B. Dinesha, V. Manib, R. Saraswathia and S. M. Chen, Direct electrochemistry of cytochrome c immobilized on a graphene oxide-carbon nanotube composite for pico-molar detection of hydrogen peroxide, RSC Adv., 2014, 4, 28229–28237 RSC.
- Q. Chen, S. Ali, X. Zhu, H. Yin, Q. Ma and Y. Qiu, A nitrite biosensor based on the immobilization of Cytochrome c on multi-walled carbon nanotubes–PAMAM–chitosan nanocomposite modified glass carbon electrode, Biosens. Bioelectron., 2009, 24, 2991–2996 CrossRef CAS.
- G. Zhao, Y. Lei, Y. Zhang, H. Li and M. Liu, Growth and Favorable Bioelectrocatalysis of Multishaped Nanocrystal Au in Vertically Aligned TiO2 Nanotubes for Hemoprotein, J. Phys. Chem. C, 2008, 112, 14786–14795 CrossRef CAS.
- Y. Haldorai, S. K. Hwang, A. I. Gopalan, Y. S. Huh, Y. K. Han and W. Voit, Direct electrochemistry of cytochrome c immobilized on titanium nitride/multi-walled carbon nanotube composite for amperometric nitrite biosensor, Biosens. Bioelectron., 2016, 79, 543–552 CrossRef CAS.
- W. C. A. Koh, M. A. Rahman, E. S. Choe, D. K. Lee and Y. B. Shim, A cytochrome c modified-conducting polymer microelectrode for monitoring in vivo changes in nitric oxide, Biosens. Bioelectron., 2008, 23, 1374–1381 CrossRef CAS.
- S. Nabi, F. A. Sofi, N. Rashid, P. P. Ingole and M. A. Bhat, Metal-organic framework functionalized sulfur doped graphene: a promising platform for selective and sensitive electrochemical sensing of acetaminophen, dopamine and H2O2, New J. Chem., 2022, 46, 1588–1600 RSC.
- S. Jian, X. Liu, H. Sun and S. Hou, The electrochemical studies of cytochrome c incorporated in 3D porous calcium alginate films on glassy carbon electrodes, RSC Adv., 2014, 4, 6165 RSC.
- S. Huang, M. Lu and L. Wang, Cytochrome c-multiwall carbon nanotube and cobalt metal organic framework/gold nanoparticle immobilized electrochemical biosensor for nitrite detection, RSC Adv., 2021, 11, 501–509 RSC.
- R. Geng, G. Zhao, M. Liu and M. Li, A sandwich structured SiO(2)/cytochrome c/SiO(2) on a boron-doped diamond film electrode as an electrochemical nitrite biosensor, Biomaterials, 2008, 29, 2794–2801 CrossRef CAS.
- J. Lei, N. S. Trofimova and O. Ikeda, Selective Oxidation of Nitric Oxide against Nitrite by Oxo-iron(IV) Porphyrin at an ITO Electrode, Chem. Lett., 2003, 32, 610–611 CrossRef CAS.
- E. Pashai, G. Najafpour, M. Jahanshahi, F. Yazdian and M. Rahimnejad, An electrochemical nitric oxide biosensor based on immobilized cytochrome c on a chitosan-gold nanocomposite modified gold electrode, Int. J. Biol. Macromol., 2018, 108, 250–258 CrossRef CAS PubMed.
- S. F. Ding, W. Wei and G. C. Zhao, Direct electrochemical response of cytochrome c on a room temperature ionic liquid, N-butylpyridinium tetrafluoroborate, modified electrode, Electrochem. Commun., 2007, 9, 2202–2206 CrossRef CAS.
- W. Zhang and G. Li, Third generation biosensors based on the DET of proteins, Anal. Sci., 2004, 20, 4 Search PubMed.
- G. Zhang, Z. A. Lan, L. Lin, S. Lin and X. Wang, Overall water splitting by Pt/g-C3N4 photocatalysts without sacrificial agents, Chem. Sci., 2016, 7, 3062–3066 RSC.
- Z. Peng, J. Liu, B. Hu, Y. Yang, Y. Guo, B. Li, L. Li, Z. Zhang, B. Cui, L. He and M. Du, Surface engineering on Ni-Ru Nanoalloys attached defective carbon sites as superior bifunctional electrocatalysts for overall water splitting, ACS Appl. Mater. Interfaces, 2020, 12, 13842–13851 CrossRef CAS.
- S. Zaman, L. Huang, A. I. Douka, H. Yang, B. You and B. Y. Xia, Oxygen reduction electrocatalysts towards practical fuel cells- progress and perspectives, Angew. Chem., Int. Ed., 2021, 60, 17832–17852 CrossRef CAS.
- X. Tian, X. F. Lu, B. Y. Xia and X. W. (D) Lou, Advanced electrocatalysts for ORR in energy conversion technologies, Joule, 2020, 4, 45–68 CrossRef CAS.
- K. Wandelt, Ebook, Encyclopedia of Interfacial chemistry: Surface science and electrochemistry, 2018, pp. 596–606, ISBN: 9780128098943 Search PubMed.
- A. C. Alkire, D. M. Kolb, J. Lipkowski and P. N. Ross, Bioelectrochemistry: Fundamentals, applications and recent developments, Wiley, 2013, pp. 413, Ebook. ISBN: 9783527644124 Search PubMed.
- Y. Wang, E. Laborda and R. G. Compton, Electrochemical oxidation of nitrite: Kinetic, mechanistic and analytical study by square wave voltammetry, J. Electroanal. Chem., 2012, 670, 56–61 CrossRef CAS.
- J. C. Hoogvliet, L. C. Lievense, C. V. Dijk and C. Veeger, Electron transfer between the hydrogenase from Desulfovibrio vulgaris, (Hildenborough) and Viologens, Eur. J. Biochem., 1988, 174, 281–285 CrossRef CAS.
- A. I. Gopalan, K. P. Lee and S. Komathi, Bioelectrocatalytic determination of nitrite ions based on polyaniline grafted nanodiamond, Biosens. Bioelectron., 2010, 26, 1638–1643 CrossRef CAS.
- F. Liang, M. Jia and J. Hu, Pt-implanted indium tin oxide electrodes and their amperometric sensor applications for nitrite and hydrogen peroxide, Electrochim. Acta, 2012, 75, 414–419 CrossRef CAS.
- S. Komathi, G. Sai-Anand, A. I. Gopalan, H. G. Lee, H. K. Yeo, S. W. Kang and K. P. Lee, Direct electrochemistry of cytochrome c with three-dimensional nanoarchitectured multicomponent composite electrode and nitrite biosensing, Sens. Actuators, B, 2016, 228, 737–747 CrossRef.
- M. R. Majidi and S. Ghaderi, Hydrogen bubble dynamic template fabrication of nanoporous Cu film supported by graphene nanaosheets: A highly sensitive sensor for detection of nitrite, Talanta, 2017, 175, 21–29 CrossRef CAS.
- T. Wang, X. Xu, C. Wang, Z. Li and D. Li, A Novel Highly Sensitive Electrochemical Nitrite Sensor Based on a AuNPs/CS/Ti3C2 Nanocomposite, Nanomaterials, 2022, 12, 397 CrossRef CAS PubMed.
- M. A. Rashed, M. Faisal, M. Alsaiari, S. A. Alsareii and F. A. Harraz, MWCNT-Doped Polypyrrole-Carbon Black Modified Glassy Carbon Electrode for Efficient Electrochemical Sensing of Nitrite Ions, Electrocatalysis, 2021, 12, 650–666 CrossRef CAS.
- Y. Ge, R. Jamal, R. Zhang, W. Zhang, Z. Yu, Y. Yan, Y. Liu and T. Abdiryim, Electrochemical synthesis of multilayered PEDOT/PEDOT-SH/Au nanocomposites for electrochemical sensing of nitrite, Microchim. Acta, 2020, 187, 248 CrossRef CAS PubMed.
- A. Mahmoodi, A. A. Ensafi and B. Rezaei, Fabrication of Electrochemical Sensor Based on CeO2−SnO2 Nanocomposite Loaded on Pd Support for Determination of Nitrite at Trace Levels, Electroanalysis, 2020, 32, 1025–1033 CrossRef CAS.
- C. Du, Y. Gao, H. Chen, P. Li, S. Zhu, J. Wang, Q. He and W. Chen, A Cu and Fe dual-atom nanozyme mimicking cytochrome c oxidase to boost the oxygen reduction reaction, J. Mater. Chem. A, 2020, 8, 16994–17001 RSC.
- S. Alan, A. S. Haas, D. L. Pilloud, K. S. Reddy, G. T. Babcock, C. C. Moser, J. K. Blasie and P. L. Dutton, Cytochrome c and Cytochrome c Oxidase: Monolayer Assemblies and Catalysis, J. Phys. Chem. B, 2001, 105, 11351–11362 Search PubMed.
- I. G. Denisov, T. M. Makris, S. G. Sligar and I. Schlichting, Structure and Chemistry of Cytochrome P450, Chem. Rev., 2005, 105, 2253–2278 CrossRef CAS PubMed.
- C. Cao, L. Wei, M. Su, G. Wang and J. Shen, Spontaneous bubble template assisted metal–polymeric framework derived N/Co dual-doped hierarchically porous carbon/Fe3O4 nanohybrids: Superior electrocatalyst for ORR in biofuel cells, J. Mater. Chem. A, 2016, 4, 9303–9310 RSC.
- Z. Wen, S. Ci, F. Zhang, X. Feng, S. Cui, S. Mao, S. Luo, Z. He and J. Chen, Nitrogen-enriched core-shell structured Fe/Fe3C-C nanorods as advanced electrocatalysts for oxygen reduction reaction, Adv. Mater., 2012, 24, 1399–1404 CrossRef CAS PubMed.
- E. H. Yu, S. Cheng, B. E. Logan and K. Scott, Electrochemical reduction of oxygen with iron phthalocyanine in neutral media, J. Appl. Electrochem., 2009, 39, 705–711 CrossRef CAS.
- S. Mukherjee, A. Mukherjee, A. Bhagi-Damodaran, M. Mukherjee, Y. Lu and A. Dey, A biosynthetic model of cytochrome c oxidaseas an electrocatalyst for oxygen reduction, Nat. Commun., 2015, 6, 8467 CrossRef CAS PubMed.
- H. Zhang, L. Huang, J. Chen, L. Liu, X. Zhu, W. Wu and S. Dong, Bionic design of cytochrome c oxidase-like single-atom nanozymes for oxygen reduction reaction in enzymatic biofuel cells, Nanoenergy, 2021, 83, 105798 CAS.
- M. Hanana, H. Arcostanzo, P. K. Das, M. Bouget, S. L. Gac, H. Okuno, R. Cornut, R. Jousselme, V. Dorcet, B. Boitrel and S. Campidelli, Synergic effect on oxygen reduction reaction of strapped iron porphyrins polymerized around carbon nanotubes, New J. Chem., 2018, 42, 19749 RSC.
- P. James, L. F. Collman, C. H. Paul and Z. Xumu, A Functional Model Related to Cytochrome c Oxidase and Its Electrocatalytic Four-Electron Reduction of O2, Science, 1997, 275, 949–951 CrossRef.
- H. Qinggang, W. Gang, L. Ke, K. Samson, L. Qing, M. Tawanda, D. Elise, N. Tebello and W.-C. Shaowei, Effects of Redox Mediators on the Catalytic Activity of Iron Porphyrins towards Oxygen Reduction in Acidic Media, ChemElectroChem, 2014, 1, 1508–1515 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr03451h |
| This journal is © The Royal Society of Chemistry 2024 |