
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Excited state dynamics of homoleptic Zn(II)dipyrrin complexes and their application in photocatalysis†
Julia
Leier
a,
Pascal
Rauthe
a,
Roberta
Tabone
b,
Claudia
Bizzarri
 *b and
Andreas-Neil
Unterreiner
*a
*b and
Andreas-Neil
Unterreiner
*a
aKarlsruhe Institute of Technology, Institute of Physical Chemistry, 76131 Karlsruhe, Germany. E-mail: andreas.unterreiner@kit.edu
bKarlsruhe Institute of Technology, Institute of Organic Chemistry, 76131 Karlsruhe, Germany. E-mail: claudia.bizzarri@kit.edu
First published on 10th July 2024
Abstract
We present photophysical studies on homoleptic zinc-dipyrrin complexes representing intramolecular donor–acceptor systems. Intramolecular charge transfer depends on ligand variation and intermolecular environmental influences. In particular, we focus on steady-state and transient absorption methods to compare the response of the ligand dynamics with that of the complexes after photoexcitation. For the first time, we successfully tested zinc bis(dipyrrinato) complexes as photocatalysts in two different aerobic photooxidation reactions. Early-time dynamics after 100 fs in dichloromethane and in ethanol can be assigned to charge transfer states, while the subsequent response constitutes broad-band excited state dynamics, most likely of overlapping singlet and triplet states when probed in a relatively wide spectral window from 350 to 1400 nm and up to a delay time of 1.6 ns. In addition, a strong correlation between steady-state and time-resolved experiments makes this experimental approach a versatile tool for future investigations of this class of molecules, especially with respect to photocatalysis. The results of this study underpin the need for further development for quantifying amplitudes of the excited state population of electronically excited singlet and triplet states.
1. Introduction
Dipyrromethane derivatives exhibit strong absorption bands in the visible wavelength range.1–4 The best-known are boron dipyrromethene (BODIPY) derivatives, characterized by particularly high quantum yields.5–7 Dipyrrinato metal complexes, on the other hand, have received less attention than their boron-containing counterparts because their molecular structure can lead to various non-radiative relaxation pathways and thus, lower quantum yields.8–10 Bis(dipyrrinato) zinc(II) complexes typically absorb in the blue to green spectral range (480–550 nm) with high extinction coefficients.11,12 In addition, rigid complexes are generally similar to the already well-studied BODIPY complexes with comparable quantum yields.13–15 One possibility to further increase the fluorescence is to fine-tune the ligands. It can be increased by choosing heteroleptic instead of homoleptic complexes and by substitution with large ligands.11,16 In addition, intermolecular interactions with the solvent are also crucial.17Current studies focus on the investigation of photoinduced processes, especially the fluorescence properties13,18–20 and the non-radiative relaxation pathway of charge transfer (CT).17,19,21–24 A zinc-based CT is unlikely due to a d10 configuration and redox inertness.18,25 Both dipyrrin ligands, however, allow an intramolecular CT (ICT) to enable a charge separated (CS) state. The formation of a charge separated state is relevant for various applications where a charge transfer process is involved, as in the case of solar energy conversion.26,27 The symmetric zinc dipyrrins are particularly interesting, which can subvert photoinduced ICT by symmetry breaking (SBCT). SBCTs are generally dependent on the polarity of the solvent and are stabilized in polar solvents.18,21,22 In addition, further non-radiative deactivations are known, such as a rotation of the phenyl ring of the dipyrrin ligand28 or a thermal energy release of a locally excited (LE) state of a single dipyrrin ligand, leading to a non-radiative state.11,19
In polar environments, the S1 state exhibits a slight charge separation, so it stands to reason that this is present in the polar S1 state. In the nonpolar solvent, however, molecular symmetry is preserved.19 In addition, a CS state is known to form a long-lived triplet state.18 This is made possible by charge recombination.17,19 In the literature, this new state is called the CS11,19 or the CT state.17,21,23 A method to circumvent the population of the nonemissive CT state is usually achieved by breaking the symmetry of the complex in its ground state, that is by using two different ligands (heteroleptic complexes),11,13,16,17,21,22,29 or specific ligand design to allow an intraligand charge transfer,30,31 allowing intense luminescence (quantum yields up to 90%) also in polar solvents.
Although zinc complexes have been attracted an increasing number of recent works to understand their photophysical behaviour, their potential role as photocatalysts (PCs) has been quite under evaluated32,33 in comparison to BODIPY, for which several publications vouched for a vast employment in this regard.7,34–37
In the present work, the behaviour of zinc-dipyrrin complexes was investigated as a function of intra- and intermolecular environmental influences. Fine adjustment of the dipyrrin ligands affects the corresponding photophysical properties of the zinc complexes. First, analyses of two homoleptic complexes (3–4) and two ligands (1–2) (see Fig. 1) allowed determination of the steady-state absorption and emission behavior. The compounds showed a mesitylene (M) as the aryl group in the meso-position and a functionalisation with methyl and/or vinyl naphthalene. Second, transient absorption spectroscopy gave further insight in their relaxation properties after photoexcitation into the first and higher excited singlet states. Probe windows spanned the UV-Vis and NIR regions. Homoleptic zinc dipyrrins show their best photochemical performance in nonpolar solvents. Nevertheless, in the majority of photocatalytic reactions occur in solvents that have a certain value of polarity.38 Therefore, different solvents such as ethanol (EtOH, polarity 0.654), dichloromethane (DCM, polarity 0.309)39 were used to determine possible solvent effects, compared with those in and cyclohexane (CHX, polarity 0.006).40,41 In particular, we chose to investigate their behaviour in photocatalytic oxidative transformations, as they represent a substantial contribution towards sustainable chemistry.42
 | ||
| Fig. 1 Structural representation of the ligands 1–2 and the homoleptic zinc complexes 3–4 investigated herein. | ||
2. Experimental section
2.1. Materials and methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v), dissolving two equivalents of the ligand and one equivalent of zinc diacetate Zn(OAc)2, where OAc is acetate, at room temperature. Further details are available in the ESI.†
:1, v/v), dissolving two equivalents of the ligand and one equivalent of zinc diacetate Zn(OAc)2, where OAc is acetate, at room temperature. Further details are available in the ESI.†
2.2. Photocatalytic experiments
The photocatalytic aerobic oxidative hydroxylation of phenylboronic acids was done by modifying a published procedure.44 In 5 mL CH2Cl2 as solvent (or the deuterated solvent CD2Cl2) phenylboronic acid (0.1 mmol), the zinc bis(dipyrrinato) derivative (0.4 mol%), and N,N-diisopropylamine (DIPA, 1.0 mmol, 10 equiv.) were placed in a flask, equipped with a septum. The solution was irradiated by white light using a solar simulator (LOT Quantum Design) for one to three hours under air. A solution of NaNO2 (50 g L−1) was used as a short-wavelength cut-off filter. The reaction was monitored via GC-MS and/or via1H NMR.The photooxidation of thioanisole (50 μL, 0.47 mmol) was performed in an aerated EtOH solution, containing 0.5 mol% of the photosensitizer, zinc bis(dipyrrinato) complex. White light, generated by the solar simulator, LOT from the Quantum Design, irradiated the solutions. Also in this case, the NaNO2 solution was used as a short-wavelength cut-off filter. The reaction stirred for diverse hours and was monitored by GC-MS. When the reaction was stopped, evaporation of the solvent allowed a quantification of the product conversion. Quenching experiments were performed by adding either para-benzoquinone (5 equiv.) or 1,4-diazabicyclo[2.2.2]octane (5 equiv.) to the freshly prepared photocatalytic solutions.
3. Results and discussion
3.1. Stationary absorption spectra
000 L mol−1 cm−1. The extinction coefficients of the lower absorption bands are ε (448 nm) ≈ 22000 L mol−1 cm−1 (1) and ε (572 nm) ≈ 27000 L mol−1 cm−1 (2). The bands in the visible region can be assigned to 1(π–π*) transitions11,12,20 of the extended π system of the dipyrrinato unit. The absorption band around 448 nm is primarly from the dipyrrin structure, while the extension of the π-system via vinyl-naphthalene (covalently attached as R and R′ in Fig. 1) leads to a considerable red-shift peakting at 572 nm in the absorption spectrum.45 Excitation at lower wavelengths (λ < 400 nm) corresponds to higher excited states.
 | ||
| Fig. 2 UV-Vis absorption spectra in terms of extinction coefficients, ε, of the ligands 1 (yellow) and 2 (blue) in DCM. | ||
To compare the influence of the solvent, the absorption spectra of the ligands were measured also in CHX and EtOH. The respective absorption maxima are given in Table 1 and show only minor solvent dependent shifts of 1–8 nm.
| DCM λmax, abs./nm | EtOH λmax, abs./nm | CHX λmax, abs./nm | |
|---|---|---|---|
| 1 | 448 | 446 | 444 |
| 2 | 575 | 570 | 574 |
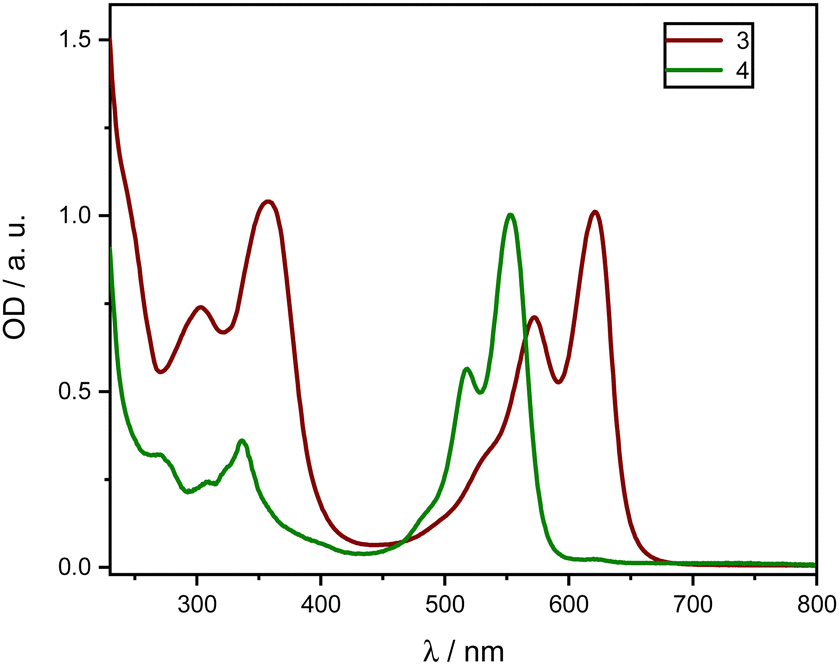 | ||
| Fig. 3 Normalized absorption spectra of homoleptic zinc complexes 3 (red) 4 (green) in DCM between 230 and 800 nm. | ||
Both complexes exhibit a first absorption band between 550 and 625 nm in the visible range. Moreover, other absorption transitions to higher electronic singlet states in the UV range below 400 nm exist. The Zn compounds show notably high extinction coefficients in the visible range (450–700 nm). These bands are described by a ligand-centered 1(π–π*) transition11,12,20 of the extended π-system of the dipyrrinato unit and were observed in all the tested solvents. In comparison to Fig. 2, the addition of Zn leads to a further red-shift of the absorption spectra, which is generally referred to intramolecular CT.22,23,46 Comparing compounds 3 and 4 reveals that the elongated π-system of 3 due to a second vinyl naphthalene moiety introduces an additional red-shift compared to 4.
It is clearly noticeable that with increasing polarity, ε drastically decreases. In particular, for the complex 4 the extinction coefficient in CHX is approximately five times higher than in polar solvents (CHX: ε (553 nm) ≈ 170000 L mol−1 cm−1, compared to EtOH: ε (551 nm) ≈ 34000 L mol−1 cm−1), while the band structure remains intact. However, a slight shift of the maxima by 2–5 nm around the maximum is observed (Table 2).
| DCM λmax, abs./nm | EtOH λmax, abs./nm | CHX λmax, abs./nm | |
|---|---|---|---|
| 3 | 623 | 618 | 620 |
| 4 | 554 | 551 | 553 |
Emission spectra exhibit maxima between 550 and 650 nm, depending on the compound, and can be attributed to the ligand-centered 1(π–π*) transition.11,12,16 The Stokes shift, exceeding 450 cm−1, is significant and indicates an excited state relaxation of the chromophores to occur before photon emission. Such relaxation processes can be induced by conformational changes, vibrations, or electronic transitions.28 The quantum yields of homoleptic complexes vary significantly depending on their functionalization. In comparison to the reference complexes without functionalization (16%),17 they are drastically increased for both substitutions. This suggests that functionalization prevents a structural change in the excited state, reducing non-radiative relaxation processes.
The most significant difference in quantum yield is observed by varying the solvent. In fact, Φ is higher in the nonpolar CHX, while in polar solvents, Φ drastically decreases to lower values than 1%, with negligible deviation within EtOH and DCM (see Table 3). This behavior is extensively described in the literature for similar homoleptic complexes.11,16–18,21,22,32 As indicated above, the presence of an energetically favorable CS state, favored in polar solvents, has been postulated.17,22,23 Theoretical calculations suggest that a thermal transition from the radiative photoexcited 1(π–π*) state (D-Zn-D*) to non-emissive CS state (D˙+-Zn-D˙−) can occur.11 This transition may involve an intramolecular charge transfer (ICT). Unlike heteroleptic complexes, this CS state is favored for homoleptic complexes in polar solvents.11 Since the ligands in homoleptic complexes are symmetric, this is sometimes called a symmetry-breaking ICT (SBCT).21,23,47
| DCM Φ | EtOH Φ | CHX Φ | |
|---|---|---|---|
| 3 | 0.008 | 0.004 | 0.250 |
| 4 | 0.005 | 0.010 | 0.580 |
The fluorescence lifetime (τ) of the zinc complexes at the emission maxima around 570 nm was measured using time-correlated single-photon counting and fitted with a monoexponential function. In CHX, the lifetime for all complexes is in the range of 4 to 5 ns. With increasing solvent polarity, the fluorescence lifetime decreases down to around 2 ns. This finding emphasizes the importance of the solvent, while a correlation between functional groups and lifetime is much less obvious.
3.2. Transient absorption spectra
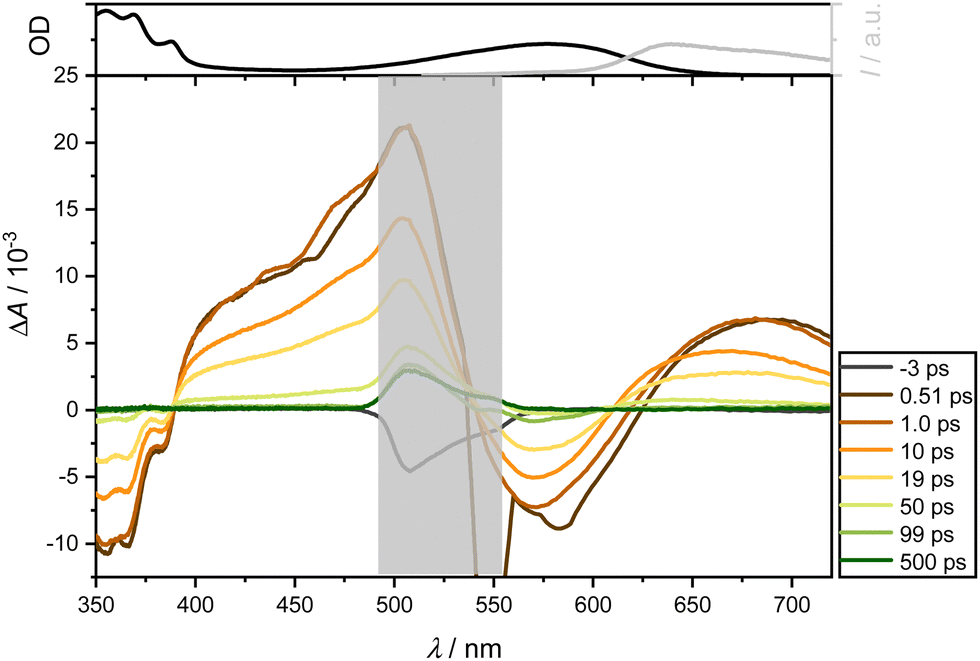 | ||
| Fig. 4 Transient absorption spectra of the ligand 2 in DCM following excitation at 575 nm (OD = 0.2) were probed between 350 and 720 nm at the indicated delay times (bottom). A distinct grey area, located around 530 nm, is included in the figure to mask the spectral region affected by scattering effects from the excitation pulse. The top section shows the normalized steady-state absorption (black) and emission spectra (λex = 500 nm, slit width 4 nm, grey). | ||
Furthermore, the ligands were excited at 400 nm, allowing a direct comparison of the impact of functionalization on the transient responses of 2 with those of 1—given that the latter do not absorb at 575 nm. More TA spectra following 400 nm excitation in the solvents CHX, EtOH, and DCM can be found in the Supporting Information. For illustrative purposes, 1 is analyzed in DCM and presented in Fig. 5.
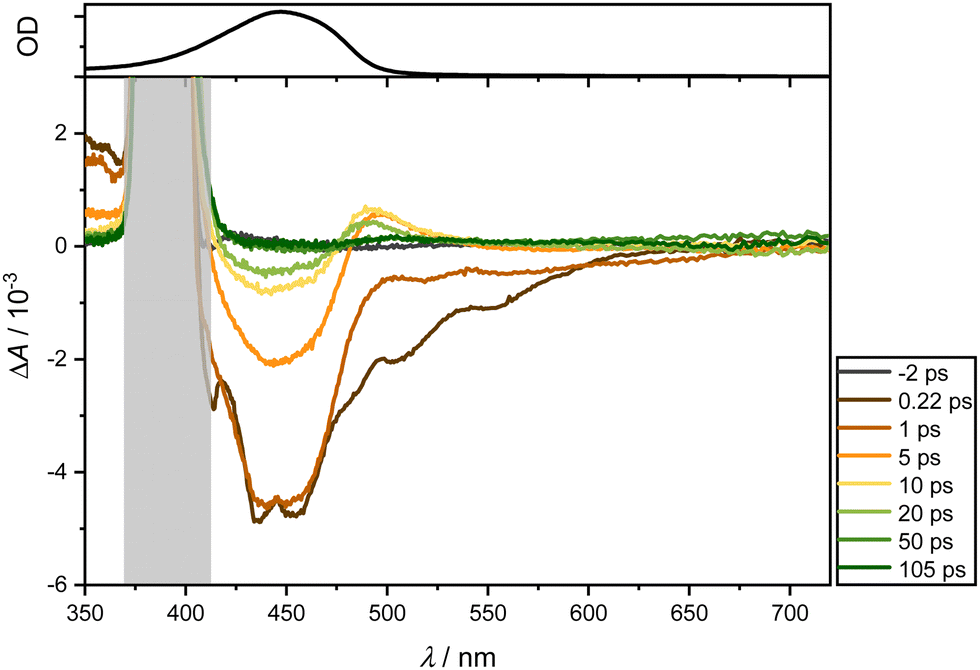 | ||
| Fig. 5 Transient absorption spectra of ligand 1 in DCM following excitation at 400 nm (OD = 0.2) were recorded between 350 and 720 nm at the specified delay times (bottom). A distinct grey area, located around 400 nm, is included in the figure to mask the spectral region affected by scattering effects from the excitation pulse. The top section displays the normalized stationary absorption spectrum. | ||
Immediately after photoexcitation, two bleaching bands emerge around 450 and, as a longer tail, towards 600 nm. These bands can be ascribed to GSB and ultrafast Stimulated Emission SE. Concurrently, two positive absorption bands appear: one around 360 nm, manifesting instantly after photoexcitation, and another around 480 nm, emerging after a few picoseconds. The latter band likely results from the superposition of the ESA band around 360 nm and the GSB. The overall spectrum exhibits short-lived responses, dissipating completely after approximately 100 ps. Unlike the dominant absorption bands in the visible region observed for ligand 2, no such dominance is evident here. Consequently, the dominant band can be attributed to the vinyl naphthalene moiety.
In summary, a short-lived excited state dynamic is observed for both ligands, relaxing within approximately 100 ps (see Supporting Information for further examples in different solvents). Across all examined solvents, a GSB manifests around 450 nm, SE around 600 nm, as well as an ESA below 380 nm and a subtly pronounced ESA around 470 nm for ligand 1. In contrast, ligand 2 and complexes lack SE, as they are dominated by the ESA, which is around 600 nm and also overshadows the GSB. Additional bleaching below 380 nm aligns with the absorption spectrum of the mesitylene skeleton.
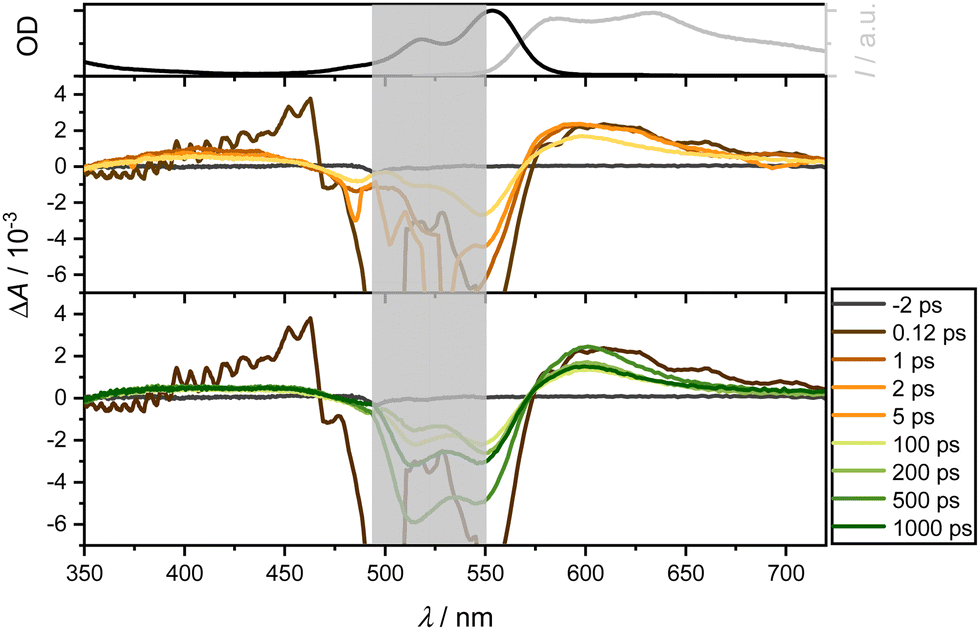 | ||
| Fig. 6 Transient absorption spectra of 4 in DCM following excitation at 550 nm (OD = 0.21) and probing between 350 and 720 nm at specified delay times (middle and bottom panels). A distinct grey area, located around 530 nm, is included in the figure to mask the spectral region affected by scattering effects from the excitation pulse. Top: Normalized steady-state absorption spectra (black) and emission spectra (grey). | ||
Following excitation of 4 at 550 nm in DCM, ground state depopulation results in a GSB in the spectral range of 520 to 570 nm. At longer wavelengths, an ESA overlays the spectrum, exhibiting relative constancy around 700 nm with slight attenuation. A comparison with the steady-state fluorescence spectrum reveals an additional overlap with SE in the range between 550 and 700 nm. Below 470 nm, a second, less dominant ESA band is observed. Notably, these contributions persist throughout the measurement window of over 1.6 ns and are assignable to both singlet and triplet state dynamics. Additionally, an ultrafast positive amplitude around 460 nm is observed (see Fig. 6, middle panel). The TA spectra exhibit two isosbestic points around 470 and 570 nm, corresponding to the relaxation channels of ESA and GSB, while SE and ESA compete around 570 nm.
The band around 470 nm can be described by an intermediate state with an ultrafast spectral signature, decaying within 150 fs. Thompson et al.17,22,47 have postulated a CT state in similar complexes with lifetimes from 1 to 16 ps for CS state formation. Although this state is supposed to be unstable in nonpolar solvents, Fig. 7 and, in particular, Fig. S10 (using a slightly higher pump wavelength of 510 nm instead of 550 nm, ESI†) show an ultrafast reminiscent of a CT state in EtOH on a timescale of 100 fs. As expected for polar solvents such as EtOH (Fig. 7), the ultrafast dynamics around 470 nm are also observable. In contrast to measurements in CHX, it is significantly more pronounced, corroborating that polar solvents favor the formation of CT states. This suggests efficient ultrafast internal conversion from higher excited singlet to lower excited states. Taking the solvent CHX as an example, even the isosbestic points at 500 and 575 nm are blue-shifted by only ca. 5 nm after UV excitation.
 | ||
| Fig. 7 Transient broadband absorption spectra of complex 4 in EtOH (λex = 560 nm, OD = 0.07). Probing between 350 and 720 nm was conducted at specified delay times (middle and bottom panels). A distinct grey area, located around 530 nm, is included in the figure to mask the spectral region affected by scattering effects from the excitation pulse. The top panel displays normalized steady-state absorption spectra (in black) and emission spectra (in grey). | ||
For the recording of the NIR transient response spanning the range of 900 to 1400 nm, complexes 3 and 4 were probed in EtOH following excitation 258 nm (Fig. 8). Despite their high extinction coefficients, the TA spectra were surprisingly not well analyzable due to poor signal-to-noise ratios. For all complexes in both solvents, a spectrally broad absorption band between 900 and 1250 nm is observed immediately after photoexcitation. This band persists within the temporal measurement window of several hundred pico-seconds without complete decay.
 | ||
| Fig. 8 Transient broadband absorption spectra of 3 (left, OD = 0.84) and 4 (right, OD = 1.03) in EtOH after excitation at 258 nm and probing between 900 and 1400 nm at the specified delay times. | ||
From the bi-exponential fitting, two time-constants can be obtained: a time constant τ2 ≫ 500 ps outside the measurement window in both solvents that can be attributed to the lifetime of an excited state; a shorter time constant, τ1, on the order of few ps is more related to vibrational motion in the excited state. As a result, long-lasting residual absorption can be found at all probed wavelengths between 350 and 720 nm as well as 900 and 1400 nm. Since there is simply a decay of the amplitudes after 0.5 ps, these dynamics could be assigned to singlet or triplet dynamics. Experiments on a ns to ms timescale could potentially resolve this issue.
3.3. Photocatalytic oxidations
We explored the photocatalytic behavior of 3 and 4 in two different aerobic photocatalytic reactions for the oxidation of (a) phenyl boronic acid to phenol and (b) phenyl methyl thioether to the corresponding sulfoxide. (see Scheme 1).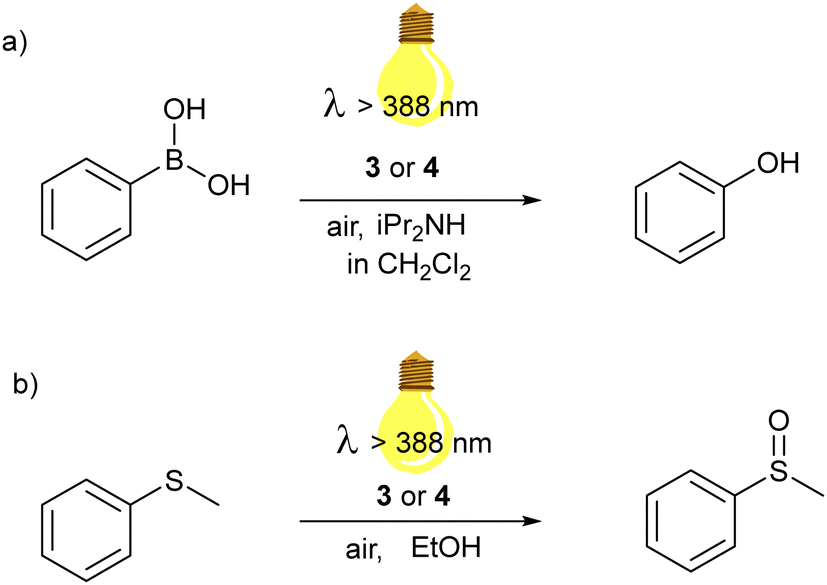 | ||
| Scheme 1 Aerobic photooxidation reactions catalyzed by Zn(dipyrrin) complexes of this work: (a) hydroxylation of phenylboronic acid; (b) oxidation of thioanisole to methyl phenyl sulfoxide. | ||
Visible-light-driven photooxidations have been explored to contribute towards a more sustainable synthesis of important chemicals, without the requirement of strong oxidants or catalysts based on expensive 4d and 5d transition metals. Among the PCs, BODIPY derivatives and organic dyes have been largely used, while examples with Zn(II) dipyrrins are not present, to the best of our knowledge. Still, this class of compounds is also attractive, as a cost-effective and sustainable alternative. Bearing two dipyrrin ligands, the extinction coefficient of the Zn(II) bis(dipyrrinato) complexes has generally higher values than that one of the corresponding BODIPY, allowing a lower concentration. In particular, we selected these two well-known photooxidation reactions with our zinc complexes to investigate which possible reactive oxygen species (ROS) is generated by our dye after irradiation. Specifically, the fate of the PC in the excited state may follow two pathways: a photoinduced electron transfer or energy transfer (Scheme 2). In the first case, the ROS is the superoxide anion O2−˙, generated from the redox reaction between molecular oxygen and the radical anion of the PC, which in turn underwent a reductive quenching from another species in solution. In the second case, the ROS is 1O2, formed by triplet–triplet annihilation between the triplet excited state of the PC and the triplet state of O2.
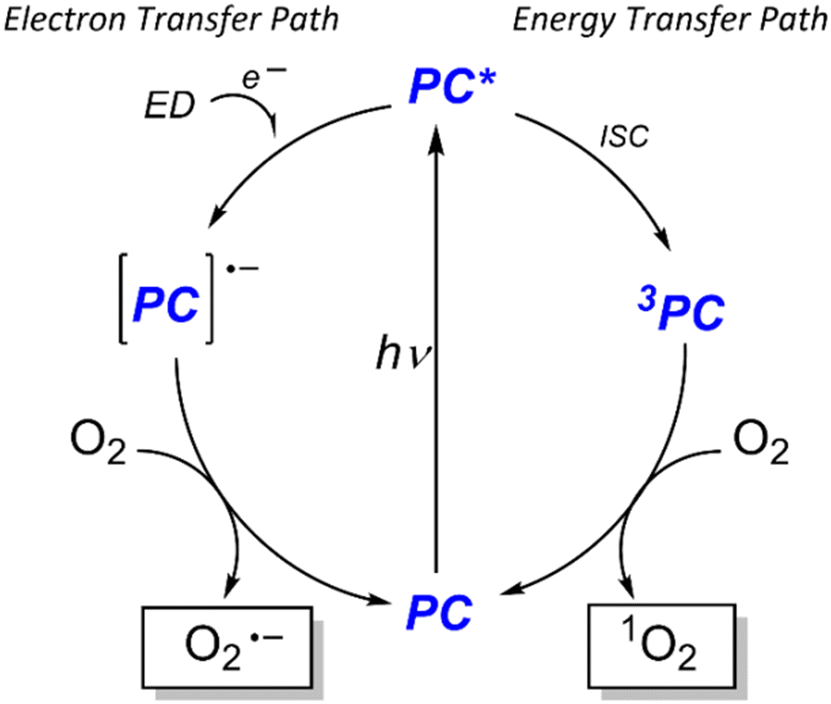 | ||
| Scheme 2 Possible photoinduced paths to generate ROS from a photocatalyst (PC). Left: The electron transfer path requires an electron donor (ED) to form the reduced PC species that generates the superoxide. Right: In the energy transfer path, after Franck–Condon excitation, the PC* undergoes intersystem crossing (ISC) to populate the triplet excited state which generates 1O2, via triplet–triplet annihilation. | ||
The proposed mechanism for the photocatalytic oxidative hydroxylation of phenylboronic acid to phenol follows the photoinduced electron transfer pathway. We used 10 equivalents of N,N-dipropylamine (DIPA) as sacrificial electron donor and a 0.4 mol% concentration of 3 in DCM. The reaction was placed behind a NaNO2 solution, working as a 388 nm cut-off filter, and irradiated by a Hg-lamp solar simulator. The formation of phenol was first monitored via gas chromatography, equipped with a mass spectrometer as a detector. After one hour, the starting material was completely converted. We repeated the reaction in deuterated DCM, to follow the reaction also via1H NMR.
The complete conversion within one hour was confirmed (see ESI,† Fig. S1). A full transformation of the phenyl boronic acid was also obtained using 4 as a PC. This outcome is very interesting since other systems that photoconvert aryl boronic acids in their phenols report a longer time scale (higher than 4 hours).44,48–51 We demonstrated that the generated ROS is the superoxide anion by performing the reaction in the presence of five equivalents of para-benzoquinone (p-BQ), an O2˙− quencher.52 No product could be detected, even after a prolonged period (3 hours) (see Fig. S3 in ESI†). To exclude a contribution of the ROS that could be formed by photoinduced energy transfer (singlet oxygen), the reaction was repeated in the presence of five equivalents of 1,4-diazabicyclo[2.2.2]octane (DABCO), which is a known 1O2 quencher.53 In this case, the product was developed independently of the presence of DABCO (see Fig. S4 in ESI†).
Thus, although the SBCT state formation of the complexes is detrimental for the luminescence in polar solvents, such as dichloromethane; it is decisive for the electron transfer pathway enabling photocatalysis. A suggested mechanism is shown in the supporting information (Fig. S8, ESI†), where the excited Zn(II) bis(dipyrrinato) complexes are reductively quenched by the amine and molecular oxygen closes the photocatalyst cycle, forming the superoxide anion. This ROS reacts further with phenyl boronic acid, which bears a negative charge on the boron, and upon rearrangement and following hydrolysis, phenol is produced.
On the contrary, we tested the energy transfer pathway from the triplet excited state of our zinc complexes through the aerobic visible-light-induced oxidation of thioanisole to the corresponding sulfoxide in EtOH (Fig. S5–S7, ESI†). Developing environmentally friendly protocols for the controlled oxidation of sulfides is also significant since sulfoxides are important starting materials in organic synthesis, polymers and active molecules in pharmaceutics.54–58 The reactions were performed in ethanol, dissolving 0.5 mmol of thioanisole and 0.5 mol % of 3 or 4. The reaction is not as fast as the photooxidation of the phenylboronic acid tested previously. After 3 h irradiation by white light generated by the solar simulator (λ > 388 nm), the methyl phenyl sulfoxide was isolated with a 93% yield when 3 was used as the photocatalyst. Further oxidation to sulfone was not detected besides a faint signal in the GC MS. Complex 4 also turned out in this photocatalytic process less efficient. In fact, the reaction needed three more hours of irradiation. The isolated product was obtained after column chromatography and was 67%. The efficiency of this photooxidation is similar to other systems employing BODIPY derivatives as PCs.59,60 Previous studies showed that also the aerobic visible-light oxidation of sulfides may occur through different pathways, involving 1O2 or O2˙−, depending on the PC used. Therefore, we performed the reactions in the presence of quenching agents: DABCO and p-BQ. In both cases, the yield of the desired sulfoxide was strongly affected, and a considerable amount of the starting thioanisole was recovered. In particular, with DABCO the yield of the sulfoxide was 8.8%, while with p-BQ 24.4% of yield could be obtained. This reaction inhibition suggested that the ROS involved in the oxidation of thioanisole are both the superoxide anion and the singlet oxygen. A possible mechanism was indicated based on the literature (Fig. S9, ESI†). In particular, the two possible pathways are shown: the photoinduced electron transfer path generates the ROS superoxide anion, which form as intermediate a three-member ring with the sulfur atom of the thioanisole radical cation, and the photoinduced energy transfer to generates the 1O2 which interacts with the starting material, oxidizing the sulphur atom to sulfoxide. Nevertheless, further insights should be possible only after additional optimization experiments, which are planned as future subjects of study in our lab. Finally, we demonstrated that the triplet dynamics suggested in the time-resolved spectroscopic experiments might be decisive for the photoinduced energy transfer pathways to catalyze photooxidation reactions such as the formation of sulfoxides from sulfides.
Conclusions
The presented zinc-dipyrrin complexes represent an intramolecular donor–acceptor system that forms an additional state through an intramolecular CT, which is sensitive to intermolecular environmental influences. This behaviour has been systematically analysed using steady-state and transient methods. In addition, intermolecular environmental influences were investigated by varying the ligands and substitutions. The dynamics of the ligands after photoexcitation are short-lived, decaying completely within a few hundred ps. In contrast, the complexes with the zinc ion show long-lived responses in the ns range, suggesting singlet and triplet state dynamics. The complexes show a bleaching band around 575 nm and SE around 600 nm, superimposed by a dominant ESA both above 620 nm and below 500 nm. The lifetime of each state is a few ns outside the time measurement range. The homoleptic complexes also show ultrashort dynamics on a scale of about 100 fs in CHX, which is enhanced in EtOH. This may be indicative of a CT state. The homoleptic complexes show a broad absorption band in the spectral range from 900 to 1250 nm with a lifetime of over 500 ps. Furthermore, we tested these zinc complexes for the first time as photocatalysts in aerobic oxidation reactions. The efficiency of the reactions should be ascribed to the population of the excited state that is quenched by iPr2NH or molecular oxygen to form the reactive species: the superoxide radical anion O2−˙ or the singlet oxygen 1O2. Specifically, the photooxidative hydroxylation of phenylboronic acid proceeds through an electron transfer pathway, while the photoinduced sulfide oxidation by these zinc complexes seems to advance mainly via triplet energy transfer, although the electron transfer pathway cannot be excluded at this stage. Thus, TA spectra from the fs to one ns regime correlate with stationary spectra, but leave room for further investigation towards quantitative assignment and tailor-made synthesis with specific excited state properties, not only for a controlled fluorescence quantum yield in dependent of solvents and ligands, but also to develop new environmentally friendly photocatalysts.Author contributions
Conceptualization: C. B. and A.-N. U.; methodology: A.-N. U.; software: J. W.; validation: J. L., P. R. and C. B.; formal analysis: P. R., J. L., C. B. and A.-N. U.; investigation: J. L., P. R., R. T. and C. B.; resources: C. B and A.-N. U.;writing – original draft, J.L., P.R., C.B. and A.-N.U.; writing – review & editing, P. R., A.-N. U. and C.B.; visualization: A.-N. U.; supervision: C. B. and A.-N. U.; project administration: A.-N.U. and C.B.; funding acquisition: C.B. and A.-N.U. All authors have read and agreed to the published version of the manuscript.Data availability
Data are available in the ESI† file and original files are available upon request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the GRK 2039 “Molecular Architectures for Fluorescent Cell Imaging” for financial support and continued key support from the Karlsruhe Institute of Technology (KIT). Open Access was enabled by the existing “Publish & Read Agreement” between KIT and the Royal Society of Chemistry (RSC).Notes and references
- F. Odobel, Y. Pellegrin and J. Warnan, Energy Environ. Sci., 2013, 6, 2041–2052 RSC.
- J. Warnan, Y. Pellegrin, E. Blart and F. Odobel, Chem. Commun., 2012, 48, 675–677 RSC.
- R. Ziessel, G. Ulrich, A. Haefele and A. Harriman, J. Am. Chem. Soc., 2013, 135, 11330–11344 CrossRef CAS PubMed.
- F. Cucinotta, B. P. Jarman, C. Caplan, S. J. Cooper, H. J. Riggs, J. Martinelli, K. Djanashvili, E. La Mazza and F. Puntoriero, ChemPhotoChem, 2018, 2, 196–206 CrossRef CAS.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS.
- M. Poddar and R. Misra, Coord. Chem. Rev., 2020, 421, 213462 CrossRef CAS.
- E. Bassan, A. Gualandi, P. G. Cozzi and P. Ceroni, Chem. Sci., 2021, 12, 6607–6628 RSC.
- R. Sakamoto, T. Yagi, K. Hoshiko, S. Kusaka, R. Matsuoka, H. Maeda, Z. Liu, Q. Liu, W.-Y. Wong and H. Nishihara, Angew. Chem., Int. Ed., 2017, 56, 3526–3530 CrossRef CAS.
- R. Sakamoto, T. Iwashima, M. Tsuchiya, R. Toyoda, R. Matsuoka, J. F. Kögel, S. Kusaka, K. Hoshiko, T. Yagi, T. Nagayama and H. Nishihara, J. Mater. Chem. A, 2015, 3, 15357–15371 RSC.
- F. Zhang, S. A. Baudron and M. W. Hosseini, New J. Chem., 2018, 42, 6997–7004 RSC.
- S. Kusaka, R. Sakamoto, Y. Kitagawa, M. Okumura and H. Nishihara, Chem. – Asian J., 2012, 7, 907–910 CrossRef CAS.
- L. Yu, K. Muthukumaran, I. V. Sazanovich, C. Kirmaier, E. Hindin, J. R. Diers, P. D. Boyle, D. F. Bocian, D. Holten and J. S. Lindsey, Inorg. Chem., 2003, 42, 6629–6647 CrossRef CAS PubMed.
- R. Sakamoto, T. Iwashima, J. F. Kögel, S. Kusaka, M. Tsuchiya, Y. Kitagawa and H. Nishihara, J. Am. Chem. Soc., 2016, 138, 5666–5677 CrossRef CAS PubMed.
- M. Tsuchiya, R. Sakamoto, S. Kusaka, Y. Kitagawa, M. Okumura and H. Nishihara, Chem. Commun., 2014, 50, 5881–5883 RSC.
- H. L. Kee, C. Kirmaier, L. Yu, P. Thamyongkit, W. J. Youngblood, M. E. Calder, L. Ramos, B. C. Noll, D. F. Bocian, W. R. Scheidt, R. R. Birge, J. S. Lindsey and D. Holten, J. Phys. Chem. B, 2005, 109, 20433–20443 CrossRef CAS PubMed.
- J. F. Kögel, S. Kusaka, R. Sakamoto, T. Iwashima, M. Tsuchiya, R. Toyoda, R. Matsuoka, T. Tsukamoto, J. Yuasa, Y. Kitagawa, T. Kawai and H. Nishihara, Angew. Chem., Int. Ed., 2016, 55, 1377–1381 CrossRef.
- C. Trinh, K. Kirlikovali, S. Das, M. E. Ener, H. B. Gray, P. Djurovich, S. E. Bradforth and M. E. Thompson, J. Phys. Chem. C, 2014, 118, 21834–21845 CrossRef CAS PubMed.
- N. Z. Alqahtani, T. G. Blevins and C. E. McCusker, J. Phys. Chem. A, 2019, 123, 10011–10018 CrossRef CAS PubMed.
- M. Asaoka, Y. Kitagawa, R. Teramoto, K. Miyagi, Y. Natori, R. Sakamoto, H. Nishihara and M. Nakano, Polyhedron, 2017, 136, 113–116 CrossRef CAS.
- M. Tsuchiya, R. Sakamoto, S. Kusaka, J. Kakinuma and H. Nishihara, Electrochemistry, 2013, 81, 337–339 CrossRef CAS.
- S. Das, W. G. Thornbury, A. N. Bartynski, M. E. Thompson and S. E. Bradforth, J. Phys. Chem. Lett., 2018, 9, 3264–3270 CrossRef CAS PubMed.
- A. N. Bartynski, M. Gruber, S. Das, S. Rangan, S. Mollinger, C. Trinh, S. E. Bradforth, K. Vandewal, A. Salleo, R. A. Bartynski, W. Bruetting and M. E. Thompson, J. Am. Chem. Soc., 2015, 137, 5397–5405 CrossRef CAS PubMed.
- M. Kellogg, A. Akil, D. S. Muthiah Ravinson, L. Estergreen, S. E. Bradforth and M. E. Thompson, Faraday Discuss., 2019, 216, 379–394 RSC.
- K. Hanson, A. Tamayo, V. V. Diev, M. T. Whited, P. I. Djurovich and M. E. Thompson, Inorg. Chem., 2010, 49, 6077–6084 CrossRef CAS.
- S. Wang, Coord. Chem. Rev., 2001, 215, 79–98 CrossRef CAS.
- L. Hammarström, Acc. Chem. Res., 2015, 48, 840–850 CrossRef.
- A. Bessette and G. S. Hanan, Chem. Soc. Rev., 2014, 43, 3342–3405 RSC.
- I. V. Sazanovich, C. Kirmaier, E. Hindin, L. Yu, D. F. Bocian, J. S. Lindsey and D. Holten, J. Am. Chem. Soc., 2004, 126, 2664–2665 CrossRef CAS.
- R. Tabone, D. Feser, E. D. Lemma, U. Schepers and C. Bizzarri, Front. Chem., 2021, 9, 754420 CrossRef CAS PubMed.
- J. A. Kübler, B. Pfund and O. S. Wenger, JACS Au, 2022, 2, 2367–2380 CrossRef.
- V. Giuso, E. Jouaiti, C. Cebrián, S. Parant-Aury, N. Kyritsakas, C. Gourlaouen and M. Mauro, ChemPhotoChem, 2023, 7, e202300092 CrossRef CAS.
- S. A. Baudron, Dalton Trans., 2013, 42, 7498–7509 RSC.
- Y. Rio, D. Sánchez-García, W. Seitz, T. Torres, J. L. Sessler and D. M. Guldi, Chem. – Eur. J., 2009, 15, 3956–3959 CrossRef CAS.
- P. De Bonfils, L. Péault, P. Nun and V. Coeffard, Eur. J. Org. Chem., 2021, 1809–1824 CrossRef CAS.
- F. Dumur, Eur. Polym. J., 2023, 196, 112241 CrossRef CAS.
- C. Coutant, J. Fischer, P. Nun and V. Coeffard, ChemistrySelect, 2023, 8, e202303316 CrossRef CAS.
- D. Wang, X. Wang, S. Zhou, P. Gu, X. Zhu, C. Wang and Q. Zhang, Coord. Chem. Rev., 2023, 482, 215074 CrossRef CAS.
- M. A. Bryden, F. Millward, O. S. Lee, L. Cork, M. C. Gather, A. Steffen and E. Zysman-Colman, Chem. Sci., 2024, 15, 3741–3757 RSC.
- C. Reichardt and T. Welton, Solvents and Solvent Effects in Organic Chemistry, Wiley-VCH, Weinheim, 4nd edn, 2011 Search PubMed.
- D. Tungulin, J. Leier, A. B. Carter, A. K. Powell, R. Q. Albuquerque, A. N. Unterreiner and C. Bizzarri, Chem. – Eur. J., 2019, 25, 3816–3827 CrossRef CAS.
- U. Sani, D. Tungulin, C. Bizzarri and F. Cucinotta, RSC Adv., 2020, 10, 2841–2845 RSC.
- L. Luo, T. Zhang, M. Wang, R. Yun and X. Xiang, ChemSusChem, 2020, 13, 5173–5184 CrossRef CAS.
- N. C. Michenfelder, C. Gienger, M. Dilanas, A. Schnepf and A.-N. Unterreiner, Molecules, 2020, 25, 2639 CrossRef CAS.
- L. Huang, X. Cui, B. Therrien and J. Zhao, Chem. – Eur. J., 2013, 19, 17472–17482 CrossRef CAS.
- M. Buyuktemiz, S. Duman and Y. Dede, J. Phys. Chem. A, 2013, 117, 1665–1669 CrossRef CAS.
- W. Rettig, Angew. Chem., Int. Ed. Engl., 1986, 25, 971–988 CrossRef.
- M. T. Whited, N. M. Patel, S. T. Roberts, K. Allen, P. I. Djurovich, S. E. Bradforth and M. E. Thompson, Chem. Commun., 2012, 48, 284–286 RSC.
- P.-F. Wei, M.-Z. Qi, Z.-P. Wang, S.-Y. Ding, W. Yu, Q. Liu, L.-K. Wang, H.-Z. Wang, W.-K. An and W. Wang, J. Am. Chem. Soc., 2018, 140, 4623–4631 CrossRef CAS PubMed.
- Y. Chen, J. Hu and A. Ding, RSC Adv., 2020, 10, 7927–7932 RSC.
- A. Ding, Y. Zhang, Y. Chen, R. Rios, J. Hu and H. Guo, Tetrahedron Lett., 2019, 60, 660–663 CrossRef CAS.
- B. Luo, Y. Zhang, Y. Chen and J. Huo, Mater. Adv., 2022, 3, 4699–4706 RSC.
- M.-T. Maurette, E. Oliveros, P. P. Infelta, K. Ramsteiner and A. M. Braun, Helv. Chim. Acta, 1983, 66, 722–733 CrossRef CAS.
- C. Ouannes and T. Wilson, J. Am. Chem. Soc., 1968, 90, 6527–6528 CrossRef CAS.
- M. Forchetta, F. Valentini, V. Conte, P. Galloni and F. Sabuzi, Catalysts, 2023, 13, 220 CrossRef CAS.
- C. Ye, Y. Zhang, A. Ding, Y. Hu and H. Guo, Sci. Rep., 2018, 8, 2205 CrossRef.
- E. A. Ilardi, E. Vitaku and J. T. Njardarson, J. Med. Chem., 2014, 57, 2832–2842 CrossRef CAS.
- E. Skolia, P. L. Gkizis, N. F. Nikitas and C. G. Kokotos, Green Chem., 2022, 24, 4108–4118 RSC.
- M. Forchetta, F. Sabuzi, L. Stella, V. Conte and P. Galloni, J. Org. Chem., 2022, 87, 14016–14025 CrossRef CAS PubMed.
- W. Li, Z. Xie and X. Jing, Catal. Commun., 2011, 16, 94–97 CrossRef CAS.
- W. Li, L. Li, H. Xiao, R. Qi, Y. Huang, Z. Xie, X. Jing and H. Zhang, RSC Adv., 2013, 3, 13417–13421 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nj02527j |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |