
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural diversity and chemical logic underlying the assembly of monoterpene indole alkaloids oligomers†‡
Pierre
Le Pogam
 * and
Mehdi A.
Beniddir
*
* and
Mehdi A.
Beniddir
*
Équipe, Chimie des Substances Naturelles, Université Paris-Saclay, CNRS, BioCIS, 17 avenue des Sciences, 91400 Orsay, France. E-mail: pierre.le-pogam-alluard@universite-paris-saclay.fr; mehdi.beniddir@universite-paris-saclay.fr
First published on 12th September 2024
Abstract
Covering: up to 2024
This review aims to draw a parallel between all known oligomers of monoterpene indole alkaloids (MIAs) by illustrating the chemical logic underlying their assembly. For this purpose, oligomeric MIAs were first comprehensively listed and organized according to the names of the backbones of their constitutive monomers and the binding sites. From this extensive list, an oligomer network was generated and unprecedented MIA statistics were mined and shared herein. Subsequently, oligomeric MIAs were categorized according to the number of connections instigated between their monomeric components (single, double, triple, and mixed tethering), then subdivided according to the uniqueness or combination of oligomerization assembly reactions. This effort outlined oligomerization trends in a scaffold-specific manner, and established binding reactivity patterns facilitating the comprehension of the associated biosynthetic processes. At last, this review illustrates a unique initiative in crafting a comprehensive repository of machine-readable metadata for MIA oligomers that could be leveraged for chemoinformatic purposes.
 Mehdi A. Beniddir | Mehdi Beniddir is a Full Professor of natural products chemistry at the Faculty of Pharmacy of Paris-Saclay University. He graduated in pharmacy and received his MSc degree from Paris-Sud University in 2009. He obtained his PhD under the guidance of Dr Françoise Guéritte and Dr Marc Litaudon at the Institut de Chimie des Substances Naturelles (ICSN-CNRS) in 2012. He was subsequently a postdoctoral fellow of Prof. Erwan Poupon at Paris-Saclay University. His research interests include the streamlined discovery of intricate natural substances from plants, marine invertebrates, and micro-organisms using prioritization strategies integrating the principles of decision theory to mimic the chemist's intuition in targeting natural substances. |
1 Introduction
From an evolutionary standpoint, oligomerization is an energetically economical biosynthetic strategy that nature commonly adopts to generate complex natural product architectures in short order.1,2 These compounds arise biosynthetically from the union of a monomeric building block with itself or another moiety that, sometimes, does not belong to the genuine oligomer family.With presumably more than 4000 members, Monoterpene Indole Alkaloids (MIAs) represent one of the most structurally diverse groups of alkaloids. Owing to their hybrid biosynthetic origin and to the inherent reactivity of strictosidine (1) (Fig. 1) as their nearly unique precursor and to that of its early derivatives, MIA compounds arise as an impressive array of structural variants disclosing complex, polycyclic scaffolds. This catalogue of intriguing molecules has been tantalizing the interest of both natural product and organic chemists' communities since the 1950s. The diverse bioactivities of many MIAs also contributed to the stern research endeavors geared towards this iconic structural class, now comprising sustained efforts to unveil the biosynthetic gene clusters3 leading to pharmacologically-relevant structures to secure their supply using engineered hosts.4–6
 | ||
| Fig. 1 Overview on MIA oligomers. | ||
The outstanding chemodiversity of MIAs relies on the oligomerization of monomeric units, using a wealth of different assembly modes to bridge their constitutive components (Fig. 1). Notably, little is known about the biochemical basis of those bridging reactions, despite their high potential. For instance, more than 700 MIA dimers, anhydrovinblastine (2) (Fig. 1), a biosynthetic precursor of the anticancer drugs vinblastine (3) and vincristine, is the sole oligomer for which the dimerization step has been deciphered.7 Remarkably, the chemical basis underpinning these associations has not been comprehensively considered so far. As a result of the interest represented by these structures, the topic of bisindole alkaloids has been reviewed four times with three of them being a contribution to different issues of the Alkaloids: Chemistry and Biology series. A first review, published in 1969, reveals a more pronounced emphasis on the dimerization modes of MIAs among the different reviews proposed in the field, although it is limited to asymmetrical dimers and is considered obsolete today due to the number of MIA dimers isolated since this review was published.8 Since then, three further reviews have been devoted to updating the diversity of bisindole alkaloids, with varying degrees of emphasis on monoterpene indole alkaloids.9–11 Thus, all these contributions covered the subject in a similar way but across different periods of time, with an emphasis on the monomeric components and few details regarding the chemical logic underlying their dimerization.
In stark contrast with these former contributions, this review aims to draw a parallel between all known MIA oligomers by illustrating the chemical logic underlying their assembly. For this purpose, oligomeric MIAs will first be classified according to the connections instigated between their monomeric components. At this level, a distinction is envisaged between compounds with a single intermonomeric connectivity and those with two or more intermonomeric connectivities. A further benefit of this review will be the ability to establish the dominant patterns of heterodimeric MIAs, which were only considered in Szabó’s 2008 review (Scheme 10),12 but on a limited set of compounds and with a limited structural accuracy. Outlining these dimerization trends in a scaffold-specific manner established binding reactivity patterns and facilitated the comprehension of the associated biosynthetic processes.
Additionally, our bibliographic survey also covered pseudodimeric structures (viz. combining both a MIA and either a tryptamine or an iridoid unit) and even hybrid structures including both a MIA and a biosynthetically unrelated partner insofar as these latter often capitalize on the same reactivity logic (Fig. 2). Although the partners likely to couple with MIAs have therefore been considered very broadly, we have adhered to a strict biosynthetic definition of the notion of MIA, limiting ourselves to molecules derived from strictosidine, as the supposedly universal precursor of all members of this phytochemical class.13 Interested readers are referred to a specialized literature for further insight into such tryptamine–iridoid alkaloids, obtained apart from the canonical strictosidine pathway.14,15
 | ||
| Fig. 2 Examples of pseudodimeric and hybrid structures. | ||
2 Comprehensive analysis of oligomeric MIA chemical space
2.1 Mining monoterpene indole alkaloid oligomers literature
The comprehensive listing of the impressive number of MIA oligomers required extensive database explorations, including Reaxys, SciFinder Scholar, and the Dictionary of Natural Products. Initially, the four above mentioned reviews were merged and used as a starting point. Next, well-tailored structural queries, based on constitutive generic skeletons, (See ESI‡) allowed for the completion of this review effort. To the best of our knowledge, the latest attempt to document the basic MIA scaffolds can be found in the Dictionary of Alkaloids where 42 skeletons had been distinguished. Building upon this effort, 78 additional skeletons were identified, defined structurally (see ESI‡) and used to chart the MIA oligomers listing. Ultimately, an up-to-date, metadata-enriched comprehensive listing of more than 950 MIA oligomers was obtained (see ESI‡) and organized according to the skeleton names of the constitutive monomers and the bridging sites. Notably, this listing has been uploaded to LOTUS16 to share this knowledge with the community.2.2 MIA oligomers statistics
The data gathered in the MIA oligomer listing allowed, for the first time, the drawing of a global interconnection landscape for this family of natural products. As such, an oligomer network (Fig. 3) was crafted from this data and interesting statistics were obtained. As of 2024, 703 dimers, 13 trimers and only one tetramer have been reported. Accordingly, the 703 known dimers seem to be dominated by 25 MIA skeletons. Some MIA subtypes are notably prevalent to capture the diversity of MIA dimers with no less than 222 such compounds comprising an aspidospermane unit, 187 molecules containing a vobasane unit and 162 revealing an ibogane-type component. The predominance of aspidospermane is in fact even greater than it may seem at first glance since almost half of aspidospermane-comprising MIA dimers are of the bis-aspidospermane subtype (102 entries), whereas bis-vobasane and bis-ibogane type dimers are far less represented (7 and 6 entries, respectively). Other prevalent contributors to the chemical space of MIA dimers include macrolane-based compounds (67 macrolane units in strict MIA dimers), eburnane (46 such compounds), corynantheane (42 entries) and pleiocarpamane (39 strict MIA dimers). It is interesting to note that this predominance of MIA scaffolds is also encountered to some extent for trimers and the unique tetramer with the aspidospermane scaffold vastly dominating this chemical space (20 units out of 43 building blocks when considering all 13 trimers and alasmontamine (A) followed by the vobasane unit (8 monomers out of 43)). One hundred pseudodimers have been identified in the literature, two-thirds of them incorporating a second tryptamine unit (74 entries). A huge majority of compounds comprising an additional tryptamine unit derive from a corynantheane-type MIA (70) with the few others being related to vobasane-type MIAs. The 26 pseudodimers featuring an additional iridoid unit are mostly found in Gelsemium-type scaffolds (mainly gelsedane and humanteninane) and, to a lesser extent, are associated with a few aspidospermane or strictosidine-type MIAs. To complete this overview, an array of extraneous units that have been involved in interunit connections were unveiled during our literature survey, including methylene group,18 urea group,19 acetonic group,20 pyrocatechuic group,21 thioether bridge,22,23 sugars24 or in the diversification of MIAs through the incorporation of unrelated biosynthetic units such as various products related to the shikimic acid pathways,25,26 gallic acid derivatives,27,28 anthranilic acids29 derivatives; (104 entries), flavonoids (6 compounds), canthinones (8 compounds) and various other miscellaneous products. Before getting to the heart of the matter, we feel it is important to mention that some MIA skeletons incorporate a biosynthetic element other than tryptamine and secologanin in their core constitution. O'Connor's30 recent elucidation of the biosynthetic pathways of strychnine demonstrated that this scaffold relies on the incorporation of a malonyl-CoA unit, which had been assumed for nearly 60 years on the basis of chemical reactivity considerations and radiolabelling studies.31,32 | ||
| Fig. 3 MIA oligomer network (trimers and tetramer are excluded) generated with Cytoscape 3.8.0.17 Self-loops refer to dimers comprising two components having the same MIA skeleton. Numbers on the edges define the number of oligomers pertaining to specific skeletons. Purple nodes are non-MIA counterparts. A machine-readable version of this figure is accessible as a ESI.‡ | ||
3 Intermonomeric connection analysis in oligomeric MIAs
During our bibliographic survey, it became apparent that many oligomerization sequences were, in some way, stringently associated with the biosynthesis of strict MIA dimers, whereas some others are geared towards the incorporation of alternative biosynthetic units. To highlight these considerations, a comprehensive list of simplified models of all MIA skeletons is provided as a ESI‡ indicating bridging sites and involved coupling partners (S1, ESI‡). Moreover, to follow the logic of this review, a reactivity table is also provided as a ESI (S2, ESI‡). This table classifies all33 of the MIA oligomers covered by our study according to the putative sequence of reactivity that led to their final scaffold with a distinct colour code to instantly highlight their gross constitution (viz. strict MIA constitution, pseudodimer with additional tryptamine unit, pseudodimer with additional iridoid unit, MIA with extraneous phenylpropanoid or gallate unit (sensu lato) and MIA with various other units). Inspired by the first classification system proposed by Kunesch et al.8 for monoterpene bisindole alkaloids in 1969, a deep analysis of the wealth of the MIA oligomers led us to classify this fascinating family according to the number of bridges instigated between the constituting units (single, double, triple, and mixed tethering) as illustrated in Fig. 4. Then, in each tethering mode, some cornerstone oligomers were addressed to illustrate a unique chemical logic or a combination of assembly reactions (See S2, ESI‡, for a comprehensive list of MIA related to a chemical logic). To make it easier for readers to follow the numbering system used in this review, we have decided to use the Le Men and Taylor MIA biosynthetic numbering.34 This section will detail the above-mentioned assembly reactions.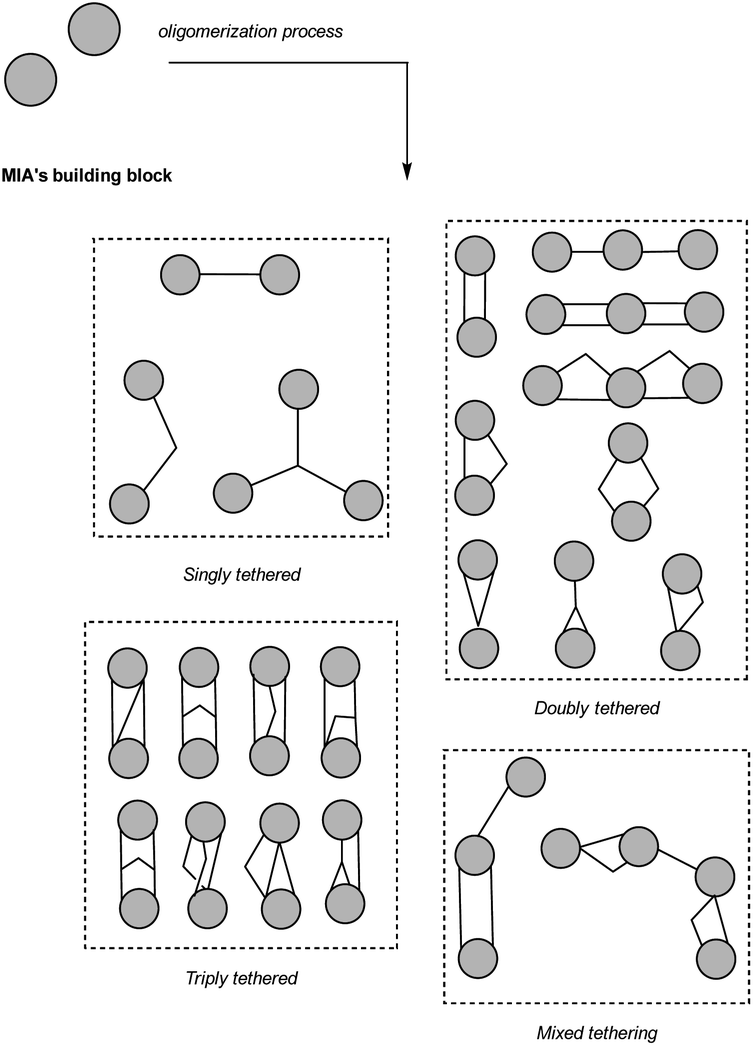 | ||
| Fig. 4 Overview of reported bridging modes in MIA oligomers. | ||
3.1 Singly-tethered oligomers
A retro-oligomerization analysis conducted on the singly-tethered MIA oligomers revealed the implication of four assembly mechanisms.3.1.1.1 Indoleninium and quinone methide. The electrophilic nature of vobasinol C-3 provides an explanation for the wealth of MIA dimers disclosing a 3-vobasinyl residue. Indeed, a vast array of MIA subtypes are known to react with this monomer, in a seemingly indistinct manner. Our literature survey indicated that no less than 150 vobasane-ibogane (Fig. 1, see S2, ESI‡) dimers have been reported to date, all of which derive from a conjugate 1,6-addition on the electrophilic C-3 position of the vobasane scaffold, with the possibility of involving additional reactions in the context of more sophisticated coupling strategies. This mere reactivity scheme is illustrated through the examples of the vallesamane/vobasane-type pseudovobparicine (4)37 and the recently reported vobasane/tryptamine pseudodimer pyrrovobasine (5) (Fig. 5).38 This reactivity is highly predominant when considering the vobasane building block as a whole since vobasane-comprising dimers involving akuammicine (1 entry), aspidospermane (2 entries), sarpagane (5 entries), vallesamane (5 entries), vincorinane (2 entries) and bis-vobasane dimers (7 entries) use its C-3 position as an electrophilic center, even though a few of them instigate partner-dependent multiple connections as will be developed later on. Quite remarkably, it seems that only the recently described ligustrinine (114) (Fig. 35) enters a dimerization process that does not involve the vobasinyl electrophilic C-3 position.
 | ||
| Fig. 5 Singly-tethered oligomers resulting from an electrophilic aromatic substitution on an indoleninium (by 1,6-conjugate addition) or a quinone methide. Note that cleavamine-derived scaffolds resulting from this reactivity are outlined in Fig. 6. | ||
The aspidospermane/cleavamine-type, exemplified by the illustrious anticancer agents vinblastine (3) and vincristine, are presumed to be obtained following a related biosynthetic scenario, although this implies more preliminary rearrangements to install a reactive indoleninium moiety compared with vobasinyl-comprising dimers. An oxidase or peroxidase-mediated oxidation of an ibogane-type MIA would first avail a 7-hydroperoxyindolenine intermediate (Fig. 6). Different oxidation mechanisms have been proposed for this purpose, involving either a radicalar mechanism (darkness, triplet oxygen) or a ionic mechanism (light, singlet oxygen).39 The easy elimination of this peroxy group is inferred to initiate a rearrangement of the ibogane scaffold to both disrupt the C-16–C-21 bond and establish a Δ2,16 function, thereby installing a reactive indoleninium moiety as outlined in Fig. 5. This structural motif is easily amenable to an electrophilic aromatic substitution involving the C-10 site of the aspidospermane-type vindoline unit on the C-16 site of the cleavamine unit via 1,6-addition, instigating the canonical C-10–C-16 connectivity, represented by 29 natural derivatives.40 This bridging mode largely dominates the diversity of cleavamine-comprising MIA dimers reported to date, especially when having in mind that further structures are presumed to derive from these MIA dimers through rearrangement of their cleavamine unit. Most rearrangements affecting the cleavamine component were reported to proceed from derivatives bearing a 15–20 epoxide function. Transannular cyclizations could then occur to create either a 7–15 or a 7–20 bond to yield either vincathicine (6) or the recently reported isovincathicine (7) (Fig. 6).41 As per vincathicine (6), this biosynthetic path has been experimentally validated from leurosine (8).42,43 A last unique example of MIA dimer stemming from this intermediate is roseadine (9).44 Again, this scaffold would require a 15–20-epoxycleavamine derivative to undertake an alternative 2–15 transannular cyclization. Rearomatization of the indolic system through fragmentation of the C-2–C-16 bond and simultaneous installment of the Δ16,17 double bond would then avail roseadine (9). Very recently, vincazalidine A (10)45 was reported as a new MIA dimer from Catharanthus roseus, apparently corresponding to a new rearrangement of the cleavamine subunit of leurosine (8). This rearrangement would result from an unprecedented mechanism initiated by the heteronucleophilic attack of the alicyclic nitrogen N-4 onto the carbonylic position of the methylester moiety at C-22 to install a remarkable site being simultaneously connected to a charged nitrogen atom and to two oxygenated functionalities (a methylether and an alcohol). The newly obtained alcohol group at C-22 could then undertake an heteronucleophilic attack at the C-20 epoxidated site to afford the final, trans-diol containing vincazalidine A (10).45 The most obvious biosynthetic derivatives of cleavamine/aspidospermane dimers are pandoline/aspidospermane type dimers (6 such products are described that also disclose a C-10 to C-16 connection). These compounds were found to co-occur in Catharanthus species, further supporting the hypothesis of a biosynthetic interrelationship.46 Oxidation-based sequences have been supported by various organic chemistry reports,42,47 apparently involving nucleophilic attack of the indolic Δ2,7 enamine on a Δ3,4 iminium functionality to install the additional C-3–C-7 connectivity, also accounting for the indolenine status of the pandoline component as in cycloleurosine (11).46 To provide a complete overview of cleavamine reactivity, the occurrence of MIA dimers disclosing other connections should be pointed out. Firstly, three vobasane/cleavamine dimers, capuvosine and its derivatives,48 involve an aromatic electrophilic substitution of cleavamine C-11 on the C-3 electrophilic position of a vobasane residue, bypassing the usual cleavamine-driven reactivity to follow the expected reaction with a vobasane-like residue, as outlined at the beginning of this section. Analogous pandoline/aspidospermane dimers have been described from the same plant source (capuvosidine).49 Remarkably, one sole monomeric cleavamine seems to be known to date: capuronine.49 Conversely, pandoline-type MIAs often occur as monomeric MIAs. Quite remarkably, monomeric pandolines are not supposed to arise from an aspidospermane precursor as described earlier but would instead be based on a Diels–Alder reaction proceeding from a seco-stemmadeninane precursor.50 As a consequence, monomeric pandolines display a Δ2,16 function51 instead of an indolenine nucleus as it appears in dimeric MIAs such as cycloleurosine (11).
 | ||
| Fig. 6 Proposed biosynthetic path to cleavamine-containing MIA dimers and possible rearrangements into cleavamine-derived MIA subtypes. Note that most of the derived scaffolds (cleaisovincathicane, cleavincathicane, cleavincaroseane and cleavincazalidane) are exclusive to the dimeric condition and are all known so far from a unique compound. | ||
Unsurprisingly, the cleavamine-derived cleavincathicane/cleaisovincathicane/cleavincarosean/cleavincazalidane have not been identified as monomeric MIAs to date. Finally, 1,6-additions may be directed towards coniferyl/p-coumaryl alcohol-derived quinone methides (Fig. 5), availing hybrid MIA/hydroxycinnamyl conjugates such as conomicidines A (12) and B,52 or towards a lignan-derived quinone methide to provide conoliferine (13) and isoconoliferine.53
3.1.1.2 Iminium. A large number of MIA dimers are obtained through a coupling of indolic sites to the C-16 position of an eburnane residue (see S1, ESI‡) as in the eburnane/aspidofractane-type pleiomutine (14) (Fig. 7).54 Such dimers arise as a consequence of a nucleophilic attack on a C-16 carbinolamine/Δ1,16 iminium and account for the constitution of an important number of MIA dimers (more than 40 entries). The electrophilicity of eburnane C-16 position is a key to understand the constitution of most eburnane-containing dimers, although a few of them instigate several connections in the end. This logic also extends to the eburnane-like constitution of the meloyuninane subtype, accounting for the C-10–C-16 connection appearing in angustifonines A (15)55 (Fig. 7) and B. It seems that only one eburnane-containing dimer, namely vobtusamine (124)56 (Fig. 37), does not involve the eburnane C-16 position. In a vast majority of cases, the nucleophilic attack is triggered by aromatic sites of the other MIA component. Biomimetic strategies have been undertaken to access such derivatives, sometimes failing to reach the sought-after dimeric structure as in the example of leucophyllidine (16)57 (Fig. 7) where a regioisomer had been obtained instead (disclosing a C-12–C-16 connectivity instead of the awaited C-10–C-16 bonding).58 A parallel can easily be drawn with the pseudodimeric structures of bonafousine (17)59 (Fig. 7)/isobonafousine associating an ibogane subunit to a building block of as yet unknown biogenetic origin, somehow evocative of a tetracyclic eburnane analogue. The α-oxygenated C-12 position of the ibogane subunit could trigger a nucleophilic attack on an iminium involving any of the two nitrogen atoms of the structure to afford these isomers. Notably, this reactivity involving an electrophilic aromatic substitution and an iminium is part of a much wider ensemble and can account for the constitution of many MIA dimers, spanning across a considerable number of scaffolds. A few illustrations of structurally diverse MIA dimers arising from this reactivity are given in Fig. 3. For example, plumocraline (18) (Fig. 7) relies on the C-10-triggered electrophilic aromatic substitution of an akuammilane component on a Δ1,2 indoleninium-containing pleiocarpamane. Some dimers are supposed to arise from electrophilic aromatic substitution on iminium ions involving the alicyclic nitrogen.60 As such, criophylline (19)61–63 (Fig. 7) involves a Δ3,4 iminium moiety of the aspidospermane-type pachysiphine being attacked by the C-10 position of a seco-schizozygane-type andrangine subunit. Melosuavine C (20)64 (Fig. 7) seemingly relies on the electrophilic aromatic substitution of the C-10 site of an aspidospermane unit on a Δ4,5 iminium-containing Melodinus-type monomer whereas uncaramine (21)65 (Fig. 7) and callophyllines66 derive from a Δ4,21 iminium-containing yohimbinoid unit. Structural oddities may rely on similar reactivities from different substrates. The partner initiating the nucleophilic attack may be structurally different at first. One such example is the electrophilic aromatic substitution triggered by a flavonoid aromatic position on a Δ4,21-containing corynantheane in epicatechocorynantheine A (22) (Fig. 7),67 a reactivity also stepping in the biosynthesis of further rubiaceous flavoalkaloids known as uncariagambiriines.68,69 Another possibility is that of an alternative iminium-containing structure as the electrophilic partner, as observed in the case of the aspidospermane-containing melofusinine A (23) (Fig. 7),70 that involves the Δ1,2 iminium function of nitrogenated iridoid unit.
 | ||
| Fig. 7 Singly-tethered oligomers resulting from an electrophilic aromatic substitution on an iminium. | ||
The unique C-2–C-10 tethering encountered in the bis-aspidospermane scaffold of tabernaebovine (24) (Fig. 7) deserves to be developed in the context of electrophilic aromatic substitution.71 Plausible mechanisms to provide this unique connectivity rely on conjugate nucleophilic additions from C-10 to a N-methylquebrachamine half. A 21-keto-containing quebrachamine unit had been selected by Movassaghi to reach tabernaebovine scaffold.72 A Δ4,21 quebrachamine iminium can also be envisaged to afford the ring closure leading to the bis-aspidospermane constitution of the second aspidospermane unit. Finally, an indoleninium-type aspidospermane scaffold cannot be ruled out either.
3.1.1.3 α,β-Unsaturated carbonyl and imine. This category of electrophilic motifs is associated with more stringent structural requirements and is therefore associated with a limited set of MIA subtypes. As such, the monoterpenic component of macrolane-type MIAs is prone to different scaffold-specific electrophilic aromatic substitutions. This scaffold name derives from macroline, a key entry to macrolane-based MIA dimers, comprising a highly reactive α,β-unsaturated carbonylic group. Its characteristic constitution had been suggested to be biosynthetically related to sarpagane-type MIAs and notably comprises two distinct forms which can be rationalized with regards to a freely-rotatable, open dialdehydic intermediate.73 An etherification occurring between an oxygenated substituent at C-19 and the –CH2OH group leads to the so-called type-A macroline (e.g. macroline) whereas a similar reaction involving an oxygenated substituent at C-21 and the same –CH2OH moiety rather leads to a type-B macroline (e.g. ring E seco-talcarpine) (Fig. 8).74 It can be noted that macroline itself does not seem to have been isolated yet as a natural product so that it only exists as a putative biosynthetic cornerstone towards dimeric MIAs. Notably, macroline has a limited shelf life as it spontaneously cyclizes into the more stable dihydroalstonerine.75 A 1,4-Michael addition on the C-21 position of the enone group of a type A-macroline supposedly gives access to perhentinine-type MIA dimers that may undertake subsequent hemiketalization to afford alstomacrophylline (25)-type derivatives (Fig. 9),76 featuring a diagnostic connection between an aromatic site of the MIA partner initiating the electrophilic aromatic substitution and the macrolane C-21 site. A type B macroline reactive unit reveals an alternative, α,β-unsaturated aldehyde-based Michael acceptor granting access to angustilongine A (26)-type dimers (Fig. 9). This second classical type of macrolane-based MIA dimers discloses a connection between an aromatic site and macrolane position C-19. One can note that an alternative mechanism has been proposed for the coupling of ring A-oxygenated macroline-type alkaloids, based on a Friedel–Crafts alkylation process stabilized by an oxonium ion.75
 | ||
| Fig. 8 Structural features of interest to decipher the dimerization trends of macrolanes and scaffolds of the so-called types A and B macrolanes. Electrophilic sites are highlighted in blue for both scaffold subtypes. Note that anhydromacrosalhine methine, a key nucleophilic partner for the dimerization of some macrolanes, differs from the gross structure given to type B macroline by the existence of two unsaturations (viz. Δ18,19 and Δ20,21). | ||
 | ||
| Fig. 9 Singly-tethered oligomers resulting from an electrophilic aromatic substitution on an α,β-unsaturated carbonyle or imine. | ||
The monoterpenic component of the akuammicine scaffold is associated with a specific reactivity giving rise to specific singly and doubly-tethered MIA dimers when it reveals an exomethylene–indolenine conjugate susceptible to nucleophilic attacks. Such oligomers comprise the kopsane/akuammicine-type arbolodinine C (27) that derives from an electrophilic aromatic substitution triggered by the C-10 site of the kopsane subunit on the electrophilic C-22 exomethylenic position of the akuammicine component (Fig. 9).77
Dimeric MIAs featuring diaryl connections are often presumed to derive from a radical coupling and, as such, the topic will be mostly covered in the next section. A dozen compounds link the C-10 position of an indoline-containing MIA, a nucleophilic site of great generality, to a recurring C-11 position of a vincorinane component featuring a constant α-oxygenated functionality. An oxidative coupling has been proposed to step in the dimerization of such compounds, without much detail (see next section for further discussion).78 Alternatively, an indolenine-containing akuammilane could install an electrophilic quinone-imine. This hypothesis is supported by the occurrence of quinone-imine containing dimers (e.g. vincarubine (28),59 rausutrine, rausutranine79).
Bis-akuammicine dimers also comprise very unusual connectivities involving their ethylidenic/ethylic side chain. As such, panganensines X/Y (29)80 feature a C-10 to C-19 connection. While the C-10 position is a very common nucleophilic site, a Δ19,20 conjugated-Δ4,21 iminium can be proposed to account for C-19 electrophilicity that is prone to undergo a 1,4-Michael addition. A similar mechanism has been proposed to incorporate a third nitrogen during the biosynthesis of arboflorine.81 Likewise, a conjugated iminium-type electrophilic partner probably steps in the biosynthesis of suadimin I (30).82 A related mechanism involving a conjugated indoleninium as the electrophilic moiety can account for the C-6–C-11 connectivity appearing in the nor-seco-stemmadeninane/ibogane taberdisines A (31)-B (Fig. 9).83 Finally, a unique intermonomeric connection appears in undulatine (32).84 While the C-10 position of the akuammilane component acts as a classical electron-rich partner, the involvement of the C-6 position of the sarpagane unit as a nucleophilic site is highly unusual. Here again, an α,β-unsaturated indoleninium system could explain the electrophilicity of C-6. Alternatively, it was suggested that the coupling reaction should proceed from a 6-hydroxylated sarpagane derivative (6-hydroxylated sarpagane85 analogues had seldom been described). An hemisynthetic investigation using DDQ as a coupling agent gave support to this putative reactivity.86
3.1.1.4 Formaldehyde. As stated in Section 2.2, extraneous units such as formaldehyde have been involved as the electrophilic partner in such reactions, as exemplified by pleiokomenine A (33) and kopoffine C (34) (Fig. 10), among many others. Such a reaction has been proposed to provide a para iminoquinone methide moiety that could further be prone to undergoing different kinds of nucleophilic attack. As per pleiokomenine A (33), the electron-rich C-10 site of an additional aspidofractane unit is presumed to trigger this electrophilic aromatic substitution.18 Kopoffine C (34) seems to arise as a consequence of an heteronucleophilic attack instigated by the N-1 nitrogen of the quebrachamine unit on a similar para iminoquinone methide of the aspidofractane counterpart.87 Methylene-bridged dimeric natural products as a whole have been recently reviewed.88
 | ||
| Fig. 10 Putative biosynthetic path to singly-tethered methylene-bridged MIA dimers. | ||
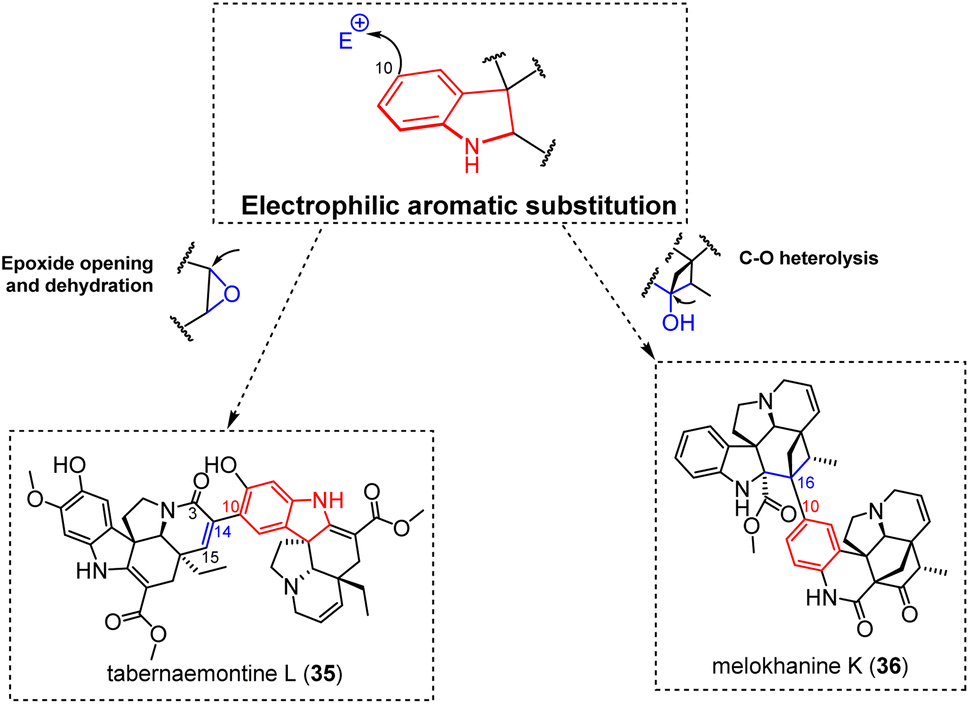
3.1.1.5 Epoxide-ring opening and nucleophilic attacks towards a hydroxylated site. The recently reported bis-aspidospermane-type tabernaemontine L (35) (Fig. 11) appears to reveal a unique example of connectivity among electrophilic aromatic substitution-driven and singly-tethered MIA dimers.89 Although unique, the C-10–C-14 connection appearing in this dimer is relatable to an electrophilic aromatic substitution targeting the C-14 position of a 14,15-epoxide-containing aspidospermane unit. Subsequent dehydration could then install the Δ14,15 functionality appearing in tabernaemontine L (35). From a biosynthetic point of view, this dimerization mechanism is supported by the known presence of such 3-oxo-14,15-epoxide-containing aspidospermanes,90,91 and analogous building blocks are proposed to step in the biosynthesis of various aspidospermane-based doubly-tethered dimers (e.g. conophylline (90)) and trimers (e.g. taberdivarine A (91)) (Fig. 25), as discussed in more detail below. The meloscandoninane-meloyinane type melokhanine K (36)92 represents a markedly different situation (Fig. 11). Its intermonomeric connectivity implies an electrophilic aromatic substitution into the meloyinane C-16 position. For this purpose, the C-16 hydroxylated monomeric precursor was reported from a plant pertaining to the same genus a few years ago, namely meloyine A.93 Even though the hydroxy group is not generally accepted as a good leaving group per se, an array of different hydroxy activation strategies has been reported to step in natural products biosynthesis.
 | ||
| Fig. 11 Singly-tethered oligomers resulting from an electrophilic aromatic substitution on an epoxide or a carbocation. | ||
 | ||
| Fig. 12 Singly-tethered oligomers resulting from ortho/ortho diphenol coupling and O/ortho phenol coupling. Color coding has been added for clarity but it does not indicate any chemical sense. | ||
 | ||
| Fig. 13 Singly-tethered oligomers resulting from diazo-coupling. Color coding has been added for clarity but it does not indicate any chemical sense. | ||
 | ||
| Fig. 14 Singly-tethered haplophyton alkaloids. Color coding has been added for clarity but it does not indicate any chemical sense. | ||
 | ||
| Fig. 15 Singly-tethered leuconolam-derived dimers. Color coding has been added for clarity but it does not indicate any chemical sense. | ||
3.1.2.1 Ortho–ortho diphenol and O-ortho phenol coupling products. The 9,9-bonded bis-ibogane type pendulifloramine (37),94 the 11,12-bonded aspidospermane-ibogane type tetrastachynine (38),95 the 10,10-bonded bis-aspidospermane type N-acetyl-16,17-dihydroxyaspidospermidine (39)96 and the 9,12-bonded bis-hunteracinane type blumeanine (40) are canonical ortho–ortho phenolic oxidative coupling products (Fig. 12). Notably, the O/ortho regioisomer of tetrastachynine (38), namely tetrastachyne (41), was co-isolated with it.95 Such MIA dimers appear to be very unusual, although a closely related analogue was later isolated.97
The connection mode of the bis-sarpagane-type dispegatrine (42)98 is consistent with a canonical ortho–ortho phenolic oxidative coupling. Nevertheless, the phenolic oxidative coupling of its monomeric component, spegatrine, could provide dispegatrine (42), but with a very modest yield of 0.25%. Conversely, Cook et al. were able to complete the synthesis of dispegatrine (42) with a much higher yield using a non-phenolic oxidative dimerization process.99 Intermolecular non-phenolic oxidative dimerizations of complex aryl substrates are known to be very rare but is documented for some indolic substrates.100 Notably, the formation of a single atropodiastereomer during Cook synthesis could finalize the structure assignment of dispegatrine (42) as its axial chirality had been overlooked in the original phytochemical report. The P-configuration of dispegatrine (42) biaryl axis would be related to an internal asymmetric induction originating from the steric hindrance related to the sarpagane cage-like structure.99 A few analogues reveal a biaryl junction contiguous to other oxygenated functionalities. One such example is the 12,12-bonded bis-ibogane-type obovatine (43),101 where the ortho-arranged substituents to the biaryl axis are a phenol and a methoxy group. In this context, it can be envisaged that the methoxylation occurs after the dimerization process. This assumption is further supported by the co-isolation of its diphenolic analogue, namely bis[11-hydroxycoronaridinyl-12-yl].101 More generally speaking, such biosynthetic assumptions (methoxylation after coupling) have received support from biosynthetic investigations carried out on fungal dimeric polyketides deriving from oxidative phenolic coupling.102 Likewise, the vincorinane/isopleiocarpamane-type ceylanine (44)103 or the bis-vincorinane-type peceylanine feature two methoxy groups being ortho-disposed to the biaryl connection, these methoxylations presumably occurring after dimerization. Again, this hypothesis is supported by the co-isolation of related dimers disclosing a furanic connection (e.g. peceyline (153)) that suppose the existence of dimeric intermediates revealing phenolic groups instead of the methoxy groups finally appearing in ceylanine (44) and peceylanine (see Fig. 49 for doubly-tethered oligomers resulting from radical coupling). Similarly, the spectroscopic data of geleganamide (45) indicated a dimeric structure of two gelsemamidane-type MIA alkaloids connected through a C-10–C-10 bond.104 A possible biogenetic access to this compound could also benefit from an amino radical. Such a rare substituent for a MIA tryptaminic benzene originates from the hydrolytic cleavage of the indolic pyrrole, as sometimes observed for gelsedane-type MIAs.105,106 An amino-derived radical could result in a spin density at the para-amino C-10 position, accounting for the intermonomeric connectivity of geleganamide (45).107
3.1.2.2 Diazo-coupled MIA dimers. Alternatively, such secondary amine-containing gelsemamidane-type MIAs can lead to aromatic azo compounds such as geleganidine B (46)19 and 11-demethoxygelsemazonamide (47) (Fig. 13).106 Although corroborated by several reactivity reports dealing with structurally diverse aniline derivatives,108 natural azo products are extremely rare. Prior to geleganidine B (46), it seems that only one natural azo compound had been reported (from the macromycete Agaricus xanthodermus), whose artefactual nature has been suggested since its first isolation and structure elucidation.109 These N,N-coupled MIA dimers are somehow evocative of some N,N-coupled indolosesquiterpene dimers known as dixiamycins, that were proved to originate from a radical-based mechanism.110 Such singly bonded N,N-bonded MIA dimers (viz. radical processes occurring from a secondary amine) still have to be reported.
3.1.2.3 Haplophyton alkaloids. Apocynaceous plants of the genus Haplophyton produce unique heterodimers featuring inter-indolic connectivities invariably associating an aspidospermane-type MIA monomer with a canthinone-type nucleus through a C-10–C-3 connection.111 These iconic structures, including haplophytine (48) and cimiciphytine (49) (Fig. 14), were first isolated from Haplophyton cimicidum.112,113 Interestingly, the canthinone-type component sometimes occurs as a quinolinone derivative and the ability of these two forms to interconvert has been experimentally validated as a semipinacol-type rearrangement,114 as evidenced since the early report of haplophytine (48) by Yates, Cava and co-workers.111 The biosynthetic relevance of this rearrangement is emphasized by the co-occurrence of hybrid dimers associating an identical aspidospermane unit and both forms of the canthinone/quinolinone counterpart within producing plants.111,115 The total synthesis of haplophytine (48) represented an important synthetic challenge owing to the steric impediments of the coupling process between the aspidospermane component and the C-3 position of the canthinone unit. Synthetic attempts by Corey et al. emphasized the trend of different mono- and bicyclic aspidospermane mimics to initiate electrophilic aromatic substitution α to the iminium nitrogen instead of the desired γ-position. Likewise, electrophilic aromatic substitution strategies targeting 2,3-epoxide-containing canthinone analogues resulted in the formation of C-10–C-2 bonded analogues (electrophilic aromatic substitution) or a 11-O–C-2 connected dimer (heteronucleophilic attack) but failed to establish the desired C-10–C-3 connectivity.116 Later attempts of electrophilic aromatic substitution proved successful but these synthetic works used a tricyclic indolenine-containing canthinone-mimic with a leaving group at C-3, which can hardly be related to a biosynthetic coupling process.117,118 Nicolaou suggested that the coupling between the canthinone and aspidospermane components of haplophytine (48) should result from an oxidative coupling reaction. The desired junction could be experimentally formed from simplified analogues, giving further support to this biosynthetic hypothesis.119
3.1.2.4 Leuconolam-derived dimer. Finally, the bis-rhazinilam constitution of bousangustine C (50) (Fig. 15), involving a C-6–C-6 connection, is seemingly unique as a MIA dimer as it does not involve the benzenoid ring of the indolic system. Even in the broader context of natural products, it seems difficult to find molecules deriving from a similar reactivity. Tryptamine-based dimers involving a similar coupling position have been repeatedly described in the group of bisindolylmaleimide/indolocarbazoles, but the coupling mechanism involving an enamine radical120 is difficult to reconcile with the specific example of bousangustine C (50). Likewise, the joined motifs of bousangustine C (50) are structurally evocative of bipyrrole alkaloids like isochrysohermidin121 or speranberculatin A.122 Nevertheless, elegant synthetic works by Boger et al. established that these compounds derived from a well-precedented addition-elimination sequence involving the uptake of singlet oxygen by a suitable bipyrrole precursors, that have been co-isolated in both cases,123 so that these compounds can hardly be related to bousangustine C (50) either. An oxidative single electron transfer at N-1 followed by deprotonation at the N-1 position of leuconolam has been proposed to avail a neutral radical. This radical could then relocate to C-6 to account for the connection appearing in this intriguing compound.124
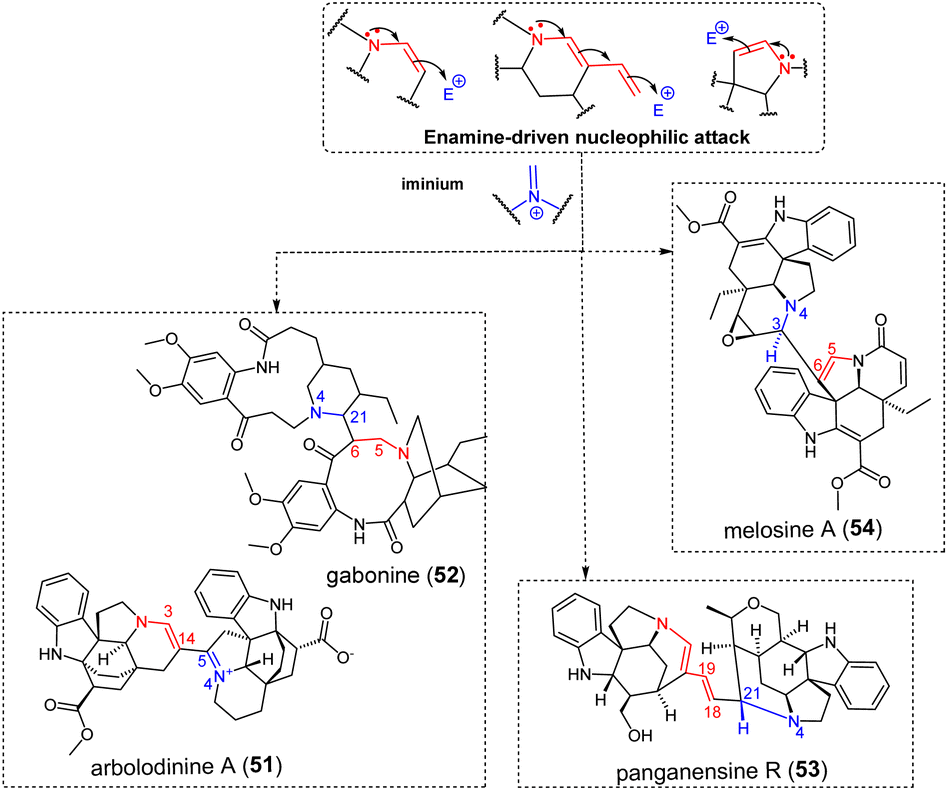
3.1.3.1 Iminium. A first group of electrophilic partners for enamine-driven dimerization reactions comprises iminium-containing metabolites (Fig. 16), as can be produced through Polonovski-type reaction.125 Although simple, this mechanism enables straightforward access to a large number of structurally diverse MIA dimers. The C-14–C-5 connectivity of the bis-aspidofractane type arbolodinine A (51)77 is easily relatable to a nucleophilic attack of the tetrahydropyridine-type Δ3,14 enamine of a first unit to the Δ4,5 iminium function of another aspidofractane-type component. The structurally-intriguing gabonine (52),126 featuring two ibogane-derived units, is supposed to dimerize upon nucleophilic attack of a Δ5,6 enamine on a Δ4,21 iminium of the other subunit. It seems that this dimer stands among the very scarce number of MIA dimers incorporating Witkop–Winterfeldt oxidized127 subunits (along with melotenuine C128), although several examples of such monomeric MIAs exist.129,130 With this in mind, it can also be assumed that the nucleophilicity of the α-carbonylated position C-6 could be sufficient to trigger the nucleophilic attack. A related mechanism can be invoked to decipher the intermonomeric connection appearing in panganensine R (53).80 This rare scaffold could be obtained considering a nucleophilic attack instigated by a dienamine function involving the Δ18,19 vinylic function of a first akuammicine-type monomer on a Δ4,21 iminium-containing akuammicine derivative, resulting in a rare C-18–C-21 junction. Alternatively, 2-pyrroline-type enamine partners could be involved. This reactivity accounts for the intermonomeric connection appearing in the bis-aspidospermane-type melosine A (54),131 that results from the nucleophilic attack of a Δ5,6 enamine-containing unit to a Δ3,4 iminium-containing coupling partner. This reaction is somewhat reminiscent of the coupling mode evoked in the coupling of the two vobtusine blocks as it appears in alasmontamine A (159) (Fig. 52).132
 | ||
| Fig. 16 Singly-tethered MIA oligomers resulting from an enamine-driven nucleophilic attack on an iminium. | ||
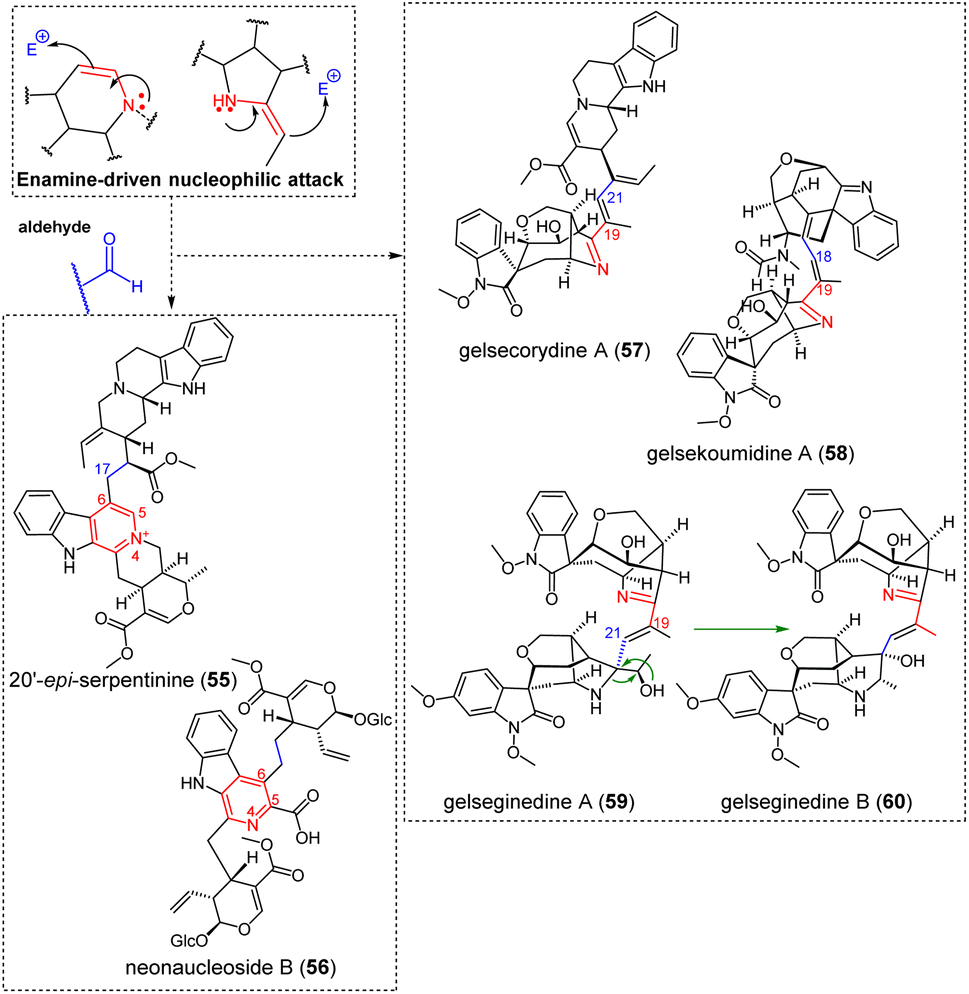
3.1.3.2 Aldehyde. Enamine-containing MIAs can also target aldehyde functions as electrophilic partners (Fig. 17). The tetrahydropyridine-derived Δ5,6 enamine, as in the case of serpentinine derivatives (e.g. 20′-episerpentinine (55)), can enable such reactivities. An enamine-driven aldolization of ajmalicine C-6 to the aldehydic C-17 position of the corynantheane northern unit is presumed to assemble the complete scaffold of serpentinines/moandaensines,133–135 resulting in the final compound after dehydration and a thermodynamically-favoured rearomatization of ajmalicine C-ring. A similar process from the Δ5,6 enamine of a strictosidine-type MIA to the aldehydic moiety of secologanin readily accounts for the pseudodimeric constitution of neonaucleoside B (56).136 Gelsedane-type MIAs are prevalent contributors to enamine reactivity-driven dimerizations. These dimerization trends can be explained by the presence of pyrrole rings and of 2-ethylidene-substituted pyrrolidines in the monoterpenic part of several such MIAs, structural features that are rare for any MIA. With regard to dimers obtained through an enamine-driven nucleophilic attack on an aldehydic function, the case of gelsecorydine A (57)137 is worth being highlighted. The nucleophilic attack would be triggered by the enamine (
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) 2-ethylidenepyrrolidine) form of a gelsedane-type gelsenicine derivative, accounting for the nucleophilicity of its C-19 position, towards the C-21 aldehydic function of vallesiachotamine. The nucleophilic attack of a similar gelsedane 2-ethylidenepyrrolidine function (at C-19) on a putative α,β-unsaturated aldehyde-containing seco-koumine at C-18 is presumed to afford the intriguing gelsedane-seco-koumine constitution of gelsekoumidine A (58).138 The seco-koumine appendage does not appear to have been reported from a monomeric MIA so far. It can be hypothesized that the different constitution of the recently reported gelseginedine A (59)139 stems from the enamine-induced nucleophilic attack of the C-19 position of the gelsedane component on the C-21 aldehyde site of a gelseleginane homologue, a MIA subtype previously assumed to correspond to a biosynthetic intermediate between humanteninane and gelsedane scaffolds.140 It has been proposed that co-isolated gelseginedine B (60) corresponds to a derivative of this earlier dimer by ring expansion of its gelselegine pyrrole in the corresponding piperidine, resulting in a hitherto unprecedented MIA scaffold.
2-ethylidenepyrrolidine) form of a gelsedane-type gelsenicine derivative, accounting for the nucleophilicity of its C-19 position, towards the C-21 aldehydic function of vallesiachotamine. The nucleophilic attack of a similar gelsedane 2-ethylidenepyrrolidine function (at C-19) on a putative α,β-unsaturated aldehyde-containing seco-koumine at C-18 is presumed to afford the intriguing gelsedane-seco-koumine constitution of gelsekoumidine A (58).138 The seco-koumine appendage does not appear to have been reported from a monomeric MIA so far. It can be hypothesized that the different constitution of the recently reported gelseginedine A (59)139 stems from the enamine-induced nucleophilic attack of the C-19 position of the gelsedane component on the C-21 aldehyde site of a gelseleginane homologue, a MIA subtype previously assumed to correspond to a biosynthetic intermediate between humanteninane and gelsedane scaffolds.140 It has been proposed that co-isolated gelseginedine B (60) corresponds to a derivative of this earlier dimer by ring expansion of its gelselegine pyrrole in the corresponding piperidine, resulting in a hitherto unprecedented MIA scaffold.
 | ||
| Fig. 17 Singly-tethered MIA oligomers resulting from an enamine-driven nucleophilic attack on an aldehyde and the rearrangement of gelseginedine A into gelseginedine B. | ||
3.1.3.3 Formaldehydes, α,β-unsaturated carbonyls and indoleninium. Extraneous units may also be incorporated after nucleophilic attack of formaldehyde by an enamine function. As in the former subsection, this reactivity often steps in the dimerization of several MIA obtained from gelsemiaceous source. Such scaffold-specific reactivity can rely on the 2-ethylidenepyrroline system (nucleophilic C-19 position) to provide a side chain including an exomethylene function, possibly amenable to epoxidation. An epoxide ring-opening initiated by another 2-ethylidenepyrroline nucleophilic attack system (again from C-19) of a second gelsedane subunit can be presumed to yield the connectivity observed in geleganimine A (61) (Fig. 18).104 Aside from MIA partners, electrophilic sites appearing on a wealth of different phytochemicals are prone to such dimerization processes, mostly aldehydic-containing substances but also α,β-unsaturated exomethylenes as encountered in iridoid monoterpenes, leading to compounds such as gelsamydine (62) (Fig. 18).141 Likewise, rhazinilam-type MIAs are prone to a dienamine-triggered nucleophilic attack on formaldehyde that can readily afford derivatives revealing either an aldehydic or an exomethylenic function at C-5 position. This exomethylenic function is prone to undergoing a nucleophilic attack involving different possible partners. For example, an heteronucleophilic attack initiated by an indolinic N-1 position can be invoked to account for the constitution of the aspidofractane/rhazinilam-based kopoffine A (63) (Fig. 18) and of the kopsane/rhazinilam-based kopoffine B.87 Alternatively, a dienamine nucleophilic attack triggered from a first rhazinilam monomer on a C-5 exomethylene-substituted rhazinilam can lead to the dimeric scaffold of bousangustine B. An additional nucleophilic attack initiated by the dienamine function of a third rhazinilam unit can yield the trimeric bousangustine A (64) (Fig. 18).124 The vallesamane/vobasane dimer vobparicine (65) (Fig. 18),142 can be assembled following a conjugate 1,6-addition of the conjugated system appearing in the Δ16,22-exomethylene-containing apparicine on the electrophilic C-3 of vobasinol.
 | ||
| Fig. 18 Singly-tethered MIA oligomers resulting from an enamine-driven nucleophilic attack on formaldehyde or on an α,β-unsaturated carbonyl. | ||
3.1.3.4 Quinone methide. Finally, the unique structures of inaequalisines A (66) (Fig. 19) and B introduced a C-7 located phenylpropene unit that acted as a dearomatizing unit, a structural trait having no precedent in the wide phytochemical class of MIAs. More generally, such phenylpropene-pyrroloindolines are very rare in the plant kingdom with only two other occurrences: angelicastigmin143 and utilisin.144 Although rare, it is quite straightforward to understand the constitution of this unusual MIA that seems to rely on an enamine attack on a feruloyl-type para-quinone methide.25 It should be noted that the proposed biosynthesis pathway of ineaqualisines would proceed from an indole-containing quebrachamine precursor which would subsequently rearrange into the final rhazidine appendage.25
 | ||
| Fig. 19 Singly-tethered MIA oligomers resulting from an enamine-driven nucleophilic attack on a conjugated quinone methide. | ||
3.1.4.1 Formaldehyde. Some of these dimerization sequences require precise structural features, which probably explains the relatively low number of MIA dimers obtained in this way. Several such bis-aspidospermanes seem to depend on both (i) the existence of a secondary alcohol function at C-18 and (ii) the occurrence of a Δ14,15 functionality (see for example fusiformine A for a relevant monomeric constituent).145 Aspidospermane-type MIAs disclosing these substituents may trigger nucleophilic attack of the 18-OH group on the Δ14,15 functionality to trigger nucleophilic attack on an extraneous formaldehyde, to install an electrophilic site at C-14 along with a classical hexacycle framework featuring an additional tetrahydrofuran ring (relatable to modestanine monomers).146 This reactivity can yield MIA dimers such as voacinol (67)147 or voacandimine C (68)148 (Fig. 20) type structures, only deriving from the bis-tetrahydrofuran containing dimer by the reductive ring opening of one or two such rings. Regarding voacinol (67), it is also possible that an extraneous water unit triggers the 1,4-Michael addition, which would be consistent with the occurrence of a Δ14,15 moiety after dehydration (see sungucine (76) in the next section for a related mechanism). The synthesis of voacinol (67) and voacandimine C (68) by Movassaghi et al. supported this putative biosynthetic scenario, although an enamine-type reactivity was retained as the second dimerization step.149 The aspidospermane/ibogane biscarpamontamine A (69) (Fig. 20) is supposed to be obtained as a consequence of a similar, enol ether-like scheme, but followed by a nucleophilic attack initiated by the N-1-position of an ibogane unit on the C-14 electrophilic site of the aspidospermane unit.128 MIA dimers incorporating extraneous methylenic units can also be related to a direct heteronucleophilic attack by a nitrogen atom. The bis-aspidospermane melofusine I (70)150 (Fig. 20) can be inferred to derive from an heteronucleophilic attack on a formaldehyde unit, subsequent dehydration and further heteronucleophilic attack of the resulting iminium by a further aspidospermane unit. Alternatively, heteronucleophilic attack on a formaldehyde unit may be initiated from the alicyclic nitrogen, as in the case of geleganidine C (71) (Fig. 20).19 In this latter dimer, this extraneous unit is later oxidized to an urea group.
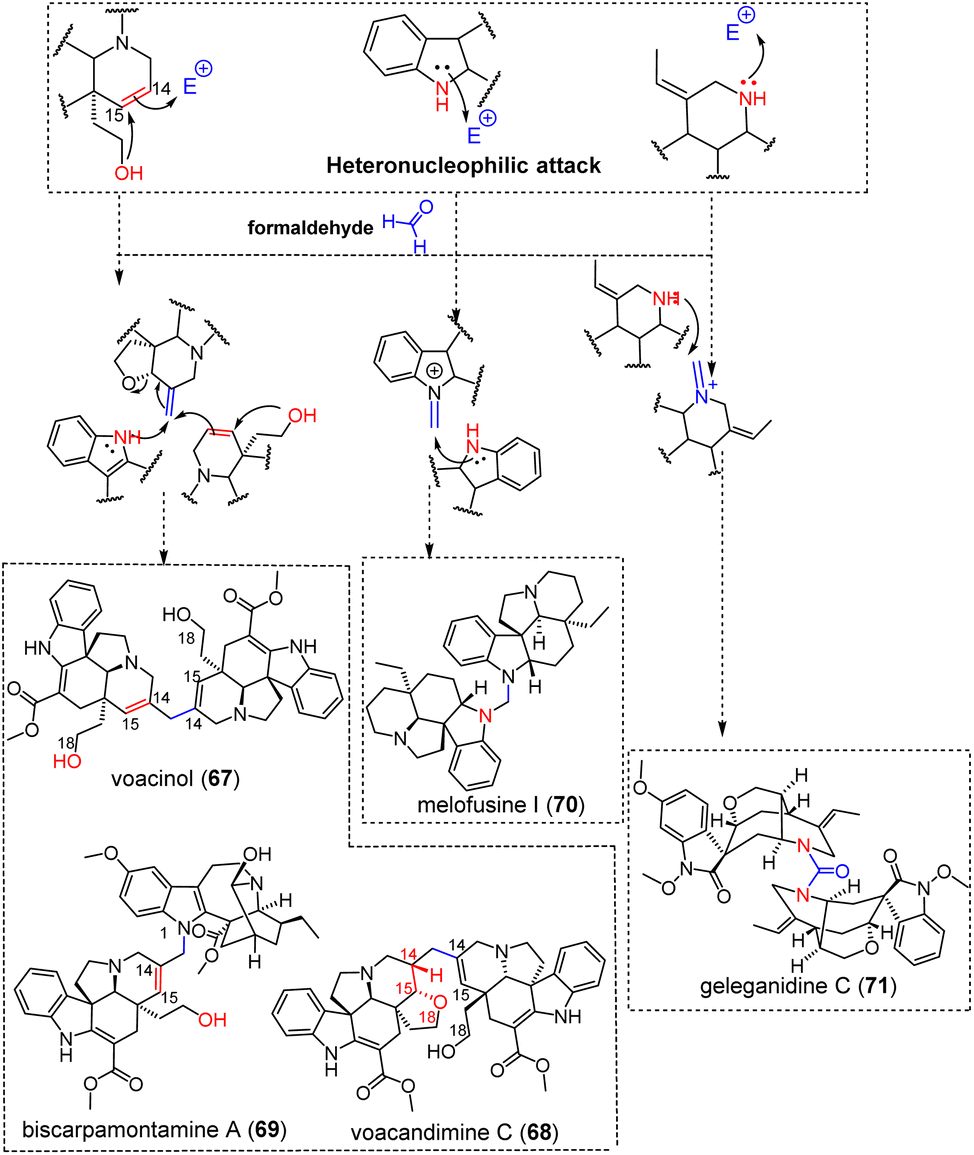 | ||
| Fig. 20 Singly-tethered oligomers resulting from an heteronucleophilic attack on formaldehyde (see S2‡ for a comprehensive listing, red- and blue-colored moieties refer to nucleophilic and electrophilic sites, respectively). | ||
3.1.4.2 Iminium. Diverse (hetero)nucleophilic attacks targeting iminium functions have been reported. Here again, the structural specificities of certain MIA subtypes are sometimes involved in these reactive pathways. Enol ether-containing macrolanes are one such example. A putative macrolane monomer of salient biosynthetic interest, anhydromacrosalhine-methine (Fig. 8), is presumed to trigger a nucleophilic attack on a Δ3,4-containing aspidospermane to afford the macrolane/aspidospermane-type pandicine (72)151 (Fig. 21) (as demonstrated by Cook et al. based on biomimetic synthesis152) or on a pleiocarpamane indoleninium to yield macrocarpamine.153 Likewise, Gan and Cook could synthesize macrocarpamine from pleiocarpamine and anhydromacrosalhine methine (Fig. 8).154 Remarkably, the dimerization process of strychnogucine A (73)155 (Fig. 21) may result from the heteronucleophilic annulation of a 18-OH function on the Δ17,23 functionality of a seco-F ring strychnine precursor initiating a nucleophilic attack on a Δ3,4 iminium-containing akuammicine electrophilic partner. This is somehow reminiscent of that having led to voacinol (67) and voacandimine C (68). In some cases, the driving force to initiate a nucleophilic attack does not rely on a heteronucleophilic attack based on an oxygen functionality already located on the reactive monomer, but rather seems to depend on the involvement of an extraneous water unit. One such example is that of the bis-aspidospermane 3-oxovoafrine B (74) (Fig. 21).69 Again, a monomeric aspidospermane unit containing a Δ14,15 functionality should be considered to undergo an heteronucleophilic attack by a water molecule that could be combined with the coupling to an iminium-containing electrophilic partner. This readily accounts for the constitution of 3-oxovoafrine B (74)69 but also for the bis-Melodinus-type suadimin A (75), or the bis-akuammicine sungucine (76) (Fig. 21).155,156 In all these cases, the hydroxy group having initiated this sequence is ultimately eliminated by dehydration, resulting in Δ14,15 functions. This double bound can be subsequently epoxidized, as observed in suadimin F (77) (Fig. 21).82
 | ||
| Fig. 21 Singly-tethered oligomers resulting from an (hetero)nucleophilic attack on an iminium (see S2‡ for a comprehensive listing, red- and blue-colored moieties refer to nucleophilic and electrophilic sites, respectively). | ||
The bis-aspidospermane-type kisantine (78) (Fig. 21) enters a unique category as the nucleophilic attack seems to result from a phenate function at C-10 on a Δ3,4 iminium-containing aspidospermane as an electrophilic partner.157 Likewise, the constitution of the melonine/eburnane-type celastromeline (79)158 (Fig. 21) is straightforward to delineate as it merely results from an heteronucleophilic attack triggered by melonine N-1 on the C-16 of an eburnane unit, previously presented as an electrophilic site of considerable generality. It should be noted that this structure is at high risk of being erroneous as we revised the structure of melonine in 2021.159
Tabercrassine A (80) (Fig. 21) is an intriguing dimer disclosing two ibogane-type units bridged by an extraneous acetone unit connecting their C-3 positions. Such compounds are presumed to be artifacts arising from the nucleophilic attack of an acetone methyl group on a Δ3,4-iminium containing ibogane-type MIA. Accordingly, ibogane monomers disclosing a 3-oxopropyl group (e.g. 3-(2′-oxopropyl)-coronaridine)20 are in line with this speculative biosynthetic scenario.160,161 A nucleophilic attack triggered by the remaining methyl group of acetone on another Δ3,4 iminium-containing ibogane unit can give access to tabercrassine A (80)/3,3′-(oxopropyl)dicoronaridine dimers.
3.1.4.3 Indoleninium. The constitution of the bis-vobasane-type bisnicalaterine A (81)162 denotes a 1,6-addition triggered by an indolic nitrogen N-1 on the electrophilic C-3 residue of another vobasine molecule. Other bis-vobasane dimers obtained through an analogous reactivity are the so-called theionbrunonines A (82)-C that feature a unique S heteroatom connecting the C-3 site of two vobasinyl residues (Fig. 22).22,23 These structures were speculated to result from the nucleophilic attack of a 3-SH containing vobasane residue to the C-3 position of vobasinol. The occurrence of a very unusual thiol substituent in this tentative substrate was presumed to arise from a 1,6-addition of cysteine on the electrophilic C-3 site of vobasine. Cysteinyl-comprising MIAs have seldom been reported to occur comprising mappiodoside I,26 and, mostly, the related pagisulfine,163 that could afford the desired precursor under the action of a cysteine C-S lyase. This assumption found later support through the isolation of the sought-after monomer, hemitheion, from the same plant a few years later.164
 | ||
| Fig. 22 Singly-tethered oligomers resulting from an heteronucleophilic attack on an indoleninium (see S2‡ for a comprehensive listing). | ||
3.1.4.4 α,β-Unsaturated carbonyl and imine. Heteronucleophilic attacks can also proceed through an aza-Michael-based dimerization (Fig. 23). The constitution of some dimers obtained following these reactions is easy to explain, such as the akuammicine/aspidofractane-type arbolodinine B (83).77 This dimer presumably derives from the nucleophilic attack of the N-1 position of an aspidofractane on the C-22 position of the conjugated exomethylene-indolenine system of the akuammicine-type valparicine. Conversely, angustiphylline (84), comprising two unusual building blocks, viz. an uleane and a nor-seco-stemmadeninane unit, requires some reactivity prior to dimerization.165 The biosynthetic pathway to angustiphylline (84) would be initiated from a N-4-oxidized stemmadeninane followed by a Potier–Polonovski fragmentation and excision of C-5 to afford a ring-opened conjugated indoleninium. A nucleophilic attack by the N-4-nitrogen of the uleane-type alstilobanine C on this open conjugated indoleninium ion would then provide angustiphylline (84).165 Although unusual as a dimerization mechanism, it is worth noting that alstilobanine C itself was proposed to derive from a Potier–Polonovski-initiated fragmentation and recyclization proceeding from stemmadenine.166 Potier and Janot proposed that the apparicine scaffold is also biosynthetically derived from stemmadeninane by this mechanism, and several examples of monomeric MIAs from Alstonia indicated that this reactivity (i.e., fragmentation-excision(s)) played an important role in the structural diversification of stemmadeninane-type MIAs.167–169 It can be assumed that the recently reported scholaphylline derives from a similar reactivity, with the monomeric block initiating the heteronucleophilic attack lacking two carbon units compared with stemmadenine.170 Again, monomeric scaffolds of this constitution have already been reported, and a biosynthetic link to stemmadenine has been suggested elsewhere.171 As a last example of aza-Michael derived MIA pseudodimer, gelseiridone (85) is a further example of gelsedane-type MIA bonded to an additional iridoid unit. This biosynthesis would proceed from a gelsedane-type gelsenicine derivative having underwent an imine hydrolytic cleavage to release a secondary amine group, able to initiate an aza-Michael addition on the conjugated α,β-unsaturated carbonyl function of 7-deoxygelsemide as an iridoid.172 Finally, the ring opening of the dihydropyran moiety of this iridoid unit provides the definite structure of gelseiridone (85).
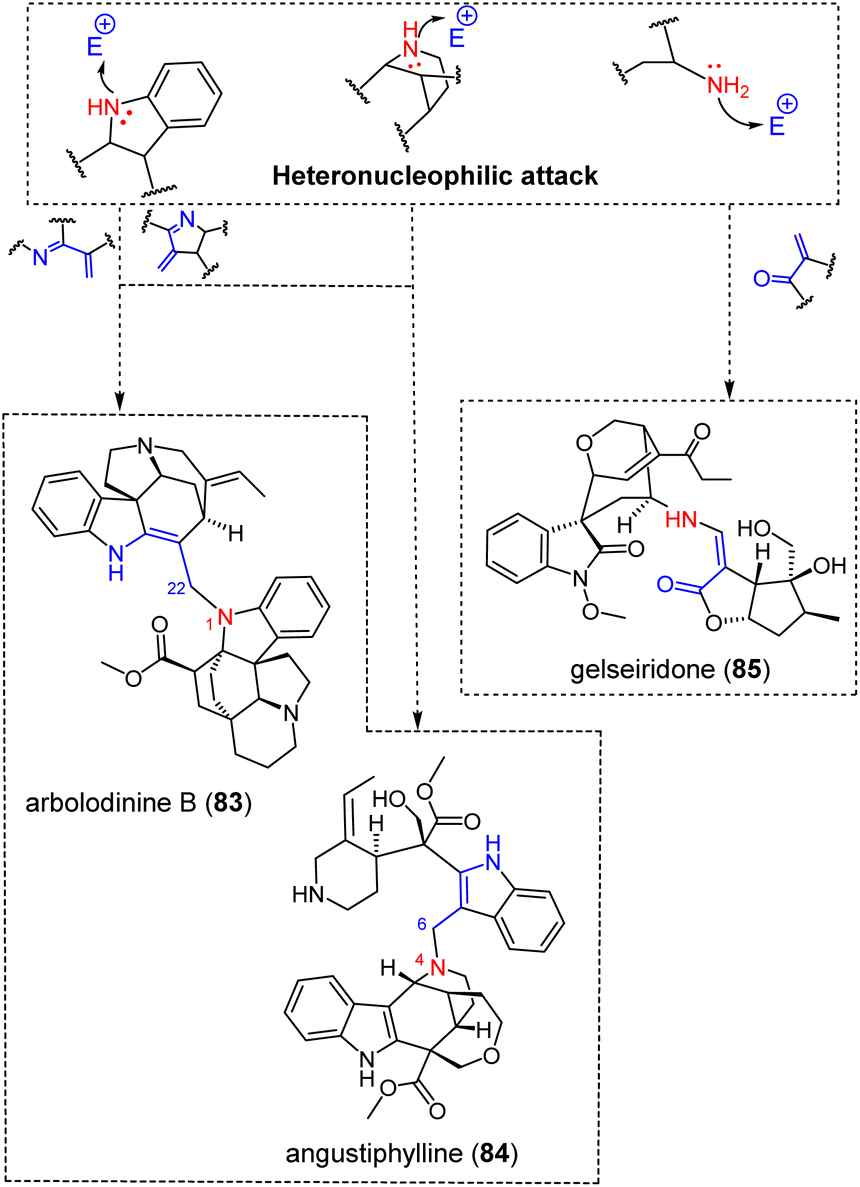 | ||
| Fig. 23 Singly-tethered oligomers resulting from an heteronucleophilic attack on an α,β-unsaturated carbonyl/imine (see S2‡ for a comprehensive listing). | ||
3.1.4.5 Carboxylic acid derivatives and aldehyde function. A very large number of MIA derivatives result from the condensation of different MIAs with diverse shikimic acid-derived building blocks. Our bibliographic survey enabled us to locate 98 adducts between MIAs and such gallic acid/cinnamic acid derivatives and, seldom, anthranilic acid derivatives.29 While some remarkable, C–C or multiply bonded adducts with shikimic acid derivatives have already been covered (conomicidine A (12), conoliferine (13) and inaequalisine A (66)) or will be developed in the dedicated section (kanluaengoside C (93), voacalgine A (146), bipleiophylline (147), and pleiomaltinine (148)), a vast majority of these hybrid natural substances (almost 9 out of 10) arise from the esterification of a MIA alcohol group on a phenolic carboxylic acid group or, in a few cases, from an amidification initiated by the indole/indoline nitrogen for some aspidospermane-type173 or ajmaline-type MIA (e.g. alstiphyllanine I (86)) (Fig. 24).28 Although the structural requirements are seemingly scarce for the MIA component to initiate such reactions, it is intriguing to note that these esterification-derived adducts are limited to a few MIA subtypes with the most prevalent contributors being yohimbinoids (24 esters on either 17-OH or 18-OH, with raugustine (87)27 as an illustration), ajmalane (15 esters on 17-OH and 7 amides on N-1), akuammilanes (15 esters on 17-OH) and aspidospermanes (11 esters on 19-OH and 4 amides on N-1). To a lesser extent, adducts on vallesiachotamane and strictosidine have also been reported. In mappiodoside J (88) the alcohol group of a sugar unit esterifies the carboxylic acid group of an extraneous cinnamate unit.26
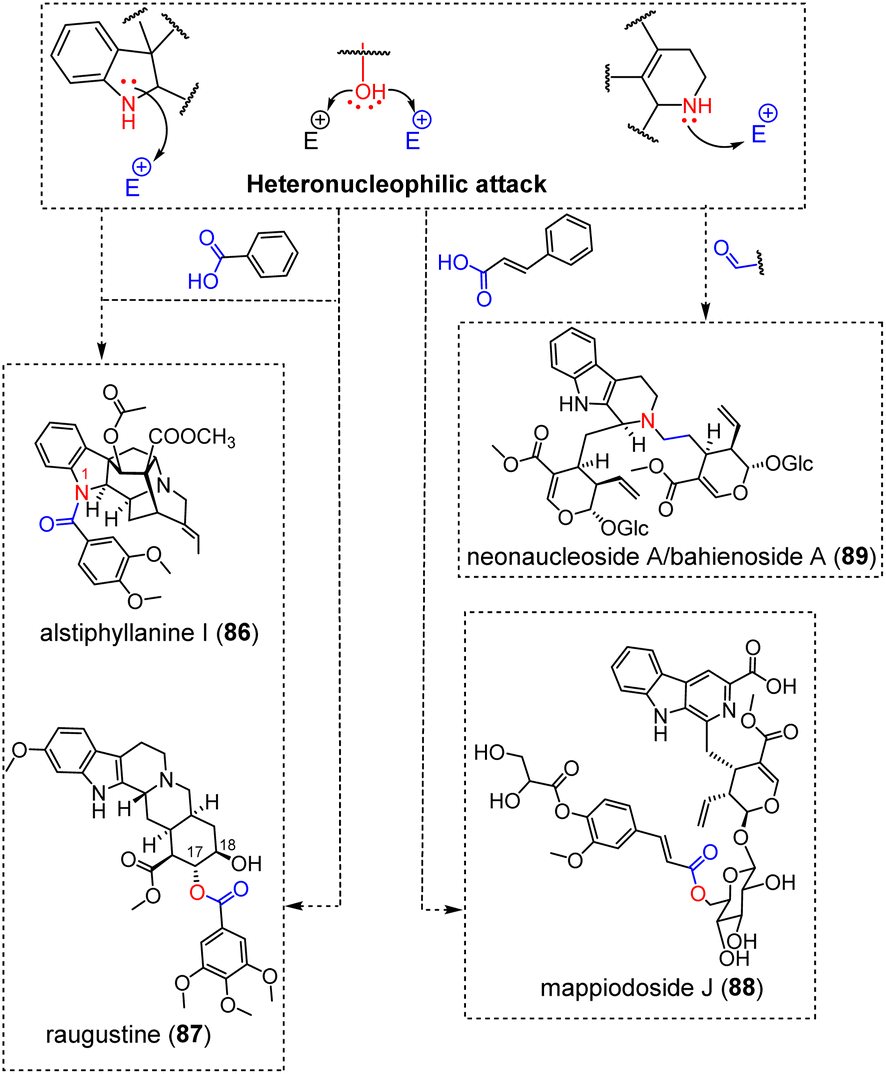 | ||
| Fig. 24 Singly-tethered oligomers resulting from an heteronucleophilic attack on carboxylic acids or aldehydes (see S2‡ for a comprehensive listing). | ||
Heteronucleophilic attack from the alicyclic nitrogen of a strictosidine derivative on the aldehydic group of a second secologanin unit can readily account for the constitution of the pseudodimeric neonaucleoside A (89), later shown to be structurally equivalent to bahienoside A through total synthesis.174
3.2 Doubly-tethered oligomers
A retro-oligomerization analysis conducted on the doubly-tethered MIA oligomers revealed the implication of five reactive sequences.3.2.1.1 Iminium. As already seen above, electrophilic aromatic substitutions often involve iminium-type electrophilic partners (Fig. 7). Such products may lead to doubly-tethered MIA dimers provided that some structural requirements are met. Furan-bridged MIA dimers (e.g. conophylline (90)) (Fig. 25) would require two additional structural features, i.e. a phenol group contiguous to the site initiating the electrophilic aromatic substitution and an epoxide moiety next to the site of the electrophilic partner. In the specific example of the bis-aspidospermane conophylline (90) (Fig. 25), this further nucleophilic attack is conducted by the 11-OH group on the epoxide unit at C-14 to install this furanic junction.47 The tris-aspidospermane-type taberdivarine A (91)175 (Fig. 25) features two such connections (it should be noted that this name had been formerly given to an unrelated vobasane/quebrachamine dimer).176 A unique instance of doubly-tethered eburnane/aspidospermane dimer is represented by bisleuconothine B (92) (Fig. 25).177 The first connection between the two building blocks befits the usual connection mode involving eburnane-type MIAs, i.e. an electrophilic aromatic substitution on an Δ1,16 iminium resulting in a typical C-16–C-10 connection. The final scaffold could be reached from this singly-tethered intermediate, with the second connection being established after nucleophilic attack of a C-9 phenol function on the C-2 site of an indoleninium-type eburnane presumed to be obtained after an enamine-driven hydroxylation at C-7.
 | ||
| Fig. 25 Doubly-tethered oligomers resulting from an electrophilic aromatic substitution on an iminium associated to an heteronucleophilic attack (see S2‡ for a comprehensive listing). | ||
3.2.1.2 Quinone methide. Likewise, the structure of the cinnamyl-vallesiachotomane-type kanluaengoside C (93)178 (Fig. 26) is reminiscent of that of electrophilic aromatic substitution-derived conjugates such as conomicidine A (12) (Fig. 5). The first connection again relies on an electrophilic aromatic substitution between the vallesiachotamane C-9 position and a cinnamyl-derived para-quinone methide. The second connection depends on both the presence of an ortho-phenolic group to the site having initiated the electrophilic aromatic substitution and the occurrence of a carboxylic acid group on the cinnamyl conjugate enabling a subsequent lactonization.
 | ||
| Fig. 26 Doubly-tethered oligomers resulting from an electrophilic aromatic substitution on a quinone methide associated to an heteronucleophilic attack (see S2‡ for a comprehensive listing). | ||
3.2.1.3 Formaldehyde. The strained pleiocarpamane scaffold is associated with an inherent electrophilicity at C-7 along with a pronounced nucleophilicity at C-2, accounting for the propensity of this MIA subtype to afford doubly-tethered MIA dimers involving these two positions.179 One such example is that of the pleiocarpamane/rhazidine type goniomedine A (94) (Fig. 27).180,181 Electrophilic aromatic substitution by the C-10 site of the rhazidine component is first proposed to install a para-iminoquinone methide group, that could be prone to undergo a Δ2,7 enamine nucleophilic attack from the pleiocarpamane to afford an indoleninium-comprising pleiocarpamane as a singly-tethered intermediate. The occurrence of a C-11 phenolic group on the rhazidine component enables an heteronucleophilic annulation on a pleiocarpamane indoleninium to afford the dihydropyran intermonomeric junction. A bis-aspidospermane dimer, melomorsine (95) (Fig. 27),182 seems to be obtained through a comparable mechanism. In this case, an exomethylene-type N-1 iminium could be added by the ortho-oxygenated C-10 position of the other subunit. The resulting indoleninium ion might then undergo a nucleophilic attack by the phenate at C-11 to install the oxazole core appearing in melomorsine (95).
 | ||
| Fig. 27 Doubly-tethered oligomers resulting from an electrophilic aromatic substitution on a formaldehyde unit associated to an heteronucleophilic attack (see S2‡ for a comprehensive listing). | ||
3.2.1.4 α,β-Unsaturated carbonyl. Lumutinines (herein exemplified by lumutinine A (96)) are doubly-tethered bis-macrolane-type MIA dimers and macralstonidine (97)183 is a macrolane/sarpagane dimer (Fig. 28). These compounds disclose a tetrahydropyranic junction and are presumed to derive from the 1,4-Michael addition of the electron-rich C-10 position of a macrolane or sarpagane residue on the exomethylenic position C-21 of a type A-macrolane (Fig. 8) to afford an hydroxyketone, prone to ring closure via hemiketal formation, and final ketalization triggered by a phenolic group at C-11.184 Biomimetic synthesis of macralstonidine (97) from macroline and Na-methylsarpagine gave support to this dimerization scenario.185 As formerly noted for some singly-tethered bis-macrolane dimers, an alternative dimerization mechanism to lumutinine A (96) relying on a Friedel–Crafts-based sequence has been suggested elsewhere.186
 | ||
| Fig. 28 Doubly-tethered oligomers resulting from an electrophilic aromatic substitution on an unsaturated carbonyl associated to an heteronucleophilic attack (see S2‡ for a comprehensive listing). | ||
3.2.1.5 Indoleninium. The constitution of the ibogane/vobasane/vobasane trimer divaricamine A (98)187 (Fig. 29) is easily relatable to the electrophilic nature of the C-3 position of vobasane-type MIAs. The middle vobasane unit undergoes an electrophilic aromatic substitution by an ibogane C-10, instigating the usual 1,6-conjugate addition when a vobasane is involved as an electrophilic partner. The connection between the vobasane building blocks results from a similar 1,6-conjugate addition but results from an heteronucleophilic attack triggered by the N-1 atom of the middle vobasane unit on the C-3 site of the other vobasane component.
 | ||
| Fig. 29 Doubly-tethered oligomers resulting from an electrophilic aromatic substitution on an indoleninium associated to an heteronucleophilic attack. | ||
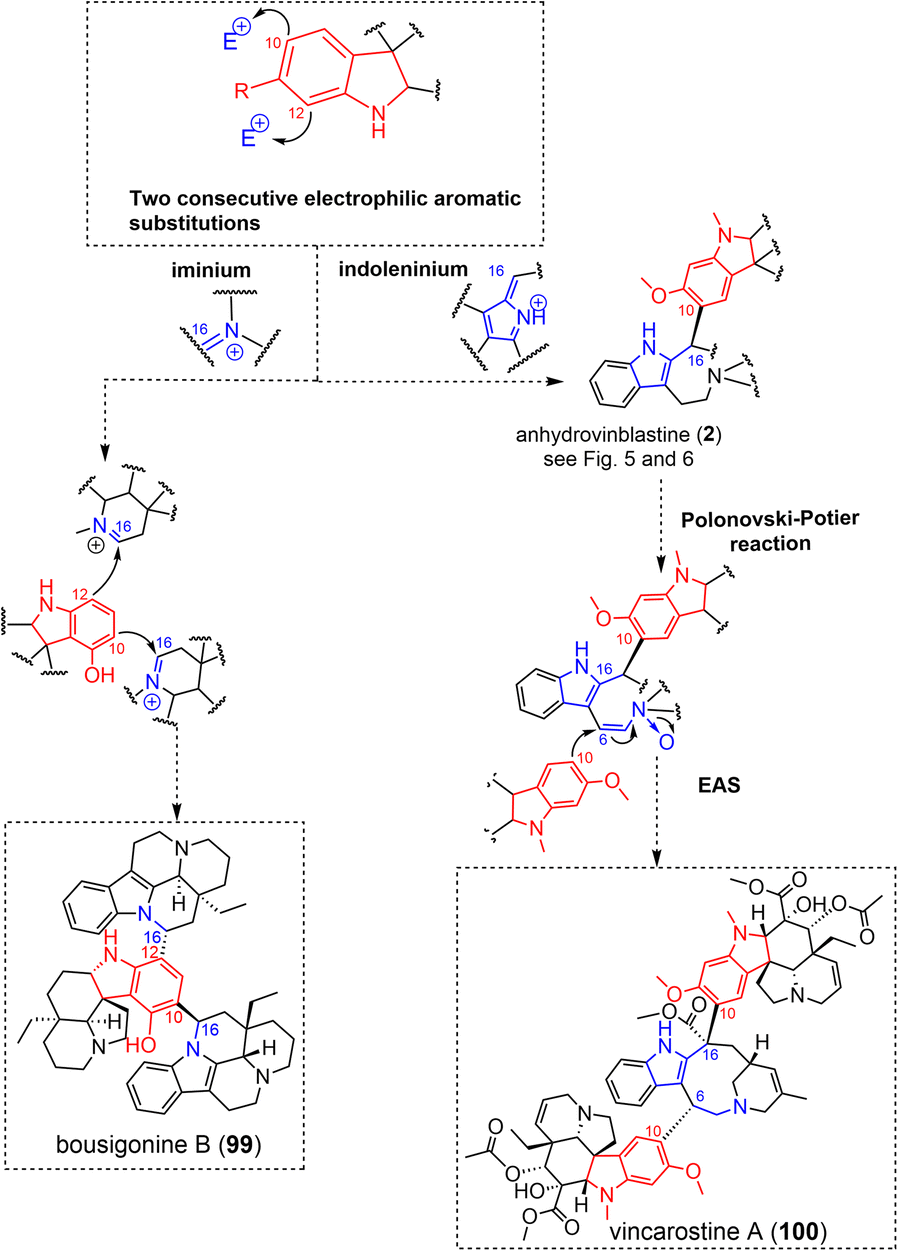 | ||
| Fig. 30 Doubly-tethered oligomers resulting from two electrophilic aromatic substitutions (EAS) on iminium or indoleninium (see S2‡ for a comprehensive listing). | ||
The recently isolated aspidospermane/cleavamine/aspidospermane MIA trimer, vincarostine A (100) (Fig. 30),189 incorporates an anhydrovinblastine substructure linked to another aspidospermane unit (vindoline) via an unprecedented aspidospermane 10-cleavamine 6 bond. This unique bond was assumed to be generated by the Polonovski reaction of anhydrovinblastine N-oxide with the introduction of vindoline as nucleophile at C-6 (through its electron-rich C-10 site).
3.2.3.1 Aza-Michael addition. The only ellipticine-comprising MIA dimer described to date, the bis-ellipticine-type strellidimine (101) (Fig. 31) obtained from Strychnos dinklagei, features a fused oxazole system as an intermonomeric junction. A putative biosynthetic scenario to this structure proceeds from an oxidized 10-hydroxyellipticine derivative (i.e. oxidized to a para quinone imine), thereby revealing an α,β-unsaturated indolenine prone to undergo heteronucleophilic attack at C-9 initiated by the dihydropyridine nitrogen N-4 of the other subunit. The quaternary adduct could then rearrange via heteronucleophilic annulation to a thermodynamically-favoured system featuring the final oxazole junction, with the rearomatization of the 10-hydroxyellipticine being a likely driving force.190 Likewise, the hexacyclic constitution of the recently reported alstoscholarinine A191 features an oxazolidine nucleus that is presumed to arise from two consecutive Mannich-type condensations initiated by glycine on (i) the C-17 alcohol group of the corynantheane-type isositsirikine and then (ii) the C-19 position of an oxidized derivative, resulting in an intermediate dihydropyridinium derivative. Subsequent dihydroxylation would then set the stage for the final dehydrative etherification of the reduced glycine analogue affording the hexacyclic, oxazolidine-containing structure.
 | ||
| Fig. 31 Doubly-tethered oligomers involving an aza-Michael addition on a conjugated iminium. | ||
The doubly-tethered bis-akuammicine leucoridine A (102) (Fig. 31) is an interesting case first involving an aza-Michael type 1,4 addition from the N-1 position of the anhydropereirine unit on the C-22 position of the conjugated exomethylene-indolenine system of the dihydrovalparicine electrophilic partner (see arbolodinine B (83) for an analogous singly-tethered MIA dimer (Fig. 23)).192 A subsequent enamine-driven nucleophilic attack on the C-22 site of the conjugated exomethylene-indoleninium unit of the anhydropereirine component yields the doubly-tethered constitution of leucoridine A (102). A biomimetic dimerization of dihydrovalparicine to leucoridine A has been accomplished a few years later. DFT calculations favoured a stepwise aza-Michael sequence rather than an alternative hetero-Diels–Alder cycloaddition.193 A similar stepwise Aza-Michael reaction can readily account for the structure of the bis-condylocarpane-type leucofoline (103) (Fig. 31).194 The bis-aspidospermane type anhydrohazuntiphyllidine reveals a similar intermonomeric connectivity with its hydroxylated derivative, hazuntiphyllidine (104) (Fig. 31), revealing an additional OH group at the C-2 position of the southern aspidospermane unit.195 Interestingly, hazuntiphyllidine (104) was shown to exist in two distinct structural forms in solution, depending on the solvent used. The analysis of hazuntiphyllidine (104) in DMSO-d6 revealed a leucofoline-like spiranic junction. Conversely, NMR analysis in C6D6 determined an additional ether connectivity resulting from heteronucleophilic attack of the 2-OH function on the C-2 position of the southern indolenine-containing component. To the best of our knowledge, this solvent-dependent number of intermonomeric connections is the unique documented example within MIA oligomers.
The bis-akuammicine type bisleucocurine A (105)196 (Fig. 31) can be related to a similar dimerization mechanism. As already envisaged in the frame of leucoridine A (102), a singly-tethered intermediate could result from the aza-Michael type 1,4-addition from anhydropereirine N-1 into the C-17 position of the conjugated exomethylene-indoleninium of the electrophilic partner. Ring closure would then result from a C-12 initiated electrophilic aromatic substitution on the C-2 position of the indoleninium-oxidized electrophilic motif. It can be noted that an equally satisfactory biosynthetic scenario could rely on a Mannich-type heteronucleophilic attack from anhydropereirine N-1 on the aldehydic function of 18-deoxy Wieland–Gumlich aldehyde.
The dimerization of the akuammicine/strychnane-type strychnobaillonine (106)197 (Fig. 32) can be supposed to proceed via an aza-Michael addition of an akuammicine indoline nitrogen into the C-17 strychnane position, taking benefit of the signature α,β-unsaturated lactame system (Δ17–23 functionality) of such analogues (the use of this functionality in the frame of a dimerization is somehow reminiscent of the path to strychnogucine A (73) and sungucine (76)). The second connection between the two units would then be related to an aldolization reaction directed towards the C-17 position of the akuammicine unit.
 | ||
| Fig. 32 Doubly-tethered oligomers involving an aza-Michael addition on an α,β-unsaturated carbonyl. | ||
The recently reported ajmalane/macrolane-type alsmaphyline A (107) (Fig. 32) features an original intermonomeric connectivity.198 The dimerization would first involve an aza-Michael addition of ajmaline N-1 on the C-21 position of an alstonerine α,β-unsaturated ketone system (cyclized, Δ20,21-containing form of a type B macrolane) (Fig. 8). The unconjugated C-19 ketone group from the macroline unit could then be prone to undergo an electrophilic aromatic substitution from the ajmalane unit C-12 position. This dimerization scheme is highly reminiscent of that proposed to yield the structurally-related alstonisidine (108) (Fig. 32).199 This molecule was proposed to result from the aza-Michael addition of an ajmaline indoline N-1 on the C-21 site of the conjugated exomethylene of the type A-macroline (Fig. 8) to yield an alternative singly-tethered intermediate that could lead to an hemiketal group with the nearby CH2OH group (alstomacroline-type intermediate). A subsequent electrophilic aromatic substitution by the C-12 position of the ajmalane on this hemiketalic C-19 position can then install the second intermonomeric bond to provide the final frame of alstonisidine (108). This assumption has been supported by early biomimetic syntheses.200,201
3.2.3.2 Pictet–Spengler. Pseudodimeric structures such as usambarine (109) (Fig. 33), tchibangensine and cinchophyllamine are presumed to derive from a Pictet–Spengler condensation between the aldehydic function at the C-17 position of a corynantheal/corynantheidal-like precursor and a tryptaminic derivative.109 As an illustration, Seguin and Koch achieved the hemisynthesis of usambarine by Pictet–Spengler condensation between corynantheidal and N-methyltryptamine.202 Likewise, strychnofolines111 and barterines112 involve the same reaction but proceed from a spiro-oxindole-containing MIA. Although this reaction had not been considered earlier in this review, Pictet–Spengler reaction is of extreme importance to the wide MIA structural class. Strictosidine (1), as the universal precursor to MIAs, derives from a Pictet–Spengler condensation between tryptamine and the aldehyde-containing monoterpene, secologanin. This reactivity is highly reminiscent of the biosynthetic steps leading to the emetine-type ipecac alkaloids, although incorporating a dopamine instead of tryptamine in that case.203
 | ||
| Fig. 33 Example of doubly-tethered pseudodimer resulting from a Pictet–Spengler reaction. | ||
3.2.3.3 Mannich reaction combined to enamine and electrophilic aromatic substitution reactivities. Stepwise reactions related to enamine reactivity and electrophilic aromatic substitution are often proposed to step in the dimerization of pleiocarpamane-containing compounds (e.g. pycnanthine (110), pycnanthinine (111) and contortarine A (112)204) (Fig. 34). The first step to initiate such dimerization relies on the heteronucleophilic attack of formaldehyde by diverse indolines (pycnanthine = a vindolinine, pycnanthinine205 = an aspidospermane and contortarine A = an aspidofractane) to install a nucleophilic exomethylene-type N-1-iminium. This functional group can undergo an enamine nucleophilic attack to yield a C-7 substituted singly-tethered intermediate disclosing a pleiocarpamane indoleninium. The second inter-unit bonding is obtained through an electrophilic aromatic substitution triggered by the C-12 position on the pleiocarpamane C-2 indoleninium.205
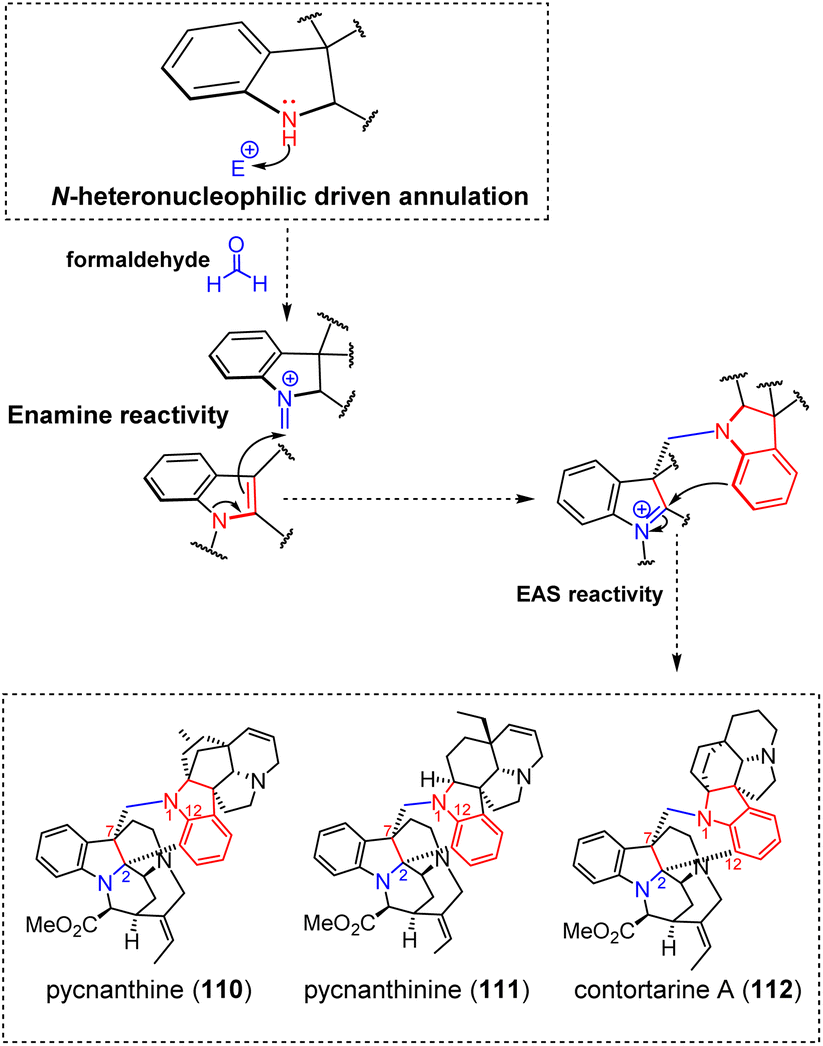 | ||
| Fig. 34 Doubly-tethered oligomers resulting from a Mannich reaction combined with enamine reactivity and electrophilic aromatic substitution. | ||
3.2.3.4 N,O-ketalization. A few MIA dimers feature a characteristic junction composed of an 1,3-oxazinane such as geissospermine (113),206 geissolosimine,207 ligustrinine (114)208 (Fig. 35) and strychnobiline.209 This junction is implemented thanks to an heteronucleophilic attack from the akuammicine component N-1 atom to the C-17′ aldehydic function of the corynantheane counterpart to give rise to a N,O-hemiketal function. Further heteronucleophilic attack of this group by the C-17 OH group of the akuammicine would then yield the N,O-ketal function, thereby installing the final 1,3-oxazinane ring. As an example, the akuammicine/corynantheane-based geissospermine (113) involves the 17-hydroxylated akuammicine-type geissoschizoline. A similar process is supposed to step in the biosynthesis of ligustrinine (114) via an heteronucleophilic attack of the vobasine N-4 into the C-17′ aldehydic site of the sarpagane component. The resulting N,O-hemiketal may then evolve into a N,O-ketal after an heteronucleophilic attack by the vobasane 17-OH group. The illustrious bis-akuammicine-type caracurine V (115)210 (Fig. 35) probably arises through a similar dimerization mechanism. Two distinct hemiaminalization reactions may be instigated in a symmetric manner from the indoline nitrogen of an akuammicine unit to the C-17 aldehydic function of the other subunit. The seven-membered oxygenated ring of caracurine V (115) would then be obtained through heteronucleophilic attack of a C-18 OH group on this N,O-hemiketal. It should be noted that ketal groups sometimes occur in this specific position for some monomeric akuammicines belonging to the so-called diaboline subtype.211 Gardmultine (116) (Fig. 35) and demethoxygardmultine had been simultaneously described from Gardenia multiflora.212 The structure of these MIA dimers features an unusual, spiro-oxazolidine-type intermonomeric junction besides incorporating two unusual spiro-sarpagane-type components. While the southern unit strictly pertains to the spiro-sarpagane (also referred to as chitosenine) subtype213 (16-hydroxygardneramine oxindole), the seemingly different constitution of the iminoether-containing southern component is equivalent to a masked oxindole214 (see the example of gardneramine for related monomeric examples).215 A first intermonomeric connection could be established by a Mannich-type heteronucleophilic attack of the N-1 atom of the gardneramine-like subunit into the oxygenated position C-17. An heteronucleophilic annulation triggered by the 16-OH on gardneramine indolenine C-2 site may then install the final spiro-oxazolidine intermonomeric junction. At last, this oxazolidine-type intermonomeric junction is reminiscent of melomorsine (95) that had been addressed in the former section (Fig. 27). Although a sequence initiated by an electrophilic aromatic substitution could readily account for its original constitution, an alternative path closer to that shown for geissospermine (113)/ligustrinine (114)/gardmultine (116) can also be proposed. In that case, the incorporation of the extraneous formaldehyde unit would result from an heteronucleophilic attack by an indolinic nitrogen. Subsequent electrophilic aromatic substitution on the corresponding N1-exomethylene-type iminium would then avail a singly-tethered Δ1,2-indoleninium-containing intermediate prone to heteronucleophilic annulation, as already shown in Fig. 27.
 | ||
| Fig. 35 Doubly-tethered oligomers resulting from N,O-ketalization. | ||
3.2.3.5 Mannich-type reactivity. The dimerization path to the akuammicine/corynantheane-type longicaudatine F (117) and longicaudatine (118) (Fig. 36) is presumed to be initiated by a Mannich-like addition of an akuammicine type indolinic nitrogen N-1 on the C-17′ aldehydic function of a corynantheane subunit, resulting in a Δ16′,17′ enamine function. The second connection between the two building blocks depends on the nature of the substituent undergoing the Δ16′,17′ enamine-triggered nucleophilic attack at the C-17 position of the akuammicine unit. An aldehydic group (desacetylretulinal-type akuammicine) would result in a longicaudatine F (117)-type scaffold.216 Alternatively, an hemiketal function, as observed in the akuammicine-type Wieland–Gumlich aldehyde residue, would result in a longicaudatine (118)-type dimer.217 In the same spirit, the double dehydration of the bis N,O-hemiketal-containing intermediate could avail the signature diazacyclooctaediene junction of toxiferine I (119) (Fig. 36).218
 | ||
| Fig. 36 Doubly-tethered oligomers resulting from Mannich-type reactivity. | ||
Finally, doubly-tethered MIA hybrids incorporating a serine-derived component have recently been reported from Gelsemium elegans, namely gelsechizines A (120) and B (121) (Fig. 36).219 Quite remarkably, these hybrid MIAs would result from an heteronucleophilic attack triggered by the non-MIA component, viz. the nitrogen atom of the serine derivative, targeting either the C-21 aldehydic group of a vallesiachotamane-residue (gelsechizine A (120)), or the C-17 aldehydic function of a corynantheane residue (gelsechizine B (121)). The chemical environment of this aldehyde group determines the additional reactivity that yields the final doubly-tethered structure. As per gelsechizine A (120), the second intermonomeric connection seems to imply an enamine nucleophilic attack on the C-19 position of the Δ19,20 functionality. Regarding gelsechizine B (121), an aza-Michael reaction would be triggered by the addition of the nitrogen atom on the C-19 position of a conjugated Δ19,20/Δ4,21 iminium system analogous to that encountered in panganensine X (29) (Fig. 9).
3.2.4.1 Ring closure-driven nucleophilic attack. A variety of bis-aspidospermane dimers such as vobtusine (122) (Fig. 37) and voacandimine A (123) (Fig. 38) reveal a tetrahydrofuranic ring evocative of some previously reported singly-tethered compounds like voacinol (67) or voacandimine C (68) (Fig. 20). In that case too, a ring-closure driven heteronucleophilic attack can be supposed to connect C-14 to an electrophilic partner. Vobtusine (122)220 and voacandimine A (123)148 derivatives may first depend on a nucleophilic attack directed on the C-22 position of a conjugated exomethylene–indolenine system to yield a singly-tethered dimeric system disclosing a C-14–C-22 bond. Further oxidation reactions would then be required to enable the formation of the second intermonomeric connection. The spiranic structure of vobtusine (122) seemingly requires the existence of a Δ3,14 enamine function on the tetrahydrofuran-containing aspidospermane unit to trigger a nucleophilic attack on an exomethylene-type N-1 iminium on the other unit. Alternatively, oxidation to a Δ3,4 iminium function from a similar singly-tethered intermediate could enable a direct heteronucleophilic attack by the indolinic nitrogen N-1 of the other subunit to access the scaffold of voacandimine A (123). A unique example of eburnane/aspidospermane dimer, namely vobtusamine (124) (Fig. 37),221 could be obtained following the same sequence from a Δ3,14 enamine-containing eburnane. Interestingly, the authors inferred that this compound could rather be obtained following an aspidospermane to eburnane rearrangement, as proposed by Wenkert and Wickberg,222 and subsequently validated experimentally.223–225 In line with this hypothesis, O'Connor, Courdavault and co-workers had identified an enzyme capable of converting an aspidospermane-type MIA into the corresponding eburnane.226 In their seminal report on vobtusamine (124), the authors could indeed transform vobtusine (122) into vobtusamine (124). The cyclopentane junction observed in callichiline (125) (Fig. 38) is structurally unique, and an elegant biosynthetic path to it had been suggested by Wenkert.227 Although callichiline (125) discloses an heterodimeric structure comprising an aspidospermane (namely tabersonine) component and a rare seco-schizozygane-type subunit, its biosynthesis was proposed to proceed from two aspidospermane-type building blocks, remarkably identical to those formerly postulated to step in the biosynthesis of vobtusine (122), as it would only differ by the existence of a Δ1,2 functionality for one of them (instead of the original Δ2,16 unsaturation). In this indolenine configuration, a sigmatropic transposition could then convert the C-22-bonded aspidospermane-unit into the corresponding seco-schizozygane (namely andrangine) by dislocating the C-7–C-21 bond and installing a C-2–C-21 bond,228 as experimentally demonstrated by Lévy et al. As formerly observed with vobtusine, an oxidation of the remaining aspidospermane unit to the corresponding Δ3,14 enamine would then be required to trigger a nucleophilic attack on the electrophilic C-7 position of the indoleninium-type andrangine, thereby availing the cyclopentanic core encountered in the structurally-unique callichiline (125) (Fig. 38).132
 | ||
| Fig. 37 Doubly-tethered oligomers resulting from ring-closure driven nucleophilic attack: putative biosynthetic pathway to vobtusine and vobtusamine (see S2‡ for a comprehensive listing). | ||
 | ||
| Fig. 38 Doubly-tethered oligomers resulting from ring-closure driven nucleophilic attack: putative biosynthetic pathway leading to voacandimine A and callichiline (see S2‡ for a comprehensive listing). | ||
3.2.4.2 Aldolization. A particular case is that of strychnofuranine (126) (Fig. 39),216 disclosing a unique furanic connection between two akuammicine components (viz. two geissoschizal components). Condensing these two aldehydic sites into the final 2,4-disubstituted furan would rely on a first sequence of aldolization/crotonization from the C-16 site of a first geissoschizal on the C-17 aldehydic site of the second unit to first provide an intermediate with a single intermonomeric bond, prone to allylic oxidation. Hemiketalization and final dehydration could afford the furanic junction appearing in strychnofuranine (126). Aldol-type reactions are mostly related to polyketides biosynthesis229 and as such hybrid compounds comprising both a MIA and a polyketide component were supposed to be obtained following this kind of reactivity. Gelsepyrrodine A (127) (Fig. 39) represents one such example of gelsedane/polyketide-type hybrid. An aldolization reaction between acetyl-CoA and the C-19 position of the gelsedane-type 19-oxo-gelsenicine and would afford a singly-bonded gelsedane-polyketide adduct.230 Subsequent heteronucleophilic attack of the alicyclic nitrogen N-4 on the carbonylic thioester would then give access to a transient carbinolamine function, also connected to a CoA thioester. This site could undergo a Claisen-type nucleophilic attack from an additional acetylCoA unit. Two dehydration reactions could provide the final pyrrole ring of gelsepyrrodine A (127). Finally, the decarboxylation of the polyketide-type side chain and its subsequent oxidation into an aldehyde leads to the final product. A related sequence can be proposed to account for the constitution of alstoscholarines (128) (Fig. 39). At first, an extraneous malonyl CoA unit can be supposed to undertake an aldolization/crotonization into the C-5 aldehyde function of a rearranged akuammilane subunit (this reactivity will be further developed in the frame of alstoniasidine A (134) biosynthesis (Fig. 41)).231 It can be assumed that the subsequent hydration of this singly-tethered intermediate enables a subsequent heteronucleophilic annulation initiated by the alicyclic nitrogen N-4 of the MIA, installing the final pyrrole ring after dehydration of the hemiaminal.232 Aldolization/crotonization-based dimerization sequences are also presumed to step in the biosynthesis of some hybrid macrolane-based oligomers. Dimerization trends related to macrolane-type MIAs have already been addressed in this document, mostly relying on the occurrence of Michael acceptors in their monoterpenic part (Fig. 8). Nevertheless, a few compounds incorporating a macrolane-type MIA reveal an alstomicine-type constitution,233viz. without a carbon residue at C-20, thereby revealing a nucleophilic enol function. Even though the constitution of such macrolane-type MIAs is not canonical, other such structures were reported, including alstonoxines A and B.234 Acid hydrolysis proved capable of converting a B-type macroline into the corresponding macroline without a carbon substitute at C-20.235 Alstopirocine (129) (Fig. 39) reveals an intriguing hybrid structure combining a macrolane but also a biosynthetically puzzling pyrrole substituted with a polyketide side chain. Kam et al. envisaged that the enolic side chain of alstomicine would initiate an aldol condensation/crotonization on an aldehyde-containing glycine analogue, presumed to evolve into a pyrrole after a Mannich-type reaction. The pyrrolic side chain would then be obtained based on a Claisen-type reaction triggered by the dienamine function of the pyrrole ring on a poly-β-ketoester.236 Likewise, the intriguing bispiranic structures of macrodasine A (130) (Fig. 39) have been suggested to derive from alstomicine-type precursors.237,238 For such compounds, the C-20 position would be alkylated by a C6 fragment which would enable a tandem hemiketalization.239 Singly-tethered analogues such as voacalgine D (131) (Fig. 39) and E are presumed to derive from a similar intermediate through different reactivity sequences. Voacalgine D (131) would be obtained after a hemiketalization and an aldolization/crotonization sequence to afford the furanic cycle. Alternatively, voacalgine E would derive from an oxa-Michael 1,4-addition with a similar aldolization/crotonization affording the furanic ring.240
 | ||
| Fig. 39 Doubly-tethered oligomers resulting from aldolization–crotonization reactions (see S2‡ for a comprehensive listing). | ||
 | ||
| Fig. 40 Doubly-tethered oligomer resulting from an hydration-driven nucleophilic attack. | ||
 | ||
| Fig. 41 Doubly-tethered oligomers resulting from acetalization reactivity. | ||
3.2.4.3 Hydration-driven nucleophilic attack. The oxazinane-containing bis-Melodinus MIA dimer suadimin C (132) (Fig. 40) would be obtained based on a reactivity analogous to that having led to the formerly detailed suadimin A (75) (Fig. 21). Again, a hydration-driven nucleophilic attack triggered by the Δ14,15 bond on the Δ3,4 iminium function of the other subunit would afford a C-3–C-14 bonded intermediate. An oxidation is then presumed to occur on the C-3 bonded unit to yield the corresponding Δ4,5 iminium that can undergo an heteronucleophilic attack by the 15-OH group of the other subunit, installing the final 1,3-oxazinane unit.241
3.2.4.4 Acetalization. The unique MIA/triterpene hybrid known to date, namely latifolianine A (133) (Fig. 41), features a 1,3-dioxane based junction. An acetalization reaction involving the C-21 aldehydic function of naucleidinal (vallesiachotamane-type MIA) and two hydroxy groups of the ursane triterpene 3β,19α,23,24-tetrahydroxyurs-12-en-28-oic acid can easily account for the constitution of the final scaffold.242 A few years ago, alstoniasidines A (134)24 (Fig. 41) and B revealed an unprecedented sugar bridge between two MIA units. The sugar moiety instigating the intermonomeric connection belongs to the vallesiachotamane-type strictosamide. The other subunit corresponds to a thoroughly modified akuammilane-type component. Such scaffolds should be related to picrinine as a common precursor, benefitting from its characteristic ether connection between C-2 and C-5. An array of picrinine derivatives result from a disruption of N-4–C-5 connectivity. Some analogues, often referred to as Hofmann degradation243-derived compounds, disclose a more or less oxidized furanoindoline scaffold.244,245 Alternatively, a concerted mechanism jointly disrupting both C-2-O and N-4–C-5 bonds might lead to reactive species containing both an indolenine core and an aldehydic function at C-5 has been proposed to step in the biosynthesis of various, highly-rearranged picrinine derivatives.231,246 Back to alstoniasidines, such an aldehyde-containing analogue can initiate an acetalization reaction implying the 6-OH residue of strictosamide glucose.
3.2.4.5 Enolization-driven aminalization. Hybrid compounds were also proposed to proceed from an aldehyde-containing picrinine derivative. As such, alstoscholarisine K (135) (Fig. 42) is an intriguing hybrid incorporating a pyrrolic unit presumed to derive from spermine or putrescine. A nucleophilic attack would be initiated by the α-carbonylated C-6 position on a Δ4,21 functionality which would then trigger a second nucleophilic attack on the C-2′ position of an extraneous Δ1-pyrroline. A Mannich-type nucleophilic addition by the pyrroline nitrogen into the aldehydic group would then lead to alstoscholarisine K (135).247
 | ||
| Fig. 42 Plausible enolization-driven aminalization path to alstoscholarisine K (135). | ||
3.2.5.1 Michael addition followed by heteronucleophilic annulation. The specific constitution of the monoterpenic component of gelsedane-type MIA was already shown to disclose a peculiar reactivity towards a broad range of electrophilic partners. Through the specific example of the pseudodimeric gelsedane-iridoid type gelsamydine (62) (Fig. 18), the possibility for the 2-ethylidenepyrroline system to undertake 1,4-Michael addition (i.e. from the C-19 position) has already been covered in this review. From such singly-tethered intermediates, the access to gelserancine B (136) (Fig. 43) is straightforward. An heteronucleophilic attack of the alicyclic nitrogen N-4 on the carbonylic lactone of the additional iridoid unit could install the final tetrahydropyridinone nucleus of gelserancine B (136).248 The pyridinium-containing gelsedane/corynantheane gelsecorydine B (137) (Fig. 43) can also be related to the biosynthesis of the singly-tethered gelsecorydine A (57) (Fig. 17). An enamine-driven nucleophilic attack initiated from the gelsedane C-19 position on the C-19′ position of the α,β-unsaturated aldehyde function of the vallesiachotamane unit in addition to an heteronucleophilic attack from the alicyclic nitrogen N-4 of the gelsemium subunit into the aldehydic component at the vallesiachotamane C-21′ position could yield the tetrahydropyridinium ring of gelsecorydine B (137)249 after dehydration.
 | ||
| Fig. 43 Doubly-tethered oligomer resulting from enamine reactivity (Michael addition + heteronucleophilic addition). | ||
The aspidospermane/aspidofractane-type meloyine I (138)250 and the recently reported ajmalicine/aspidofractane-based rupestrisines A (139) (Fig. 43) and B251 feature an unprecedented tetrahydropyridine-based intermonomeric junction. None of the seminal phytochemical reports proposed a dimerization mechanism to account for their unusual intermonomeric connectivity. In both cases, a nucleophilic attack initiated by an enamine function (involving the alicyclic nitrogen in the case of the Δ3,14-containing rupestrisine A (139) ajmalicine unit or the indolinic nitrogen for the Δ2,16-containing aspidospermane unit of meloyine I (138)) into the C-15′ position of a conjugated iminium-containing aspidofractane can be presumed to avail a singly-tethered dimeric intermediate. Considering the Δ3′,4′ iminium analogue of the enamine intermediate would allow final ring closure through an heteronucleophilic annulation instigated by the indolinic nitrogen N-1. Notably, the relative configurations of rupestrisines A (139) and B had been later revised by Kutateladze and co-workers using a machine-learning improved DU8-ML DFT-NMR workflow.252 The structure shown for rupestrisine A (139) (Fig. 43) takes into account this structure revision, differing from the initial one by the configuration of C-3′.
3.2.5.2 Unconjugated iminium addition in combination with heteronucleophilic annulation. Husson, Lounasmaa et al. reported on the intriguing structures of ervafolidine (140) and ervafolidene (141) (Fig. 44) in the early 1980s.253 Beyond the dimerization mechanism, the MIA subtype of the southern counterpart raised some biosynthetic questions since these atypical dimers were first described. The dimerization would rely on the condensation of a Δ2,16 enamine derived from the aspidospermane unit on a Δ4,21 iminium-containing 20-epi-pandoline. This reactivity would be followed by the heteronucleophilic annulation triggered by the 20-OH group of the 20-epi-pandoline component on the C-2 position of the indolenine-type aspidospermane setting up the intermonomeric ether connection. To complete the biosynthesis of ervafolidine (140) and ervafolidene (141), its southern component must now rearrange itself into the final isopandolinane-type constitution, i.e. rearrange the piperidine ring into the corresponding ketone-containing spiropyrrolidine (Fig. 44). For this purpose, Henriques et al. suggested to consider an oxidized pandoline component revealing a Δ3,4 iminium. Nucleophilic attack by an OH- on this functionality could then lead to a carbinol amine and then to an aziridinium intermediate that can open to yield the corresponding ketone.253 Various organic synthesis studies support this presumed biosynthesis sequence.254,255
 | ||
| Fig. 44 Top: doubly-tethered oligomer resulting from enamine reactivity (iminium addition + heteronucleophilic addition). Bottom: putative rearrangement from pandolinane to isopandolinane. | ||
3.2.5.3 Conjugate 1,6-addition in combination with aza 1,6-addition. Monogagaine (142)256 and flabellipparicine (143)257 (Fig. 45) are rare MIA dimers featuring a vallesamane building block, along with a more or less transformed vobasane constituent. The C-3 site of the vobasanoid component has already been described as an electrophilic site of considerable generality, as it can undergo a conjugate 1,6-addition from a wealth of nucleophilic partners. As per monogagaine (142) and flabellipparicine (143), this usual 1,6-addition would be triggered by the exomethylene function of the conjugated Δ16,22 exomethylenic functionality of the vallesamane unit to afford a singly-bonded intermediate. In both cases, the driving force for this nucleophilic attack would be an heteronucleophilic attack by the indolic nitrogen of the vobasanoid component on the C-16 position of the vallesamane (viz. apparicine) counterpart.258
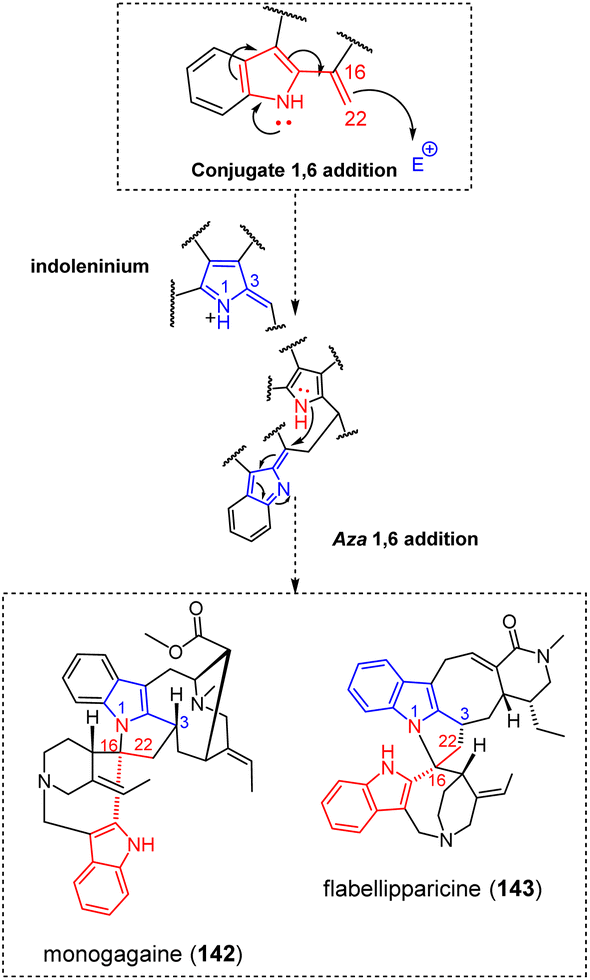 | ||
| Fig. 45 Doubly-tethered oligomer resulting from conjugated 1,6-addition and aza 1,6-addition. | ||
3.2.5.4 Nucleophilicity and electrophilicity of the pleiocarpamane skeleton. Some salient reactivity trends of the pleiocarpamane-type MIAs had been outlined earlier in this review through the examples of pycnanthine (110), pycnanthinine (111) and contortarine A (112) that shed light on both the nucleophilicity of pleiocarpamane scaffold C-7 position and the electrophilicity of C-2 (Fig. 34). The pleiocarpamane/vobasane-type hunterizeyline F (144)259 (Fig. 46) results from a nucleophilic attack triggered by the enamine function comprised in the strained indolic system of the pleiocarpamane-type MIA on the C-3 electrophilic site of a vobasinyl residue. Heteronucleophilic annulation by the vobasane indolic nitrogen on the C-2 position of the indoleninium-containing pleiocarpamane would then avail the five-membered nitrogenated heterocycle connecting the pleiocarpamane and vobasane components of hunterizeyline F (144) (Fig. 46). Although structurally different, the biosynthesis of villalstonine (145)201 (Fig. 46) proceeds via a similar mechanism. The nucleophilic attack of the pleiocarpamane enamine function at C-7 should be directed on the exomethylenic C-21 site of the α,β-unsaturated ketone system of macroline (formally a type A macroline Fig. 8) to perform a 1,4-Michael addition (see alstomacrophylline (25) (Fig. 9) for an analogous reactivity). An hemiketalization reaction should then take place in the macrolane unit from the C-17 OH group on the no longer-conjugated ketone carbonyl at C-19. Finally, heteronucleophilic annulation by the hemiketalic OH group on pleiocarpamane C-2 indoleninium provides the final structure of villalstonine (145). The early biomimetic synthesis of villalstonine (145) substantiates this putative biosynthetic pathway.201
 | ||
| Fig. 46 Doubly-tethered MIA dimers resulting from the characteristic reactivity of the pleiocarpamane skeleton. | ||
Pleiocarpamane derivatives were shown to react with diverse phenolic substances. Voacalgine A (146) (Fig. 47) represents a pleiocarpamane-type MIA fused with a pyrocatechuic-type benzoic acid moiety. First erroneously elucidated after its first isolation in 2015,240 its structure was revised to the disclosed regioisomer a few years later, in the frame of its hemisynthesis.260 The postulated biosynthesis of voacalgine A (146) involves the oxidation of the catechol moiety of pyrocatechuic acid into the corresponding ortho-quinone. A 1,4-Michael addition initiated by the pleiocarpamane enamine function on the electrophilic site contiguous to the carboxylic acid function of this oxidized unit could yield a singly-tethered indoleninium, prone to further heteronucleophilic annulation through attack of the carboxylic acid OH group on the C-2 electrophilic position of the indoleninium.260 Oxidation of the catechol unit of voacalgine A (146) would enable a subsequent 1,6-addition by a second pleiocarpamane component through a similar enamine-driven nucleophilic attack. A heteronucleophilic attack from the contiguous phenolic group on the C-2 indoleninium site of this second pleiocarpamane unit would lead to bipleiophylline (147) (Fig. 47),261 the sole MIA dimer known to date comprising two MIA components anchored on an aromatic spacer platform. Again, this speculative biosynthetic scenario received support from synthetic reports.260,262 A slightly different, yet related MIA-phenolic hybrid is represented by the pleiocarpamane-γ-pyrone-type pleiomaltinine (148) (Fig. 47). Along with its original report, Kam et al. suggested that the biosynthesis of pleiomaltinine should proceed from an oxidized form of the co-isolated maltol to reveal a conjugated exomethylenic system prone to undergo a nucleophilic attack by the pleiocarpamine Δ2,7 enamine moiety. An heteronucleophilic annulation by the contiguous phenolic site on pleiocarpamine indoleninium C-2 could then yield pleiomaltinine (148).236 Again, this putative scenario was further supported by synthesis, using 3-siloxy-4-pyrone derivatives that disclose a reactivity analogous to that of the putative oxidized pyrone.263 In a similar spirit of reactivity, pleiomalicine (149) (Fig. 47) oligomerization assembly may start with the oxidation of the enamine part of pleiocarpamine into a 7-hydroxy-indoleninium precursor. It is worth noting that this oxygen addition at C-7, enabling dearomatization of indole, can be encountered in other MIAs such as terengganensine A264 and rhazidine.265 At last, an heteronucleophilic attack by the 7-OH group on a carbamoyl phosphate unit could install a singly-tethered intermediate prone to yield the final scaffold by heteronucleophilic annulation on the indoleninium C-2 position.236
 | ||
| Fig. 47 Doubly-tethered heterodimers resulting from the characteristic reactivity of the pleiocarpamane skeleton. | ||
3.2.5.5 Aldehyde addition combined to a Mannich-type reaction. As a unique trimeric instance in this section, trirosaline (150) (Fig. 48) represents an intriguing tris-ajmalicine-type MIA and the first trimer ever reported from the emblematic plant, Catharanthus roseus.266 Its oligomerization process would rely on the condensation of a Δ5,6 enamine, which is derived from a first ajmalicine unit on the retro-Mannich-derived C-5′ aldehydic function of a seco-ajmalicine unit. The second intermonomeric N–C connection could be obtained through a Mannich-type reaction from the seco-ajmalicine N-4′ on the C-6′′ oxidized ajmalicine of a third ajmalicine residue. This latter reactivity is, in some ways, reminiscent of undulatine (32) (Fig. 9) bridging mode.
 | ||
| Fig. 48 Plausible biosynthetic path to trirosaline (aldehyde addition and Mannich-type reaction). | ||
3.2.6.1 Phenolic coupling. Pleiocraline (151)267 and pleiocorine (152)268 are two pleiocarpamane–akuammilane MIA dimers that share a common benzofuroindoline junction (Fig. 49). This connection mode is presumed to derive from an oxidative coupling between the C-7 site of the pleiocarpamane unit with the C-11 ortho-phenolic site of the akuammilane counterpart. Such a reactivity had already been considered for the singly-tethered haplophytine (48) and cimiciphytine (49) (Fig. 14), where the canthinone component reveals some degree of structural analogy with the pleiocarpamane scaffold. The singly-tethered C-7–C-11 intermediate can undergo a final heteronucleophilic annulation on the C-2 pleiocarpamane indoleninium to install the sought-after benzofuroindoline motif. Notably, in the context of their synthesis of haplophytine (48),119 Nicolaou and Chen accessed doubly-tethered intermediates analogous to pleiocraline (151) and pleiocorine (152), further strengthening the analogy between these two kinds of MIA dimers. The isopleiocarpamane/vincorinane—type peceyline (153) (Fig. 49) features a dibenzofuran junction involving the benzene ring of both indoline systems.269 The biaryl C–C linkage between C-10 and C-11′ is first presumed to arise through a radicalar process (see ceylanine (44), Fig. 14), probably benefitting from the occurrence of ortho-phenolic functionalities increasing the spin density at the connected sites. Subsequently, a dehydrative etherification would cause a ring closure and install the final furanic junction by forming an ether bond between C-11 and C-10′. This process is of considerable generality in the biosynthesis of dibenzofuran-type natural products.270
 | ||
| Fig. 49 Doubly-tethered oligomers resulting from phenolic couplings (see S2‡ for a comprehensive listing, pink color indicates groups involved in the radical coupling reactivities). | ||
3.2.6.2 (Hetero)-Diels–Alder. Dimeric MIAs obtained through a Diels–Alder reaction are quite scarce. A first interesting case is represented by secamines, rare MIA dimers mainly described in the 1970s with partial stereochemical assignments. Secodine-type alkaloids are prone to dimerization owing to their acrylic ester system. Therefore, presecamine (154)271 (Fig. 50) is a dimeric secodine obtained through a Diels–Alder reaction involving the Δ16,17 functionality of a first secodine as the dienophile and the Δ2′,7′ and Δ16′,17′ of a second secodine building block as the dienic system. Presecamine (154) was shown to rearrange quantitatively to secamine (155) (Fig. 50) in dilute HCl at room temperature.271 Cordell et al. proposed a rearrangement mechanism giving rise to a conjugated indoleninium as shown in Fig. 50, that could undergo 1,6-addition from the indolic nitrogen on the C-16′ position of the other secodine subunit.271 A further case of Diels-Alder derived MIA dimer is the stemmadeninane/strychnane-based kopsiyunnanine M (156) (Fig. 50).272
 | ||
| Fig. 50 Doubly-tethered oligomers resulting from Diels–Alder cycloadditions (see S2‡ for a comprehensive listing, pink color refers to structural elements related to Diels–Alder reaction). | ||
The dienic system is represented by the Δ20,21 and the vinylic Δ18,19 functionalities of the stemmadeninane unit, reacting on the characteristic Δ17,23 functionality of the strychnane counterpart. Another last example of cycloaddition-related dimerization process is that of kopsiyunnanine A (157) (Fig. 51).273 This condylocarpane/vallesiachotamane-based MIA dimer could be obtained through a hetero-Diels–Alder reaction involving the α,β-unsaturated aldehyde of vallesiachotamane as the diene (i.e. Δ19,20 and the aldehydic group at C-21) and the vinylic Δ18′,19′ functionality of an oxidized derivative of 18,19-dehydrotubotaiwine as the dienophile.273 Although unique in the broad group of MIA oligomers, hetero-Diels–Alder reaction appears to be more common in the bis-diterpenoids group274 and is also invoked to access dimeric triterpenes and sterols,275,276 affording the signature dihydropyran-type intermonomeric junction. Diels–Alder-based dimerization reactions are also involved in the biosynthesis of borreverine/isoborreverine-type dimeric structures that proceed from borrerine.277 Enamine reactivity followed by heteronucleophilic annulation (borreverine) or heteronucleophilic attack (isoborreverine) further step in the biosynthesis of these compounds that lie outside of the scope of the current review.278,279
 | ||
| Fig. 51 Doubly-tethered oligomer resulting from an hetero-Diels–Alder cycloaddition. (see S2‡ for a comprehensive listing, pink color indicates structural features involved in hetero-Diels–Alder reaction). | ||
3.3 Oligomers featuring a mixed tethering mode
A limited number of oligomers result from a combination of the two above mentioned tethering modes. They are disclosed in Fig. 52.A remarkable example of oligomeric MIA with mixed tethering modes is alasmontamine A (159) (Fig. 52),132 the sole example of tetrameric MIA known to date, comprising four aspidospermane units. More precisely, this structure should be considered as a dimeric vobtusine (122). The biosynthetic path to vobtusine (122) supposedly steps through a ring closure-driven nucleophilic attack from the Δ14,15 functionality on a conjugated exomethylene indolenine system (as reported to occur in the biosynthesis of both voacandimine A (123) and voatriafricanine A (160)). The spiranic junction of vobtusine would then derive from the enamine nucleophilic attack of a newly installed Δ3,14 moiety into an exomethylene-type N-1 iminium of the other aspidospermane unit (Fig. 37). The connection between the two vobtusine building blocks can easily be related to an enamine (Δ5,6)-driven nucleophilic attack into a Δ4′,5′ iminium function of the other vobtusine counterpart. As of 2024, a MIA dimer disclosing this precise connectivity does not seem to have been reported, but this reactivity can be related to that having led to the bis-aspidofractane arbolodinine A (51) that involved a similar Δ4,5 iminium electrophilic partner but involved a different Δ3,14 enamine (Fig. 16).
 | ||
| Fig. 52 Oligomers featuring mixed tethering modes (see S2‡ for a comprehensive listing, red and blue-colored moieties refer to a first set of nucleophilic and electrophilic sites, respectively; orange and green colored moieties refer to a second set of nucleophilic and electrophilic sites, respectively). | ||
3.4 Triply-tethered oligomers
A few dimeric MIAs feature three intermonomeric connections. Most often, these junctions can be related to a doubly-bonded dimeric structure but involve an additional reaction. | ||
| Fig. 53 Triply-tethered oligomers resulting from heteronucleophilic annulation-driven oligomerization. | ||
Finally, the diazacyclooctadiene junction of bis-akuammicine type MIA as it appears in toxiferine I (119) (Fig. 54) was experimentally shown to represent an entry point into many triply-tethered MIA dimers.284 These dimeric compounds can readily undergo further chemical change, shown to depend on the conditions of pH, heat and the presence of oxygen.80 In these conditions, the central ring of these dimeric structures can undergo changes in oxygenation level (Fig. 54). The triple connection appearing in matopensine (164)285 can be related to the non-dehydrated precursor of the diazacyclooctadiene scaffold, from which it could derive by dehydrative etherification. Divarine (165)286 features a similar junction but distinct tethering sites compared with matopensine (164). An intermediate revealing both a N,O-hemiketalic group and an epoxide function can be supposed to step in the biosynthesis of this MIA dimer as shown in Fig. 54. The epoxide ring opening at C-16 initiated by the N,O-hemiketalic OH group at the C-17 position of the other component can be presumed to yield the intermonomeric connectivity appearing in divarine (165), only requiring subsequent O methylation to obtain the final product. Toxiferine IX (166)/caracurine II (167)-type triply-tethered akuammicine dimers (viz. featuring an additional C-16 to C-16′ bond) has been investigated by Battersby and co-workers in the late 1960s.287 It was noted that the dimerization of toxiferin I into caracurine II (167) depended on acid and oxygen. A tentative mechanism has been proposed that would be initiated by an enamine nucleophilic attack of O2 at C-16, hinting a plausible artefactual origin for such compounds. A second nucleophilic attack triggered by the second enamine function of this eight-membered junction into the peroxyhemiketal, could then yield the complex triple intermonomeric connection appearing in toxiferine IX (166)/caracurine II (167).
 | ||
| Fig. 54 Plausible biosynthetic path to diazacyclooctadiene-containing MIA dimers and related compounds. | ||
Photooxidation reactions were shown to lead to another subtype of triply-tethered akuammicine dimers. Since the late 1950s,288,289 Bernauer and co-workers reported on unusual, photosensitized oxygen transfer reactions to several calabash curares. The diazacyclooctadiene-containing C-dihydrotoxiferine I and toxiferine I (119) were respectively shown to transform into calebassine (168) and C-alkaloid A when irradiated in aerated solutions in the presence of eosin as sensitizer. Exposed to light and air in the absence of sensitizer, alternative scaffolds containing an additional ether bond (viz. respectively C-curarine (169) and C-alkaloid E) would be obtained instead.288,289 A radical hydroxylation at C-2 can be envisaged for C-curarine (169), which could evolve into the sought-after ether connectivity by heteronucleophilic attack to the C-2 oxidized indoleninium akuammicine counterpart. In neither case could a definite reaction mechanism be established although transannular reactions analogous to that converting toxiferine I to C-alkaloid A did not seem to have been reported prior to these photooxidation studies.290,291
The diazacyclooctane junction and the ether connection between the two aspidospermane components appearing in hazuntiphylline (170)292 (Fig. 55) can be presumed to be analogous with the biosynthetic scheme leading to leucofoline (103)-like (viz. stepwise aza-Michael, Fig. 31). Two distinct aza-Michael reactions from the indolinic N-1 of both components on the C-22 exomethylene of the indolenine-exomethylene system of the other counterpart can be supposed to install the eight-membered intermonomeric junction. A possible hydroxylation at the C-2 position of both units could set the stage for the final dehydrative etherification yielding the three connections between both aspidospermane units. It is also plausible that this ether connection results from an heteronucleophilic attack from this OH-2 group on an indoleninium.
 | ||
| Fig. 55 Plausible biosynthetic chemical logic for the assembly of hazuntiphylline. | ||
Our literature survey resulted in locating a single example of triply-tethered MIA-containing entity not involving two MIAs or a MIA and a pseudodimer: melohenine A (171) (Fig. 56).129 This example is remarkable given the modest size of the extraneous unit instigating these three connections, viz. glyceraldehyde. The biosynthesis is presumed to proceed from an oxidized form of the strychnane-like dihydrodeoxyisostrychnidine, which is capable of initiating aldolization into glyceraldehyde phosphate from C-22, followed by an hemiketalization reaction at C-23 to afford a doubly-tethered intermediate. An heteronucleophilic annulation on a Δ1,2- indoleninium could then lead to the final scaffold.
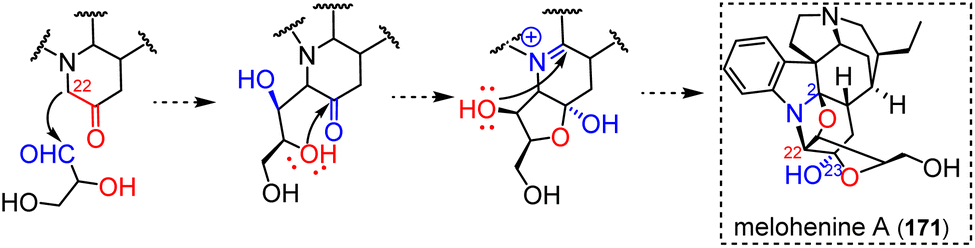 | ||
| Fig. 56 Plausible biosynthetic path to melohenine A. | ||
 | ||
| Fig. 57 Plausible biosynthetic path to triply-tethered MIA pseudodimers. | ||
4 Outlook and concluding remarks
The body of work highlighted here demonstrates that numerous oligomerization steps follow a limited number of assembly reactions.299 For instance, in the case of electrophilic aromatic substitutions, eight electrophilic motifs have been enumerated and identified throughout this endeavour. In addition, MIA skeletons reactivity mappings have been generated during this work and are depicted in ESI (S1).‡ Moreover, an exhaustive list of non-MIA adducts could have been compiled and a MIA oligomer network (Fig. 3) has been built. We foresee that this global overview will be used by the community as a unique assistance for structure elucidation and anticipation of MIA oligomers. Ultimately, the comprehensive repository of MIA oligomers machine-readable metadata (ESI‡) associated to this review has been uploaded on LOTUS16 and could be leveraged for chemoinformatics purposes. Altogether, we hope that our literature survey will provide a versatile platform to assist natural product chemists in assessing the structural novelty of future oligomeric MIAs.Although some efforts have been made to determine the biosynthesis of these complex natural products,300 it is important to point out that most of the biosynthetic pathways covered here are highly speculative. In this context, several biosynthetic routes are possible to reach certain dimers discussed in this manuscript and it is hardly possible to favour one over the others. As an illustration, different mechanistic scenarios leading to a few dimers have been developed in this manuscript, among many other available examples (see melomorsine (95) and bisleucocurine A (105) for some such examples).
Prior to concluding our review, we were willing to compare the wealth of processes involved in MIA oligomerization with other groups of alkaloids. Few alkaloid families can rival MIA in terms of numbers of representatives, hampering a fair comparison with most phytochemical classes. The main such example that came to our mind was the group of bisbenzylisoquinoline alkaloids, that comprises a bit more than 500 entries.301,302 Nevertheless, this diversity relies on a much more limited set of assembly modes that is relatable to a combination of diphenyl ether linkages and/or benzylphenyl ether (methyleneoxy) linkage. Plausible mechanisms of formation for this latter bridging mode, not known to the best of our knowledge in the realm of MIA, have been thoroughly addressed elsewhere.303 Number (up to 3) and sites of connections lead to further refine the classification of these dimeric compounds. As N-methylcoclaurine is the gateway to benzylisoquinolines, most bisbenzylisoquinolines share its oxygenation pattern, probably limiting available dimerization mechanisms. Although very limited in relation to the current topic, these bridging modes can lead to a few junctions that have not yet been observed within MIAs, due to certain specific combinations of coupling modes and assembly sites, especially when the methyleneoxy bridge specific to bisbenzylisoquinolines is involved. Two such examples are benzodioxin-comprising bisbenzylisoquinolines (obtained as a consequence of two contiguous diphenyl ether couplings) that have repeatedly been reported.304 The seven-membered 1,4-dioxepine junction in racemosidine A results from the combination of an ether coupling and a contiguous methyleneoxy bond.305 Notably, cissampereine-type dimers, revealing a simple methyleneoxy connection, are presumed to derive from such intermediates.303 Triply-tethered repanduline-type bisbenzylisoquinolines,306 revealing a spiro-1,4-dioxane junction would imply an ether-bonded precursor that would enable the subsequent installation of a methyleneoxy functionality through an original dearomatizing sequence.307 These assembly logics can be extended to most homo- and heterodimeric bisbenzylisoquinoline-derived scaffolds (e.g. aporphine, protoberberines, pavines, seco-phenanthrenes etc.). However, some dimers revealing more unusual connectivities are obtained by mechanisms that are strictly analogous to those encountered in this review. As such, several bis-aporphines (ovigeridimerine,308 ovihernangerine, oviisocorydine309) are being bridged through a urea junction that is exactly similar to that being reported in geleganidine C (71) (Fig. 20), presumably involving an heteronucleophilic attack of an extraneous formaldehyde unit. Likewise, the methylene-bridged lagesianine B310 is strongly reminiscent of the putative dimerization reaction having led to melofusine I (70) (Fig. 20). The co-isolated bisbenzylisoquinoline-type isopyruthaldine and isopythaldine311 are the unique such dimers linked through a methylene group, which guided some authors to assume that these compounds should arise through an electrophilic aromatic substitution with formaldehyde312 (see the example of pleiokomenine A (33) (Fig. 10). As the last example of extraneous methylene unit-containing dimer, the unique case of the bisbenzylisoquinoline medelline, isolated by Cavé in 1986,313 deserved to be mentioned here. This compound features a methylenedioxy bridge between its two components, that can be traced back to an heteronucleophilic attack initiated from two O atoms instead of a melofusine I-type sequence initiated by two N atoms. The presence of a nearby ether connectivity results in a 1,3,6-oxacine intermonomeric junction. Such a reactivity has yet to be described in the realm of MIA. Another disturbing link between the world of isoquinolines and that of MIAs lies in the tendency of dopamine to undergo Pictet–Spengler reactions with secologanine to produce the so-called monoterpenoid ipecac alkaloids.314 These reactions produce a hybrid structure that is very reminiscent of strictosidine, namely deacetyllipecoside giving a straight access to a series of alkaloids analogous to type I MIAs albeit lacking their pyrrole nucleus due to the amine-containing partner being dopamine instead of tryptamine. Among many others, one can quote the examples of the “corynantheane-like” protoemetine,315 and of the “vallesiachotamane-like” alangiside316 and various azaberberines often referred to as alamarine derivatives317,318 (these latter being reminiscent of angustine). Due to their structural similarity, these protoemetine-type ipecac alkaloids show strikingly similar reactivity to corynantheane-type alkaloids, as shown by the constitution of cephaeline/emetine which denotes Pictet–Spengler-type condensation with an additional dopamine unit.203 Closer still to the MIA, various tubulosine319,320 derivatives condense with a tryptamine unit, which is extremely similar to usambarine (109)-type corynantheane-tryptamine pseudodimers (Fig. 33). Ipecac alkaloids had also been shown to instigate phenylpropene adducts through an amide connection,321 as observed for some MIAs like cylindrocarpine322 (see the example of alstiphyllanine I (86) for a related compound including a gallate derivative instead (Fig. 24)). Similarly, aporphine-gallate adducts linked by an amide bond have been described very recently.323 It is interesting to note that benzylisoquinoline alkaloids, mostly aporphines, tend to react with carbamic acid too.324,325 Nevertheless, these adducts constantly arise through a N-triggered nucleophilic attack that does not seem to be reported for any MIA so far. The tertiary nature of the alicyclic nitrogen of most MIA probably accounts for this, even though some alkaloids isolated from Gelsemium sources disclose secondary amines that could afford such hybrid structures in the future. To draw a final parallel with the world of MIAs, benzylisoquinolines are also about to enter the era of trimeric compounds, with the first example of such a compound published in 2018: neoliensinine.326
It is interesting to note that some specific alkaloid series are associated with typical substituents that may step in some specific bridging modes. One such example is that of tropanic alkaloids that tend to be substituted with diverse organic acids (e.g. mesaconic acid, itaconic acid, angelic acid or senecic acid).327 Both carboxylic acid ends of mesaconic acid can be esterified by tropanic alkaloids units to yield a wealth of different dimers.328 Ditropane diesters of itaconic acid have also been described.329 Likewise, tropane alkaloids substituted with a tropic acid residue are prone to spontaneous dimerization through a Diels-Alder-type cyclization to obtain belladonnine/scopadonnine derivatives.330 Photoinduced [2 + 2] cycloadditions, unknown to date for MIA and benzylisoquinolines families, produce cyclobutane-centered tropane dimers. These dimers rely on the unsaturations occurring on an organic acid-type substituent and/or of a phenylpropene residue.331 Truxillin-type332 MIA dimers could be envisaged from the numerous MIA dimers disclosing a phenylpropene residue, but such compounds had not been described so far. Remarkable trimers have made use of several of these reactivities, such as grahamine, which requires both a mesaconic acid bridging group and a [2 + 2] cycloaddition.333 Other trimers are biosynthesized by mechanisms close to MIA chemistry, such as the recently reported trissaccardine, based on two aza-Michael additions.334 Tropane dimers being bridged through an urea function had also been reported, as formerly known from MIA and benzylisoquinolines.335
It appeared clearly throughout this review that numerous oligomerization chemical logics extend beyond MIA skeleton boundaries (see hazuntiphylline (170) (aspidospermane) and C-curarine (169) (akuammicine) Fig. 54) and often serve as the starting points to several scaffolds either through highly evolved enzymatic pathways or diversification opportunities through spontaneous reaction or both.336 Among those diversification opportunities, Potier-Polonovski fragmentation and retro Mannich often lead to structural rearrangements of certain MIA skeletons resulting in unexpected oligomerizations (see, trirosaline (150), angustiphylline (84)). Moreover, a glance at the reactivity table (S2, ESI‡) shows, for example, that dimerizations based on the Pictet–Spengler reaction have so far been strictly linked to the biosynthesis of pseudodimeric MIAs incorporating an additional tryptamine unit. Similarly, esterification and amidification reactions appear to have been exclusively involved in the incorporation of phenylpropanoids/gallates/anthranilates.
Advances in the characterization, isolation techniques, and synthesis of this fascinating class of phytochemicals have significantly enriched the understanding of their chemistry. The landmark advances in the fields of mass spectrometry, NMR, and chemoinformatics provide natural chemists with ever-expanding capacities to pinpoint minor, structurally new metabolites since the very first steps of phytochemical processing and to target their isolation through a hypothesis-driven pipeline. MIA in particular strongly benefitted from the molecular networking strategies thanks to the upload of MS/MS data related to 172 structurally diverse MIAs337 to the GNPS338 repositories. We foresee that the number of newly described MIA oligomers, trimers in particular, will increase in the coming years enabling, hopefully, the discovery of new assembly patterns.
We hope that this global overview of the unified chemical logic underlying the assembly of MIA oligomers will provide new insights on the biosynthesis and inspiring prospects for biomimetic total syntheses.
5 Data availability
The data supporting this article have been included as part of the ESI.‡ The comprehensive repository of MIA oligomers machine-readable metadata (InChI and SMILES) associated to this review has been uploaded on LOTUS and is accessible at https://www.wikidata.org/.6 Author contributions
Pierre Le Pogam: conceptualization, writing – original draft, review & editing, supervision, validation, Mehdi A. Beniddir: conceptualization, writing – original draft, review & editing, supervision, validation.7 Conflicts of interest
There are no conflicts to declare.8 Acknowledgement
M. A. B. and P. L. P. were supported by the National French Agency (ANR grant 20-CE43-0010). M.A.B. warmly thanks all his students (past and present) who have been involved in MIA discovery program for their enthusiasm and their efforts, rendering these discoveries a true pleasure to pursue. The authors warmly thank Adriano Rutz for the upload of the MIA oligomer biosource to LOTUS.9 Notes and references
- S. A. Snyder, A. M. ElSohly and F. Kontes, Nat. Prod. Rep., 2011, 28, 897–924 RSC.
- Q. T. Khong, D. Li, B. A. P. Wilson, K. Ranguelova, M. Dalilian, E. A. Smith, A. Wamiru, E. I. Goncharova, T. Grkovic, D. Voeller, S. Lipkowitz, M. J. Schnermann, B. R. O'Keefe and L. Du, Org. Lett., 2022, 24, 9468–9472 CrossRef CAS PubMed.
- C. Cuello, E. Stander, H. Jansen, T. Dugé De Bernonville, A. Oudin, C. Birer Williams, A. Lanoue, N. Giglioli Guivarc’h, N. Papon, R. Dirks, M. Jensen, S. O’Connor, S. Besseau and V. Courdavault, F1000Research, 2022, 11, 1541 CAS.
- J. Zhang, L. G. Hansen, O. Gudich, K. Viehrig, L. M. M. Lassen, L. Schrübbers, K. B. Adhikari, P. Rubaszka, E. Carrasquer-Alvarez, L. Chen, V. D'Ambrosio, B. Lehka, A. K. Haidar, S. Nallapareddy, K. Giannakou, M. Laloux, D. Arsovska, M. A. K. Jørgensen, L. J. G. Chan, M. Kristensen, H. B. Christensen, S. Sudarsan, E. A. Stander, E. Baidoo, C. J. Petzold, T. Wulff, S. E. O'Connor, V. Courdavault, M. K. Jensen and J. D. Keasling, Nature, 2022, 609, 341–347 CrossRef CAS PubMed.
- M. J. Dror, J. Misa, D. A. Yee, A. M. Chu, R. K. Yu, B. B. Chan, L. S. Aoyama, A. P. Chaparala, S. E. O'Connor and Y. Tang, J. Ind. Microbiol. Biotechnol., 2023, kuad047 Search PubMed.
- S. A. Bradley, B. J. Lehka, F. G. Hansson, K. B. Adhikari, D. Rago, P. Rubaszka, A. K. Haidar, L. Chen, L. G. Hansen, O. Gudich, K. Giannakou, B. Lengger, R. T. Gill, Y. Nakamura, T. D. de Bernonville, K. Koudounas, D. Romero-Suarez, L. Ding, Y. Qiao, T. M. Frimurer, A. A. Petersen, S. Besseau, S. Kumar, N. Gautron, C. Melin, J. Marc, R. Jeanneau, S. E. O'Connor, V. Courdavault, J. D. Keasling, J. Zhang and M. K. Jensen, Nat. Chem. Biol., 2023, 19, 1551–1560 CrossRef CAS PubMed.
- M. Sottomayor and A. Ros Barceló, Protoplasma, 2003, 222, 97–105 CrossRef CAS PubMed.
- J. Garnier, M. Koch and N. Kunesch, Phytochemistry, 1969, 8, 1241–1254 CrossRef CAS.
- G. A. Cordell and J. E. Saxton, The Alkaloids: Chemistry and Physiology, 1981, 20, pp. 1–295 Search PubMed.
- T.-S. Kam and Y.-M. Choo, in The Alkaloids: Chemistry and Biology, ed. G. A. Cordell, Academic Press, 2006, vol. 63, pp. 181–337 Search PubMed.
- M. Kitajima and H. Takayama, The Alkaloids: Chemistry and Biology, 2016, 76, pp. 259–310 Search PubMed.
- L. Szabó, Molecules, 2008, 13, 1875–1896 CrossRef.
- X.-H. Cai, M.-F. Bao, Y. Zhang, C.-X. Zeng, Y.-P. Liu and X.-D. Luo, Org. Lett., 2011, 13, 3568–3571 CrossRef CAS PubMed , In 2011, the description of alstrostines A and B revealed a potential new precursor towards monoterpene indole alkaloids that would include a tryptophane unit and two seco-iridoid components, which may thus fall into the category of pseudodimers..
- C. Kornpointner, A. Berger, F. Traxler, A. Hadžiabdić, M. Massar, J. Matek, L. Brecker and J. Schinnerl, Phytochemistry, 2020, 173, 112296 CrossRef CAS PubMed.
- A. Berger, K. Valant-Vetschera, J. Schinnerl and L. Brecker, Phytochem. Rev., 2022, 21, 915–939 CrossRef CAS.
- A. Rutz, M. Sorokina, J. Galgonek, D. Mietchen, E. Willighagen, A. Gaudry, J. G. Graham, R. Stephan, R. Page, J. Vondrášek, C. Steinbeck, G. F. Pauli, J.-L. Wolfender, J. Bisson and P.-M. Allard, Elife, 2022, 11, e70780 CrossRef CAS PubMed.
- P. Shannon, A. Markiel, O. Ozier, N. S. Baliga, J. T. Wang, D. Ramage, N. Amin, B. Schwikowski and T. Ideker, Genome Res., 2003, 13, 2498–2504 CrossRef CAS PubMed.
- E. Otogo N'Nang Obiang, G. Genta-Jouve, J.-F. Gallard, B. Kumulungui, E. Mouray, P. Grellier, L. Evanno, E. Poupon, P. Champy and M. A. Beniddir, Org. Lett., 2017, 19, 6180–6183 CrossRef PubMed.
- W. Zhang, X.-J. Huang, S.-Y. Zhang, D.-M. Zhang, R.-W. Jiang, J.-Y. Hu, X.-Q. Zhang, L. Wang and W.-C. Ye, J. Nat. Prod., 2015, 78, 2036–2044 CrossRef CAS.
- E. Okuyama, L.-H. Gao and M. Yamazaki, Chem. Pharm. Bull., 1992, 40, 2075–2079 CrossRef CAS.
- T.-S. Kam, S.-J. Tan, S.-W. Ng and K. Komiyama, Org. Lett., 2008, 10, 3749–3752 CrossRef CAS.
- E. Otogo N'Nang, G. Bernadat, E. Mouray, B. Kumulungui, P. Grellier, E. Poupon, P. Champy and M. A. Beniddir, Org. Lett., 2018, 20, 6596–6600 CrossRef.
- E. Otogo N'Nang, J. F. Gallard, P. Champy, P. Le Pogam and M. A. Beniddir, Magn. Reson. Chem., 2022, 60, 1178–1184 CrossRef.
- X. Wei, Z. Dai, J. Yang, A. Khan, H.-F. Yu, Y.-L. Zhao, Y.-F. Wang, Y.-P. Liu, Z.-F. Yang, W.-Y. Huang, X.-H. Wang, X.-D. Zhao and X.-D. Luo, Bioorg. Med. Chem., 2018, 26, 1776–1783 CrossRef CAS PubMed.
- G. Cauchie, E. O. N'Nang, J. J. J. van der Hooft, P. Le Pogam, G. Bernadat, J.-F. Gallard, B. Kumulungui, P. Champy, E. Poupon and M. A. Beniddir, Org. Lett., 2020, 22, 6077–6081 CrossRef CAS.
- J.-J. Wei, W.-Q. Wang, W.-B. Song, J. Li and L.-j. Xuan, Phytochem. Lett., 2018, 23, 1–4 CrossRef CAS.
- J. M. Müller, Experientia, 1957, 13, 479–481 CrossRef PubMed.
- H. Arai, K. Zaima, E. Mitsuta, H. Tamamoto, A. Saito, Y. Hirasawa, A. Rahman, I. Kusumawati, N. C. Zaini and H. Morita, Bioorg. Med. Chem., 2012, 20, 3454–3459 CrossRef CAS PubMed.
- E. Davioud, C. Kan, J.-C. Quirion, B. C. Das and H.-P. Husson, Phytochemistry, 1989, 28, 1383–1387 CrossRef CAS.
- B. Hong, D. Grzech, L. Caputi, P. Sonawane, C. E. R. López, M. O. Kamileen, N. J. Hernández Lozada, V. Grabe and S. E. O'Connor, Nature, 2022, 607, 617–622 CrossRef CAS.
- C. Schlatter, E. E. Waldner, H. Schmid, W. Maier and D. Gröger, Helv. Chim. Acta, 1969, 52, 776–789 CrossRef CAS.
- C. Schlatter, E. E. Waldner, H. Schmid, D. Gröger, K. Stolle and K. Mothes, Helv. Chim. Acta, 1966, 49, 1714–1715 CrossRef CAS.
- We inform the readers that this review effort outlined only one unclassified MIA, named tenuiphylline. This puzzling case is addressed in S3, ESI‡.
- J. Le Men and W. I. Taylor, Experientia, 1965, 21, 508–510 CrossRef CAS PubMed.
- M. E. Tanner, Nat. Prod. Rep., 2015, 32, 88–101 RSC.
- C. T. Walsh, ACS Chem. Biol., 2014, 9, 2718–2728 CrossRef CAS.
- T. A. van Beek, R. Verpoorte and P. Q. Kinh, Planta Med., 1985, 51, 277–279 CrossRef.
- H. Fouotsa, P. Mkounga, A. M. Lannang, J. Vanheuverzwijn, Z. Zhou, K. Leblanc, S. Rharrabti, A. E. Nkengfack, J.-F. Gallard, V. Fontaine, F. Meyer, E. Poupon, P. Le Pogam and M. A. Beniddir, Org. Biomol. Chem., 2022, 20, 98–105 RSC.
- B. González, N. Veiga, G. Hernández, G. Seoane and I. Carrera, J. Nat. Prod., 2023, 86, 1500–1511 CrossRef.
- J. P. Kutney, Nat. Prod. Rep., 1990, 7, 85–103 RSC.
- M. Hiroki, A. Abulikemu, C. Totsuka, Y. Hirasawa, T. Kaneda and H. Morita, J. Nat. Med., 2024, 78, 216–225 CrossRef CAS PubMed.
- A. Brossi and M. Suffness, The Alkaloids: Antitumor Bisindole Alkaloids from Catharanthus roseus (L.), Academic Press, 1990 Search PubMed.
- N. Neuss and S. S. Tafur, United States Pat., US3968113A, 1977 Search PubMed.
- A. El-Sayed, G. A. Handy and G. A. Cordell, J. Nat. Prod., 1983, 46, 517–527 CrossRef CAS PubMed.
- Y. Hirasawa, A. Kase, A. Okamoto, K. Suzuki, M. Hiroki, T. Kaneda, N. Uchiyama and H. Morita, J. Nat. Med., 2024, 78, 382–392 CrossRef CAS PubMed.
- C.-H. Wang, G.-C. Wang, Y. Wang, X.-Q. Zhang, X.-J. Huang, D.-M. Zhang, M.-F. Chen and W.-C. Ye, Fitoterapia, 2012, 83, 765–769 CrossRef CAS PubMed.
- C. Szántay, Pure Appl. Chem., 1990, 62, 1299–1302 CrossRef.
- I. Chardon-Loriaux and H.-P. Husson, Tetrahedron Lett., 1975, 16, 1845–1848 CrossRef.
- I. Chardon-Loriaux, M.-M. Debray and H.-P. Husson, Phytochemistry, 1978, 17, 1605–1608 CrossRef CAS.
- F. Reuß and P. Heretsch, Nat. Prod. Rep., 2021, 38, 693–701 RSC.
- J. Bruneton, A. Cavé, E. W. Hagaman, N. Kunesch and E. Wenkert, Tetrahedron Lett., 1976, 17, 3567–3570 CrossRef.
- K.-H. Lim and T.-S. Kam, Helv. Chim. Acta, 2009, 92, 1895–1902 CrossRef CAS.
- K.-H. Lim and T.-S. Kam, Tetrahedron Lett., 2009, 50, 3756–3759 CrossRef CAS.
- D. W. Thomas, H. Achenbach and K. Biemann, J. Am. Chem. Soc., 1966, 88, 1537–1544 CrossRef CAS PubMed.
- Y.-H. Fu, Y.-T. Di, H.-P. He, S.-l. Li, Y. Zhang and X.-J. Hao, J. Nat. Prod., 2014, 77, 57–62 CrossRef CAS.
- The specific case of vobtusamine is, however, very particular. It was indeed suggested that the eburnane half of this compound would occur as a consequence of an aspidospermane to eburnane rearrangement as experimentally validated by Wenkert from vobtusine.
- C.-Y. Gan, W. T. Robinson, T. Etoh, M. Hayashi, K. Komiyama and T.-S. Kam, Org. Lett., 2009, 11, 3962–3965 CrossRef CAS.
- G. Pandey, A. Mishra and J. Khamrai, Org. Lett., 2017, 19, 3267–3270 CrossRef CAS.
- M. Damak, A. Ahond and P. Potier, Bull. Soc. Chim. Fr., 1980, 9, 490–495 Search PubMed.
- C.-E. Nge, K.-S. Sim, S.-H. Lim, N. F. Thomas, Y.-Y. Low and T.-S. Kam, J. Nat. Prod., 2016, 79, 2709–2717 CrossRef CAS PubMed.
- E. Otogo N'Nang, G. Cauchie, P. Retailleau, S. T. Agnandji, J.-F. Gallard, E. Mouray, P. Grellier, P. Champy, P. Le Pogam and M. A. Beniddir, J. Nat. Prod., 2023, 86, 1202–1210 CrossRef.
- J. Bruneton, A. Bouquet and A. Cavé, Phytochemistry, 1974, 13, 1963–1967 CrossRef CAS.
- A. Cavé, J. Bruneton, A. Ahond, A. M. Bui, H. P. Husson, C. Kan, G. Lukacs and P. Potier, Tetrahedron Lett., 1973, 14, 5081–5084 CrossRef.
- Y.-P. Liu, Y.-L. Zhao, T. Feng, G.-G. Cheng, B.-H. Zhang, Y. Li, X.-H. Cai and X.-D. Luo, J. Nat. Prod., 2013, 76, 2322–2329 CrossRef CAS PubMed.
- A. Arnone, G. Nasini and L. Merlini, J. Chem. Soc., Perkin Trans. 1, 1987, 571–575 RSC.
- T.-S. Kam, K.-H. Lee and S.-H. Goh, Phytochemistry, 1991, 30, 3441–3444 CrossRef CAS.
- T. Kouamé, A. T. Okpekon, N. F. Bony, A. D. N'Tamon, J.-F. Gallard, S. Rharrabti, K. Leblanc, E. Mouray, P. Grellier, P. Champy, M. A. Beniddir and P. Le Pogam, Molecules, 2020, 25, 2654 CrossRef PubMed.
- N. Yoshikado, S. Taniguchi, N. Kasajima, F. Ohashi, K. I. Doi, T. Shibata, T. Yoshida and T. Hatano, Heterocycles, 2009, 77, 793–800 CrossRef CAS PubMed.
- M. Oshima, H. Aoyama, Y. Shimozu, S. Taniguchi, T. Miura and T. Hatano, Heterocycles, 2019, 98, 804–812 CrossRef CAS.
- Z.-W. Liu, M. Song, J.-Y. Wang, D.-Z. Wang, B. Sun, L. Shi, R.-W. Jiang, M. Ma and X.-Q. Zhang, Phytochemistry, 2023, 211, 113678 CrossRef CAS.
- T. P. Lien, C. Kamperdick, T. Van Sung, G. Adam and H. Ripperger, Phytochemistry, 1998, 49, 1797–1799 CrossRef CAS.
- J. W. Medley and M. Movassaghi, Angew. Chem., Int. Ed., 2012, 51, 4572–4576 CrossRef CAS.
- P. Obitz, S. Endreß and J. Stöckigt, Phytochemistry, 1995, 40, 1407–1417 CrossRef CAS.
- M. T. Rahman and J. M. Cook, in Studies in Natural Products Chemistry, ed. Atta-Ur-Rahman, Elsevier, 2020, vol. 64, pp. 1–57 Search PubMed.
- M. T. Rahman, V. V. N. P. B. Tiruveedhula and J. M. Cook, Molecules, 2016, 21, 1525 CrossRef PubMed.
- J. S.-Y. Yeap, H. M. Saad, C.-H. Tan, K.-S. Sim, S.-H. Lim, Y.-Y. Low and T.-S. Kam, J. Nat. Prod., 2019, 82, 3121–3132 CrossRef CAS PubMed.
- S.-K. Wong, J. S.-Y. Yeap, C.-H. Tan, K.-S. Sim, S.-H. Lim, Y.-Y. Low and T.-S. Kam, Tetrahedron, 2021, 78, 131802 CrossRef CAS.
- A. Jossang, P. Fodor and B. Bodo, J. Org. Chem., 1998, 63, 7162–7167 CrossRef CAS PubMed.
- S. Subhadhirasakul, H. Takayama, N. Aimi, D. Ponglux and S.-I. Sakai, Chem. Pharm. Bull., 1994, 42, 1427–1431 CrossRef CAS.
- J.-M. Nuzillard, P. Thépenier, M.-J. Jacquier, G. Massiot, L. Le Men-Olivier and C. Delaude, Phytochemistry, 1996, 43, 897–902 CrossRef CAS.
- K.-H. Lim and T.-S. Kam, Org. Lett., 2006, 8, 1733–1735 CrossRef CAS.
- X.-H. Gao, P.-Q. Wu, Y.-Y. Fan, B. Zhou and J.-M. Yue, Chin. J. Chem., 2023, 41, 2296–2304 CrossRef CAS.
- J. Chen, S. Kongkiatpaiboon and X.-H. Cai, Phytochemistry, 2024, 222, 114075 CrossRef CAS PubMed.
- J. M. Nuzillard, T. M. Pinchon, C. Caron, G. Massiot and L. Le Men-Olivier, C. R. Acad. Sci., 1989, 309, 195–198 CAS.
- V. M. Malikov, M. R. Sharipov and S. Y. Yunusov, Chem. Nat. Compd., 1972, 8, 741–742 CrossRef.
- G. Massiot, J. M. Nuzillard, B. Richard and L. Le Men-Olivier, Tetrahedron Lett., 1990, 31, 2883–2884 CrossRef CAS.
- C. Chen, J.-W. Liu, L.-L. Guo, F. Xiong, X.-Q. Ran, Y.-R. Guo, Y.-G. Yao, X.-J. Hao, R.-C. Luo and Y. Zhang, Phytochemistry, 2022, 203, 113392 CrossRef CAS PubMed.
- Y. Fan, J. Shen, Z. Liu, K. Xia, W. Zhu and P. Fu, Nat. Prod. Rep., 2022, 39, 1305–1324 RSC.
- Y. Yu, S.-M. Zhao, M.-F. Bao and X.-H. Cai, Org. Chem. Front., 2020, 7, 1365–1373 RSC.
- R. Torrenegra, J. A. P. Pedrozo, H. Achenbach and P. Bauereiß, Phytochemistry, 1988, 27, 1843–1848 CrossRef CAS.
- L. Zhang, Z. Hua, Y. Song and C. Feng, Fitoterapia, 2014, 97, 142–147 CrossRef CAS PubMed.
- F.-R. Li, L. Liu, Y.-P. Liu, J.-T. Wang, M.-L. Yang, A. Khan, X.-J. Qin, Y.-D. Wang and G.-G. Cheng, Phytochemistry, 2021, 184, 112673 CrossRef CAS PubMed.
- B.-J. Zhang, C. Liu, M.-F. Bao, X.-H. Zhong, L. Ni, J. Wu and X.-H. Cai, Tetrahedron, 2017, 73, 5821–5826 CrossRef CAS.
- A. B. Nama, T. Paululat, G. R. Ebede, P. H. D. Betote, D. E. Pegnyemb, J. N. Mbing, J. T. Ndongo, H. Ihmels and H. Laatsch, Phytochem. Lett., 2023, 54, 63–69 CrossRef CAS.
- M. Damak, A. Ahond and P. Potier, Bull. Soc. Chim. Fr., 1981, 2, 213–216 Search PubMed.
- E. C. Miranda and B. Gilbert, Experientia, 1969, 25, 575–576 CrossRef CAS PubMed.
- J. Abaul, P. Bourgeois, E. Philogene, M. Damak, A. Ahond, C. Poupat and P. Potier, C. R. Acad. Sci. Ser. 2, 1984, 298, 627–629 CAS.
- M. Lin, B. Yang and D. Q. Yu, Acta Pharm. Sin., 1986, 21, 114–118 CAS.
- C. R. Edwankar, R. V. Edwankar, J. R. Deschamps and J. M. Cook, Angew. Chem., Int. Ed., 2012, 51, 11762–11765 CrossRef CAS PubMed.
- P. A. Keller, N. R. Yepuri, M. J. Kelso, M. Mariani, B. W. Skelton and A. H. White, Tetrahedron, 2008, 64, 7787–7795 CrossRef CAS.
- E. Valencia, A. Madinaveitia, J. Bermejo and A. G. Gonzalez, J. Nat. Prod., 1995, 58, 134–137 CrossRef CAS.
- W. Hüttel and M. Müller, ChemBioChem, 2007, 8, 521–529 CrossRef PubMed.
- A. Pervin and A. Muzaffar, Heterocycles, 1988, 27, 2051–2057 CrossRef.
- J. Qu, L. Fang, X.-D. Ren, Y. Liu, S.-S. Yu, L. Li, X.-Q. Bao, D. Zhang, Y. Li and S.-G. Ma, J. Nat. Prod., 2013, 76, 2203–2209 CrossRef CAS PubMed.
- L.-Z. Lin, G. A. Cordell, C.-Z. Ni and J. Clardy, Tetrahedron Lett., 1989, 30, 1177–1180 CrossRef CAS.
- M.-X. Sun, Y. Cui, Y. Li, W.-Q. Meng, Q.-Q. Xu, J. Zhao, J.-C. Lu and K. Xiao, Phytochemistry, 2019, 162, 232–240 CrossRef CAS PubMed.
- J. C. Quirion, H. P. Husson, C. Kan and I. R. C. Bick, Nat. Prod. Lett., 1993, 2, 41–48 CrossRef CAS.
- S. Laha and R. G. Luthy, Environ. Sci. Technol., 1990, 24, 363–373 CrossRef CAS.
- M. Gill and R. J. Strauch, Z. Naturforsch. C, 1984, 39, 1027–1029 CrossRef CAS PubMed.
- M. Baunach, L. Ding, T. Bruhn, G. Bringmann and C. Hertweck, Angew. Chem., Int. Ed., 2013, 52, 9040–9043 CrossRef CAS PubMed.
- P. Yates, F. N. MacLachlan, I. D. Rae, M. Rosenberger, A. G. Szabo, C. R. Willis, M. P. Cava, M. Behforouz, M. V. Lakshmikantham and W. Zeiger, J. Am. Chem. Soc., 1973, 95, 7842–7850 CrossRef CAS.
- E. F. Rogers, H. R. Snyder and R. F. Fischer, J. Am. Chem. Soc., 1952, 74, 1987–1989 CrossRef CAS.
- A. Adesomoju, M. Lakshmikantham and M. Cava, Heterocycles, 1983, 20, 1511–1517 CrossRef CAS.
- Z.-L. Song, C.-A. Fan and Y.-Q. Tu, Chem. Rev., 2011, 111, 7523–7556 CrossRef CAS PubMed.
- A. A. Adesomoju, V. H. Rawal, M. V. Lakshmikantham and M. P. Cava, J. Org. Chem., 1983, 48, 3015–3017 CrossRef CAS.
- P. D. Rege, Y. Tian and E. J. Corey, Org. Lett., 2006, 8, 3117–3120 CrossRef CAS PubMed.
- K. C. Nicolaou, S. M. Dalby, S. Li, T. Suzuki and D. Y. K. Chen, Angew. Chem., Int. Ed., 2009, 48, 7616–7620 CrossRef CAS PubMed.
- H. Satoh, K.-I. Ojima, H. Ueda and H. Tokuyama, Angew. Chem., Int. Ed., 2016, 55, 15157–15161 CrossRef CAS PubMed.
- K. C. Nicolaou, S. M. Dalby, S. Li, T. Suzuki and D. Y.-K. Chen, Angew. Chem., Int. Ed., 2009, 48, 7616–7620 CrossRef CAS PubMed.
- Y.-L. Du and K. S. Ryan, Curr. Opin. Chem. Biol., 2016, 31, 74–81 CrossRef CAS PubMed.
- Y. Masui, C. Kawabe, K. Mastumoto, K. Abe and T. Miwa, Phytochemistry, 1986, 25, 1470–1471 CrossRef CAS.
- J.-G. Shi, H.-Q. Wang, M. Wang, Y.-C. Yang, W.-Y. Hu and G.-X. Zhou, J. Nat. Prod., 2000, 63, 782–786 CrossRef CAS PubMed.
- H. H. Wasserman, R. W. DeSimone, D. L. Boger and C. M. Baldino, J. Am. Chem. Soc., 1993, 115, 8457–8458 CrossRef CAS.
- B.-B. Shi, J.-S. Lu, J. Wu, M.-F. Bao and X.-H. Cai, Org. Chem. Front., 2021, 8, 2601–2607 RSC.
- D. Grierson, in Organic Reactions, 2004, pp. 85–295 Search PubMed.
- D. F. Dickel, C. L. Holden, R. C. Maxfield, L. E. Paszek and W. I. Taylor, J. Am. Chem. Soc., 1958, 80, 123–125 CrossRef CAS.
- M. Mentel and R. Breinbauer, Curr. Org. Chem., 2007, 11, 159–176 CrossRef CAS.
- J.-W. Liu, Z.-Q. Huo, Q. Zhao, X.-J. Hao, H.-P. He and Y. Zhang, Fitoterapia, 2019, 138, 104347 CrossRef CAS PubMed.
- T. Feng, X.-H. Cai, Y. Li, Y.-Y. Wang, Y.-P. Liu, M.-J. Xie and X.-D. Luo, Org. Lett., 2009, 11, 4834–4837 CrossRef CAS PubMed.
- M. Wu, P. Wu, H. Xie, G. Wu and X. Wei, Planta Med., 2011, 77, 284–286 CrossRef CAS PubMed.
- S. Shao, H. Zhang, C.-M. Yuan, Y. Zhang, M.-M. Cao, H.-Y. Zhang, Y. Feng, X. Ding, Q. Zhou, Q. Zhao, H.-P. He and X.-J. Hao, Phytochemistry, 2015, 116, 367–373 CrossRef CAS PubMed.
- Y. Hirasawa, S. Miyama, T. Hosoya, K. Koyama, A. Rahman, I. Kusumawati, N. C. Zaini and H. Morita, Org. Lett., 2009, 11, 5718–5721 CrossRef CAS PubMed.
- C. F. Alcover, G. Bernadat, F. A. Kabran, P. Le Pogam, K. Leblanc, A. E. Fox Ramos, J.-F. Gallard, E. Mouray, P. Grellier, E. Poupon and M. A. Beniddir, J. Nat. Prod., 2020, 83, 1207–1216 CrossRef CAS PubMed.
- R. Verpoorte, M. Frédérich, C. Delaude, L. Angenot, G. Dive, P. Thépenier, M.-J. Jacquier, M. Zèches-Hanrot, C. Lavaud and J.-M. Nuzillard, Phytochem. Lett., 2010, 3, 100–103 CrossRef CAS.
- H. Irie, K. Ishizuka, S. Kawashima, N. Masaki, K. Osaki, T. Shingu, S. Uyeo, H. Kaneko and S. Naruto, J. Chem. Soc., Chem. Commun., 1972, 871 RSC.
- A. Itoh, T. Tanahashi, N. Nagakura and T. Nishi, Phytochemistry, 2003, 62, 359–369 CrossRef CAS PubMed.
- N.-P. Li, M. Liu, X.-J. Huang, X.-Y. Gong, W. Zhang, M.-J. Cheng, W.-C. Ye and L. Wang, J. Org. Chem., 2018, 83, 5707–5714 CrossRef CAS PubMed.
- W. Zhang, W. Xu, G.-Y. Wang, X.-Y. Gong, N.-P. Li, L. Wang and W.-C. Ye, Org. Lett., 2017, 19, 5194–5197 CrossRef CAS PubMed.
- J. Lin, J. Wu, M.-F. Bao, S. Kongkiatpaiboon and X.-H. Cai, Phytochemistry, 2024, 222, 114077 CrossRef CAS PubMed.
- H. Takayama, Y. Tominaga, M. Kitajima, N. Aimi and S. Sakai, J. Org. Chem., 1994, 59, 4381–4385 CrossRef CAS.
- L. Lin, G. A. Cordell, C. Ni and J. Clardy, J. Org. Chem., 1989, 54, 3199–3202 CrossRef CAS.
- T. A. van Beek, R. Verpoorte and A. B. Svendsen, Tetrahedron Lett., 1984, 25, 2057–2060 CrossRef CAS.
- P. Pachaly, A. Kroll-Horstmann and K. Sin, Die Pharmazie, 2000, 55, 777 CAS.
- T. T. Nguyen, D. H. Nguyen, B. T. Zhao, D. D. Le, D. H. Choi, Y. H. Kim, T. H. Nguyen and M. H. Woo, Bioorg. Chem., 2017, 74, 221–227 CrossRef CAS PubMed.
- Y.-C. Li, J. Yang, X.-R. Zhou, X.-H. Liang and Q.-Y. Fu, Z. Naturforsch. B, 2016, 71, 193–195 CrossRef CAS.
- A.-M. Bui, B. C. Das and P. Potier, Phytochemistry, 1980, 19, 1473–1475 CrossRef CAS.
- T. R. Govindachari, G. Sandhya, S. Chandrasekharan and K. Rajagopalan, J. Chem. Soc., Chem. Commun., 1987, 1137–1138 RSC.
- M. Kitajima, M. Iwai, N. Kogure, R. Kikura-Hanajiri, Y. Goda and H. Takayama, Tetrahedron, 2013, 69, 796–801 CrossRef CAS.
- K. M. Flynn, I.-S. Myeong, T. Pinto and M. Movassaghi, J. Am. Chem. Soc., 2022, 144, 9126–9131 CrossRef CAS PubMed.
- X.-H. Cai, H. Jiang, Y. Li, G.-G. Cheng, Y.-P. Liu, T. Feng and X.-D. Luo, Chin. J. Nat. Med., 2011, 9, 259–263 CAS.
- C. Kan-Fan, G. Massiot, B. C. Das and P. Potier, J. Org. Chem., 1981, 46, 1481–1483 CrossRef CAS.
- T. Gan and J. M. Cook, Tetrahedron Lett., 1996, 37, 5037–5038 CrossRef CAS.
- F. Mayerl and M. Hesse, Helv. Chim. Acta, 1978, 61, 337–351 CrossRef CAS.
- T. Gan and J. M. Cook, J. Org. Chem., 1998, 63, 1478–1483 CrossRef CAS.
- M. Frédérich, M.-C. De Pauw, C. Prosperi, M. Tits, V. Brandt, J. Penelle, M.-P. Hayette, P. DeMol and L. Angenot, J. Nat. Prod., 2001, 64, 12–16 CrossRef PubMed.
- J. Lamotte, L. Dupont, O. Dideberg, K. Kambu and L. Angenot, Tetrahedron Lett., 1979, 20, 4227–4228 CrossRef.
- W. I. Taylor, J. Org. Chem., 1965, 30, 309–310 CrossRef CAS.
- H. Mehri, S. Baassou and M. Plat, J. Nat. Prod., 1991, 54, 372–379 CrossRef CAS.
- T. Kouamé, G. Bernadat, V. Turpin, M. Litaudon, A. T. Okpekon, J.-F. Gallard, K. Leblanc, S. Rharrabti, P. Champy, E. Poupon, M. A. Beniddir and P. Le Pogam, Org. Lett., 2021, 23, 5964–5968 CrossRef PubMed.
- Y. Zhang, Y.-X. Yuan, M. Goto, L.-L. Guo, X.-N. Li, S. L. Morris-Natschke, K.-H. Lee and X.-J. Hao, J. Nat. Prod., 2018, 81, 562–571 CrossRef CAS PubMed.
- Y. Zhang, L. Guo, G. Yang, F. Guo, Y. Di, S. Li, D. Chen and X. Hao, Fitoterapia, 2015, 100, 150–155 CrossRef CAS PubMed.
- A. E. Nugroho, Y. Hirasawa, N. Kawahara, Y. Goda, K. Awang, A. H. A. Hadi and H. Morita, J. Nat. Prod., 2009, 72, 1502–1506 CrossRef CAS PubMed.
- M. Bert, G. Baudouin, F. Tillequin and M. Koch, Heterocycles, 1986, 24, 1567–1570 CrossRef CAS.
- E. Otogo N'Nang, P. Le Pogam, T. Ndong Mba, C. Sima Obiang, E. Mouray, P. Grellier, B. Kumulungui, P. Champy and M. A. Beniddir, J. Nat. Prod., 2021, 84, 1409–1413 CrossRef PubMed.
- W.-F. Ku, S.-J. Tan, Y.-Y. Low, K. Komiyama and T.-S. Kam, Phytochemistry, 2011, 72, 2212–2218 CrossRef CAS PubMed.
- K. Koyama, Y. Hirasawa, K. Zaima, T. C. Hoe, K.-L. Chan and H. Morita, Bioorg. Med. Chem., 2008, 16, 6483–6488 CrossRef CAS PubMed.
- P. Potier and M. Janot, C. R. Acad. Sci. Paris Ser. C, 1973, 276, 1727–1729 CAS.
- X.-H. Zhong, M.-F. Bao, C.-X. Zeng, B.-J. Zhang, J. Wu, Y. Zhang and X.-H. Cai, Phytochem. Lett., 2017, 20, 77–83 CrossRef CAS.
- P. Krishnan, F.-K. Lee, K.-W. Chong, C.-W. Mai, A. Muhamad, S.-H. Lim, Y.-Y. Low, K.-N. Ting and K.-H. Lim, Org. Lett., 2018, 20, 8014–8018 CrossRef CAS PubMed.
- K.-H. Lim, C.-W. Mai, S.-K. Wong, Y.-Y. Low, S.-H. Lim and P. Krishnan, Phytochem. Lett., 2024, 61, 97–103 CrossRef CAS.
- A. P. G. Macabeo, K. Krohn, D. Gehle, R. W. Read, J. J. Brophy, G. A. Cordell, S. G. Franzblau and A. M. Aguinaldo, Phytochemistry, 2005, 66, 1158–1162 CrossRef CAS PubMed.
- N. Kogure, N. Ishii, M. Kitajima, S. Wongseripipatana and H. Takayama, Org. Lett., 2006, 8, 3085–3088 CrossRef CAS PubMed.
- B. V. Milborrow and C. Djerassi, J. Chem. Soc. C, 1969, 417–424 RSC.
- J. Sakamoto, Y. Umeda, K. Rakumitsu, M. Sumimoto and H. Ishikawa, Angew. Chem., Int. Ed., 2020, 59, 13414–13422 CrossRef CAS PubMed.
- J. Chen, Y. Yu, J. Wu, M.-F. Bao, S. Kongkiatpaiboon, J. Schinnerl and X.-H. Cai, Bioorg. Chem., 2021, 116, 105314 CrossRef CAS PubMed.
- B.-J. Zhang, X.-F. Teng, M.-F. Bao, X.-H. Zhong, L. Ni and X.-H. Cai, Phytochemistry, 2015, 120, 46–52 CrossRef CAS PubMed.
- A. E. Nugroho, W. Zhang, Y. Hirasawa, Y. Tang, C. P. Wong, T. Kaneda, A. H. A. Hadi and H. Morita, J. Nat. Prod., 2018, 81, 2600–2604 CrossRef CAS PubMed.
- T. Kanchanapoom, P. Sahakitpichan, N. Chimnoi, C. Petchthong, W. Thamniyom, P. Nangkoed and S. Ruchirawat, Phytochem. Lett., 2021, 41, 83–87 CrossRef CAS.
- A. Mauger, M. Jarret, C. Kouklovsky, E. Poupon, L. Evanno and G. Vincent, Nat. Prod. Rep., 2021, 38, 1852–1886 RSC.
- M. A. Beniddir, M.-T. Martin, M.-E. Tran Huu Dau, P. Grellier, P. Rasoanaivo, F. Guéritte and M. Litaudon, Org. Lett., 2012, 14, 4162–4165 CrossRef CAS PubMed.
- M. A. Beniddir, M.-T. Martin, M.-E. Tran Huu Dau, P. Rasoanaivo, F. Guéritte and M. Litaudon, Tetrahedron Lett., 2013, 54, 2115–2119 CrossRef CAS.
- Y.-L. He, W.-M. Chen and X.-Z. Feng, J. Nat. Prod., 1994, 57, 411–414 CrossRef CAS.
- E. E. Waldner, M. Hesse, W. I. Taylor and H. Schmid, Helv. Chim. Acta, 1967, 50, 1926–1939 CrossRef CAS PubMed.
- S.-H. Lim, S.-J. Tan, Y.-Y. Low and T.-S. Kam, J. Nat. Prod., 2011, 74, 2556–2562 CrossRef CAS PubMed.
- S. Zhao, X. Liao and J. M. Cook, Org. Lett., 2002, 4, 687–690 CrossRef CAS PubMed.
- K. P. Pandey, M. T. Rahman and J. M. Cook, Molecules, 2021, 26, 3459 CrossRef CAS PubMed.
- Y. Hirasawa, R. Yasuda, W. Minami, M. Hirata, A. E. Nugroho, T. Tougan, N. Uchiyama, T. Hakamatsuka, T. Horii and H. Morita, Tetrahedron Lett., 2021, 83, 153423 CrossRef CAS.
- Y.-L. Wang, C.-R. Guo, Y. Mu, Y.-L. Lin, H.-J. Yan, Z.-W. Wang and X.-J. Wang, Tetrahedron Lett., 2019, 60, 151042 CrossRef CAS.
- Y. Hirasawa, Y. Kakizoe, T. Tougan, N. Uchiyama, T. Horii and H. Morita, J. Nat. Med., 2024, 78, 768–773 CrossRef CAS PubMed.
- S. Michel, F. Tillequin and M. Koch, J. Chem. Soc. Chem. Commun., 1987, 229–230 RSC.
- F. Zhang, K. Yang, H. Liu, T. Yang, R. Zhou, X. Zhang, G. Zhan and Z. Guo, Phytochemistry, 2023, 209, 113610 CrossRef CAS PubMed.
- C.-Y. Gan, T. Etoh, M. Hayashi, K. Komiyama and T.-S. Kam, J. Nat. Prod., 2010, 73, 1107–1111 CrossRef CAS PubMed.
- P. Kokkonda, K. R. Brown, T. J. Seguin, S. E. Wheeler, S. Vaddypally, M. J. Zdilla and R. B. Andrade, Angew. Chem., Int. Ed., 2015, 54, 12632–12635 CrossRef CAS PubMed.
- C.-Y. Gan, Y.-Y. Low, W. T. Robinson, K. Komiyama and T.-S. Kam, Phytochemistry, 2010, 71, 1365–1370 CrossRef CAS PubMed.
- A.-M. Bui, B. C. Das, E. Guittet and P. Potier, J. Nat. Prod., 1991, 54, 514–518 CrossRef CAS.
- A. E. Nugroho, Y. Hirasawa, T. Hosoya, K. Awang, A. H. A. Hadi and H. Morita, Tetrahedron Lett., 2010, 51, 2589–2592 CrossRef CAS.
- A. T. Tchinda, O. Jansen, J.-N. Nyemb, M. Tits, G. Dive, L. Angenot and M. Frédérich, J. Nat. Prod., 2014, 77, 1078–1082 CrossRef CAS PubMed.
- Z.-W. Li, C.-L. Fan, B. Sun, L. Huang, Z.-Q. Wang, X.-J. Huang, S.-Q. Zhang, W.-C. Ye, Z.-L. Wu and X.-Q. Zhang, Chem.–Eur. J., 2024, 30, e202303519 CrossRef CAS PubMed.
- J. M. Cook and P. W. Le Quesne, J. Org. Chem., 1971, 36, 582–586 CrossRef CAS.
- D. E. Burke, J. M. Cook and P. W. Le Quesne, J. Chem. Soc. Chem. Commun., 1972, 697 RSC.
- D. E. Burke, J. M. Cook and P. W. Le Quesne, J. Am. Chem. Soc., 1973, 95, 546–552 CrossRef CAS.
- E. Seguin and M. Koch, Planta Med., 1979, 37, 175–177 CrossRef CAS.
- N. Nagakura, G. Höfle, D. Coggiola and M. H. Zenk, Planta Med., 1978, 34, 381–389 CrossRef CAS.
- J. T. Ndongo, J. N. Mbing, M. F. Tala, A. Monteillier, D. E. Pegnyemb, M. Cuendet and H. Laatsch, Phytochemistry, 2017, 144, 189–196 CrossRef CAS PubMed.
- A. A. Gorman and H. Schmid, Monatshefte für Chemie und verwandte Teile anderer Wissenschaften, 1967, 98, pp. 1554–1566 Search PubMed.
- M.-M. Janot, Tetrahedron, 1961, 14, 113–125 CrossRef CAS.
- H. Rapoport and R. E. Moore, J. Org. Chem., 1962, 27, 2981–2985 CrossRef CAS.
- H. M. de Sousa, A. B. da Silva, M. K. A. Ferreira, A. W. da Silva, J. E. S. A. de Menezes, E. S. Marinho, M. M. Marinho, H. S. dos Santos and O. D. L. Pessoa, Planta Med., 2023, 89, 979–989 CrossRef CAS PubMed.
- M. J. G. Tits and L. Angenot, Planta Med., 1978, 34, 57–61 CrossRef CAS.
- R. Verpoorte and A. B. Svendsen, J. Pharm. Sci., 1978, 67, 171–174 CrossRef CAS PubMed.
- F. C. Ohiri, R. Verpoorte and A. B. Svendsen, J. Ethnopharmacol., 1983, 9, 167–223 CrossRef CAS PubMed.
- S.-I. Sakai, N. Aimi, K. Yamaguchi, E. Yamanaka and J. Haginiwa, J. Chem. Soc., Perkin Trans. 1, 1982, 1257–1262 RSC.
- S.-I. Sakai, N. Aimi, K. Yamaguchi, H. Ohhira, K. Hori and J. Haginiwa, Tetrahedron Lett., 1975, 16, 715–718 CrossRef.
- N. Aimi, S.-I. Sakai and Y. Ban, in The Alkaloids: Chemistry and Pharmacology, ed. A. Brossi, Academic Press, 1990, vol. 36, pp. 1–68 Search PubMed.
- N. Aimi, S. Sakai, Y. Iitaka and A. Itai, Tetrahedron Lett., 1971, 12, 2061–2064 CrossRef.
- G. Massiot, B. Massoussa, M.-J. Jacquier, P. Thépénier, L. Le Men-Olivier, C. Delaude and R. Verpoorte, Phytochemistry, 1988, 27, 3293–3304 CrossRef CAS.
- G. Massiot, M. Zeches, C. Mirand, L. Le Men-Olivier, C. Delaude, K. H. C. Baser, R. Bavovada, N. G. Bisset and P. J. Hylands, J. Org. Chem., 1983, 48, 1869–1872 CrossRef CAS.
- A. R. Battersby, Pure Appl. Chem., 1963, 6, 471–482 CAS.
- N.-P. Li, J.-S. Liu, J.-W. Liu, H.-Y. Tian, H.-L. Zhou, Y.-R. Zheng, X.-J. Huang, J.-Q. Cao, W.-C. Ye and L. Wang, Bioorg. Chem., 2021, 107, 104624 CrossRef CAS PubMed.
- N. Kunesch, Y. Rolland, J. Poisson, P. L. Majumder, R. Majumder, A. Chatterjee, V. C. Agwada, J. Naranjo, M. Hesse and H. Schmid, Helv. Chim. Acta, 1977, 60, 2854–2859 CrossRef CAS.
- B. Danieli, G. Lesma, G. Palmisano, S. Tollari and B. Gabetta, J. Org. Chem., 1983, 48, 381–383 CrossRef CAS.
- E. Wenkert and B. Wickberg, J. Am. Chem. Soc., 1965, 87, 1580–1589 CrossRef CAS PubMed.
- G. Hugel, G. Massiot, J. Lévy and J. Le Men, Tetrahedron, 1981, 37, 1369–1375 CrossRef CAS.
- B. Danieli, G. Lesma, G. Palmisano and B. Gabetta, J. Chem. Soc. Chem. Commun., 1981, 908–909 RSC.
- L. Calabi, B. Danieli, G. Lesma and G. Palmisano, J. Chem. Soc., Perkin Trans. 1, 1982, 1371–1379 RSC.
- F. Kellner, F. Geu-Flores, N. H. Sherden, S. Brown, E. Foureau, V. Courdavault and S. E. O'Connor, Chem. Commun., 2015, 51, 7626–7628 RSC.
- A. T. McPhail, E. W. Hagaman, N. Kunesch, E. Wenkert and J. Poisson, Tetrahedron, 1983, 39, 3629–3637 CrossRef CAS.
- J. Lévy, P. Maupérin, M. D. de Maindreville and J. Le Men, Tetrahedron Lett., 1971, 12, 1003–1006 CrossRef.
- B. J. Rawlings, Nat. Prod. Rep., 1997, 14, 523–556 RSC.
- M.-X. Sun, H.-H. Gao, J. Zhao, L. Zhang and K. Xiao, Tetrahedron Lett., 2015, 56, 6194–6197 CrossRef CAS.
- X.-H. Cai, Z.-Z. Du and X.-D. Luo, Org. Lett., 2007, 9, 1817–1820 CrossRef CAS PubMed.
- J. M. Wood, D. P. Furkert and M. A. Brimble, Nat. Prod. Rep., 2019, 36, 289–306 RSC.
- T.-S. Kam and Y.-M. Choo, J. Nat. Prod., 2004, 67, 547–552 CrossRef CAS PubMed.
- T.-S. Kam and Y.-M. Choo, Tetrahedron, 2000, 56, 6143–6150 CrossRef CAS.
- T. Kishi, M. Hesse, C. W. Gemenden, W. I. Taylor and H. Schmid, Helv. Chim. Acta, 1965, 48, 1349–1362 CrossRef CAS PubMed.
- S.-J. Tan, Y.-M. Choo, N. F. Thomas, W. T. Robinson, K. Komiyama and T.-S. Kam, Tetrahedron, 2010, 66, 7799–7806 CrossRef CAS.
- S.-J. Tan, W. T. Robinson, K. Komiyama and T.-S. Kam, Tetrahedron, 2011, 67, 3830–3838 CrossRef CAS.
- T.-S. Kam and Y.-M. Choo, Tetrahedron Lett., 2003, 44, 8787–8789 CrossRef CAS.
- T.-S. Kam, Y.-M. Choo and K. Komiyama, Tetrahedron, 2004, 60, 3957–3966 CrossRef CAS.
- Y. Hirasawa, H. Arai, A. Rahman, I. Kusumawati, N. C. Zaini, O. Shirota and H. Morita, Tetrahedron, 2013, 69, 10869–10875 CrossRef CAS.
- X.-H. Gao, Y.-Y. Fan, Q.-F. Liu, S.-H. Cho, G. F. Pauli, S.-N. Chen and J.-M. Yue, Org. Lett., 2019, 21, 7065–7068 CrossRef CAS.
- J. J. Kezetas Bankeu, D. U. Kenou Kagho, Y. S. Fotsing Fongang, R. M. Kouipou Toghueo, B. M. Mba’ning, G. R. Tchouya Feuya, F. Boyom Fekam, J. C. Tchouankeu, S. A. Ngouela, N. Sewald, B. N. Lenta and M. S. Ali, J. Nat. Prod., 2019, 82, 2580–2585 CrossRef CAS.
- K. Jewers and J. McKenna, J. Chem. Soc., 1958, 2209–2217 RSC.
- X.-W. Yang, X.-J. Qin, Y.-L. Zhao, P. K. Lunga, X.-N. Li, S.-Z. Jiang, G.-G. Cheng, Y.-P. Liu and X.-D. Luo, Tetrahedron Lett., 2014, 55, 4593–4596 CrossRef CAS.
- B.-Y. Hu, Y.-L. Zhao, Y.-J. He, Y. Qin and X.-D. Luo, Phytochemistry, 2024, 217, 113926 CrossRef CAS PubMed.
- G.-Y. Zhu, X.-J. Yao, L. Liu, L.-P. Bai and Z.-H. Jiang, Org. Lett., 2014, 16, 1080–1083 CrossRef CAS PubMed.
- H.-F. Yu, C.-F. Ding, L.-C. Zhang, X. Wei, G.-G. Cheng, Y.-P. Liu, R.-P. Zhang and X.-D. Luo, Org. Lett., 2021, 23, 5782–5786 CrossRef CAS.
- J.-H. Gu, W. Zhang, W.-Y. Cai, X.-X. Fu, H.-L. Zhou, N.-P. Li, H.-Y. Tian, J.-S. Liu, W.-C. Ye and L. Wang, Org. Chem. Front., 2021, 8, 1918–1925 RSC.
- N.-P. Li, M. Liu, X.-J. Huang, X.-Y. Gong, W. Zhang, M.-J. Cheng, W.-C. Ye and L. Wang, J. Org. Chem., 2018, 83, 5707–5714 CrossRef CAS PubMed.
- X.-H. Cai, Y. Li, Y.-P. Liu, X.-N. Li, M.-F. Bao and X.-D. Luo, Phytochemistry, 2012, 83, 116–124 CrossRef CAS PubMed.
- Z.-W. Wang, J.-P. Zhang, Q.-H. Wei, L. Chen, Y.-L. Lin, Y.-L. Wang, T. An and X.-J. Wang, Tetrahedron Lett., 2021, 87, 153525 CrossRef CAS.
- I. M. Novitskiy and A. G. Kutateladze, J. Org. Chem., 2022, 87, 4818–4828 CrossRef CAS PubMed.
- A. Henriques, C. Kan, A. Chiaroni, C. Riche, H. P. Husson, S. K. Kan and M. Lounasmaa, J. Org. Chem., 1982, 47, 803–811 CrossRef CAS.
- G. Costa, C. Riche and H. P. Husson, Tetrahedron, 1977, 33, 315–320 CrossRef CAS.
- L. Duhamel, J. M. Poirier and P. Granger, J. Org. Chem., 1979, 44, 3576–3578 CrossRef CAS.
- T. A. v. Beek, P. P. Lankhorst, R. Verpoorte, G. Massiot, R. Fokkens, C. Erkelens, P. Perera and C. Tibell, Z. Naturforsch. B, 1985, 40, 693–701 CrossRef.
- Y.-S. Cai, A. M. Sarotti, T.-L. Zhou, R. Huang, G. Qiu, C. Tian, Z.-H. Miao, A. Mándi, T. Kurtán, S. Cao and S.-P. Yang, J. Nat. Prod., 2018, 81, 1976–1983 CrossRef CAS PubMed.
- It can also be envisaged that an heteronucleophilic attack initiated by the vobasane N-1 position onto the C-16 position of the Δ16,22 function of the vallesamane component could be the driving force for its 1,6-conjugate addition onto the C-3 site of the vobasane component.
- M.-F. Bao, C.-X. Zeng, Y.-P. Liu, B.-J. Zhang, L. Ni, X.-D. Luo and X.-H. Cai, J. Nat. Prod., 2017, 80, 790–797 CrossRef CAS PubMed.
- D. Lachkar, N. Denizot, G. Bernadat, K. Ahamada, M. A. Beniddir, V. Dumontet, J.-F. Gallard, R. Guillot, K. Leblanc, E. O. N'Nang, V. Turpin, C. Kouklovsky, E. Poupon, L. Evanno and G. Vincent, Nat. Chem., 2017, 9, 793–798 CrossRef CAS PubMed.
- T.-S. Kam, S.-J. Tan, S.-W. Ng and K. Komiyama, Org. Lett., 2008, 10, 3749–3752 CrossRef CAS PubMed.
- K. Okada, K.-i. Ojima, H. Ueda and H. Tokuyama, J. Am. Chem. Soc., 2023, 145, 16337–16343 CrossRef CAS PubMed.
- R. E. Ziegler, S.-J. Tan, T.-S. Kam and J. A. Porco Jr, Angew. Chem., Int. Ed., 2012, 51, 9348–9351 CrossRef CAS PubMed.
- S. Uzir, A. M. Mustapha, A. H. A. Hadi, K. Awang, C. Wiart, J.-F. Gallard and M. Païs, Tetrahedron Lett., 1997, 38, 1571–1574 CrossRef CAS.
- S. Markey, K. Biemann and B. Witkop, Tetrahedron Lett., 1967, 8, 157–160 CrossRef PubMed.
- S. Szwarc, A. Jagora, S. Derbré, K. Leblanc, S. Rharrabti, C. Said-Hassane, C. El Kalamouni, J.-F. Gallard, P. Le Pogam and M. A. Beniddir, Org. Lett., 2024, 26, 274–279 CrossRef CAS PubMed.
- B. C. Das, J. P. Cosson and G. Lukacs, J. Org. Chem., 1977, 42, 2785–2786 CrossRef CAS.
- B. C. Das, J. P. Cosson, G. Lukacs and P. Potier, Tetrahedron Lett., 1974, 15, 4299–4302 CrossRef.
- N. Kunesch, A. Cavé, E. W. Hagaman and E. Wenkert, Tetrahedron Lett., 1980, 21, 1727–1730 CrossRef CAS.
- M. Millot, A. Dieu and S. Tomasi, Nat. Prod. Rep., 2016, 33, 801–811 RSC.
- G. A. Cordell, G. F. Smith and G. N. Smith, J. Chem. Soc. D, 1970, 191–192 RSC.
- M. Kitajima, M. Nakazawa, Y. Wu, N. Kogure, R.-P. Zhang and H. Takayama, Tetrahedron, 2016, 72, 6692–6696 CrossRef CAS.
- Y. Wu, M. Kitajima, N. Kogure, R. Zhang and H. Takayama, Tetrahedron Lett., 2008, 49, 5935–5938 CrossRef CAS.
- Y. Chang, C. Sun, C. Wang, X. Huo, W. Zhao and X. Ma, Nat. Prod. Rep., 2022, 39, 2030–2056 RSC.
- M. Kobayashi, K. Kawazoe, T. Katori and I. Kitagawa, Chem. Pharm. Bull., 1992, 40, 1773–1778 CrossRef CAS.
- Y. Inouye, Y. Sugo, T. Kusumi and N. Fusetani, Chem. Lett., 1994, 23, 419–420 CrossRef.
- F. Tillequin, M. Koch, M. Bert and T. Sevenet, J. Nat. Prod., 1979, 42, 92–95 CrossRef CAS PubMed.
- F. Tillequin, M. Koch, J.-L. Pousset and A. Cave, J. Chem. Soc. Chem. Commun., 1978, 826–828 RSC.
- L. S. Fernandez, M. S. Buchanan, A. R. Carroll, Y. J. Feng, R. J. Quinn and V. M. Avery, Org. Lett., 2009, 11, 329–332 CrossRef CAS PubMed.
- G. Philippe, E. Prost, J.-M. Nuzillard, M. Zèches-Hanrot, M. Tits, L. Angenot and M. Frédérich, Tetrahedron Lett., 2002, 43, 3387–3390 CrossRef CAS.
- H. Fouotsa, P. Le Pogam, P. Mkounga, A. M. Lannang, G. Bernadat, J. Vanheuverzwijn, Z. Zhou, K. Leblanc, S. Rharrabti, A. E. Nkengfack, J. F. Gallard, V. Fontaine, F. Meyer, E. Poupon and M. A. Beniddir, J. Nat. Prod., 2021, 84, 2755–2761 CrossRef CAS PubMed.
- V. C. Agwada, J. Naranjo, M. Hesse, H. Schmid, Y. Rolland, N. Kunesch, J. Poisson and A. Chatterjee, Helv. Chim. Acta, 1977, 60, 2830–2853 CrossRef CAS.
- M. Zeches, G. Lukacs, G. M. L. Le Men-Olivier and M.-M. Debray, J. Nat. Prod., 1982, 45, 707–713 CrossRef CAS.
- N. G. Bisset, Acta Amazonica, 1988, 18, 255–290 CrossRef.
- G. Massiot, B. Massoussa, P. Thepenier, M.-J. Jacquier and L. Le Men-Olivier, Heterocycles, 1983, 20, 2339–2342 CrossRef CAS.
- R. Mukherjee, B. C. Das, P. A. Keifer and J. N. Shoolery, Heterocycles, 1994, 38, 1965–1970 CrossRef CAS.
- A. R. Battersby, H. F. Hodson, G. V. Rao and D. A. Yeowell, J. Chem. Soc. C, 1967, 2335–2339 RSC.
- K. Bernauer, H. Schmid and P. Karrer, Helv. Chim. Acta, 1957, 40, 1999–2003 CrossRef CAS.
- K. Bernauer, F. Berlage, H. Schmid and P. Karrer, Helv. Chim. Acta, 1958, 41, 1202–1206 CrossRef CAS.
- A. Schönberg, in Preparative Organic Photochemistry, ed. A. Schönberg, Springer Berlin Heidelberg, Berlin, Heidelberg, 1968, pp. 373–406 Search PubMed.
- S. P. Singh, V. I. Stenberg and S. S. Parmar, Chem. Rev., 1980, 80, 269–282 CrossRef CAS.
- A.-M. Bui, B. C. Das, E. Guittet, J.-Y. Lallemand and P. Potier, J. Nat. Prod., 1986, 49, 321–325 CrossRef CAS.
- G. Massiot, P. Thépenier, M.-J. Jacquier, L. Le Men-Olivier, R. Verpoorte and C. Delaude, Phytochemistry, 1987, 26, 2839–2846 CrossRef CAS.
- L. Merlini, R. Mondelli, G. Nasini and M. Hesse, Tetrahedron, 1970, 26, 2259–2279 CrossRef CAS PubMed.
- E. Winterfeldt, Chem. Ber., 1964, 97, 2463–2468 CrossRef CAS.
- H. Riesner and E. Winterfeldt, J. Chem. Soc. Chem. Commun., 1972, 786–787 RSC.
- A. R. Battersby and J. C. Turner, J. Chem. Soc., 1960, 717–725 RSC.
- E. E. Van Tamelen, P. E. Aldrich and J. B. Hester, Jr., J. Am. Chem. Soc., 1959, 81, 6214–6221 CrossRef CAS.
- B. R. Lichman, Nat. Prod. Rep., 2021, 38, 103–129 RSC.
- L. Caputi, J. Franke, S. C. Farrow, K. Chung, R. M. E. Payne, T.-D. Nguyen, T.-T. T. Dang, I. Soares Teto Carqueijeiro, K. Koudounas, T. Dugé de Bernonville, B. Ameyaw, D. M. Jones, I. J. C. Vieira, V. Courdavault and S. E. O'Connor, Science, 2018, 360, 1235–1239 CrossRef CAS PubMed.
- P. L. Schiff, in Alkaloids: Chemical and Biological Perspectives, ed. S. W. Pelletier, Pergamon, 1999, vol. 14, pp. 1–284 Search PubMed.
- C. Weber and T. Opatz, in The Alkaloids: Chemistry and Biology, ed. H.-J. Knölker, Academic Press, 2019, vol. 81, pp. 1–114 Search PubMed.
- M. Shamma and J. Moniot, Heterocycles, 1975, 3, 297–300 CrossRef CAS.
- I. R. C. Bick and S. Sotheeswaran, Aust. J. Chem., 1978, 31, 2077–2083 CrossRef CAS.
- J.-Z. Wang, Q.-H. Chen and F.-P. Wang, J. Nat. Prod., 2010, 73, 1288–1293 CrossRef CAS PubMed.
- C. D. Critchett, H. R. W. Dharmaratne, S. Sotheeswaran, A. M. Galal, P. L. Schiff and I. R. C. Bick, Aust. J. Chem., 1989, 42, 2043–2046 CrossRef CAS.
- J. Harley-Mason, A. S. Howard, W. I. Taylor, M. J. Vernengo, I. R. C. Bick and P. S. Clezy, J. Chem. Soc. C, 1967, 1948–1951 RSC.
- C. Jih-Jung, T. Ian-Lih, T. Ishikawa, W. Chyl-Jia and C. Ih-Sheng, Phytochemistry, 1996, 42, 1479–1484 CrossRef.
- J.-J. Chen, T. Ishikawa, C.-Y. Duh, I.-L. Tsai and I.-S. Chen, Planta Med., 1996, 62, 528–533 CrossRef CAS PubMed.
- M. L. R. Ferreira, I. C. de Pascoli, I. R. Nascimento, J. Zukerman-Schpector and L. M. X. Lopes, Phytochemistry, 2010, 71, 469–478 CrossRef CAS PubMed.
- R. S. Istatkova and S. A. Philipov, Phytochemistry, 2000, 54, 959–964 CrossRef CAS PubMed.
- K. W. Bentley, Nat. Prod. Rep., 2002, 19, 332–356 RSC.
- D. Cortes, J. Saez, R. Hocquemiller, A. Cavé and A. Cavé, Heterocycles, 1986, 24, 607–610 CrossRef CAS.
- N. Nagakura, G. Höfle and M. H. Zenk, J. Chem. Soc. Chem. Commun., 1978, 896–898 RSC.
- Y.-S. Cai, C. Wang, C. Tian, W.-T. Sun, L. Chen, D. Xiao, S.-Y. Zhou, G. Qiu, J. Yu, K. Zhu and S.-P. Yang, J. Nat. Prod., 2019, 82, 2645–2652 CrossRef CAS PubMed.
- A. Itoh, T. Tanahashi and N. Nagakura, Phytochemistry, 1991, 30, 3117–3123 CrossRef CAS.
- S. C. Pakrashi, B. Achari, E. Ali, P. P. Ghosh Dastidar and R. R. Sinha, Tetrahedron Lett., 1980, 21, 2667–2670 CrossRef CAS.
- A. Bhattacharjya, R. Mukhopadhyay and S. C. Pakrashi, Tetrahedron Lett., 1986, 27, 1215–1216 CrossRef CAS.
- P. Brauchli, V. Deulofeu, H. Budzikiewicz and C. Djerassi, J. Am. Chem. Soc., 1964, 86, 1895–1896 CrossRef CAS.
- A. Itoh, T. Tanahashi and N. Nagakura, J. Nat. Prod., 1995, 58, 1228–1239 CrossRef CAS.
- N. Nagakura, A. Itoh and T. Tanahashi, Phytochemistry, 1993, 32, 761–765 CrossRef CAS.
- C. Djerassi, A. A. P. G. Archer, T. George, B. Gilbert, J. N. Shoolery and L. F. Johnson, Experientia, 1960, 16, 532–534 CrossRef CAS PubMed.
- P. Teerapongpisan, V. Suthiphasilp, P. Kumboonma, T. Maneerat, T. Duangyod, R. Charoensup, P. Promnart and S. Laphookhieo, Phytochemistry, 2024, 220, 114020 CrossRef CAS PubMed.
- Y.-W. Ge, S. Zhu, M.-Y. Shang, X.-Y. Zang, X. Wang, Y.-J. Bai, L. Li, K. Komatsu and S.-Q. Cai, Phytochemistry, 2013, 86, 201–207 CrossRef CAS PubMed.
- Y.-G. Guo, Y.-H. Ding, G.-J. Wu, S.-L. Zhu, Y.-F. Sun, S.-K. Yan, F. Qian, H.-Z. Jin and W.-D. Zhang, Fitoterapia, 2018, 127, 96–100 CrossRef CAS PubMed.
- G.-M. Yang, J. Sun, Y. Pan, J.-L. Zhang, M. Xiao and M.-S. Zhu, Fitoterapia, 2018, 124, 58–65 CrossRef CAS PubMed.
- W. J. Griffin and G. D. Lin, Phytochemistry, 2000, 53, 623–637 CrossRef CAS PubMed.
- O. Muñoz, R. Hartmann and E. Breitmaier, J. Nat. Prod., 1991, 54, 1094–1096 CrossRef.
- A. San-Martín, C. Labbé, O. Muñoz, M. Castillo, M. Reina, G. De La Fuente and A. González, Phytochemistry, 1987, 26, 819–822 CrossRef.
- H. W. Voigtländer and W. Rosenberg, Arch. Pharmazie, 1959, 292, 632–642 CrossRef.
- S. Cretton, T. A. Bartholomeusz, M. Humam, L. Marcourt, Y. Allenbach, D. Jeannerat, O. Muñoz and P. Christen, J. Nat. Prod., 2011, 74, 2388–2394 CrossRef CAS PubMed.
- M. Lounasmaa and T. Tamminen, in The Alkaloids: Chemistry and Pharmacology, ed. G. A. Cordell, Academic Press, 1993, vol. 44, pp. 1–114 Search PubMed.
- R. Hartmann, A. San-Martin, O. Muñoz and E. Breitmaier, Angew. Chem., Int. Ed., 1990, 29, 385–386 CrossRef.
- Z.-Y. Chan, P. Krishnan, S. M. Modaresi, L.-W. Hii, C.-W. Mai, W.-M. Lim, C.-O. Leong, Y.-Y. Low, S.-K. Wong, K.-T. Yong, A. Z.-X. Leong, M.-K. Lee, K.-N. Ting and K.-H. Lim, J. Nat. Prod., 2021, 84, 2272–2281 CrossRef CAS PubMed.
- S. F. Aripova, E. G. Sharova and S. Y. Yunusov, Chem. Nat. Compd., 1982, 18, 606–607 CrossRef.
- S. A. Snyder, A. Gollner and M. I. Chiriac, Nature, 2011, 474, 461–466 CrossRef CAS PubMed.
- A. E. Fox Ramos, P. Le Pogam, C. Fox Alcover, E. Otogo N'Nang, G. Cauchie, H. Hazni, K. Awang, D. Bréard, A. M. Echavarren, M. Frédérich, T. Gaslonde, M. Girardot, R. Grougnet, M. S. Kirillova, M. Kritsanida, C. Lémus, A.-M. Le Ray, G. Lewin, M. Litaudon, L. Mambu, S. Michel, F. M. Miloserdov, M. E. Muratore, P. Richomme-Peniguel, F. Roussi, L. Evanno, E. Poupon, P. Champy and M. A. Beniddir, Sci. Data, 2019, 6, 15 CrossRef PubMed.
- M. Wang, J. J. Carver, V. V. Phelan, L. M. Sanchez, N. Garg, Y. Peng, D. D. Nguyen, J. Watrous, C. A. Kapono, T. Luzzatto-Knaan, C. Porto, A. Bouslimani, A. V. Melnik, M. J. Meehan, W.-T. Liu, M. Crusemann, P. D. Boudreau, E. Esquenazi, M. Sandoval-Calderon, R. D. Kersten, L. A. Pace, R. A. Quinn, K. R. Duncan, C.-C. Hsu, D. J. Floros, R. G. Gavilan, K. Kleigrewe, T. Northen, R. J. Dutton, D. Parrot, E. E. Carlson, B. Aigle, C. F. Michelsen, L. Jelsbak, C. Sohlenkamp, P. Pevzner, A. Edlund, J. McLean, J. Piel, B. T. Murphy, L. Gerwick, C.-C. Liaw, Y.-L. Yang, H.-U. Humpf, M. Maansson, R. A. Keyzers, A. C. Sims, A. R. Johnson, A. M. Sidebottom, B. E. Sedio, A. Klitgaard, C. B. Larson, C. A. Boya P, D. Torres-Mendoza, D. J. Gonzalez, D. B. Silva, L. M. Marques, D. P. Demarque, E. Pociute, E. C. O'Neill, E. Briand, E. J. N. Helfrich, E. A. Granatosky, E. Glukhov, F. Ryffel, H. Houson, H. Mohimani, J. J. Kharbush, Y. Zeng, J. A. Vorholt, K. L. Kurita, P. Charusanti, K. L. McPhail, K. F. Nielsen, L. Vuong, M. Elfeki, M. F. Traxler, N. Engene, N. Koyama, O. B. Vining, R. Baric, R. R. Silva, S. J. Mascuch, S. Tomasi, S. Jenkins, V. Macherla, T. Hoffman, V. Agarwal, P. G. Williams, J. Dai, R. Neupane, J. Gurr, A. M. C. Rodriguez, A. Lamsa, C. Zhang, K. Dorrestein, B. M. Duggan, J. Almaliti, P.-M. Allard, P. Phapale, L.-F. Nothias, T. Alexandrov, M. Litaudon, J.-L. Wolfender, J. E. Kyle, T. O. Metz, T. Peryea, D.-T. Nguyen, D. VanLeer, P. Shinn, A. Jadhav, R. Muller, K. M. Waters, W. Shi, X. Liu, L. Zhang, R. Knight, P. R. Jensen, B. O. Palsson, K. Pogliano, R. G. Linington, M. Gutierrez, N. P. Lopes, W. H. Gerwick, B. S. Moore, P. C. Dorrestein and N. Bandeira, Nat. Biotechnol., 2016, 34, 828–837 CrossRef CAS PubMed.
Footnotes |
| † We dedicate this manuscript to Nicole Kunesch in recognition of her seminal contribution to the classification of MIA oligomers. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4np00011k |
| This journal is © The Royal Society of Chemistry 2024 |