
Using a cyclocarbon additive as a cyclone separator to achieve fast lithiation and delithiation without dendrite growth in lithium-ion batteries†
Jiacheng
Gong
a,
Jiabao
Zhu
a,
Xiao
He
 *ab and
Jinrong
Yang
*a
*ab and
Jinrong
Yang
*a
aShanghai Engineering Research Center of Molecular Therapeutics and New Drug Development, Shanghai Frontiers Science Center of Molecule Intelligent Syntheses, School of Chemistry and Molecular Engineering, East China Normal University, Shanghai, 200062, China. E-mail: jryang@chem.ecnu.edu.cn; xiaohe@phy.ecnu.edu.cn
bNew York University–East China Normal University Center for Computational Chemistry, New York University Shanghai, Shanghai, 200062, China
First published on 13th November 2023
Abstract
Carbon materials are widely used for reversible lithium uptake in the anode of lithium-ion batteries. Nevertheless, the challenge of uncontrollable dendrite deposition during fast charge–discharge cycles remains a grand hurdle. Various strategies have been explored to prevent detrimental heterogeneous dendrite metal deposits, such as interface engineering and electrolyte modification, but they often compromise the reverse diffusion freedom of Li+ ions during discharging and are incompatible with the most mainstream use of graphite as an anode material. Here, we propose the incorporation of a novel carbon allotrope of cyclocarbon as a potential additive in the anode. In contrast to conventional carbon materials, density functional theory calculations reveal that cyclocarbon has a much higher affinity for Li atoms than Li+ ions, even surpassing the inherent cohesion of Li atoms, due to the charge transfer from the 2s orbital of Li atoms to the unique in-plane π orbital of cyclocarbon. Furthermore, ab initio molecular dynamics simulations show that Li+ ions can shuttle freely back and forth across cyclocarbon, whereas the lithiation process for Li atoms occurs rapidly within picoseconds. The delithiation of Li atoms within cyclocarbon follows a voltage-gated mechanism that is effectively controlled by an external electric field of 3 V nm−1. Remarkably, cyclocarbon exhibits potential compatibility with commercialized graphite electrodes via the π–π interaction and also can be extended to sodium-ion and potassium-ion batteries. These distinct compatibility, scalability and electrochemical properties of cyclocarbon provide a new avenue to realize both safety and ultrafast rechargeable performance of ion batteries.
1. Introduction
Lithium-ion batteries (LIBs), whose commercialization has gained widespread adoption in eco-friendly electric vehicles, primarily utilize carbon allotrope materials as anodes1–6 ranging from graphite, porous carbon to carbon nanotubes (CNTs). These carbon-based anodes not only contribute to environmentally friendly batteries, but also offer a relatively high theoretical capacity and tunable electrical conductivity. For example, commercial LIBs using graphite-based anodes can achieve a maximum capacity of 1.1 A h,7 and a carbon nanotube–graphene hybrid fiber has a considerable electrical conductivity of about 12 S cm−1 at room temperature.8 To meet the growing demand for enhancing vehicle convenience and endurance, the charging rate needs to be boosted,9 accompanied by a mass of Li atoms that will accumulate on the surface of the graphite electrode.10 This accumulation poses a challenge of reversibility arising from uneven deposition in the form of dendrites on the electrode surface11 during the rapid charge and discharge of LIBs. The formation of dendrite structures jeopardizes the performance of LIBs by creating electrically isolated “dead” or “orphaned” Li atoms12–14 that lose their connection to the current-collecting electrode. Moreover, the continuous dendrite growth leads to internal short circuits when the anode and cathode directly contact each other by piercing through the cell separator. These defects impair cell performance such as the cycle efficiency and life15,16 and pose serious safety hazards including catastrophic risks of thermal instability.17,18 Therefore, advancing the next generation of LIBs in the commercial market hinges on effectively suppressing Li dendrite growth to achieve both high energy density and safety.In recent decades, substantial progress has been made in developing strategies to achieve more uniform Li deposition, including doped and surface functionalized electrodes,19–21 artificial solid electrolyte interphases (SEIs),22–24 highly concentrated and solid-state electrolytes25–27 and separator modification engineering.28–30 However, these strategies still face a difficult balance between delivery and capacitance. For instance, the use of a heptafluorobutyrate acid–Li SEI22 and the high-concentration lithium bis(fluorosulfonyl)imide with 1,2-dimethoxyethane electrolyte resulted in a uniform morphology, minimizing the curvature of the Li-deposited microstructure.25 Although these strategies have shown advantages in achieving more uniform Li deposition during charging, some strategies ignored the losses in the discharging process, including compromising the reversible freedom of both Li+ ion diffusion and already formed “dead” Li atoms. For example, oxygen-functionalized graphene with the abundance of carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) groups showed large binding energies for both Li+ ions and Li atoms to mitigate dendrite deposition, via a high forward Li+ ion surface diffusion rate and the assistance of Li nucleation during charging.31 But the irreversible diffusion of Li+ ions and the sacrificial “dead” Li atoms combined with oxidation groups during discharging can be seen from a decreased response current from −0.50 to −0.75 mA under a reverse scan of 1.1 V in the first cycle. This decrease is attributed to the oxidation groups, which act as powerful rivals of the graphene base in Li+ ion affinity and only offer assistance in the initial phase of Li dendrite deposition. Moreover, most of these strategies need to replace already widely used graphite electrodes with alternative materials,32,33 incurring high monetary and environmental costs. To retain the advantages of graphite anodes, carbon additives have been long sought for nondestructive modification that simultaneously realize unimpeded Li+ ion diffusion and lead to Li dendrite decomposition.
O) groups showed large binding energies for both Li+ ions and Li atoms to mitigate dendrite deposition, via a high forward Li+ ion surface diffusion rate and the assistance of Li nucleation during charging.31 But the irreversible diffusion of Li+ ions and the sacrificial “dead” Li atoms combined with oxidation groups during discharging can be seen from a decreased response current from −0.50 to −0.75 mA under a reverse scan of 1.1 V in the first cycle. This decrease is attributed to the oxidation groups, which act as powerful rivals of the graphene base in Li+ ion affinity and only offer assistance in the initial phase of Li dendrite deposition. Moreover, most of these strategies need to replace already widely used graphite electrodes with alternative materials,32,33 incurring high monetary and environmental costs. To retain the advantages of graphite anodes, carbon additives have been long sought for nondestructive modification that simultaneously realize unimpeded Li+ ion diffusion and lead to Li dendrite decomposition.
Inspired by the experimental synthesis of the novel carbon allotrope cyclocarbon34 and its large-scale production yielding up to 64%,35 we propose the use of cyclocarbon as an additive to effectively suppress dendrite deposition in LIBs without dragging Li+ ion diffusion. Through density functional theory (DFT) calculation on a set of cyclocarbon systems, including C14, C18 and C22, we found that the interaction between a Li atom and C14 exhibits the strongest adsorption, even surpassing the cohesion of the Li atom. Interestingly, in contrast to conventional carbon materials with positive cell voltages, the adsorption energy of a Li+ ion with C14 is much higher than that of a Li atom, resulting in a negative cell voltage of −1.0 V. This distinct phenomenon is attributed to the large orbital delocalization property of the unique in-plane π orbital in cyclocarbon, promoting the transfer of charge from a Li atom to the π orbital. Furthermore, ab initio molecular dynamics (AIMD) simulations demonstrate that a Li+ ion can shuttle freely back and forth across C14, and the lithiation process of the carbon ring for a Li atom can be observed within a short time frame of 4.0 ps. The potential energy reaction path associated with the voltage-assisted Li switching reveals that an electric field of 3 V nm−1 can reverse the stability of a Li atom with cyclocarbon, leading to the delithiation of the Li atom from the interior to the exterior of cyclocarbon. In the case of sodium-ion batteries (SIBs) and potassium-ion batteries (PIBs), host–guest interactions between cyclocarbon and an atom highlight the addition of C18 to facilitate the decomposition of dendrites. Additionally, cyclocarbon is compatible with commercialized graphite electrodes via the π–π interaction. Our findings suggest that cyclocarbon can act as an effective drain cleaner in regulating the lithiation and delithiation, mitigating the detrimental effects of Li dendrite deposition, thereby addressing the safety and life needs of batteries.
2. Computational details
Previous study36 has demonstrated that the level of ωB97XD/6-311+G(2d) can be enabled to accurately describe the geometry structure and electronic orbitals of cyclocarbon under an external electric field. Therefore, the same exchange–correlation functional37 and basis set38 were chosen to optimize the structure in the gas phase, implemented in the Gaussian 16 (A.03) program.39 To obtain electron density difference (EDD) maps, Multiwfn code40 was utilized based on the above geometry and wavefunction. AIMD simulations were conducted for cyclocarbon systems in the presence of alkali metal ions or atoms, calculated by using ORCA 5.0.141 software. The simulations were performed using the ωB97XD functional with a medium-sized basis set (def-TZVP)42 and a time step of 1 fs. To maintain the temperature at 300 K, the Nose–Hoover chain thermostat43 was employed.3. Results and discussion
3.1. Li dendrite deposition on carbon-based electrodes
Li dendrite deposition can occur on the surface of over-charged and fast-charged graphite electrodes, even when the charging rate is as low as 0.7 mA for 30 minutes44 or as high as 8.5 mA for only 1 minute.45 To prevent dendrite growth, previous experiments have focused on making the Li+ ion flux more uniform over surfaces and promoting uniform Li nucleation during charging.46 As depicted in Fig. 1a, the introduction of uniformly doped atoms expedites Li+ forward diffusion by increasing the adsorption with Li+ ions, surpassing that of graphene-based electrodes. But the strong affinity of these doped atoms leads to the retention of partial Li+ ions,47,48 subsequently impeding the Li+ ion reverse diffusion during discharging and effectively acting as formidable competitors against carbon-based electrodes. Conferred by ref. 47, the relatively low initial coulombic efficiency of around 50% is mainly attributed to the irreversible trapping of Li+ ions at the active sites of the graphene-like matrix. Conversely, some approaches aim to utilize evenly distributed doped atoms to facilitate the formation of uniform crystal nuclei composed of covalent compounds,49–51 while they do not adequately address the issue of pre-existing “dead” Li dendrites, which can form when anodes are rapidly charged and discharged. In essence, the interplay between carbon-based electrodes and Li atoms, which directly contributes to dendrite deposition, has been largely overlooked during both the charging and discharging processes.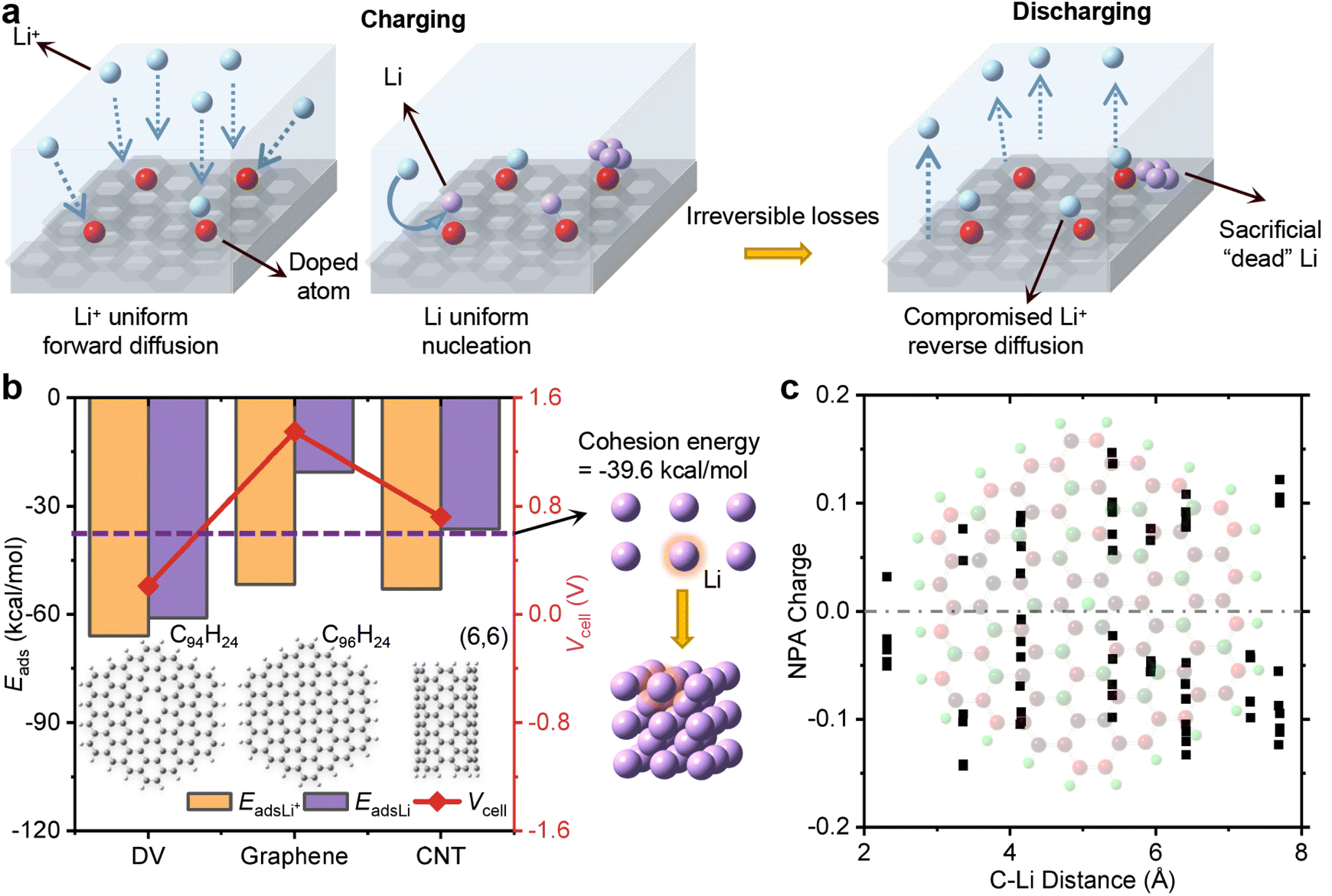 | ||
| Fig. 1 Li deposited on different carbon-based materials. (a) Advantages and disadvantages of previous strategies in preventing Li dendrite deposition. General experiments employ evenly distributed doped atoms to expedite the forward diffusion of Li+ ions (left) and aid in Li uniform nucleation (middle) during charging. However, in the process of discharging (right), the strong interaction of doped atoms would compromise the reverse diffusion of Li+ ions and cannot cope with the already formed ‘dead’ Li dendrite. (b) The formation of Li dendrite deposition on double-vacant graphene (DV), graphene and carbon nanotubes (CNTs). Dendrite deposition is explained by considering the Li+/Li adsorption energy with the cell voltage of these carbon-based materials (left) and the cohesion energy of the Li atom in a cell (middle). The value of the adsorption energy between the Li atom and carbon-based materials (EadsLi) is far lower than that between the Li+ ion and these materials (EadsLi+), exhibiting a positive cell voltage (Vcell), and the adsorption energy of the Li atom on graphene or CNTs is higher than the cohesion energy of a Li atom (purple dotted line). (c) Natural population analysis (NPA) charge of C atoms in Li@graphene. The background is the NPA charge map of Li@graphene, in which the darker the red of the atom, the more negative the charge. The negative charges in graphene transferred from the Li atom are dispersed by the presence of numerous C atoms with delocalized π orbitals, resulting in a weak electrostatic attraction between the Li atom and conventional carbon-based electrodes. | ||
To explore the difference of Li deposition with Li+ diffusion on various carbon-based electrodes, we conducted a comparative analysis of the adsorption energy of a Li atom and a Li+ ion on conventional carbon materials. These materials are graphene, defective graphene (DV) and CNTs since their function in guiding dendrite-free Li deposition as a Li storage skeleton has been widely proved.52–57 To save computing resources without being affected by the boundary effect, a hexagonal fragment of graphene consisting of 96 carbon atoms, with a sufficiently large size,58,59 was constructed with the boundary passivated with 24 hydrogen atoms (C96H24). Two connected carbon atoms can be removed to form a double-vacant graphene (C94H24).60,61 Additionally, a [6,6] carbon nanotube (CNT) saturated with 24 hydrogen atoms [CNT(6,6)], whose interaction with a Li+ ion or a Li atom has been previously calculated,62 was included for comparative and validation purposes in this work. These structures were optimized using a cost-efficient 6-311G(d) basis set38,63 since all carbon atoms exhibit sp2 hybridization and all C–C bonds maintain a uniform length,64 obviating the necessity for a more precise description. The adsorption energy is given by eqn (1):
| Eads = ELi+/Li@C − EC − ELi+/Li | (1) |
The adsorption energy and the cell voltage for Li+/Li on various carbon-based materials are illustrated in the left chart in Fig. 1b. The adsorption energies between a Li+ ion and DV, graphene, and CNT(6,6) are −66.0 kcal mol−1, −51.8 kcal mol−1 and −52.9 kcal mol−1, respectively, suggesting no significant difference among these materials due to the presence of cation–π interactions. Inversely, the adsorption energy between a Li atom and these conventional carbon materials is much higher compared with that between a Li+ ion and these materials, resulting in a positive cell voltage, in agreement with previous theoretical results (see Table S1†). In the case of DV, the strong adsorption of a Li atom is observed with a much lower value of −60.0 kcal mol−1 compared to the adsorption energy between a Li+ ion and DV. This disparity is attributed to the exposure of flaws within DV, which facilitates the formation of new chemical bonds between unpaired electrons and Li atoms.67 But the similar adsorption energies between a Li+ ion and a Li atom on DV result in a cell voltage of only 0.2 V. Graphene and CNT(6,6) systems exhibit larger cell voltages of 1.4 and 0.7 V, respectively, consistent with the previous theoretical result of 0.9 V.62 Their corresponding adsorption energies for an isolated Li atom are −20.5 and −36.4 kcal mol−1, higher than the cohesion energy of a Li atom (−39.6 kcal mol−1) as indicated by the purple dotted line in the middle of Fig. 1b. The inadequate competitiveness of conventional carbon-based materials for Li atoms can be verified again by comparing this adsorption energy with that of a Li atom bound by other Li atoms in a cluster (−34.0 kcal mol−1) in Fig. S1.† As the natural population analysis (NPA) charge distributions of graphene depicted in Fig. 1c, delocalized π orbitals contribute to the relevant weak adsorption between the Li atom and conventional carbon materials. The NPA charge of the Li atom is 0.951, indicating that it turns into a Li+ cation with a positive charge. But the negative charges gained in graphene are dispersed by the presence of numerous C atoms with delocalized π orbitals, weakening the electrostatic attraction in Li@graphene, which can be seen from the slightly high negative charge (−0.05) of the C atoms closest to the Li atom in graphene. Consequently, newly deposited Li atoms tend to accumulate on existing Li deposition rather than dispersing on the exposed surface of carbon-based electrodes.
By understanding and manipulating the adsorption properties of the Li atom on carbon-based materials, the key to controlling dendrite formation hinges on the anode additive facilitating the dispersal of Li atoms.68 Strategies aimed at enhancing the dispersal of Li atoms and preventing their preferential accumulation can help in mitigating the formation of dendrite structures. This, in turn, improves the overall performance and safety of lithium-ion batteries, enabling their successful deployment in various applications.
3.2. Host–guest interactions between cyclocarbon and Li atoms
To resolve the issue of Li dendrite deposition, it is crucial to select a suitable size of cyclocarbon for Li+/Li. In our theoretical calculations, we initially examined the geometrical structures of C14, C18 and C22, which are aromatic compounds with similar distorted ring structures of D(n/2)h symmetry,69 possessing out-plane with in-plane conjugated orbitals and high flexibility in their conjugated structures.70 The inner diameters of these cyclocarbons range from 5.7 to 9.0 Å, as depicted in Fig. 2a. As the size of the cyclocarbons increases, their structures undergo a transformation from a cumulenic ring to a polyynic ring, primarily due to symmetry breaking triggered by a second-order Jahn–Teller distortion.40 The small size of cyclocarbon structures usually tends to maintain uniform all bond angles and bond lengths. However, the overlap between the in-plane p orbitals of C14 is large to ensure a stable structure with slight alternated bond lengths but more apparent alternated angles, conforming to previous theoretical calculations.71,72 The relative bond length alternation is measured at 3.1%, while the relative bond angle alternation is 16.9% (see Fig. S2a†). Additionally, this unique structure contributes to the distribution of electrostatic potential, where most areas in C14 are positive (see Fig. S2†). Understanding these structural characteristics and the electrostatic potential is crucial in determining the interaction between the cyclocarbons and Li+/Li.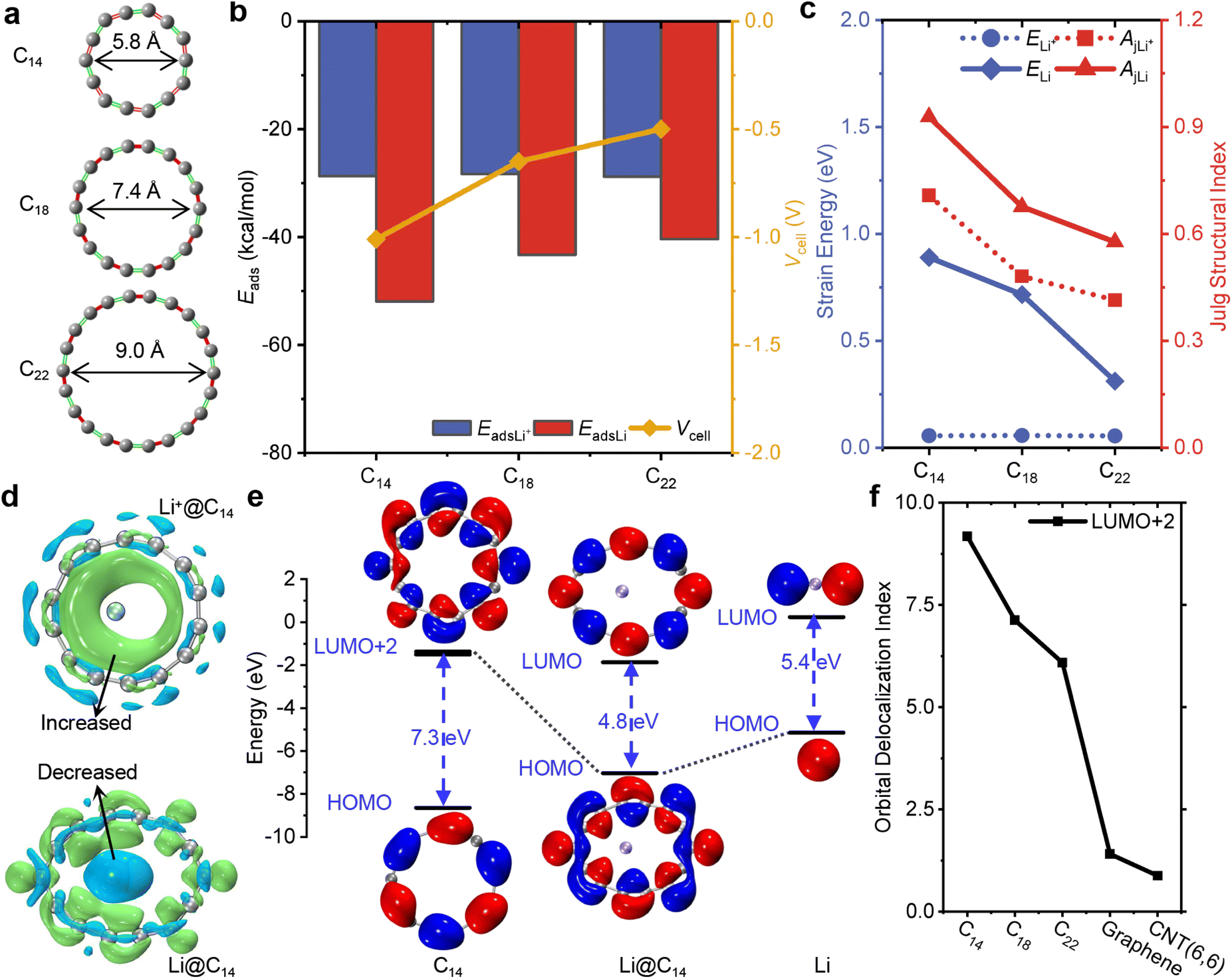 | ||
| Fig. 2 Abnormal static adsorption in Li@C14 more stable than in Li+@C14. (a) Different geometries of C14, C18 and C22. The diameter (d) of the cyclocarbon increases with an increase in the number of carbon atoms. (b) Li+/Li adsorption energies and cell voltages of C14, C18 and C22. The more negative or lower the adsorption energy, the stronger the interaction. The absolute value of adsorption energy of the Li atom in the cyclocarbon (EdasLi), distinctly decreases with the size of the ring increases, is far higher than that between the Li+ ion and the cyclocarbon (EdasLi+), exhibiting a negative cell voltage (Vcell). (c) Strain energy and the Julg structure index of the cyclocarbon in the Li+/Li@Cn complex. The larger the energy or the index, the more drastic the deformation of the cyclocarbon. The strain energy of the cyclocarbon interacting with the Li atom (ELi) sharply decreases with the size of the ring increases and is far higher than that of the carbon ring interacting with the Li+ ion (ELi+). The Julg structure index of the cyclocarbon interacting with the Li atom (AjLi) is larger than that of the carbon ring interacting with the Li+ ion (AjLi+). (d) Electron density difference maps of Li+/Li@C14. Green and blue isosurfaces (isovalue = 0.0015 a.u.) represent the regions where the electron density is increased and decreased, respectively, after complexation between Li+/Li and C14. The electron is transferred from the Li atom (green isosurfaces) to C14 (blue isosurfaces), while there is no charge transfer between the Li+ ion and C14. (e) Frontier molecular orbitals of C14, Li@C14 and Li. Red and blue regions (isovalue = 0.025 a.u.) denote the positive and negative orbital phases. The electron from the 2s orbital (HOMO) of the Li atom is transferred to the in-plane π orbitals (LUMO+2) of C14 to form the HOMO in the complex of Li@C14. (f) The orbital delocalization index (ODI) of LUMO+2 in C14, C18, C22, graphene and CNT(6,6). The ODI was calculated by the Hirshfeld orbital analysis method with built-in sphericalized atomic densities in free states, and its value can range between 0 and 100. The larger the value of the ODI, the more localized the orbital is. The ODI of LUMO+2 in the cyclocarbon is far larger than that of LUMO+2 in graphene and CNT(6,6). | ||
Furthermore, we conducted a thorough analysis of the static configurations and adsorption energy of cyclocarbon for the guest inclusion of Li+/Li (Li+/Li@Cn) to assess their stability and cell voltage. The structures of Li+/Li inside and outside the ring are shown in Fig. S3.† The adsorption energies of Li+/Li outside cyclocarbon are consistently larger than those of Li+/Li inside cyclocarbon (see Table S2†), suggesting that Li+/Li can be stabilized within cyclocarbon. Particularly, C14 exhibits a remarkable capability to redistribute and extract a Li atom from the outside, as evidenced by the adsorption energy of the Li atom outside C14 (−19.7 kcal mol−1) far larger than that of the Li atom inside C14 (−51.9 kcal mol−1). In Fig. 2b, the adsorption energies of Li+@Cn all remain at around −28.0 kcal mol−1, which is higher than that of Li@Cn, revealing that cyclocarbon shows a higher affinity for a Li atom. Meanwhile, the adsorption energy of Li@Cn experiences a notable increase from −51.9 to −41.4 kcal mol−1 as the ring size increases. Cyclocarbon enables the retention of the reversible freedom of Li+ ion diffusion on carbon-based electrodes, owing to the weaker cation–π interaction between C14 and a Li+ ion compared with that between graphene and a Li+ ion (see Fig. S4†). Also, the weak interaction mainly consolidates the self-assembly inclusion of the Li+/Li guest into the cyclocarbon host. The static attractions conform to the distribution of the electrostatic potential that the positive Li+ ion is adsorbed by the negative short bonds (see Fig. S5†). Moreover, among the series of new cyclocarbon systems, C14 possesses the largest negative voltage, exceeding −1.0 V. As a result, C14 is highly favorable for LIBs, displaying a stronger tendency to adsorb Li atoms than Li+ ions. This characteristic sets it apart from other carbon-based materials, such as graphene.
The deformation of C14 with a stretched diameter in Li@C14 is more pronounced compared with Li+@C14 (see Table S3†). This adaptive guest inclusion of flexible C14 can be attributed to its symmetry breaking with stretched geometry. To quantify the energetic difference caused by distortion, we calculated the strain energy and used the Julg structure index73 to compare the breaking of aromaticity in an asymmetric structure. The calculation details can be found in Note S2.† As shown in Fig. 2c, both the strain energy and Julg structure index of cyclocarbon in Li@Cn decrease significantly as the ring size increases, revealing that the more compact the cyclocarbon, the stronger the distortion after interacting with the guest inclusion of Li atoms. The strain energy of C14 is 0.9 eV, higher than that of C18 (0.7 eV) and C22 (0.3 eV). Similarly, the Julg structure index of C14 is also the largest (0.9) compared with that of C18 (0.7) and C22 (0.6). This trend further confirms the considerable reduction in the adsorption energy for Li@Cn. In contrast, there is no significant difference in the strain energy of cyclocarbon in Li+@Cn. The underlying mechanism of the deformation in the C14 structure is revealed by the EDD maps of the Li+/Li@C14 complex shown in Fig. 2d. The EDD map clearly reveals the transfer and reorganization of electrons from the 2s orbital of the Li atom to the in-plane π orbital of C14, conforming to an earlier theoretical calculation of charge-separated states between the guest of the Li atom and the host of the carbon ring.74 When the Li+ ion is embedded in C14, the increased electron density accumulates at the inner side of the ring because of the cation–π interaction. Therefore, the uneven distribution of electronic charge leads to structural distortion, as C14 gains partial electrons from a Li atom.
To determine the transferred electron between orbitals, Fig. 2e illustrates the highest occupied molecular orbitals (HOMOs) of C14, Li@C14 and Li, as well as the lowest unoccupied molecular orbital (LUMOs) of Li@C14 and Li, and LUMO+2 of C14. Notably, both LUMO+2 of C14 and LUMO of Li@C14 are in a similar in-plane π orbital, proving the transfer charge from the Li atom to LUMO+2 of C14 rather than its LUMO (see Fig. S6†). The energy gap between the HOMO and LUMO+2 in C14 is 7.2 eV, which decreases to 4.8 eV upon combining with the Li atom. To highlight the inherent distinction between cyclocarbon and conventional carbon materials, the degree of delocalization of orbitals is characterized by the orbital delocalization index (ODI).75 A smaller value of ODI indicates a higher degree of delocalization. As depicted in Fig. 2f, the ODI of LUMO+2 for cyclocarbon slightly decreases as the ring size increases. The ODI value of cyclocarbon is significantly greater than those of graphene and CNT(6,6), allowing them to concentrate electrons obtained from Li atoms. The ODI of LUMO+2 in C14 is 9.2, much larger than that of graphene (1.4), indicating that in-plane π orbitals in C14 are highly localized on each carbon atom. As verified by the details in Fig. S7,† the delocalized orbitals of graphene dispersed the transferred negative electrons, weakening the electrostatic attraction, in line with its weaker adsorption with a Li atom than that between C14 and the Li atom. Therefore, the C14 monomer possesses unique in-plane π orbitals with the highest degree of localization, which will not disperse negative electrons and thus exhibit the strong electrostatic attraction to the positive Li+ cation converted from an electron-losing atom, leading to strong adsorption.
3.3. Lithiation and delithiation of cyclocarbon
Building on the above static analysis, we carried out AIMD simulations to investigate the diffusion of a Li+ ion and the lithiation process of a Li atom in the presence of C14 at room temperature. The distance between the Li+ ion or the Li atom and the mass center is defined as Dc1. The shortest perpendicular distance from the Li+ ion or the Li atom to the plane of C14 is represented by Dp, where the plane is fitted by using the least-squares method, ensuring that it passes through the mass center. During the simulations, the Li+ ion approaching C14 from the outside vicinity is quickly captured and released back to the external area within picoseconds. Initially, the Li+ ion is located at a perpendicular distance of 5.6 Å under the plane of C14. As the trend lines at the bottom of Fig. 3a, Dc1 rapidly decreases from 6.2 Å to approximately 0 Å at 0.4 ps, while Dp quickly increases from −5.6 Å to 0 Å. This indicates that a Li+ ion can be effectively embedded within the carbon ring in the second snapshot at the top. Then, the Li+ ion shuttles freely across cyclocarbon and back to the ring exterior, as evidenced by different variation trends of values between Dp and Dc1. From 0.4 to 1.7 ps, Dp increases to approximately 2.0 Å and then decreases back to 0 Å, while Dc1 exhibits a wavelike pattern, increasing to 5.3 Å. Additionally, the initial placement location of the Li+ ion does not impede its free shuttle behavior (see Fig. S8†).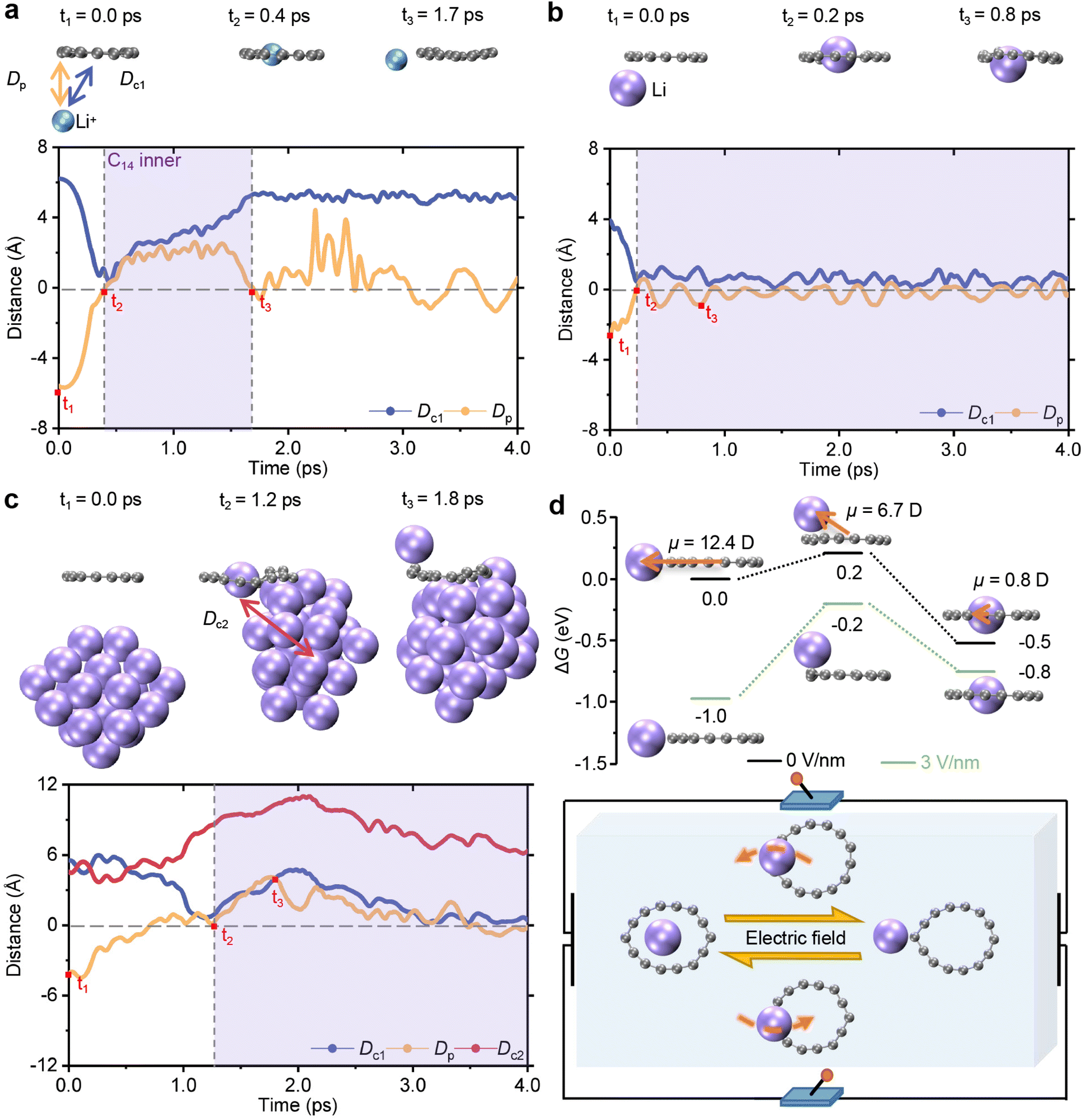 | ||
| Fig. 3 Ab initio molecular dynamics simulations for the lithiation process and the voltage-controllable delithiation pathway in C14. (a) The dynamic course of the outside Li+ ion interacting with C14, described by Dp (blue) and Dc1 (yellow). The purple region indicates that the ion or the atom is inside C14, whose Dp and Dc1 values are at around 0 Å. The outside Li+ ion can enter the inside of C14 and then free back to the outside. (b) The dynamic course of the sole outside Li atom adsorbed by C14, described by Dp (blue) and Dc1 (yellow). The outside Li atom can enter the inside of C14 and then keep moving up and down the inside area. (c) The dynamic course of the outside Li atom in a cluster adsorbed by C14, described by Dp (blue), Dc1 (yellow) and Dc2 (red). The outside Li atom in a cluster, consisting of 30 Li atoms, can be separated from other Li atoms by entering the inside of C14 and then keeps moving up and down the inside plane. (d) External Electric Field (EEF) influence on relevant free energies (ΔG) of the Li atom complex outside C14 (left), the transition state (middle) and the Li atom dimer inside C14 (right) at 298.15 K (upper) and EEF usage (under). Without an EEF (black lines), the free energy of the Li atom complex outside C14 is higher than that of the inside one. However, after adding an EEF of 3 V nm−1 (green lines), their stability is reversed with the energy of the Li atom complex inside C14 is higher than that of the outside one, subjecting to the voltage-gated mechanism. The dipole moment (μ) in the orange arrows of the Li atom dimer inside C14 is far smaller than that of the Li atom complex outside C14 whose free energy is easier to influence by the EEF. | ||
The adsorption behavior of a single Li atom and C14 was simulated to study the initial phase of Li dendrite deposition. Unlike the Li+ ion, the Li atom outside C14 can be grabbed by the ring. As illustrated by the trend lines in Fig. 3b, both the absolute values of Dp and Dc1 decrease to 0 Å and then fluctuate around the same value. Initially, the Li atom is located beneath C14, where Dp in the vertical direction is 2.5 Å with a Dc1 of 3.9 Å. Over time, both Dp and Dc1 approach 0 Å at around 0.2 ps, indicating that the Li atom has entered the interior of C14 due to the stronger adsorption of the Li atom inside C14. Subsequently, Dp and Dc1 fluctuate with minimal motion, indicating that the Li atom moves within a confined area of C14, as shown in the third snapshot above.
Furthermore, we investigated the adaptation between C14 and the Li cluster following dendrite deposition formation. In addition to Dp and Dc1, we also calculated the distance between the separated Li atom and the geometric center of the remaining Li clusters (Dc2). As shown in Fig. 3c, one Li atom in the Li cluster composed of 30 Li atoms is captured by C14 and begins moving around the inner area, as evident from the simultaneous decrease of the values for Dp and Dc1 to 0 Å. Initially, the Li atom is stably bound by the other Li atoms in a Li cluster of 30 Li atoms, exhibiting a regular crystal structure located away from the C14 ring. The absolute values of Dp and Dc1 decrease simultaneously and reach approximately 0 Å, indicating the Li atom captured by C14. Concurrently, Dc2 increases to its maximum value and then decreases before stabilizing, demonstrating that the Li atom is separated to some extent from the other Li atoms in the cluster. Subsequently, until approximately 2.0 ps, Dc2 continues to increase to 11.0 Å. Afterward, Dp and Dc1 decrease back to about 0 Å, and Dc2 slightly decreases but remains larger than the initial 5.5 Å, revealing that the separated Li will be drawn back into C14 while maintaining an appropriate degree of separation. Similarly, the initial location of the atom (see Fig. S9†) or the cluster (see Fig. S10†) does not impair the fast lithiation of C14 for a Li atom. These results demonstrate that C14 can separate one Li atom from the cluster and allow it to move around within the vicinity of the inner area of the ring instead of being rigidly fixed.
To simulate the electric field that existed in practical LIBs, an external electric field (EEF) was introduced in the simulation to examine its effect on the relevant Gibbs free energy (ΔG) among different geometries. In addition to the two stable configurations of the coplanar Li atom outside and inside C14, the transition state is also identified, in which the Li atom is bound above the carbon ring, as presented in Fig. 3d. In the absence of an EEF, the free energy of the Li atom complex inside C14 is 0.5 eV lower than that of the Li atom complex outside C14. A forward diffusion energy barrier of 0.2 eV implies that the outside Li atom can easily transform into the inside Li atom. The dipole moment of the Li atom complex outside C14 is significantly larger with 12.4 D, compared to 0.8 D of the inside one. A 3 V m−1 of EEF opposite to the orientation of the dipole moment leads to the energy of the Li atom complex outside C14 being lower than that of the inside one. A reverse diffusion energy barrier of 0.6 eV indicates the delithiation of cyclocarbon that the inside Li atom can transfer to the vicinity outside C14. Therefore, as illustrated in the sketch at the bottom of Fig. 3d, the binding site of the Li atom and C14 can be switched by the direction or value of the EEF, subjecting to the voltage-gated mechanism.
3.4. Compatibility and scalability of cyclocarbon
In current LIBs, graphene-based electrodes are widely utilized. To disperse the accumulation of Li deposition, C14 exhibits compatibility to be employed as a coating material on the surface of graphene. As depicted in the left bar diagram of Fig. 4a (see Note S3† for calculation details), C14 can be adsorbed onto the graphene surface (see Fig. S11b†). The interaction energy between C14 and graphene is −17 kcal mol−1, lower than that between C14 and C14 (−6 kcal mol−1), confirming the stable adsorption of C14. Furthermore, the presence of C14 enhances the adsorption of Li atoms (see Fig. S11a†). The interaction energy of the Li atom adsorbed by the combination of C14 and graphene is −48 kcal mol−1, lower than that adsorbed by sole C14 or graphene. The adsorption energy difference from −31 kcal mol−1 of the Li atom interacting with graphene to −43 kcal mol−1 of the Li atom interacting with C14 is consistent with the DFT result that C14 has a higher affinity for capturing the Li atom compared to graphene (see Table S4†). By coating commercial graphene anodes with C14, as shown on the right side of Fig. 4a, C14 can effectively capture dissociated Li atoms and prevent the formation of dendrite deposition, offering a potential solution for addressing the issue of lithium dendrite formation in LIBs.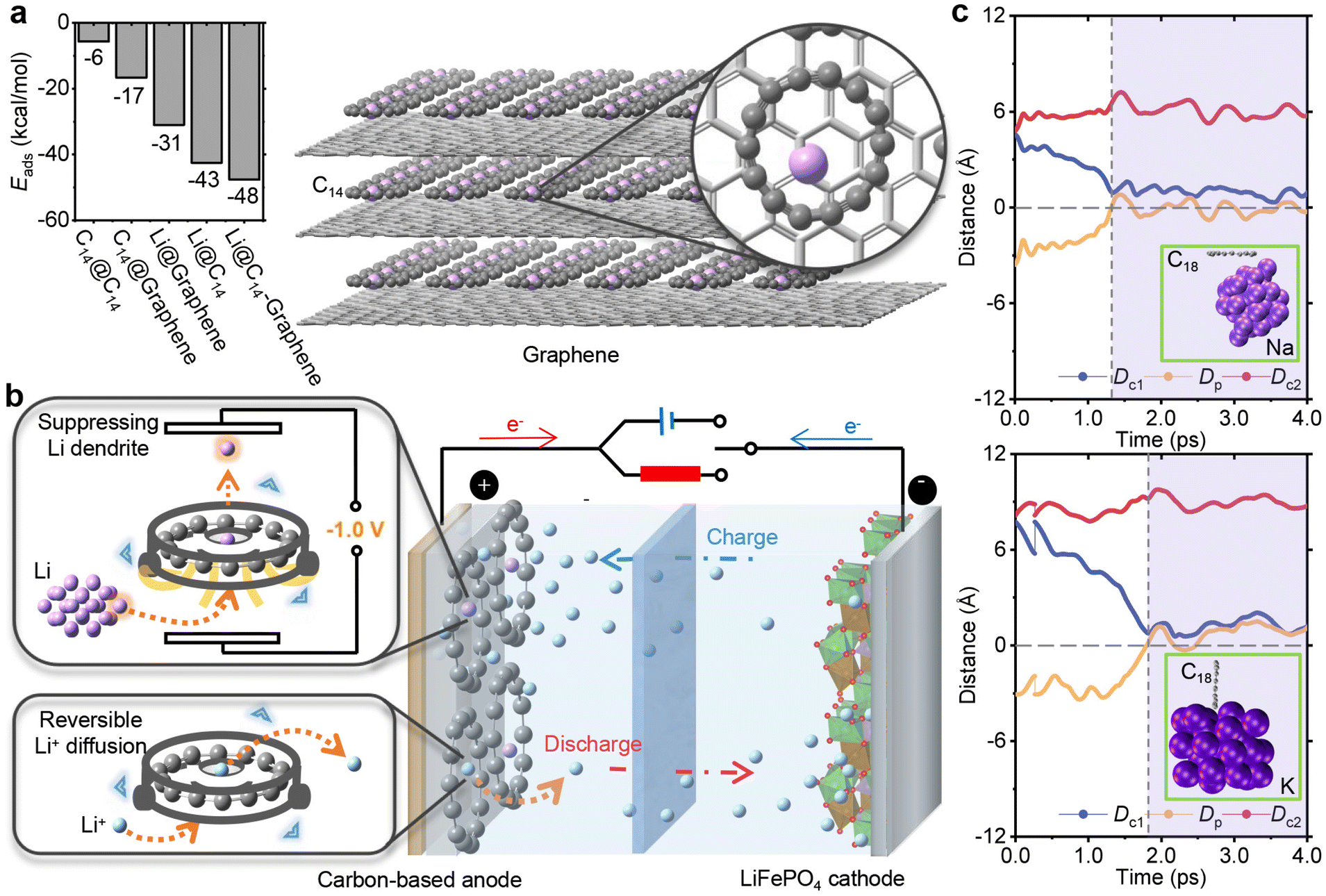 | ||
| Fig. 4 C14 acting as a cyclone separator integrated in graphite. (a) Adsorption energies of periodical C14@C14, C14@graphene, Li@graphene, Li@C14 and Li@C14-graphene (left), and the periodical structure of Li atoms adsorbed by C14 coated on graphene (right). The more negative or lower the adsorption energy, the stronger the interaction. The adsorption energy between C14 and C14 (C14@C14) is higher than that between C14 and graphene (C14@graphene), exhibiting that C14 has the potential to uniformly cover the surface of the electrode. (b) C14 acting as a deposition drain cleaner on the surface of the LIB anode. C14 exhibits a negative cell voltage of −1.0 V to decompose dendrite deposition, capturing the Li atom by lithiation. The reversible freedom of Li+ diffusion during charging and discharging would not be impeded by C14, as seen from the free shuttle of the Li+ ion across the cyclocarbon inner. (c) 4.0 ps trajectory of ab initio molecular dynamics simulations of the outside Na atom (upper) and K atom (under), interacting with C18, described by Dp (blue), Dc1 (yellow) and Dc2 (red). The outside Na or K atom in a cluster, consisting of 30 atoms, can be separated from other atoms by entering the inside of C18 and then keeps moving up and down the inside plane when the values of both Dp and Dc1 are close to 0 Å in the purple region. | ||
The diverse potential functions of C14 in LIBs are shown in Fig. 4b, highlighting its unique ability to serve as an independent mechanism for cleaning Li dendrite deposition without impeding reversible Li+ diffusion. Firstly, C14 demonstrates exceptional mobility towards the sites of Li deposition, acting as a drain cleaner for Li dendrites. As a physically coated additive on the surface, C14 can be easily applied without altering the inherent stability and capacity of graphene, serving as an undamaging pretreatment. In contrast to doping atoms such as carbonyl groups strongly binding Li+ ions,31 C14 exhibits unimpeded freedom of Li+ ion diffusion. Thereby, crucial parameters with reversibility like forward and reverse electrical conductivity,76 coulombic efficiency77 and charge–discharge rate78 can be preserved during both charging and discharging. Moreover, the use of carbon allotropes as additives in electrodes is more environmentally friendly and cost-effective compared to introducing non-carbon atoms, such as precious metals.79 The adsorption strength between a Li atom and C14 is even greater than the interaction between the outermost Li atom and other Li atoms in the cluster, enabling C14 to disassemble irregular dendrite depositions. This unique function sets C14 apart from conventional carbon-based materials like graphene. Furthermore, the processes of capturing Li atoms and releasing Li+ ions result in a significant embedding voltage of −1.0 V. The captured Li atom also possesses a flexible movement along the ring plane, allowing for the regulation of the nonplanar structure through an EEF. This gated mechanism facilitates the retention and controlled release of Li atoms, promoting the formation of harmless flat Li depositions. Overall, C14 in LIBs demonstrates exceptional capabilities as an independent Li dendrite deposition cleaning mechanic, providing mobility, selectivity and regulation while preserving the essential properties of the electrode material.
Cyclocarbon also shows a scalable application to suppress other alkali metal dendrite depositions without impeding the reversible freedom of ion diffusion. As seen from the details in ESI section S2,† C18 is better suited for SIBs and PIBs due to its optimal host–guest interactions with Na+/Na and K+/K, which exhibits the second-largest values of negative cell voltages with −0.9 V and −1.7 V. This capacity also enables that C18 does not impede the reversible Na+ and K+ ion diffusions observed from the ion freely shuttling across the carbon ring in relevant AIMD processes. Additionally, when addressing the critical issue of decomposing dendrite deposition as depicted in Fig. 4c, C18 demonstrates outstanding performance by selectively capturing only one Na or K atom while maintaining adjustable conservation or release of the atom. Incorporating into the previous theoretical calculations that C18 can act as an electron transport material,80 a regulation of switching the location of the inside atom81 and a uniform coating adsorbed onto graphene,64 C18 also exhibits outstanding electrochemical properties as C14 had. Therefore, cyclocarbon is a highly promising carbon-based additive for advanced battery technologies, exhibiting superior scalability for both SIBs and PIBs to suppress dendrite deposition.
4. Conclusions
In this work, we propose an innovative approach to effectively suppress dendrite deposition in LIBs by introducing a cyclocarbon additive. By conducting DFT calculations on various carbon-based systems, such as conventional carbon materials with graphene, double-vacant graphene, carbon nanotubes, and a series of new cyclocarbon systems, including C14, C18 and C22, we found that C14 is particularly effective in redistributing deposited Li atoms due to its lower adsorption energy of a Li atom, even surpassing the cohesion energy of the Li atom. Unlike other conventional carbon materials that typically show a positive cell voltage from 0.2 to 1.4 V because of their higher affinity for Li+ ions, C14 exhibits a negative cell voltage of −1.0 V to suppress dendrite deposition. This distinction in interaction arises from the large orbital delocalization property of unique in-plane π orbitals in cyclocarbon, which enables charge transfer from the Li atom to the π orbital, but other carbon materials possess out-plane π orbitals that form cation–π interactions with Li+ ions. Ab initio molecular dynamics simulations have demonstrated the reversible diffusion of a Li+ ion shuttling across C14 and its lithiation of a Li atom occurred within 4 ps, followed by a Li atom of oscillatory motion weaving up and down around the cyclocarbon structure. Furthermore, the potential energy profile of the diffusion path for a Li atom from the outside to the inside of cyclocarbon reveals that an external electric field of 3 V nm−1 can reverse the stability of Li with a cyclocarbon complex, suggesting the voltage-gated mechanism for delithiation within cyclocarbon.Remarkably, cyclocarbon exhibits potential compatibility with commercialized graphite electrodes via the π–π interaction and also can be extended to SIBs and PIBs. In the context of SIBs and PIBs, host–guest interactions between cyclocarbon and alkali atoms show that the addition of C18 is more favorable for decomposing dendrite deposition. These distinct compatibility, scalability and electrochemical properties of cyclocarbon hold promise for the application of cyclocarbon as a novel electrode additive to enhance the lifespan and safety of alkali metal batteries and may also extend to other metals prone to dendrite deposition. We anticipate that these results will inspire experimental chemists to explore the potential applications of cyclocarbon and further advance the field of metal dendrite suppression.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (Grant No. 2019YFA0905200), the National Natural Science Foundation of China (Grant No. 22273023, 22203032, and 21922301), the Shanghai Municipal Natural Science Foundation (Grant No. 23ZR1418200), the Shanghai Frontiers Science Center of Molecule Intelligent Syntheses, and the Fundamental Research Funds for the Central Universities. We thank the Supercomputer Center of East China Normal University (ECNU Multifunctional Platform for Innovation 001) for providing computer resources.References
- H. Kwon, J.-H. Lee, Y. Roh, J. Baek, D. J. Shin, J. K. Yoon, H. J. Ha, J. Y. Kim and H.-T. Kim, Nat. Commun., 2021, 12, 5537 CrossRef CAS PubMed.
- R. Mo, F. Li, X. Tan, P. Xu, R. Tao, G. Shen, X. Lu, F. Liu, L. Shen, B. Xu, Q. Xiao, X. Wang, C. Wang, J. Li, G. Wang and Y. Lu, Nat. Commun., 2019, 10, 1474 CrossRef PubMed.
- C. Liang, Y. Chen, M. Wu, K. Wang, W. Zhang, Y. Gan, H. Huang, J. Chen, Y. Xia, J. Zhang, S. Zheng and H. Pan, Nat. Commun., 2021, 12, 119 CrossRef CAS PubMed.
- Y. Zhou, X. Zhang, Y. Ding, L. Zhang and G. Yu, Adv. Mater., 2020, 32, 2005763 CrossRef CAS.
- J. C. Abrego-Martinez, Y. Wang, V. Vanpeene and L. Roué, Carbon, 2023, 209, 118004 CrossRef CAS.
- I. Zeferino González, H.-C. Chiu, R. Gauvin, G. P. Demopoulos, M. Miki-Yoshida, A. M. Valenzuela-Muñiz and Y. Verde-Gómez, Carbon, 2022, 199, 486–496 CrossRef.
- S. Schindler, M. Bauer, M. Petzl and M. A. Danzer, J. Power Sources, 2016, 304, 170–180 CrossRef CAS.
- H. Cheng, Z. Dong, C. Hu, Y. Zhao, Y. Hu, L. Qu, N. Chen and L. Dai, Nanoscale, 2013, 5, 3428 RSC.
- C.-Y. Wang, T. Liu, X.-G. Yang, S. Ge, N. V. Stanley, E. S. Rountree, Y. Leng and B. D. McCarthy, Nature, 2022, 611, 485–490 CrossRef CAS PubMed.
- Z. M. Konz, B. M. Wirtz, A. Verma, T.-Y. Huang, H. K. Bergstrom, M. J. Crafton, D. E. Brown, E. J. McShane, A. M. Colclasure and B. D. McCloskey, Nat. Energy, 2023, 8, 450–461 CrossRef CAS.
- M. J. Zachman, Z. Tu, S. Choudhury, L. A. Archer and L. F. Kourkoutis, Nature, 2018, 560, 345–349 CrossRef CAS PubMed.
- C. Fang, J. Li, M. Zhang, Y. Zhang, F. Yang, J. Z. Lee, M.-H. Lee, J. Alvarado, M. A. Schroeder, Y. Yang, B. Lu, N. Williams, M. Ceja, L. Yang, M. Cai, J. Gu, K. Xu, X. Wang and Y. S. Meng, Nature, 2019, 572, 511–515 CrossRef CAS PubMed.
- P. P. Paul, E. J. McShane, A. M. Colclasure, N. Balsara, D. E. Brown, C. Cao, B. R. Chen, P. R. Chinnam, Y. Cui, E. J. Dufek, D. P. Finegan, S. Gillard, W. Huang, Z. M. Konz, R. Kostecki, F. Liu, S. Lubner, R. Prasher, M. B. Preefer, J. Qian, M. T. F. Rodrigues, M. Schnabel, S. B. Son, V. Srinivasan, H. G. Steinrück, T. R. Tanim, M. F. Toney, W. Tong, F. Usseglio-Viretta, J. Wan, M. Yusuf, B. D. McCloskey and J. Nelson Weker, Adv. Energy Mater., 2021, 11, 2100372 CrossRef CAS.
- F. Liu, R. Xu, Y. Wu, D. T. Boyle, A. Yang, J. Xu, Y. Zhu, Y. Ye, Z. Yu, Z. Zhang, X. Xiao, W. Huang, H. Wang, H. Chen and Y. Cui, Nature, 2021, 600, 659–663 CrossRef CAS PubMed.
- C. Jin, T. Liu, O. Sheng, M. Li, T. Liu, Y. Yuan, J. Nai, Z. Ju, W. Zhang, Y. Liu, Y. Wang, Z. Lin, J. Lu and X. Tao, Nat. Energy, 2021, 6, 378–387 CrossRef CAS.
- Y. Xiang, M. Tao, G. Zhong, Z. Liang, G. Zheng, X. Huang, X. Liu, Y. Jin, N. Xu, M. Armand, J.-G. Zhang, K. Xu, R. Fu and Y. Yang, Sci. Adv., 2021, 7, eabj3423 CrossRef CAS PubMed.
- X. Liu, L. Yin, D. Ren, L. Wang, Y. Ren, W. Xu, S. Lapidus, H. Wang, X. He, Z. Chen, G.-L. Xu, M. Ouyang and K. Amine, Nat. Commun., 2021, 12, 4235 CrossRef CAS PubMed.
- Y. Ye, L.-Y. Chou, Y. Liu, H. Wang, H. K. Lee, W. Huang, J. Wan, K. Liu, G. Zhou, Y. Yang, A. Yang, X. Xiao, X. Gao, D. T. Boyle, H. Chen, W. Zhang, S. C. Kim and Y. Cui, Nat. Energy, 2020, 5, 786–793 CrossRef CAS.
- A. Jamaluddin, Y.-Y. Sin, E. Adhitama, A. Prayogi, Y.-T. Wu, J.-K. Chang and C.-Y. Su, Carbon, 2022, 197, 141–151 CrossRef CAS.
- Z. Ju, J. Nai, Y. Wang, T. Liu, J. Zheng, H. Yuan, O. Sheng, C. Jin, W. Zhang, Z. Jin, H. Tian, Y. Liu and X. Tao, Nat. Commun., 2020, 11, 488 CrossRef CAS PubMed.
- Z. Liang, K. Yan, G. Zhou, A. Pei, J. Zhao, Y. Sun, J. Xie, Y. Li, F. Shi, Y. Liu, D. Lin, K. Liu, H. Wang, H. Wang, Y. Lu and Y. Cui, Sci. Adv., 2019, 5, eaau5655 CrossRef CAS.
- Y. Xie, Y. Huang, Y. Zhang, T. Wu, S. Liu, M. Sun, B. Lee, Z. Lin, H. Chen, P. Dai, Z. Huang, J. Yang, C. Shi, D. Wu, L. Huang, Y. Hua, C. Wang and S. Sun, Nat. Commun., 2023, 14, 2883 CrossRef CAS PubMed.
- X. Sun, S. Yang, T. Zhang, Y. Shi, L. Dong, G. Ai, D. Li and W. Mao, Nanoscale, 2022, 14, 5033–5043 RSC.
- D. Luo, L. Zheng, Z. Zhang, M. Li, Z. Chen, R. Cui, Y. Shen, G. Li, R. Feng, S. Zhang, G. Jiang, L. Chen, A. Yu and X. Wang, Nat. Commun., 2021, 12, 186 CrossRef CAS PubMed.
- Y. Chen, M. Li, Y. Liu, Y. Jie, W. Li, F. Huang, X. Li, Z. He, X. Ren, Y. Chen, X. Meng, T. Cheng, M. Gu, S. Jiao and R. Cao, Nat. Commun., 2023, 14, 2655 CrossRef CAS PubMed.
- R. Weber, M. Genovese, A. J. Louli, S. Hames, C. Martin, I. G. Hill and J. R. Dahn, Nat. Energy, 2019, 4, 683–689 CrossRef CAS.
- T. Ma, Y. Ni, Q. Wang, W. Zhang, S. Jin, S. Zheng, X. Yang, Y. Hou, Z. Tao and J. Chen, Angew. Chem., Int. Ed., 2022, 61, e202207927 CrossRef CAS.
- C. Li, S. Liu, C. Shi, G. Liang, Z. Lu, R. Fu and D. Wu, Nat. Commun., 2019, 10, 1363 CrossRef PubMed.
- Y. Ma, F. Wu, N. Chen, Y. Ma, C. Yang, Y. Shang, H. Liu, L. Li and R. Chen, Chem. Sci., 2022, 13, 9277–9284 RSC.
- X. Luo, X. Lu, Y. Chen, X. Chen, H. Guo, C. Song, N. Wang, D. Su, G. Wang, L. Cui and Y. Liu, Mater. Adv., 2020, 1, 3449–3459 RSC.
- Y. Yu, W. Huang, X. Song, W. Wang, Z. Hou, X. Zhao, K. Deng, H. Ju, Y. Sun, Y. Zhao, Y.-C. Lu and Z. Quan, Electrochim. Acta, 2019, 294, 413–422 CrossRef CAS.
- D. Lin, Y. Liu, Z. Liang, H. W. Lee, J. Sun, H. Wang, K. Yan, J. Xie and Y. Cui, Nat. Nanotechnol., 2016, 11, 626–632 CrossRef CAS PubMed.
- W. Zou, Q. Li, Z. Zhu, L. Du, X. Cai, Y. Chen, G. Zhang, S. Hu, F. Gong, L. Xu and L. Mai, Nanoscale, 2021, 13, 3027–3035 RSC.
- K. Kaiser, L. M. Scriven, F. Schulz, P. Gawel, L. Gross and H. L. Anderson, Science, 2019, 365, 1299–1301 CrossRef CAS.
- L. M. Scriven, K. Kaiser, F. Schulz, A. J. Sterling, S. L. Woltering, P. Gawel, K. E. Christensen, H. L. Anderson and L. Gross, J. Am. Chem. Soc., 2020, 142, 12921–12924 CrossRef CAS PubMed.
- T. Lu and Q. Chen, ChemPhysChem, 2021, 22, 386–395 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615 RSC.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian16, Revision A.03, Gaussian Inc., Wallingford CT, 2016.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- G. J. Martyna, M. L. Klein and M. Tuckerman, J. Chem. Phys., 1992, 97, 2635–2643 CrossRef.
- W. Lu, C. M. López, N. Liu, J. T. Vaughey, A. Jansen and D. W. Dennis, J. Electrochem. Soc., 2012, 159, A566–A570 CrossRef CAS.
- H. Honbo, K. Takei, Y. Ishii and T. Nishida, J. Power Sources, 2009, 189, 337–343 CrossRef CAS.
- J. W. Choi and D. Aurbach, Nat. Rev. Mater., 2016, 1, 16013 CrossRef CAS.
- J. Xu, J. Mahmood, Y. Dou, S. Dou, F. Li, L. Dai and J.-B. Baek, Adv. Mater., 2017, 29, 1702007 CrossRef PubMed.
- X. Shen, Y. Li, T. Qian, J. Liu, J. Zhou, C. Yan and J. B. Goodenough, Nat. Commun., 2019, 10, 900 CrossRef.
- P. Zhai, T. Wang, W. Yang, S. Cui, P. Zhang, A. Nie, Q. Zhang and Y. Gong, Adv. Energy Mater., 2019, 9, 1804019 CrossRef.
- C. Chen, J. Guan, N. W. Li, Y. Lu, D. Luan, C. H. Zhang, G. Cheng, L. Yu and X. W. D. Lou, Adv. Mater., 2021, 33, 2100608 CrossRef CAS.
- B. Tian, Z. Huang, X. Xu, X. Cao, H. Wang, T. Xu, D. Kong, Z. Zhang, J. Xu, J. Zang, X. Li and Y. Wang, J. Mater. Sci. Technol., 2023, 132, 50–58 CrossRef CAS.
- D. Lin, Y. Liu, Z. Liang, H.-W. Lee, J. Sun, H. Wang, K. Yan, J. Xie and Y. Cui, Nat. Nanotechnol., 2016, 11, 626–632 CrossRef CAS PubMed.
- D. Han, X. Wang, Y.-N. Zhou, J. Zhang, Z. Liu, Z. Xiao, J. Zhou, Z. Wang, J. Zheng, Z. Jia, B. Tian, J. Xie, Z. Liu and W. Tang, Adv. Energy Mater., 2022, 12, 2201190 CrossRef CAS.
- N. Kim, H. Cha, S. Chae, T. Lee, Y. Lee, Y. Kim, J. Sung and J. Cho, Energy Environ. Sci., 2023, 16, 2505–2517 RSC.
- W. Deng, X. Zhou, Q. Fang and Z. Liu, Adv. Energy Mater., 2018, 8, 1703152 CrossRef.
- Y. Zhang, B. Liu, E. Hitz, W. Luo, Y. Yao, Y. Li, J. Dai, C. Chen, Y. Wang, C. Yang, H. Li and L. Hu, Nano Res., 2017, 10, 1356–1365 CrossRef CAS.
- Z. Y. Wang, Z. X. Lu, W. Guo, Q. Luo, Y. H. Yin, X. B. Liu, Y. S. Li, B. Y. Xia and Z. P. Wu, Adv. Mater., 2021, 33, 2006702 CrossRef CAS PubMed.
- S. Sangavi, N. Santhanamoorthi and S. Vijayakumar, Mol. Phys., 2018, 117, 462–473 CrossRef.
- S. Shanmugam, S. Nachimuthu and V. Subramaniam, J. Mater. Sci., 2020, 55, 5920–5937 CrossRef CAS.
- Y. Zhou, W. Chu, F. Jing, J. Zheng, W. Sun and Y. Xue, Appl. Surf. Sci., 2017, 410, 166–176 CrossRef CAS.
- D. Datta, J. Li, N. Koratkar and V. B. Shenoy, Carbon, 2014, 80, 305–310 CrossRef CAS.
- S. Gao, G. Shi and H. Fang, Nanoscale, 2016, 8, 1451–1455 RSC.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS.
- T. Lu, Z. Liu and Q. Chen, Mater. Sci. Eng., B, 2021, 273, 115425 CrossRef CAS.
- M. K. Aydinol, A. F. Kohan, G. Ceder, K. Cho and J. Joannopoulos, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 1354–1365 CrossRef CAS.
- G. Ceder, M. K. Aydinol and A. F. Kohan, Comput. Mater. Sci., 1997, 8, 161–169 CrossRef CAS.
- Y. Okamoto, J. Phys. Chem. C, 2016, 120, 14009–14014 CrossRef CAS.
- Y. Xu, H. Zheng, H. Yang, Y. Yu, J. Luo, T. Li, W. Li, Y.-N. Zhang and Y. Kang, Nano Lett., 2021, 21, 8664–8670 CrossRef CAS.
- T. Torelli and L. Mitas, Phys. Rev. Lett., 2000, 85, 1702–1705 CrossRef CAS.
- Z. Liu, T. Lu and Q. Chen, Carbon, 2020, 165, 468–475 CrossRef CAS.
- M. Li, Z. Gao, Y. Han, Y. Zhao, K. Yuan, S. Nagase, M. Ehara and X. Zhao, Phys. Chem. Chem. Phys., 2020, 22, 4823–4831 RSC.
- N. D. Charistos and A. Muñoz-Castro, Phys. Chem. Chem. Phys., 2020, 22, 9240–9249 RSC.
- A. Julg and P. Franois, Theor. Chim. Acta, 1967, 8, 249–259 CrossRef CAS.
- Y. F. Yang and L. S. Cederbaum, Angew. Chem., Int. Ed., 2021, 60, 16649–16654 CrossRef CAS PubMed.
- T. Lu, Multiwfn Manual, version 3.7(dev), Section 4.8.5, available at https://sobereva.com/multiwfn (accessed 2022) Search PubMed.
- M. Chintapalli, K. Timachova, K. R. Olson, S. J. Mecham, D. Devaux, J. M. DeSimone and N. P. Balsara, Macromolecules, 2016, 49, 3508–3515 CrossRef CAS.
- J. Xiao, Q. Li, Y. Bi, M. Cai, B. Dunn, T. Glossmann, J. Liu, T. Osaka, R. Sugiura, B. Wu, J. Yang, J.-G. Zhang and M. S. Whittingham, Nat. Energy, 2020, 5, 561–568 CrossRef CAS.
- R. Tian, S.-H. Park, P. J. King, G. Cunningham, J. Coelho, V. Nicolosi and J. N. Coleman, Nat. Commun., 2019, 10, 1933 CrossRef.
- S. Zhang, S. Xiao, D. Li, J. Liao, F. Ji, H. Liu and L. Ci, Energy Storage Mater., 2022, 48, 172–190 CrossRef.
- L. Zhang, H. Li, Y. P. Feng and L. Shen, J. Phys. Chem. Lett., 2020, 11, 2611–2617 CrossRef CAS.
- Z. Liu, X. Wang, T. Lu, A. Yuan and X. Yan, Carbon, 2022, 187, 78–85 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr04649d |
| This journal is © The Royal Society of Chemistry 2024 |