
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring nature's battlefield: organismic interactions in the discovery of bioactive natural products
Yuyang
Wang
a,
Yan-Ni
Shi
 b,
Hao
Xiang
ac and
Yi-Ming
Shi
*ac
b,
Hao
Xiang
ac and
Yi-Ming
Shi
*ac
aKey Laboratory of Quantitative Synthetic Biology, Center for Synthetic Biochemistry, Shenzhen Institute of Synthetic Biology, Shenzhen Institute of Advanced Technology, Chinese Academy of Sciences, Shenzhen 518055, China. E-mail: ym.shi@siat.ac.cn
bInstitute of Chemical Biology, Shenzhen Bay Laboratory, Shenzhen 518132, China
cUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 24th September 2024
Abstract
Covering: up to March 2024.
Microbial natural products have historically been a cornerstone for the discovery of therapeutic agents. Advanced (meta)genome sequencing technologies have revealed that microbes harbor far greater biosynthetic capabilities than previously anticipated. However, despite the application of CRISPR/Cas-based gene editing and high-throughput technologies to activate silent biosynthetic gene clusters, the rapid identification of new natural products has not led to a proportional increase in the discovery rate of lead compounds or drugs. A crucial issue in this gap may be insufficient knowledge about the inherent biological and physiological functions of microbial natural products. Addressing this gap necessitates recognizing that the generation of functional natural products is deeply rooted in the interactions between the producing microbes and other (micro)organisms within their ecological contexts, an understanding that is essential for harnessing their potential therapeutic benefits. In this review, we highlight the discovery of functional microbial natural products from diverse niches, including those associated with humans, nematodes, insects, fungi, protozoa, plants, and marine animals. Many of these findings result from an organismic-interaction-guided strategy using multi-omic approaches. The current importance of this topic lies in its potential to advance drug discovery in an era marked by increasing antimicrobial resistance.
 Yan-Ni Shi | Yan-Ni Shi earned a PhD in Medicinal Chemistry from the University of Chinese Academy of Sciences. She was a postdoctoral fellow in Prof. Helge Bode's lab in the Department of Biosciences at Goethe University Frankfurt. Simultaneously, she served as a Guest Scientist in the Department of Natural Products in Organismic Interactions at the Max Planck Institute for Terrestrial Microbiology. Currently, she is an assistant professor at the Institute of Chemical Biology, Shenzhen Bay Laboratory. Her research focuses on the use of automated facilities in the discovery of bioactive natural products and the development of methodologies for compound isolation and structural elucidation. |
 Hao Xiang | Hao Xiang earned his Bachelor's degree in Bioinformatics from Chongqing University of Posts and Telecommunications. While pursuing his Master's degree in Chemical Biology, he received the National Graduate Scholarship. Afterwards, he became a PhD student at the Institute of Synthetic Biology, Shenzhen Institute of Advanced Technology, Chinese Academy of Sciences. He specializes in data-driven genome mining for natural product discovery and the application of Retrieval-Augmented Generation techniques using large language models. |
 Yi-Ming Shi | Yi-Ming Shi obtained a PhD in Medicinal Chemistry from the University of Chinese Academy of Sciences under the supervision of Prof. Han-Dong Sun. After completing postdoctoral training in Chemical Biology and Chemical Ecology supported by the Alexander von Humboldt Foundation in the Bode lab at Goethe University Frankfurt, he became a Project Group Leader at the Max Planck Institute for Terrestrial Microbiology. Within the Excellent Overseas Young Scientists Fund Program, he is a professor at the Institute of Synthetic Biology, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences. His research is centered on drug-lead discovery by synergizing bioinformatics, synthetic biology, and natural product chemistry, driven by microbe–microbe and microbe-higher eukaryote interactions. |
1 Introduction
Microbial natural products have been constituting a prolific source for medicinal therapies, beginning with the discovery of penicillin, which initiated the Golden Age of Antibiotics. This period, spanning from the 1940s to the 1960s, was marked by the discovery and development of numerous antibiotics, such as streptomycin, tetracycline, and vancomycin, many of which remain frontline treatments in clinical use today.1 These bioactive compounds were mostly discovered through screening extracts of actinomycetes, which are readily isolated from soil.2 However, sustained implementations of such a bioactivity-guided strategy have led to a significant decrease in the hit rates of drug leads, a trend further complicated by the spread of drug-resistant strains and the emergence of highly pathogenic infectious diseases.3Advances in sequencing technologies and assembly algorithms have revealed that the number of biosynthetic gene clusters (BGCs) responsible for synthesizing microbial natural products far exceeds the number of compounds identified to date.4 This discrepancy presents an unparalleled opportunity to explore the biosynthetic potential of microorganisms for new drug candidates in the post-genomic era. Nevertheless, to avoid being overwhelmed by the deluge of BGC data and to harness such big data resources, it is imperative to prioritize the most promising BGCs via multi-omics technologies and a multidisciplinary strategy. The challenge of identifying new entities from vast data by integrating bioinformatic prediction with experimental translation likely stems from a lack of understanding of the ecological functions of microbial natural products.
In response to organismic interactions in ecological environments, microorganisms produce compounds with a variety of physiological and biological functions,5–8 including metal chelators for nutrient uptake, antioxidants for protection against oxidative damage, signaling molecules to coordinate microbial community behaviors, and antimicrobials for defense against other competing microbes. Furthermore, symbiotic and commensal microorganisms produce growth factors to support their hosts, along with immunosuppressants and cytotoxins to assist hosts in killing other higher eukaryotes.7,8 These microbial natural products confer adaptive advantages to the producers in environments with intense organismic competition and limited food and nutrient availability. In return, ecology serves as a driving force for microorganisms to select practical BGCs across taxonomic lineages.9 These intricate relationships also drive microorganisms to evolve and select resistance genes to counteract the effects of toxic compounds produced by the microorganisms themselves.10 Therefore, identifying self-resistance genes within BGCs could help identify the putative molecular targets of a natural product(s) and prioritize BGCs for downstream synthetic biology efforts, such as gene cluster refactoring and heterologous expression.
Here, we highlight the potential of organismic interaction-guided strategies in discovering new bioactive microbial natural products for drug-lead discovery. While traditional microbiology methods involving microorganism isolation and cultivation were employed and remain essential and indispensable, understanding the ecological context and interactions in natural habitats was critical in guiding and promoting these discoveries. By leveraging the extensive biosynthetic capabilities of microbes in microbe–microbe and microbe–eukaryote relationships, organismic interaction-guided strategies can effectively contribute to combating the growing issue of antimicrobial resistance and the urgent need for new drugs.
2 Human–microbe interactions: bioactive compounds from an invisible organ of the human body
The human microbiota, communities of microbes living inside and on our bodies, are primarily inherited from the mother and subsequently shaped by environmental influences. Although our microbiota only weigh 500 g, the ratio of the microbial cells to human cells is approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1,11 and the gene content of the microbiota is 150-fold higher than that of the human chromosomes.12 The human microbiota functioning collectively as one of the most critical organs are instrumental in the processes of digestion, nutrient synthesis, immune regulation, protection of gastrointestinal and vagina against pathogens, drug metabolism, and mental health modulation.13 The microbiota encoding thousands of pathways shared across diverse strains,14 produce small molecules from dietary and host-derived substrates, as well as through de novo synthesis.15
:1,11 and the gene content of the microbiota is 150-fold higher than that of the human chromosomes.12 The human microbiota functioning collectively as one of the most critical organs are instrumental in the processes of digestion, nutrient synthesis, immune regulation, protection of gastrointestinal and vagina against pathogens, drug metabolism, and mental health modulation.13 The microbiota encoding thousands of pathways shared across diverse strains,14 produce small molecules from dietary and host-derived substrates, as well as through de novo synthesis.15
Many of the above biological and physiological processes are either governed by or associated with small molecules.16 Linking and elucidating their biosynthetic pathways in the microbiota, particularly via big data analysis,17–20 allows us to prioritize BGCs for structural identification and functional characterization of bioactive compounds. These compounds might be promising in targeting and modulating humans' biological pathways and protecting the human host.
2.1 Lugdunin and epifadin: guardians of the human nasal cavity
The human microbiota regulate colonization by competitors using antimicrobials, thereby aiding colonized microbes to block invasion. The human nasal cavity is notably deficient in nutrients, indicating that resident bacteria likely employ efficient strategies for mutual competition.21 Two staphylococci isolates, Staphylococcus lugdunensis IVK28 and Staphylococcus epidermidis IVK83 from the nose, produce the broad-spectrum antibiotics lugdunin (1)22 and epifadin (2),23 respectively (Fig. 1). Compounds produced in a species-specific manner have the potential to modulate the local microbial community dynamics. Therefore, understanding the ecological roles of these bioactive compounds in their natural environments is necessary for assessing their potential therapeutic applications. Lugdunin (1) and epifadin (2) were tested against the nasal opportunistic pathogen Staphylococcus aureus to evaluate their effectiveness in reducing colonization of this common pathogen, as S. aureus is carried by approximately 1/3 of the human population and can cause illnesses ranging from minor skin infections to life-threatening systemic infectious diseases.24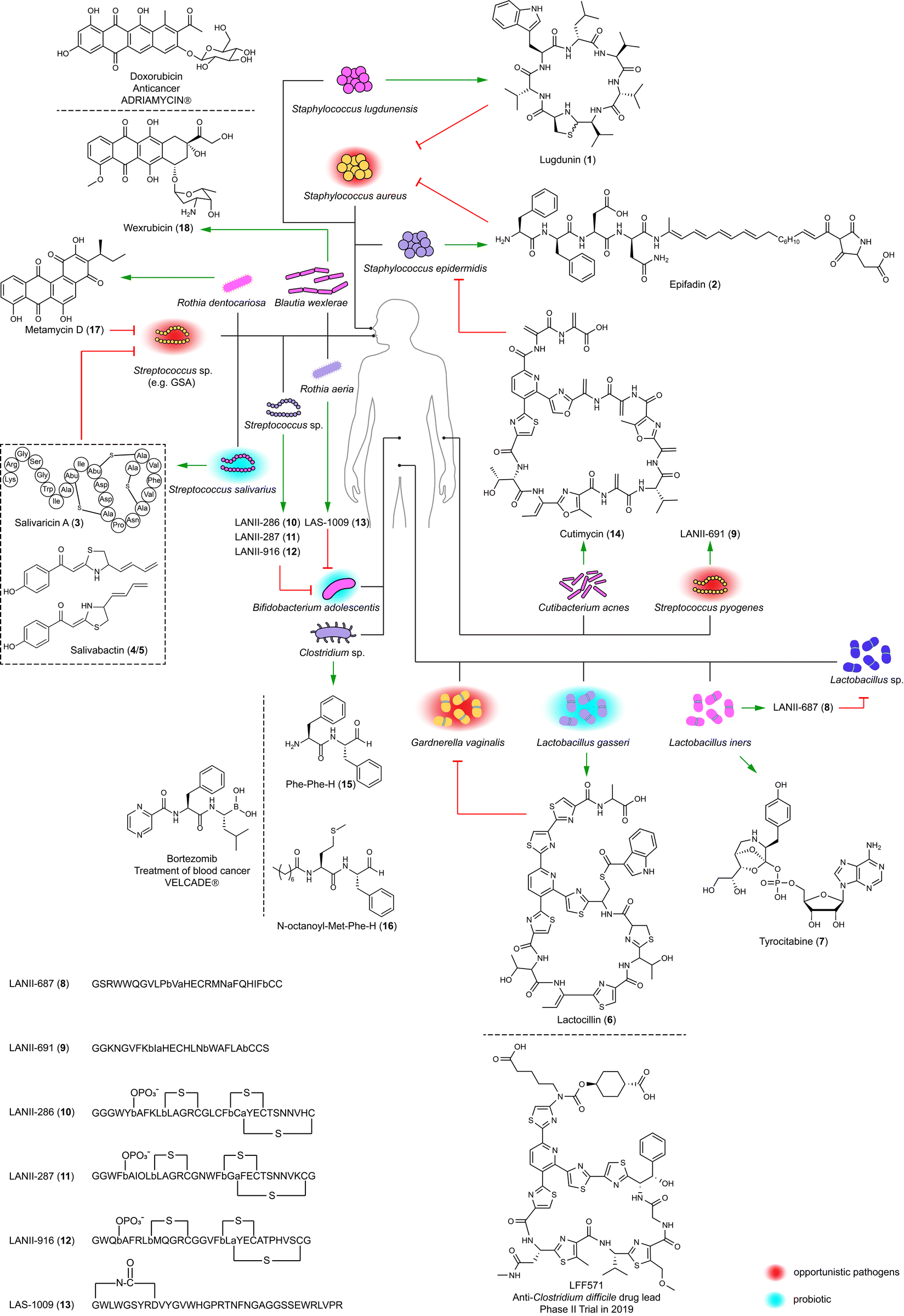 | ||
| Fig. 1 Antibiotics (1–6, 8–14, and 17–19), inhibitor of protein synthesis (7), and protease inhibitors (15 and 16) identified within the human microbiota across various body sites (nose, mouth, gut, vagina, and skin) and their interactions with microbes (green arrow, production; red arrow, inhibition). The drug molecules mirroring the bioactive compounds are also shown. (a) and (b) in compounds 8–13 represent dehydrated serine and threonine residues, respectively. | ||
Lugdunin (1), a non-ribosomal cyclopeptide natively produced by S. lugdunensis IVK28, is characterized by a thiazolidine moiety. This unique structural feature is formed by a terminal reductase domain that triggers head-to-tail thiazolidination.22 Structure–activity relationship (SAR) studies have shown that tryptophan, leucine, and thiazolidine residues as well as its secondary amine are indispensable for antimicrobial activity.25 This activity, particularly against difficult-to-treat methicillin-resistant S. aureus (MRSA) with a minimum inhibitory concentration (MIC) of 1.5 μg mL−1, involves the dissipation of the bacterial cell membrane potential. Notably, lugdunin (1) maintains its potency against MRSA without developing resistance over a 30 day period. Moreover, it enhances the human host defense by promoting the expression and release of LL-37 and CXCL8 and by recruiting phagocytic cells. This action thereby amplifies the innate immune response in primary human keratinocytes mediated by commensal microbes.26
Epifadin (2) is natively produced by S. epidermidis,23 the most abundant commensal bacterium in the nasal mucus of healthy humans.27 The compound is a hybrid of peptide, polyene, and tetramic acid, which is synthesized by a polyketide synthase-non-ribosomal peptide synthetase (PKS-NRPS) gene cluster located on a 55-kb plasmid. Interestingly, epifadin (2) has a broad antimicrobial activity by perturbing membrane integrity of bacteria and fungi, but it is highly unstable under standard lab conditions, as well as in simulating skin or nasopharyngeal habitats. The transient life span of epifadin (2), lasting only a few hours, may serve to limit collateral damage to mutualistic bacteria and host cells. This might suggest a previously unrecognized antimicrobial strategy, distinct from narrow-target and contact-dependent approaches.23
2.2 Salivaricins and salivabactin: shields for the human oral cavity
The human oral cavity serves as a direct interface for the exchange of substances between humans and external environments. It harbors diverse microbial communities in sites such as saliva, gingiva, palate, tonsils, and teeth, where many kinds of microbes coexist and interact with each other and with the human host.28,29 Understanding these microbes, especially Streptococcus, is essential for comprehending human health and diseases. This includes conditions like dental caries associated with Streptococcus mutans, as well as diseases like pharyngitis and impetigo linked to Streptococcus pyogenes (Group A Streptococcus; GAS).28–30Streptococcus salivarius K12 is a probiotic originally isolated from the oral cavity of a healthy infant. Its antibacterial activity largely relies on its 190-kb megaplasmid, pSsK12, which houses two BGCs encoding two different families of bacteriocins.31 Some strains of this species produce lantibiotics, namely salivaricins (3, Fig. 1) and their phosphorylated products,32 to penetrate oral biofilms and kill specific disease-associated bacteria including S. pyogenes, and also enhance host immunity by priming neutrophil and boosting phagocytosis.32,33 The production of salvaricins is controlled by the sal operon located on pSsK12. Also located on this plasmid is a trans-AT PKS-NRPS BGC (sar) directing the discovery of salivabactin (4/5, Fig. 1) from its salAB (responsible for the biosynthesis of salivaricins) deletion mutant with the observation of preserved anti-GAS activity of this mutant. Salivabactin (4/5), produced by the wild-type strain, is a mixture of two geometric isomers and represents a novel scaffold of antibiotics for the presence of 2-ylidene-1,3-thiazolidines moiety.34 Salivabactin (4/5) is active against GAS with an MIC of ∼2 μg mL−1 and other clinical Gram-positive pathogens, without inhibiting against Gram-negative pathogens. Its in vivo protection against GAS is comparable to penicillin G. The findings illustrate that the megaplasmid pSsK12 endows S. salivarius K12 with potent antibacterial properties, underlining its importance in the probiotic's ability to combat pathogens and enhance host immunity.
2.3 Big data driving the discovery of drug-like molecules
The examples discussed showcase that peptides and peptide-polyketide hybrids enable human-colonized bacteria to set up barriers for blocking pathogenic invasions.35 Therefore, structural and functional characterization of these compounds can yield new antimicrobials against pathogens with minimal impact on healthy commensal strains, and assist in developing therapeutic bacteria, the efficacy of which depends on human host colonization. This concept is illustrated by analyzing the biosynthetic capabilities of the human microbiota from the Human Microbiome Project (HMP).17–20 The HMP was initiated to enhance our understanding of the human microbiota and their roles in health and disease,36 which generates a comprehensive genome resource repository from various microbial communities in and on the human body.Detailed investigations originated from a systematic examination of 752 metagenomes from phase 1 of HMP36 which led to the identification of 3118 BGCs in healthy individuals.17 These BGCs are predominantly identified in bacterial genera typically associated with the human microbiome, such as Bacteroides, Parabacteroides, Corynebacterium, Rothia, and Ruminococcus. These genera are not commonly found in non-human environments, highlighting the unique biosynthetic capabilities of the human microbiota. The >3000 BGCs include classes of saccharides, non-ribosomal peptides, polyketides, terpenes, ribosomally synthesized and post-translationally modified peptides (RiPPs), and hybrids. The gut and oral cavity are particularly rich in BGCs due to their high microbial abundance. For example, a typical gut sample harbors 599 BGCs, while an oral cavity sample contains 1061 BGCs. In contrast, the skin, airways, and urogenital tract have fewer BGCs, reflecting their lower microbial diversity. BGCs encoding RiPPs are modestly enriched across all body sites.17,29 A prevalent NRPS family with up to four modules and a terminal reductase domain exclusively exists in the gut, while polyketide synthases (PKS) are relatively uncommon.17,19 These findings highlight the diversity and uniqueness of the human microbiota.
Lactocillin (6) with a relatively narrow-spectrum antibacterial activity disrupts protein synthesis in pathogens like S. aureus and Enterococcus faecalis with MICs of 42 and 425 nM, respectively. It also inhibits the vaginal pathogen like Gardnerella vaginalis with an MIC of 212 nM, yet preserves vaginal commensal Lactobacillus species. Its structure is similar to the antibiotic LFF571 that underwent Phase II clinical trials conducted by Novartis for treating Clostridium difficile infection diarrheal.38 The BGC for lactocillin (6) has also been detected in oral metatranscriptomic reads, suggesting its mobility and active transcription in the human microbiota. The discovery of lactocillin (6) might explain the molecular mechanism of L. gasseri in maintaining vaginal health.
Lactobacillus iners LEAF 2052A-d is an isolate from the vagina of a bacterial vaginosis patient and encodes a widely distributed BGC (tybA-E) in the human microbiota. This BGC was heterologously expressed in Pseudomonas putida under an orthogonal T7 promoter with computer-aided design of synthetic genetic elements inserted in front of each gene. This led to the identification of tyrocitabine (7), a nucleotide featuring an orthoester linkage at its phosphate moiety. The Similarity Ensemble Approach, which computationally predicts potential targets by assessing chemical similarities between tryocitabine (7) and known ligands, led to the identification of components involved in protein translation. Tryocitabine (7) inhibited the translation of a GFP reporter with a half-maximal inhibition (IC50) of 13 mM (Fig. 1), while the acylated tyrocitabine lost this bioactivity, indicating a prodrug mechanism.39
LANII-687 (8) from a vaginal strain of Lactobacillus iners LEAF 2053A-b showed broad-spectrum activity, notably against vancomycin-resistant Enterococcus faecium (VRE) and S. aureus, but also targeted commensal Lactobacillus species, implicating L. iners in vaginal microbiota dysbiosis. LANII-691 (9) from the skin-associated Streptococcus pyogenes GA19702 had a similar antibiotic spectrum with less potency.
Lanthipeptides (e.g. LANII-286 (10), LANII-287 (11), and LANII-916 (12)) and lasso peptides (e.g. LAS-1009 (13)) from oral Streptococcus sp. M344 and Rothia aeria F0474 specifically inhibited the gastrointestinal commensal Bifidobacterium adolescentis. Oral bacteria translocate to the gut through saliva, ingestion of contaminated food, or bloodstream infection following periodontal disease. Once in the gut, these bacteria colonize the intestinal environment and interact with resident microbial communities. This potentially leads to the reduction of B. adolescentis, a beneficial gut commensal. B. adolescentis produces short-chain fatty acids, which help it compete with pathogens and support the immune system. The reduction in B. adolescentis can increase gut permeability and create a pro-inflammatory state, which are risk factors for inflammatory bowel disease. Furthermore, the altered microbial environment can promote the growth of pathogenic bacteria and oncogenic processes, contributing to the development of colorectal cancer and liver cirrhosis.41–43 Lanthipeptides (10–12) and lasso peptides (13) from oral bacteria targeting B. adolescentis suggest that RiPPs are involved in pathogenic processes, including gut microbiota dysbiosis, primary sclerosing cholangitis, and ulcerative colitis. This finding partially explains the molecular mechanism of dysbiosis caused by microbial translocation.20
The human skin with hair follicles and glands rich in lipids and proteins serves as more than a mere physical and chemical barrier. It creates a niche with acidic, high-salt, desiccated, and aerobic conditions for the growth of commensal bacteria, while also potentially harboring opportunistic pathogens such as S. aureus.44,45 Based on the initial analysis of large-scale BGCs,17 a follow-up study focused on a RiPP BGC present in some isolates of Cutibacterium acnes, one of the most common species within the human skin microbiota. The BGC is linked to the biosynthesis of cutimycin (14), whose structure resembles that of lactocillin (6). S. aureus and S. epidermidis are susceptible to cutimycin (14) with MICs ranging from 0.2 to 3.2 μM.45 This might partially explain that the overgrowth of C. acnes, possibly due to the inhibition of S. epidermidis by cutimycin (14), leads to the development of acne, while in healthy skin, S. epidermidis controls the proliferation of C. acnes.46 The cutimycin (14) BGC is also present in the nasal microbiome metagenomic data, indicating its protective role in niche competition by inhibiting the colonization of pathogens across body sites.45
 | ||
| Scheme 1 Conversion of dihydropyrazinones and pyrazinones from dipeptide aldehydes. | ||
The structural resemblance of dipeptide aldehydes to Bortezomib, a proteasome inhibitor as an anticancer drug for treating multiple myeloma and mantle cell lymphoma, and therefore, compounds such as Phe-Phe-H (15), Val-Phe-H, Leu-Phe-H, and N-acyl Met-Phe-H (16) were tested for their in vitro activity against lysosomal cysteine proteases (Fig. 1). Dipeptide aldehydes with a free amino group (e.g.15) exhibited potent activity against cathepsin L with an IC50 of 5 nM but significantly less against cathepsin B with an IC50 of 9.4 μM. Conversely, N-acyl dipeptide aldehyde (e.g.16) displayed no detectable activity against cathepsin L but showed nanomolar efficacy against cathepsin S with an IC50 of 13 nM, indicating a distinctive selectivity profile. Isotopic tandem orthogonal proteolysis-activity-based protein profiling revealed that Phe-Phe-H (15) inhibits the catalytic cysteine of cathepsin L with significant selectivity over other cathepsins.47
3 Bacteria–nematode–insect interactions: drug leads from the gut of nematodes
Entomopathogenic nematodes that carry specific endosymbiotic bacteria, Xenorhabdus and Photorhabdus, in their gut, canonically belong to the genera of Steinernema and Heterorhabditis. The bacteria–nematode pair actively hunts insect larvae in the soil by detecting signals emitted from plant roots damaged by insects. When the nematodes invade insect prey, the endosymbiotic bacteria are released into the insect hemolymph, where they proliferate and synthesize toxins, lytic enzymes, and natural products. These products aid in promoting the development of the nematode host (isopropylstilbene, 19),48 killing the insect prey,49,50 decomposing the insect cadaver, and protecting the cadaver (e.g. odilorhabdins, evybactin, darobactin, and dynobactin, 20–25)51–56 against other soil-living prokaryotic and eukaryotic organisms (Fig. 2).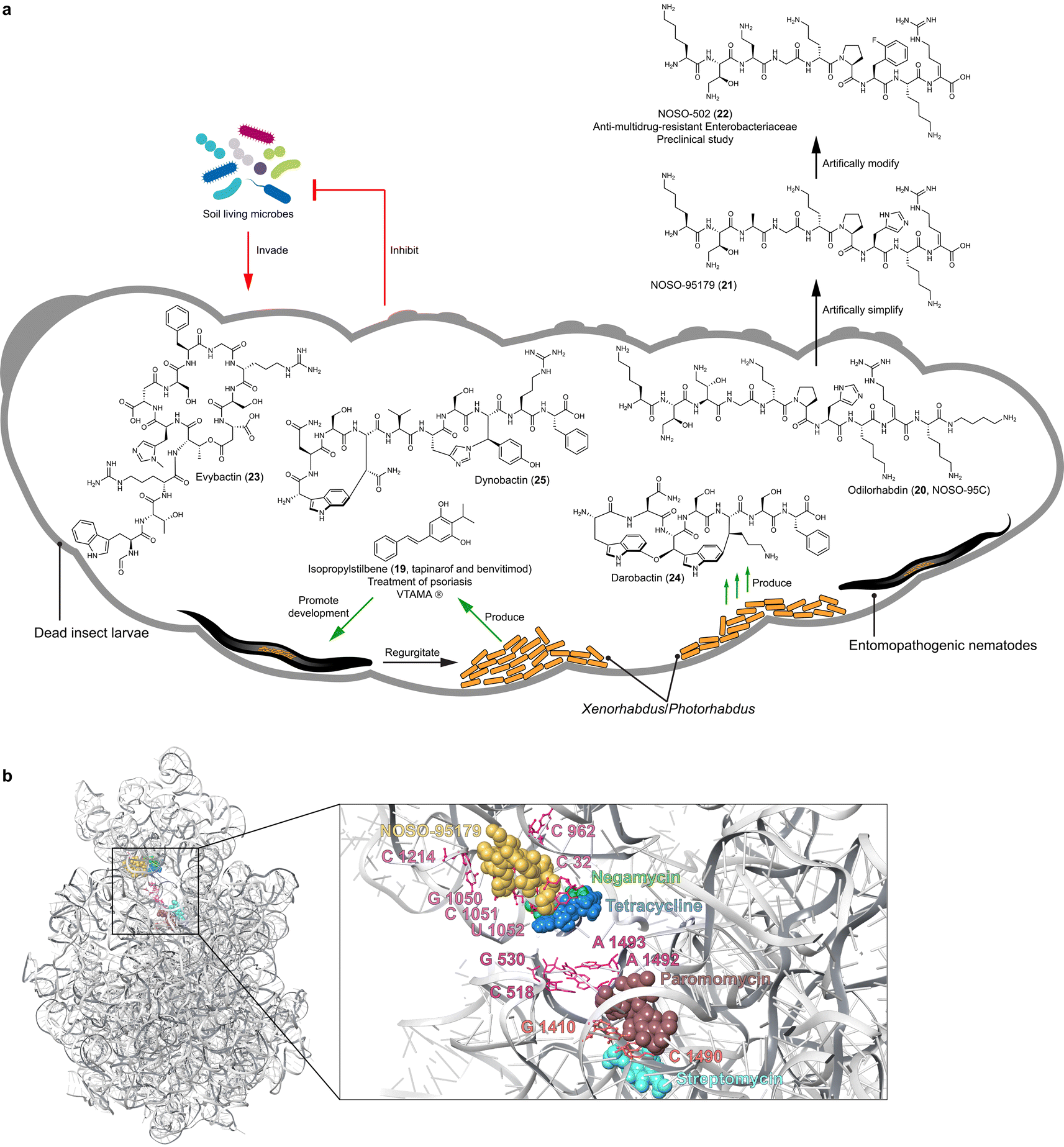 | ||
| Fig. 2 (a) Growth factor (19) and promising antibiotics (20 and 23–25) identified from entomopathogenic bacteria, Xenorhabdus and Photorhabdus. (b) Overview and close-up views of the NOSO-95179 (21) binding site at the decoding center of the small ribosomal subunit, relative to the binding sites of negamycin, tetracycline, paromomycin, and streptomycin. | ||
Such bacteria–nematode–insect interactions offer a promising model for understanding the ecological functions of the bacterial natural products, which could dramatically inspire the discovery of drug leads. This is because: (1) the ecosystem is constructible in laboratory environments.57,58 The endosymbiotic bacteria, entomopathogenic nematodes, and insects can be efficiently cultured, enabling imitation of the interactions under standard laboratory conditions. (2) The endosymbiotic bacteria are genetically tractable.59 This allows elucidating the ecological function of individual bacterial natural products. (3) Xenorhabdus and Photorhabdus, belonging to γ-Proteobacteria, harbor vast biosynthetic potential of natural products, which is comparable to the prolific Actinomycetia in both quantity and diversity. Additionally, the adaptation of Xenorhabdus and Photorhabdus to harsh environments and competition with other soil organisms likely drives the selection of valuable BGCs that yield highly efficacious natural products.49,60 (4) Over 40% of genes in nematodes are highly similar to human genes,61 and both nematodes and insects have been widely used as model organisms in the study of human diseases and drug screening. Assuming that the bacterial natural products, which are non-toxic at effective concentrations towards the nematode host, are tailored for specific ecological niches, we might already have solutions for diseases from the tripartite relationship. Based on the functional assignment of the bacterial natural products on the ecosystem, we could apply analogous usage for treating human diseases. Here we stress three prominent compound classes as exemplary cases.
3.1 Isopropylstilbene: a signaling molecule for treating psoriasis
Isopropylstilbene (19), known as tapinarof and benvitimod and first discovered from Photorhabdus luminescens, is a multipotent compound and an essential growth factor of dauer-stage nematodes (Fig. 2).48 In 2022, isopropylstilbene (19), acting as an agonist of the aryl hydrocarbon receptor (AhR) and marketed under the brand name VTAMA®, was approved as a clinical drug by the FDA for treating psoriasis.62Stilbenes are well-known natural products in plants, whose biosynthesis involves type III PKS systems.63 Interestingly, the efficacy that distinguishes isopropylstilbene (19) from other stilbenes is attributed to the isopropyl group,64 which is installed by the cross-talk between type II PKS and fatty acid biosynthesis, remarkably different from the pathway in plants.48,65,66 The biosynthetic genes are highly conserved across all Photorhabdus strains;49 however, unlike a canonical BGC, these biosynthetic genes are scattered throughout the genome as individual genes and small operons.48
Despite the unknown mode of action of isopropylstilbene (19) in the development of entomopathogenic nematodes, it binds to AhR,67,68 reminiscent of the interaction between Pseudomonas aeruginosa and eukaryotic hosts.69 The AhR of eukaryotic hosts monitors the signaling molecules of P. aeruginosa, such as phenazines, homoserine lactones, and quinolones, to regulate the scale and intensity of eukaryotic host immune defense.69 Therefore, it is reasonable to hypothesize that entomopathogenic nematode hosts sense isopropylstilbene (19) produced by Photorhabdus to defend against other pathogens through fine-tuning immune responses.
3.2 Odilorhabdins and evybactin, non-ribosomal peptides for multidrug-resistant infections
Insect cadavers in the soil are rich in proteins, lipids, carbohydrates, and vitamins, which must be protected from competitors to allow the entomopathogenic nematodes to continue their life cycle. Consequently, it is not surprising that entomopathogenic bacteria biosynthesize natural products that are active against bacteria, fungi, and protozoa, which are abundant in the soil surrounding the insect cadaver. Comprehensive BGC analysis revealed that the odl NRPS BGC responsible for the biosynthesis of odilorhabdins was a unique gene cluster family in Xenorhabdus and predominantly Photorhabdus.49 Odilorhabdins, also known as NOSO-95A–C (e.g.20) initially identified from Xenorhabdus nematophila CNCM I-4530 exhibited broad-spectrum activity against carbapenem-resistant Enterobacteriaceae (CRE) and MRSA with MICs ranging from 8 to 16 μg mL−1 (Fig. 2).56 The antibiotics are linear cationic peptides with non-proteinogenic amino acid residues and a C-terminal putrescine. A N-acetyltransferase confers self-detoxification by primarily acetylating Dab(βOH)2.70Inspired by the investigation of the detoxification mechanism, Sarciaux et al. synthesized NOSO-95C (20) and undertook in-depth SAR studies through alanine-scanning.71 This approach identified the essentiality of the primary amino group in Lys1, Dab(βOH)2, and Orn5, leading to the development of NOSO-95179 (21). This modified compound, with improved inhibition of bacterial translation and reduced toxicity, paved the way for the development of the preclinical antibiotic candidate NOSO-502 (22). NOSO-502 exhibited potent activity against multidrug-resistant strains with MICs ranging from 0.5 to 4 μg mL−1 and a favorable safety profile, making it a promising candidate for treating serious hospital-acquired infections. Odilorhabdins bind the small 30S subunit and form multiple hydrogen bonds with 16S rRNA residues. This binding site differs from other antibiotics targeting the 30S subunit, such as aminoglycosides and tetracyclines, highlighting odilorhabdins' unique mechanism of action.56
Evybactin (23) produced by Photorhabdus noenieputensis DSM25462 was discovered by differential screening of 58 Xenorhabdus and Photorhabdus strains for antitubercular potential.53 A type I NRPS mega BGC with 12 modules, spanning 49.6 kb, encodes the biosynthesis of the cyclic depsipeptide. This BGC appears to be a singleton, which has only been found in P. noenieputensis so far. Evybactin (23) exhibits high selectivity against Mycobacterium tuberculosis H37Rv (MICs 0.0625–0.250 μg mL−1) without affecting S. aureus and other commensal bacteria, and has non-toxicity to human cells. It gains entry into Mycobacterium tuberculosis cells via the relatively rare BacA-type transporter, not found in human gut commensals, which explains the selectivity of evybactin (23). The compound inhibits an allosteric site of DNA gyrase with an IC50 of 0.25 μg mL−1, which is different from the binding sites of drugs like moxiflxacin and similar to that of a new class of synthetic thiophene antibiotics.53 Thus, evybactin (23) emerges as a promising drug candidate, mitigating the risk of antibiotic resistance and safeguarding the health of humans and microbiota.
3.3 Darobactin and dynobactin: RiPPs revolutionizing the combat against pathogenic Gram-negative bacteria
Darobactin (24) initially identified from Photorhabdus khanii HGB1456 by screening a panel of Xenorhabdus and Photorhabdus strains for activity against E. coli is a heptapeptide belonging to RiPPs (Fig. 2).55 It is biosynthesized by a five-gene operon (darA-E), with heterologous expression studies72,73 confirming that DarA (precursor peptide) and DarE (radical SAM enzyme) are sufficient for darobactin (24) production in E. coli.73 DarE catalyzes the formation of a unique aromatic–aliphatic ether linkage between two tryptophan residues, as well as an aromatic–aliphatic carbon–carbon bond between tryptophan and lysine residues.74,75 This endows darobactin (24) with a rigid β-strand conformation.Darobactin (24) has selective antibiotic activity against multiple Gram-negative bacteria, both in vitro and in animal infection models. This includes strains resistant to polymyxin, such as Pseudomonas aeruginosa with MICs of 2 to 16 μg mL−1, and those resistant to β-lactam, including Klebsiella pneumoniae with MICs of 2 to 4 μg mL−1 and E. coli with an MIC of 2 μg mL−1. Importantly, it remains safe for human cells and commensal gut bacteria at therapeutically effective concentrations.55 The rigid β-strand conformation of darobactin (24) mimics the recognition signal of native BamA substrates, enabling 24 to specifically target BamA. BamA is a bacterial insertase located on the outer membrane facilitating the assembly and insertion of outer membrane proteins. Consequently, darobactin (24) compromises the integrity of the outer membrane without penetrating the bacterial cells.55,76 Interestingly, BamA analogous in gut commensals such as Bacteroides have a single point mutant G424D in the key β-strand, different from pathogenic Gram-negative bacteria. This mutation may partially explain the insensitivity of gut commensals to darobactin.76
The intriguing structure and unique mode of action of darobactin have sparked tremendous follow-up studies in metabolic engineering,77–79 chemical synthesis,80,81 SAR analysis,72,77–79 and genome mining82,83 to enhance production yields and identify new analogs. A study using the radical SAM enzyme DarE as a probe, whose homologs are distributed across the bacteria kingdom, led to the identification of dynobactin (25) from Photorhabdus australis (Fig. 2).52 The structure of dynobactin (25) with a carbon–carbon bond linking tryptophan to asparagine residue, as well as a carbon–nitrogen bond connecting tyrosine to histidine, was elucidated by cryo-electron microscopy and micro-electron diffraction. This endows dynobactin with a flexible conformation in contrast with the rigidity of darobactin. Similar to darobactin, dynobactin also targets BamA with an IC50 of 16 ± 2 nM, but with higher affinity to BamA and better water solubility—probably owing to its flexible conformation and additional ionizable sites.52
4 Other eukaryotes and microbe interactions
After discussing the interplay between humans and their resident microbiota (Fig. 1), as well as the interactions between the endosymbiotic bacteria, nematode, and insect (Fig. 2), we extend our focus to a wider spectrum of eukaryotes and their microbial partners. Given the ubiquity of microbes across all earthly environments, from terrestrial to marine, and their adaptation to hosts of varying sizes, from protozoa to plants and insects, we aim to understand how these eukaryotic organisms not only survive but also thrive with the contributions of their microbial partners. This not only broadens our understanding of microbial versatility but also enhances our ability to leverage these relationships for developing innovative therapeutic and environmental solutions.4.1 Bacteria–fungi interactions: microorganisms cooperatively fend off enemies
The nests of fungus-growing ants have been pivotal in pioneering studies in chemical ecology, providing an exceptionally informative system for investigating bacteria–fungi interactions, as well as for discovering lead compounds. This ecological phenomenon governed by natural products has been the subject of many extensive reviews.84 This sets a foundation on which implementation of cell imaging has enhanced our knowledge of bacteria–fungi interactions into a new dimension.Fungi are increasingly recognized for hosting endobacteria, as revealed through imagining and sequencing techniques. These symbiotic interactions often induce or enhance the production of natural products, serving as defense mechanisms against other (micro)organisms. For example, certain soil fungi, upon preyed by soil-dwelling nematodes, have evolved to produce nematocidal compounds as a survival strategy. This adaptation is notably demonstrated by the symbiotic relationships between the fungus Podila verticillata and its endosymbiont Candidatus Mycoavidus necroximicus, as well as between the fungus Rhizopus microsporus and its endosymbiont Mycetohabitans rhizoxinica. The Ca. M. necroximicus produces polyketides termed necroximes (e.g.26) against Caenorhabditis elegans with an IC50 of 24.66 μM and a cyclic lipopeptide symbiosin (27), which synergistically protect the fungal host from nematodes. Although symbiosin alone is ineffective, it acts as a biosurfactant, influencing the permeability of substances into the nematodes (Fig. 3).85,86 The symbiosin BGC encodes a seven-module NRPS generating a hexapeptide backbone, indicating a module-skipping mechanism.86 The endosymbiont M. rhizoxinica produces rhizoxins S1 and S2 (e.g.28) to safeguard its fungus host R. microsporus from the amoeba Planoprotostelium aurantium (IC50 = 65 nM for 28 in liquid survival assay) and nematode predation (IC50 = 248 μM for 28 in liquid feeding inhibition assay) (Fig. 3).87 A bacterium–fungus complex contains the bacterial endosymbiont Alcaligenes faecalis SCSIO B001, which predominantly resides within the mycelia of its fungal host Spiromastix sp. SCSIO F190 and is also presented in the spores of the fungal host. The symbiont was isolated from a marine sediment sample collected in the Northern South China Sea by streaking serial dilutions onto fucose-proline agar plates containing trimethoprim and nystatin. The fungal host produces the polyketide spiromarmycin (29, Fig. 3), a compound exhibiting broad-spectrum antibacterial and antifungal activity against pathogenic bacteria with MICs ranging from 4 to 128 μg mL−1 and (phyto)pathogenic fungi with MICs ranging from 1 to 16 μg mL−1 against different Candida albicans strains. Notably, the presence of the endobacterium A. faecalis significantly up-regulates the yield of spiromarmycin (29).88
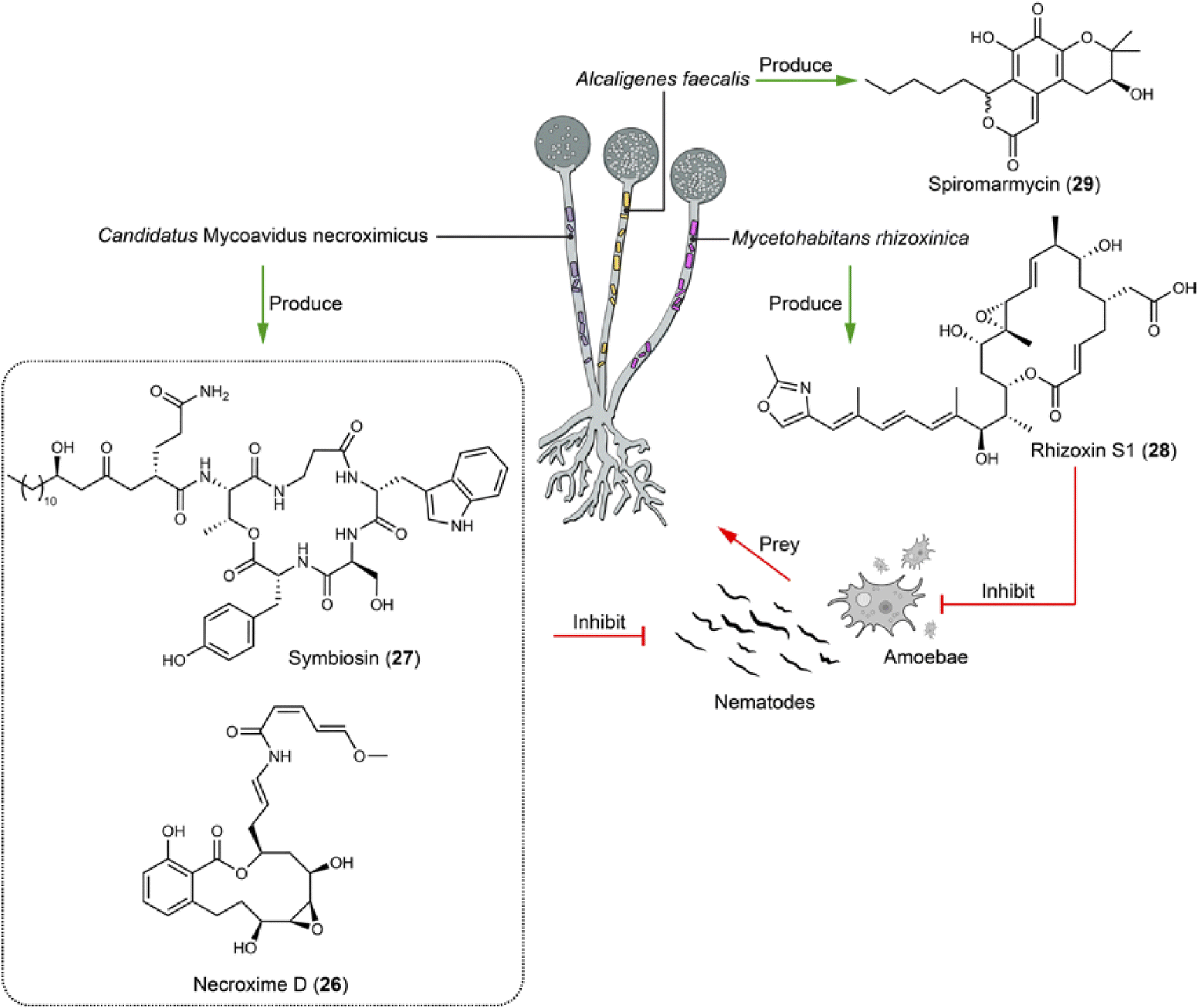 | ||
| Fig. 3 Fungal endobacteria produce compounds (26–29) that defend their fungal host against the predation of nematodes and amoebae, as well as against the infection of various pathogenic microbes. | ||
Fusarium graminearum is the primary phytopathogenic fungus responsible for Fusarium head blight in cereals. During the sexual stage, a microbiome comprising various bacterial species is formed on the surface of perithecia, affording a specific microecological niche that facilitates disease establishment through interactions spanning from mutualism to antagonism.89,90 Nonetheless, the molecular mechanisms underlying antagonistic interactions are obscure, because bacteria playing an antagonistic role often do not dominate under natural conditions. Pantoea agglomerans ZJU23 within this bacterial community notably stands out due to its highest antagonistic activity towards F. graminearum. The bacterium produces herbicolin A (30), a glycosylated lipopeptide compound that exhibits potent antifungal activity against F. graminearum with an EC50 of 1.72 μg mL−1 and a wide array of phytopathogens, while being non-toxic to plant leaves and fruits (Fig. 4). The mechanism by which herbicolin A (30) functions involves impeding ergosterol biosynthesis in fungal pathogens, thus disrupting fungal lipid rafts.89 Therefore, herbicolin A (30) has potential for application in agricultural and clinical settings.
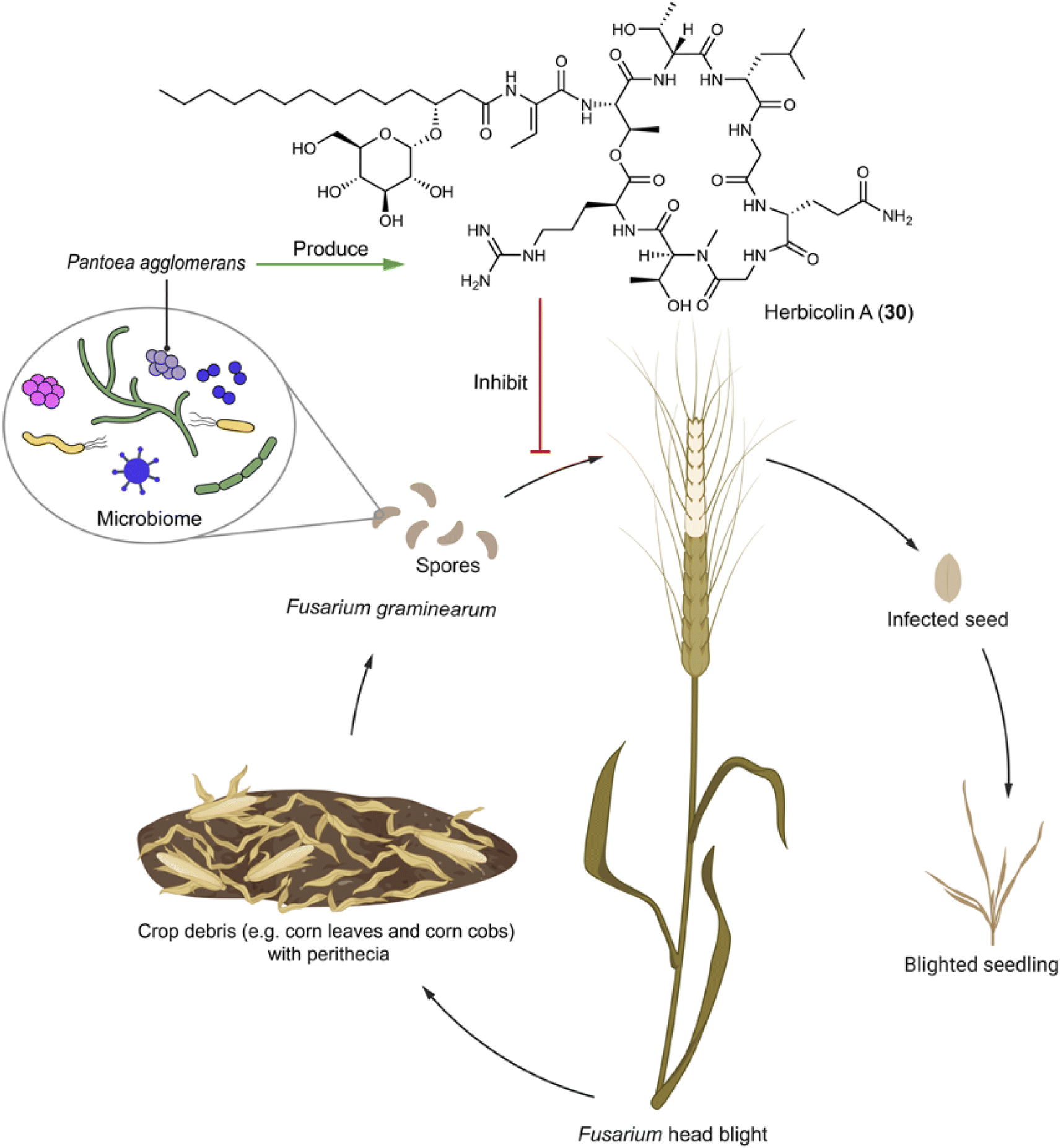 | ||
| Fig. 4 Microbiome on the spores of F. graminearum has an antagonizer that produces herbicolin A (30) to inhibit the phytopathogenic fungus. | ||
4.2 Bacteria–plants interactions: FR900359 against pathogenic nematodes holds promise for treating GqPCR-related diseases
FR900359 (31), a cyclic depsipeptide characterized by unusual amino acid units like N-methyldehydroalanine and N,O-dimethylthreonine, was originally isolated from Ardisia crenata, a plant widely used in traditional Chinese medicine found across tropic and subtropical areas.91 From a biosynthetic perspective, the presence of non-proteogenic amino acid residues suggests that 31 likely originates from microbes that establish a symbiosis relationship with the plant. The frs BGC encoded in Candidatus Burkholderia crenata, an obligate bacterium in A. crenata leaves, is linked to FR900359 (31) production (Fig. 5). In the ecological system, Ca. B. crenata adapts by reducing its genome to focus on 31 production, benefiting A. crenata by deterring insect predation, while A. crenata provides Ca. B. crenata with nutrients.92 Additionally, a culturable bacterium Chromobacterium vaccinii isolated from roots of cranberry is otherwise able to produce FR900359 (31), offering protection against pathogenic nematodes as demonstrated both in vivo and in vitro experiments (Fig. 5), by inhibiting the signal transduction of their Gq protein.93 The biosynthetic logic of FR900359 (31) reveals novel features, such as an atypical thioesterase (Fig. 5b) domain catalyzing intermolecular side chain transesterification.94 This highlights the potential for attaching the macrocyclic core with different side chains (propionyl in FR900359 and acetyl in its analog YM-254890) that can lead to crucial pharmacological differences, for example, FR900359 metabolized significantly faster and has a slower dissociation kinetic compared to YM-254890.95 FR900359 (31) is the most potent known inhibitor of the heterotrimeric Gq family proteins, engaging its molecular target with unparalleled selectivity.96 Thus, the compound holds significant promise for treating Gq-related diseases such as asthma and uveal melanoma.97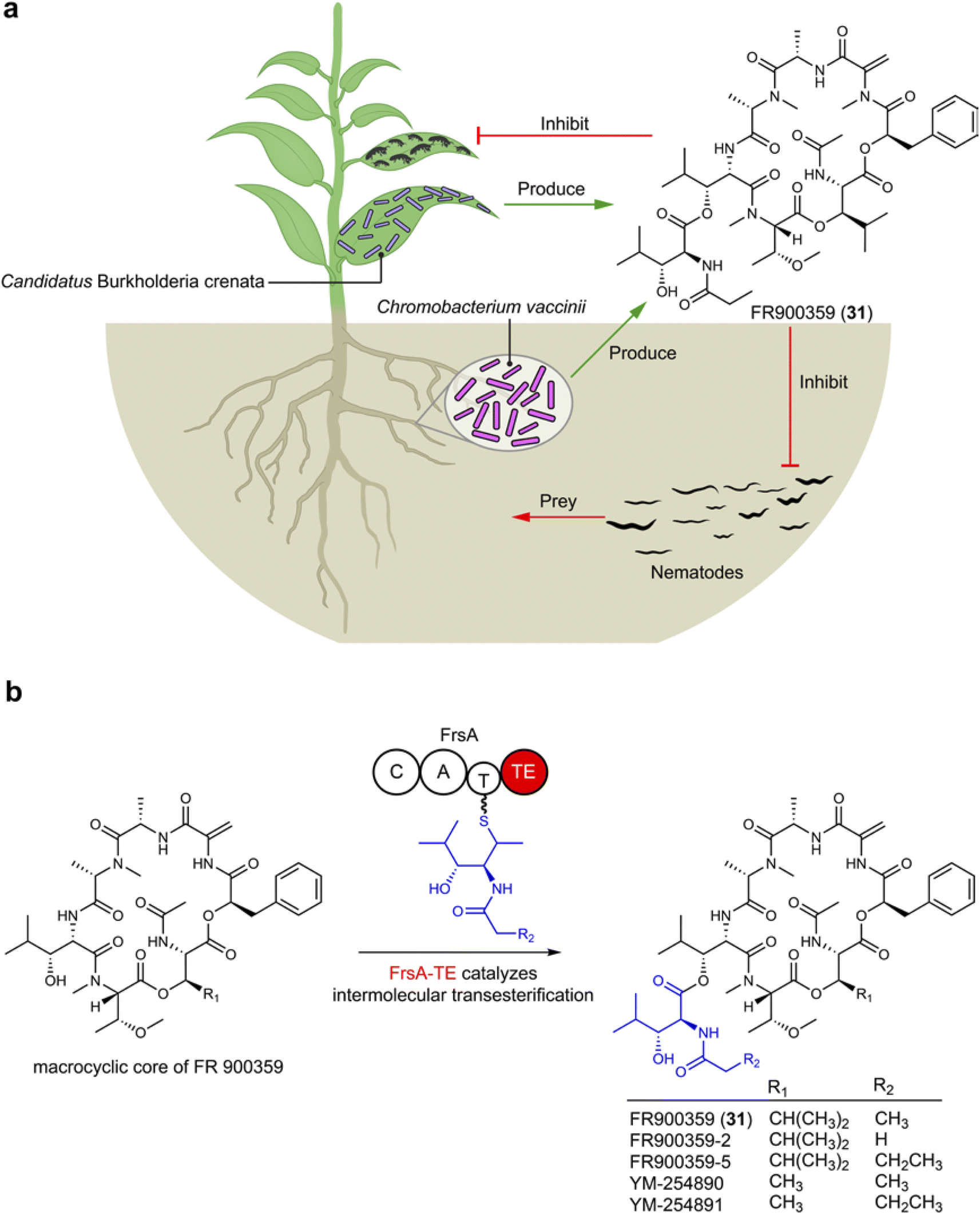 | ||
| Fig. 5 (a) Plant microbes produce FR900359 (31) to protect the plant host against the predation of nymphs of bean bugs and pathogenic nematodes. (b) FrsA-TE catalyzes intermolecular transesterification of side chains to the macrocyclic core of FR900359 (31). | ||
4.3 Bacteria–amoebae interactions: polyketides and non-ribosomal peptides defending the amoeba host as amoebicidals and antifungals
Amoebae are a diverse group of single-celled organisms found in soil, water, and within host organisms. Some species of amoebae are harmless, while others are pathogenic to humans, causing neglected tropical diseases and other conditions like amoebic dysentery, Acanthamoeba keratitis, and primary amoebic meningoencephalitis (brain eating).In soil environments, the interaction between the model eukaryote Dictyostelium discoideum and Pseudomonas sp. QS1027 interaction has been extensively studied.98–100 This interest stems from the isolation of Pseudomonas sp. QS1027 strain from D. discoideum, alongside the discovery that D. discoideum uses other Pseudomonas as a food source. This relationship indicates that Pseudomonas when associated with the amoeba could produce compounds lethal to other competitors (amoebicidals) and antimycotic agents. Amoebicidal and antimycotic activities are interconnected to some extent, as the social amoeba D. discoideum shares certain characteristics with fungi, including aspects of their life cycles like spore formation and the ability to aggregate and form multicellular structures under specific conditions. Exploiting this ecological relationship has led to the isolation and identification of various natural products from Pseudomonas sp. QS1027, including the polyketide mupirocin (32) and the non-ribosomal peptides jessenipeptins (e.g.33) and keanumycins (e.g.34).98,99 Mupirocin (32) and jessenipeptin (33) with an IC50 of 4 μg mL−1 against D. discoideum demonstrate synergistic effects against amoebae, Bacillus subtilis, and MRSA (Fig. 6).99,100 Keanumycin A (34) shows strong amoebicidal activity with IC50 values of 4.4, 2.0, and 3.1 nM against D. discoideum, Acanthamoeba castellanii, and Acanthamoeba comandoni, as well as potent activity against different fungal pathogens of humans and plants.98
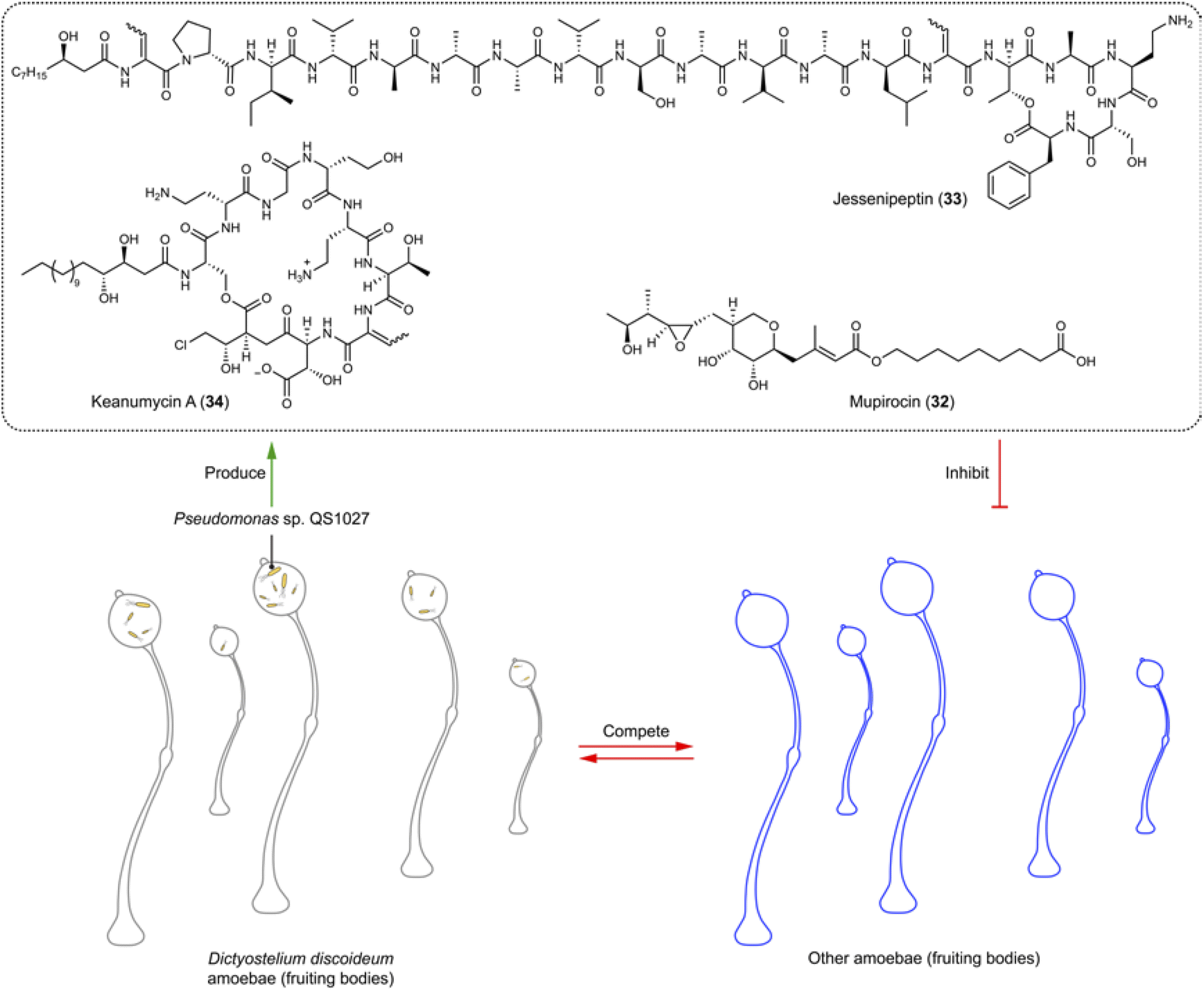 | ||
| Fig. 6 Amoebae store Pseudomonas sp. that produces various amoebicidals and antifungals (32–34). | ||
4.4 Bacteria–bee interactions: symbiotic defenses against fatal pathogenic threats
Bees, as essential pollinators, are critical in ecological balance and economic activities. They support the biodiversity of natural habitats through the pollination of a vast array of plants and contribute to agricultural productivity and food security by pollinating crops. Honeybee larvae and pupae may die from epizootic America Foulbrood or European Foulbrood, caused by the super pathogens Paenibacillus larvae and Melissococcus plutonius, respectively. To fight against these fatal pathogenic bacteria, symbiotic or gut bacteria associated with bees may be employed to produce bioactive molecules for host protection. For example, from stingless bee Melipona scutellaris, the symbiotic Actinobacteria strains Streptomyces sp. ICBG1323 which produces lobophorins (e.g.35), and Micromonospora sp. ICBG1321 which produces kosinostatin (36), show selective and high inhibition against P. larvae with MICs ranging from 0.040 to 0.19 μM (Fig. 7).101 The gut microbiota of bees are also a potential source for antibiotic discovery. A systematic investigation of 477 bee gut microbiota genomes from honeybees and bumblebees identified a RiPP from Gilliamella apis active against M. plutonius. Although the exact structure remains unidentified, this finding underscores the potential of bee gut microbiota in producing therapeutic agents for bee disease control.102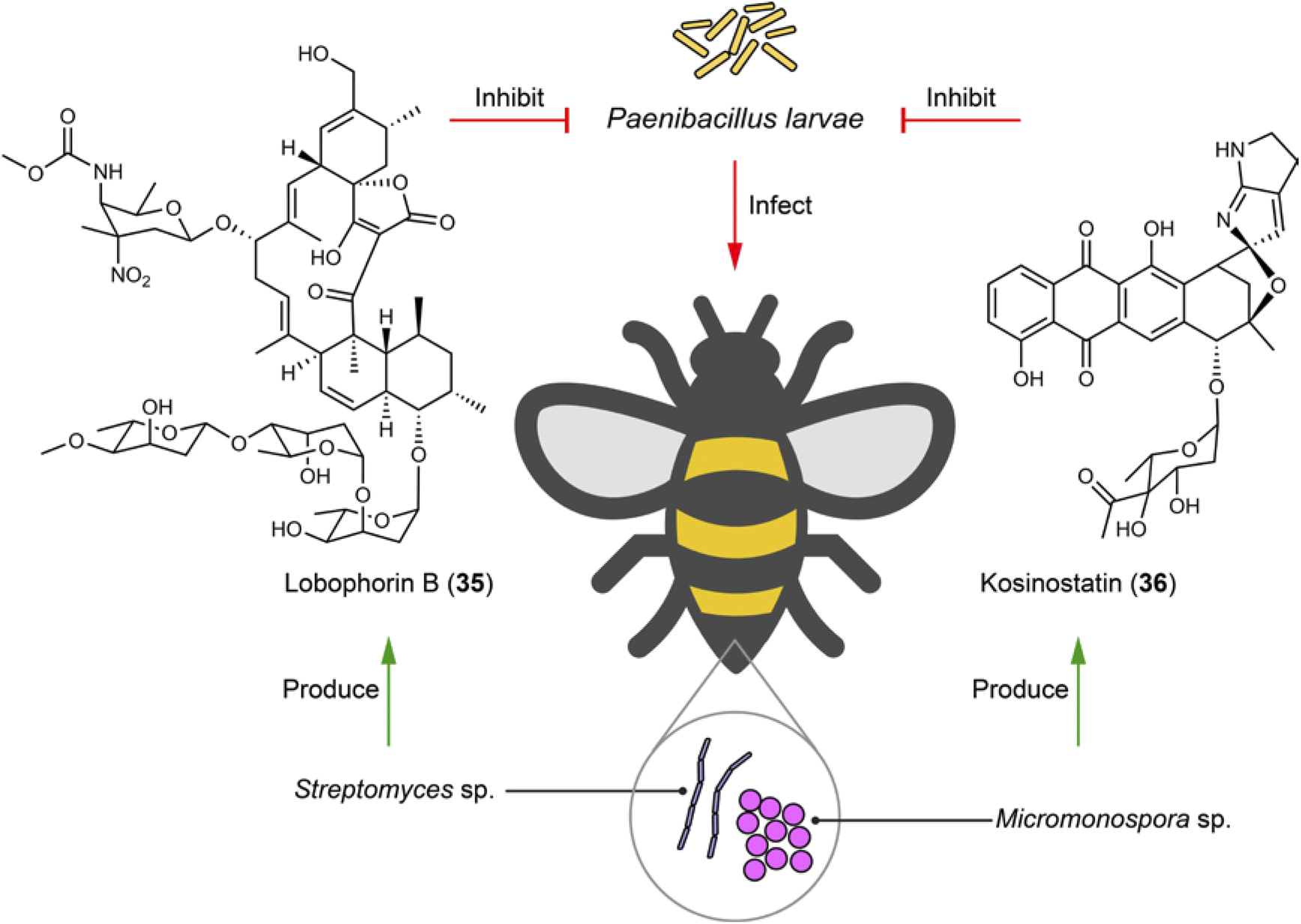 | ||
| Fig. 7 Microbiome on bees helps with fighting against fatal pathogenic bacteria by producing antibiotics (35 and 36). | ||
4.5 Bacteria–beetle interactions: unleashing polyketide arsenals against fungal threats
Beetles undergo metamorphosis development stages from egg to adult, facing threats from numerous entomopathogenic fungi before reaching maturity. Lagria villosa has evolved unique “back pockets” in the larval and pupal stages to house bacterial symbiont Burkholderia gladioli Lv-StB HKI0739.103 The symbiotic bacteria protect the immature life stages against the entomopathogen Purpureocillium lilacinum by producing antifungal agents. Two polyketides, lagriamide (37) and gladiofungin (38), have been identified to showcase such a defensive function. For example, 38 inhibits the growth of the insect pathogen P. lilacinum already at 5 μg mL−1 (Fig. 8).103,104 Interestingly, B. gladioli Lv-StB HKI0739 with a reduced genome that renders the bacteria unculturable under lab conditions, prioritizes antibiotic production to protect the immature life stages.105 The polyketide lagriamide (37) shows structural similarity with the potent antitumor agent bistramide A from marine ascidians, indicating that such a compound class for chemical defense is universally used by symbionts associated with different animal species.103 Adult beetles also employ symbionts to tackle possible pathogenic fungal infections. For example, the pine beetle Dendroctonus frontalis uses Streptomyces sp. SPB74, which produces a polyene peroxide mycangimycin (39) to selectively inhibit its fungal antagonist, Ophiostoma minus with an MIC of 1.2 μg mL−1 (Fig. 8). Mycangimycin (39) shows comparable efficacy to amphotericin B.106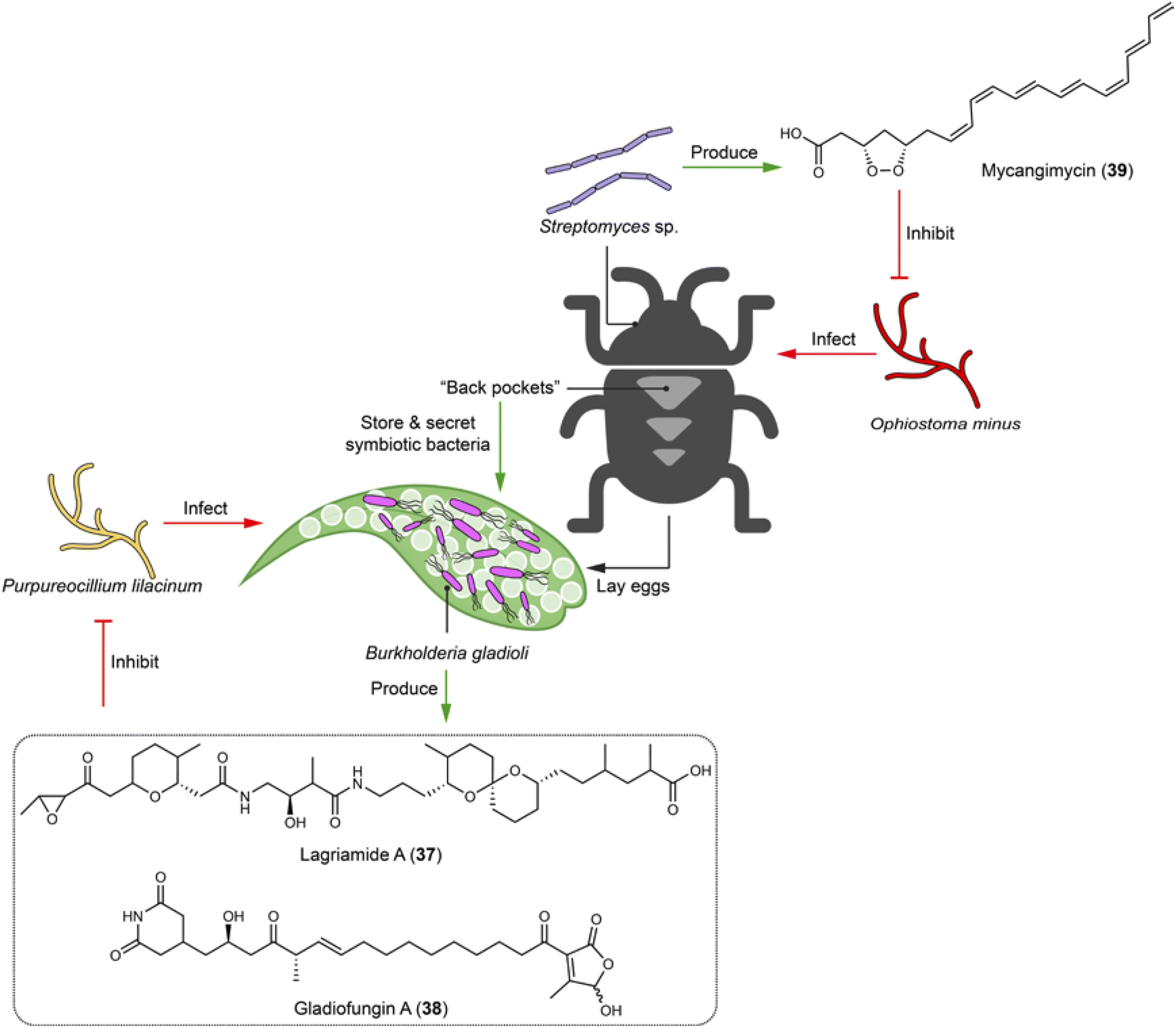 | ||
| Fig. 8 Symbiotic bacteria of beetles use chemical arsenals (37–39) to protect the insect host and offspring against fungal infection. | ||
4.6 Bacteria–mosquito interactions: non-ribosomal peptides preventing malaria transmission
Mosquitoes are vectors in spreading infectious diseases, such as malaria, Zika fever, dengue fever, and chikungunya fever. Within the guts of certain mosquitoes (e.g. Aedes and Anopheles), microbial communities generate natural products to establish and mediate the association with the mosquitoes and Plasmodium parasites that cause malaria.107,108Serratia, the most abundant genus within laboratory- and field-caught Anopheles stephensi mosquitoes, includes a strain that can be isolated from both midguts and salivary glands being capable of producing stephensiolides A–K. Among them, stephensiolide F (40) moderately inhibits entomopathogenic Bacillus subtilis with an IC50 of 15 μg mL−1, suggesting a protective role for mosquitoes from being infected and killed by entomopathogens (Fig. 9). The structure of stephensiolides is characterized by hydrophobic tails and hydrophilic head groups that may function as biosurfactants. This might enhance the swarming capability of Serratia sp. to facilitate colonization and migration across mosquito tissues, coincident with the strain isolated from different tissues.108 Analysis of the BGCs of the mosquito microbiome led to the discovery of eight types of siderophores, including serratiochelin A (41) and pyochelin (42). Compounds 41 and 42 serve not only as metal chelators, but also (i) for female mosquitoes, reduce blood-feeding propensity and overall fecundity, thereby decreasing the possibility of Plasmodium parasites spread; and (ii) for Plasmodium parasites, inhibit multiple species and life stages of Plasmodium, thereby blocking malaria transmission (Fig. 9).107 The discovery of stephensiolides in Serretia elucidates their ecological roles and can help understand its pathogenesis in humans. Furthermore, this finding suggests the potential of genetically engineering Serratia with selective elements for controlling population of mosquitoes, thus reducing the spread of serious infectious diseases.109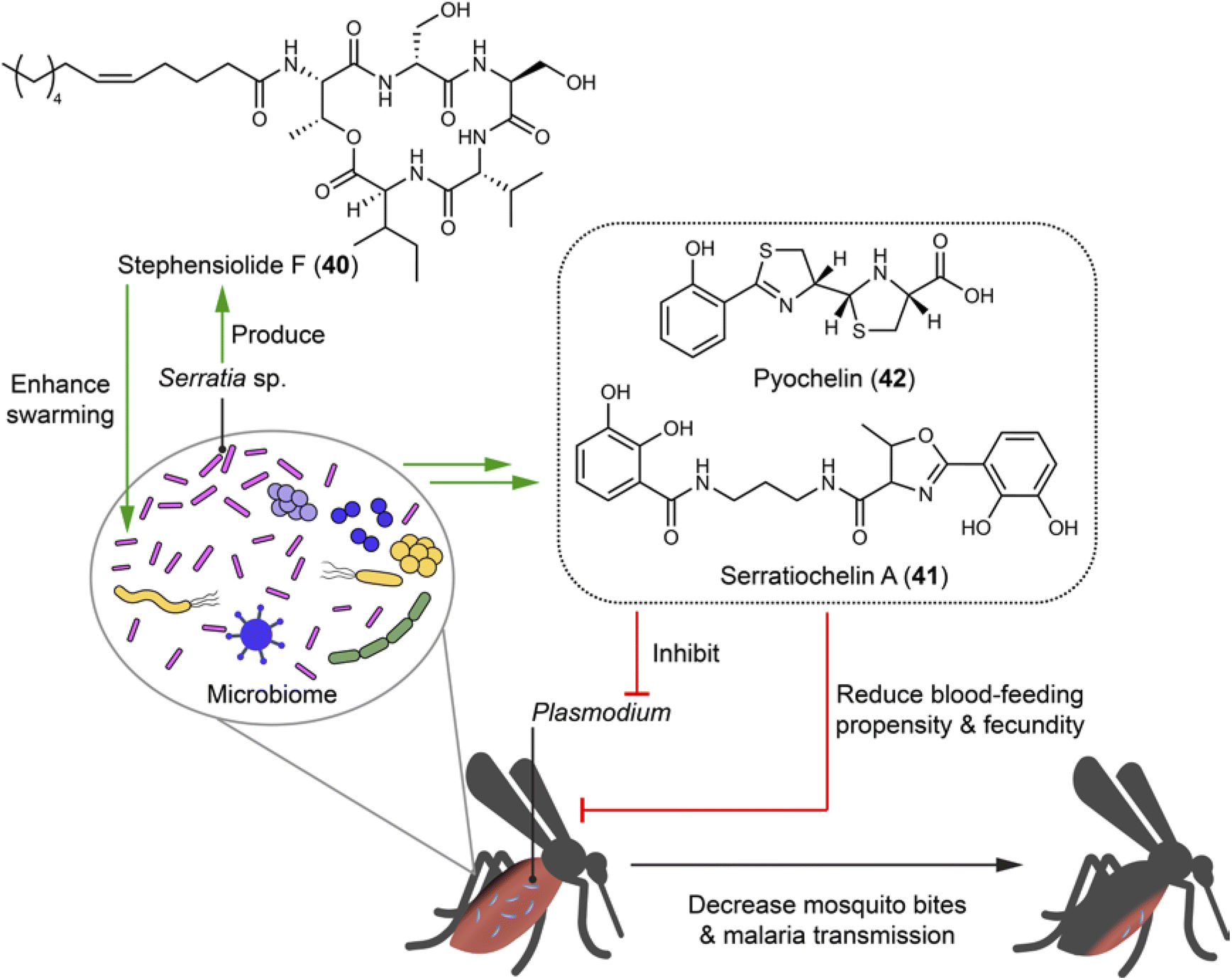 | ||
| Fig. 9 Microbiome of mosquitoes generates compounds (40–42) that enhance swarming capability and inhibit entomopathogenic bacteria. The microbiome of mosquitoes produces siderophores to antagonize the blood-feeding habits of mosquitoes and block the transmission of malaria parasites. | ||
4.7 Bacteria–sponge interactions: symbionts yielding defensive drug-like compounds
Sponges hosting diverse symbiotic microbes represent important sources of novel natural products. Many natural products initially isolated from sponges have later been reattributed to bacterial origins. For example, Haliclona sponges house the obligate symbiont Candidatus Endohaliclona renieramycinifaciens in their specialized cellular reservoirs called chemobacteriocytes. The bacteria engage in mutualistic symbiosis and produce the chemical defense compounds renieramycins (e.g.43), which exert antimicrobial and cytotoxic activities of ecological and therapeutical relevance (Fig. 10). Their production depends on the sponges hosts for substrates like angelic acid and tyrosine, as well as cofactors needed for the biosynthetic enzyme activities. This necessity arises because the symbiotic bacteria have lost central metabolic pathways, including those for amino acids, cofactors, prosthetic group, and nucleoside biosynthesis.110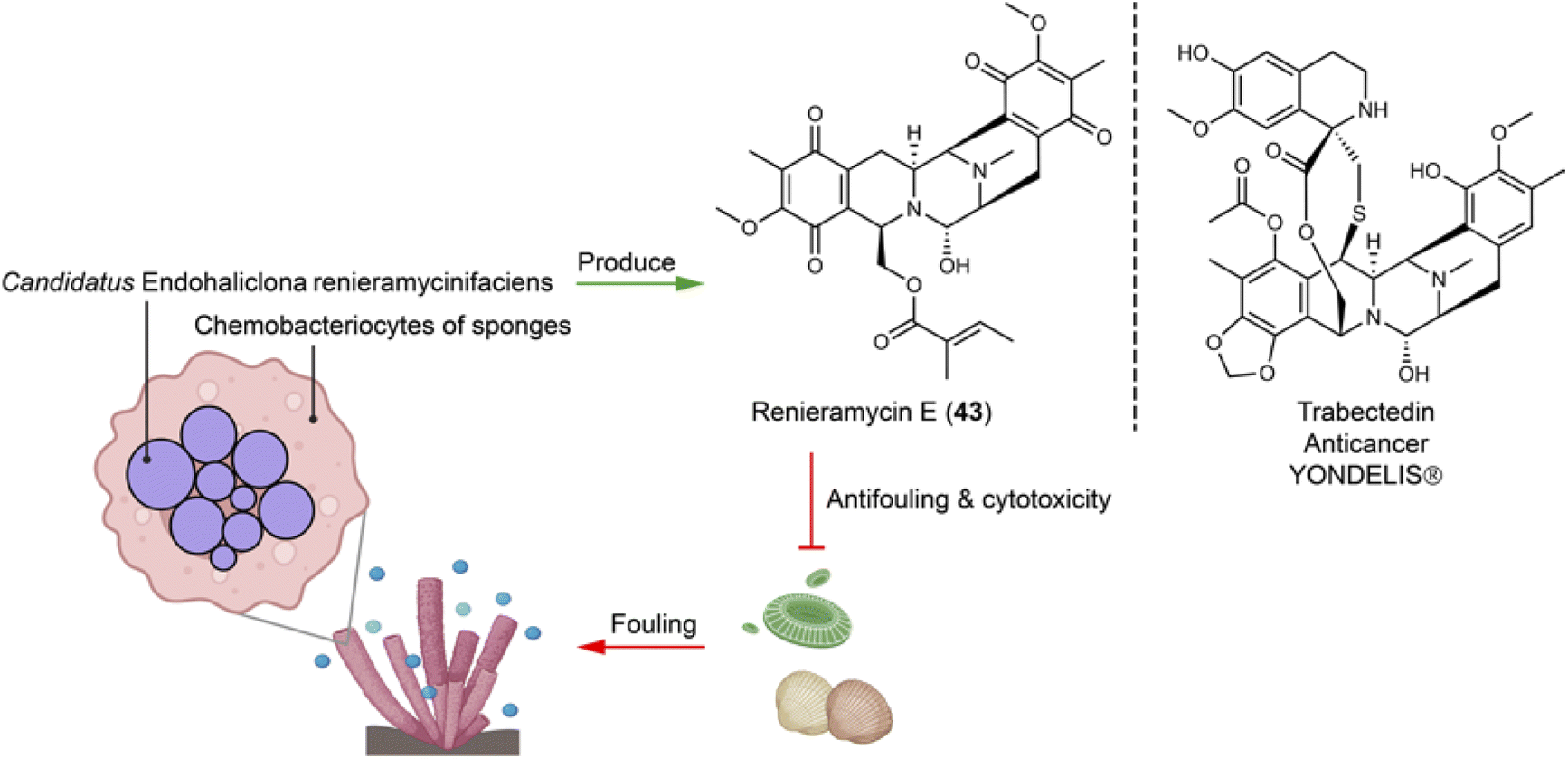 | ||
| Fig. 10 Symbiotic bacteria residing in special compartments of sponges produce defensive chemicals (e.g.43) that are structurally similar to a clinical anticancer drug. | ||
4.8 Bacteria–shipworms interactions: non-ribosomal peptides and polyketides targeting multidrug-resistant pathogens
Shipworms (Teredinidae) have adapted to a lifestyle of feeding on wood and rely on their symbiotic bacteria partner, Teredinibacter turnerae. This culturable species living in gills is isolated from various shipworm species. It helps with wood degradation and nitrogen fixation, while the shipworms provide shelter and nutrients. This shipworms-T. turnerae relationship has advantages for studying compounds of ecological significance due to: (1) wood digestion of T. turnerae, which provide abundant glucose, a precursor for natural product biosynthesis;111 and (2) the presence of conserved BGCs regulated by N-decanoyl-L-homoserine lactone112 in T. turnerae isolates worldwide suggesting potential ecological functions for the compounds related to these BGCs.113 Following these clues, four lipopeptides turnercylamycins (e.g.44 and 45) related to a conversed NRPS BGC were isolated from T. turnerae T7901.111 Turnercylamycins A (44) and B (45) selectively inhibit Gram-negative bacteria, including E. coli (MIC = 1 μg mL−1), Klebsiella pneumoniae (MIC = 2 μg mL−1), and colistin-resistant Acinetobacter baumanni (MIC = 8 μg mL−1), without cytotoxicity (Fig. 11). Both turnercyclamycins and colistin interact with the LPS pathway, but turnercyclamycins remain effective against clinical isolates resistant to colistin. This suggests that turnercyclamycins may act through a different mechanism or target different sites within the LPS pathway compared to colistin. Additionally, turnercyclamycins do not exert their antibacterial effect through mechanisms mediated by mcr-1, thus displaying a distinct resistance profile from colistin.111,114 These differences underscore the potential of turnercyclamycins as promising new candidates for treating multidrug-resistant bacterial infections.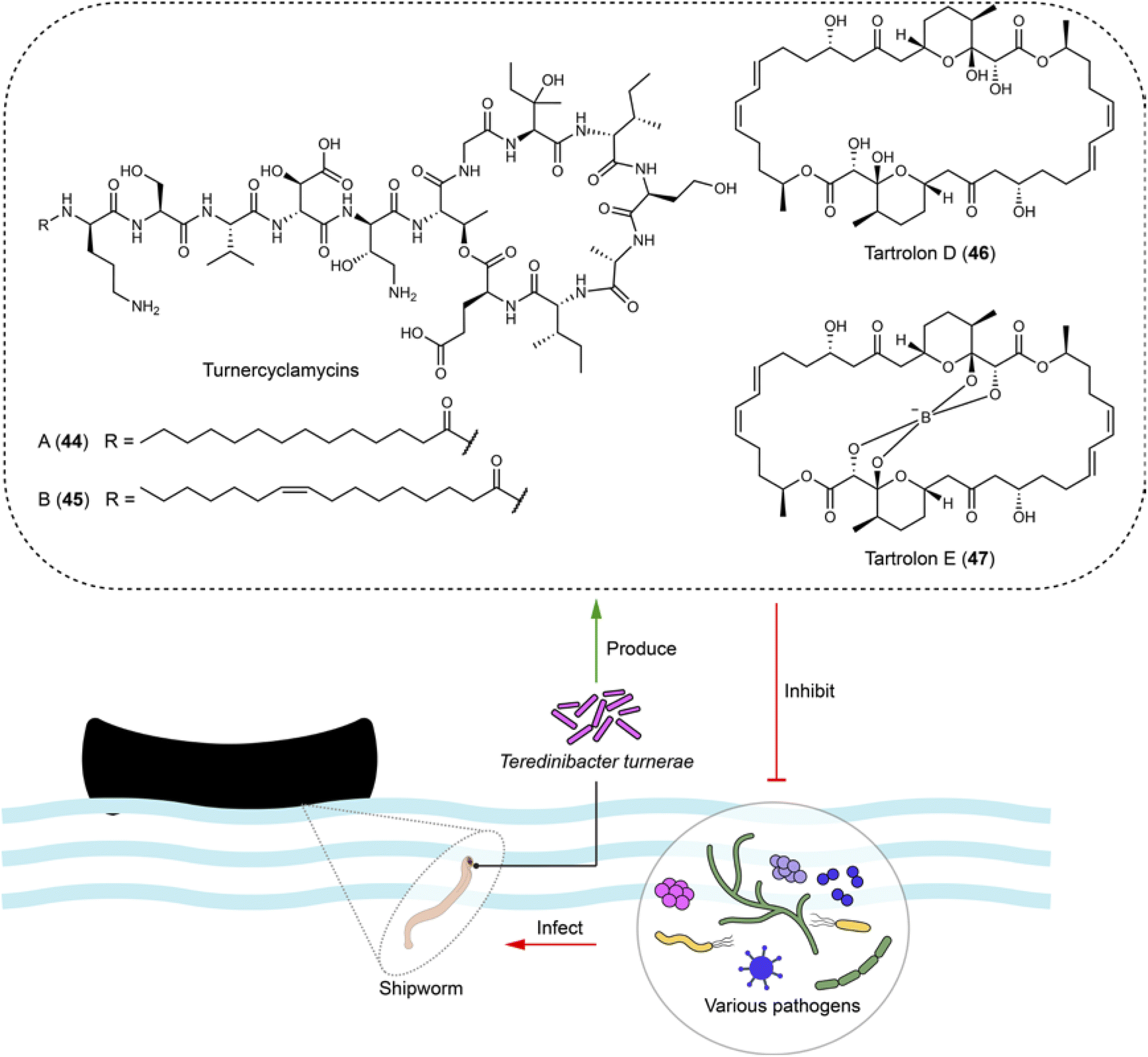 | ||
| Fig. 11 Culturable bacteria from the gills of shipworms produce broad-spectrum antibiotics (44–47), which are active against various bacterial pathogens, including those in the ESKAPE group. | ||
The macrodiolides tartrolons, including tartrolon D (46) and its boronated derivative tartrolon E (47), synthesized by the trans-AT PKS in T. turnerae exhibit broad bioactivities against competing gill symbionts and transient microbes, such as Bacillus subtili, Pseudomonas aeruginosa (MIC = 0.36 μM for 47), Vibrio anguillarum, and MRSA (MIC = 1.14 μM for 47), as well as apicomplexan parasites (EC50 = 3.1 nM for 47), potentially due to their ability to bind to boron (Fig. 11). This serves both boron detoxification in the ocean and boron transportation within the shipworm–bacteria symbiosis, and also offers a defensive mechanism against bacteria and Plasmodium.115,116
4.9 Bacteria–algae–slug triple interactions: anticancer and defensive non-ribosomal peptides delivered by predation
Kahalalides comprising over 20 variants117–122 are depsipeptides initially discovered from the sea slug Elysia rufescens. Kahalalide F (48),122 a notable member, underwent clinical trials for the treatment of esophageal and xenograft-associated cancers dominantly in 2012, with its cyclopeptide backbone being primarily responsible for the bioactivity.123 The structure feature, a hybrid of a fatty acid and a cyclic peptide with several D- and non-proteinogenic amino acid residues, led to the hypothesis that kahalalides are produced by a bacterial or fungal symbiont. Integrative analyses using metagenomic, metatranscriptomic, chemical analysis, and fluorescence microscopy have identified the intracellular bacterium Cadidatus Endobryopsis kahalalidefaciens as the authentic producer. This bacterium lives obligately within the algae Bryopsis sp., which is the diet of the slug E. rufescens. The bacterial symbiont has a reduced genome, in which 20 NRPSs account for 20%, a configuration likely to prioritize the production of kahalalides. These NRPSs are each linked to kahalalides of varying peptide chain-lengths and amino acid compositions.124 Through predating the algae Bryopsis sp., the slug accumulated kahalalides, thereby gaining a chemical defense (Fig. 12).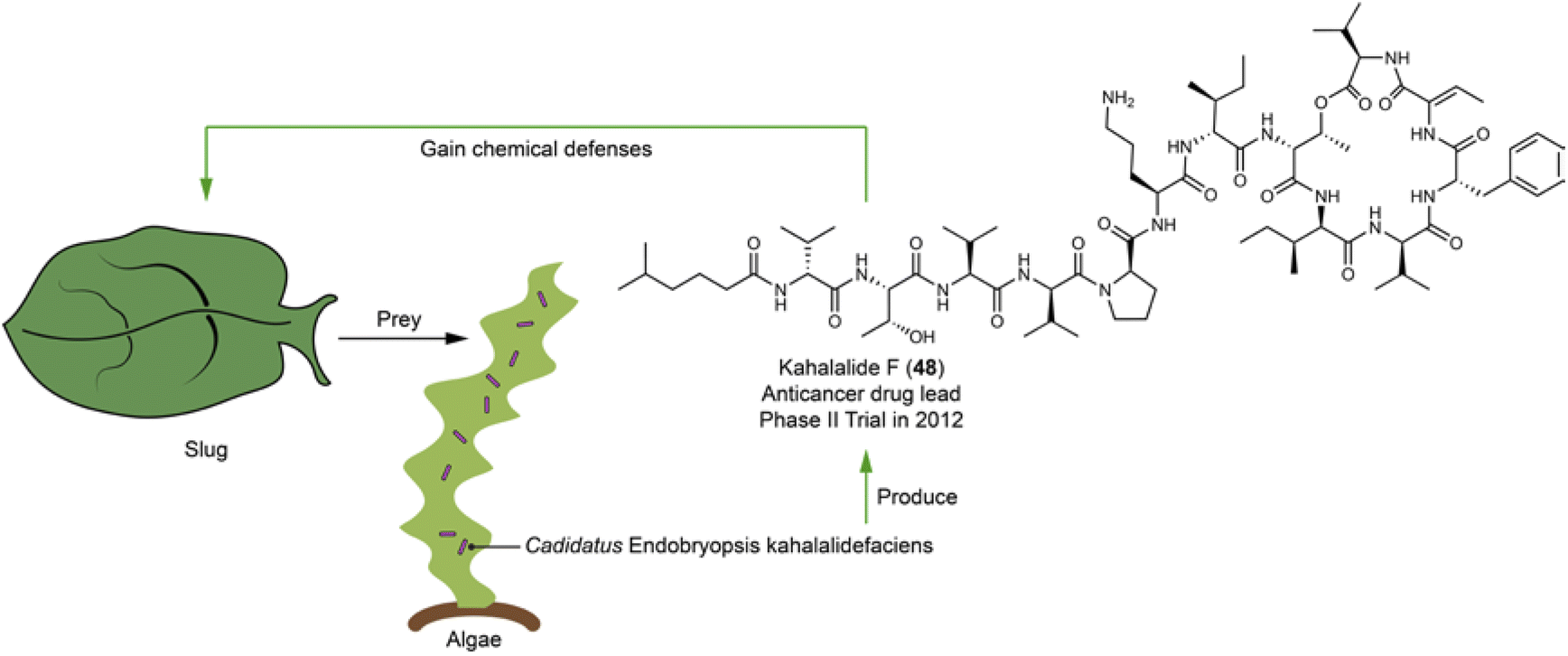 | ||
| Fig. 12 Slugs acquire chemical defensive compounds (e.g.48) by eating algae that harbor symbiotic bacterium producing kahalalides. | ||
5 Summary and outlook
The traditional approach for discovering microbial natural products involves isolating culturable microbes, often from soil or extreme environments, to identify compounds expressed in wild-type strains through bioassay-guided fractionation. This inevitably leads to the frequent rediscovery of bioactive compounds. Even in the post-genomic era, where the genome mining strategy has been extensively applied, the prevailing paradigm continues to focus on characterizing a certain BGC class across different taxa or individual BGCs within a single sequenced genome. A major limitation of this paradigm, particularly when resistance genes are not prioritized, is the lack of insights into biological and physiological functions of the biosynthesized compound(s).In contrast, the generation of functional microbial natural products necessitates interactions between the microbe and either other microbes or higher eukaryotes. This phenomenon, which demonstrates the true essence of natural products, is underappreciated yet highly likely to be ubiquitous, especially in environmental niches characterized by complex interactions among multiple organisms, such as commensalism, mutualism, exploitation, and competition. These interactions can specifically influence the diversity and evolution of biosynthetic pathways, for example, by facilitating gene transfer or promoting selective pressures that lead to new metabolic capabilities. Additionally, interactions play a critical role in determining the ecological functions and distributions of natural products, such as by affecting the survivability and competitive advantages of producer microbes within various habitats. Studying natural products within the context of organismic interactions offers unparalleled insights into the survival strategies of microbial producers with their eukaryotic hosts and against predators or competitors. It also reveals unique metabolic pathways tailored for specific purposes. These findings not only deepen our understanding of ecological dynamics but also provide new clues for drug discovery as demonstrated in this review.
To better understand and exploit the natural products from multi-organism systems, several challenges and opportunities need to be addressed. One challenge is to decipher the molecular basis of the communication and regulation between different organisms. This may require developing new methods and tools for co-cultivation, microbiome engineering, and synthetic biology. Another challenge is to identify natural products that are only expressed under certain ecological conditions or interactions, which may escape detection by conventional screening methods. This may require applying multi-omic-based approaches, including large-scale comparison of biosynthetic gene clusters, proteomics, transcriptomics, and metabolomics, to capture the dynamic changes of natural product expression and function in response to different stimuli or environments. By integrating multiple disciplines and perspectives, such as ecology, microbiology, chemistry, and bioinformatics, the study of natural products from multi-organism systems can open up new avenues for understanding and harnessing the chemical diversity and complexity of nature.
6 Data availability
No primary research results, software, or code have been included and no new data were generated or analyzed as part of this review.7 Author contributions
Yuyang Wang: investigation and writing – original draft. Yan-Ni Shi: visualization and writing – original draft. Hao Xiang: investigation and visualization. Yi-Ming Shi: conceptualization, funding acquisition, project administration, supervision, validation, writing – review & editing8 Conflicts of interest
There are no conflicts to declare.9 Acknowledgements
The authors are grateful to Prof. Helge B. Bode (Max Planck Institute for Terrestrial Microbiology) for constructive discussion. This work was supported by the National Natural Science Foundation of China (32470066).10 References
- M. I. Hutchings, A. W. Truman and B. Wilkinson, Curr. Opin. Microbiol., 2019, 51, 72–80 CrossRef CAS.
- J. Bérdy, J. Antibiot., 2005, 58, 1–26 CrossRef.
- K. Lewis, Cell, 2020, 181, 29–45 CrossRef CAS.
- A. Gavriilidou, S. A. Kautsar, N. Zaburannyi, D. Krug, R. Muller, M. H. Medema and N. Ziemert, Nat. Microbiol., 2022, 7, 726–735 CrossRef CAS.
- D. A. van Bergeijk, B. R. Terlouw, M. H. Medema and G. P. van Wezel, Nat. Rev. Microbiol., 2020, 18, 546–558 CrossRef CAS PubMed.
- E. B. Van Arnam, C. R. Currie and J. Clardy, Chem. Soc. Rev., 2018, 47, 1638–1651 RSC.
- Y.-M. Shi and H. B. Bode, Nat. Prod. Rep., 2018, 35, 309–335 RSC.
- K. Scherlach and C. Hertweck, Annu. Rev. Microbiol., 2020, 74, 267–290 CrossRef CAS.
- R. Schmidt, D. Ulanova, L. Y. Wick, H. B. Bode and P. Garbeva, ISME J., 2019, 13, 2656–2663 CrossRef PubMed.
- Y. Yan, N. Liu and Y. Tang, Nat. Prod. Rep., 2020, 37, 879–892 RSC.
- A. W. Walker and L. Hoyles, Nat. Microbiol., 2023, 8, 1392–1396 CrossRef CAS PubMed.
- J. Qin, R. Li, J. Raes, M. Arumugam, K. S. Burgdorf, C. Manichanh, T. Nielsen, N. Pons, F. Levenez, T. Yamada, D. R. Mende, J. Li, J. Xu, S. Li, D. Li, J. Cao, B. Wang, H. Liang, H. Zheng, Y. Xie, J. Tap, P. Lepage, M. Bertalan, J. M. Batto, T. Hansen, D. Le Paslier, A. Linneberg, H. B. Nielsen, E. Pelletier, P. Renault, T. Sicheritz-Ponten, K. Turner, H. Zhu, C. Yu, S. Li, M. Jian, Y. Zhou, Y. Li, X. Zhang, S. Li, N. Qin, H. Yang, J. Wang, S. Brunak, J. Dore, F. Guarner, K. Kristiansen, O. Pedersen, J. Parkhill, J. Weissenbach, H. I. T. C. Meta, P. Bork, S. D. Ehrlich and J. Wang, Nature, 2010, 464, 59–65 CrossRef CAS PubMed.
- J. A. Gilbert, M. J. Blaser, J. G. Caporaso, J. K. Jansson, S. V. Lynch and R. Knight, Nat. Med., 2018, 24, 392–400 CrossRef CAS.
- A. Almeida, A. L. Mitchell, M. Boland, S. C. Forster, G. B. Gloor, A. Tarkowska, T. D. Lawley and R. D. Finn, Nature, 2019, 568, 499–504 CrossRef CAS PubMed.
- L. Wu, Q. Zhang, Z. Deng and Y. Yu, Nat. Prod. Rep., 2022, 39, 919–925 RSC.
- M. S. Donia and M. A. Fischbach, Science, 2015, 349, 1254766 CrossRef PubMed.
- M. S. Donia, P. Cimermancic, C. J. Schulze, L. C. Wieland Brown, J. Martin, M. Mitreva, J. Clardy, R. G. Linington and M. A. Fischbach, Cell, 2014, 158, 1402–1414 CrossRef CAS.
- G. Aleti, J. L. Baker, X. Tang, R. Alvarez, M. Dinis, N. C. Tran, A. V. Melnik, C. Zhong, M. Ernst, P. C. Dorrestein and A. Edlund, mBio, 2019, 10, e00319–e00321 CrossRef.
- Y. Sugimoto, F. R. Camacho, S. Wang, P. Chankhamjon, A. Odabas, A. Biswas, P. D. Jeffrey and M. S. Donia, Science, 2019, 366, eaax9176 CrossRef CAS.
- A. M. King, Z. Zhang, E. Glassey, P. Siuti, J. Clardy and C. A. Voigt, Nat. Microbiol., 2023, 8, 2420–2434 CrossRef CAS PubMed.
- B. Krismer, M. Liebeke, D. Janek, M. Nega, M. Rautenberg, G. Hornig, C. Unger, C. Weidenmaier, M. Lalk and A. Peschel, PLoS Pathog., 2014, 10, e1003862 CrossRef PubMed.
- A. Zipperer, M. C. Konnerth, C. Laux, A. Berscheid, D. Janek, C. Weidenmaier, M. Burian, N. A. Schilling, C. Slavetinsky, M. Marschal, M. Willmann, H. Kalbacher, B. Schittek, H. Brotz-Oesterhelt, S. Grond, A. Peschel and B. Krismer, Nature, 2016, 535, 511–516 CrossRef CAS PubMed.
- B. O. Torres Salazar, T. Dema, N. A. Schilling, D. Janek, J. Bornikoel, A. Berscheid, A. M. A. Elsherbini, S. Krauss, S. J. Jaag, M. Lammerhofer, M. Li, N. Alqahtani, M. J. Horsburgh, T. Weber, J. M. Beltran-Belena, H. Brotz-Oesterhelt, S. Grond, B. Krismer and A. Peschel, Nat. Microbiol., 2024, 9, 200–213 CrossRef CAS.
- H. F. Wertheim, D. C. Melles, M. C. Vos, W. van Leeuwen, A. van Belkum, H. A. Verbrugh and J. L. Nouwen, Lancet Infect. Dis., 2005, 5, 751–762 CrossRef.
- N. A. Schilling, A. Berscheid, J. Schumacher, J. S. Saur, M. C. Konnerth, S. N. Wirtz, J. M. Beltran-Belena, A. Zipperer, B. Krismer, A. Peschel, H. Kalbacher, H. Brotz-Oesterhelt, C. Steinem and S. Grond, Angew. Chem., Int. Ed., 2019, 58, 9234–9238 CrossRef CAS.
- K. Bitschar, B. Sauer, J. Focken, H. Dehmer, S. Moos, M. Konnerth, N. A. Schilling, S. Grond, H. Kalbacher, F. C. Kurschus, F. Gotz, B. Krismer, A. Peschel and B. Schittek, Nat. Commun., 2019, 10, 2730 CrossRef.
- H. J. Kim, A. Jo, Y. J. Jeon, S. An, K. M. Lee, S. S. Yoon and J. Y. Choi, Microbiome, 2019, 7, 80 CrossRef.
- Y. Lin, X. Liang, Z. Li, T. Gong, B. Ren, Y. Li and X. Peng, Int. J. Oral Sci., 2024, 16, 2 CrossRef PubMed.
- J. L. Baker, J. L. Mark Welch, K. M. Kauffman, J. S. McLean and X. He, Nat. Rev. Microbiol., 2024, 22, 89–104 CrossRef CAS.
- S. Brouwer, T. Rivera-Hernandez, B. F. Curren, N. Harbison-Price, D. M. P. De Oliveira, M. G. Jespersen, M. R. Davies and M. J. Walker, Nat. Rev. Microbiol., 2023, 21, 431–447 CrossRef CAS PubMed.
- O. Hyink, P. A. Wescombe, M. Upton, N. Ragland, J. P. Burton and J. R. Tagg, Appl. Environ. Microbiol., 2007, 73, 1107–1113 CrossRef CAS PubMed.
- A. Barbour, L. Smith, M. Oveisi, M. Williams, R. C. Huang, C. Marks, N. Fine, C. Sun, F. Younesi, S. Zargaran, R. Orugunty, T. D. Horvath, S. J. Haidacher, A. M. Haag, A. Sabharwal, B. Hinz and M. Glogauer, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2219392120 CrossRef CAS.
- A. Barbour, P. Wescombe and L. Smith, Trends Microbiol., 2020, 28, 578–593 CrossRef CAS.
- H. Do, Z. R. Li, P. K. Tripathi, S. Mitra, S. Guerra, A. Dash, D. Weerasekera, N. Makthal, S. Shams, S. Aggarwal, B. B. Singh, D. Gu, Y. Du, R. J. Olsen, C. LaRock, W. Zhang and M. Kumaraswami, Nat. Microbiol., 2024, 9, 502–513 CrossRef PubMed.
- M. Sassone-Corsi and M. Raffatellu, J. Immunol., 2015, 194, 4081–4087 CrossRef CAS.
- P. J. Turnbaugh, R. E. Ley, M. Hamady, C. M. Fraser-Liggett, R. Knight and J. I. Gordon, Nature, 2007, 449, 804–810 CrossRef CAS.
- W. J. Y. Chee, S. Y. Chew and L. T. L. Than, Microb. Cell Fact., 2020, 19, 203 CrossRef.
- J. A. Leeds, Cold Spring Harbor Perspect. Med., 2016, 6, a025445 CrossRef.
- J. R. Patel, J. Oh, S. Wang, J. M. Crawford and F. J. Isaacs, Cell, 2022, 185, 1487–1505 CrossRef CAS.
- H. M. P. R. N. C. Integrative, Nature, 2019, 569, 641–648 CrossRef.
- Y. Ismail, V. Mahendran, S. Octavia, A. S. Day, S. M. Riordan, M. C. Grimm, R. Lan, D. Lemberg, T. A. Tran and L. Zhang, PLoS One, 2012, 7, e38217 CrossRef CAS PubMed.
- J. J. Farrell, L. Zhang, H. Zhou, D. Chia, D. Elashoff, D. Akin, B. J. Paster, K. Joshipura and D. T. W. Wong, Gut, 2012, 61, 582–588 CrossRef CAS PubMed.
- B. Li, Y. Ge, L. Cheng, B. Zeng, J. Yu, X. Peng, J. Zhao, W. Li, B. Ren, M. Li, H. Wei and X. Zhou, Int. J. Oral Sci., 2019, 11, 10 CrossRef.
- T. Nakatsuji, T. H. Chen, S. Narala, K. A. Chun, A. M. Two, T. Yun, F. Shafiq, P. F. Kotol, A. Bouslimani, A. V. Melnik, H. Latif, J.-N. Kim, A. Lockhart, K. Artis, G. David, P. Taylor, J. Streib, P. C. Dorrestein, A. Grier, S. R. Gill, K. Zengler, T. R. Hata, D. Y. M. Leung and R. L. Gallo, Sci. Transl. Med., 2017, 9, eaah4680 CrossRef PubMed.
- J. Claesen, J. B. Spagnolo, S. F. Ramos, K. L. Kurita, A. L. Byrd, A. A. Aksenov, A. V. Melnik, W. R. Wong, S. Wang, R. D. Hernandez, M. S. Donia, P. C. Dorrestein, H. H. Kong, J. A. Segre, R. G. Linington, M. A. Fischbach and K. P. Lemon, Sci. Transl. Med., 2020, 12, eaay5445 CrossRef CAS.
- J.-P. Claudel, N. Auffret, M.-T. Leccia, F. Poli, S. Corvec and B. Dréno, Dermatology, 2019, 235, 287–294 CrossRef PubMed.
- C.-J. Guo, F.-Y. Chang, T. P. Wyche, K. M. Backus, T. M. Acker, M. Funabashi, M. Taketani, M. S. Donia, S. Nayfach, K. S. Pollard, C. S. Craik, B. F. Cravatt, J. Clardy, C. A. Voigt and M. A. Fischbach, Cell, 2017, 168, 517–526 CrossRef CAS.
- S. A. Joyce, A. O. Brachmann, I. Glazer, L. Lango, G. Schwär, D. J. Clarke and H. B. Bode, Angew. Chem., Int. Ed., 2008, 47, 1942–1945 CrossRef CAS.
- Y.-M. Shi, M. Hirschmann, Y.-N. Shi, S. Ahmed, D. Abebew, N. J. Tobias, P. Grün, J. J. Crames, L. Pöschel, W. Kuttenlochner, C. Richter, J. Herrmann, R. Müller, A. Thanwisai, S. J. Pidot, T. P. Stinear, M. Groll, Y. Kim and H. B. Bode, Nat. Chem., 2022, 14, 701–712 CrossRef CAS.
- Y.-M. Shi, M. Hirschmann, Y.-N. Shi and H. B. Bode, ACS Chem. Biol., 2022, 17, 2221–2228 CrossRef CAS PubMed.
- Y.-M. Shi, J. J. Crames, L. Czech, K. A. J. Bozhüyük, Y.-N. Shi, M. Hirschmann, S. Lamberth, P. Claus, N. Paczia, C. Rückert, J. Kalinowski, G. Bange and H. B. Bode, Angew. Chem., Int. Ed., 2022, 61, e202206106 CrossRef CAS.
- R. D. Miller, A. Iinishi, S. M. Modaresi, B.-K. Yoo, T. D. Curtis, P. J. Lariviere, L. Liang, S. Son, S. Nicolau, R. Bargabos, M. Morrissette, M. F. Gates, N. Pitt, R. P. Jakob, P. Rath, T. Maier, A. G. Malyutin, J. T. Kaiser, S. Niles, B. Karavas, M. Ghiglieri, S. E. J. Bowman, D. C. Rees, S. Hiller and K. Lewis, Nat. Microbiol., 2022, 7, 1661–1672 CrossRef CAS.
- Y. Imai, G. Hauk, J. Quigley, L. Liang, S. Son, M. Ghiglieri, M. F. Gates, M. Morrissette, N. Shahsavari, S. Niles, D. Baldisseri, C. Honrao, X. Ma, J. J. Guo, J. M. Berger and K. Lewis, Nat. Chem. Biol., 2022, 18, 1236–1244 CrossRef CAS.
- Y.-M. Shi, A. O. Brachmann, M. A. Westphalen, N. Neubacher, N. J. Tobias and H. B. Bode, Nat. Chem. Biol., 2019, 15, 331–339 CrossRef CAS PubMed.
- Y. Imai, K. J. Meyer, A. Iinishi, Q. Favre-Godal, R. Green, S. Manuse, M. Caboni, M. Mori, S. Niles, M. Ghiglieri, C. Honrao, X. Ma, J. Guo, A. Makriyannis, L. Linares-Otoya, N. Bohringer, Z. G. Wuisan, H. Kaur, R. Wu, A. Mateus, A. Typas, M. M. Savitski, J. L. Espinoza, A. O'Rourke, K. E. Nelson, S. Hiller, N. Noinaj, T. F. Schaberle, A. D'Onofrio and K. Lewis, Nature, 2019, 576, 459–464 CrossRef CAS.
- L. Pantel, T. Florin, M. Dobosz-Bartoszek, E. Racine, M. Sarciaux, M. Serri, J. Houard, J. M. Campagne, R. M. de Figueiredo, C. Midrier, S. Gaudriault, A. Givaudan, A. Lanois, S. Forst, A. Aumelas, C. Cotteaux-Lautard, J. M. Bolla, C. Vingsbo Lundberg, D. L. Huseby, D. Hughes, P. Villain-Guillot, A. S. Mankin, Y. S. Polikanov and M. Gualtieri, Mol. Cell, 2018, 70, 83–94 CrossRef CAS.
- D. Abebew, F. S. Sayedain, E. Bode and H. B. Bode, J. Agric. Food Chem., 2022, 70, 498–506 CrossRef CAS.
- R. A. R. Machado, L. Thönen, C. C. M. Arce, V. Theepan, F. Prada, D. Wüthrich, C. A. M. Robert, E. Vogiatzaki, Y.-M. Shi, O. P. Schaeren, M. Notter, R. Bruggmann, S. Hapfelmeier, H. B. Bode and M. Erb, Nat. Biotechnol., 2020, 38, 600–608 CrossRef CAS.
- E. Bode, A. K. Heinrich, M. Hirschmann, D. Abebew, Y.-N. Shi, T. D. Vo, F. Wesche, Y.-M. Shi, P. Grün, S. Simonyi, N. Keller, Y. Engel, S. Wenski, R. Bennet, S. Beyer, I. Bischoff, A. Buaya, S. Brandt, I. Cakmak, H. Çimen, S. Eckstein, D. Frank, R. Fürst, M. Gand, G. Geisslinger, S. Hazir, M. Henke, R. Heermann, V. Lecaudey, W. Schäfer, S. Schiffmann, A. Schüffler, R. Schwenk, M. Skaljac, E. Thines, M. Thines, T. Ulshöfer, A. Vilcinskas, T. A. Wichelhaus and H. B. Bode, Angew. Chem., Int. Ed., 2019, 58, 18957–18963 CrossRef CAS.
- N. J. Tobias, H. Wolff, B. Djahanschiri, F. Grundmann, M. Kronenwerth, Y.-M. Shi, S. Simonyi, P. Grün, D. Shapiro-Ilan, S. J. Pidot, T. P. Stinear, I. Ebersberger and H. B. Bode, Nat. Microbiol., 2017, 2, 1676–1685 CrossRef CAS.
- J. Parkinson, M. Mitreva, C. Whitton, M. Thomson, J. Daub, J. Martin, R. Schmid, N. Hall, B. Barrell, R. H. Waterston, J. P. McCarter and M. L. Blaxter, Nat. Genet., 2004, 36, 1259–1267 CrossRef PubMed.
- Drug Trials Snapshots: VTAMA, https://www.fda.gov/drugs/drug-approvals-and-databases/drug-trials-snapshots-vtama Search PubMed.
- H. Morita, H. Noguchi, J. Schroder and I. Abe, Eur. J. Biochem., 2001, 268, 3759–3766 CrossRef CAS.
- M. Kronenwerth, A. O. Brachmann, M. Kaiser and H. B. Bode, Chembiochem, 2014, 15, 2689–2691 CrossRef CAS PubMed.
- A. O. Brachmann, D. Reimer, W. Lorenzen, E. Augusto Alonso, Y. Kopp, J. Piel and H. B. Bode, Angew. Chem., Int. Ed., 2012, 51, 12086–12089 CrossRef CAS PubMed.
- S. Kavakli, G. L. C. Grammbitter and H. B. Bode, Tetrahedron, 2022, 128, 133116 CrossRef CAS.
- V. Aoki and R. L. Orfali, J. Eur. Acad. Dermatol. Venereol., 2023, 37, 1093–1094 CrossRef CAS PubMed.
- S. H. Smith, C. Jayawickreme, D. J. Rickard, E. Nicodeme, T. Bui, C. Simmons, C. M. Coquery, J. Neil, W. M. Pryor, D. Mayhew, D. K. Rajpal, K. Creech, S. Furst, J. Lee, D. Wu, F. Rastinejad, T. M. Willson, F. Viviani, D. C. Morris, J. T. Moore and J. Cote-Sierra, J. Invest. Dermatol., 2017, 137, 2110–2119 CrossRef CAS PubMed.
- P. Moura-Alves, A. Puyskens, A. Stinn, M. Klemm, U. Guhlich-Bornhof, A. Dorhoi, J. Furkert, A. Kreuchwig, J. Protze, L. Lozza, G. Pei, P. Saikali, C. Perdomo, H. J. Mollenkopf, R. Hurwitz, F. Kirschhoefer, G. Brenner-Weiss, J. Weiner 3rd, H. Oschkinat, M. Kolbe, G. Krause and S. H. E. Kaufmann, Science, 2019, 366, eaaw1629 CrossRef CAS.
- A. Lanois-Nouri, L. Pantel, J. Fu, J. Houard, J.-C. Ogier, S. Polikanov Yury, E. Racine, H. Wang, S. Gaudriault, A. Givaudan and M. Gualtieri, mBio, 2022, 13, e02821–e02826 CrossRef.
- M. Sarciaux, L. Pantel, C. Midrier, M. Serri, C. Gerber, R. Marcia de Figueiredo, J. M. Campagne, P. Villain-Guillot, M. Gualtieri and E. Racine, J. Med. Chem., 2018, 61, 7814–7826 CrossRef CAS PubMed.
- N. Bohringer, R. Green, Y. Liu, U. Mettal, M. Marner, S. M. Modaresi, R. P. Jakob, Z. G. Wuisan, T. Maier, A. Iinishi, S. Hiller, K. Lewis and T. F. Schaberle, Microbiol. Spectrum, 2021, 9, e0153521 CrossRef.
- Z. G. Wuisan, I. D. M. Kresna, N. Bohringer, K. Lewis and T. F. Schaberle, Metab. Eng., 2021, 66, 123–136 CrossRef CAS.
- S. Guo, S. Wang, S. Ma, Z. Deng, W. Ding and Q. Zhang, Nat. Commun., 2022, 13, 2361 CrossRef CAS.
- H. Nguyen, I. D. Made Kresna, N. Bohringer, J. Ruel, E. Mora, J. C. Kramer, K. Lewis, Y. Nicolet, T. F. Schaberle and K. Yokoyama, J. Am. Chem. Soc., 2022, 144, 18876–18886 CrossRef CAS PubMed.
- H. Kaur, R. P. Jakob, J. K. Marzinek, R. Green, Y. Imai, J. R. Bolla, E. Agustoni, C. V. Robinson, P. J. Bond, K. Lewis, T. Maier and S. Hiller, Nature, 2021, 593, 125–129 CrossRef CAS PubMed.
- C. E. Seyfert, C. Porten, B. Yuan, S. Deckarm, F. Panter, C. D. Bader, J. Coetzee, F. Deschner, K. Tehrani, P. G. Higgins, H. Seifert, T. C. Marlovits, J. Herrmann and R. Muller, Angew. Chem., Int. Ed., 2023, 62, e202214094 CrossRef CAS PubMed.
- S. Gross, F. Panter, D. Pogorevc, C. E. Seyfert, S. Deckarm, C. D. Bader, J. Herrmann and R. Muller, Chem. Sci., 2021, 12, 11882–11893 RSC.
- C. E. Seyfert, A. V. Müller, D. J. Walsh, J. Birkelbach, A. M. Kany, C. Porten, B. Yuan, D. Krug, J. Herrmann, T. C. Marlovits, A. K. H. Hirsch and R. Müller, J. Med. Chem., 2023, 66, 16330–16341 CrossRef CAS PubMed.
- Y. C. Lin, F. Schneider, K. J. Eberle, D. Chiodi, H. Nakamura, S. H. Reisberg, J. Chen, M. Saito and P. S. Baran, J. Am. Chem. Soc., 2022, 144, 14458–14462 CrossRef CAS PubMed.
- M. Nesic, D. B. Ryffel, J. Maturano, M. Shevlin, S. R. Pollack, D. R. Gauthier Jr, P. Trigo-Mourino, L. K. Zhang, D. M. Schultz, J. M. McCabe Dunn, L. C. Campeau, N. R. Patel, D. A. Petrone and D. Sarlah, J. Am. Chem. Soc., 2022, 144, 14026–14030 CrossRef CAS.
- N. Böhringer, J.-C. Kramer, E. de la Mora, L. Padva, Z. G. Wuisan, Y. Liu, M. Kurz, M. Marner, H. Nguyen, P. Amara, K. Yokoyama, Y. Nicolet, U. Mettal and T. F. Schäberle, Cell Chem. Biol., 2023, 30, 943–952 CrossRef PubMed.
- S. Ma, W. Xi, S. Wang, H. Chen, S. Guo, T. Mo, W. Chen, Z. Deng, F. Chen, W. Ding and Q. Zhang, J. Am. Chem. Soc., 2023, 145, 22945–22953 CrossRef CAS PubMed.
- S. Schmidt, S. Kildgaard, H. Guo, C. Beemelmanns and M. Poulsen, Nat. Prod. Rep., 2022, 39, 231–248 RSC.
- S. P. N. Hannah Büttnera, K. Vandelannoote, Z. Cseresnyés, B. Dose, I. Richter, R. Gerst, M. Thilo Figge, T. P. Stinear, S. J. Pidot and a. C. Hertweck, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2110669118 CrossRef PubMed.
- H. Buttner, S. J. Pidot, K. Scherlach and C. Hertweck, Chem. Sci., 2022, 14, 103–112 RSC.
- S. R. Ingrid Richter, Z. Cseresnyés, I. Ferling, H. Büttner, S. P. Niehs, R. Gerst, K. Scherlach, M. Thilo Figge, F. Hillmann and C. Hertweck, mBio, 2022, 13, e01440 Search PubMed.
- M. Shao, C. Sun, X. Liu, X. Wang, W. Li, X. Wei, Q. Li and J. Ju, Commun. Biol., 2020, 3, 527 CrossRef CAS PubMed.
- S. Xu, Y. X. Liu, T. Cernava, H. Wang, Y. Zhou, T. Xia, S. Cao, G. Berg, X. X. Shen, Z. Wen, C. Li, B. Qu, H. Ruan, Y. Chai, X. Zhou, Z. Ma, Y. Shi, Y. Yu, Y. Bai and Y. Chen, Nat. Microbiol., 2022, 7, 831–843 CrossRef CAS PubMed.
- Y. Chen, J. Wang, N. Yang, Z. Wen, X. Sun, Y. Chai and Z. Ma, Nat. Commun., 2018, 9, 3429 CrossRef PubMed.
- M. Fujioka, S. Koda, Y. Morimoto and K. Biemann, J. Org. Chem., 2002, 53, 2820–2825 CrossRef.
- M. Crusemann, R. Reher, I. Schamari, A. O. Brachmann, T. Ohbayashi, M. Kuschak, D. Malfacini, A. Seidinger, M. Pinto-Carbo, R. Richarz, T. Reuter, S. Kehraus, A. Hallab, M. Attwood, H. B. Schioth, P. Mergaert, Y. Kikuchi, T. F. Schaberle, E. Kostenis, D. Wenzel, C. E. Muller, J. Piel, A. Carlier, L. Eberl and G. M. Konig, Angew. Chem., Int. Ed., 2018, 57, 836–840 CrossRef PubMed.
- W. Hanke, J. Alenfelder, J. Liu, P. Gutbrod, S. Kehraus, M. Crüsemann, P. Dörmann, E. Kostenis, M. Scholz and G. M. König, J. Chem. Ecol., 2023, 49, 549–569 CrossRef CAS.
- C. Hermes, R. Richarz, D. A. Wirtz, J. Patt, W. Hanke, S. Kehraus, J. H. Voss, J. Kuppers, T. Ohbayashi, V. Namasivayam, J. Alenfelder, A. Inoue, P. Mergaert, M. Gutschow, C. E. Muller, E. Kostenis, G. M. Konig and M. Crusemann, Nat. Commun., 2021, 12, 144 CrossRef CAS.
- J. G. Schlegel, M. Tahoun, A. Seidinger, J. H. Voss, M. Kuschak, S. Kehraus, M. Schneider, M. Matthey, B. K. Fleischmann, G. M. Konig, D. Wenzel and C. E. Muller, ACS Pharmacol. Transl. Sci., 2021, 4, 888–897 CrossRef CAS.
- R. Schrage, A. L. Schmitz, E. Gaffal, S. Annala, S. Kehraus, D. Wenzel, K. M. Bullesbach, T. Bald, A. Inoue, Y. Shinjo, S. Galandrin, N. Shridhar, M. Hesse, M. Grundmann, N. Merten, T. H. Charpentier, M. Martz, A. J. Butcher, T. Slodczyk, S. Armando, M. Effern, Y. Namkung, L. Jenkins, V. Horn, A. Stossel, H. Dargatz, D. Tietze, D. Imhof, C. Gales, C. Drewke, C. E. Muller, M. Holzel, G. Milligan, A. B. Tobin, J. Gomeza, H. G. Dohlman, J. Sondek, T. K. Harden, M. Bouvier, S. A. Laporte, J. Aoki, B. K. Fleischmann, K. Mohr, G. M. Konig, T. Tuting and E. Kostenis, Nat. Commun., 2015, 6, 10156 CrossRef CAS PubMed.
- E. Kostenis, E. M. Pfeil and S. Annala, J. Biol. Chem., 2020, 295, 5206–5215 CrossRef CAS PubMed.
- S. Gotze, R. Vij, K. Burow, N. Thome, L. Urbat, N. Schlosser, S. Pflanze, R. Muller, V. G. Hansch, K. Schlabach, L. Fazlikhani, G. Walther, H. M. Dahse, L. Regestein, S. Brunke, B. Hube, C. Hertweck, P. Franken and P. Stallforth, J. Am. Chem. Soc., 2023, 145, 2342–2353 CrossRef.
- S. Zhang, R. Mukherji, S. Chowdhury, L. Reimer and P. Stallforth, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2013759118 CrossRef CAS.
- J. Arp, S. Gotze, R. Mukherji, D. J. Mattern, M. Garcia-Altares, M. Klapper, D. A. Brock, A. A. Brakhage, J. E. Strassmann, D. C. Queller, B. Bardl, K. Willing, G. Peschel and P. Stallforth, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 3758–3763 CrossRef CAS.
- D. Rodríguez-Hernández, W. G. P. Melo, C. Menegatti, V. B. Lourenzon, F. S. do Nascimento and M. T. Pupo, New J. Chem., 2019, 43, 10109–10117 RSC.
- H. Lang, Y. Liu, H. Duan, W. Zhang, X. Hu and H. Zheng, Nat. Commun., 2023, 14, 7650 CrossRef CAS PubMed.
- L. V. Florez, K. Scherlach, I. J. Miller, A. Rodrigues, J. C. Kwan, C. Hertweck and M. Kaltenpoth, Nat. Commun., 2018, 9, 2478 CrossRef.
- S. P. Niehs, J. Kumpfmuller, B. Dose, R. F. Little, K. Ishida, L. V. Florez, M. Kaltenpoth and C. Hertweck, Angew. Chem., Int. Ed., 2020, 59, 23122–23126 CrossRef CAS PubMed.
- S. C. Waterworth, L. V. Florez, E. R. Rees, C. Hertweck, M. Kaltenpoth and J. C. Kwan, mBio, 2020, 11(1), e02430 CrossRef CAS.
- D. C. Oh, J. J. Scott, C. R. Currie and J. Clardy, Org. Lett., 2009, 11, 633–636 CrossRef CAS.
- J. G. Ganley, A. Pandey, K. Sylvester, K. Y. Lu, M. Toro-Moreno, S. Rutschlin, J. M. Bradford, C. J. Champion, T. Bottcher, J. Xu and E. R. Derbyshire, Cell Chem. Biol., 2020, 27, 817–826 CrossRef CAS PubMed.
- J. G. Ganley, G. Carr, T. R. Ioerger, J. C. Sacchettini, J. Clardy and E. R. Derbyshire, Chembiochem, 2018, 19, 1590–1594 CrossRef CAS PubMed.
- S. Wang, A. L. A. Dos-Santos, W. Huang, K. C. Liu, M. A. Oshaghi, G. Wei, P. Agre and M. Jacobs-Lorena, Science, 2017, 357, 1399–1402 CrossRef CAS PubMed.
- M. D. Tianero, J. N. Balaich and M. S. Donia, Nat. Microbiol., 2019, 4, 1149–1159 CrossRef CAS.
- B. W. Miller, A. L. Lim, Z. Lin, J. Bailey, K. L. Aoyagi, M. A. Fisher, L. R. Barrows, C. Manoil, E. W. Schmidt and M. G. Haygood, Cell Chem. Biol., 2021, 28, 1628–1637 CrossRef CAS.
- J. M. D. Robes, M. A. Altamia, E. G. Murdock, G. P. Concepcion, M. G. Haygood and A. W. Puri, Appl. Environ. Microbiol., 2022, 88, e00222–e00270 CrossRef.
- M. A. Altamia, Z. Lin, A. E. Trindade-Silva, I. D. Uy, J. R. Shipway, D. V. Wilke, G. P. Concepcion, D. L. Distel, E. W. Schmidt and M. G. Haygood, mSystems, 2020, 5(3), e00261 CrossRef CAS PubMed.
- A. L. Lim, B. W. Miller, Z. Lin, M. A. Fisher, L. R. Barrows, M. G. Haygood, E. W. Schmidt and A. Kumar, Microbiol. Spectrum, 2023, 11, e02306 Search PubMed.
- R. M. O'Connor, V. F. Nepveux, J. Abenoja, G. Bowden, P. Reis, J. Beaushaw, R. M. Bone Relat, I. Driskell, F. Gimenez, M. W. Riggs, D. A. Schaefer, E. W. Schmidt, Z. Lin, D. L. Distel, J. Clardy, T. R. Ramadhar, D. R. Allred, H. M. Fritz, P. Rathod, L. Chery and J. White, PLoS Pathog., 2020, 16, e1008600 CrossRef.
- S. I. Elshahawi, A. E. Trindade-Silva, A. Hanora, A. W. Han, M. S. Flores, V. Vizzoni, C. G. Schrago, C. A. Soares, G. P. Concepcion, D. L. Distel, E. W. Schmidt and M. G. Haygood, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E295–E304 CrossRef CAS PubMed.
- S. Komori, S. Yamabe, R. Matsuta, Y. Yamazaki, M. Fukuoka, S. Sato and K. Takada, Biosci., Biotechnol., Biochem., 2024, 88, 399–404 CrossRef.
- M. A. Albadry, K. M. Elokely, B. Wang, J. J. Bowling, M. F. Abdelwahab, M. H. Hossein, R. J. Doerksen and M. T. Hamann, J. Nat. Prod., 2013, 76, 178–185 CrossRef CAS PubMed.
- M. Serova, A. de Gramont, I. Bieche, M. Riveiro, C. Galmarini, M. Aracil, J. Jimeno, S. Faivre and E. Raymond, Mar. Drugs, 2013, 11, 944–959 CrossRef CAS.
- J. Gao and M. T. Hamann, Chem. Rev., 2011, 111, 3208–3235 CrossRef CAS.
- M. L. Ciavatta, P. Devi, M. Carbone, V. Mathieu, R. Kiss, A. Casapullo and M. Gavagnin, Tetrahedron, 2016, 72, 625–631 CrossRef CAS.
- M. T. Hamann and P. J. Scheuer, J. Am. Chem. Soc., 1993, 115, 5825–5826 CrossRef CAS.
- J. C. Jimenez, A. Lopez-Macia, C. Gracia, S. Varon, M. Carrascal, J. M. Caba, M. Royo, A. M. Francesch, C. Cuevas, E. Giralt and F. Albericio, J. Med. Chem., 2008, 51, 4920–4931 CrossRef CAS PubMed.
- J. Zan, Z. Li, M. D. Tianero, J. Davis, R. T. Hill and M. S. Donia, Science, 2019, 364, eaaw6732 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |