
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIsolation, biological activity, and synthesis of isoquinoline alkaloids†
Xiaorong
Yang
ab,
Xiaolou
Miao
ab,
Lixia
Dai
ac,
Xiao
Guo
d,
Janar
Jenis
e,
Jiyu
Zhang
ab and
Xiaofei
Shang
 *abcd
*abcd
aKey Laboratory of Veterinary Pharmaceutical Development of Ministry of Agriculture, Key Laboratory of New Animal Drug Project, Lanzhou Institute of Husbandry and Pharmaceutical Sciences, Chinese Academy of Agricultural Sciences, Lanzhou 730050, Gansu Province, PR China. E-mail: shangxiaofei@caas.cn; Tel: (+86) 931-2115262
bChina-Kazakh Joint Research Center for Natural Veterinary Drug, Lanzhou 730050, P. R. China
cCollege of Veterinary Medicine, Gansu Agricultural University, Lanzhou 730070, China
dTibetan Medicine Research Center of Qinghai University, Qinghai University Tibetan Medical College, Qinghai University, Xining 810016, P. R. China
eThe Research Center for Medicinal Plants, Al-Farabi Kazakh National University, Almaty 050040, Kazakhstan
First published on 2nd October 2024
Abstract
Covering: 2019 to 2023
Isoquinoline alkaloids, an important class of N-based heterocyclic compounds, have attracted considerable attention from researchers worldwide. To follow up on our prior review (covering 2014–2018) and present the progress of this class of compounds, this review summarizes and provides updated literature on novel isoquinoline alkaloids isolated during the period of 2019–2023, together with their biological activity and underlying mechanisms of action. Moreover, with the rapid development of synthetic modification strategies, the synthesis strategies of isoquinoline alkaloids have been continuously optimized, and the total synthesis of these classes of natural products is reviewed critically herein. Over 250 molecules with a broad range of bioactivities, including antitumor, antibacterial, cardioprotective, anti-inflammatory, neuroprotective and other activities, are isolated and discussed. The total synthesis of more than nine classes of isoquinoline alkaloids is presented, and thirteen compounds constitute the first total synthesis. This survey provides new indications or possibilities for the discovery of new drugs from the original naturally occurring isoquinoline alkaloids.
 Xiaorong Yang | Xiaorong Yang received his BS in Applied Chemistry from Tiangong University in 2012 and PhD in Organic Chemistry from Lanzhou University in 2020. He is currently a assistant professor of Lanzhou Institute of Husbandry and Pharmaceutical Sciences, CAAS. His research interests include the methodological study of visible light catalysis, structural modification of bioactive compounds from natural products. He has published about 13 articles, applied for more than 10 patents and received several projects. |
 Xiaolou Miao | Xiaolou Miao received his BS in Pharmacy of Shenyang Pharmaceutical University in 1996. He is currently a associate professor at the Key Laboratory of Veterinary Pharmaceutical Development of Ministry of Agriculture, Lanzhou Institute of Husbandry and Pharmaceutical Sciences, CAAS. His research interests include the design and synthesis of bioactive compounds and natural products, as well as the discovery of antiparasitic lead compounds. He has published about 30 articles, applied for more than 10 patents and received several projects. |
 Lixia Dai | Lixia Dai received her BS degree in Animal science from Gansu Agricultural University in 2013 and her MS degree in Animal nutrition and feed science from Northwest Agriculture and Forestry University in 2016, and she is currently a PhD student in College of Veterinary Medicine of Gansu Agricultural University. Her research interests include the discovery and mechanism of action of anti parasitic lead compounds, and the comprehensive utilization of non medicinal parts of medicinal materials. |
 Xiao Guo | Xiao Guo received her BS degree in Bioscience from Northwest Normal University in 2010 and her MS degree in Plant sciences from Northwest Normal University in 2013, and her PhD degree in Veterinary Science from Chinese Academy of Agricultural Science in 2017. Now she is currently an associate professor of Tibetan Medicine College in Qinghai University. Her research interests include the isolation, structural elucidation, and bioactivity of natural products. |
 Janar Jenis | Janar Jenis received her BS in applied chemistry from Xinjiang University in 1999 and her PhD in organo-metalic chemistry, catalyze and plant chemistry from Al-Farabi Kazakh National University in 2009. She is currently a Professor at Al-Farabi Kazakh National University. Her research interests include the isolation and identify of bioactive compounds from natural source. She has published more than 80 research articles, applied for more than 10 patents, and directed several projects. |
 Jiyu Zhang | Jiyu Zhang received his BS in veterinary science from Beijing Agricultural University in 1991 and his PhD in basic veterinary medicine from Jilin University in 2002. He is currently a Professor at Lanzhou Institute of Husbandry and Pharmaceutical Sciences, CAAS. His research interests include the design and synthesis of bioactive compounds as veterinary medicines, as well as drug resistance and safety evaluation. He has published more than 100 research articles, applied for more than 30 patents, and directed several projects. |
 Xiaofei Shang | Xiaofei Shang received his BS degree in Bioscience from Londong University in 2007, his MS degree in Biochemistry from Lanzhou University in 2010, and his PhD degree in Medicinal Chemistry Biology from Lanzhou University in 2019. He worked as a postdoctoral scholar in Beijing Youan Hospital of Capital Medical University in 2020–2023. He is currently a professor of Lanzhou Institute of Husbandry and Pharmaceutical Sciences, CAAS. His research interests include the isolation, structural elucidation, and structural modification of bioactive compounds from natural products. He has published about 50 articles, applied for more than 10 patents and received several projects, including the National Youth Talent Support Program, National Key Research and Development Program of China. |
1. Introduction
Isoquinoline alkaloids, an important class of N-heterocyclic bioactive natural products, are common throughout living organisms, predominantly in the plant kingdom.1 Derived from tyrosine or phenylalanine building blocks, isoquinoline alkaloids are thought to be highly conserved metabolites in ancient vascular plants at the chemotaxonomic level.2,3 Since the first bioactive isoquinoline alkaloid and nitrogen-containing natural product, morphine, was isolated from the opium plant Papaver somniferum in the early 19th century,4 increasing numbers of isoquinoline alkaloids, such as benzylisoquinolines, aporphines, berberines, protopines, etc., have been identified from natural sources over the past 200 years, resulting in a wide range of structural diversity (Fig. 1).5–8 Most of these compounds and their derivatives exhibit significant bioactivities, such as antitumor, antidiabetic, metabolic, anti-inflammatory, antibacterial, antiparasitic, cardioprotective, neuroprotective and other effects (Fig. 1).9–11 | ||
| Fig. 1 The structural diversity and bioactivities of isoquinoline alkaloids in nature source. | ||
Although there is a long history and large gap between pharmacological research and the clinical use of natural products, isoquinoline alkaloids have high probabilities of success in the drug discovery and development process.12 Many of them and their derivatives are used as pharmaceutical drugs to treat various diseases,13–16 such as the analgesic morphine, the antitussive codeine,13 the antibacterial berberine, the antihuntingtonian tetrabenazine,14 the antirheumatic sinomenine,15 the muscle relaxant agent cisatracurium,14 and the antiparkinsonian apomorphine.14 In the past five years, duvelisib, a PI3K inhibitor approved by the FDA, has been used to treat adult patients with relapsed or refractory CLL/SLL;17 roxadustat, which is approved in China, has been used to treat anaemia;18 and tenapanor, which was approved by the FDA in 2019, has been used to treat constipated irritable bowel syndrome (IBS-C). Recently, lurbinectedin and ensifentrine have also been approved to treat adult patients with metastatic small cell lung cancer (SCLC) disease progression after platinum-based chemotherapy and chronic obstructive pulmonary disease (COPD), respectively. SJ733 and JNJ-74856665 have advanced into the phase 1a/b trials, showing antimalarial efficacy and anti-acute myeloid leukemia (AML) activity, respectively (Fig. 2).19 Moreover, isoquinoline alkaloids from Macleaya cordata exhibit growth-promoting effect, and are widely applied in agricultural fields.20 Therefore, the search for novel isoquinolines as promising drug leads remains an active area in natural product chemistry.
 | ||
| Fig. 2 The development history of some commercially isoquinoline alkaloids and their analogs. | ||
In view of the importance and significant biological activities of isoquinoline alkaloids, thousands of publications have been released over the past 200 years. We previously reviewed the developments in this field from the perspective of biological activities (covering 2014–2018).6 Over the past five years, along with novel technology applications, many new studies have been widely performed, and more new alkaloids have been isolated from plants and microorganisms as new leads in the discovery of useful chemotherapeutic agents. Owing to their wide structural diversity and biological activity, isoquinoline alkaloids have always been an important synthetic target for organic synthesis, and chemical synthesis has made considerable progress in the past five years.14 Moreover, their novel pharmacological activities and comprehensive mechanisms of action have been explored. In 2020, 242 publications were released in the Scopus database, and 105 articles were published in the areas of pharmacology, toxicology, and pharmaceutics.9 The increasing number of publications reflects the research intensity of this class of compounds, as well as their importance for drug development. A more comprehensive and up-to-date review is merited.
To show the progress of this class of compounds from 2019 to 2023, this review covers for two aspects, (i) the chemical structures and biological activities of newly isolated isoquinoline alkaloids; and (ii) the updated total synthesis and biosynthesis of this class of natural products and the application of new synthesis methodologies and strategies. We hope that this review provides new clues for the discovery of new drugs from naturally occurring isoquinoline alkaloids.
2. Structures and bioactivities of isolated isoquinoline alkaloids
In the past five years, in addition to classical phytochemical methods, modern chromatographic technologies have been widely used to identify isoquinoline alkaloids. A total of 253 new compounds with varied chemical spaces and structural diversities have been isolated and identified from microorganisms and plants, which are present mainly in primitive angiosperms of the families Ranunculaceae, Berberidaceae, Papaveraceae, and Fumariaceae (Table S1†).2.1. Simple isoquinoline alkaloids
Simple isoquinoline alkaloids are distributed mainly in the genera Papaver, Corydalis, Thalictrum and others, and thirty of these kinds of compounds (1–30) were isolated from marine microorganisms and plants from 2019 to the early of 2023 (Fig. 3 and 4). | ||
| Fig. 3 The chemical structure of compounds 1–16. | ||
 | ||
| Fig. 4 The chemical structure of compounds 17–30. | ||
In 2019, a new simple isoquinoline alkaloid, mucroniferanine M (1) was isolated from Corydalis mucronifera.21 In another study conducted in the following year, a new antiviral isoquinoline alkaloid, 1-(6-hydroxy-7-methylisoquinolin-1-yl) ethanone (2) from Thalictrum glandulosissimum, was found to have prominent anti-tobacco mosaic virus (TMV) activity, with an inhibition rate of 28.4% at 20 μM, which was close to that of the positive control (30.2%).22 Then, litcubanine A (3) with an unusual C-1 methyl group and an N → O group with significant anti-inflammatory activity, was subsequently isolated from Litsea cubeba.23
More chemicals were found in 2022. From the roots of Thalictrum cultratum and T. baicalense, a new simple alkaloid, dehydrothalflavine (4), was found along with other known compounds.24 A new 9-phenylisoquinoline alkaloid, (aS)-7,8-dimethoxy-9-(2-carboxy-4,5-dimethoxyphenyl)-3,4-dihydroisoquinoline-1(2H)-one (5), was isolated from the lateral roots of Aconitum carmichaelii and was shown to have cardioprotective effects against doxorubicin-induced toxicity in H9c2 cells.25 Bungeanoline D (6), as well as five known benzylisoquinoline alkaloids, namely, (S)-norjuziphine, (S)-laudanidine, (S)-reticuline and (S)-armepavine, were isolated from the whole herbs of Corydalis bungeana. Among them, (S)-reticuline has been shown to have antagonistic effect on the D2 receptor with an IC50 value of 2.04 μM![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 26 (Fig. 3).
26 (Fig. 3).
Since the first natural isoquinoline isolated from bacteria was reported by Fukum et al. in 197727 and then by Kubo et al. in 1988,28 more isoquinoline alkaloids have been found in microorganisms, particularly in marine microorganisms. In 2019, a new isoquinoline alkaloid, 6-methylisoquinoline (7), was identified from white button mushrooms (Agaricus bisporus),29 and three compounds TMC-120A (8), TMC-120B (9) and TMC-120 C (10) were isolated from Aspergillus insuetus. 8 and 9 were shown to significantly reduce PTZ-induced seizures and epileptiform brain activity.30 Three new simple isoquinolinequinones containing a 5,8-dioxo-5,8-dihydroisoquinoline moiety, namely, methyl O-demethylrenierate (11), O-demethylrenierol (12), and 1,6-dimethyl-7-hydroxy-5,8-dihydroisoquinoline-5,8-dione (13), were isolated from the sponge Haliclona sp.31 Moreover, two new compounds, spathullins A (14) and B (15) were isolated from culture broths of Penicillium spathulatum Em19, and are considered promising sources for finding new antibacterial secondary metabolites. In particular, 15 was the most potent against all the tested pathogens, with an MIC as low as 1 μg mL−1 (5 μM) against S. aureus32 (Table S3†). Mansouramycin F (16), which has significant tumor selectivity against 36 tumor cell lines, was identified from the marine-derived Streptomyces sp. isolate B184833 (Fig. 3). In the past five years, in the search for find potential antitumor agents from natural products, the cytotoxicities newly isolated isoquinoline alkaloids have been commonly studied (Table S2†).
Puniceusines A–N (17–30) (Fig. 4) were subsequently isolated from a deep-sea-derived fungus Aspergillus puniceus SCSIO z021. Among them, 19 and 20 showed selective inhibitory activity against the protein tyrosine phosphatase CD45, with IC50 values of 8.4 and 5.6 μM (Table S4†), respectively; 20 had moderate cytotoxicity toward H1975 with an IC50 value of 11.0 μM (Table S2†); and 30 contained an active center –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N+, and exhibited antibacterial activity (Table S3†). The authors also reported that puniceusines C–E and H–M contained an isoquinolinyl, a polysubstituted benzyl or a pyronyl at position C-7 of the isoquinoline nucleus, and substitutions at C-7 of the isoquinoline nucleus will affect their bioactivity.34
N+, and exhibited antibacterial activity (Table S3†). The authors also reported that puniceusines C–E and H–M contained an isoquinolinyl, a polysubstituted benzyl or a pyronyl at position C-7 of the isoquinoline nucleus, and substitutions at C-7 of the isoquinoline nucleus will affect their bioactivity.34
2.2. Benzylisoquinoline alkaloids
In 2020, using HPTLC-DESI-MSn five known benzylisoquinoline alkaloids, namely, reticuline, magnocurarine, armepavine and coclaurine, were identified from Ocotea spixiana.35 Lima et al.36 identified thirty-one isoquinoline alkaloids from Annona salzmannii; however, only N,O-dimethylcoclaurine N-oxide (31) is an unprecedented alkaloid that functions as a ligand for acetylcholinesterase. Linderine A (32),37, (+)-O,O-dimethylautumnaline (33),38 and a new isoquinoline alkaloid glycoside, phellodendronoside A (34)39 were obtained from Lindera aggregata, Androcymbium palaestinum and Phellodendron chinense. In 2020 and 2022, two new compounds, hypeontine (35) and hypecocarpinine (36), were identified in Hypecoum ponticum,40,41 respectively, and compound 35 presented an inhibitory effect against Pseudomonas aeruginosa (MIC of 64.0 μg mL−1) (Fig. 5).
 | ||
| Fig. 5 The chemical structure of compounds 31–50. | ||
As noted,6 the genus Corydalis contains many benzyl isoquinoline alkaloids. In 2022, a rare N-benzyl isoquinoline alkaloid, 3,4-2H-tomentelline C (37), was isolated from C. tomentella and showed strong cytotoxicity against HepG2 cells, with an IC50 value of 7.42 μM.42 In the same year, 9-demethylmucroniferanine A (38) and hendersine B ethyl ester (39) were isolated from the Tibetan Medicine C. hendersonii; 38 presented the best anti-gastric cancer activity in vivo and in vitro through topoisomerase I activity, with IC50 values of 5.1 μM for MGC-803 cells and 7.6 μM for HGC-27 cells. Further study revealed that it attenuated proliferative capacity, caused G2/M phase arrest, inhibited cell migration and invasion, induced cell apoptosis and increased the Bax/Bcl-2 ratio.43 In addition, from C. yanhusuo, five new pairs of isoquinoline alkaloid enantiomers with a rare 9-methyl moiety, designated as yanhusanines B and D–F (39, 41–43), exhibited selective inhibitory activities against human carboxylesterase (hCE2), with IC50 values ranging from 2.0–13.2 μM (Table S4†).44 Mucroniferanines I (43) and J (44) were isolated from in C. mucronifera; however, they do not present cholinesterase inhibition.21,45 Subsequently, bracteatinine (45), which is representative of the rare 2-benzylisoquinoline alkaloid subclass and a close derivative of corgoine, was subsequently isolated from the aerial parts of C. bracteata,46 and edulisine J (46), which significantly triggers the secretion of insulin in the HIT-T15 cells at a concentration of 40 μM, was identified from C. edulis, as were edulisines C (47) and K (48).47
From Thalictrum foliolosum, Sun and Han isolated 5,6,7,12-tetramethoxy-2-methyl-13-hydroxy-11-(4′-methoxycarbonylphenoxy)-benzylisoquinoline (49) and 5,6,7,12-tetramethoxy-2-methyl-13-hydroxy-11-(4′-carbonylphenoxy)benzylisoquin-oline (50) (Fig. 5). These compounds exhibited cytotoxic activities against H460, H23, HTB-58, A549, H441, and H2170 cells, with IC50 values less than 20 μM.48 From another species, T. baicalense, a benzylisoquinoline bearing a formyl group at C-3, baicalensine B (51), was isolated and presented moderate antiproliferative activities against Caco-2 and HL-60 cells.49 Dehydrooxonorreticuline(3,4-dihydro-7-hydroxy-6-methoxy-1-isoquinoliny-l)(3-hydroxy-4-methoxyphenyl)-methanone (52) (Fig. 6) was isolated from Diclinanona calycina.50
 | ||
| Fig. 6 The chemical structure of compounds 51–72. | ||
In 2022, from the rhizomes of Menispermum dauricum, seventeen benzylisoquinoline alkaloids were isolated and named menisperdaurines A–Q (53–69). As glycosidic benzylisoquinolines, menisperdaurines A–E (53–57) have glucose moieties attached at the C-12 position. Menisperdaurine H (60) is the first example in which benzylisoquinoline and an aromatic unit are connected by a sugar bridge. Menisperdaurine A (53) exhibits the highest antagonistic activity (IC50 = 1.0 μM).51 Two aporphine-benzylisoquinoline alkaloids, (6aS,1′R)-apormenidaurine A (70) and (6aS,1′S)-apormenidaurine B (71), were also isolated from this species; the former compound exhibited potent D1 receptor antagonistic activity (IC50 = 8.4 μM).52 Additionally, a new compound, muraricine (72) (Fig. 6), was isolated from the root bark of Berberis vulgaris.53 In 2023, Chen et al.54 isolated four new alkaloids, (1R,1′R) isoliensinine N′-oxide (73), (1R,1′R) isoliensinine N-oxide (74), (1R,1′R) liensinine N′-oxide (75), and (1R,1′R) neferine N-oxide (76), as well as 10 known compounds from Plumula nelumbinis, the green embryo of a lotus seed. 76 at 0.5 μg mL−1 significantly reduced melanogenesis in α-MSH-stimulated B16F10 cells, and the tyrosinase (TYR) activity was inhibited by 78.7% at 4 μg mL−1, which was greater than that of α-arbutin (41.3%). Additionally, it exhibited superior antimelanogenic effects compared with α-arbutin in a zebrafish model probably by inhibiting key proteins involved in melanin production, such as the microphthalmia-associated transcription factors TYR, TRP-1, and TRP-2. This result indicates that 76 could be used as a potential drug for treating hyperpigmentation. Two new benzylisoquinoline alkaloids, sinotumitones A (77) and B (78), were isolated from the stems of Sinomenium acutum,55 (S)-1,2,3,4-tetrahydro-7-methoxy-8-hydroxy-2-methyl-13-methoxybenzylisoquinoline (79) was obtained from Fissistigma polyanthum and exhibited discernible AChE (IC50 of 25.6 μM) and BChE (IC50 of 33.0 μM) inhibition (Table S4†).56
In addition to muraricine, two new bisbenzylisoquinoline alkaloids, bersavine (80) and berbostrejdine (81) (Fig. 7) were isolated from B. vulgaris in 2019.53 In addition, Ali et al. isolated a novel bisbenzylisoquinoline alkaloid, 13-nitrochondrofoline (82), from another species, B. brevissima, which showed antitrypanosomal activity in vitro.57 In the same year, two new bisbenzylisoquinolines, (1R,1′S)-N-formylcepharanthine (83) and (1R,1′S)-N-formylisotetrandrine (84) were identified from Stephania cepharantha. These compounds present the significant inhibitory effects against NO production in overactivated BV2 cells with the IC50 values of 12.0 and 12.6 μM, respectively.58 In the same year, mucroniferanine L (85) the first natural amide bond-linked isoquinoline alkaloid dimer from Corydalis mucronifera, was reported.45
 | ||
| Fig. 7 The chemical structure of compounds 73–89. | ||
Bisbenzylisoquinoline alkaloids are also found in Menispermum dauricum. (1′R)-Pavermenidaurine (86), consisting of a benzylisoquinoline and a special benzylisoquinoline analog carrying an extra methylene group at C-10, was identified.52 Moreover, three bisbenzylisoquinoline alkaloids with constructed C–C bonds, (1R,1′R)-dauricisoline H (87), (1R,1′R)-dauricisoline I (88) and (1R,1′R)-dauricisoline J (89) (Fig. 7), were isolated.52
In 2020, from an aqueous extract of Corydalis yanhusuo tubers, a spirobenzylisoquinoline alkaloid with a rare 1-oxa-6-azaspiro[4.5]decane core containing an N-CHO, named groupyanhusanine A, was identified (90) (Fig. 8).44 In 2022, (±)-hypeisoxazole A (91), a racemic pair of rearranged alkaloids possessing an unprecedented diindeno [2,1-c:2′,1′-d] isoxazole scaffold, was subsequently isolated from Hypecoum erectum, which presented neuronal excitability activity in a spontaneous calcium oscillation model.59 Finally, a new alkaloid, chelidoniumine A (92), was isolated from the roots and rhizomes of Hylomecon japonica,60 and (13S,14S)-tomentelline E (93) (Fig. 8) from Corydalis tomentella showed anti-neuroinflammatory activity at 25 μM against LPS-induced BV2 microglia cells.61
 | ||
| Fig. 8 The chemical structure of compounds 90–93. | ||
2.3. Aporphine isoquinoline alkaloids
Aporphine alkaloids can be classified into subtypes, including simple aporphines, oxoaporphines, dehydroaporphines, proaporphines, and dimeric aporphinoid alkaloids.62,63 | ||
| Fig. 9 The chemical structure of compounds 94–114. | ||
In 2020, two oxide isoquinoline alkaloids, 6R,6aS-N-nantenine Nβ-oxide (102) and 6S,6aS-N-nantenine Nα-oxide (103), were subsequently isolated from the seeds of Nandina domestica.67 Six aporphine alkaloids, corybungines F–K (104–109) (Fig. 9), have been isolated from Corydalis bungeana. Among them, corybungines H presented moderate D2 antagonism in CHO-D2 cells, with an IC50 value of 9.12 μM.26 Additionally, (R)-1,2-methylenedioxy-9-methoxy-11-hydroxyaporphine (110), (6S,6aR)-N-methylcalycinine-Nα-oxide (111), and (6R,6aR)-N-methylcalycinine-Nβ-oxide (112) were also isolated from Fissistigma polyanthum.56
In 2019, dactylicapnosines A (113) and B (114) (Fig. 9), two reconstructed aporphine alkaloids with unprecedented five-membered carbon rings, were isolated from the stems of Dactylicapnos scandens.68113 exerts significant in vivo anti-inflammatory and analgesic effects by inhibiting the expression of TNF-α, IL-1β, and PGE2, whose activities are superior to those of the control drug isocorydine.
More oxoaporphines, such as 3-methoxy-2′-carbonyl-oxohernandalin (115) (from T. tenue), were isolated from the genus Thalictrum.65 Two new isoquinoline alkaloids, 9-(2′-formyl-5′,6′-dimethoxyphenoxy)-1,2,3,10-tetramethoxy oxoaporphine (116) and 3-methoxy-2′-formyloxohernandalin (117), were found in the roots of T. foetidum.64
 | ||
| Fig. 10 The chemical structure of compounds 115–123. | ||
In 2019, three new alkaloids with unprecedented oxahomoaporphine and 8-oxohomoaporphine skeletons were isolated from the bark of D. surinamensis, and named duguetinine (119), duguesuramine (120) and 11-methoxyduguesuramine (121) (Fig. 10). Among them, duguetinine has an unusual oxahomoaporphine skeleton bearing an oxepane moiety, and duguesuramine and 11-methoxyduguesuramine contain an extra carbon between the tetrahydroisoquinoline and the pendant aromatic rings.63 In addition, a newly reported cularine alkaloid (S)-2,3,12,12a-tetrahydro-5,6,9,10-tetramethoxy-1-methyl-1H-[1]benzoxepino[2,3,4-ij]-isoquinoline (122), along with four known alkaloids were isolated from the roots of Stephania cepharantha; however, they were not cytotoxic to three human cancer cell lines (A549, MCF-7, and SW480).69 In addition to three simple aporphines, an oxalyl-fused dehydroaporphine alkaloid, 8,9-dimethoxylettowianthine (123) (Fig. 10), and the known compound lettowianthine, were isolated from Fissistigma polyanthum.56
2.4. Berberine and protoberberine isoquinoline alkaloids
These kinds of compounds, such as berberine, exhibit various pharmacological effects. In the past five years, more compounds have been identified from the genus Corydalis. In 2019, mucroniferanine H (124) from Corydalis mucronifera presented the inhibitory effects on AChE and BuChE, with IC50 values of 2.31 μM and 36.71 μM, respectively (Table S4†),45 as a new compound, hendersine G (125) was isolated from the whole plant of C. hendersonii, as were other known hendersines B–F.70 In 2020, yanhusanine F (126) was identified from an aqueous extract of C. yanhusuo tubers.44 A new class of alkaloid dimer, baicalensine A (127) (Fig. 11), which contains berberine conjugated to a ring-opened isoquinoline, was isolated from Thalictrum baicalense; it presented moderate antiproliferative activities against Caco-2 and HL-60 cells with IC50 values of 9.24 and 10.15 μM, respectively, while those of positive control 5-FU were 20.24 and 3.15 μM, respectively (Table S2†).49 | ||
| Fig. 11 The chemical structure of compounds 124–150. | ||
Five new isoquinoline alkaloids, (8R,13R,14R)-8-methoxycarbonylmethyl thalictrifoline (128), (13R,14R)-13-hydroxy-13-methyl-8-oxosinactine (129), tomentelline F (130) and isotomentelline F (131) (Fig. 11) were isolated from Corydalis tomentella, and 129 showed good anti-neuroinflammatory activity.61 Additionally, 13R-tomentelline (132) was also isolated.42 Moreover, corybungines A–E (133–137) including a protoberberine-type alkaloid (133), with a unique 6-norprotoberberine skeleton (134), one 13,14-seco-protoberberine-type alkaloid containing a carboxyl group at C-12 (135), and two 1a,14-seco-protoberberine-type alkaloids with a 4-(hydroxymethyl)phenoxy moiety (136, 137) have been isolated from the whole herb extract of C. bungeana.71 Subsequently, (±)-decumicorine A (138) and (±)-epi-decumicorine A (139), two pairs of enantiomeric isoquinoline alkaloids featuring a novel phenylpropanoid-conjugated protoberberine skeleton, were isolated from the rhizomes of C. decumbens. 138 exhibited an antiviral entry effect on SARS-CoV-2 pseudovirus by blocking spike binding to the ACE2 receptor on HEK-293T-ACE2h host cells within their maximum nontoxic concentration of 100 μM.72
Dai et al.,73via the HSQC-based small molecule accurate recognition technology (SMART) strategy, discovered 19 compounds from C. saxicola in 2023, among which corydalisines A (140) and B (141) were new isoquinoline alkaloids. Unfortunately, none of them presented cytotoxicity against six human cancer cell lines. Isogroenlandicine (142), which does not affect platelet function, was isolated from C. bracteata.46 An undescribed isoquinoline alkaloid, edulisine A (143), which has a unique coupled pattern of coptisine and ferulic acid via Diels–Alder [4 + 2] cycloaddition, was isolated from C. edulis.47
Moreover, from Hypecoum leptocarpum, a new compound leptocaramine (144) along with five known compounds, leptopidine, corydamine, protopine, dihydroprotopine and oxohydrastinine, were found, which presented strong activity against A549 cells and moderate cytotoxicity against MGC-803 cells with IC50 values of 6.68 and 27.98 μg mL−1, respectively (Table S2†).41 From the rhizomes of Menispermum dauricum, five protoberberine analogs, menisperdaurines R–V (145–149) (Fig. 11), were also isolated.51 Finally, (7R,14S)-N-methylthaipetaline hydroxide (150) with a pentaoxygenated N-methyltetrahy droprotoberberine core structure was identified from Fissistigma polyanthum without anti-AChE and anti-BChE activities.56
2.5. Naphthylisoquinoline alkaloids
Naphthylisoquinolines are a group of structurally diverse secondary metabolites containing both naphthalene and isoquinoline bicyclic systems connected by a C,C or C,N biaryl axis, and dimeric representatives. These compounds presented outstanding antitumor and antiparasitic activities, especially against leishmaniasis and trypanosomiasis. Since 1970, the first C,C-coupled alkaloid, ancistrocladine, was discovered from the Indian plant Ancistrocladus heyneanus, and more than 250 structurally divergent monomeric and dimeric naphthylisoquinoline alkaloids have been identified. Most of these compounds have been isolated from only the tropical plant families Ancistrocladaceae and Dioncophyllaceae of Asian and African lianas and are classified as dioncophyllaceae-type alkaloids and ancistrocladaceae-type alkaloids. Dioncophyllaceae-type alkaloids have an R-configuration at C-3 and lack an oxygen function at C-6. Structurally similar Ancistrocladaceae-type alkaloids have been found in closely related Ancistrocladaceae plant families.8,74 Over the past two decades, extensive studies on the isolation and bioactivity evaluation of naphthylisoquinoline alkaloids have been carried out by Bringmann's group and other groups.75–88In the past five years, additional compounds have been identified from the tropical liana Ancistrocladus sp. by this group, and their antiplasmodial or cytotoxic activities have also been explored. In 2019, from the twigs and leaves of the Central African liana A. ealaensis, ten rarely unprecedented 7,8′-coupled naphthylisoquinoline alkaloids were isolated, including eight new compounds, named ealamines A–H (151–158) (Fig. 12), and two known compounds, 6-O-demethylancistrobrevine A and yaoundamine A. Among them, ealamines A–G (151–157) are the first 7,8′-linked “hybrid-type” naphthylisoquinoline alkaloids, i.e., 3R-configured and 6-oxygenated in the tetrahydroisoquinoline part, and ealamine H (158) is a typical Ancistrocladaceae-type alkaloid, with a 3S-configuration at C-3 and an oxygen function at C-6. 151–157 exhibited specific antiplasmodial activities against Plasmodium, Trypanosoma and Leishmania and displayed preferential cytotoxic effects toward PANC-1 cells; ealamine C was the most potent agent, with a PC50 value of 9.9 μM.89
 | ||
| Fig. 12 The chemical structure of compounds 151–174. | ||
Moreover, ancistrobreveines A–D (159–162) (Fig. 12) with five coupling types (5,1′, 7,1′, 7,8′, and 5,8′) were identified from A. abbreviatus. 161 is the first example of a 7,8′-linked fully dehydrogenated naphthylisoquinoline discovered in nature that is configurationally stable at the biaryl axis, and that exhibited antiproliferative activities against drug-sensitive acute lymphoblastic CCRF-CEM cells and the multidrug resistant (MDR) subline, CEM/ADR5000.90 This group subsequently discovered seven new ancistrocladaceae-type seco-naphthylisoquinoline alkaloids, ancistrosecolines A–F (163–168), along with 1-nor-8-O-demethylancistrobrevine H (169) (Fig. 12). The tetrahydroisoquinoline ring of 163–168 is cleaved, with loss of C-1, and 169 is the first naturally occurring naphthylisoquinoline lacking the otherwise generally present methyl group at C-1. Among them, ancistrosecoline D exhibited strong cytotoxicity against HeLa cells with an IC50 value of 11.2 μM.91 In addition, the first N-methylated, cationic naphthylisoquinoline alkaloid, ancistrobrevinium A (170) was discovered in the root bark extract of A. abbreviatus, and showed weak cytotoxic activity against A549 cells (IC50 = 50.6 μM) (Table S2†).92 Five related 5,8′-linked monomeric alkaloids, named ikelacongolines A–D (171–174) (Fig. 12), and two constitutionally unsymmetric dimers, mbandakamines B3 (175) and B4 (176) (Fig. 13), were identified from A. korupensis and A. ealaensis. The dimers 177 and 176 are structurally unusual quateraryls comprising two 5,8′-coupled monomers linked via a sterically strongly constrained 6′,1′′-connection between their naphthalene units.93
 | ||
| Fig. 13 The chemical structure of compounds 175–186. | ||
In 2019, a series of unusual dimeric naphthylisoquinoline alkaloids, namely, cyclombandakamine A (177), 1-epi-cyclombandakamine A (178), and cyclombandakamines A3–7 (179–183) (Fig. 13), were isolated from the leaves of the tropical liana A. ealaensis and have a chemically thrilling structural array consisting of a twisted dihydrofuran-cyclohexenone-isochromene system.94 Then, spirombandakamine A3 (184) and cyclombandakamines A8 (185) and A9 (186) (Fig. 13), three polycyclic naphthylisoquinoline dimers, along with mbandakamines C and D, were found in the leaves of a botanically as of yet unidentified Congolese Ancistrocladus plant, which is morphologically closely related to A. ealaensis. 184 is only the third known naphthylisoquinoline dimer with a novel spiro-fused novel molecular framework and the first such representative to possess a relative trans-configuration at the two chiral centers in both tetrahydroisoquinoline subunits.95
Three new Dioncophyllaceae-type naphthylisoquinoline dimers, jozibrevines A–C (187–189) (Fig. 14), were subsequently isolated from A. abbreviatus, along with the known dimer jozimine A2. These compounds have the same constitutions and identical absolute configurations at the five stereogenic centers but differ in their axial chirality. Two typical Ancistrocladaceae-type monomeric compounds, ancistrobrevines K (190) and L (191), with the S-configuration at C-3 and an oxygen function at C-6 were identified. A new hybrid-type alkaloid, ancistrobrevine M (192) was identified, which is 3R-configured and 6-oxygenated, and an “inverse hybrid-type” counterpart, dioncoline A (193) (Fig. 14), which is a 3S-configured naphthylisoquinoline lacking an O-functionality at C-6, were isolated. They showed the pronounced antiplasmodial activities in the submicromolar range.96
 | ||
| Fig. 14 The chemical structure of compounds 187–193. | ||
2.6. Phenanthridine alkaloids
Phenanthridine isoquinoline compounds occur in higher plants and show a wide spectrum of nonspecific biological activities.6 Sanguinarine is one of the most famous compounds in this class. In 2020, one benzophenanthridine type, dehydroambiguanine A (194) and one isoquinoline type, ambiguanine J (the C ring of a benzophenanthridine-type alkaloid was opened) (195) (Fig. 15), were isolated from the n-butanol fraction of Corydalis ambigua subsp. amurensis. Among them, 194 exhibited anti-proliferative activity on HCT 116 cells, with an IC50 value of 49.8 μM, and ambiguanine J showed activity against A549 cells with an IC50 value of 60.2 μM (Table S2†).97 Three new compounds, bungeanoline A (196), menisperdaurine W (197) and narciclasine-4-O-β-D-xylopyranoside (198), were subsequently isolated from C. bungeana,71Menispermum dauricum,51 and Zephyranthes minuta6 (Fig. 15), respectively.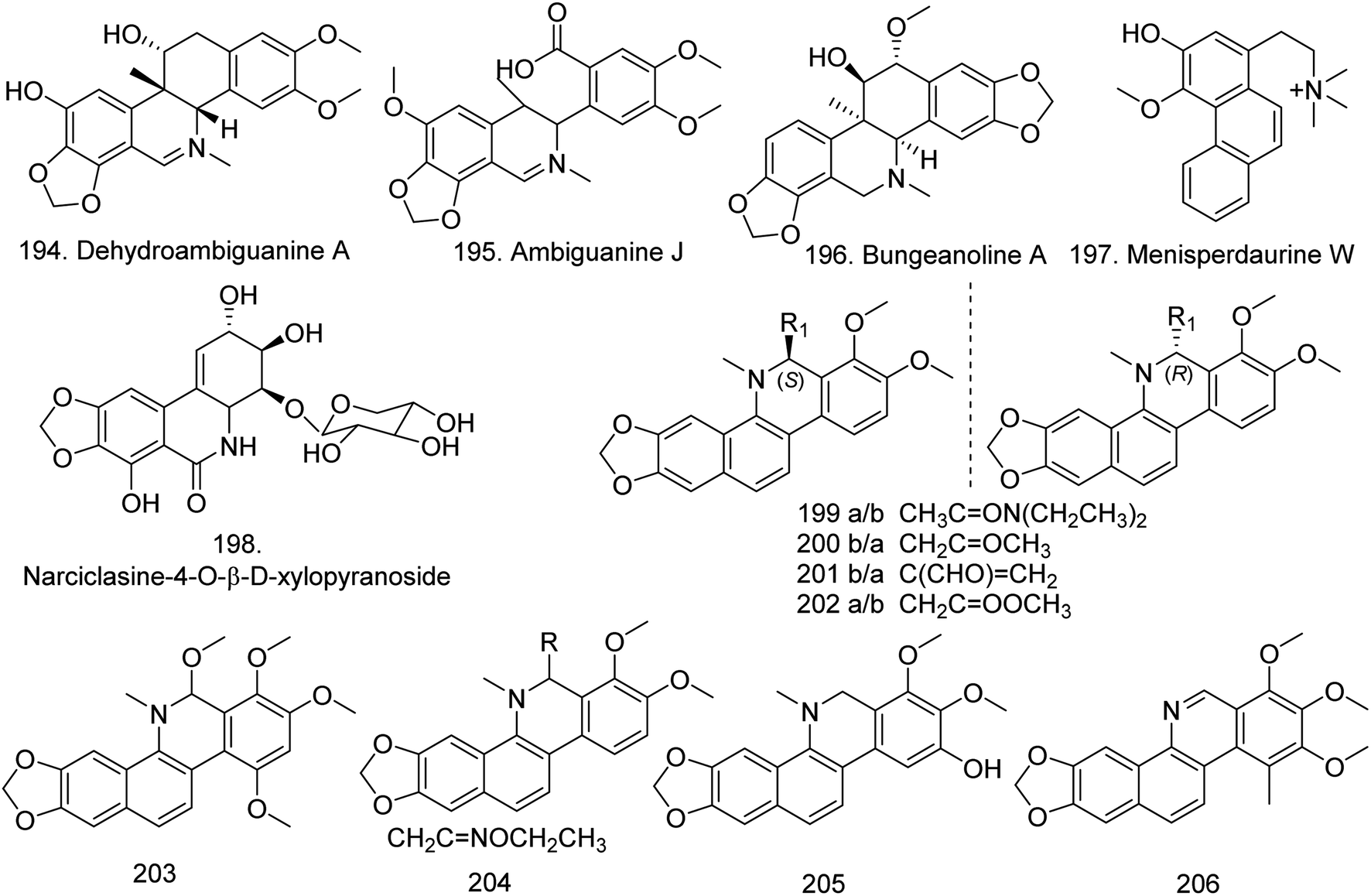 | ||
| Fig. 15 The chemical structure of compounds 194–206. | ||
Six pairs of enantiomeric isoquinoline alkaloids 6S/R-(N,N-diethylacetamido)yl-dihydrochelerythrine (199), 6R/S-acetonyl-9-hydroxy-dihydrochelerythrine (200), 6S/R-acroleinyl-dihydrochelerythrine (201), 6S/R-acetatemethyl-dihydrochelerythrine (202), 6,10-dimethoxydihydrochelerythrine (203), 6-ethoxy-ethaniminyl dihydroche-landine (204), 9-hydroxy-dihydrochelerythrine (205), and 9-methoxy-10-hydroxy-norchelerythrine (206) (Fig. 15) were isolated from the roots and rhizomes of Hylomecon japonica. Among them, 201, 203, 204, 206 and the known alkaloids 6-methoxydihydrosanguinarine, 6-acetaldehyde-dihyrochelerythrine, dihydrosanguina-line and 10-methoxy boconoline presented inhibitory effects on MCF-7 cells, with IC50 values lower than 20 μM (Table S2†).60
2.7. Morphine isoquinoline alkaloids
Morphine alkaloids have a 1-benzylisoquinoline skeleton with one additional ring closure. In 2020, four new isoquinoline alkaloids, (6S,7S,9R,13S)-6,7-di-O-acetyl-N-formylsinococuline (207), (6S,7S,9R,13S)-N-formylsinococuline (208), (6S,7S,9R,13-S)-6-O-acetyl-N-formylsinococuline (209), (6S,7S,9R,13S)-7-O-acetyl-N-formylsino-coculine (210)58 and (6S,9S,13R,14S)-6-O-acetyl-7,8-didehydro-4-hydroxy-3,7-dimethoxymorphinan-6-ol (211)69 (Fig. 16), were identified from Stephania cepharantha; however, they did not present the anti-inflammatory activity against NO production or cytotoxicity in three human cancer cell lines (A549, MCF-7, SW480). In 2022, a new morphine derivative, bungeanoline B (212) together with three known compounds, protostephanone, salutaridine, and salutaridine N-oxide, were isolated from the whole herb extract of Corydalis bungeana.71 In 2023, Zeng et al.55 isolated four new morphine isoquinoline alkaloids from Sinomenium acutum, including three N-oxide alkaloids, sinotuminins A (213), B (214) and C (215), and sinotuminin D; however, they did not have NO or AChE production inhibitory effects.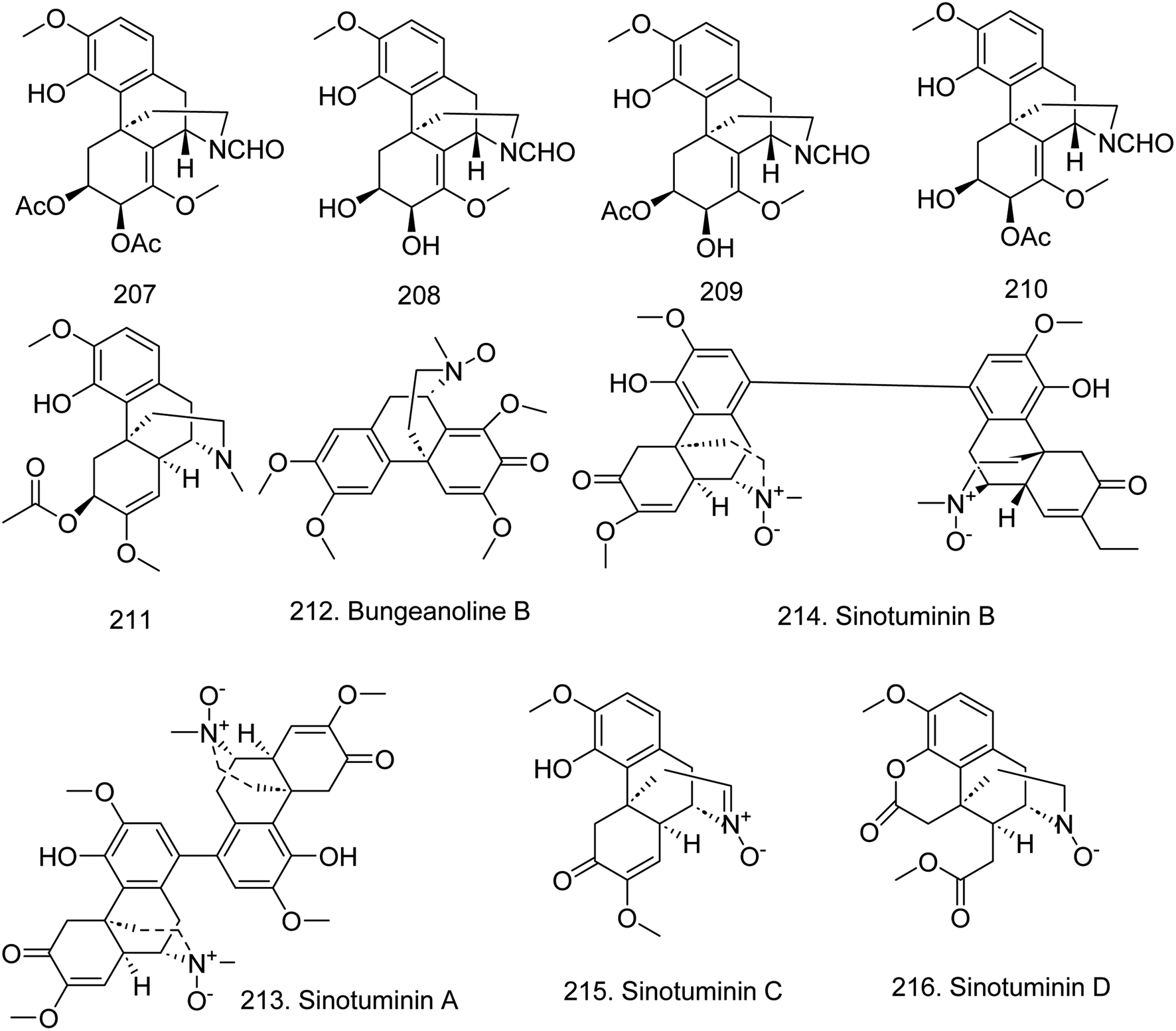 | ||
| Fig. 16 The chemical structure of compounds 207–216. | ||
2.8. Phthalideisoquinoline alkaloids
Since the first prototypical phthalideisoquinoline hemiacetal alkaloid, (+)-egenine, was isolated from Fumaria vaillantii in 1983, this kind of compound has attracted considerable scientific attention, more than 30 compounds with two asymmetric centers in the molecule at C-1 and C-9 have been identified from the families Fumariaceae, Papaveraceae, Berberidaceae, and Ranunculaceae. They can be divided into two classes according to their chemical features: typical phthalideisoquinoline alkaloids with an intact tetracyclic skeleton and seco-phthalideisoquinoline alkaloids with a cleaved B ring that results in the formation of a dimethylaminoethyl side chain.98,99In 2019, six new phthalideisoquinoline hemiacetal alkaloids, namely corydecumines B–G (217–222) (Fig. 17), as well as the known compounds corydecumine A and corydecumbine, were isolated from the bulbs of Corydalis decumbens. All new compounds inhibited neuron excitability at low micromolar concentrations.100 This group subsequently isolated two new phthalideisoquinoline hemiacetal alkaloid derivatives, corybensines A (223) and B (224). 224 inhibited neuron excitability with an IC50 value of 10.0 μM in the suppression of SCO frequency, indicating its antiepileptic and analgesic properties, and the methylenedioxy group at the at C-5 and C-6 and the oxygen-containing substituent at C-7 are important contributors to its activity.101 Mucroniferanine K (225) was isolated from C. mucronifera.45 Two novel benzylisoquinoline alkaloids, namely, 9S-alkterlactone B (226) and (1S,9S)-1-hydroxy-9-methyl-corlumine (227) (Fig. 17), were isolated from C. tomentella.42 Moreover, four undescribed phthalideisoquinoline alkaloids, namely, edulisine B (228) with a benzo [1,2-d:3,4-d]bis[1,3]dioxole moiety, and edulisines D–F (229–231) (Fig. 17), were isolated from the whole plants of C. edulis, and (+)-edulisine B, (−)-edulisine B and (−)-edulisine F all exhibited insulinotropic action in vitro.47
 | ||
| Fig. 17 The chemical structure of compounds 217–231. | ||
2.9. Various isoquinoline alkaloids
In 2020, three undescribed alkaloids, namely, gingantelline (232), gigantellinine (233), and gigancrinine (234) (Fig. 18), together with the well-known sanguinine, cherylline, lycorine, crinine and flexinine, were isolated from Crinum jagus. However, only sanguinine was remarkably effective against AChE, with an IC50 value of 1.83 μM.102 Cryptowrayines A (235) and B (236) (Fig. 18), which have approximately 1.4-fold and 1.2-fold quinone reductase-inducing activity in Hepa 1c1c7 cells at 3.125 μM, respectively, were subsequently isolated from the twigs of Cryptocarya wrayi (Table S2†).103 A new narceine-type alkaloid, bungeanoline C (237), was obtained from the whole herb extract of Corydalis bungeana.26 | ||
| Fig. 18 The chemical structure of compounds 232–247. | ||
(1′R,2′S)-Coptichine B (238) from C. tomentella exhibited strong antibacterial activities against two Gram-positive bacteria, Staphylococcus aureus and Bacillus subtilis, and two negative bacteria, Escherichia coli and Pseudomonas aeruginosa with MIC values of 3.12, 3.12, 12.5 and 6.25 μg mL−1, respectively42 (Table S3†). Moreover, mahimbrine A (239), which possesses a rare benzotropolone framing scaffold, was isolated from the endemic plant of Mahonia imbricata. However, it did not have antimicrobial activity at 1 mg mL−1.104 Two phenethyisoquinoline alkaloids, (+)-O-methylkreysigine-N-oxide (240) and (+)-1-demethylandrocine (241) (Fig. 18), were subsequently isolated from the leaves of Androcymbium palaestinum.38
Six new lamellarin sulfates, lamellarin K-20-sulfate (242), lamellarin E-20-sulfate (243), lamellarin A3-20-sulfate (244), lamellarin B1-20-sulfate (245), lamellarin D-8-sulfate (246), and lamellarin B2-20-sulfate (247) (Fig. 18), were isolated from the methanolic extract of the Pacific tunicate Didemnum ternerratum, and 245 exhibited moderate cytotoxicity against HCT-116 with an IC50 of 9.7 μM (Table S2†).105
Among sponges, more isoquinoline alkaloids were found. In 2019, a new bistetrahydroisoquinolinequinone, jorunnamycin A (248), with antimetastatic activity, was isolated from a Thai blue sponge Xestospongia sp.106 Mansouramycins E (249) and G (250) (Fig. 19) were found in the marine-derived Streptomyces sp. isolate B1848.33 A new isoquinolinequinones, 21-dehydroxyrenieramycin F (251), was isolated from the sponge Haliclona sp.31 Citronamine A (252) (Fig. 19), an isoquinoline alkaloid containing an unprecedented pentacyclic ring system was isolated from the Australian marine sponge Citronia astra.107 Turbinmicin (253) (Fig. 19), which has antibacterial activity, was identified from the marine microbiome (Table S3†).108
 | ||
| Fig. 19 The chemical structure of compounds 248–253. | ||
3. Synthesis of isoquinoline alkaloids
3.1. Chemical syntheses of isoquinoline alkaloids
Isoquinoline alkaloids are natural products with complex and diverse chemical structures that are widely used in medicine, pesticides, and other fields. However, the direct extraction and separation of these alkaloids from nature are often costly and inefficient, making synthetic methods a promising alternative. Developing new synthetic strategies to increase the efficiency of isoquinoline alkaloid synthesis is crucial for advancing the research and application of these compounds.109–111 As global environmental and energy issues become increasingly prominent, the development of green, efficient, and economically sustainable synthetic strategies has taken on particular importance. In recent years, photoredox catalysis,112–116 electrochemical synthesis,117 and continuous flow reaction118–120 strategies have garnered significant attention in the field of organic synthetic chemistry, and are seen as key technologies driving the transformation of organic synthesis. Here, the first total synthesis of isoquinoline alkaloid natural products, new synthetic strategies, and the application of new technologies in the synthesis of isoquinoline alkaloids will be introduced.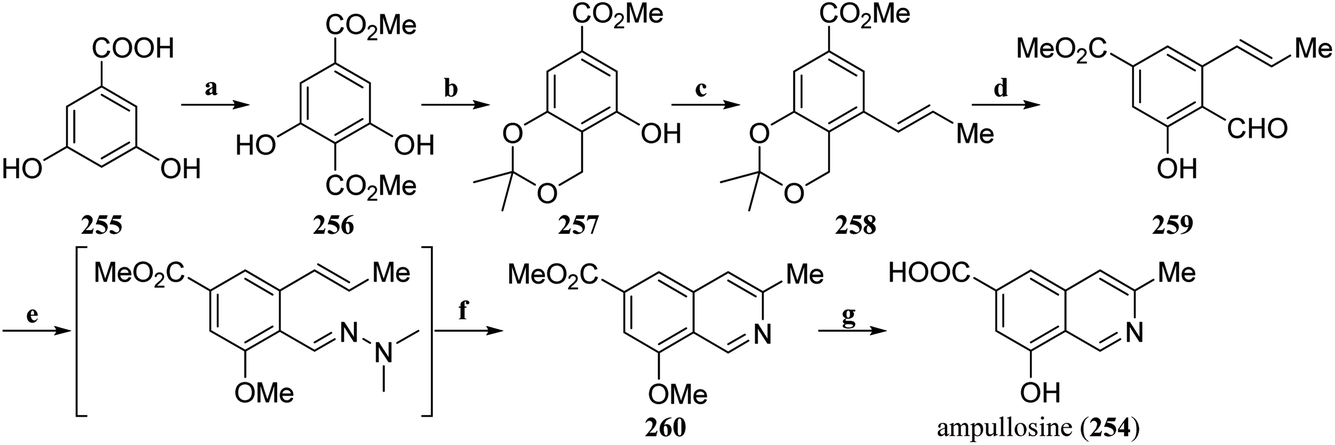 | ||
| Scheme 1 Total synthesis of ampullosine (254) by Kaufman's group. Reagents and conditions: (a) (i) CO2, KHCO3, 1,2-propanediol, 180 °C, (ii) Me2SO4, KHCO3, acetone, reflux; (b) (i) NaBH4, THF/H2O, 0.1 M phosphate buffer pH 7.5, 0 °C-rt, (ii) 2,2-DMP, TsOH, rt; (c) (i). PhNTf2, DMAP, NEt3, DCM, rt, (ii) PdCl2(PPh3)2, DavePhos, THF/H2O, 80 °C; (d) (i) TsOH, THF/H2O, 60 °C, (ii) MnO2, EtOAc, 78 °C; (e) (i) MeI, K2CO3, DMF, 0 °C-rt, (ii) Me2N-NH2, AcOH, PhCF3, rt; (f) MW, 160 °C; (g) Al0, I2, DMSO, MeCN, 80 °C. | ||
6,8-Dimethoxy-1,3-dimethylisoquinoline (261) is a simple isoquinoline alkaloid natural product and a fragment of many complex natural products or drugs. In 2019, Kaufman et al. reported a concise and efficient synthesis strategy that achieved a total synthesis of 261 in just three steps with a total yield of 27.3% (Scheme 2).124 First, Ce-catalyzed methoximation of acetophenone (262) was used to obtain 263, and a ruthenium-catalyzed allyl reaction with methyloxime as the guide group was carried out to generate 264. Finally, ruthenium-catalyzed allyl isomerization and microwave-facilitated 6π-azaelectrocylization were carried out via a one-pot method to complete the total synthesis of 6,8-dimethoxy-1,3-dimethylisoquinoline (261).
 | ||
| Scheme 2 Total synthesis of 6,8-dimethoxy-1,3-dimethylisoquinoline (261) by Kaufman's group. Reagents and conditions: (a) MeONH2·HCl, CeCl3·7H2O, NaOAc, EtOH, 50 °C; (b) allyl acetate, [RuCl2(p-cymene)]2, AgSbF6, DCE, rt; (c) (i) RuHCl(CO)(PPh3)3, 90 °C, (ii) PhMe, 150–160 °C. | ||
In 2020, Bracher et al. refined the total synthesis strategy for 1-oxoisoquinoline alkaloids, introducing the C-3 and C-4 units of the isoquinoline core using 2-ethoxyvinyl boronate as the C2 structural unit. Starting with commercially available ortho-bromobenzoic acid, which is activated with thionyl chloride and then amidated with ammonia or aqueous methylamine solution to obtain benzamide derivatives, only two to three convenient operations are needed to construct 1-oxo-, 1-oxo-3,4-dihydro-, and 1,3,4-trioxoisoquinoline alkaloids, establishing a concise and efficient general synthetic method. Based on this synthetic strategy, the first total synthesis of the dimer alkaloids berbanine and berbidine was completed (Scheme 3).125 Starting from N-methylbenzamide 265, the 1-oxoisoquinoline alkaloid 266 was constructed according to an established synthetic method, followed by Pd/C reduction to obtain 1-oxo-3,4-dihydroisoquinoline alkaloid 267. Subsequently, it was combined with 268via the Ullman coupling reaction to generate the intermediate 269, and then the berbanine (270) was obtained through the Pomeranz–Fritsch reaction, followed by N-methylation and reduction to give the berbidine (271).
 | ||
| Scheme 3 Total synthesis of berbanine (270) and berbidine (271) and by Bracher's group. Reagents and conditions: (a) trans-2-ethoxyvinylboronic acid pinacol ester, Pd(Ph3P)4, Cs2CO3; (b) TFA; (c) H2, Pd/C, 20–40 bar; (d) CuBr·Me2S, Cs2CO3; (e) (i) aminoacetaldehyde dimethyl acetal, (ii) TFAA, BF3·(AcOH)2; (f) (i) MeI, (ii) NaBH4. | ||
In 2021, Ramapanicker et al. designed a new method for the synthesis of enantioselective C-1-substituted tetrahydroisoquinoline natural products with proline-catalyzed asymmetric α-hydrazination as a key step,126 achieving asymmetric total synthesis of six different natural products (Scheme 4). The commercially available 2-(3,4-dimethoxyphenyl)ethan-1-ol 278 was converted into the aldehyde 279 through a simple two-step process. The aldehyde 279 was subjected to asymmetric α-hydrazination via dibenzyl azodicarboxylate (DBAD) in the presence of L-proline, and after reduction with NaBH4, β-hydrazino alcohol 280 was obtained. RANEY®-Ni was used to hydrogenate 280, followed by in situ cyclization to produce (−)-calycotomine (272). The hydroxyl group in Boc-protected 281 was converted to an unstable toluenesulfonyl derivative and then reduced to 282via LiAlH4. Deprotection afforded (−)-salsolidine (273) and methylation produced (−)-carnegine (274). The hydroxyl group in 281 was oxidized to form the aldehyde 283, followed by a Wittig reaction to generate the olefin 284. Hydrogenation reduced the number of double bonds, deprotection and methylation to obtain (+)-homolaudanosine (275). Cbz protected 272 to obtain 285, which was oxidized to the aldehyde 286, and then reacted by Wittig reaction to obtain the olefin 287. Hydroreduction followed by a Pictet–Spengler reaction produced (+)-homoprotoberberine (276). Aldehyde 279 was treated with dibenzyl azodicarboxylate in the presence of D-proline, followed by a Wittig reaction to obtain the unsaturated ester 288. The one-pot method reduced and cyclized to produce 289. Finally, the lactam 289 was reduced to obtain (+)-crispine A (277).
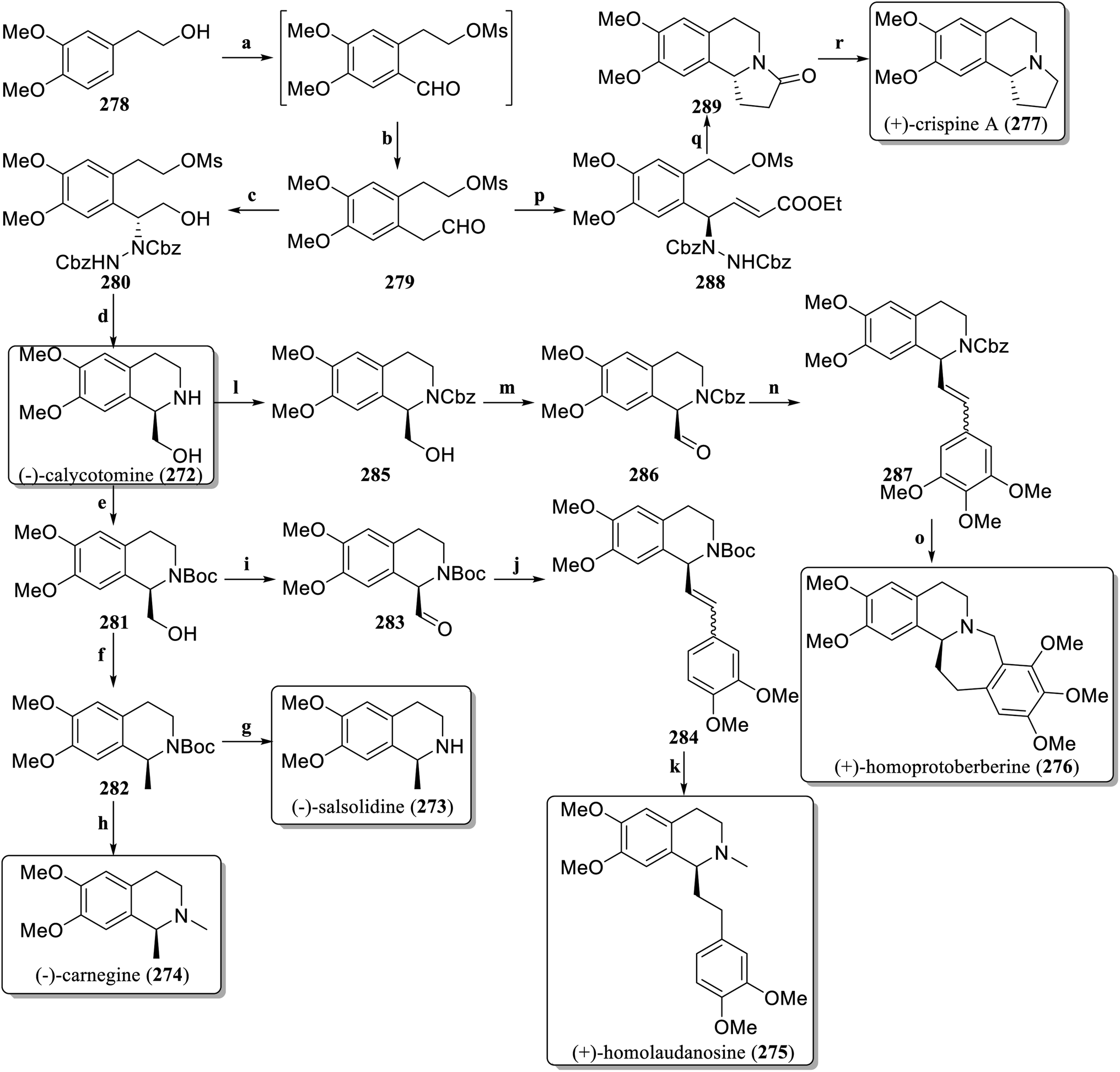 | ||
| Scheme 4 Total synthesis of C-1 substitutes tetrahydroisoquinoline alkaloids by Ramapanicker's group. Reagents and conditions: (a) (i) MsCl, Et3N, DCM, 0 °C to rt, (ii) dichloromethyl methyl ether, SnCl4, CH2Cl2, −20 °C-rt; (b) (i). MeOCH2PPh3Br, t-BuOK, THF, 0 °C to rt, (ii) THF, 2 N HCl, 0 °C-rt; (c) (i) DBAD, L-proline, CH3CN, 0 °C-rt, (ii) NaBH4, EtOH; (d) RANEY®-Ni, MeOH, H2, rt; (e) Boc2O, NaHCO3, THF, rt; (f) (i) TsCl, pyridine, CH2Cl2, 0 °C-rt, (ii) LiAlH4, THF, 0 °C-rt; (g) TFA, CH2Cl2, 0 °C; (h) LiAlH4, THF, reflux; (i) IBX, DMSO, rt; (j) (3,4-dimethoxybenz-yl)triphenylphosphonium bromide, t-BuOK, THF, 0 °C; (k) (i) Pd/C, EtOAc, H2, rt, (ii) LiAlH4, THF, reflux; (l) CbzCl, NaHCO3, THF, 0 °C; (m) IBX, DMSO, rt; (n) triphenyl(3,4,5-trimethoxybenzyl)phosphonium bromide, t-BuOK, THF, 0 °C; (o) (i) Pd/C, EtOAc, H2, rt, (ii) HCHO, HCl, EtOH, reflux; (p) (i) DBAD, L-proline, CH3CN, 0 °C to rt, (ii) Ph3PCHCO2Et, 0 °C to rt; (q) (i) H2, RANEY®-Ni, MeOH, rt, (ii) K2CO3, MeOH, reflux; (r) LiAlH4, THF, reflux. | ||
In 2022, Clausen et al. optimized the synthesis method of TMC-120B (9) to achieve its total synthesis with only a 7-step reaction (9% overall yield),127 a significant improvement over the previously reported 11-step reactions (2.9% overall yield)128 (Scheme 5). Starting from the commercially available 2-bromo-6-hydroxybenzaldehyde 291, 292 was obtained through Sonogashira coupling with 290. The MOM protected the phenolic hydroxyl group to generate 293, which constructed the isoquinoline scaffold 294 through imination and intramolecular ethynyl-imino cyclization. Deprotection under acidic conditions led to the formation of 295, which subsequently underwent alkylation with ethyl bromoacetate to generate 296. In contrast to previous synthesis strategies, the core scaffold 297 was constructed via intramolecular Friedel–Crafts acylation. Finally, under acidic conditions, an aldol condensation reaction occurred between 297 and acetone to form TMC-120B (9).
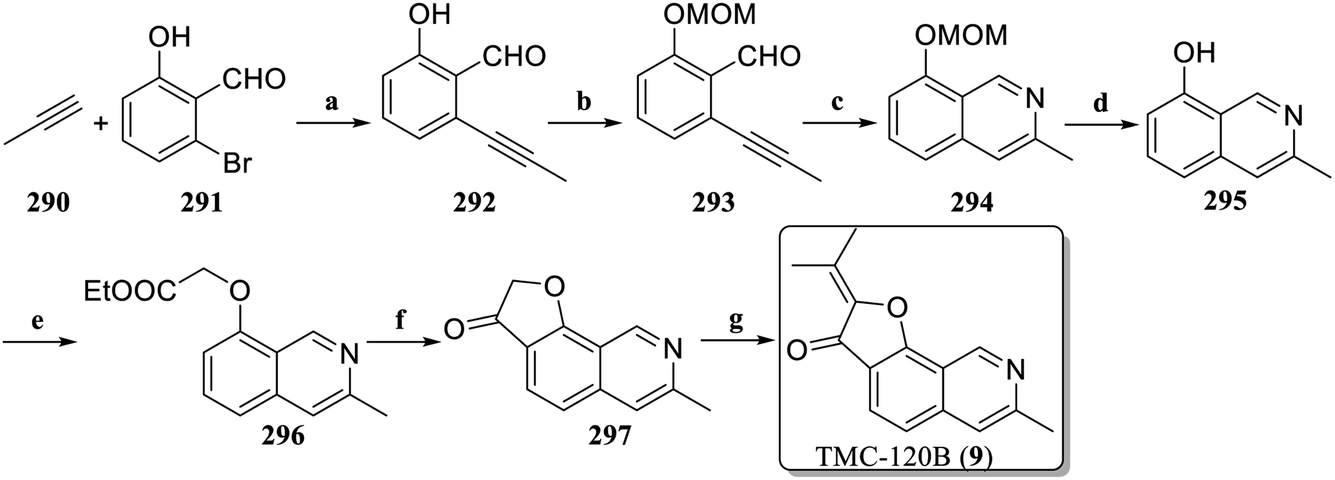 | ||
| Scheme 5 Total synthesis of TMC-120B (9) by Clausen's group. Reagents and conditions: (a) PdCl2(PPh3)2, Et3N, CuI, 55 °C, 24 h; (b) MOMCl, DIPEA, CH2Cl2, 40 °C, 16 h; (c) NH3, 80 °C, 7 h; (d) HCl, MeOH, reflux, 3 h; (e) ethyl bromoacetate, NaOEt, DMF, 12 h; (f) (i) NaOH, EtOH, 1 h, (ii) SOCl2, reflux, 1 h, (iii) AlCl3, 1 h; (g) p-TsOH·H2O, acetone, DCE, reflux, 5 h. | ||
 | ||
| Scheme 6 Total synthesis of rac-muraricine (72) by Bracher's group. Reagents and conditions: (a) (methoxymethyl)triphenylphosphonium chloride, LDA; (b) ethyl chloroformate, Et3N, NaOH; (c) TFA, CH2Cl2; (d) CH3NO2, NH4AcO, CH3COOH; (e) Zn, HCl, MeOH; (f) ethyl chloroformate, Et3N, CH2Cl2; (g) (i) CuBr·Me2S, Cs2CO3, (ii) Pd/C, H2, MeOH; (h) paraformaldehyde, TFA, CH2Cl2, 0 °C-rt; (i) LiAlH4, THF, 50 °C. | ||
Bracher and coworkers improved the N-acyl-Pictet–Spengler condensation reaction in 2021, enabling the efficient synthesis of 1-benzyltetrahydroisoquinolines.130 This strategy uses ω-methoxystyrene (303) as a convenient alternative to aryl acetaldehyde to rapidly construct the compound skeleton via an N-acyl-Pictet–Spengler reaction with N-ethoxycarbonyl phenethylamines (304). After removing the ethoxycarbonyl groups from 305, N-methylated isoquinoline alkaloids could be obtained via route A, or the corresponding alkaloids with free N–H could be generated via route B. In this synthetic strategy, the dual utilization of ethoxycarbonyl significantly simplified the synthesis of hydroxylated 1-benzyltetrahydroisoquinolines. Through this strategy, 10 racemic benzyl isoquinoline alkaloids were efficiently synthesized (Scheme 7).
 | ||
| Scheme 7 Total synthesis of racemic 1-benzyltetrahydroisoquinoline alkaloids by Bracher's group. Reagents and conditions: (a) TFA, DCM; (b) CH3Li, THF, 0 °C; (c) LiAlH4, THF, reflux. | ||
The asymmetric catalytic Pictet–Spengler reaction represents an ideal strategy for the synthesis of tetrahydroisoquinoline (THIQ) and related natural products. In 2022, List et al. employed imidodiphosphorimidate (IDPi) as the asymmetric organocatalyst to promote the asymmetric Pictet–Spengler reaction between the aldehydes 307 and the N-methoxycarbonyl phenethylamines 306 (Scheme 8).131 This approach efficiently afforded the tetrahydroisoquinoline derivatives 308 with excellent enantioselectivity. Through this strategy, key intermediates for the synthesis of isoquinoline alkaloid natural products could be obtained in a concise and efficient manner. Simple chemical transformations then allow for the efficient synthesis of natural products. By reducing or removing the carbamate functional group, asymmetric THIQs such as laudanosine, romneine, and lophocerine with N-methylation or N–H could be obtained. Removing the carbamate and treating it with a base could effectively cyclize it to form the corresponding γ-lactam, which could be further synthesized into the tetrahydroisoquinoline alkaloids oleracein E and crispine A. After reductive methylation, oxidative phenol–phenol coupling could construct an androcymbine skeleton. The intermediate with the carbamate functional group removed could undergo a Pictet–Spengler reaction with formaldehyde to construct the tetrahydroberberine natural product xylopinine. The transition metal-promoted intramolecular coupling reaction efficiently synthesized the aporphine natural product glaucine. Through the constant current electrolysis (CCE) conditions developed by Opatz and Waldvogel,132 the morphinan skeleton amurine could be efficiently obtained.
 | ||
| Scheme 8 Catalytic asymmetric Pictet–Spengler platform and application by List's group. Reagents and conditions: (a) (S,S)-IDPi, CHCl3, rt. | ||
In 2022, Baskar's group established a visible light-mediated organophoto-redox catalyzed method for the α-benzylation of tertiary amines (Scheme 9a).133 The organic nonmetallic photocatalyst 4-CzIPN was used to achieve the green and efficient synthesis of compound N-aryl-substituted tertiary amines and benzyl bromide under visible light irradiation, and a series of tetrahydroisoquinoline derivatives were constructed. Furthermore, this method could be used to synthesize biologically active natural products of isoquinoline alkaloids (Scheme 9b). The tetrahydroisoquinoline derivative (309) obtained as the photocatalysis reaction product was converted to important alkaloids rac-norlaudanosine (310), rac-laudanosine (311) and rac-xylopinine (312) through simple chemical transformations.
 | ||
| Scheme 9 Metal-free photoredox catalyzed α-benzylation and application by Baskar's group. Reagents and conditions: (a) 4-CzIPN, K2CO3, MeCN, Ar, rt, blue LED; (b) CAN, MeCN/H2O, 0 °C; (c) HCHO, NaBH4, MeCN/H2O, rt; (d) HCHO, HCOOH, H2O, 90 °C. | ||
The enantioselective C–H functionalization strategy catalyzed by transition metals has provided a new approach for the retrosynthetic analysis of natural products, revolutionizing the traditional logic of synthesizing natural compounds.134 Recently, Shi and Yao et al. established a cobalt-catalyzed enantioselective C–H/N–H cyclization reaction between picolinamides (313) and alkynes (314), providing a direct and efficient synthetic strategy to prepare C1-chiral 1,2-dihydroisoquinolines (315) with good yields and excellent enantioselectivity (Scheme 10).135 Based on this method, the N-benzhydrylpicolinamide 316 underwent enantioselective C–H/N–H cyclization with trimethylsilylacetylene to produce 317. After the TMS was removed and the dihydroisoquinoline was hydrogenated and deprotected, (S)-1-phenyl-1,2,3,4-tetrahydroisoquinoline (318), which is the key intermediate for the synthesis of (+)-solifenacin and FR115427, was obtained. According to this strategy, picolinamides 319 and 320 underwent kinetic resolution reactions with trimethylsilylacetylene to produce the enantioselective C–H/N–H cyclization products 321 and 322, respectively. After the TMS was removed, the dihydroisoquinolines were hydrogenated and deprotected, and (S)-norlaudanosine (324) and 323 were formed. 324 could be further converted into (S)-laudanosine (325), (S)-xylopinine (326), and (S)-sebiferine (327) with excellent enantioselectivity. After N-methylation, 323 was converted to (S)-cryptostyline II (328). The synthesis of the above natural products can be carried out on a gram scale, efficiently obtaining hundreds of milligrams of target natural products or drug candidate molecules, which further demonstrates the important application value of this strategy in the field of synthesis chiral isoquinoline alkaloids.
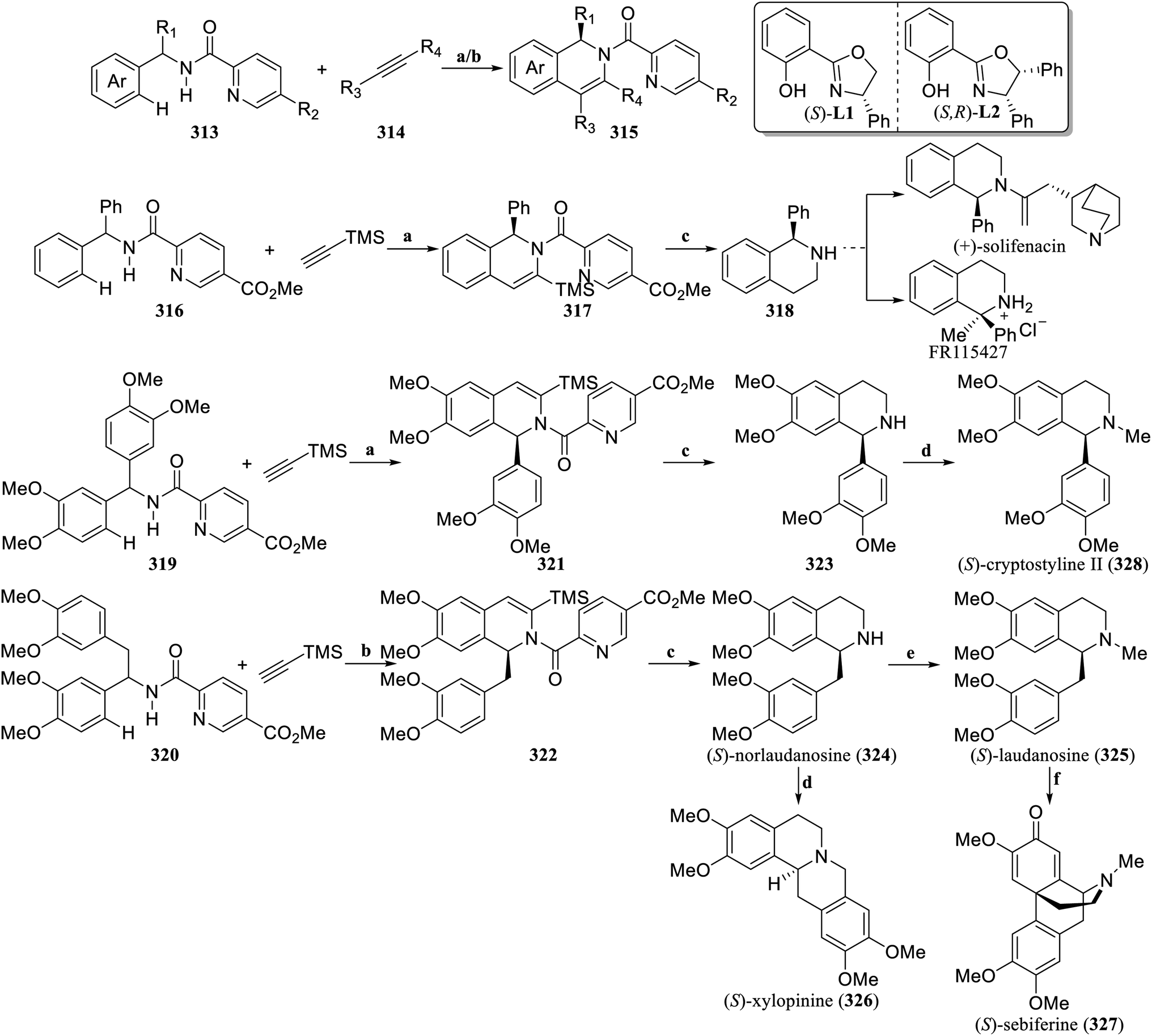 | ||
| Scheme 10 Cobalt-catalyzed enantioselective C–H/N–H cyclization by Shi's group. Reagents and conditions: (a) Co(OAc)2·4H2O, (S)-L1, Mn(OAc)2·4H2O, PivONa·H2O, MeOH, O2, 90 °C; (b) Co(OAc)2·4H2O, (S,R)-L2, Mn(OAc)2·4H2O, PivONa·H2O, MeOH, O2, 90 °C; (c) (i) CF3COOH, DCM, −5 °C-rt, (ii) Pd/C, H2, MeOH, 50 °C, (iii) LiAlH4, THF, −78 °C-rt; (d) HCHO, HCO2H, 90 °C; (e) HCHO, MeOH, then NaBH4, rt; (f) PIFA, HPA, BF3·Et2O, MeCN, −20–0 °C. | ||
Lumb's group has established a new method for the selective catalytic aerobic cross-dehydrogenative coupling reaction of phenols and catechols to construct aryl ether compounds, which are difficult to synthesize via traditional Ullman coupling. Based on this method, the simple and efficient total synthesis of the tetrahydroisoquinoline alkaloid natural products (S,S)-thalicarpine (329)136 and (S,S)-tetramethylmagnolamine (330)137 could be achieved in total yields of 20% and 21%, respectively. Compared with the previous synthesis methods (with total yields of 1.5% for 329 and 14% for 330), the new method can synthesize hundreds of milligrams of natural products in a single batch, which holds higher application value for studying their biological activities or synthesizing analogs.
Through the amidation of 3,4-dimethoxyphenethylamine (331) with 3-hydroxy-4-methoxyphenylacetic acid (332) and 4-hydroxyphenylacetic acid (333) respectively, followed by Bischler–Napieralski cyclization, the asymmetric tetrahydroisoquinoline skeletons 334 and 335 were constructed via asymmetric Noyori hydrogenation. Based on Fagnou's aporphine synthesis method,138334 was further converted into aporphine 336. Boc protected 335 and further oxidized it to obtain 337. Finally, under the established standard conditions, catalytic aerobic CDC reaction was performed on 336 and 337 to generate the aryl ether 338. After deprotection and methylation, (S,S)-thalicarpine (329) was obtained (Scheme 11). After Boc protection of 335, aerobic oxidative coupling was achieved under the established standard reaction conditions, and the corresponding aryl ether 339 was obtained after reduction. After methylation of the phenolic hydroxyl groups and reduction of N-Boc, (S,S)-tetramethylmagnolamine (330) was subsequently obtained (Scheme 12).
 | ||
| Scheme 11 Total synthesis of (S,S)-thalicarpine (329) by Lumb's group. Reagents and conditions: (a) (i) neat, 200 °C, (ii) POCl3, MeCN, reflux, (iii) (R,R)-Ru(Ts-DPEN)(p-cymene)Cl, HCO2H/Et3N (5:2), DMF, rt; (b) (i) Br2, HCl, AcOH, then Boc2O, MeOH, (ii). BnBr, Cs2CO3, DMSO, (iii) Pd(OAc)2, K2CO3, PhDavePhos, DMA, 130 °C, then Pd/C, MeOH, H2; (c) (i) Boc2O, MeOH, (ii) IBX, DMF; (d) (i) O2, CuCl, 4-MeO-Py, rt, (ii) NaBH4, MeOH; (e) (i) Cs2CO3, MeI, DMSO, (ii) LiAlH4, THF, reflux. | ||
 | ||
| Scheme 12 Total synthesis of (S,S)-tetramethylmagnolamine (330) by Lumb's group. Reagents and conditions: (a) Boc2O, MeOH, rt; (b) [Cu(MeCN)4](PF6), DBED, 4 Å MS, DCM, O2, rt; (c) Na2S2O4 workup; (d) (i) MeI, Cs2CO3, DMSO, (ii) LiAlH4, THF, reflux. | ||
In 2020, Bracher's group established a new method for the synthesis of bisbenzylisoquinoline alkaloids,139 which could efficiently construct rac-tetrandrine (340) and its diastereomer isotetrandrine (341) by replacing the Bischler–Napieralski reaction with the N-acyl Pictet–Spengler cyclization and combined with the Ullmann reaction (Scheme 13).
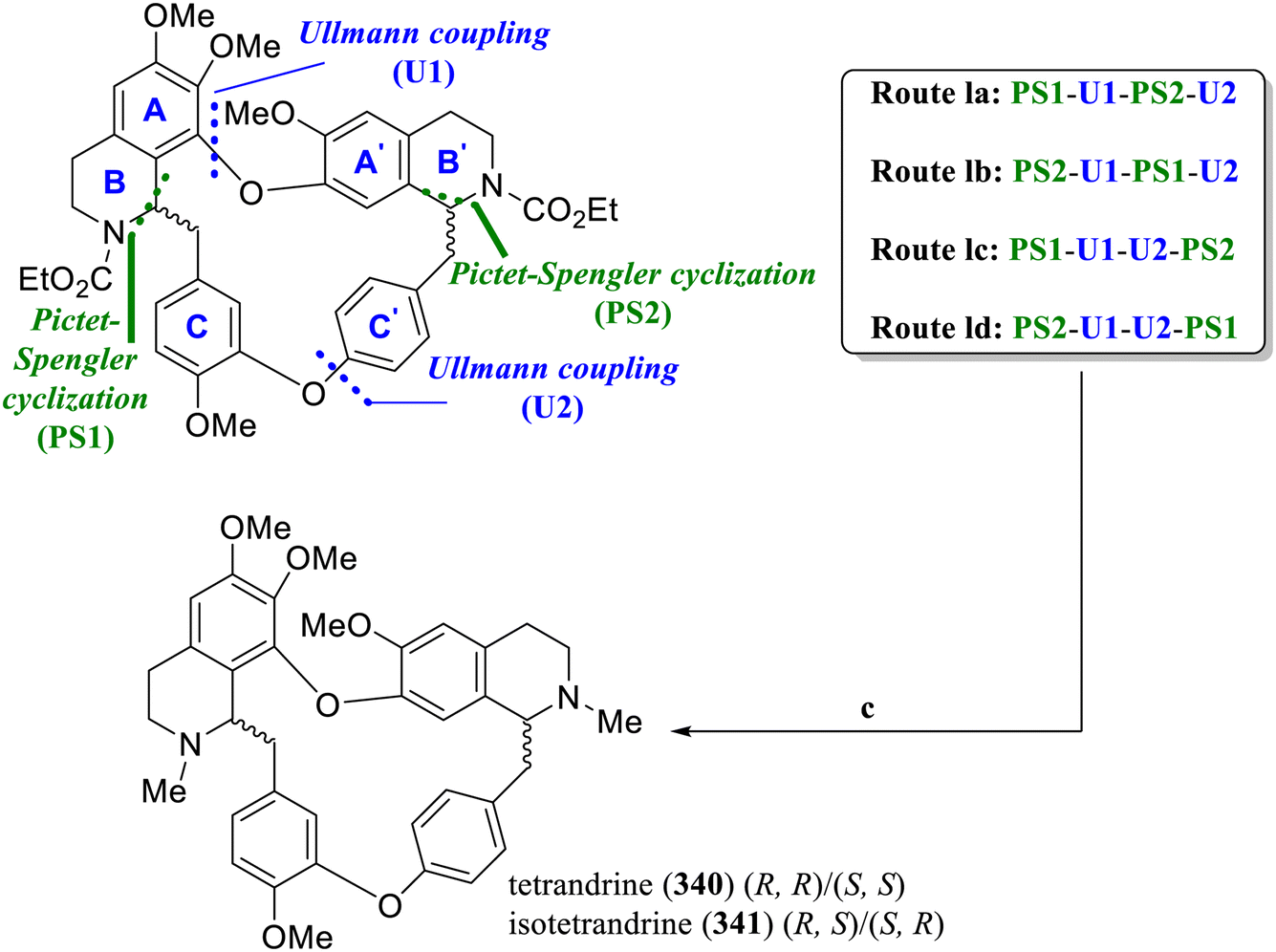 | ||
| Scheme 13 Total synthesis of tetrandrine (340) and isotetrandrine (341) by Bracher's group. Reagents and conditions: (a) Ullmann coupling: CuBr·Me2S, Cs2CO3, pyridine, 110 °C; (b) Pictet–Spengler cyclization: TFA, CH2Cl2, 0 °C-rt; (c) LiAlH4, THF, 80 °C. | ||
The N-acyl Pictet–Spengler cyclization reaction directly provides a tetrahydroisoquinoline skeleton, and the required N-methyl group can be obtained simply by reducing the carbamate group, significantly reducing the number of synthetic steps. In routes 1a and 1b, the two 1-benzyl tetrahydroisoquinoline moieties were constructed in the early stages through an intermolecular N-acyl Pictet–Spengler reaction, and the macrocycle was subsequently constructed through intramolecular diaryl ether synthesis. Conversely, in routes 1c and 1d, the intramolecular N-acyl Pictet–Spengler cyclization was the core step for constructing the macrocycle in the later stages. All four routes enable the total synthesis of rac-tetrandrine (340) and its diastereomer isotetrandrine (341) through more efficient synthetic steps (12 steps), with route 1d achieving the highest total yield of 19.2% among the four routes. In terms of stereoselectivity, routes 1a and 1b do not result in good stereoselectivity, producing an almost equimolar mixture of diastereomers, whereas routes 1c and 1d show good stereoselectivity for one or the other diastereomer.
 | ||
| Scheme 14 Total synthesis of dactylicapnosine A (342) by Zhang's group. Reagents and conditions: (a) (i) BnBr, K2CO3, acetone, 65 °C, (ii) O3, DCM-MeOH, then Me2S, −78 °C; (b) (i) NBS, AcOH, DCM, rt, (ii) CF3COOH, DCM, rt, (iii) NaOH, (Boc)2O, rt; (c) Pd(OAc)2, PPh3, K2CO3, DMA, 150 °C, MW; (d) (i) Pd/C, H2, MeOH-EtOAc, rt, (ii) IBD, HFIP-H2O, 0 °C; (e) O2, K2CO3, THF-MeOH, rt; (f) NaIO4, MeOH, 100 °C; (g) (i) MeCN, 170 °C, MW, (ii) MeI, K2CO3, THF, 100 °C. | ||
Later, in 2020, Zhang and Luo et al. reported a new synthetic method that employed acid-induced ortho-quinone isomerization, cobalt-mediated regioselective cyclocontraction of p-quinone, and oxidative methoxylation of enone as key steps. This strategy enabled a more efficient synthesis of dactylicapnosine A and achieved the first total synthesis of dactylicapnosine B.140 This approach may also provide new insights for the synthesis of other medicinally valuable dactylicapnosine-like analogs (Scheme 15). Starting from 2,3,4-trimethoxybenzaldehyde 351, the corresponding phenol was obtained through Baeyer–Villiger oxidation. The subsequent allyl ether compound underwent a Claisen rearrangement to produce 344, which followed the same synthetic path to obtain the aporphine derivative 347. After removal of the Boc-protecting group, the amine 352 was oxidized to the ortho-quinone 353. Acid-induced isomerization of the ortho-quinone gave the p-quinone 354. Then, through cobalt-mediated regioselective cyclocontraction of p-quinone, the key intermediate 355 was obtained. After dehydration to obtain 356, oxidative methoxylation of the enone was performed to obtain the key precursor 357. Finally, deprotection and methylation afforded dactylicapnosine A (342) with a 12% overall yield. By using boron trichloride for demethylation, an unstable diketone intermediate was obtained. After methoxylation with DMP, the first total synthesis of dactylicapnosine B (343) was achieved.
 | ||
| Scheme 15 Total synthesis of dactylicapnosine A (342) and dactylicapnosine B (343) by Zhang's group. Reagents and conditions: (a) H2O2, H2SO4, MeOH, rt; (b) allyl bromide, K2CO3, acetone, reflux; (c) DMF, 190 °C; (d) TFAA, DCM, rt; (e) (i) NCS, THF, rt, (ii) t-BuONa, ClCO2Me, rt; (f) (i) Pd/C, H2, MeOH, rt, (ii) IBX, DMSO, rt; (g) H2SO4, MeCN, rt; (h) CoCl2, MeOH-THF, 80 °C; (i) Et3N, (CF3CO)2O, DCM, rt; (j) H2O2, Na2CO3, MeOH, rt; (k) (i) CH3ONa, MeOH, rt, (ii) MeI, K2CO3, THF, 100 °C; (l) BCl3, DCM, −78 °C; (m) DMP, TsOH, MeOH, 80 °C. | ||
In 2020, Anderson et al. established a modular strategy for synthesizing enantioselective aporphine (Scheme 16).141 The phenylethylamine 331 and 358, which obtained from vanillin and piperonal through Henry reaction and reduction, were coupled with the carboxylic acid 359 to prepare the amides 360 and 361. Bischler–Napieralski cyclization followed by direct reduction by Noyori asymmetric transfer hydrogenation produced the chiral tetrahydroisoquinolines 362 and 363, respectively. After the amino group was protected, palladium catalyzed the ortho-arylation reaction to construct the aporphine backbone 364–367. The synthesis of (+)-norglaucine (368) and (+)-nordicentrine (369) was achieved through the deprotection of the Boc group. (+)-glaucine (370) and (+)-dicentrine (371) were obtained through reductive methylation.
 | ||
| Scheme 16 Total synthesis of (+)-norglaucine (368), (+)-nordicentrine (369), (+)-glaucine (370) and (+)-dicentrine (371) by Anderson's group. Reagents and conditions: (a) CH3NO2, ethylenediamine, AcOH, 70 °C; (b) LiAlH4, THF, 70 °C; (c) EDC·HCl, NMM, HOBt, DMF, 0 °C to rt; (d) (i) 2-chloropyridine, Tf2O, DCM, −78 °C to 0 °C, (ii) (R,R)-Ru(Ts-DPEN)(p-cymene)Cl, HCO2H/Et3N (5:2), 0 °C to rt; (e) (i) Boc2O, i-Pr2EtN, DMAP, CH2Cl2, rt, or (ii) MeOCOCl, i-Pr2EtN, DMAP, CH2Cl2, rt; (f) Pd(OAc)2, (t-Bu)2PMeHBF4, K2CO3, DMA, 130 °C; (g) (i) ZnBr2, CH2Cl2, rt, or (ii) LiAlH4, THF, 0 °C to rt. | ||
In 2020, Raminelli's group described the stereoselective total synthesis of (S)- and (R)-nuciferine (Scheme 17).142 The key to this synthesis was the preparation of the chiral intermediate 373via the triethylamine-mediated coupling of the dihydroisoquinoline 372 and the chiral acid chloride 375. The dihydroisoquinoline 372 was obtained from the 3,4-dimethoxyphenethylamine 331 by acetylation, Bischler–Napieralski cyclization, and enamidation reactions. Subsequently, the intermediate 373 underwent a [4 + 2] cyclization reaction with the benzyne precursor 374 to obtain diastereoselective intermediates 376 and 377. Then, the hydrolysis reaction was carried out under microwave heating conditions, and 378 and 379 were obtained. Finally, the N-methylation reaction achieved (S)-nuciferine (380) and (R)-nuciferine (381) with total yields of 13% and 4%, respectively, in the 6-step reaction.
 | ||
| Scheme 17 Total synthesis of (S)-nuciferine (380) and (R)-nuciferine (381) by Raminelli's group. Reagents and conditions: (a) Ac2O, pyridine, 90 °C; (b) (i) POCl3, toluene, reflux, (ii) 40% NaOH, H2O, rt; (c) 375, Et3N, DCM, 0 °C to rt; (d) CsF, MeCN, rt; (e) LiOH, EtOH/H2O (2:1), MW 180 °C; (f) (i) 37% CH2O, DCM, rt, (ii) NaBH4, rt. | ||
Qin's group unified the total synthesis routes for benzo[d][1,3]dioxole-type aporphines, coptisines, and dibenzopyrrocolines. By utilizing Noyori asymmetric hydrogenation and diastereoselective resolution to achieve excellent enantioselectivity, and combining palladium-catalyzed arylation as a key step, they accomplished the first asymmetric total synthesis of benzo[d][1,3]dioxole aporphine alkaloids (6aS,6a′S)-(+)-ovigeridi-merine (382), (S)-(+)-ovigerine (383), and (S)-(+)-N-formylovigerine (384) (Scheme 18).143 With slight modifications to the synthetic route, and by combining it with aza-Michael addition, the Bischler–Napieralski reaction, and N-arylation, the total synthesis of benzo[d][1,3]dioxole-type benzylisoquinoline alkaloids of coptisines and dibenzopyrrocolines was also achieved. The key intermediate, substituted phenylacetic acid, was constructed starting from commercially available piperonylic acid (385). Through a series of reactions, which encompassed the formation of precursor oxazoline (386), aryl modification, and hydrolysis of the oxazoline to obtain the aldehyde 387, followed by a Wittig reaction, hydrolysis, and Pinnick oxidation, the key carboxylic acid 388 was synthesized. The 388 was amidated with homopiperonylamine to give 389, which underwent Bischler–Napieralski cyclization to generate the air-sensitive 390. The enantiomer 391 was obtained via Noyori asymmetric hydrogenation.
 | ||
| Scheme 18 Total synthesis of (S)-(+)-ovigerine (383), (S)-(+)-N-formylovigerine (384) and (6aS,6a′S)-(+)-ovigeridimerine (382) by Qin's group. Reagents and conditions: (a) (i) HATU, 2-amino-2-methylpropan-ol, DMF, rt, (ii) p-TsCl, Et3N, DMAP, CH2Cl2, rt, (iii) NaOH, MeOH, rt; (b) (i) n-BuLi (2.4 M), THF, −50 °C, (ii) I2, THF, −78 to −50 °C; (c) oxalic acid, THF/H2O (4/1), rt; (d) NaHMDS (1 M), Ph3P+CH2OMeCl−, THF, 0 °C to rt; (e) (i) TsOH·H2O, THF, reflux, (ii) NaClO2, NaH2PO4, 2-methyl-2-butene, 0 °C; (f) homopiperonylamine, EDCI, HOBt, DIPEA, DMF, 0 °C to rt; (g) POCl3, MeCN, reflux; (h) (i) RuCl[(R,R)-TsDPEN](p-cymene), HCO2H/Et3N (5/2), DMF, 0 °C to rt, (ii) N-acetyl-L-leucine, i-Pr2O/MeOH (1/2), reflux; (i) 10% NaOH, pH = 12–13, rt; (j) (Boc)2O, DIPEA, THF, 40 °C; (k) triphosgene, Et3N, CH2Cl2, 0 °C; (l) (i) K2CO3, Pd(OAc)2, PhDave-Phos, DMF, 110 °C, (ii) ZnBr2, CH2Cl2, rt; (m) ethyl formate, Ar, 75 °C; (n) 391, DIPEA, MeCN, 40 °C; (o) K2CO3, Pd(OAc)2, X-Phos, DMF, 110 °C; (p) Pd(OAc)2, TBAC, tert-butyl acrylate, DMF, 120 °C; (q) MeSO3H, CH2Cl2/t-BuOAc (1/3), 0 °C to rt; (r) t-BuOH, reflux; (s) ZnBr2, CH2Cl2, rt. | ||
After Boc protection and N-acylation, 392 and 393 were generated, and 393 was further acylated to give the intermediate 394. Palladium-catalyzed intramolecular arylation of 392 and 394 produced (S)-(+)-ovigerine (383), (S)-(+)-N-formylovigerine (384), and (6aS, 6a′S)-(+)-ovigeridimerine (382). In addition, based on this synthesis strategy, the first asymmetric total synthesis of the impatiens 395 and 396 was completed, but the compound spectra were different from those of impatien B.
Through detailed analysis of the reaction mechanism144 of 311 radical cyclization intermediate 397 rearranged to rac-glaucine by erythrinadienone intermediate 398 and hydrolyzed into rac-sebiferine, Felpin's group established a flow-controlled divergent synthesis of aporphine and morphinandienone alkaloids based on biomimetic oxidative cyclization using a high-valent iodine(III) reagent in 2022 (Scheme 19).145 To stabilize the aromatic radical cations formed by PIFA and BF3·Et2O, hexafluoroisopropanol (HFIP) was used as the mobile phase solvent to selectively generate aporphine-type natural products. The use of PIDA and TMSOTf in wet CH3CN allowed the synthesis to diverge into morphinedienone-type natural products. Mixing the two streams in a T-mixer significantly improved mixing efficiency due to the high viscosity of HFIP. Under optimized conditions, this method could provide five natural aporphine products in good to moderate yields depending on the substrate used. Changing the solvent to wet CH3CN could effectively provide five morphinedienone alkaloids. This strategy offers a new approach for the large-scale production of such natural products.
 | ||
| Scheme 19 Flow chemistry divergency between aporphine and morphinandione alkaloids by Felpin's group. | ||
Based on the benzyne cyclization strategy146 combined with an oxidative dehydrogenation reaction, Raminelli's group successfully constructed the key dehydroaporphine intermediate, achieving the first total synthesis of (±)-urbaine (399), the asymmetric total synthesis of (S)-nuciferine (380), and the second-generation total synthesis of lisicamine (400).147 Isoquinoline 401 was obtained from the 3,4-dimethoxyphenethylamine 331 by acetylation, Bischler–Napieralski cyclization, and enamidation reactions. Then, the isoquinoline 401 underwent a [4 + 2] cyclization reaction with the benzyne precursor 374 to construct the aporphine skeleton, which was selectively oxidized to obtain the key dehydroaporphine intermediate 402. The first total synthesis of the bisaporphine alkaloid rac-urbaine (399) was completed with a total yield of 11% after dimerization of 402 (Scheme 20). After asymmetric hydrogenation and N-methylation, (S)-nuciferine (380) was obtained. Oxidation with Fremy's salt produced lisicamine (400).
 | ||
| Scheme 20 Total synthesis of rac-urbaine (399), (S)-nuciferine (380) and lisicamine (400) by Raminelli's group. Reagents and conditions: (a) (i) (MeCO)2O, pyridine, 90 °C, (ii) POCl3, PhMe, NaOH (40%), H2O, rt, (iii) (CF3CO)2O, pyridine, DCM, −50 °C; (b) CsF, MeCN, 80 °C, (ii) NaBH4, EtOH, rt, (iii) NCS, DCM, rt; (c) N2, DCM, 50 °C; (d) [Ir(COD)Cl]2, (Sa,R,R)-MomoPhos, I2, H2, 50 °C; (e) Fremy's salt, Na2CO3, MeOH, rt. | ||
Based on the previously established strategy of using three components to construct the 1-methylene-THIQ skeleton,148,149 Zhou's group optimized the Catellani reaction conditions involving aryl iodide, azetidine, and terminal alkynes, and established NBS-mediated cyclization reaction conditions. The modular synthesis of 1-bromomethylene-THIQ scaffold for the multipurpose intermediate in synthetic chemistry was realized. Suzuki–Miyaura coupling reactions and the Heck reaction, among others, could quickly convert 1-bromomethylene-THIQ scaffolds into synthetic intermediates with various structures (Scheme 21).150 On the basis of this new method, the concise synthesis of rac-cularine (403), and 8-oxopseudopalmatine (404) was completed, and dactyllactone A (405) was total synthesized for the first time. The intermediate compound 408 was hydrogenated to remove the benzyl protecting groups and simultaneously reduce enamide to obtain 409. Palladium-catalyzed intramolecular coupling produced 410. Removal of Ts, followed by reductive amination with formaldehyde solution generated rac-cularine (403). Starting from the intermediate compound 411, catalytic hydrogenation and a detoluenesulfonyl group were used to obtain 412. Subsequently, 412 was reacted with formalin in AcOH via the Mannich reaction to yield the lutenine 413. According to the reported procedure,151 8-oxopseudopalmatine (404) was obtained by oxidation. The intermediate compound 414 and the alkenyliodide 415 were coupled via Suzuki–Miyaura coupling to obtain the key intermediate 416. Photocyclization of 416 with a 365 nm UV lamp at room temperature to obtain 417. Finally, after removal of Ts, followed by simple reductive N-methylation, the first total synthesis of dactyllactone A (405), was realized.
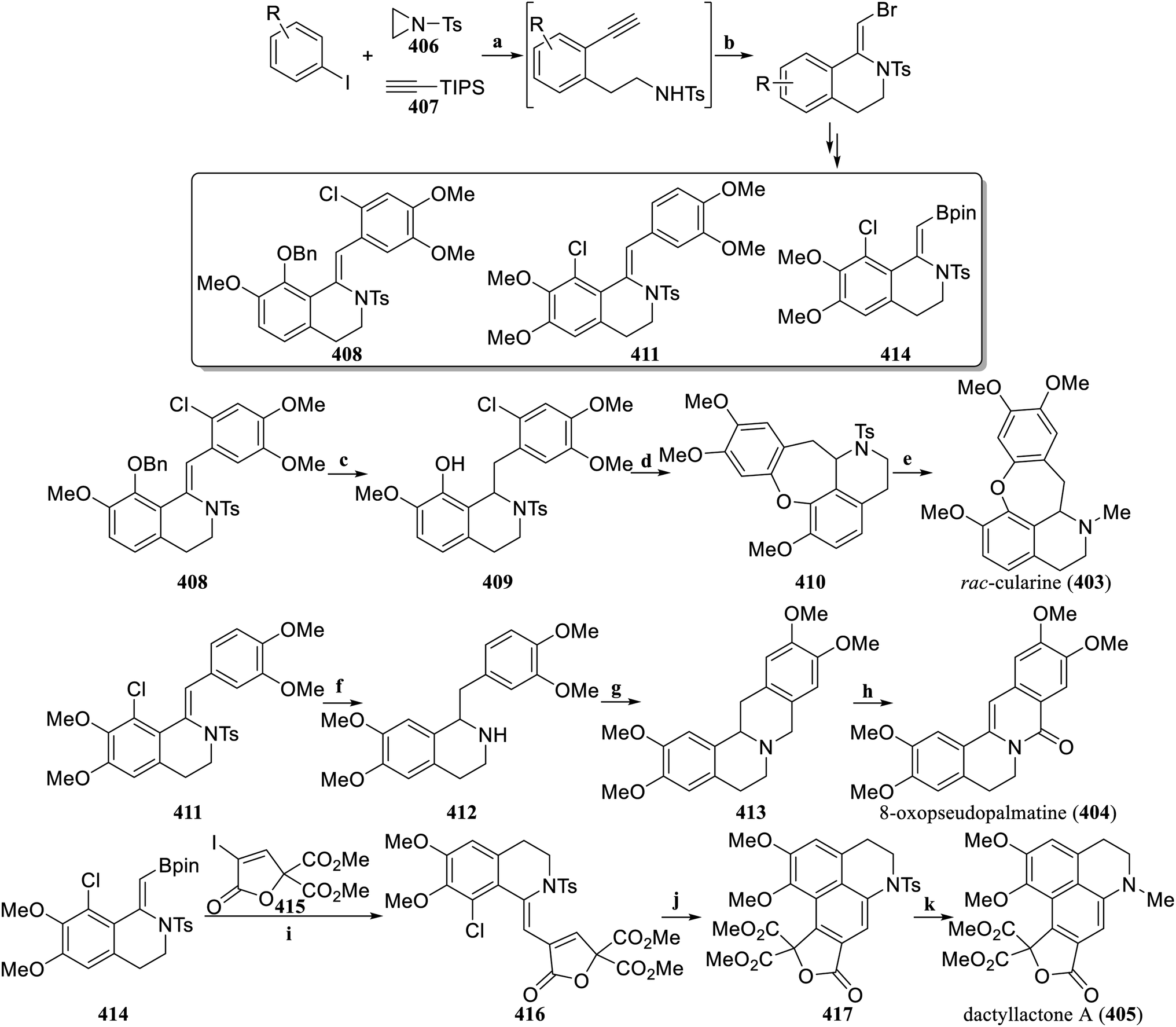 | ||
| Scheme 21 Total synthesis of rac-cularine (403), 8-oxopseudopalmatine (404) and dactyllactone A (405) by Zhou's group. Reagents and conditions: (a) (i) K2CO3, Pd(OAc)2, Dave-Phos, NBE derivative, MeCN, 60 °C, (ii) TBAF, THF, rt; (b) NBS, DBU, MeCN, rt; (c) PtO2, H2, MeOH:DCM = 1:1, TFA, Et3SiH, DCM, rt; (d) Pd(OAc)2, tert-butyl-XPhos, NaOH, toluene, 140 °C; (e) (i) Na, naphthalene, DME, −78 °C, (ii) HCHO (37%), NaBH3CN, acetic acid, MeCN, 0 °C; (f) Pd/C (10%), H2, MeOH/DCM (1:1), Mg powder, MeOH, sonication, rt; (g) HCHO (37%), acetic acid, 100 °C; (h) Pd(OAc)2, Cu(OAc)2, O2, toluene, 120 °C; (i) Pd2(dba)3, AsPh3, Ag2O, THF, rt; (j) hv 365 nm, MeOH, rt; (k) (i) H2SO4, DCM, rt, (ii) HCHO (37%), NaBH3CN, acetic acid, MeCN, 0 °C. | ||
Zhou's group subsequently reported a concise total synthesis method for rac-stepharine (418) and rac-pronuciferine (419) (Scheme 22).152 Through the established Pd/NBE cooperative catalysis of a three-component Catellani reaction and the Suzuki–Miyaura coupling reaction, the key intermediate 420 was prepared, and then coupled with 421via the Suzuki–Miyaura reaction to produce 422. The N-tosyl group was removed, and Au/Ag-catalyzed 6-exo-dig cyclization was performed to produce 1-methylene-THIQ 423. The N-protecting group was replaced, and then 424 underwent oxidative dearomatization promoted by PIDA, followed by reduction with NaBH4 to obtain rac-stepharine (418). Finally, rac-pronuciferine (419) was generated through reductive amination.
 | ||
| Scheme 22 Total synthesis of rac-stepharine (418) and rac-pronuciferine (419) by Zhou's group. Reagents and conditions: (a) (i) K2CO3, Pd(OAc)2, Dave-Phos, 5-norbornene-2-carbonitrile, MeCN, 60 °C, (ii) (Boc)2O, DMAP, CH2Cl2, rt; (b) (i) NaOH, Pd(OAc)2, SPhos, toluene, 95 °C, (ii) TBAF, THF, 50 °C; (c) Mg powder, MeOH, sonication; (d) AuPPh3Cl, AgNTf2, 4 Å molecular sieves, EtOH, CH2Cl2, 30 °C; (e) (i) CF3COOH, rt, (ii) (CF3CO2)2O, Et3N, CH2Cl2, 0 °C; (f) CF3CH2OH, PIDA, NaBH4, 0 °C; (g) HCHO (37%), NaBH3CN, AcOH, MeCN, 0 °C. | ||
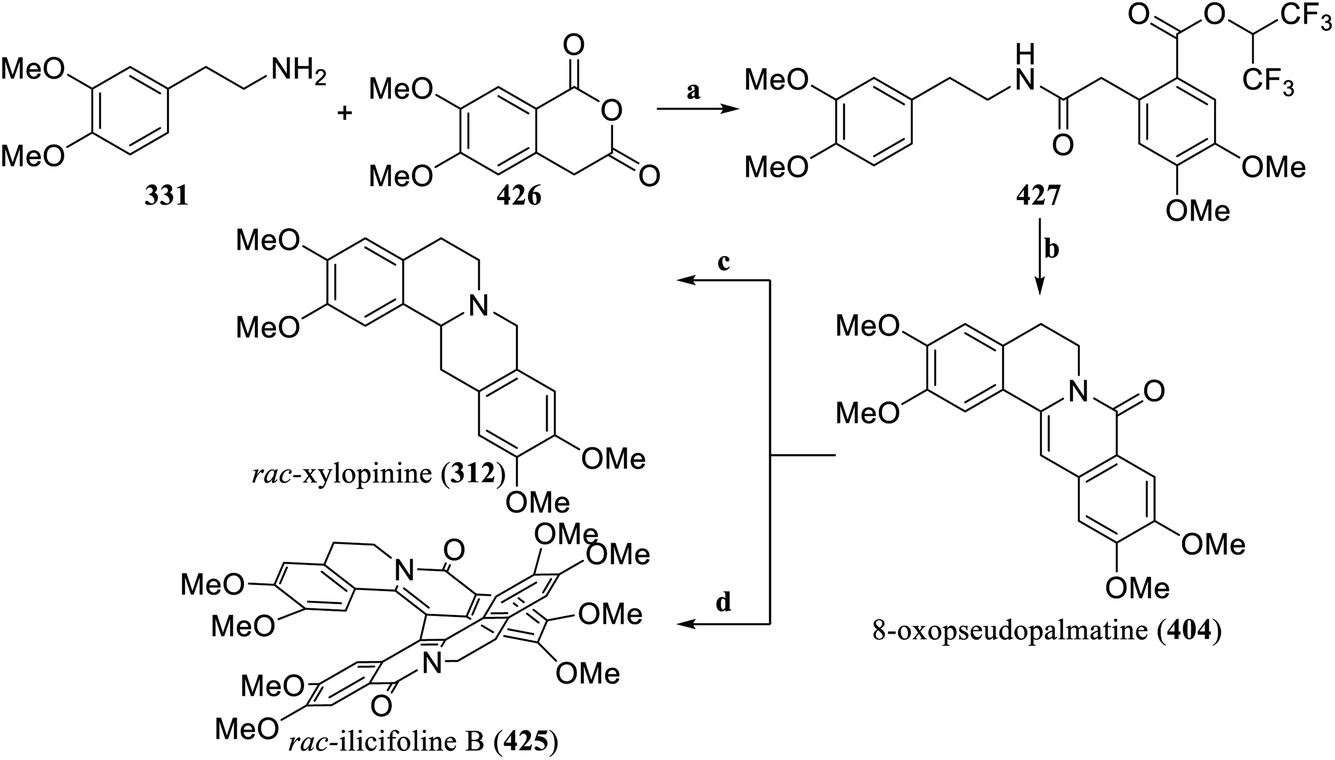 | ||
| Scheme 23 Total synthesis of 8-oxopseudopalmatine (404), rac-ilicifoline B (425) and rac-xylopinine (312), by Christmann's group. Reagents and conditions: (a) EDC, DMAP, HFIP, DCM; (b) (i) POCl3, toluene, 110 °C, (ii) K2CO3, nBu4NBr, MeOH, 80 °C; (c) (i) LiAlH4, AlCl3, Et2O, reflux, (ii) RuCl[(S,S)-TsDPEN](mesitylene), HCOOH, Et3N, rt; (d) PIFA, BF3·Et2O, DCM, −78 °C. | ||
Yamamoto et al. achieved the asymmetric total synthesis of the C8-substituted tetrahydroproberberine natural product (−)-javaberine A (428) and its epimer through asymmetric hydroamination and Bischler–Napieralski cyclization in 2021.156 First, the cyclization precursor was constructed, which obtained 432 by the Heck reaction of 2-bromo-4,5-dimethoxybenzaldehyde (430) and 3,4-dimethylstyrene (431), and then 433 was obtained via the Henry reaction, reduction and methylation reaction. The lithium amide-chiral bisoxazo-line catalyzed the intramolecular asymmetric hydroamination reaction of 433, which was demethylated by Polonovski-type reaction to obtain 434. Condensation of 434 with homoveratric acid afforded the amide 435. Bischler–Napieralski cyclization of 435 produced 436, followed by LiAlH4 reduction and BBr3 demethylation to afford (−)-javaberine A (428) and epi-javaberine A (429) (Scheme 24).
 | ||
| Scheme 24 Total synthesis of (−)-javaberine A (428) and epi-javaberine A (429), by Yamamoto's group. Reagents and conditions: (a) Pd(OAc)2, K3PO4, DMA, 140 °C; (b) (i) MeNO2, NH4OAc, 100 °C, (ii) LiAlH4, THF-Et2O, rt, (iii) HCOOH, Ac2O, rt, (iv) LiAlH4, THF, rt; (c) (i) chiral box ligand, n-BuLi, i-Pr2NH, toluene, −30 °C, (ii) mCPBA, DCM, rt, (iii) HCl, FeSO4·7H2O, rt; (d) EDC, HOBt, Et3N, DCM, rt; (e) POCl3, MeCN, reflux; (f) LiAlH4, THF, −78 °C, (ii) BBr3, DCM, 0 °C. | ||
In 2021, Chen's group completed the asymmetric total synthesis of tetrahydro-protoberberine alkaloids (Scheme 25).157 First, the efficient and sustainable synthesis of the secondary amine hydrochlorides 437a–d was realized by using disubstituted phenethylamine 331, 358 and disubstituted benzaldehyde as raw materials through a continuous flow system. The key Pictet–Spengler reaction/Friedel–Crafts hydroxyalkylation/dehydration cascade cyclization reaction conditions were subsequently optimized to further convert 437a–d to dihydroprotoberberines 438a–d. By screening the chiral ligands, the Ir-catalyzed enantioselective hydrogenation was successfully realized, the chiral center of C-14 was constructed, the efficient asymmetric total synthesis of (−)-canadine (439), (−)-rotundine (440), (−)-sinactine (441), and (−)-xylopinine (326) were completed.
 | ||
| Scheme 25 Total synthesis of (−)-canadine (439), (−)-rotundine (440), (−)-sinactine (441) and (−)-xylopinine (326) by Chen's group. Reagents and conditions: (a) 4 Å MS powder, 10% Pd(OH)2/C-SiO2, H2, MeOH, continuous flow synthesis; (b) B(OH)3, NaCl, 98% HCOOH, 80 °C; (c) [Ir(COD)Cl]2, (S,S)-f-BINAPHANE, KBr, H2, DCM:AcOH = 9:1, rt. | ||
Subsequently, based on this synthetic strategy,157 Chen's group achieved the first fully continuous flow asymmetric synthesis method for natural tetrahydroproto-berberine alkaloids in 2022 (Scheme 26).158 Using commercialized homopiperony-lamine and disubstituted benzaldehyde as starting materials, four reaction steps and work-up processing were realized through six different continuous flow devices integrated in a flow platform, including the reductive amination of secondary amines, the formation of secondary amine hydrochlorides, Pictet–Spengler reaction/Pomeranz–Fritsch reaction cascade cyclization to form the dihydroprotoberberine core skeleton, and enantioselective catalytic hydrogenation. Without any intermediate purification, this new fully continuous process ultimately synthesized (−)-isocanadine (442), (−)-tetrahydropseudocoptisine (443), (−)-stylopine (444) and (−)-nandinine (445) with an enantiomeric excess (ee) of over 84% and a total yield of over 43%. Moreover, the total reaction residence time was reduced from 100 hours to 32.5 minutes. Compared with traditional batch operations, this fully continuous flow synthesis process offers advantages such as high production efficiency, good time economy, and high route sustainability; this provides a new scalable and efficient continuous production technology for the total synthesis of tetrahydroprotoberberine alkaloids. This further demonstrates the potential application of continuous flow reaction technology in the total synthesis of natural products.
 | ||
| Scheme 26 Continuous-flow total synthesis of (−)-isocanadine (442), (−)-tetrahydropseudocoptisine (443), (−)-stylopine (444) and (−)-nandinine (445) by Chen's group. | ||
In 2022, Cuny's group achieved the first total synthesis of pallimamine (446) and its epimer (447) (Scheme 27).159 Vanillin (448) was selectively iodinated and hydroxylated at the 5-position, followed by the method of Banwell et al.160 to obtain 449. The benzaldehyde 449 was condensed with nitromethane, and after reduction of the olefin and nitro groups, the desired phenylethylamine derivative 450 was obtained. The Bischler–Napieralski cyclization then gave the key fragment imine 451. The reaction of the 4-bromo-1,2-dimethoxybenzene (452) with dimethylmalonic acid in the presence of 2,2,6,6-tetramethylpiperidine lithium (TMP) gave the diester 453, followed by methylation and ester hydrolysis to obtain 454. Dehydration cyclization gave another key fragment, anhydride 455. The formal [4 + 2] cycloaddition reaction of 451 and 455 obtained the epimers 456 and 457. After LiAlH4 reduction, intramolecular Mitsunobu reaction achieved the first total synthesis of the pallimamine (446) and epi-pallimamine (447) with total yields of 0.19% and 1.1%, respectively, laying the foundation for pharmacological studies of this interesting and unique tetrahydroberberine natural product.
 | ||
| Scheme 27 Total synthesis of pallimamine (446) and epi-pallimamine (447), by Cuny's group. Reagents and conditions: (a) (i) I2, KI, NaHCO3, H2O, rt, (ii) CuSO4·5H2O, 20% NaOH, reflux, (iii) Me2SO4, Na2CO3, acetone, reflux; (b) (i) MeNO2, NH4OAc, AcOH, 90 °C, (ii) NaBH4, MeOH, 0 °C, (iii) Pt/C, H2, EtOAc, rt; (c) (i) HCO2Et, Et3N, reflux, (ii) POCl3, CHCl3, 55 °C; (d) MeO2CCH2CO2Me, TMP, n-BuLi, THF, −78 °C; (e) (i) LDA, MeI, THF, −78 °C-rt, (ii) KOH, MeOH:H2O (1:2), reflux; (f) AcCl, reflux; (g) (i) DCM, 0 °C-rt, (ii) SOCl2, MeOH, reflux; (h) (i) LAH, THF, 0 °C-rt, (ii) DIAD, n-Bu3P, DCM, rt. | ||
In 2023, Chen's group established an efficient and environmentally friendly green dehydrogenation reaction method. Through I2-mediated electrochemical acceptorless dehydrogenation (ECAD) could eco-efficiently construct aromatized of protoberberine (PB) and 13-methylprotoberberine (13-MePB) in last-stage (Scheme 28).161 This new method for aromatization of the tetracyclic framework was applied to divergent syntheses of ten PBs and eight 13-MePBs in high yields. Importantly, this I2-ECAD protocol can be conducted on a gram scale and applied successfully in a continuous flow microreactor successfully.
 | ||
| Scheme 28 Synthesis of protoberberine (PB) and 13-methylprotoberberine (13-MePB) by Chen's group. | ||
 | ||
| Scheme 29 Total synthesis of O,N-dimethylhamatine (458) by Lipshutz's group. Reagents and conditions: (a) (i) NBS, acetone, rt, (ii) diethoxyphosphorylacetic acid ethyl ester, DBU, MeCN, rt, (iii) 90/10 TFA/H2O, rt; (b) Ac2O, NaOAc, rt; (c) (i) NaOH, EtOH, rt, (ii) Pd/C, H2, EtOH, rt, (iii) MeI, K2CO3, acetone, reflux, (iv) LAH, THF, rt; (d) (i) TBAB, DCM, rt, (ii) PPh3, 1,2-dibromotetrachloroe-thane, DCM, rt, (iii) L-selectride, 0 °C; (e) (i) triisopropylsilyl chloride (TIPSCl), imidazole, DCM, 0 °C, (ii) 3,5-dimethoxybromobenzene, Mg, CuI, THF, −35 °C; (f) (i) PPh3, diisopropyl azodicarboxy-late (DIAD), phthalimide, THF, 0 °C, (ii) (H2N)2, EtOH, reflux, (iii) methyl formate, CH3CO2H, rt; (g) (i) POCl3, 2-methylpyrazine, DCM, 0 °C, (ii) MeMgCl, Et2O, −78 °C, (iii) N-iodosuccinimide, TFA, MeCN, rt, (iv) paraformaldehyde, HCO2H, MeOH, reflux, (v) tetrabutylammonium fluoride (TBAF), THF, rt; (h) dicycloheylcarbodiimide (DCC), 1-naphthoic acid, 4-dimethylaminopyridine (DMAP), DCM, rt; (i) (i) Pd(IPr*)(cinnamyl)Cl, ZnCl2, THF, rt, (ii) NaOH, MeOH, rt; (j) DCC, DMAP, p-bromobenzoic acid, DCM, rt; (k) (i) NaOH, MeOH, rt, (ii) n-BuLi, tosyl chloride (TosCl), −78 °C, H2O, 0 °C, (iii) LiBEt3H, THF, 0 °C-rt. | ||
In 2020, Cheon's group established a new method to construct biaryl products with high atroposelectivity through the atroposelective aryl coupling reaction based on the chirality of the existing center.163 This new method does not require the use of an external chiral source, but utilizes a central chirality group for atroposelectivity control. The total synthesis of C5–C8′ coupled naphthylisoquinoline alkaloids with M-configuration was achieved (Scheme 30). By adjusting the types of ligands and bases, the Suzuki–Miyaura coupling conditions for the naphthyl pinacol boronic acid ester 471 and the chiral aryl iodide 472 were optimized to obtain biaryl coupling product 473 with the M-configuration. After debenzyl protection, ancistrotanzanine B (474) was obtained via the Bischler–Napieralski reaction. Reduction of 474 with NaBH4 gave the cis-product 1-epi-ancistrotectoriline A (475). In the presence of AlEt3, the reduction of 474 with LiAlH4 resulted in the corresponding trans-product ancistrotectoriline A (476), which was then N-methylated to obtain N-methyl-ancistrotectoriline A (477).
 | ||
| Scheme 30 Total synthesis of ancistrotanzanine B (474), 1-epi-ancistrotectoriline A (475), ancistrotectoriline A (476) and N-methyl-ancistrotectoriline A (477) by Cheon's group. Reagents and conditions: (a) Pd(OAc)2, t-BuXPhos, K3PO4, BHT, DMF, 100 °C; (b) (i) Pd/C, H2, MeOH, rt, (ii) POCl3, MeCN, 80 °C; (c) NaBH4, MeOH, rt; (d) AlEt3, LiAlH4, THF, −78 °C-rt; (e) (i) Boc2O, TEA, DCM, rt, (ii) LiAlH4, THF, 60 °C. | ||
In the same year, the total synthesis of naphthylisoquinoline alkaloids with (P)-configurations, including ancistroealaine A (479), ancistrobertsonine C (480) and 6-O-methyl-ancistrobertsonine A (481), were achieved by adjusting the ligand and base species via the same synthesis strategy used by Cheon's group (Scheme 31).164
 | ||
| Scheme 31 Total synthesis of ancistroealaine A (479), ancistrobertsonine C (480) and 6-O-methyl-ancistrobertsonine A (481) by Cheon's group. Reagents and conditions: (a) Pd(OAc)2, SPhos, Ba(OH)2, BHT, DMF, 100 °C; (b) (i) Pd/C, H2, MeOH, rt, (ii) POCl3, MeCN, 80 °C; (c) (i) NaBH4, MeOH, rt, (ii) ClCO2Me, TEA, DCM, 0 °C, (iii) LiAlH4, THF, 60 °C; (d) (i) AlEt3, LiAlH4, THF, −78 °C-rt, (ii) ClCO2Me, TEA, DCM, 0 °C, (iii) LiAlH4, THF, 60 °C. | ||
Zhou et al. established a novel modular method for efficiently constructing 1,3-trans-disubstituted tetrahydroisoquinolines through a three-component Catellani reaction and an Au-catalyzed cyclization/reduction cascade reaction, providing a new synthetic strategy for the total synthesis of naphthylisoquinoline alkaloids and related drugs.149 In this method (Scheme 32), simple aryl iodide 482, aziridine 483 and (triisopropylsilyl)acetylene 407 were used as the building blocks to promote the Catellani reaction through a Pd-ligand/NBE derivative catalytic system, and the Au-catalyzed intramolecular 6-exo-dig cyclization and reduction reaction was used in tandem to construct the 1,3-trans-disubstituted tetrahydroisoquinoline 484 in a concise and efficient manner. The iodo product 485 was then coupled with naphthalene boronic acid 486via a Suzuki–Miyaura coupling reaction to assemble the axially chiral intermediate 487(M + P). Catalytic hydrogenation simultaneously removed benzyl protection and Cbz protection, completing the total synthesis of korupensamine A (488) and B (489). This is the shortest (requiring only five reaction steps) and most efficient (with a total yield of 30%) synthetic route to date.
 | ||
| Scheme 32 Total synthesis of korupensamine A (488) and B (489) by Zhou's group. Reagents and conditions: (a) (i) Pd(OAc)2, DavePhos, NBE derivative, K2CO3, MeCN, 70 °C, TBAF, rt, (ii) AuCl(PPh3), AgSbF6, THF, reflux, (iii) Et3SiH, TFA, 4A MS, DCM, rt; (b) I2, CF3CO2Ag, CHCl3, rt; (c) Pd(OAc)2, XPhos, K3PO4, THF/H2O, 80 °C; (d) Pd/C, H2, EtOH/DCM, rt. | ||
Under the action of TFA, N-iodosuccinimide (NIS) iodized the key intermediate 487 (M + P) to obtain the aryl iodides 492a and 492b, respectively. Subsequently, Turbo Grignard reagent (iPrMgCl·LiCl) was used to treat 492a, forming the aryl zinc reagent 493in situ through metal transfer. It then underwent Negishi coupling with 492a and 492b, respectively. After catalytic hydrogenation to simultaneously remove benzyl and Cbz protections, the more challenging target products, namely, michellamines B (490) and C (491), were efficiently synthesized (Scheme 33).
 | ||
| Scheme 33 Total synthesis of michellamines B (490) and C (491) by Zhou's group. Reagents and conditions: (a) NIS, TFA, MeCN, rt, dark; (b) iPrMgCl·LiClI2, ZnCl2, THF, rt; (c) (i) Pd(PPh3)4, THF, 80 °C, (ii) Pd/C, H2, EtOH/DCM, rt. | ||
In 2023, van Otterlo's group established the first catalyst-controlled atroposelective cross-coupling reaction that enabled the direct synthesis of different naphthylisoquino-line alkaloids from their respective precursors (Scheme 34).165 First, northern naphthalene building blocks started with commercially available 1,8-dihydroxynaphthalene (494), naturally occurring methyl groups were selected for protection of phenol functional groups, and then regioselective OH-directed bromination at the 8′-position of the naphthalene core were performed using NBS. The naturally occurring methyl and synthetically more labile isopropyl group was then selected as the protective group of the phenol functional group in 495, and gave 496a,b. Hartwig's CuI-catalyzed methylation reaction was used to achieve regioselective methylation, and then the bromide 497a,b was converted to northern naphthalene building blocks 498a,b. From the Henry reaction of 3,5-dimethoxybenzal-dehyde (499) catalyzed by LiAlH4 with nitroethane, and then the TBS protection of free alcohols, 500 in cis/trans configuration was obtained. The secondary nitro group was then converted to an amine. The acylation of the amine provides the amide 501, which then undergoes a Bischler–Napieralski reaction to obtain one of the southern isoquinoline building block 502.
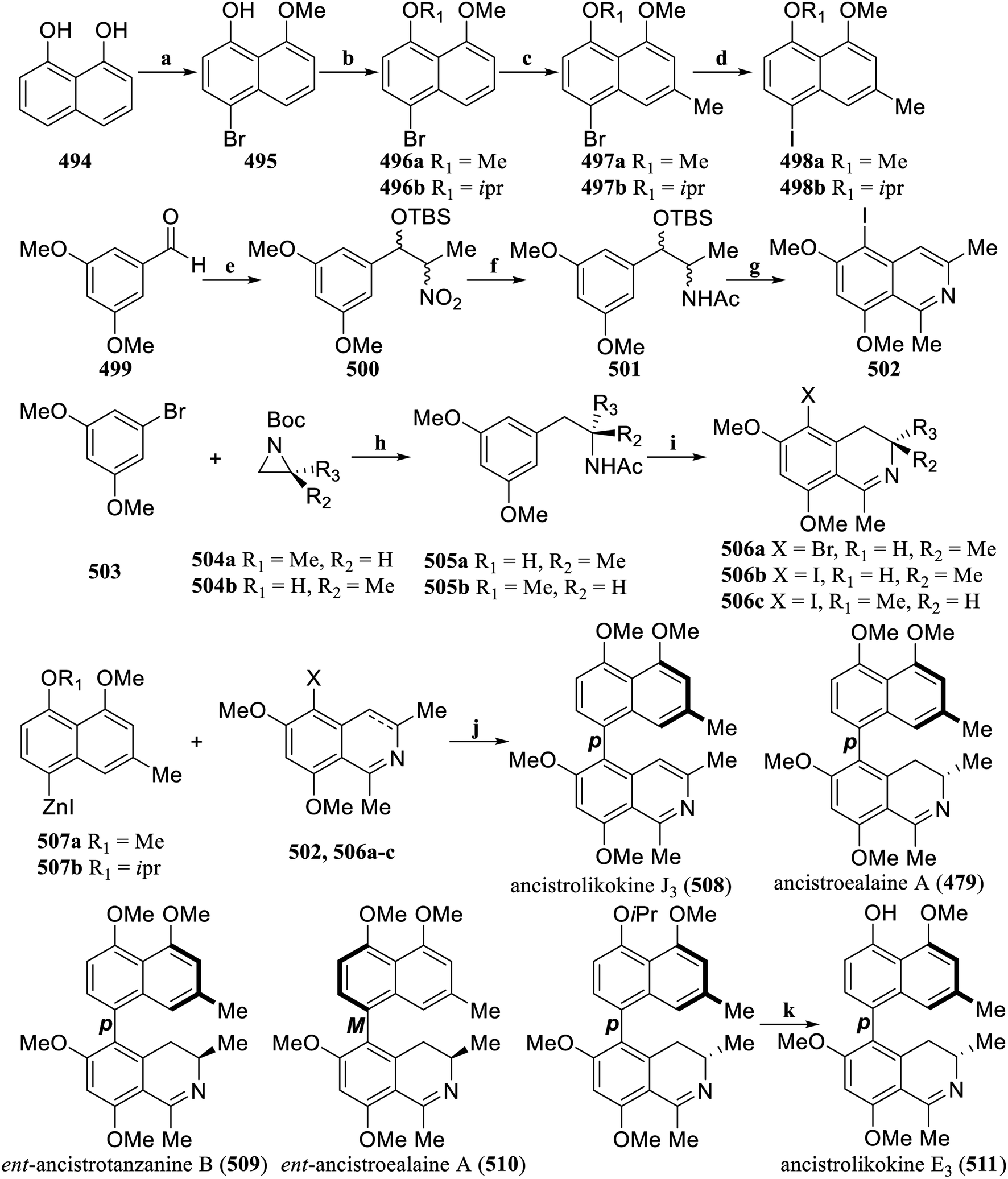 | ||
| Scheme 34 Total synthesis of 5,8′-naphthylisoquinoline alkaloids (479, 508–511) by van Otterlo's group. Reagents and conditions: (a) (i) MeI, K2CO3, acetone, rt, (ii) NBS, MeCN, rt; (b) iPrI, K2CO3, acetone, 80 °C; (c) (i) HBpin, [{Ir(cod)OMe}2], dtbpy, THF, 80 °C, (ii) (MeO)3PO, LiOtBu, CuI, LiI, DMI, 50 °C; (d) nBuLi, Et2O, I2, −78 °C-rt; (e) (i) EtNO2, LiAlH4, THF, 0 °C-rt, (ii) TBSCl, imidazole, DCM, 45 °C; (f) (i) CoCl2, NaBH4, MeOH, 0 °C-rt, (ii) AcCl, DMAP, NEt3, DCM, 0 °C-rt; (g) (i) POCl3, MeCN, 85 °C, (ii) NIS, TfOH, 0 °C; (h) (i) Mg, THF (0.5 m), rt-70 °C, then 505a,b, CuBr, THF, 0 °C-rt, (ii) TFA, DCM, 0 °C-rt, then AcCl, DMAP, NEt3, DCM, 0 °C-rt; (i) (i) NIS, TFA, MeCN, 0 °C-rt, (ii) Tf2O, 2-Cl-pyridine, DCM, −78 °C-rt; (j) NiCl2(DME), Lassaletta's N,N-ligand, THF/DMF, 60 °C; (k) BCl3, DCM, −30 °C. | ||
The format reagent generated in situ from 503 attacks the Boc-protected chiral aziridine 504a,b, and then the Boc is deprotected and acetylated to obtain the chiral amide 505a,b, followed by a iodination and a final Bischler–Napieralski reaction to obtain the southern isoquinoline building blocks 506a–c. Based on the Lassaletta's second-generation N,N-ligand, a catalyst-controlled Pd-catalyzed Suzuki cross-coupling reaction system was established, and a series of naphthalene isoquinoline alkaloids 479, 508–511 were obtained.
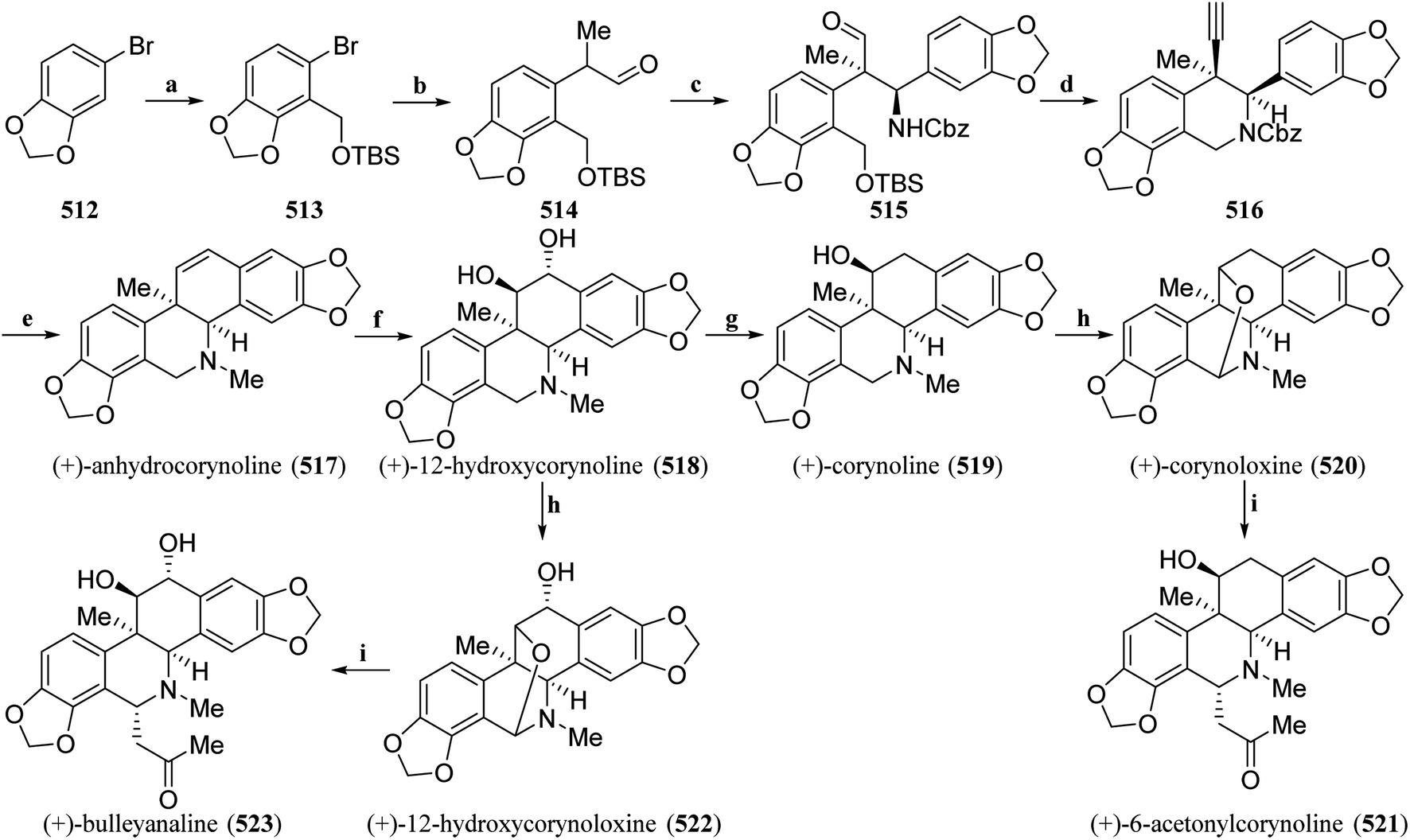 | ||
| Scheme 35 Total synthesis of isoquinoline alkaloids (517–523) from genus Corydalis by Trost's group. Reagents and conditions: (a) (i) LDA, THF, −78 °C, DMF, rt, (ii) NaBH4, MeOH, rt (iii) TBSCl, imidazole, rt; (b) (i) Pd(OAc)2, DavePhos, LiHMDS, PhMe, 70 °C, (ii) DIBAL-H, DCM, −78 °C; (c) imine, Zn-(S,S)-ProPhenol, PhMe, 3A MS, 4 °C; (d) (i) K2CO3, Ohira–Bestmann reagent, MeOH, rt, (ii) CSA, MeOH, sulfonyldiimidazole, Cs2CO3, MeCN, rt; (e) IPrAuNTf2, PhMe, rt, then Red-Al, rt-80 °C; (f) H2O2, HCOOH, then Na2S2O5, NaOH, rt; (g) Et3SiH, BF3·Et2O, DCE, rt; (h) I2, KOAc, K2CO3, EtOH, reflux; (i) KOH, MeOH, 50 °C. | ||
In 2020, Snaddon's group achieved enantioselective synthesis of chelidonium alkaloids (Scheme 36) by establishing asymmetric allylic alkylation reaction conditions catalyzed by a Lewis base and transition metal,170 combined with Hofmann rearrangement.171 Under the optimized reaction conditions, the electrophile 525 underwent asymmetric allylic alkylation with 524, followed by ammonolysis of the ester using ammonium hydroxide and reduction of the aldehyde to obtain the corresponding alcohol 526. PIDA was used to facilitate Hofmann rearrangement of 526, and then the primary alcohol was converted into the corresponding bromide before treatment with sodium hydride to promote intramolecular N-alkylation, resulting in 527. The second-generation Grubbs catalyst facilitated ring-closing metathesis, resulting in benzo[c]phenanthridine skeleton 528. N-Bromosuccinimide (NBS) mediated regioselective bromonium ion hydrolysis to obtain bromohydrin, which was then treated with potassium tert-butoxide at low temperature to obtain the critical epoxide 529. LiAlH4 reduced the epoxide 529 to give (+)-chelidonine (530). (+)-Norchelidonine (531) was prepared by treating 529 with L-selectride. Bismuth trichloride catalyzed the hydrolytic ring-opening of epoxide 529, followed by reduction with LiAlH4 to produce (+)-chelamine (532). In addition, the efficient synthesis of strychnos alkaloids and pharmaceutical lead compounds was achieved by combining Lewis base/transition metal-catalyzed asymmetric allyl alkylation with Hoffman rearrangement, indicating that this strategy has important application value in the synthesis of other alkaloids and analogs designed for drug development.
 | ||
| Scheme 36 Total synthesis of (+)-chelidonine (530), (+)-norchelidonine (531) and (+)-chelamine (532) by Snaddon's group. Reagents and conditions: (a) (i) (S)-BTM, Hartwig's (R,R,Ra)-Ir catalyst, iPr2NEt, THF, NH4OH, rt, (ii) NaBH4, MeOH, rt; (b) (i) PhI(OAc)2, KOH, MeOH/THF, rt, (ii) PPh3, CBr4, then NaH, DMF, rt; (c) Grubbs second generation catalyst, PhMe, rt; (d) (i) NBS, H2O, rt, (ii) KOtBu, THF, −78 °C; (e) LiAlH4, 1,4-dioxane, reflux; (f) L-selectride, THF, 40 °C; (g) (i) BiCl3, MeCN/H2O, 0 °C, (ii) LiAlH4, 1,4-dioxane, reflux. | ||
Murphy's group and Banwell's group completed the total synthesis of zephycandidine A (533) in 2020 and 2021, respectively. Murphy's group used haemanthamine (534), which can be obtained in large quantities, as a raw material, which can be directly converted into trispheridine (535) at high temperatures, and then placed in 1,2-dibromoethane to heat, and the key intermediate 536 can be obtained. Finally, according to the method reported by Cronin and coworkers,172 the target compound 533 was obtained by reacting with liquid ammonia to close the ring (Scheme 37a).173
 | ||
| Scheme 37 Total synthesis of zephycandidine A (533) by Murphy's and Banwell's group. Reagents and conditions: (a) heat, 190–195 °C; (b) 1,2-dibromoethane, 90 °C; (c) (i) NH3 (liq.), −78 to −33 °C, (ii) MnO2, Na2CO3, −78 °C-rt, (iii) toluene, 111 °C; (d) (i) I2, K2CO3, t-BuOH, 70 °C, (ii) PhI(OAc)2, K2CO3, DMSO, rt; (e) Pd(OAc)2, NaOAc, DMF, 110 °C. | ||
Banwell's group achieved the total synthesis of 533 from readily available starting materials via Buchwald–Hartwig and Heck reactions (Scheme 37b).174 According to the method reported by Ishihara and Togo, the 2-imidazoline derivative 538 could be synthesized using 537 and ethylenediamine as raw materials. The total synthesis of 533 was subsequently achieved via palladium-catalyzed cross-coupling of 538 with o-iodophenylboronic acid (539).
Jeganmohan's group had established a series of new methods for the synthesis of benzophenanthridines 544, cis-fused dihydrobenzo[c]phenanthridinones 545 and benz-o[c]phenanthridine alkaloids 546 by a metal-catalyzed reaction of 7-azabenzonorborna-dienes 543 with the aromatic aldoximes 540,175 aryl amides 541176 and the aryl nitrones 542,177 and applied them to the synthesis of natural products (Scheme 38).
 | ||
| Scheme 38 Synthesis methods of benzophenanthridines (544), cis-fused dihydrobenzo[c]phenanth-ridinones (545) and benzo[c]phenanthridine alkaloids (546) by Jeganmohan's group. Reagents and conditions: (a) [{Rh(CH3CN)3(Cp*)}{SbF6}2], NaOAc, DCE, 100 °C; (b) 6 N HCl, 1,4-dioxane, 80 °C; (c) [Ru(p-cymene)Cl2]2, AgSbF6, AgOTf, DCE, 100 °C; (d) [Cp*RhCl2]2, AgSbF6, Ag2O, 4A MS, DCE, 120 °C. | ||
In 2022, Liu et al. established a novel strategy for the efficient construction of benzo[c]phenanthridine structures through sequential transition metal-catalyzed reactions and condition-controlled Mannich reactions,178 achieving efficient total synthesis of eight benzo[c]phenanthridine alkaloids and two derivatized molecules (25–34% total yield).
The strategy began with the preparation of diaryl acetylenes using readily available building blocks 547 and 548via the Sonogashira coupling reaction, and then the key 1,5-enynes 549 was synthesized via a one-pot Wittig reaction. Subsequently, the 1-iodo-2-phenylnaphthalenes 550 was synthesized under the optimal reaction conditions of one-pot Au/Ag-catalyzed cyclization tandem in situ iodination. The copper-catalyzed cross-coupling reaction between 550 and methylamine was used to construct the C–N bond, and the total synthesis of benzo[c]phenanthridine alkaloids was completed through a one-pot Mannich reaction under different reaction atmospheres. The oxidative products 551a–e were synthesized under an oxygen atmosphere, whereas the reductive products 552a–e were synthesized under a nitrogen atmosphere (Scheme 39). This efficient and economical new synthetic strategy is expected to further expand the research and application of benzo[c]phenanthridine alkaloids and their derivatives.
 | ||
| Scheme 39 Total synthesis of benzo[c]phenanthrene alkaloids by Liu's group. Reagents and conditions: (a) (i) Pd(Ph3P)2Cl2, CuI, Et3N, THF, rt, (ii) methyltriphenylphosphonium bromide, NaHMDS, THF, −78 °C-rt; (b) IPrAuCl, AgSbF6, NIS, DCM, rt; (c) Cu (powder), MeNH2 (H2O), EtOH, 110 °C; (d) (HCHO)n, TFA, N2, MgSO4, EtOH, 65 °C; (e) (HCHO)n, TFA, O2, MgSO4, EtOH, 65 °C. | ||
Amaryllidaceae alkaloids such as γ-lycorane have gained great attention in the field of total synthesis.179 In recent years, many studies have investigated different synthesis strategies. In 2020, Prasad's group accomplished the total synthesis of γ-lycorane is accomplished from (S)-lactic acid (Scheme 40a).180 The known phosphonate 553 derived from (S)-lactic acid was used as the raw material and converted into the Weinreb amide 554 by Horner–Wittig olefination, Claessen rearrangement, etc. The Weinreb amide 554 reacts with vinylmagnesium bromide, and then undergoes Luche reduction, hydroxyl protection and deprotection to obtain 555. By Overman rearrangement, RCM ring closure, deprotection, oxidation and stereoselective reduction, the allyl alcohol 555 was converted to cyclohexene 556. After Claessen rearrangement, the ring was closed to obtain lactam 557, and finally, the Pictet–Spengler reaction was used to form an isoquinoline backbone, and the lactam was reduced to obtain (+)-γ-lycorane (558a).
 | ||
| Scheme 40 Total synthesis of γ-lycorane. Reagents and conditions: (a) (i) vinylmagnesium bromide, THF, 0 °C, (ii) CeCl3·7H2O, NaBH4, MeOH, rt, (iii) MOMCl, DIPEA, DMAP, DCM, reflux, (iv) TBAF, THF, 0–50 °C; (b) (i) Cl3CCN, DBU, DCM, 0 °C, then xylene, MW 160 °C, (ii) Grubbs' 2nd generation catalyst, DCM, reflux, (iii) PPTS, MeOH/H2O, 80 °C, (iv) DMP, NaHCO3, DCM, 0 °C-rt, then NaBH4, CaCl2, MeOH, 0 °C; (c) (i) CH3C(OEt)2, EtCOOH, toluene, seal tube, 150 °C, (ii) Cs2CO3, DMSO, 100 °C; (d) (i) (HCHO)n, TFA, DCE, rt, (ii) H2, Pd/C, MeOH, rt, (iii) LiAlH4, THF, reflux; (e) (i) Pd(OAc)2, DMSO, O2, 65 °C, (ii) n-Bu3P, DIPEA, 110 °C; (f) (i) H2, PtO2, EtOH, rt, (ii) LiAlH4, THF, reflux; (g) (i) MeOH, r.t, (ii) NaBH4, 0 °C; (h) I2/K2CO3, DCM, 0 °C-rt; (i) Pd(PPh3)4, Cs2CO3, 1,4-dioxone, 100 °C; (j) Br2, DCM, −17 °C; (k) (i) KOt-Bu, THF, rt, (ii) Pd(OAc)2, PPh3, i-Pr2NEt, DMF, reflux, (iii) H2, Pd/C, MeOH, rt, (iv) LiAlH4, THF, reflux. | ||
Chuang et al. developed a Pd-catalyzed aza-Wacker-Heck cyclization reaction to construct the galanthan skeleton.181 Through this one-pot sequential C–N and C–C bond-forming process, fused aza-tetracyclic compounds can be efficiently synthesized. Based on this synthetic strategy, using compound 559 as the starting material, galanthan-7-one and its isomer were concisely synthesized via a one-pot method. Subsequent reduction of the olefins and carbonyl groups produced (±)-γ-lycorane (558c) (Scheme 40b).
Wang and coworkers have successfully established a synthetic strategy with palladium-catalyzed aryl C–H alkylation as the key reaction,182 achieving the concise and efficient total synthesis of (−)-γ-lycorane (558b) (Scheme 40c). The benzylamine 561 underwent reductive amination with the aldehyde 560, and the intermediate product 562 was generated. The intermediate products underwent intramolecular iodine cyclization under the action of I2/K2CO3 to construct a cis-5,6-ring structure, and then palladium-catalyzed aryl C–H alkylation was carried out to obtain (−)-γ-lycorane (558b).
In 2022, Donohoe's group, for the first time, applied hydrogen-borrowing C–C bond formation as a key step in total synthesis (Scheme 40d), developing a hydrogen-borrowing alkylation of aziridinyl alcohol combined with Ph* (Me5C6) deprotection/cyclization reaction, which efficiently constructed fused 6,5 heterocyclic structures.183 Initially, chiral aziridine was synthesized from the cyclohexenone 563 according to the method reported in the literature.184 After reduction, deprotection, and nucleophilic substitution reactions, the key aziridinyl alcohol 564 was obtained. Research on hydrogen-borrowing C–C bond formation reactions has revealed that the hydrogen-borrowing catalytic alkylation of Ph*COMe with the aziridinyl alcohol 564 was achieved. Treatment with liquid bromine produced the dibrominated lactam 565, which was used to construct the 6,5 heterocyclic. Following regioselective elimination of the alkyl bromide, an intramolecular Heck reaction was used for cyclization to construct the galanthan skeleton. Reduction of the double bond and carbonyl group afforded (−)-γ-lycorane (558b). This synthetic strategy has potential application value in the construction of various complex polycyclic nitrogen-containing natural products and their derivatives.
Recently, Yang's group achieved the collectively asymmetric total synthesis of all members of lycorane, including (+)-α, (+)-β, (+)-γ, and (−)-δ, using Carreira's dual Ir/amine-catalyzed α-allylation of the 2-phthalimidoacetaldehyde (567) with allylic alcohol 566 as a key reaction (Scheme 41).185 Through the regulation of chiral ligands, 568a,b with different stereoselectivities could be obtained after Sakurai allylation. 568a underwent RCM cyclization and catalytic hydrogenation to obtain the alcohol 569, followed by DMP (Dess–Martin periodinane) oxidation and the Horner–Wadsworth–Emmons reaction to produce the α,β-unsaturated ester 570. Under different reduction conditions, the stereoselective products 571a and 571b are generated. Then, intramolecular nucleophilic substitution was carried out via the Mitsunobu reaction to give 572a and 572b. Pictet–Spengler cyclization of 572a with eschenmoser salt gave (+)-α-lycorane (558d). The amine 572b was converted to carbamate and then subjected to the Bischler–Napieralski reaction, followed by the reduction of the oxolycorane intermediate to obtain (+)-β-lycorine (558e).
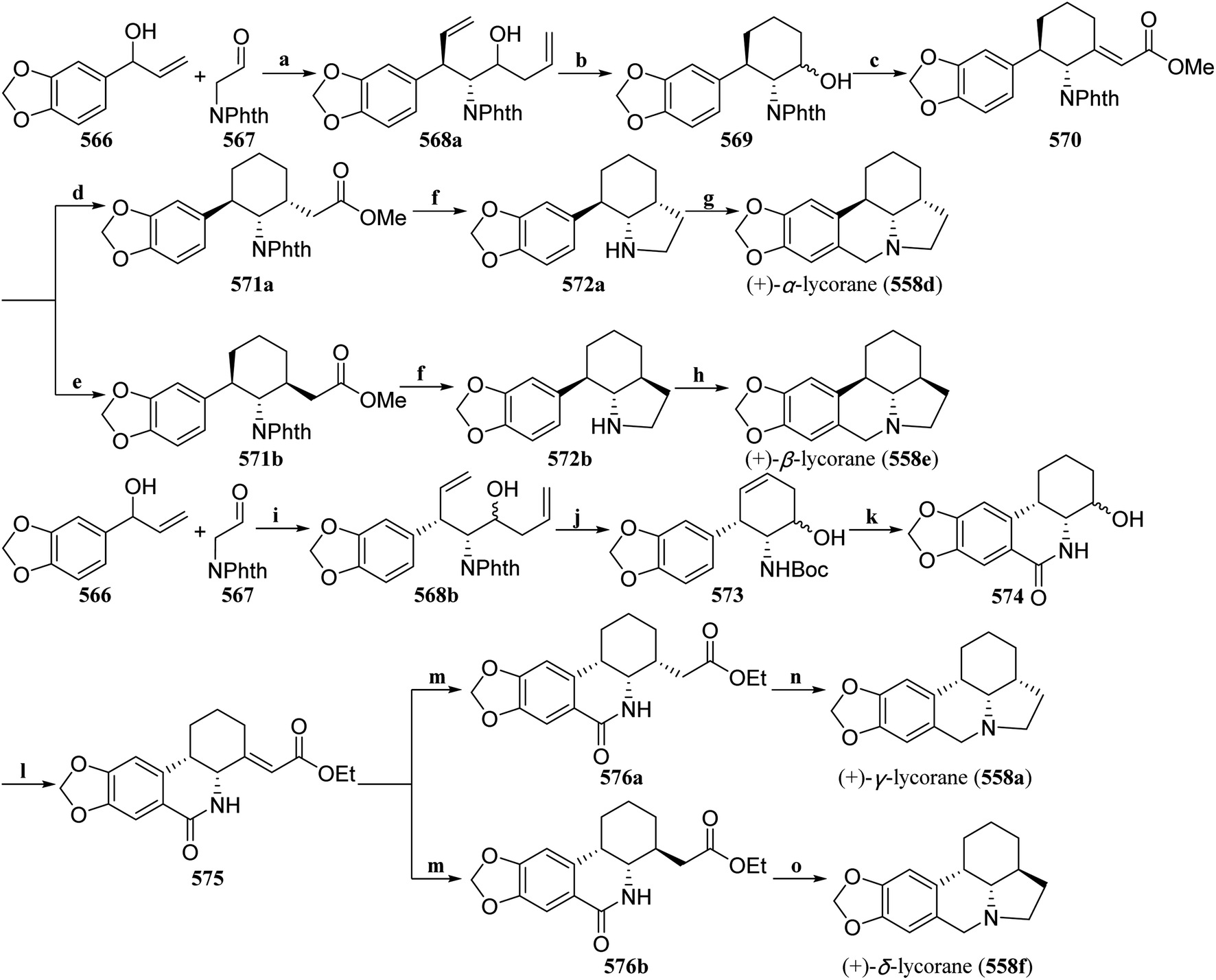 | ||
| Scheme 41 Total synthesis of γ-lycorane by Yang's group. Reagents and conditions: (a) (i) [Ir(cod)Cl]2, Carreira ligand (R)-L, proline-derived amine (S)-A, maleic acid, DCE, 0 °C, (ii) allyltrimethylsilane, BF3·Et2O, DCM, −78–0 °C; (b) (i) Grubbs' 2nd generation catalyst, DCM, reflux, (ii) H2, Pd/C, EtOH, rt; (c) (i) DMP, DCM, rt, (ii) trimethyl phosphonoacetate, t-BuOk, THF, 0 °C-rt; (d) H2, Pd/C, EtOH, rt; (e) NiBr2(DME), (S)-Me-DuPhoS, ZnCl2, DMF/H2O, 110 °C; (f) (i). NaBH4, i-PrOH/H2O, 100 °C, (ii) DIAD, PPh3, THF, 0 °C-rt; (g) Eschenmoser's salt, THF, 40 °C; (h) (i). ClCO2Me, Et3N, DCM, rt, (ii) POCl3, 90 °C, (iii) LiAlH4, THF, rt; (i) (i) [Ir(cod)Cl]2, Carreira ligand (S)-L, proline-derived amine (S)-A, Bi(OTf)3, DCE, 0 °C, (ii) ethylenediamine, MeOH/DCM, 40 °C, (iii) (Boc)2O, Na2CO3, THF/H2O, rt; (j) (i) Grubbs' 2nd generation catalyst, DCM, reflux, (ii) H2, Pd/C, EtOH, rt; (k) (i) H2, Pd/C, EtOH, rt, (ii) Ac2O, Et3N, DMAP, DCM, rt, (iii) PPh3O, BF3·Et2O, Tf2O, DCM, rt, (iv) K2CO3, MeOH, rt; (l) (i) DMP, DCM, rt, (ii) ethyl (triphenylphosphoranylidene)-acetate, DCM, rt; (m) H2, Pd/C, EtOH, rt; (n) (i) DIBAL-H, DCM, −78 °C (ii) LiAlH4, THF, 50 °C; (o) (i) LiAlH4, THF, reflux, (ii) MsCl, Et3N, DCM, 0 °C. | ||
On the other hand, 568b underwent RCM cyclization, and the phthalimide was replaced with a Boc group to give 573. The catalytic hydrogenation of 573 afforded cyclohexane, which was acetylated with Ac2O. Upon treatment with Hendrickson's reagent, the Bischler–Napieralski reaction was performed, resulting in an acetylated intermediate that was converted to 574. After DMP oxidation and then Wittig reaction, 575 was obtained. Under different reduction conditions, the stereoselective products 576a and 576b were produced. Dibal-H was added to reduce 576a, quickly close the ring, and then reduce the lactam, giving (+)-γ-lycorane (558a). 576b was reduced to an amino alcohol, which was then converted to (−)-δ-lycorane (558f) by the intramolecular displacement of methanesulfonate.
In 2022, Waldvogel and Opatz et al. efficiently constructed spirodienones via an electrochemical method with simple starting materials.186 Based on this versatile key transformation (Scheme 42), they designed a general biomimetic synthetic strategy for Amaryllidaceae alkaloids. Different aldehydes react with 577 to obtain the key reaction precursor 578, which is then converted into the corresponding spirodienone 579 through an electrocatalytic reaction. This key reaction could achieve excellent yields in both batch and continuous flow reactors. After the deprotection of 579 under alkaline conditions, the intramolecular azaza-Michael addition reaction occurred to generate the tetracyclic cores 580a–c. After removing the benzyl protection from 580b, it along with 580a and 580c, was reduced by an L-selective reducing agent to produce rac-siculine (581), rac-maritidine (582a), rac-epimaritidine (582b), rac-crinine (583a), and rac-epicrinine (583b).
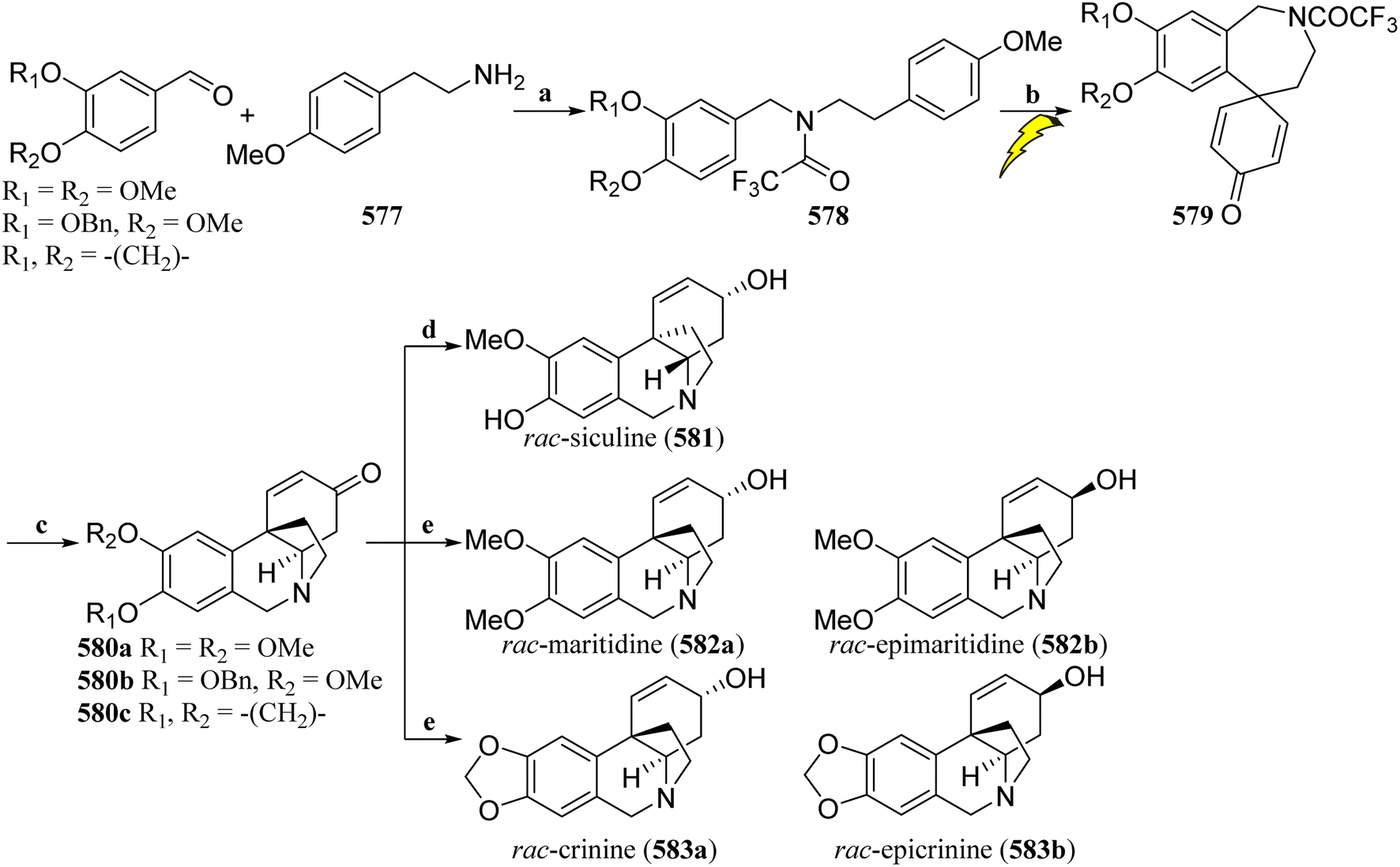 | ||
| Scheme 42 Total synthesis of Amaryllidaceae alkaloids (581, 582a,b, 583a,b) by Waldvogel and Opatz's group. Reagents and conditions: (a) (i) MeOH, 80 °C, (ii) NaBH4, 0 °C, (iii) TFAA, pyridine, 0 °C; (b) BDD/Pt, MeCN, aq. HBF4, −20 °C, 2.0 F, 1.0 mA cm−2, 200 rpm; (c) 10% KOH, MeCN, r.t; (d) (i) BCl3, DCM, −78 °C, (ii) L-selectride, THF, −78 °C; (e) L-selectride, THF, −78 °C. | ||
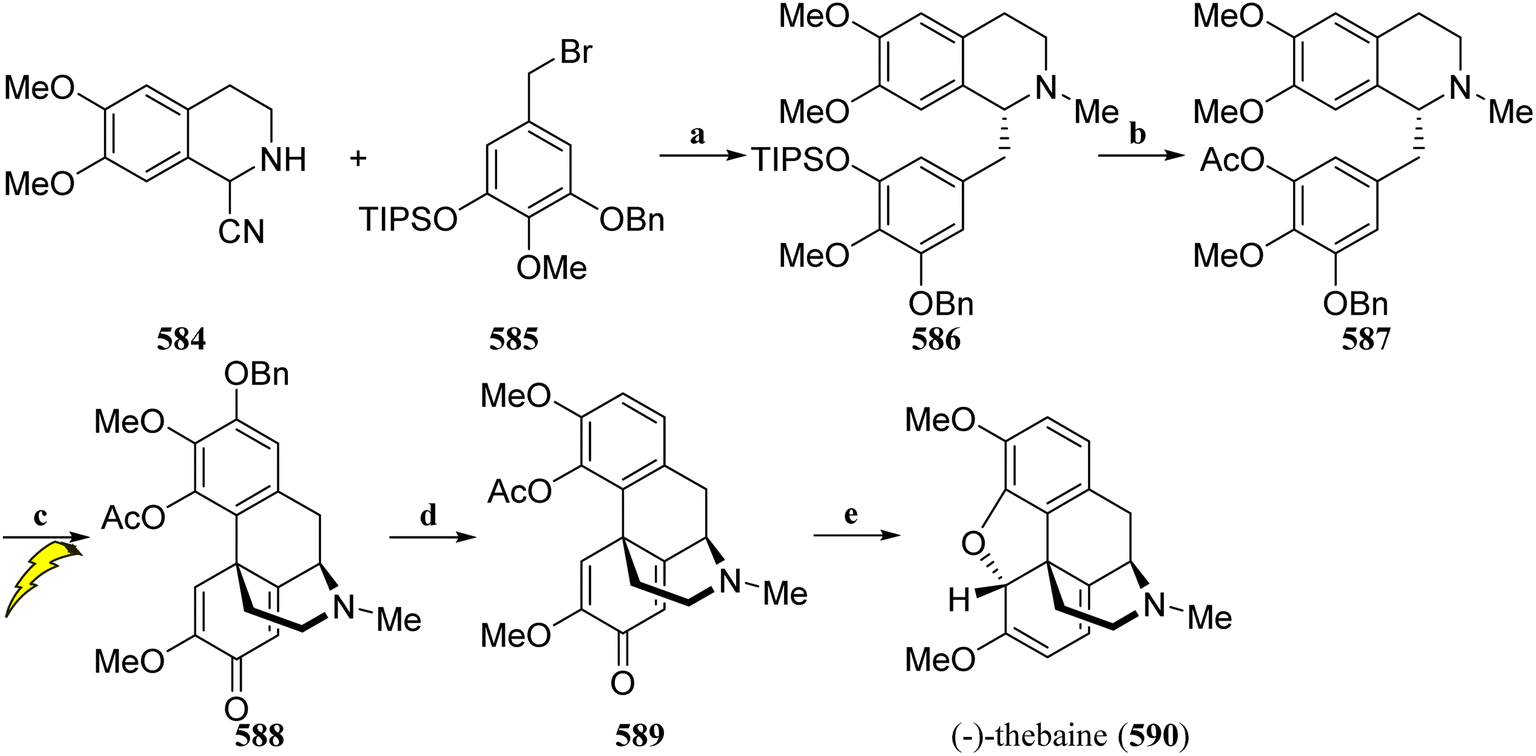 | ||
| Scheme 43 Total synthesis of (−)-thebaine (590) by Opatz's group. Reagents and conditions: (a) (i) KHMDS, THF, −78 °C, then bromide 585, −78 °C-rt, (ii) RuCl(p-cymene)[(S,S)-Ts-DPEN], HCOOH, Et3N, DMF, 0 °C-rt, (iii) HCHO, NaBH4, MeOH, 0 °C; (b) (i) TBAF, THF, rt, (ii) AcCl, Et3N, DMAP, DCM, rt; (c) constant current electrolysis, undivided cell, Pt electrodes, j = 1.5 mA cm−2, Q = 2.2 F, MeCN, HBF4, 0 °C; (d) (i) Pd/C, 1,4-cyclohexadiene, EtOH, 35 °C, (ii) Tf2O, pyridine, 0 °C, (iii) Pd(PPh3)4, Et3N, HCOOH, DMF, 60 °C; (e) (i) K2CO3, MeOH, rt, (ii) CeCl3·7H2O, NaBH4, MeOH, 0 °C, (iii) N,N-dimethylformamidedineopentyl acetal, dioxane, 80 °C. | ||
In 2021, Dong's group developed an asymmetric catalytic “cut-and-sew” strategy that efficiently constructed all-carbon fused-ring structures and quaternary stereocenters. Based on this strategy, they achieved the first asymmetric total synthesis of the morphine alkaloid (−)-thebainone A (591) (Scheme 44).189 Initially, the key ketal-containing substrate (595) was prepared through a Mitsunobu coupling reaction between the phenol 594 and the alcohol 593, which was synthesized via Birch reduction and ketal formation from the anisole 592.
 | ||
| Scheme 44 Total synthesis of (−)-thebainone A (591) by Dong's group. Reagents and conditions: (a) (i) Li/NH3, tBuOH, THF, −78 °C, (ii) PPTS, glycol, DCM, rt; (b) DEAD, PPh3, THF, rt; (c) [Rh(COD)2]NTf2, (R)-DTBM-segphos, 1,2-DFB, 130 °C; (d) (i) LiAlH4, THF, 0 °C, (ii) Ac2O, pyridine, DMAP, then 2 M HCl, 0 °C; (e) (i) BBr3, DCM, −30 °C, (ii) CH2N2, MeOH, Et2O, rt; (f) TMSOCH2CH2OTMS, TMSOTf, then pyridine, then TsNHMe, Cs2CO3, then MeOH; (g) Martin's sulfurane, DCM, rt; (h) sodium naphthalenide, THF, −30 °C, then 2 M HCl; (i) NaSEt, DMF, 100 °C; (j) TFA, MeCN, Pd(TFA)2, DMSO, 80 °C. | ||
After optimizing the conditions for the asymmetric “cut-and-sew” reaction, the key intermediate 596 was obtained with 80% yield and 97:3 er. The ketone in 596 was reduced via LiAlH4, followed by acetate protection and in situ ketal removal to yield the ketone 597. The C–O bond in the dihydropyran moiety of 597 was cleaved using BBr3 and methylated to produce 598. Afterthe ketone was protected and aminated via an SN2 reaction, followed by the removal of the Ac protecting group, compound 599 was obtained. Upon elimination of the alcohol moiety using Martin's sulfurane, a formal hydroamination process occurred in 600 using sodium naphthalenide, completing the construction of the piperidine ring and revealing the ketone moiety. The more sterically hindered methyl ether in 601 was selectively cleaved under the nucleophilic conditions to give 602. Finally, the asymmetric total synthesis of (−)-thebainone A (591) was achieved through the α,β-desaturation of the ketone 602via the Pd(TFA)2/DMSO system described by Ellman and colleagues.190 This synthetic strategy, with its deconstructive approach, provides new insights for building more complex structures.
Zhu's group established the asymmetric catalytic enantioselective construction of tricyclic enone intermediates in 2021 and developed three different intramolecular C–N bond formation processes,191 achieving the enantioselective total synthesis of three topologically distinct hasubanan alkaloids (Scheme 45). The aldehyde 603 underwent Wacker oxidation, aldol condensation, and aromatization to obtain the β-naphthol 604. After allylation of the phenolic hydroxyl group, the α-allyl-β-naphthol 605 was obtained through Claisen rearrangement. Following You's reported reaction conditions,192 the Takemoto's catalyst catalyzed a dearomatizative Michael addition reaction between the β-naphthol 605 and nitroethylene, resulting in an α,α-disubstituted β-naphthalenone. Reduction of the enone gave 606. Further reduction with LiAlH4 produced amino alcohol, which was then N-acylated under Schotten–Baumann conditions and subsequently reduced to obtain the N-methylamine 607. The amino group of 607 was protected with a Boc group, followed by in situ oxidation of the secondary alcohol to produce the key synthetic intermediate ketone 608. The Wacker oxidation of 608 gave the diketone intermediate 609, which underwent intramolecular aldol condensation, and cyclopentenone was dehydrogenated to obtain a highly π-conjugated tricyclic compound 610. After carbonyl α-bromination, the O-MOM and N-Boc protecting groups were removed and cyclized to obtain (−)-sinoracutine (611). Under the action of the second-generation Grubbs catalyst, 608 underwent a cross-metathesis reaction with isopropenylboronic acid pinacol ester, and diketone 612 was obtained after oxidation. The intramolecular aldol condensation reaction gave tricyclic enone 613, which was deprotonated and hydroxylated to form an α-hydroxy ketone, and dehydrogenated to form the highly conjugated intermediate 614. Oxidation of 614 to dione followed by removal of the N-Boc and O-MOM protecting groups and hemiaminal formation afforded (−)-cepharatine A (615). Methylation of the alcohol hydroxyl group produced (−)-cepharatine C (616).
 | ||
| Scheme 45 Total synthesis of the three sub-classes of hasubanan alkaloids (611, 615, 616 and 619) by Zhu's group. Reagents and conditions: (a) PdCl2, CuCl2, O2, DMF/H2O, rt, then NaOH, MeOH; (b) (i) allyl bromide, K2CO3, acetone, reflux, (ii) 1,2-dichlorobenzene, 180 °C; (c) (i) (1R,2R)-Takemoto thiourea catalyst, nitroethylene, 3 Å MS, rt, (ii) L-selectride, THF, −78 °C; (d) (i) LiAlH4, Et2O, 0 °C-rt, (ii) ClCO2Me, Na2CO3, THF/H2O, rt, (iii) LiAlH4, THF, reflux; (e) Boc2O, DCM, rt, then DMP, NaHCO3, 0 °C-rt; (f) PdCl2, O2, NMP/H2O; (g) (i) KOtBu, toluene/tBuOH, 0 °C-rt, (ii). DDQ, DCE, 50 °C; (h) LHMDS, NBS, −78 °C, THF, then TFA, DCM, rt; (i) isopropenylboronic acid pinacol ester, Grubbs second-generation catalyst, DCM, 50 °C, then NaBO3·4H2O, THF/H2O, rt; (j) 0.5 M NaOMe in MeOH, reflux; (k) (i) LHMDS, MoOPH, THF, −78 °C to −20 °C, (ii) DDQ, DCE, 50 °C; (l) DMP, DCM, 0 °C-rt, then TFA; (m) H2SO4 (1 M in MeOH)/CH(OMe)3/MeOH = 0.1/1/1, 65 °C; (n) TMSOTf, 2,6-lutidine, DCM, 0 °C-rt, then TBAF, 0 °C; (o) (i) KOtBu, Et2O/HCO2Et, 0 °C-rt, then trimethylene dithiotosylate, AcOK, MeOH, reflux, (ii) KOtBu, Me2SO4, DMSO, rt; (p) PIFA, TFA, MeCN/H2O, then TFA, rt. | ||
The aza-[4.4.3]-propellane structure 617 was constructed from 613 by domino-N-deprotection/intramolecular aza-Michael addition reaction, and the methyl enol ether 618 was obtained by regioselective thioneation. Hydrolysis of dithioketal and removal of the O-MOM protecting group produced (−)-cepharamine (619). This strategy is expected to be widely used in the synthesis of members of this natural product family, and it can be used for the synthesis of morphine alkaloids and their derivatives by slightly adjusting the reaction route, which is important for reference.
Qin and Zhang et al. established a new strategy in 2022 for the efficient synthesis of opioid natural products and their pharmaceutical molecules (Scheme 46), utilizing Ir-catalyzed asymmetric hydrogenation of imine and biomimetic intramolecular dearomatization coupling as the key reactions.193 The condensation of phenethylamine 620 and the carboxylic acid 621 produced the amide 622, followed by PMB protection of the phenolic hydroxyl group and subsequent Bischler–Napieralski cyclization to construct the isoquinoline skeleton, resulting in 623 after asymmetric hydrogenation. Optimization of the intramolecular dearomatization coupling reaction enabled a 50-gram-scale reaction, leading to 624 after removal of the PMB protecting group. The reduction of the carbonyl group in 624 resulted in the formation of a dihydrofuran ring, generating the key pentacyclic core skeleton 625 on a 10-gram scale. The singlet oxygen produced by photocatalysis was subjected to [4 + 2] cycloaddition reaction with 625 to generate 626. Under acidic conditions, catalytic hydrogenation cleaved the O–O bond, resulting in the tertiary hydroxyl compound 627. The reduction of 627 removed the Ts protecting group while reducing the carbonyl group, and the N-methyl group was introduced via reductive amination. Hydrogenation of the double bond, followed by IBX oxidation and acid-catalyzed dehydration elimination, produced 628. Catalytic hydrogenation reduced the alkene moiety in 628, resulting in (−)-oxycodone (629) with an overall yield of 11%.
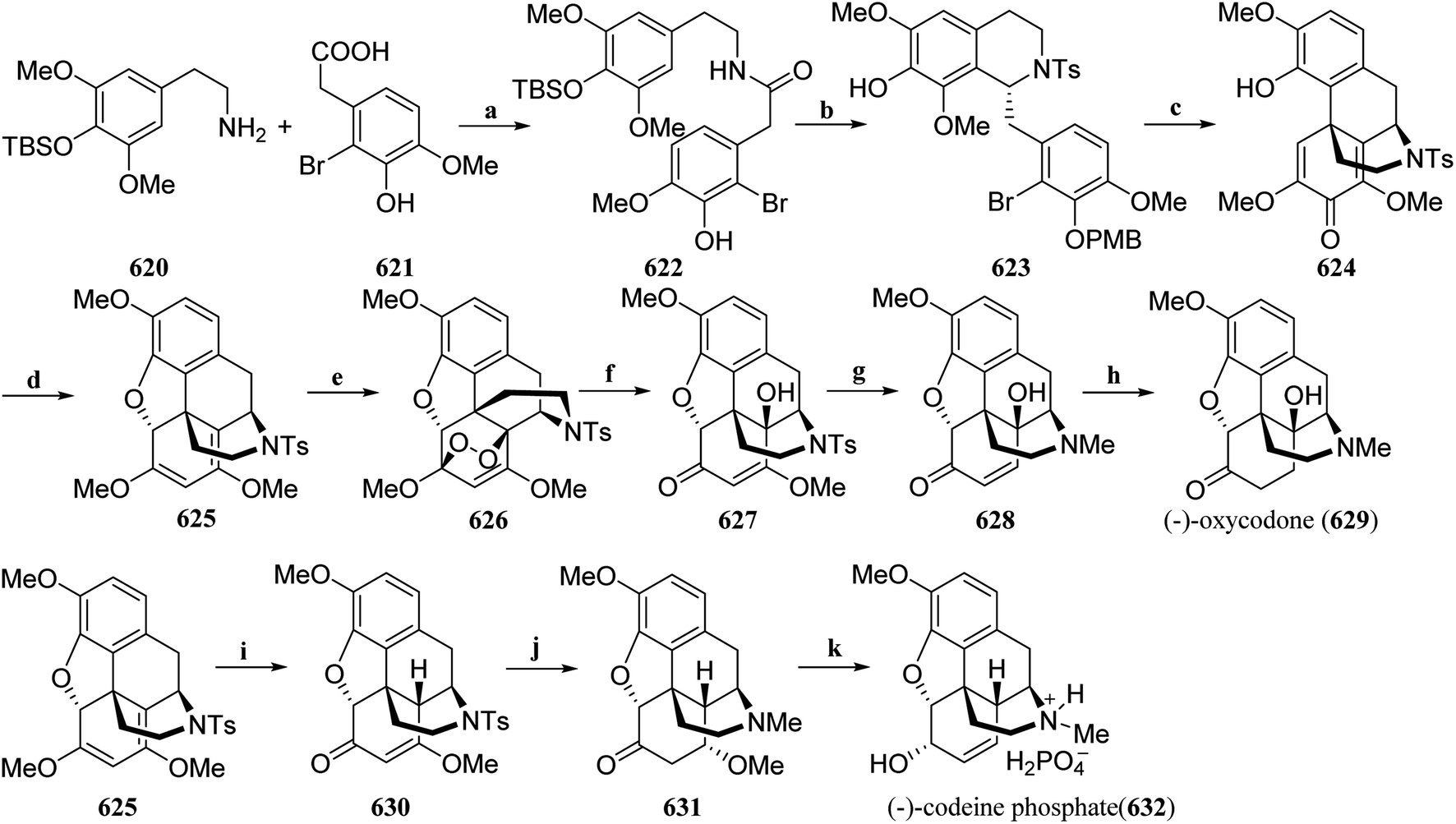 | ||
| Scheme 46 Bioinspired total synthesis of (−)-oxycodone (629) and (−)-codeine (632) by Qin's group. Reagents and conditions: (a) TBTU, Et3N, DCM, 0 °C-rt; (b) (i) K2CO3, PMBCl, MeCN, 40 °C, (ii) Tf2O, 2-fluoropyridine, DCM, −30 °C, (iii) [Ir(cod)Cl]2, (R)-BINAP, TEAI, CHCl3, H2, 0 °C, then Et3N, TsCl, (iv) KF, HBr, MeCN/H2O, 50 °C; (c) (i) Pd(PPhtBu2)2Cl2, tBuOK, DME, 85 °C, (ii) HOAc, 90 °C; (d) (i) NaBH4, MeOH/DCM, 0 °C, (ii) dimethylformamide dimethyl acetal, 1,4-dioxane, 60 °C, (e) TPP, O2, blue LED, DCM, rt; (f) Pd/C, HCO2H, iPrOH/H2O, H2, rt; (g) (i) LiAlH4, DME, 40 °C, (ii) (HCHO)n, MeOH, then NaBH4, rt, then PdCl2, H2, MeOH, 30 °C, (iii) IBX, DMSO, rt, (iv) TsOH, THF, 60 °C; (h) Pd/C, H2, MeOH, 30 °C; (i) (i) NaBH4, MeOH/DCM, 0 °C, (ii) dimethylformamide dimethyl acetal, 1,4-dioxane, 60 °C, (iii) Pd/C, HCO2H, iPrOH/H2O, H2, rt; (j) (i) LiAlH4, DME, 40 °C, (ii) (HCHO)n, MeOH, then NaBH4, rt, then PdCl2, H2, MeOH, 30 °C, (iii) IBX, DMSO, rt; (k) (i) TsOH, THF, 50 °C, (ii) NaBH4, MeOH, 0 °C. | ||
The key intermediate 625 underwent keto–enol tautomerization and alkene migration to form the ketone 630, which could be converted into 631 under similar reaction conditions. Elimination of the methoxy group in 631, followed by diastereoselective reduction of the carbonyl group, provided (−)-codeine (632) with an overall yield of 13%. This synthetic strategy can also be widely applied to the efficient synthesis of opioid drugs such as (−)-oxymorphone, (−)-naloxone, (−)-naltrexone, and (−)-nalbuphine, with costs comparable to those of the currently widely used semisynthetic methods from cultivated poppies, demonstrating significant application value. Importantly, the entire synthesis process requires only one column chromatography step, and most intermediates can be purified through simple work-up or recrystallization, greatly increasing the industrial application prospects of this reaction strategy.
Based on the biomimetic dearomatization arene coupling strategy, Qin's group realized the asymmetric total synthesis of buprenorphine and dihydroetorphine from the key intermediate, in 2022 (Scheme 47).194 According to the previous research,195 the synthesis of the intermediate 633 was realized. The Diels–Alder cycloaddition of methylvinyl ketone with the diene 633 gave the adduct, then hydrogenated the double bond to furnish 634. The carbonyl group in 634 reacted with different Grignard reagents to obtain two important intermediates 635 and 636. Next, the tosyl (Ts) group was removed, and the resulting secondary amine was directly subjected to reductive amination with cyclopropanecarbaldehyde and paraformaldehyde. Finally, the regioselective O-demethylation gave buprenorphine (637) and dihydroetorphine (638).
 | ||
| Scheme 47 Total synthesis of buprenorphine (637) and dihydroetorphine (638) by Qin's group. Reagents and conditions: (a) (i) methylvinyl ketone, PhMe, 80 °C, (ii) Pd/C, AcOH, MeOH, H2, rt; (b) t-BuMgCl, PhMe, rt; (c) (i) LiAlH4, THF, 0–60 °C, (ii) cyclopropanecarbaldehyde, MeOH, then NaBH4, rt, (iii) EtONa, tert-dodecanethiol, DMSO, 150 °C; (d) n-PrMgCl, PhMe, 50 °C; (e) (i) LiAlH4, THF, 0–60 °C, (ii). (HCHO)n, MeOH, then NaBH4, rt, (iii) EtONa, tert-dodecanethiol, DMSO, 150 °C. | ||
Based on the potential biosynthetic pathway of hasubanan alkaloids, Nagasawa et al. established a versatile synthetic strategy (Scheme 48).196 The optically pure compound 640 could be obtained through several simple transformation steps from commercially available aldehydes. Through a key diastereoselective oxidative phenolic coupling reaction, the hasubanan tricyclic dienone skeletons 641 and 642 with different substituents could be constructed in one step. 641 underwent reductive debromination to obtain 643. Under acidic conditions, 642 and 643 underwent aza-Michael reaction and silanol elimination to obtain the conjugated dienones 644 and 645. After protected the phenolic hydroxyl groups with MOM, Davis oxidation produced the corresponding α-hydroxy ketones 646 and 647. After the deprotection and reductive amination of 646, a key biosynthetic intermediate 648 was obtained, which can spontaneously undergo a retro-aza-Michael reaction and hemiaminal formation at room temperature, efficiently produced (−)-cepharatine A (615), which could be methylated to obtain (−)-cepharatine C (616). The reaction precursor 647 underwent deprotection under acidic conditions, simultaneously triggering retro-aza-Michael reaction and hemiaminal formation to form an unstable hemiaminal, which was then reductively aminated in situ to obtain (+)-cepharatine B (649). Methylation then afforded (+)-cepharatine D (650). This synthetic strategy not only provides a new synthetic pathway but also offers potential clues for elucidating the undetermined biosynthetic pathway of cepharatines.
 | ||
| Scheme 48 Total synthesis of cepharatines A–D by Nagasawa's group. Reagents and conditions: (a) PIDA, MeOH, HFIP, 0 °C; (b) HCOONa, Pd(PPh3)4, DMF, 90 °C; (c) HCl, DCM, rt; (d) (i) MOMCl, i-Pr2NEt, DCM, rt, (ii) Davis oxidation agent, KHMDS, THF, −78 °C; (e) (i) TFA, rt, (ii) HCHO aq., NaBH3CN, MeCN, rt; (f) neat, rt; (g) CH(OMe)3, H2SO4, MeOH, 65 °C; (h) TFA, rt; (i) HCHO aq., NaBH3CN, MeCN, rt; (j) TMSCHN2, PhMe/MeOH, rt. | ||
 | ||
| Scheme 49 Total synthesis of rac-β-noscapine (651) rac-β-hydrastine (652) by Zhao's group. Reagents and conditions: (a) Pd2(dba)3, DIPEA, DMF, 110 °C; (b) Pd(OAc)2, AgOAc, HOAc, 117 °C; (c) (i) oxone, NaHCO3, acetone-DCM-H2O, 0 °C, (ii) CF3COOH, DCM, 0 °C; (d) HCHO, HCOOH, rt; (e) (i) HCHO, HCOOH, rt, (ii) MeOK, MeOH, rt. | ||
Furthermore, by further optimizing the asymmetric epoxidation conditions, the key chiral phthalide tetrahydroisoquinoline skeleton 660 was successfully synthesized in one-pot. After reductive amination and N-methylation, epimerization under alkaline conditions resulted in the first asymmetric total synthesis of (−)-β-hydrastine (661). This novel synthetic strategy is expected to find widespread application in the diastereo- and enantioselective total synthesis of such natural products (Scheme 50).198
 | ||
| Scheme 50 Total synthesis of (−)-β-hydrastine (661) by Zhao's group. Reagents and conditions: (a) (i) (Boc)2O, TEA, DCM, rt, (ii) CF3COOAg, DCM, −5 °C; (b) Pd(OAc)2, AgOAc, HOAc, 117 °C; (c) Shi catalyst, oxone, K2CO3, Bu4NHSO4, CH3CN-buffer, 0 °C; (d) (i) CF3COOH, DCM, 0 °C, (ii) HCHO, HCOOH, rt, (iii) MeOK, MeOH, rt. | ||
Despite the practicality of photoredox strategies, their application in the total synthesis of natural products remains a challenge, presumably owing to the difficulty in performing stereoselective reactions with substrates laden with specific functional groups. However, the concise and efficient construction of natural product derivatives through this strategy still holds significant practical importance in areas such as drug development. For example, Hong and Sun et al. developed a strategy combining visible light catalysis and copper-catalyzed radical–radical cross-coupling method, which allowed for the one-step construction of the phthalide isoquinoline skeleton, enabling the efficient synthesis of a series of N-aryl phthalide isoquinolines containing various substituents (Scheme 51).199
 | ||
| Scheme 51 Photocatalytic efficient construction of phthalide isoquinoline skeleton by Hong and Sun's group. Reagents and conditions: (a) [Ir(dF(CF3)ppy)2(dt-bpy)]PF6, Cu(OTf)2, toluene, Ar, blue LED, rt. | ||
 | ||
| Scheme 52 Total synthesis of tylophorine (663) by Rao's group. Reagents and conditions: (a) [Ir(dF(CF3)ppy)2(dCF3bpy)]PF6, NBu4OP(O)(OBu)2, PhtSCF3, PhCF3, blue LED, rt; (b) Red-Al, dioxane, THF, rt. | ||
Ohno and Inuki et al. efficiently constructed a 6,6-spirocyclic structure through visible light-mediated photoredox catalytic stereoselective radical cyclization reaction, using it as the key intermediate to achieve the first total synthesis of zephycarinatines C (671) and D (672) (Scheme 53).201 According to the coupling method reported by Macherla et al.,202 the carboxylic acid 664 reacted with oxazolidine 665 to obtain amide 666. After hydrolysis of the ester group, the key reaction precursor 667 was generated. Under the reaction conditions of visible light-catalyzed carboxylic acid to generate the radical intermediate, 667 was smoothly cyclized to obtain the critical spirocyclic skeleton 668. In addition, the reaction conditions for the continuous flow chemistry system, which can produce cyclization products with moderate yields, were screened, laying a foundation for further expanding the synthesis scale. Under acidic conditions, the oxazolidine ring of 668 was opened to obtain the corresponding alcohol, which was then oxidized and amidated to give 669. The oxidation of 669 produced an α,β-unsaturated ketone, which underwent intramolecular 1,4-addition to obtain 670. After selective reduction of the carbonyl group, stereoinversion introduced a methoxy group, followed by N-alkylation, completing the first total synthesis of zephycarinatines C (671) and D (672).
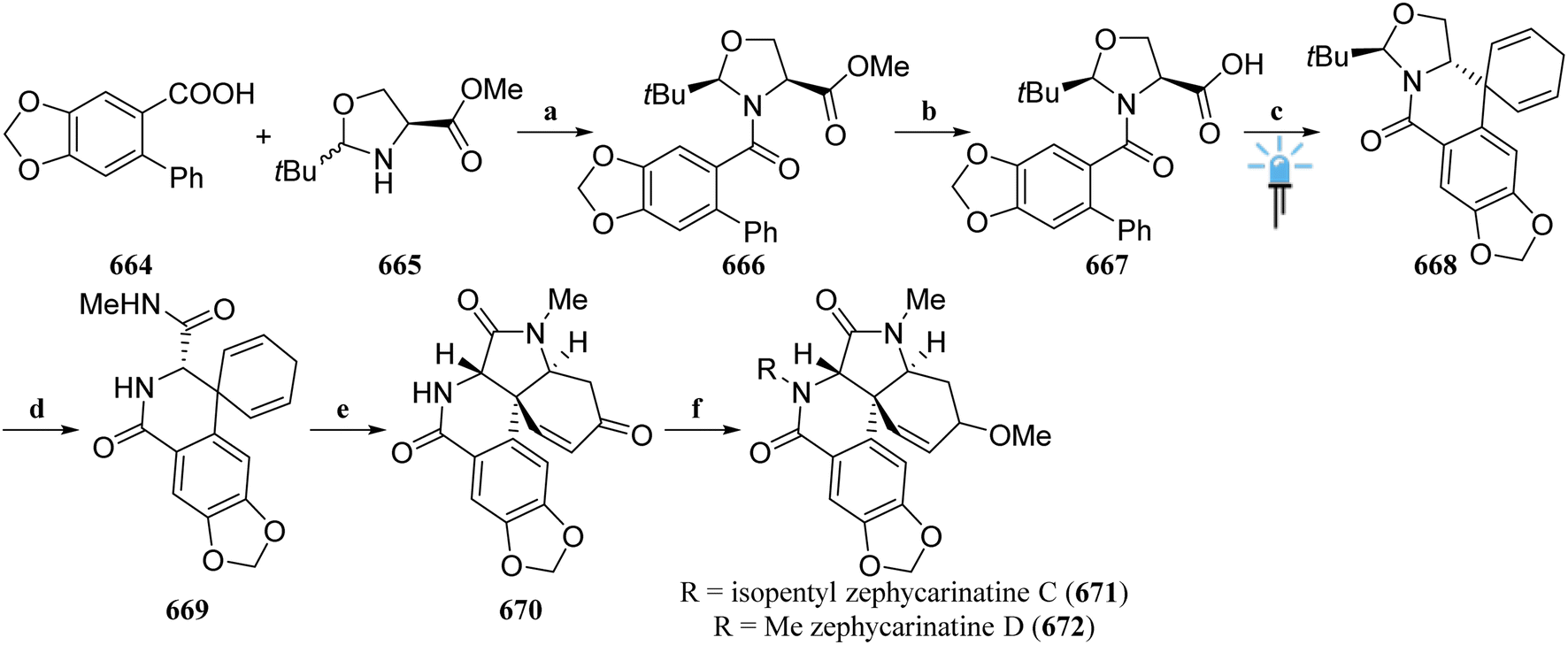 | ||
| Scheme 53 Total synthesis of zephycarinatines C (671) and D (672) by Ohno's group. Reagents and conditions: (a) Et3N, DCM, then MsCl, 0 °C-rt; (b) LiOH·H2O, MeOH, 40 °C; (c) [Ir(dF(CF3)ppy)2(dtbpy)]PF6, K2CO3, MeCN, blue LED, batch, rt, or [Ir(dF(CF3)ppy)2(dtbpy)]PF6, TMG, MeCN, blue LED, flow, rt; (d) (i) TsOH·H2O, MeCN/H2O, 90 °C, (ii) AZADOL, NaClO2, NaOCl, MeCN/pH 7 buffer, 0 °C-rt, (iii) MeNH2, EDC, HOBt, DIPEA, DMF, 0 °C-rt; (e) NMO, TPAP, MeCN, −20–0 °C; (f) (i) LiAlH4, THF, −78 °C, (ii) Ms2O, Et3N, DCM, −40 °C, then MeOH. rt, (iii) NaH, DMF, isopentyl bromide or MeI, 0 °C-rt. | ||
In 2020, Yang et al.207 (Scheme 54) efficiently constructed a chiral unnatural amino acid synthesis unit by referencing the palladium-catalyzed arylation reaction of the alanine-derived amide developed by Yu's group.203–205 Based on the amino acid ester (675), they completed the collective synthesis of a series of renieramycin-type tetrahydroisoquinolines, achieving the first asymmetric total synthesis of (−)-jorunnamycin A (248), (−)-fennebricin A (686) and (−)-renieramycin J (685) (Scheme 54).206 Under the optimal conditions reported by Yu, aryl iodide compound 673 underwent an arylation reaction with alanine derivative 674. After removing the amide directing group, TBS protecting group, and phthalimide protecting group, the key reaction precursor, amino ester 675, was obtained. From 675, two fragments 677 and 678 of the pentacyclic ring system of tetrahydroisoquinoline could be constructed, respectively. The Boc group protected the amino group on the 675, and then hydrolyzed the ester, the phenolic hydroxyl group was protected by TBS to obtain 678. The intermolecular Pictet–Spengler reaction of 675 with benzoxyacetaldehyde was used to synthesize 676, TBS protected the phenol hydroxyl group, and reduction the ester group to obtain the alcohol 677. Coupling of 677 and 678 produced the amide 679. Subsequent oxidation of the primary alcohol with DMP led to a hemiaminal, and after removing the hydroxyl protecting group, an intramolecular Pictet–Spengler reaction took place, simultaneously removing the Boc group and completing the construction of the pentacyclic backbone 680. After reductive amination, N-methylamine 681 was obtained. The reduction of 681 with LiAlH4 gave an unstable hemiaminal intermediate, which was immediately converted to a more stable compound 682 under the conditions of TMSCN and BF3·Et2O. Oxidation of 682 with DDQ produced (−)-jorunnamycin A (248). TBS then protected the alcohol hydroxyl group of (−)-jorunnamycin A, which underwent an intermolecular Mannich reaction. Removal of TBS protection gave (−)-fennebricin A (686). Acylation of 682 with angelic acid, followed by DDQ oxidation, generated (−)-renieramycin M (684), which underwent an intermolecular Mannich reaction leading to the formation of (−)-renieramycin J (685). Additionally, the acylation of 681 with angelic acid and subsequent DDQ oxidation gave (−)-renieramycin G (683).
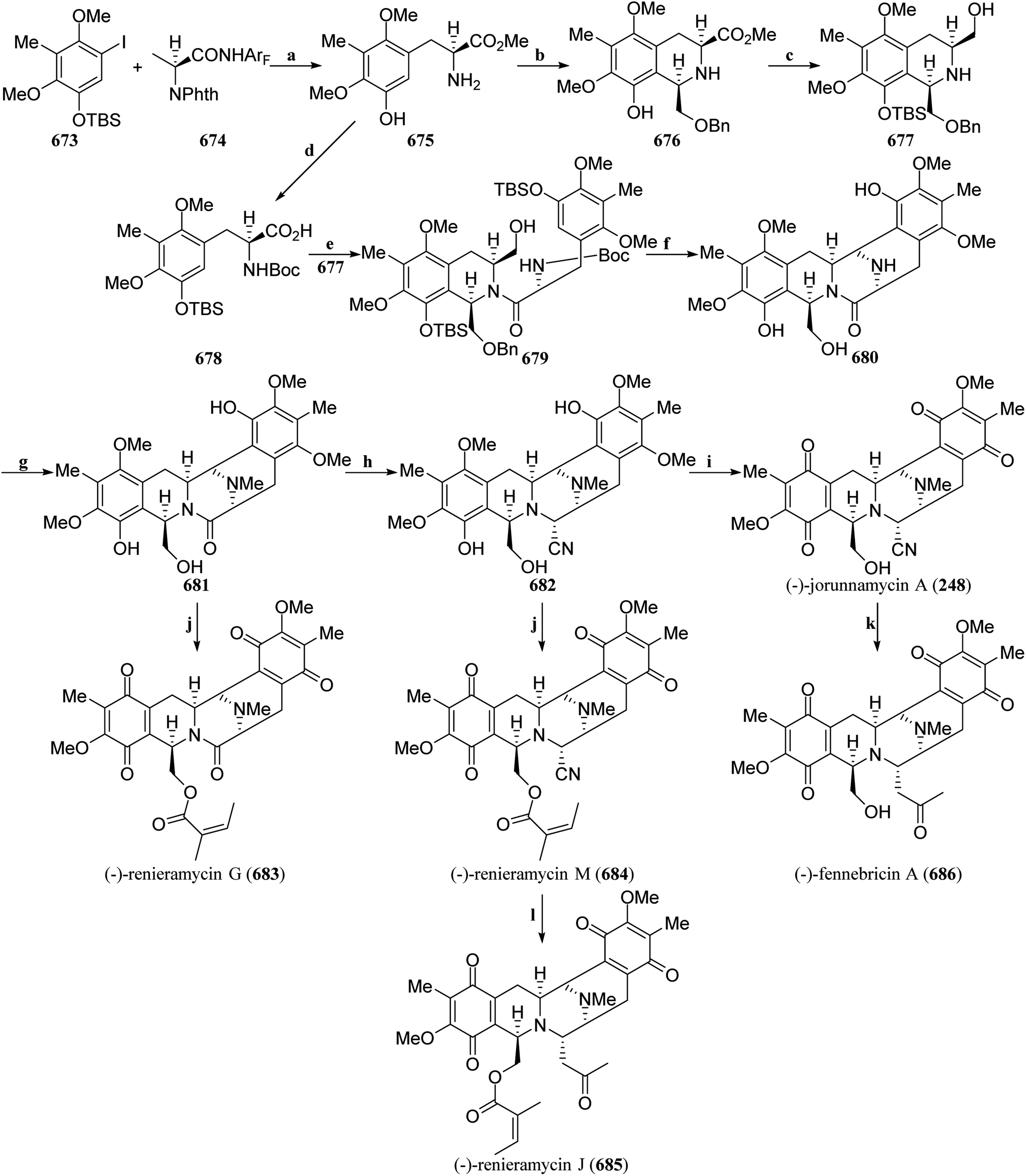 | ||
| Scheme 54 Total synthesis of (−)-fennebricin A (686), (−)-renieramycin J (685), (−)-renieramycin G (683), (−)-renieramycin M (684), and (−)-jorunnamycin A (248) by Yang's group. Reagents and conditions: (a) (i) Pd(TFA)2, Ag2CO3, 2-picoline, CF3COOH, DCE, 100 °C, (ii) BF3·Et2O, MeOH, 100 °C, (iii) ethylenediamine, DCM/MeOH, 40 °C; (b) benzoxyacetaldehyde, AcOH, 4A MS, DCM, rt; (c) (i) imidazole, TBSCl, DMF, rt, (ii) LiAlH4, THF, 0 °C; (d) (i) LiOH, MeOH/H2O, rt, (ii) imidazole, TBSCl, DMF, rt; (e) BOPCl, Et3N, DCM, 0 °C; (f) (i) DMP, DCM, 0 °C-rt, (ii) TBAF, THF, 0 °C, (iii) TFSA, 0 °C; (g) HCHO, NaCNBH3, AcOH, MeOH, rt; (h) (i) LiAlH4, THF, −20–0 °C, (ii) TMSCN, BF3·Et2O, DCM, −30 °C; (i) DDQ, acetone/H2O, rt; (j) (i) angelic acid, Et3N, 2,4,6-trichlorobenzoyl chloride, toluene, 90 °C, (ii) DDQ, acetone/H2O, rt; (k) (i) imidazole, TBSCl, DMF, rt, (ii) AgNO3, acetone, rt, (iii) HF·pyridine, THF, 0 °C-rt; (l) AgNO3, acetone, 50 °C. | ||
Okano's group established a divergent synthetic method for symmetric/asymmetric lamellarins and their homologs in 2020207 based on their recent discovery that the ester group promoted halogen dance as a key reaction.208 The nitrogen atom of ethyl pyrrole-2-carboxylate 687 was protected, followed by bromination to obtain the α,β-dibromopyrrole 688. Treatment with LDA facilitated the smooth migration of the α-bromo group, resulting in the β,β′-dibromopyrrole 689. Owing to the different reactivities of the bromo groups, the two bromo groups at the β positions could be converted into the same or different aryl derivatives 690 and 691via the Suzuki–Miyaura coupling reaction. After ester hydrolysis, Pd-catalyzed intramolecular esterification occurred. Following deprotection, N-alkylation with phenylethyl alcohol was performed under Mitsunobu conditions to obtain the N-alkylated derivatives 692 and 693. The hypervalent iodine PIFA promoted the formation of C–C bonds, constructing the lamellarin skeleton. Deprotection then led to the generation of Lamellarin S (694) and Z (695) (Scheme 55). This synthetic strategy could also be applied to the synthesis of other pyrrolidine alkaloids.
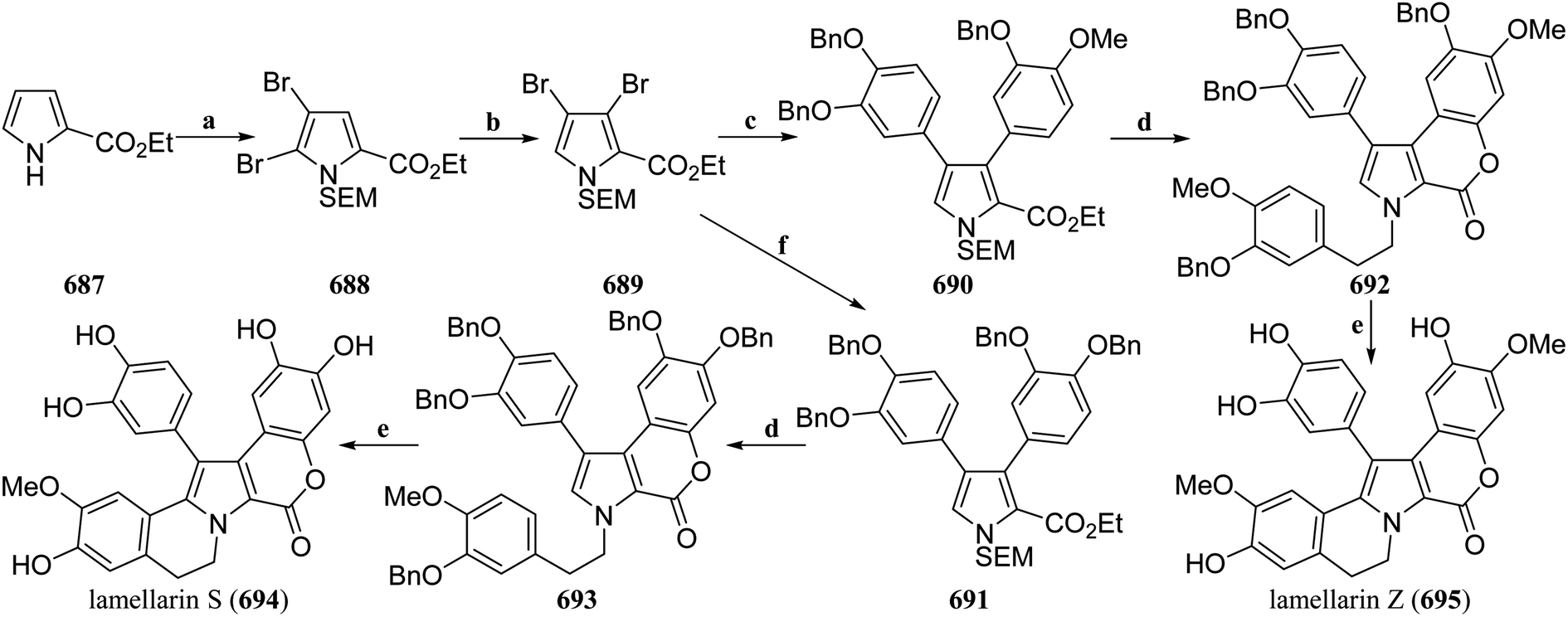 | ||
| Scheme 55 Total synthesis of lamellarin S (694) and Z (695) by Okano's group. Reagents and conditions: (a) (i) Br2, CHCl3, 0 °C-rt, (ii) NaH, SEMCl, THF, 0 °C-rt; (b) (i) LDA, THF, −78 °C, (ii) H2O, −78 °C; (c) (i) arylboronate ester, Pd(dba)3, P(4-CF3C6H4)3, Ba(OH)2·8H2O, 1,4-dioxane/H2O, reflux, (ii) arylboronate ester, Pd(PPh3)4, Ba(OH)2·8H2O, 1,4-dioxane/H2O, reflux; (d) (i) KOH, 18-crown-6, EtOH, reflux, (ii) Pd(OAc)4, EtOAc, reflux, (iii) TFA, DCM, rt, then evaporation, NH2OH· HCl, Na2CO3, THF/H2O, rt, (iv) phenylethyl alcohol, PPh3, DIAD, THF, rt; (e) (i) PIFA, BF3·Et2O, DCM, −40 °C, (ii) Pd/C, H2, EtOH/EtOAc, rt; (f) arylboronate ester, Pd(PPh3)4, Ba(OH)2·8H2O, 1,4-dioxane/H2O, reflux. | ||
In 2021, Okano's group further optimized the synthetic strategy,209 achieving a bottom-up synthesis of lamellarin alkaloids (Scheme 56). The LDA-induced migration of the α-bromo group produced the intermediate, α-pyrrolyl lithium, which could be converted into the corresponding organozinc compound. This compound could then be coupled with aryl iodide via Pd catalysis to obtain the fully substituted pyrrole derivative 696, in a one-pot. After deprotection and cyclization, the tetrahydroisoquinoline skeleton 697 was constructed. Similarly, halogen-lithium exchange was carried out according to the different reactivities of the bromine groups, which could replace the β-bromide near the ester group with the boronic acid ester group to obtain the key synthetic intermediate 698. This synthetic intermediate enables the total synthesis of the lamellarins L (699), J (700), and G (701) through stepwise arylation.
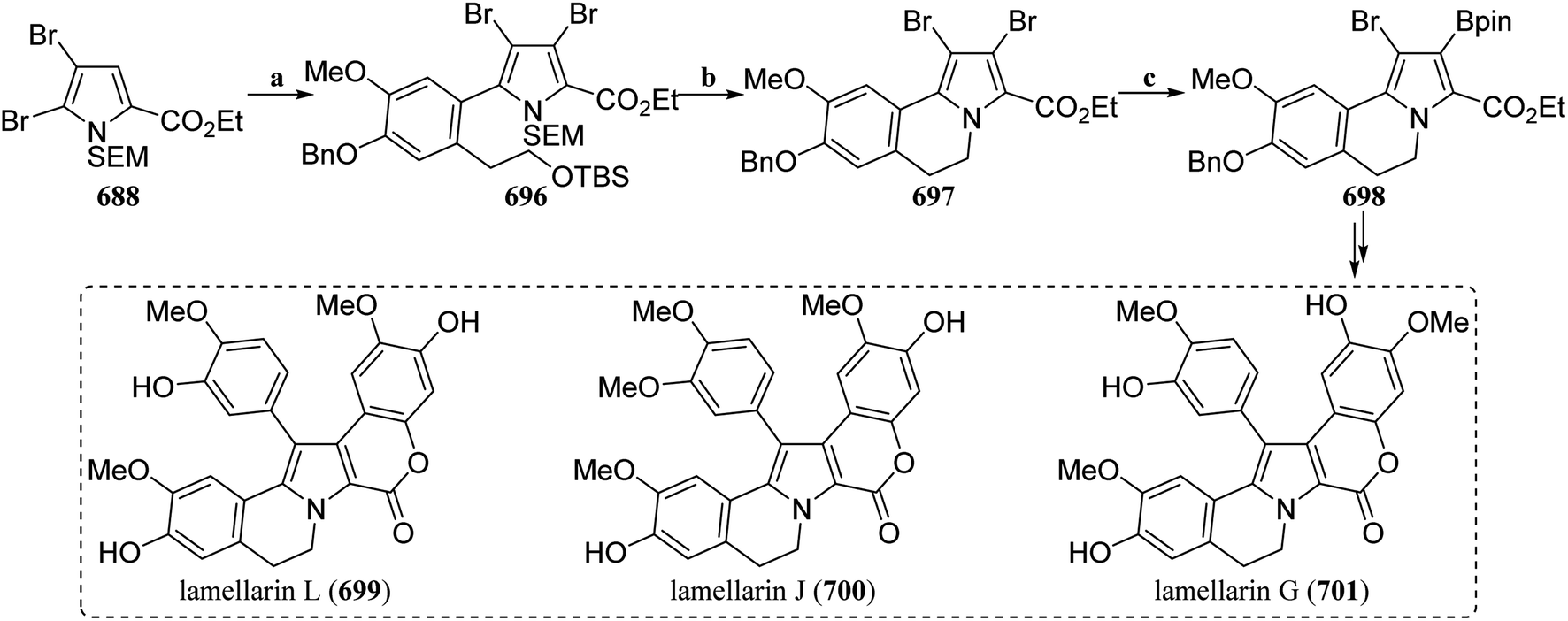 | ||
| Scheme 56 Total synthesis of lamellarin L (699), J (700) and G (701) by Okano's group. Reagents and conditions: (a) (i) LDA, THF, −78 °C, (ii) ZnCl2·TMEDA, −78 °C, (iii) Pd(PPh3)4, aryl iodide, 60 °C; (b) (i) TFA, CH2Cl2, rt, (ii) Na2CO3, THF/H2O, rt, (iii) MsCl, Et3N, CH2Cl2, 0 °C; (c) 3,5-(CF3)2C6H3Li, THF, −78 °C, then i-PrO-Bpin, −78 °C-rt. | ||
In 2022, Samanta et al. developed a cheap Ru(II)-catalyzed C–H bond arylation reaction based on quinone-type carbene migratory insertion, efficiently constructing the indolocoumarin skeleton by combining it with a Brønstead acid-promoted cyclization reaction (Scheme 57).210 This new method was applied to achieve the total synthesis of isolamellarins A and B. First, the tetrahydroisoquinoline 702 and allyl acetate derivatives 703/704 underwent Baylis–Hillman reaction to construct the tetrahydroisoquinoline-fused pyrrole skeletons 705 and 706. After amidation of the ester groups, these compounds were converted into the key precursors 707 and 708. Under optimal reaction conditions, the amides 707 and 708 reacted with o-quinonediazide to complete the total synthesis of isolamellarins A (709) and B (710).
 | ||
| Scheme 57 Total synthesis of isolamellarin A (709) and isolamellarin B (710) by Samanta's group. Reagents and conditions: (a) (i) CuBr, TBHP, m-xylene, reflux, (ii) DDQ, m-xylene, rt; (b) (i) CuBr, TBHP, m-xylene, 140 °C, (ii) Pd(OAc)2, PPh3, bromobenzene, CS2CO3, rt; (c) pyrrolidine, LiHMDS, toluene, rt; (d) o-quinonediazide, [Ru(p-cymene)Cl2]2, AgNTf2, PivOH, tAMOH, 90 °C, then AcOH, 130 °C. | ||
Wang et al. achieved the first total synthesis of sinopyrine B (711)211 using a unique formal [3 + 2] cyclization between amino-substituted malononitrile developed by their group and TMS-acetylene as the key step to construct the pyrrolo[2,1-α]isoquinoline skeleton.212 Phenylethylamine 712 was obtained via the Henry reaction with nitromethane, TBS protection of hydroxyl group and reduction reaction of the inexpensive isovanillin 639, which was cyclized with paraformaldehyde by Picket–Spengler to construct an isoquinoline backbone 713. Through the subsequent ammonolysis of ethyl formate, formamide 714 was obtained. Under established reaction conditions, formamide 714 reacted with TMSCN to generate the key intermediate 715, which underwent a formal [3 + 2] cyclization with TMS-acetylene to complete the construction of the pyrrolo[2,1-α]isoquinoline skeleton 716. Finally, DIBAL-H was used to reduce the cyano group to obtain aldehyde 717, followed by the removal of TMS and TBS groups to complete the first total synthesis of sinopyrine B (711) with a total yield of 7% (Scheme 58). This concise and efficient synthetic strategy provides new ideas for the synthesis of other biologically active natural products or pharmacological molecules containing the pyrrolo[2,1-α]isoquinoline skeleton.
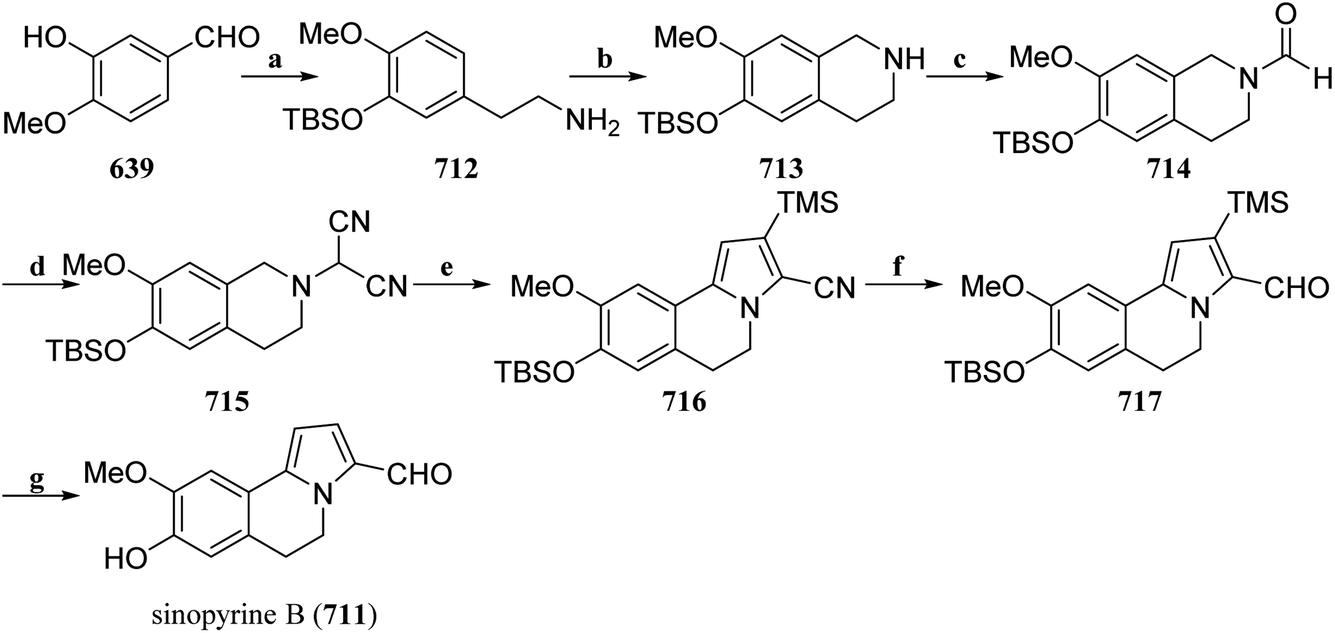 | ||
| Scheme 58 Total synthesis of sinopyrine B (711) by Wang's group. Reagents and conditions: (a) (i)CH3NO2, CH3COONH4, AcOH, 100 °C, (ii) TBSCl, Et3N, DCM, 0 °C, (iii) NaBH4, MeOH, 0 °C, (iv) Pd/C, H2, MeOH, rt; (b) (HCHO)n, TFA, DCM, 0 °C; (c) HCOOEt, HCOOH, 60 °C; (d) TMSCN, Cu(OTf)2, n-heptane, 80 °C; (e) DDQ, DMF, 80 °C; (f) DIBAL-H, Et2O, 0 °C; (g) TBAF, THF, 0 °C. | ||
Photoredox catalysis and electrochemical synthesis methods can generate highly reactive intermediates under mild and environmentally friendly reaction conditions, achieving target transformations with high selectivity and efficiency, significantly increasing substrate compatibility and expanding the scope of application. In many cases, they have become alternatives or complements to traditional organic synthesis reactions, providing more direct and unique ways of chemical bond breaking and building for organic synthesis.112–115 Furthermore, it also offers new directions for retrosynthetic analysis of complex compounds such as natural products, making the synthesis of these compounds simpler and more efficient.116,117 These novel synthetic strategies have not only been applied in the first total synthesis of isoquinoline alkaloid natural products such as dactyllactone A (405), zephycarinatines C (671) and D (672), but also optimized the synthesis strategies of isoquinoline alkaloids such as (−)-thebaine (590), (−)-oxycodone (629), Amaryllidaceae alkaloids (581, 582a,b, 583a,b), tylophorine (663), rac-norlaudanosine (310), rac-laudanosine (311), and rac-xylopinine (312), providing new synthetic pathways. Research on the combined application of continuous flow reaction technology,213–216 it is expected to further expand their potential for large-scale production applications. Currently, the efficient preparation of protoberberine (PB) and 13-methylprotoberberine (13-MePB) at the gram scale has been achieved through the combination of electrochemical synthesis methods and continuous flow reaction systems (Scheme 28).161
As an emerging reaction technology, the continuous flow reaction allows for safer, more environmentally friendly, and efficient conversions than traditional synthesis processes, while generating less waste.118 In recent years, continuous flow reactions have been widely used in synthesis research and in the industrial production of pharmaceuticals, pesticides, chemical products, and their intermediates,217,218 showing great application potential.119,120 Chen's group achieved the first fully continuous flow asymmetric synthesis method for natural tetrahydroprotoberberine alkaloids (Scheme 26).158 Compared with traditional synthesis processes, this continuous flow reaction process is more efficient (the reaction time is reduced from 100 hours to 32.5 minutes) and more environmentally friendly (no intermediate purification is needed, reducing solvent usage and waste generation). However, it is regrettable that this process still faces certain challenges in terms of scaled-up mass production, and further in-depth research is urgently needed.
3.2. Biosynthesis of isoquinoline alkaloids
Isoquinoline alkaloids are mostly extracted and isolated from plants. Through in-depth research, many natural products with potential application value have been discovered. However, the natural content of isoquinoline alkaloids in plants is relatively low, which severely limits further in-depth research and industrial applications.219 Although several high-value isoquinoline alkaloids, such as morphine, are still obtained through large-scale planting, many problems remain, such as needing to occupy a large amount of farmland, unstable yields caused by environmental impacts, and environmental pollution caused by complex separation and purification. Therefore, researchers are committed to overcoming this problem through chemical synthesis. Chemical synthesis is currently the most commonly used method to obtain isoquinoline alkaloids. A variety of isoquinoline alkaloid natural products and their analogs can be obtained through different synthesis strategies. These findings provide a rich material basis for the applied research on isoquinoline alkaloids. However, for structurally complex isoquinoline alkaloids, chemical synthesis still has serious limitations, such as multiple reaction steps, the influence of byproducts, and poor regio- and stereospecificity. Moreover, similar to plant extraction, chemical synthesis can also cause environmental pollution.With the continuous development of biotechnology, biosynthetic strategies provide a new and promising option for obtaining isoquinoline alkaloids. Through biotechnology methods, such as gene sequencing, transcriptomics, and proteomics, researchers have elucidated the biosynthetic pathways of various isoquinoline alkaloids in their natural hosts. With the help of gene editing technology, DNA synthesis technology, metabolic engineering, protein engineering, and synthetic biology, the biosynthesis of isoquinoline alkaloids and their derivatives has been achieved, offering advantages such as high efficiency, strong specificity, and environmental friendliness. Currently, enzymatic biosynthesis and microbial cell factories are the most widely used biosynthetic strategies. In this section, we will introduce the latest progress in the biosynthesis of isoquinoline alkaloids according to the two commonly used biosynthetic strategies.
Hailes and Ward et al. successfully constructed a one-pot multienzyme cascade reaction in vitro by combining the tyrosinase CnTYR, the tyrosine decarboxylase EfTyrDC, and additional enzymes including the transaminase CvTAm and the norcoclaurine synthase TfNCS. Using four different tyrosine derivatives as substrates, they achieved efficient biosynthesis of two natural and six unnatural benzylisoquinolines with high stereoselectivity and a 23–66% isolated yield (Scheme 59a).221 Moreover, this one-pot multienzyme cascade reaction could be successfully scaled up to 1 g, demonstrating the potential application value of this strategy. Subsequently, Hailes et al. combined CnTYR, EfTyrDC, CvTAm, and TfNCS in a parallel multienzyme cascade reaction system, achieving the biosynthesis of halogenated and dihalogenated benzylisoquinoline alkaloids with high enantiomeric purity, which was difficult to achieve through chemical synthesis (Scheme 59b).222 In particular, after mutagenesis to produce a tyrosinase mutant, the tolerance to halogenated tyrosine was improved, increasing the range of substrates. The combination of this multienzyme cascade strategy and an enzyme mutagenesis strategy has strong potential for use in the synthesis of chiral benzylisoquinoline alkaloids.
 | ||
| Scheme 59 (a) Benzylisoquinoline biosynthesis and (b) halo benzylisoquinoline biosynthesis by Hailes's group. | ||
Norcoclaurine synthase (NCS) promotes the Pictet–Spengler reaction between dopamine derivatives and aryl aldehydes in plants, generating a single enantiomer of benzylisoquinoline.223 However, previous reports have indicated that NCS has poor tolerance for α-methyl substituted aldehydes, making it difficult to obtain the target product.224 Hailes's group showed that wild-type T. flavum NCS (Δ33TfNCS) exhibited good tolerance to α-methyl substituted aldehydes, generating THIQ products with two defined stereocenters (Scheme 60a).225 This transformation, which is difficult to achieve through traditional chemical synthesis, provides a new strategy for the synthesis of corydaline-like isoquinoline alkaloids. Active site mutants could further improve the diastereomeric ratio and conversion rate, demonstrating their potential in enzyme catalysis engineering. Several selected NCS variants showed tolerance to aliphatic ketones, α-substituted ketones, cyclic ketones, and diketones, achieving chemically challenging 1,1′-substituted and spiro-tetrahydroisoquinoline derivatives (Scheme 60b).226
 | ||
| Scheme 60 Biosynthesis of dopamine derivatives with (a) aldehydes, (b) ketones by Hailes's group. | ||
In the biosynthetic pathway of plants, methyltransferase (MT) plays a role downstream of norcoclaurine synthase (NCS) to achieve methylation,227 which is crucial for the biosynthesis and diversification of benzylisoquinolines. However, the high cost of the cofactor S-adenosylmethionine (SAM) limits its application in larger-scale in vitro biocatalytic reactions. Based on previous studies and a modular in vitro cofactor supply system consisting of the enzymes EcMAT and EcMTAN from Escherichia coli,228 Hailes et al. established an in vitro one-pot multienzyme cascade reaction system. With two MT enzymes, RnCOMT and MxSafC, which have unique characteristics, as the core, combined with enzymes such as Cj-6-OMT and TfNCS, they achieved efficient construction of methylated THIQ alkaloid natural products and their derivatives with high enantiomeric purity (Scheme 61).229
 | ||
| Scheme 61 Biosynthesis of methylated THIQs in vitro via one-pot cascades by Hailes's group. | ||
Unlike the NCS mechanism of action, the unique catalytic mechanism of IRED does not rely on groups such as the C6-hydroxyl of the isoquinoline ring, making it tolerant to highly modified DHIQ precursors. Using the previously identified IRED as a key enzyme,230 Qu et al. designed a new multienzyme cascade reaction system that achieved phenylisoquinoline derivatives that are difficult to obtain in the NCS cascade reaction system (Scheme 62).231 First, high-yield chemically synthesized 1-phenyl and 1-benzyl-6,7-dimethoxydihydroisoquinoline precursors were constructed. These precursors were reduced by the imine reductase IR45 to obtain the corresponding (S)-THIQ, which was subsequently methylated by N-methyltransferase (CNMT). This process efficiently synthesized isoquinoline natural products and their medicinal derivatives with almost 100% yield.
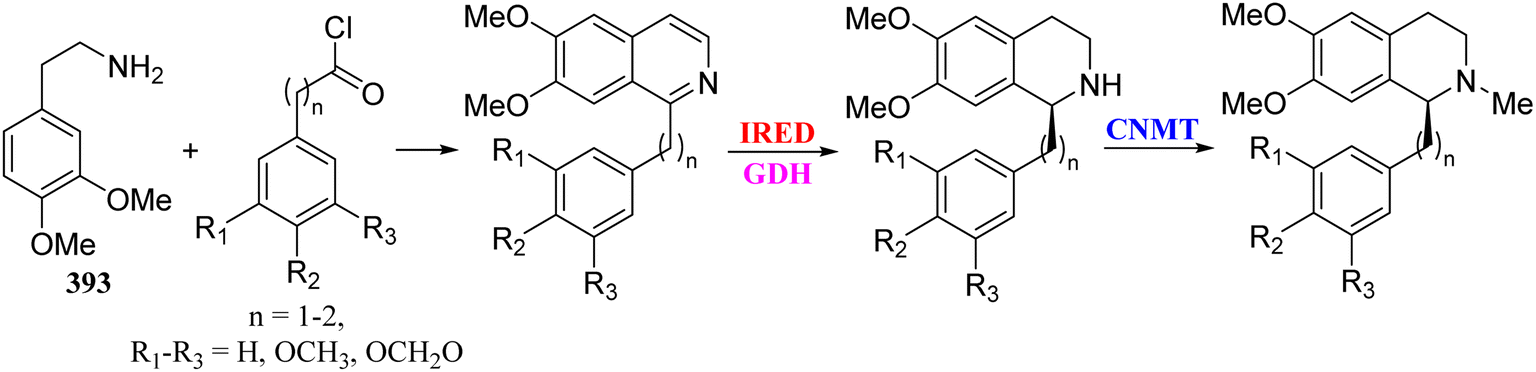 | ||
| Scheme 62 Biosynthesis of THIQs in vitro via IRED approach by Qu's group.232 | ||
Smolke's group analyzed the crystal structure of the essential enzyme (S)-scoulerine 9-O-methyltransferase of the berberine natural product biosynthesis pathway in Thalictrum flavum, and used this crystal structure information to construct a series of mutants to analyze the structure-activity relationship of TfS9OMT, and found specific mutants with regioselective and substrate-selective changes. Using these engineered TfS9OMT mutants, the first de novo yeast strains producing (S)- tetrahydropalmatine and (S)-tetrahydropalmatrubine were constructed.240 The results of this study suggest that characterizing enzyme structures to guide protein engineering can provide specific enzymes for heterologous biosynthetic pathways, which has important potential for the targeted production of difficult-to-synthesize but high-value isoquinoline alkaloids and their derivatives.
The in vivo engineering biocatalytic cascade method based on the whole-cell system has proven to be a powerful tool for synthesizing more high-value products.241 Based on the new biosynthetic method for 4-hydroxyphenylacetaldehyde,242 Xiao et al. established four enzymatic cascade reactions in Escherichia coli via the decarboxylase BLPad or AnPad, the styrene monooxygenase StyAB, styrene oxide isomerase RostyC, and the norcoclaurine synthase Δ29TfNCS. Through the cascade of four biocatalytic reactions: decarboxylation-epoxidation-isomerization-condensation, the efficient synthesis of two natural isoquinoline alkaloids and their derivatives was achieved from coumaric acid derivatives and dopamine derivatives.243 Furthermore, it can efficiently synthesize isoquinoline alkaloids using lignocellulosic biomass as the raw material, with the target compound concentration exceeding 1 g L−1.
Despite the fact that some key functional enzymes remain unidentified, a semi-biosynthetic strategy combined with mild chemical transformation conditions can help compensate for the lack of certain functional enzymes. Examples of the microbial production of artemisinin244 and vinblastine245 have demonstrated the effectiveness and feasibility of this semibiosynthetic strategy, which is expected to find widespread application in industrial production in the future. Based on the yeast biosynthesis platform established in the early stage for the production of key intermediates of tetrahydropapaverine,246,247 Smolke's group developed N-methylcoclaurine hydroxy-lase variants and scoulerine 9-O-methyltransferase variants through protein engineering, and combined with a series of genetic modifications to achieve de novo biosynthesis of tetrahydropapaverine. Subsequently, semi-biosynthesis of papaverine was achieved under chemical transformation conditions using hydrogen peroxide as an oxidant.248 Based on this semi-biosynthetic strategy, Li et al. recently used 15 enzymes to modify the initial yeast strains to obtain an engineered yeast strain and established a complete semibiosynthetic pathway for de novo production of berberine through chemical transformation by heating the bioreactor.249 Furthermore, this engineered yeast was tolerant of fluorotyrosine, enabling the biosynthesis of fluorinated benzylisoquinoline and fluorinated berberine.
Multiple platforms for the de novo heterologous biosynthesis of benzylisoquinoline alkaloids have been constructed based on engineered S. cerevisiae strains. However, developing microbial strains and biosynthetic platforms capable of producing bisbenzylisoquinoline alkaloids remains a challenge. The Smolke's group reported a breakthrough in 2021, discovering that the enzymes PbDRS-DRR and BsCYP80A1 can function in yeast, achieving epimerization of benzylisoquinoline alkaloids and facilitating the coupling of two benzylisoquinoline alkaloid monomers. Thus, they constructed an engineered yeast strain capable of producing bisbenzylisoquinoline alkaloids.250 Through strain engineering, protein engineering, and optimization of fermentation conditions, efficient biosynthesis of guattegaumerine and berbamunine was achieved.
Escherichia coli is currently one of the most commonly used heterologous microorganisms in addition to S. cerevisiae. Moreover, compared to the yeast system, E. coli has a greater capacity to produce tetrahydroprotoberberine intermediates.251,252 However, it is unfortunate that berberine bridge enzyme (BBE), as the rate-limiting enzyme in the tetrahydroprotoberberine (THPB) biosynthetic pathway, does not function in E. coli, which limits the reconstruction of the THPB biosynthetic pathway in this organism. In 2021, Liu et al. overcame the functional expression barrier of BBE in E. coli through a combination of strategies such as screening BBE from different sources, optimizing codons, protein tagging strategies, and optimizing promoters.253 They reconstructed the biosynthetic pathways of (S)-scoulerine, (S)-tetrahydropalmatine, (S)-corydalmine, and related intermediates in E. coli. After the culture conditions were optimized, efficient biosynthesis of these compounds was achieved in the corresponding engineered E. coli microbial cell factories.
Compared with traditional organic synthesis, biosynthesis excels in terms of regioselectivity and stereoselectivity. However, due to the specificity of enzymes, they have unique requirements for reaction substrates, which limits the range of substrates suitable for biosynthesis. Additionally, since the natural biosynthetic pathways or enzymes of many isoquinoline alkaloids have not been fully elucidated, the isoquinoline alkaloids that have been extensively studied and biosynthesized include mainly phenylisoquinoline alkaloids, benzylisoquinoline alkaloids, and berberine. Many complex or newly isolated isoquinoline alkaloids cannot be obtained through biosynthetic pathways in the short term. Organic total synthesis, on the other hand, has advantages in this regard, especially in terms of substrate compatibility and the synthesis of diverse derivatives. This makes traditional organic synthesis play a crucial role in the early research process of isoquinoline drug development or the discovery of novel isoquinoline alkaloid activities. With the development of new chemical synthesis techniques such as photocatalysis, electrocatalysis, and continuous flow chemistry, organic synthesis is expected to achieve the green and efficient synthesis of isoquinoline alkaloids and their derivatives.261
In recent years, the development of plant metabolic pathways, enzyme engineering, and biotechnology methods has led to a number of breakthroughs in the microbial synthesis of plant secondary metabolites. As a plant secondary metabolite with important research and medicinal value, the biosynthesis of isoquinoline alkaloids has important research significance. To date, researchers have analyzed the biosynthetic pathway of benzylisoquinoline alkaloids (BIAs) and have identified a variety of microbial BIA synthesis pathways, including the thebaine and magnoflorine pathways. On this basis, the synthesis pathways of these alkaloids in microorganisms have been reconstructed by simulating the synthesis pathway of BIA in plants, and the biosynthesis of BIA has been realized. However, owing to the complex structure of most BIAs and the lengthy metabolic pathways, the catalytic enzymes involved have wide selectivity for BIA intermediates in the main metabolic flow pathways, resulting in uncontrollable metabolic flow and a low yield of target compounds. Moreover, the structure of isoquinoline alkaloids is complex and diverse, their biosynthetic pathways are difficult to analyze, and the biosynthesis of diverse isoquinoline alkaloids in a short period of time is difficult. Chemical synthesis is highly important for the construction of diverse isoquinoline alkaloids and their derivatives and for the in-depth study of their physiological activities. Moreover, with the analysis of the biosynthesis pathway, the total synthesis strategy of isoquinoline alkaloids by modifying some enzymes instead of expensive noble metal catalysts has been widely studied. Therefore, the combination of the flexible and diverse characteristics of chemical synthesis and the potential for inexpensive and efficient biosynthesis is important for future research on isoquinoline alkaloids.
However, there is no doubt that biosynthesis, especially the establishment of microbial cell factories, has extremely attractive advantages for the green and efficient production of high-value isoquinoline alkaloids. Furthermore, advancements in technologies such as enzyme immobilization, novel screening methods, and enzyme mutagenesis are expected to gradually overcome many limitations of biocatalysis, including poor stability, low efficiency, and a narrow substrate range. This will make enzyme catalysis a more feasible and sustainable option for synthesizing small molecules.262 As research on the natural biosynthetic pathways and key functional enzymes of isoquinoline alkaloids deepens, and with the continuous development of biotechnology, the efficiency of biosynthesis is expected to be further improved, and costs will be reduced in the future.
4. Conclusion
Owing to their various chemical structures and pharmacological activities, isoquinoline alkaloids have high probabilities of success in drug discovery and development. Continuing our previous review (covering 2014–2018), this manuscript summarizes and provides updated literature on novel isoquinoline alkaloids isolated during the period of 2019–2023, and more than 250 molecules with various pharmacological activities were isolated. A new class of phenanthridine alkaloids that includes dehydroambiguanine A (194) with anti-proliferative activity,97 and the wide application of isoquinoline alkaloids was highlighted. The identification of new compounds, significant biological activities or novel mechanisms of action will undoubtedly contribute to the continual development of new drugs in the future.However, the potential of this promising and expanding platform of active natural compounds has only been partially realized by both the academic community and the pharmaceutical industry. As we critically reviewed, although the synthesis strategies have been continuously optimized and more efficient, inexpensive and environmentally friendly synthetic routes for this class of natural products have been discovered over the past five years, achieving large-scale production of isoquinoline alkaloids still faces considerable challenges, hindering further research on these promising compounds, especially in pharmacology and clinical trials. For nearly five years, only Yamada reported a scaled-up synthesis strategy for (−)-emetine in 2023,263 which is capable of synthesizing the target compound at a scale of 237.1 grams. Additionally, Qin's team established a new strategy for opioid natural products and their drug molecules in 2022 (Scheme 46),193 which can perform reactions at a maximum scale of 50 grams. The entire synthesis process requires only a one column chromatography step, and most of the intermediates can be purified through simple processing or recrystallization. This strategy is expected to achieve scaled-up production of more than 100 grams through further optimization.
Moreover, although many isoquinoline alkaloids have been discovered from plants in recent decades, only a few compounds with promising biological activity have been identified, which severely limits further drug development. Currently, marine bacteria produce a plethora of compounds with unusual chemical structures and represent a new natural resource for finding alkaloids with valuable biological functions.108 Table S1† shows that an increasing number of promising isoquinoline alkaloids have been isolated and identified from marine bacteria. For example, Zhang et al.108 reported that a promising lead, turbinmicin, from a sea squirt microbiome component, Micromonospora sp., exhibits a broad safety index, better in vitro potency by targeting Sec14 and mouse model efficacy against multidrug-resistant fungal pathogens, which could help combat devastating global fungal pathogens such as C. auris.
In summary, continued attention and long-term research on the isolation and identification of naturally occurring isoquinoline alkaloids will lead to targeted pharmacological modeling, and developing new strategies for the total synthesis of isoquinolines will provide important support for synthetic modifications, resulting in new and better drugs based on the original effects of these alkaloids.
5. Data availability
The data supporting this article have been included as part of the ESI.†6. Conflicts of interest
All authors declare that they have no competing interests.7. Acknowledgements
This work was supported financially by the National Key Research and Development Program of China (2023YFD1800803, 2023YFD1801304), the National Natural Science Foundation of China (32373055, 32302924), the Outstanding Youth Fund of Gansu Province (23JRRA563), the Innovation Project of the Chinese Academy of Agricultural Sciences (No. CAAS-ASTIP-2015-LIHPS), and China Agriculture Research System (CARS-37).8. References
- K. Bhadra and G. S. Kumar, Med. Res. Rev., 2011, 31, 821–862 CrossRef CAS PubMed.
- I. I. Koleva, T. A. van Beek, A. E. M. F. Soffers, B. Dusemund and I. M. C. M. Rietjens, Mol. Nutr. Food Res., 2012, 56, 30–52 CrossRef CAS PubMed.
- D. K. Liscombe, B. P. MacLeod, N. Loukanina, O. I. Nandi and P. J. Facchini, Phytochemistry, 2005, 66, 1374–1393 CrossRef CAS PubMed.
- D. Santoro, G. Bellinghieri and V. Savica, J. Nephrol., 2011, 24, S133–S136 CrossRef PubMed.
- J. T. Zhu, L. X. Song, S. N. Shen, W. X. Fu, Y. Y. Zhu and L. Liu, Molecules, 2023, 28, 7789 CrossRef CAS PubMed.
- X.-F. Shang, C.-J. Yang, S. L. MorrisNatschke, J.-C. Li, X.-D. Yin, Y.-Q. Liu, X. Guo, J.-W. Peng, M. Goto, J.-Y. Zhang and K.-H. Lee, Med. Res. Rev., 2020, 40, 2212–2289 CrossRef CAS PubMed.
- C. Weber and T. Opatz, The Alkaloids: Chemistry and Biology, 2019, vol. 81, pp. 1–114 Search PubMed.
- N. Tajuddeen and G. Bringmann, Nat. Prod. Rep., 2021, 38, 2154–2186 RSC.
- E. Plazas, M. C. M. Avila, D. R. Munoz and L. E. S. Cuca, Pharmacol. Res., 2022, 177, 106126 CrossRef CAS PubMed.
- L. Xu, Y. Li, Y. Dai and J. Peng, Pharmacol. Res., 2018, 130, 451–465 CrossRef CAS PubMed.
- M. Iranshahy, R. J. Quinn and M. Iranshahi, RSC Adv., 2014, 4, 15900–15913 RSC.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2016, 79, 629–661 CrossRef CAS PubMed.
- C. Freestone and R. Eccles, J. Pharm. Pharmacol., 1997, 49, 1045–1049 CrossRef CAS PubMed.
- M. E. Pyne and V. J. J. Martin, Curr. Opin. Green Sustainable Chem., 2022, 33, 100561 CrossRef CAS.
- M. Xu, L. Liu, C. Qi, B. Deng and X. Cai, Planta Med., 2008, 74, 1423–1429 CrossRef CAS PubMed.
- M. Heinrich and H. L. Teoh, J. Ethnopharmacol., 2004, 92, 147–162 CrossRef CAS PubMed.
- H. A. Blair, Drugs, 2018, 78, 1847–1853 CrossRef PubMed.
- S. Dhillon, Drugs, 2019, 79, 563–572 CrossRef CAS PubMed.
- A. H. Gaur, J. S. McCarthy, J. C. Panetta, R. H. Dallas, J. Woodford, L. Tang, A. M. Smith, T. B. Stewart, K. C. Branum, B. B. Freeman III, N. D. Patel, E. John, S. Chalon, S. Ost, R. N. Heine, J. L. Richardson, R. Christensen, P. M. Flynn, Y. Van Gessel, B. Mitasev, J. J. Moehrle, F. Gusovsky, L. Bebrevska and R. K. Guy, Lancet Infect. Dis., 2020, 20, 964–975 CrossRef CAS PubMed.
- Y.-L. Wang, C.-Y. Wu and S.-W. Zhai, Nat. Prod. Res., 2023, 37, 1867–1871 CrossRef CAS PubMed.
- J. Zhang, C. Zhang, F.-C. Xu, Quesheng, Q.-Y. Zhang, P.-F. Tu and H. Liang, Phytochemistry, 2019, 159, 199–207 CrossRef CAS PubMed.
- D. Luo, N. Lyu, L.-M. Liao, Q. Gao, Y.-K. Li, J. Li, X. Liu, X.-M. Li, G.-Y. Yang, Y.-Q. Ye, Q.-F. Hu and M. Dong, China J. Chin. Mater. Med., 2020, 45, 2568–2570 Search PubMed.
- H. Xia, Y. Liu, G. Xia, Y. Liu, S. Lin and L. Guo, Front. Immunol., 2021, 12, 685556 CrossRef CAS PubMed.
- J.-J. Xue, J.-Y. Li, B.-J. Li, Y.-T. Jin, C.-H. Wang, C.-M. Xue, H.-M. Zhang, Z.-L. Li, D.-H. Li and H.-M. Hua, China J. Chin. Mater. Med., 2022, 47, 2676–2680 Search PubMed.
- J. Liu, Q. Lin, Q.-M. Zhou, C.-W. Meng, L. Xiong, X. Wang, L.-L. Miao, L. Guo and C. Peng, China J. Chin. Mater. Med., 2022, 47, 3265–3269 Search PubMed.
- Y. Han, T. Hou, Z.-H. Zhang, Y.-D. Wang, J.-X. Cheng, H. Zhou, J.-X. Wang, J.-T. Feng, Y.-F. Liu, Z.-M. Guo and X.-M. Liang, Fitoterapia, 2022, 159, 105175 CrossRef CAS PubMed.
- H. Fukumi, H. Kurihara, T. Hata, C. Tamura, H. Mishima, A. Kubo and T. Arai, Tetrahedron Lett., 1977, 18, 3825–3828 CrossRef.
- A. Kubo, Y. Kitahara, S. Nakahara, R. Iwata and R. Numata, Chem. Pharm. Bull., 1988, 36, 4355–4363 CrossRef CAS PubMed.
- Y. Tian, W. Gui, P. B. Smith, I. Koo, L. A. Murray, M. T. Cantorna, G. H. Perdew and A. D. Patterson, J. Agric. Food Chem., 2019, 67, 9286–9294 CrossRef CAS PubMed.
- D. Copmans, S. Kildgaard, S. A. Rasmussen, M. Slezak, N. Dirkx, M. Partoens, C. V. Esguerra, A. D. Crawford, T. O. Larsen and P. A. M. de Witte, Mar. Drugs, 2019, 17, 607 CrossRef CAS PubMed.
- Y. N. Kim, Y. K. Ji, N.-H. Kim, N. Van Tu, J.-R. Rho and E. J. Jeong, Mar. Drugs, 2021, 19, 90 CrossRef CAS PubMed.
- C. Nord, J. J. Levenfors, J. Bjerketorp, C. Sahlberg, B. Guss, B. Oberg and A. Broberg, Molecules, 2019, 24, 4616 CrossRef CAS PubMed.
- M. Shaaban, K. A. Shaaban, G. Kelter, H. H. Fiebig and H. Laatsch, Mar. Drugs, 2021, 19, 715 CrossRef CAS PubMed.
- C.-M. Liu, F.-H. Yao, X.-H. Lu, X.-X. Zhang, L.-X. Luo, X. Liang and S.-H. Qi, Mar. Drugs, 2022, 20, 78 CrossRef CAS PubMed.
- R. S. Conceicao, I. M. A. Reis, A. P. M. Cerqueira, C. J. Perez, M. C. dos Santos Junior, A. Branco, D. R. Ifa and M. B. Botura, Phytochem. Anal., 2020, 31, 711–721 CrossRef CAS PubMed.
- J. M. Lima, G. M. Leme, E. V. Costa and Q. B. Cass, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2021, 1164, 122493 CrossRef CAS PubMed.
- X.-H. Duan, X.-W. Zhang, P. He, M. Qin, L. Pei, J.-C. Zhao and C.-H. Li, J. Chin. Med. Mater., 2021, 44, 76–78 Search PubMed.
- T. El-Elimat, M. B. Alhawarri, J. Rivera-Chavez, J. E. Burdette, A. Czarnecki, M. Al-Gharaibeh, A. H. Al Sharie, A. Alhusban, F. Q. Alali and N. H. Oberlies, Fitoterapia, 2020, 146, 104706 CrossRef CAS PubMed.
- Y. Si, X. Li, T. Guo, W. Wei, J. Zhang, A. Jia, Y. Wang, A. Zhao, J. Chang and S. Feng, Fitoterapia, 2021, 154, 105021 CrossRef CAS PubMed.
- T. Doncheva, N. Kostova, V. Valcheva, R. Toshkovska, V. Vutov and S. Philipov, Nat. Prod. Res., 2020, 34, 668–674 CrossRef CAS PubMed.
- H. Wen, X. Yuan, C. Li, J. Li and H. Yue, Nat. Prod. Res., 2024, 38, 1392–1397 CrossRef CAS PubMed.
- K. Du, Y. Liu, K. Zong, Y. Wang, J. Li and D. Meng, Phytochemistry, 2022, 200, 113240 CrossRef CAS PubMed.
- T. Luo, Z. Li, X.-M. Deng, K. Jiang, D. Liu, H.-H. Zhang, T. Shi, L.-Y. Liu, H.-X. Wen, Q.-E. Li and Z. Wang, Bioorg. Med. Chem., 2022, 60, 116705 CrossRef CAS PubMed.
- L.-Y. Wang, B.-L. Qiu, H. Xia, G. Xia, B.-B. Xiao, J.-F. Zhang, W.-C. Zhong and S. Lin, J. Nat. Prod., 2020, 83, 489–496 CrossRef CAS PubMed.
- J. Zhang, C. Zhang, F. C. Xu, Quesheng, Q. Y. Zhang, P. F. Tu and H. Liang, Phytochemistry, 2019, 159, 199–207 CrossRef CAS PubMed.
- A. O. Whaley, A. K. Whaley, V. Toporkova, E. Fock, N. Rukoyatkina, S. N. Smirnov, G. B. Satimov, B. A. Abduraxmanov and S. Gambaryan, Fitoterapia, 2023, 171, 105697 CrossRef CAS PubMed.
- Z.-T. Peng, H.-X. Huo, L.-H. Chao, Y.-L. Song, D.-F. Liu, Z.-W. Wang, Y. Zhang, Y.-F. Zhao, P.-F. Tu, J. Zheng and J. Li, Phytochemistry, 2023, 209, 113637 CrossRef CAS PubMed.
- N. Sun and Y. Han, J. Asian Nat. Prod. Res., 2021, 23, 1–8 CrossRef CAS PubMed.
- J.-J. Xue, C.-Y. Jiang, D.-L. Zou, B.-J. Li, J.-C. Lu, D.-H. Li, B. Lin, Z.-L. Li and H.-M. Hua, Org. Lett., 2020, 22, 7439–7442 CrossRef CAS PubMed.
- E. V. Costa, L. d. N. Soares, J. d. S. Chaar, V. R. Silva, L. d. S. Santos, H. H. F. Koolen, F. M. A. d. Silva, J. F. Tavares, G. Zengin, M. B. P. Soares and D. P. Bezerra, Molecules, 2021, 26, 3714 CrossRef CAS PubMed.
- H.-L. Wei, Y.-P. Zhao, J.-X. Wang, Y. Han, H. Li, H. Zhou, T. Hou, C.-J. Wang, Y.-M. Yao, X.-L. Zhang, Y.-F. Liu and X.-M. Liang, Bioorg. Chem., 2022, 127, 106027 CrossRef CAS PubMed.
- H.-L. Wei, Y. Han, H. Zhou, T. Hou, Y.-M. Yao, C.-M. Wen, C.-R. Wang, J.-X. Wang, A.-J. Shen, X.-L. Zhang, H. Li and Y.-F. Liu, Phytochemistry, 2022, 194, 113015 CrossRef CAS PubMed.
- A. Hostalkova, J. Marikova, L. Opletal, J. Korabecny, D. Hulcova, J. Kunes, L. Novakova, D. I. Perez, D. Jun, T. Kucera, V. Andrisano, T. Siatka and L. Cahlikova, J. Nat. Prod., 2019, 82, 239–248 CrossRef CAS PubMed.
- Y.-C. Chen, Y.-Y. Liu, L. Chen, D.-M. Tang, Y. Zhao and X.-D. Luo, J. Agric. Food Chem., 2023, 71, 16090–16101 CrossRef CAS PubMed.
- H.-L. Zeng, Y.-X. Cai, S.-S. Xu, S.-F. Wu, Y.-L. Li, X.-L. Chen, L.-Y. Kong and J.-G. Luo, Fitoterapia, 2023, 165, 105404 CrossRef CAS PubMed.
- K. Jiang, X. Liu, Y.-M. Liu, L.-N. Wang, Y.-T. Xiao and F.-C. Wu, J. Nat. Prod., 2023, 86, 2162–2170 CrossRef CAS PubMed.
- S. Ali, M. Alamzeb, M. U. Rashid and W. N. Setzer, J. Nat. Prod., 2020, 83, 1383–1393 CrossRef CAS PubMed.
- J. Xiao, J.-Y. Song, B. Lin, W. Li, Y.-Q. Yang, J.-Y. Liu, Y. Hou, G. Chen and N. Li, J. Nat. Prod., 2020, 83, 864–872 CrossRef CAS PubMed.
- P.-T. Sun, Y.-G. Cao, G.-M. Xue, M. Li, C.-L. Zhang, F. Zhao, Z.-Y. Cao, D. Wang, K. R. Gustafson, X.-K. Zheng, W.-S. Feng and H. Chen, Org. Lett., 2022, 24, 1476–1480 CrossRef CAS PubMed.
- Z. Cao, S. Zhu, Z. Xue, F. Zhang, L. Zhang, Y. Zhang, Y. Guo, G. Zhan, X. Zhang and Z. Guo, Phytochemistry, 2022, 202, 113321 CrossRef CAS PubMed.
- Y.-M. Wang, W.-Z. Ming, H. Liang, Y.-J. Wang, Y.-H. Zhang and D.-L. Meng, Bioorg. Chem., 2020, 95, 103489 CrossRef CAS PubMed.
- H. Guinaudeau, M. Leboeuf and A. Cave, J. Nat. Prod., 1979, 42, 325–360 CrossRef CAS.
- H. Guinaudeau, M. Shamma, B. Tantisewie and K. Pharadai, J. Nat. Prod., 1982, 45, 355–357 CrossRef CAS.
- C.-F. Ding, Z. Dai, H.-F. Yu, X.-D. Zhao and X.-D. Luo, Chin. J. Nat. Med., 2019, 17, 698–706 CAS.
- L. Xu, W. Yang, J. Hu, C.-M. Han and P.-F. Li, J. Asian Nat. Prod. Res., 2020, 22, 618–625 CrossRef CAS PubMed.
- T. A. Majrashi, F. Zulfiqar, A. G. Chittiboyina, Z. Ali and I. A. Khan, Nat. Prod. Res., 2019, 33, 2823–2829 CrossRef CAS PubMed.
- J. Qin, S.-Y. Zhang, Y.-B. Zhang, L.-F. Chen, N.-H. Chen, Z.-N. Wu, D. Luo, G.-C. Wang and Y.-L. Li, Nat. Prod. Res., 2021, 35, 3254–3260 CrossRef CAS PubMed.
- B. Wang, Y.-J. Zhao, Y.-L. Zhao, Y.-P. Liu, X.-N. Li, H. Zhang and X.-D. Luo, Org. Lett., 2020, 22, 257–260 CrossRef CAS PubMed.
- H. Ma, X. Jin, F. Wang, J. Jiang, L. Cheng, S. Hu, G. Zhang and H. Xu, Nat. Prod. Res., 2023, 9, 1–7 Search PubMed.
- S.-A. Wuken, X. Yin, J. Mi, S.-G. Jiao, H.-X.-G. Zhang, X.-C. Zhou, P.-F. Tu, X.-Y. Chai and C.-S. Liu, Chin. J. Exp. Tradit. Med. Formulae, 2019, 25, 172–175 Search PubMed.
- Y. Han, T. Hou, Z.-H. Zhang, Y.-H. Zhu, J.-X. Cheng, H. Zhou, J.-X. Wang, J.-T. Feng, Y.-F. Liu, Z.-M. Guo and X.-M. Liang, Phytochemistry, 2022, 199, 113209 CrossRef CAS PubMed.
- J.-G. Lu, Y. Wang, M.-R. Yang, C.-Y. Wang, J. Meng, J. Liu, Z. Yang, K. Wu, L.-P. Bai, G.-Y. Zhu and Z.-H. Jiang, Arch. Pharmacal Res., 2022, 45, 631–643 CrossRef CAS PubMed.
- L.-M. Dai, R.-Z. Huang, J. Zhu, Z.-S. Song, Z.-G. Hong, F. Qin and H. Wang, J. Mol. Struct., 2023, 1293, 136240 CrossRef CAS.
- B. K. Lombe, D. Feineis and G. Bringmann, Nat. Prod. Rep., 2019, 36, 1513–1545 RSC.
- S. Singh, N. Pathak, E. Fatima and A. S. Negi, Eur. J. Med. Chem., 2021, 226, 113839 CrossRef CAS PubMed.
- P. Patel, Phytomed. Plus, 2021, 1, 100070 CrossRef.
- J. Dickerhoff, N. Brundridge, S. A. McLuckey and D. Yang, J. Med. Chem., 2021, 64, 16205–16212 CrossRef CAS PubMed.
- S.-H. Yan, L.-M. Hu, X.-H. Hao, J. Liu, X.-Y. Tan, Z.-R. Geng, J. Ma and Z.-L. Wang, iScience, 2022, 25, 104773 CrossRef CAS PubMed.
- K. Wang, C. Gu, G. Yu, J. Lin, Z. Wang, Q. Lu, Y. Xu, D. Zhao, X. Jiang, W. Mai, S. Liu and H. Yang, Liver Res., 2022, 6, 167–174 CrossRef CAS.
- F. Xu, M. Liu, Y. Liao, Y. Zhou, P. Zhang, Y. Zeng and Z. Liu, Phytomed, 2022, 104, 154314 CrossRef CAS PubMed.
- K.-B. Wang, Y. Liu, J. Li, C. Xiao, Y. Wang, W. Gu, Y. Li, Y.-Z. Xia, T. Yan, M.-H. Yang and L.-Y. Kong, Nat. Commun., 2022, 13, 6016 CrossRef CAS PubMed.
- M. A. Leyva-Peralta, R. E. Robles-Zepeda, R. S. Razo-Hernandez, L. P. A. Berber, K. O. Lara, E. Ruiz-Bustos and J. C. Galvez-Ruiz, Anti-Cancer Agents Med. Chem., 2019, 19, 1820–1834 CrossRef CAS PubMed.
- S. Gaba, A. Saini, G. Singh and V. Monga, Bioorg. Med. Chem., 2021, 38, 116143 CrossRef CAS PubMed.
- C. Fu, G. Guan and H. Wang, Curr. Pharm. Des., 2018, 24, 2760–2764 CrossRef CAS PubMed.
- F. Bray, J. Ferlay, I. Soerjomataram, R. L. Siegel, L. A. Torre and A. Jemal, Ca-Cancer J. Clin., 2018, 68, 394–424 CrossRef PubMed.
- Q. Su, M. Fan, J. Wang, A. Ullah, M. A. Ghauri, B. Dai, Y. Zhan, D. Zhang and Y. Zhang, Cell Death Dis., 2019, 10, 939 CrossRef CAS PubMed.
- Q. Su, J. Wang, F. Liu and Y. Zhang, Toxicol. In Vitro, 2020, 66, 104840 CrossRef CAS PubMed.
- J. Wang, Q. Su, Q. Wu, K. Chen, A. Ullah, M. A. Ghauri and Y. Zhang, Arch. Pharmacal Res., 2021, 44, 1025–1036 CrossRef CAS PubMed.
- D. T. Tshitenge, T. Bruhn, D. Feineis, D. Schmidt, V. Mudogo, M. Kaiser, R. Brun, F. Wuerthner, S. Awale and G. Bringmann, J. Nat. Prod., 2019, 82, 3150–3164 CrossRef CAS PubMed.
- S. Fayez, D. Feineis, L. A. Assi, E.-J. Seo, T. Efferth and G. Bringmann, RSC Adv., 2019, 9, 15738–15748 RSC.
- S. Fayez, T. Bruhn, D. Feineis, L. A. Assi, S. Awale and G. Bringmann, J. Nat. Prod., 2020, 83, 1139–1151 CrossRef CAS PubMed.
- N. Tajuddeen, S. Fayez, P. P. Kushwaha, D. Feineis, L. Ake Assi, S. Kumar and G. Bringmann, Nat. Prod. Res., 2023, 1–5, DOI:10.1080/14786419.2023.2194648.
- J.-P. Mufusama, D. Feineis, V. Mudogo, M. Kaiser, R. Brun and G. Bringmann, RSC Adv., 2019, 9, 12034–12046 RSC.
- D. T. Tshitenge, T. Bruhn, D. Feineis, V. Mudogo, M. Kaiser, R. Brun and G. Bringmann, Sci. Rep., 2019, 9, 9812 CrossRef PubMed.
- B. K. Lombe, D. Feineis, V. Mudogo, M. Kaiser and G. Bringmann, J. Nat. Prod., 2021, 84, 1335–1344 CrossRef CAS PubMed.
- S. Fayez, J. Li, D. Feineis, L. A. Assi, M. Kaiser, R. Brun, M. A. Anany, H. Wajant and G. Bringmann, J. Nat. Prod., 2019, 82, 3033–3046 CrossRef CAS PubMed.
- Z. Liu, Z. Mi, P. Wang, S. Chang, N. Han and J. Yin, Nat. Prod. Res., 2020, 34, 3305–3312 CrossRef CAS PubMed.
- D. Katoch, D. Kumar, Y. S. Padwad, B. Singh and U. Sharma, Nat. Prod. Res., 2020, 34, 233–240 CrossRef CAS PubMed.
- D. B. MacLean, The Alkaloids: Chem Pharmacol., 1985, vol. 24, pp. 253–286 Search PubMed.
- C.-L. Zhang, Q.-L. Huang, J. Chen, W.-J. Zhang, H.-X. Jin, H.-B. Wang, C. B. Naman and Z.-Y. Cao, J. Nat. Prod., 2019, 82, 2713–2720 CrossRef CAS PubMed.
- C.-L. Zhang, Q.-L. Huang, Q. Zhu, J. Chen, F. Zhang and Z.-Y. Cao, Fitoterapia, 2020, 144, 104494 CrossRef CAS PubMed.
- S. Ka, M. Masi, N. Merindol, R. Di Lecce, M. B. Plourde, M. Seck, M. Gorecki, G. Pescitelli, I. Desgagne-Penix and A. Evidente, Phytochemistry, 2020, 175, 112390 CrossRef CAS PubMed.
- Y.-L. Liu, Y. Wang, X.-R. He, L.-S. Gan, F. Xu, Y.-J. Xu, X.-N. Wang, T. Shen and Z.-W. Zhou, Fitoterapia, 2022, 156, 105086 CrossRef CAS PubMed.
- M.-S. Zhang, Y. Deng, S.-B. Fu, D.-L. Guo and S.-J. Xiao, Molecules, 2018, 23, 1539 CrossRef PubMed.
- J. Bracegirdle, L. P. Robertson, P. A. Hume, M. J. Page, A. V. Sharrock, D. F. Ackerley, A. R. Carroll and R. A. Keyzers, J. Nat. Prod., 2019, 82, 2000–2008 CrossRef CAS PubMed.
- G. A. U. Ecoy, S. Chamni, K. Suwanborirux, P. Chanvorachote and C. Chaotham, J. Nat. Prod., 2019, 82, 1861–1873 CrossRef CAS PubMed.
- D. W. Prebble, D. C. Holland, L. P. Robertson, V. M. Avery and A. R. Carroll, Org. Lett., 2020, 22, 9574–9578 CrossRef CAS PubMed.
- F. Zhang, M. Zhao, D. R. Braun, S. S. Ericksen, J. S. Piotrowski, J. Nelson, J. Peng, G. E. Ananiev, S. Chanana, K. Barns, J. Fossen, H. Sanchez, M. G. Chevrette, I. A. Guzei, C. Zhao, L. Guo, W. Tang, C. R. Currie, S. R. Rajski, A. Audhya, D. R. Andes and T. S. Bugni, Science, 2020, 370, 974–978 CrossRef CAS PubMed.
- M. Chrzanowska, A. Grajewska and M. D. Rozwadowska, Chem. Rev., 2016, 116, 12369–12465 CrossRef CAS PubMed.
- R. Singh, S. Kumar, M. T. Patil, C. M. Sun and D. B. Salunke, Adv. Synth. Catal., 2020, 362, 4027–4077 CrossRef CAS.
- N. Tajuddeen, D. Feineis, H. Ihmels and G. Bringmann, Acc. Chem. Res., 2022, 55, 2370–2383 CrossRef CAS PubMed.
- X.-Y. Yu, J.-R. Chen and W.-J. Xiao, Chem. Rev., 2021, 121, 506–561 CrossRef CAS PubMed.
- K. P. S. Cheung, S. Sarkar and V. Gevorgyan, Chem. Rev., 2022, 122, 1543–1625 CrossRef PubMed.
- A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Chem. Rev., 2022, 122, 1485–1542 CrossRef CAS PubMed.
- L. Capaldo, D. Ravelli and M. Fagnoni, Chem. Rev., 2022, 122, 1875–1924 CrossRef CAS PubMed.
- S. P. Pitre and L. E. Overman, Chem. Rev., 2022, 122, 1717–1751 CrossRef CAS PubMed.
- C. Zhu, N. W. J. Ang, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Cent. Sci., 2021, 7, 415–431 CrossRef CAS PubMed.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed.
- H. Kim, K.-I. Min, K. Inoue, D. J. Im, D.-P. Kim and J.-i. Yoshida, Science, 2016, 352, 691–694 CrossRef CAS PubMed.
- C. Liu, J. Xie, W. Wu, M. Wang, W. Chen, S. B. Idres, J. Rong, L.-W. Deng, S. A. Khan and J. Wu, Nat. Chem., 2021, 13, 451–457 CrossRef CAS PubMed.
- D. F. Vargas, E. L. Larghi and T. S. Kaufman, Nat. Prod. Rep., 2019, 36, 354–401 RSC.
- D. F. Vargas, E. L. Larghi and T. S. Kaufman, RSC Adv., 2019, 9, 33096–33106 RSC.
- D. F. Vargas, E. L. Larghi and T. S. Kaufman, Eur. J. Org Chem., 2018, 5605–5614 CrossRef CAS.
- S. Fonzo, D. F. Vargas and T. S. Kaufman, Synthesis, 2019, 51, 3908–3914 CrossRef CAS.
- R. Schuetz, S. Schmidt and F. Bracher, Tetrahedron, 2020, 76, 131150 CrossRef CAS.
- A. Ansari, A. B. Gorde and R. Ramapanicker, Tetrahedron, 2021, 88, 132121 CrossRef CAS.
- A. K. Haidar, N. D. Kjeldsen, N. S. Troelsen, V. Previtali, K. P. Lundquist, T. O. Larsen and M. H. Clausen, Molecules, 2022, 27, 521 CrossRef CAS PubMed.
- T. Choshi, T. Kumemura, H. Fujioka, Y. Hieda and S. Hibino, Heterocycles, 2012, 84, 587–595 CrossRef CAS PubMed.
- R. Schuetz, M. Mueller, S. Gerndt, K. Bartel and F. Bracher, Arch. Pharm., 2020, 353, e2000106 CrossRef CAS PubMed.
- M. Keller, K. Sauvageot-Witzku, F. Geisslinger, N. Urban, M. Schaefer, K. Bartel and F. Bracher, Beilstein J. Org. Chem., 2021, 17, 2716–2725 CrossRef CAS PubMed.
- M. J. Scharf and B. List, J. Am. Chem. Soc., 2022, 144, 15451–15456 CrossRef CAS PubMed.
- A. Lipp, M. Selt, D. Ferenc, D. Schollmeyer, S. R. Waldvogel and T. Opatz, Org. Lett., 2019, 21, 1828–1831 CrossRef CAS PubMed.
- S. Elavarasan, J. Preety, R. Abinaya, T. Saravanan, K. K. Balasubramanian, N. Venkatramaiah and B. Baskar, Chem.–Asian J., 2022, 17, e202200878 CrossRef CAS PubMed.
- W. Liu, B. Hong, J. Wang and X. Lei, Acc. Chem. Res., 2020, 53, 2569–2586 CrossRef CAS PubMed.
- Y.-J. Wu, J.-H. Chen, M.-Y. Teng, X. Li, T.-Y. Jiang, F.-R. Huang, Q.-J. Yao and B.-F. Shi, J. Am. Chem. Soc., 2023, 145, 24499–24505 CAS.
- W. Xu, Z. Huang, X. Ji and J.-P. Lumb, ACS Catal., 2019, 9, 3800–3810 CrossRef CAS.
- Z. Huang, X. Ji and J.-P. Lumb, Org. Lett., 2019, 21, 9194–9197 CrossRef CAS PubMed.
- M. Lafrance, N. Blaquière and K. Fagnou, Chem. Commun., 2004, 2874–2875 RSC.
- R. Schuetz, M. Meixner, I. Antes and F. Bracher, Org. Biomol. Chem., 2020, 18, 3047–3068 RSC.
- Y. Zhao, Y. Li, B. Wang, J. Zhao, L. Li, X.-D. Luo and H. Zhang, J. Org. Chem., 2020, 85, 13772–13778 CrossRef CAS PubMed.
- P. Pieper, E. McHugh, M. Amaral, A. G. Tempone and E. A. Anderson, Tetrahedron, 2020, 76, 130814 CrossRef CAS.
- G. P. Perecim, V. M. Deflon, G. R. Martins, L. M. C. Pinto, G. A. Casagrande, D. Oliveira-Silva and C. Raminelli, Tetrahedron, 2020, 76, 131461 CrossRef CAS.
- J. Lv, Z.-H. Li, A.-J. Deng and H.-L. Qin, Org. Biomol. Chem., 2022, 20, 658–666 RSC.
- H. Hamamoto, Y. Shiozaki, H. Nambu, K. Hata, H. Tohma and Y. Kita, Chem.–Eur. J., 2004, 10, 4977–4982 CrossRef CAS PubMed.
- B. B. J. Tra, A. Abolle, V. Coeffard and F.-X. Felpin, Eur. J. Org Chem., 2022, 2022, e202200301 CrossRef.
- H. Takikawa, A. Nishii, T. Sakai and K. Suzuki, Chem. Soc. Rev., 2018, 47, 8030–8056 RSC.
- T. R. Cipriano da Silva, R. B. Oliveira, A. L. M. Nicastro, R. P. Ureshino and C. Raminelli, ChemistrySelect, 2023, 8, e202302821 CrossRef CAS.
- H.-G. Cheng, S. Chen, R. Chen and Q. Zhou, Angew. Chem., Int. Ed., 2019, 58, 5832–5844 CrossRef CAS PubMed.
- M. Bai, S. Jia, J. Zhang, H.-G. Cheng, H. Cong, S. Liu, Z. Huang, Y. Huang, X. Chen and Q. Zhou, Angew. Chem., Int. Ed., 2022, 61, e202205245 CrossRef CAS PubMed.
- S. Jia, M. Bai, S. Zhou, R. Sheng, H.-G. Cheng and Q. Zhou, Sci. China: Chem., 2023, 66, 3136–3140 CrossRef CAS.
- S. Nishimoto, H. Nakahashi and M. Toyota, Tetrahedron Lett., 2020, 61, 152599 CrossRef CAS.
- R. Chen, S. Jia, Y. Man, H.-G. Cheng and Q. Zhou, Synthesis, 2024, 56, 1695–1701 CrossRef CAS.
- G. He, B. List and M. Christmann, Angew. Chem., Int. Ed., 2021, 60, 13591–13596 CrossRef CAS PubMed.
- D. Stubba, G. Lahm, M. Geffe, J. W. Runyon, A. J. Arduengo III and T. Opatz, Angew. Chem., Int. Ed., 2015, 54, 14187–14189 CrossRef CAS PubMed.
- J. Yu, Z. Zhang, S. Zhou, W. Zhang and R. Tong, Org. Chem. Front., 2018, 5, 242–246 RSC.
- S. Uenishi, R. Kakigi, K. Hideshima, A. Miyawaki, J. Matsuoka, T. Ogata, K. Tomioka and Y. Yamamoto, Tetrahedron, 2021, 90, 132165 CrossRef CAS.
- W. Li, M. Jiang, W. Chen, Y. Chen, Z. Yang, P. Tang and F. Chen, J. Org. Chem., 2021, 86, 8143–8153 CrossRef CAS PubMed.
- W. Li, M. Jiang, M. Liu, X. Ling, Y. Xia, L. Wan and F. Chen, Chem.–Eur. J., 2022, 28, e202200700 CrossRef CAS PubMed.
- R. Boda and G. D. Cuny, Tetrahedron, 2022, 129, 133146 CrossRef CAS.
- A. Cervi, P. Aillard, N. Hazeri, L. Petit, C. L. L. Chai, A. C. Willis and M. G. Banwell, J. Org. Chem., 2013, 78, 9876–9882 CrossRef CAS PubMed.
- W. Li, S. Huang, M. Jiang, Y. Chen, Z. Yang, P. Tang and F. Chen, ChemCatChem, 2023, 15, e202201553 CrossRef CAS.
- E. D. Slack, R. Seupel, D. H. Aue, G. Bringmann and B. H. Lipshutz, Chem.–Eur. J., 2019, 25, 14237–14245 CrossRef CAS PubMed.
- Y.-I. Jo, C.-Y. Lee and C.-H. Cheon, Org. Lett., 2020, 22, 4653–4658 CrossRef CAS PubMed.
- Y.-I. Jo, C.-Y. Lee and C.-H. Cheon, J. Org. Chem., 2020, 85, 12770–12776 CrossRef CAS PubMed.
- D. Berthold and W. van Otterlo, Chem.–Eur. J., 2023, 29, e202302070 CrossRef CAS PubMed.
- B. M. Trost, C.-I. J. Hung and Z. Jiao, J. Am. Chem. Soc., 2019, 141, 16085–16092 CrossRef CAS PubMed.
- B. M. Trost, C.-I. Hung, T. Saget and E. Gnanamani, Nat. Catal., 2018, 1, 523–530 CrossRef CAS.
- B. M. Trost, C.-I. J. Hung and E. Gnanamani, ACS Catal., 2019, 9, 1549–1557 CrossRef CAS.
- I. Ninomiya, O. Yamamoto and T. Naito, J. Chem. Soc., Perkin Trans. 1, 1980, 212–216 RSC.
- L. S. Hutchings-Goetz, C. Yang, J. W. B. Fyfe and T. N. Snaddon, Angew. Chem., Int. Ed., 2020, 59, 17556–17564 CrossRef CAS PubMed.
- C. M. Pearson, J. W. B. Fyfe and T. N. Snaddon, Angew. Chem., Int. Ed., 2019, 58, 10521–10527 CrossRef CAS PubMed.
- A. D. C. Parenty, K. M. Guthrie, Y. F. Song, L. V. Smith, E. Burkholder and L. Cronin, Chem. Commun., 2006, 11, 1194–1196 RSC.
- P. J. Murphy, J. Tibble-Howlings, R. M. Kowalczyk and K. Stevens, Tetrahedron Lett., 2020, 61, 151785 CrossRef CAS.
- B. Sheng, C. Zeng, J. Chen, W.-C. Ye, W. Tang, P. Lan and M. G. Banwell, Eur. J. Org Chem., 2022, 2022, e202101511 CrossRef CAS.
- N. Aravindan and M. Jeganmohan, J. Org. Chem., 2021, 86, 14826–14843 CrossRef CAS PubMed.
- N. Aravindan, V. Vinayagam and M. Jeganmohan, Org. Lett., 2022, 24, 5260–5265 CrossRef CAS PubMed.
- N. Aravindan and M. Jeganmohan, Org. Lett., 2023, 25, 3853–3858 CrossRef CAS PubMed.
- J. Fu, B. Li, X. Wang, H. Wang, M. Deng, H. Yang, B. Lin, M. Cheng, L. Yang and Y. Liu, Org. Lett., 2022, 24, 8310–8315 CrossRef CAS PubMed.
- Z. Jin and G. Yao, Nat. Prod. Rep., 2019, 36, 1462–1488 RSC.
- M. B. Uphade and K. R. Prasad, Tetrahedron, 2020, 76, 131661 CrossRef CAS.
- R.-S. Tang, L.-Y. Chen, C.-H. Lai and T.-H. Chuang, Org. Lett., 2020, 22, 9337–9341 CrossRef CAS PubMed.
- Y. Pang, H. Xiao, W. Ou, X. Zhang, X. Wang and S. Huang, Tetrahedron Lett., 2020, 61, 151733 CrossRef CAS.
- C. J. J. Hall, I. S. Marriott, K. E. Christensen, A. J. Day, W. R. F. Goundry and T. J. Donohoe, Chem. Commun., 2022, 58, 4966–4968 RSC.
- Y. Menjo, A. Hamajima, N. Sasaki and Y. Hamada, Org. Lett., 2011, 13, 5744–5747 CrossRef CAS PubMed.
- T.-Y. Zhang, L.-Y. Zhang, X. Liang, K. Wei and Y.-R. Yang, Org. Lett., 2022, 24, 2905–2909 CrossRef CAS PubMed.
- D. Pollok, L. M. Grossmann, T. Behrendt, T. Opatz and S. R. Waldvogel, Chem.–Eur. J., 2022, 28, e202201523 CrossRef CAS PubMed.
- A. Lipp, D. Ferenc, C. Guetz, M. Geffe, N. Vierengel, D. Schollmeyer, H. J. Schaefer, S. R. Waldvogel and T. Opatz, Angew. Chem., Int. Ed., 2018, 57, 11055–11059 CrossRef CAS PubMed.
- A. Lipp, M. Selt, D. Ferenc, D. Schollmeyer, S. R. Waldvogel and T. Opatz, Org. Lett., 2019, 21, 1828–1831 CrossRef CAS PubMed.
- S.-H. Hou, A. Y. Prichina and G. Dong, Angew. Chem., Int. Ed., 2021, 60, 13057–13064 CrossRef CAS PubMed.
- D. Sun, B. Pedersen and J. A. Ellman, Chem. Sci., 2019, 10, 535–541 RSC.
- G. Li, Q. Wang and J. Zhu, Nat. Commun., 2021, 12, 36 CrossRef CAS PubMed.
- S.-G. Wang, X.-J. Liu, Q.-C. Zhao, C. Zheng, S.-B. Wang and S.-L. You, Angew. Chem., Int. Ed., 2015, 54, 14929–14932 CrossRef CAS PubMed.
- X. Zhou, W. Li, R. Zhou, X. Wu, Y. Huang, W. Hou, C. Li, Y. Zhang, W. Nie, Y. Wang, H. Song, X.-Y. Liu, Z. Zheng, F. Xie, S. Li, W. Zhong and Y. Qin, CCS Chem., 2021, 3, 1376–1383 CrossRef CAS.
- Y. Tang, Y. Zhang, J. Zhao, F. Xue, H. He, F. Xue, X.-Y. Liu and Y. Qin, Tetrahedron Lett., 2022, 104, 154027 CrossRef CAS.
- H. He, F. Xue, Z. Hu, P. Li, Q. Xiao, M. Zhang, F. Xue, D. Zhang, H. Song, X.-Y. Liu, Z. Zheng, S. Li, W. Zhong and Y. Qin, Org. Chem. Front., 2022, 9, 2322–2327 RSC.
- M. Odagi, T. Matoba and K. Nagasawa, J. Org. Chem., 2022, 87, 1065–1073 CrossRef CAS PubMed.
- J. Li, Y. Liu, X. Song, T. Wu, J. Meng, Y. Zheng, Q. Qin, D. Zhao and M. Cheng, Org. Lett., 2019, 21, 7149–7153 CrossRef CAS PubMed.
- J. Li, T. Wu, X. Song, Y. Zheng, J. Meng, Q. Qin, Y. Liu, D. Zhao and M. Cheng, RSC Adv., 2020, 10, 18953–18958 RSC.
- M. Zhang, M. Zhao, Y. Wang, L. Chen, B. Liu, X. You, W. Sun, L. Hong and G. Li, J. Org. Chem., 2023, 88, 1720–1729 CrossRef CAS PubMed.
- C. Zhang, Y. Wang, Y. Song, H. Gao, Y. Sun, X. Sun, Y. Yang, M. He, Z. Yang, L. Zhan, Z.-X. Yu and Y. Rao, CCS Chem., 2019, 1, 352–364 CrossRef CAS.
- H. Takeuchi, S. Inuki, K. Nakagawa, T. Kawabe, A. Ichimura, S. Oishi and H. Ohno, Angew. Chem., Int. Ed., 2020, 59, 21210–21215 CrossRef CAS PubMed.
- T. Ling, V. R. Macherla, R. R. Manam, K. A. McArthur and B. C. M. Potts, Org. Lett., 2007, 9, 2289–2292 CrossRef CAS PubMed.
- J. He, S. Li, Y. Deng, H. Fu, B. N. Laforteza, J. E. Spangler, A. Homs and J.-Q. Yu, Science, 2014, 343, 1216–1220 CrossRef CAS PubMed.
- G. Chen, T. Shigenari, P. Jain, Z. Zhang, Z. Jin, J. He, S. Li, C. Mapelli, M. M. Miller, M. A. Poss, P. M. Scola, K.-S. Yeung and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 3338–3351 CrossRef CAS PubMed.
- G. Chen, Z. Zhuang, G.-C. Li, T. G. Saint-Denis, Y. Hsiao, C. L. Joe and J.-Q. Yu, Angew. Chem., Int. Ed., 2017, 56, 1506–1509 CrossRef CAS PubMed.
- Y. Zheng, X.-D. Li, P.-Z. Sheng, H.-D. Yang, K. Wei and Y.-R. Yang, Org. Lett., 2020, 22, 4489–4493 CrossRef CAS PubMed.
- D. Morikawa, K. Morii, Y. Yasuda, A. Mori and K. Okano, J. Org. Chem., 2020, 85, 8603–8617 CrossRef CAS PubMed.
- Y. Hayashi, K. Okano and A. Mori, Org. Lett., 2018, 20, 958–961 CrossRef CAS PubMed.
- K. Morii, Y. Yasuda, D. Morikawa, A. Mori and K. Okano, J. Org. Chem., 2021, 86, 13388–13401 CrossRef CAS PubMed.
- S. Sarkar and R. Samanta, Org. Lett., 2022, 24, 4536–4541 CrossRef CAS PubMed.
- J. Li, L. M. Jiang, F. Cheng, Y.-J. Zhou, D.-S. Duan, D.-Y. Zhu, K. Zhang, Z. Xiong and S.-H. Wang, Tetrahedron Lett., 2022, 106 Search PubMed.
- X.-Q. Mou, Z.-L. Xu, L. Xu, S.-H. Wang, B.-H. Zhang, D. Zhang, J. Wang, W.-T. Liu and W. Bao, Org. Lett., 2016, 18, 4032–4035 CrossRef CAS PubMed.
- M. A. Bajada, J. Sanjose-Orduna, G. Di Liberto, S. Tosoni, G. Pacchioni, T. Noel and G. Vile, Chem. Soc. Rev., 2022, 51, 3898–3925 RSC.
- T. Noel, Y. Cao and G. Laudadio, Acc. Chem. Res., 2019, 52, 2858–2869 CrossRef CAS PubMed.
- L. Buglioni, F. Raymenants, A. Slattery, S. D. A. Zondag and T. Noel, Chem. Rev., 2022, 122, 2752–2906 CrossRef CAS PubMed.
- M. Elsherbini and T. Wirth, Acc. Chem. Res., 2019, 52, 3287–3296 CrossRef CAS PubMed.
- A. C. Brewer, P. C. Hoffman, J. R. Martinelli, M. E. Kobierski, N. Mullane and D. Robbins, Org. Process Res. Dev., 2019, 23, 1484–1498 CrossRef.
- K. P. Cole, J. M. Groh, M. D. Johnson, C. L. Burcham, B. M. Campbell, W. D. Diseroad, M. R. Heller, J. R. Howell, N. J. Kallman, T. M. Koenig, S. A. May, R. D. Miller, D. Mitchell, D. P. Myers, S. S. Myers, J. L. Phillips, C. S. Polster, T. D. White, J. Cashman, D. Hurley, R. Moylan, P. Sheehan, R. D. Spencer, K. Desmond, P. Desmond and O. Gowran, Science, 2017, 356, 1144–1150 CrossRef CAS PubMed.
- F. Ghirga, A. Bonamore, L. Calisti, I. D'Acquarica, M. Mori, B. Botta, A. Boffi and A. Macone, Int. J. Mol. Sci., 2017, 18, 2464 CrossRef PubMed.
- B. Gao, B. Yang, X. Feng and C. Li, Nat. Prod. Rep., 2022, 39, 139–162 RSC.
- Y. Wang, N. Tappertzhofen, D. Mendez-Sanchez, M. Bawn, B. Lyu, J. M. Ward and H. C. Hailes, Angew. Chem., Int. Ed., 2019, 58, 10120–10125 CrossRef CAS PubMed.
- Y. Wang, F. Subrizi, E. M. Carter, T. D. Sheppard, J. M. Ward and H. C. Hailes, Nat. Commun., 2022, 13, 5436 CrossRef CAS PubMed.
- R. Roddan, J. M. Ward, N. H. Keep and H. C. Hailes, Curr. Opin. Chem. Biol., 2020, 55, 69–76 CrossRef CAS PubMed.
- B. R. Lichman, M. C. Gershater, E. D. Lamming, T. Pesnot, A. Sula, N. H. Keep, H. C. Hailes and J. M. Ward, FEBS J., 2015, 282, 1137–1151 CrossRef CAS PubMed.
- R. Roddan, G. Gygli, A. Sula, D. Mendez-Sanchez, J. Pleiss, J. M. Ward, N. H. Keep and H. C. Hailes, ACS Catal., 2019, 9, 9640–9649 CrossRef CAS.
- J. Zhao, D. Mendez-Sanchez, R. Roddan, J. M. Ward and H. C. Hailes, ACS Catal., 2021, 11, 131–138 CrossRef CAS.
- T. Gurkok, E. Ozhuner, I. Parmaksiz, S. Ozcan, M. Turktas, A. Ipek, I. Demirtas, S. Okay and T. Unver, Front. Plant Sci., 2016, 7, 98 Search PubMed.
- J. Siegrist, S. Aschwanden, S. Mordhorst, L. Thoeny-Meyer, M. Richter and J. N. Andexer, ChemBioChem, 2015, 16, 2576–2579 CrossRef CAS PubMed.
- F. Subrizi, Y. Wang, B. Thair, D. Mendez-Sanchez, R. Roddan, M. Cardenas-Fernandez, J. Siegrist, M. Richter, J. N. Andexer, J. M. Ward and H. C. Hailes, Angew. Chem., Int. Ed., 2021, 60, 18673–18679 CrossRef CAS PubMed.
- J. Zhu, H. Tan, L. Yang, Z. Dai, L. Zhu, H. Ma, Z. Deng, Z. Tian and X. Qu, ACS Catal., 2017, 7, 7003–7007 CrossRef CAS.
- L. Yang, J. Zhu, C. Sun, Z. Deng and X. Qu, Chem. Sci., 2020, 11, 364–371 RSC.
- A. L. Meadows, K. M. Hawkins, Y. Tsegaye, E. Antipov, Y. Kim, L. Raetz, R. H. Dahl, A. Tai, T. Mahatdejkul-Meadows, L. Xu, L. Zhao, M. S. Dasika, A. Murarka, J. Lenihan, D. Eng, J. S. Leng, C.-L. Liu, J. W. Wenger, H. Jiang, L. Chao, P. Westfall, J. Lai, S. Ganesan, P. Jackson, R. Mans, D. Platt, C. D. Reeves, P. R. Saija, G. Wichmann, V. F. Holmes, K. Benjamin, P. W. Hill, T. S. Gardner and A. E. Tsong, Nature, 2016, 537, 694–697 CrossRef CAS PubMed.
- H. Shuai, M. Myronovskyi, B. Rosenkraenzer, C. Paulus, S. Nadmid, M. Stierhof, D. Kolling and A. Luzhetskyy, ACS Chem. Biol., 2022, 17, 598–608 CrossRef CAS PubMed.
- N. S. Roy, I.-Y. Choi, T. Um, M. J. Jeon, B.-Y. Kim, Y.-D. Kim, J.-K. Yu, S. Kim and N.-S. Kim, Plants, 2021, 10, 1314 CrossRef CAS PubMed.
- I. Desgagne-Penix, Phytochem. Rev., 2021, 20, 409–431 CrossRef CAS.
- C. Guo, Y. Chen, D. Wu, Y. Du, M. Wang, C. Liu, J. Chu and X. Yao, Int. J. Mol. Sci., 2022, 23, 10898 CrossRef CAS PubMed.
- Y. Yang, P. Hu, X. Zhou, P. Wu, X. Si, B. Lu, Y. Zhu and Y. Xia, Fitoterapia, 2020, 140, 104412 CrossRef CAS PubMed.
- L. M. T. Chacon, H. Leiva, I. C. Z. Vahos, D. C. Restrepo and E. Osorio, Biocatal. Agric. Biotechnol., 2023, 50, 102670 CrossRef.
- X. Liu, J. Bu, Y. Ma, Y. Chen, Q. Li, X. Jiao, Z. Hu, G. Cui, J. Tang, J. Guo and L. Huang, Plant Physiol. Biochem., 2021, 168, 507–515 CrossRef CAS PubMed.
- T. R. Valentic, J. T. Payne and C. D. Smolke, ACS Catal., 2020, 10, 4497–4509 CrossRef CAS.
- S. Wu, R. Snajdrova, J. C. Moore, K. Baldenius and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2021, 60, 88–119 CrossRef CAS PubMed.
- M. Zhao, X. Hong, Abdullah, R. Yao and Y. Xiao, Green Chem., 2021, 23, 838–847 RSC.
- M. Zhao, Z. Qin, A. Abdullah and Y. Xiao, Green Chem., 2022, 24, 3225–3234 RSC.
- D. K. Ro, E. M. Paradise, M. Ouellet, K. J. Fisher, K. L. Newman, J. M. Ndungu, K. A. Ho, R. A. Eachus, T. S. Ham, J. Kirby, M. C. Y. Chang, S. T. Withers, Y. Shiba, R. Sarpong and J. D. Keasling, Nature, 2006, 440, 940–943 CrossRef CAS PubMed.
- J. Zhang, L. G. Hansen, O. Gudich, K. Viehrig, L. M. M. Lassen, L. Schruebbers, K. B. Adhikari, P. Rubaszka, E. Carrasquer-Alvarez, L. Chen, V. D'Ambrosio, B. Lehka, A. K. Haidar, S. Nallapareddy, K. Giannakou, M. Laloux, D. Arsovska, M. A. K. Jorgensen, L. J. G. Chan, M. Kristensen, H. B. Christensen, S. Sudarsan, E. A. Stander, E. Baidoo, C. J. Petzold, T. Wulff, S. E. O'Connor, V. Courdavault, M. K. Jensen and J. D. Keasling, Nature, 2022, 609, 341–347 CrossRef CAS PubMed.
- S. Galanie, K. Thodey, I. J. Trenchard, M. F. Interrante and C. D. Smolke, Science, 2015, 349, 1095–1100 CrossRef CAS PubMed.
- I. J. Trenchard, M. S. Siddiqui, K. Thodey and C. D. Smolke, Metab. Eng., 2015, 31, 74–83 CrossRef CAS PubMed.
- O. K. Jamil, A. Cravens, J. T. Payne, C. Y. Kim and C. D. Smolke, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2205848119 CrossRef CAS PubMed.
- J. Han and S. Li, Commun. Chem., 2023, 6, 27 CrossRef CAS PubMed.
- J. T. Payne, T. R. Valentic and C. D. Smolke, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2112520118 CrossRef CAS PubMed.
- W. C. DeLoache, Z. N. Russ, L. Narcross, A. M. Gonzales, V. J. J. Martin and J. E. Dueber, Nat. Chem. Biol., 2015, 11, 465–471 CrossRef CAS PubMed.
- A. Nakagawa, E. Matsumura, T. Koyanagi, T. Katayama, N. Kawano, K. Yoshimatsu, K. Yamamoto, H. Kumagai, F. Sato and H. Minami, Nat. Commun., 2016, 7, 10390 CrossRef CAS PubMed.
- W. Zhao, M. Liu, C. Shen, K. Liu, H. Liu, C. Ou, W. Dai, X. Liu and J. Liu, Green Chem., 2021, 23, 5944–5955 RSC.
- C. E. Hodgman and M. C. Jewett, Metab. Eng., 2012, 14, 261–269 CrossRef CAS PubMed.
- S. K. Hammer and J. L. Avalos, Nat. Chem. Biol., 2017, 13, 823–832 CrossRef CAS PubMed.
- P. S. Grewal, J. A. Samson, J. J. Baker, B. Choi and J. E. Dueber, Nat. Chem. Biol., 2021, 17, 96–103 CrossRef CAS PubMed.
- S. d'Oelsnitz, W. Kim, N. T. Burkholder, K. Javanmardi, R. Thyer, Y. Zhang, H. S. Alper and A. D. Ellington, Nat. Chem. Biol., 2022, 18, 981–989 CrossRef PubMed.
- M. M. Mitchler, J. M. Garcia, N. E. Montero and G. J. Williams, Curr. Opin. Biotechnol., 2021, 69, 172–181 CrossRef CAS PubMed.
- G. S. Hossain, M. Saini, R. Miyake, H. Ling and M. W. Chang, Trends Biotechnol., 2020, 38, 797–810 CrossRef CAS PubMed.
- J. Hafner, J. Payne, H. MohammadiPeyhani, V. Hatzimanikatis and C. Smolke, Nat. Commun., 2021, 12, 1760 CrossRef CAS PubMed.
- K. Lovato, P. S. Fier and K. M. Maloney, Nat. Rev. Chem., 2021, 5, 546–563 CrossRef CAS PubMed.
- R. A. Sheldon and J. M. Woodley, Chem. Rev., 2018, 118, 801–838 CrossRef CAS PubMed.
- M. Yamada, K. Azuma, I. Takizawa, Y. Ejima, M. Yamano, K. Satoh, T. Doi, H. Ueda and H. Tokuyama, Org. Process Res. Dev., 2023, 27, 343–357 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4np00023d |
| This journal is © The Royal Society of Chemistry 2024 |