
Eco-friendly, in-water, and catalyst-free assembly of acylethenylpyrroloimidazoindoles from 3H-indoles and acylpyrrolylacetylenes†
Ludmila A.
Oparina
 ,
Kseniya V.
Belyaeva
,
Nikita A.
Kolyvanov
,
Igor A.
Ushakov
,
Denis N.
Tomilin
,
Lyubov N.
Sobenina
,
Anton V.
Kuzmin
and
Boris A.
Trofimov
*
,
Kseniya V.
Belyaeva
,
Nikita A.
Kolyvanov
,
Igor A.
Ushakov
,
Denis N.
Tomilin
,
Lyubov N.
Sobenina
,
Anton V.
Kuzmin
and
Boris A.
Trofimov
*
A. E. Favorsky Irkutsk Institute of Chemistry, Siberian Branch, Russian Academy of Sciences, 1 Favorsky Str., Irkutsk 664033, Russia. E-mail: boris_trofimov@irioch.irk.ru
First published on 17th November 2023
Abstract
The merging of fundamentally and biologically important heterocyclic structures, such as pyrroles, indoles, and imidazoles, in molecules of dihydropyrrolo[1′,2′:3,4]imidazo[1,2-a]indoles functionalized by E-acylethenyl groups was achieved in water on the platform of commercial 3H-indoles using readily available acylpyrrolylacetylenes as additional building blocks. The yields of the ensembles reached 88% (100 °C, 8 h), which is noticeably better than that produced using conventional solvents (MeCN, MeNO2, DMSO, and CF3CH2OH), wherein the reaction proceeds at a rate of approximately 10 times longer (96 h) with lower yields of the target products (not higher than 80%). The reaction presumably proceeds in a micellar-like microreactor self-assembled in a two-phase aqueous medium that secures favorable mutual orientation of the merging molecules, providing a facile [2+3] concerted cycloaddition to finally form fused polyheterocyclic systems. The performed DFT calculations are in agreement with such mechanistic considerations, particularly those underlying the crucial role of water in the studied cascade process.
Introduction
One of the most challenging areas of modern organic chemistry is the search for new synthetic methodologies to design complex molecular structures using pot-, atom-, step-economical, resource-, and energy-saving methods and available starting materials.1–3 Recently, we have reported the catalyst- and metal-free reaction of 1-pyrrolines with acylpyrrolylacetylenes to afford tetrahydrodipyrrolo[1,2-a:1′,2′-c]imidazoles, rare and fused heterocyclic systems.4,5 This reaction is of particular interest because it allows the creation of complex molecules from relatively simple substrates. In fact, acylpyrrolylacetylenes are readily available from transition metal-free Al2O3-promoted room temperature cross-coupling of acylbromoacetylenes with pyrroles.6,7 During our systematic efforts to design general one-pot and atom-economic strategies for the construction of biologically important fused heterocyclic systems, in particular pyrroloimidazolidine scaffolds, via a new reaction of pyrrolylacetylene cycloaddition to the double C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond,4,5 herein, we have focused our attention on 3H-indoles as a possible platform to assemble dihydropyrrolo[1′,2′:3,4]imidazo[1,2-a]indoles, typical representatives of the above-fused systems. Such ensembles could be built up if 3H-indoles would be capable of cyclizing with acylpyrrolylacetylenes. Close congeners of pyrroloimidazoindoles are the key structural units of the fungal metabolite quinadoline B, which suppresses lipid droplet synthesis in mouse macrophages and exhibits significant antiviral activities,8,9 for instance, against influenza virus A.10 This compound was identified in silico to have strong inhibitory activity against the five target proteins in SARS-CoV2 and can be utilized as a template or prototype for the further development of multitargeting ligands against SARS-CoV2.11,12 This dihydropyrrolo[1′,2′:3,4]imidazo[1,2-a]indole scaffold integrates the privileged entities (imidazo[1,2-a]indoles and pyrrolo[1,2-c]imidazoles), which are in the focus of modern research associated with biology, pharmacology, and health care (Fig. 1). The tetrahydroimidazo[1,2-a]indole fragment is contained in the cholecystokinin CCK1 receptor antagonist asperlicin,13 the substance P antagonist fiscalin A,14 the antifungic fumiquinazolines,15 tryptoquivalines showed NF-kB inhibition.16,17 The alkaloidal metabolite chaetominine with cytotoxic activity against the human leukemic K562 and colon cancer SW1116 cell lines, includes the imidazo[1,2-a]indole fragment.18 On the other hand, pyrrolo[1,2-c]imidazole structural motif is found in several classes of bioactive products, including aldose reductase inhibitors,19 neuropeptide S receptor antagonist,20 and demonstrated a high anticancer potency21,22 and antibacterial23 activity. Noteworthily, in the previous study, clear-cut strategies to design the above fused tetracyclic molecules were not provided, and these molecules had remained exclusively of natural origin.8,9
N bond,4,5 herein, we have focused our attention on 3H-indoles as a possible platform to assemble dihydropyrrolo[1′,2′:3,4]imidazo[1,2-a]indoles, typical representatives of the above-fused systems. Such ensembles could be built up if 3H-indoles would be capable of cyclizing with acylpyrrolylacetylenes. Close congeners of pyrroloimidazoindoles are the key structural units of the fungal metabolite quinadoline B, which suppresses lipid droplet synthesis in mouse macrophages and exhibits significant antiviral activities,8,9 for instance, against influenza virus A.10 This compound was identified in silico to have strong inhibitory activity against the five target proteins in SARS-CoV2 and can be utilized as a template or prototype for the further development of multitargeting ligands against SARS-CoV2.11,12 This dihydropyrrolo[1′,2′:3,4]imidazo[1,2-a]indole scaffold integrates the privileged entities (imidazo[1,2-a]indoles and pyrrolo[1,2-c]imidazoles), which are in the focus of modern research associated with biology, pharmacology, and health care (Fig. 1). The tetrahydroimidazo[1,2-a]indole fragment is contained in the cholecystokinin CCK1 receptor antagonist asperlicin,13 the substance P antagonist fiscalin A,14 the antifungic fumiquinazolines,15 tryptoquivalines showed NF-kB inhibition.16,17 The alkaloidal metabolite chaetominine with cytotoxic activity against the human leukemic K562 and colon cancer SW1116 cell lines, includes the imidazo[1,2-a]indole fragment.18 On the other hand, pyrrolo[1,2-c]imidazole structural motif is found in several classes of bioactive products, including aldose reductase inhibitors,19 neuropeptide S receptor antagonist,20 and demonstrated a high anticancer potency21,22 and antibacterial23 activity. Noteworthily, in the previous study, clear-cut strategies to design the above fused tetracyclic molecules were not provided, and these molecules had remained exclusively of natural origin.8,9
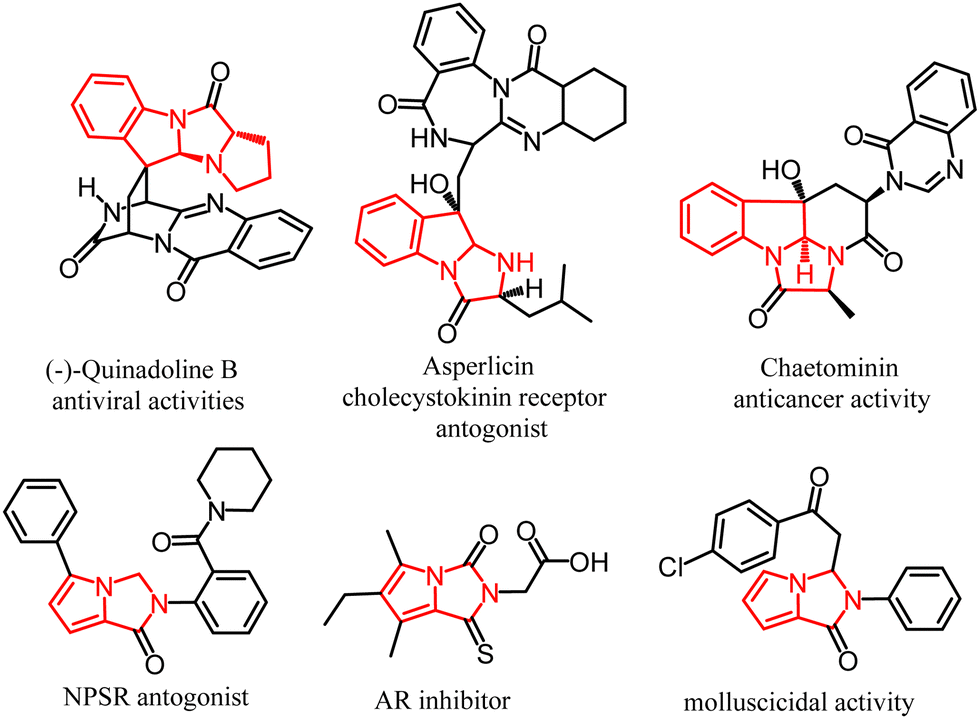 | ||
| Fig. 1 Representative bioactive molecules bearing imidazo[1,2-a]indole and pyrrolo[1,2-c]imidazole units. | ||
The merging of both structural motifs into one molecule might lead to a new group of polyheterocyclic compounds with practically important biological properties. As mentioned above, this goal could be reached on the platform of 3H-indoles provided that they would comply with the cyclization with acylpyrrolylacetylenes. As demonstrated in this communication, this expectation came true even, to our delight, in a highly eco-friendly version with water as the reaction medium.
Results and discussion
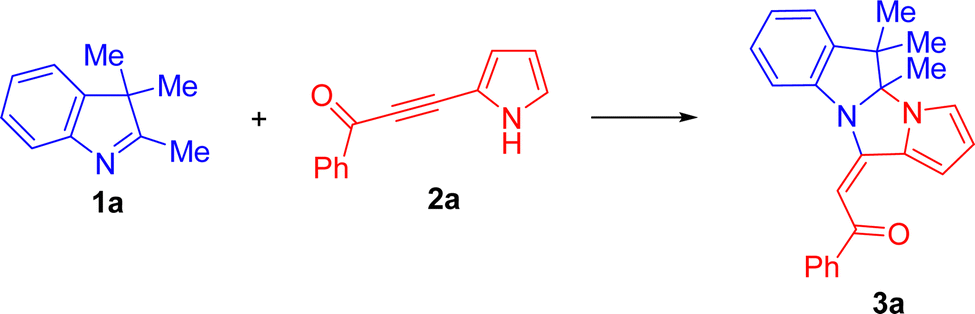
To commence our study, 3H-indole 1a and benzoylpyrrolylacetylene 2a were chosen as reference substrates to afford dihydropyrrolo[1′,2′:3,4]imidazo[1,2-a]indole 3a. Initially, we checked whether this reaction was feasible in conventional organic solvents (Table 1). The reaction was monitored using 1H NMR spectroscopy, following the disappearance of the Me-group signals of indole 1a at 2.25 and 1.28 ppm and the H-3 signal of the starting pyrrolylacetylene 2a at 6.35 ppm.|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Conditions | Conversion (2a)b % | Yield (3a)b % |
| a 3H-indole 1a (0.25 mmol), benzoylpyrrolylacetylene 2a (0.25 mmol), solvent 1 mL. b Yields determined by 1H NMR using durene as an internal standard. c The starting pyrrolylacetylene 2a was recovered nearly quantitatively. d ND = not detected. e 10 mol% t-BuOK was added. f 10 mol% DABCO was added. g 10 mol% PdCl2(MeCN)2 was added. h Under MW activation. | ||||
| 1 | MeOH | 20–25 °C, 96 h | 0c | NDd |
| 2 | MeOH | 60 °C, 48 h | 20 | 18 |
| 3 | MeCN | 60 °C, 48 h | 45 | 42 |
| 4 | MeCN | 60 °C, 72 h | 59 | 52 |
| 5 | MeCN | 80 °C, 48 h | 79 | 73 |
| 6 | MeCN | 80 °C, 96 h | 94 | 85 |
| 7 | MeNO2 | 80 °C, 96 h | 91 | 78 |
| 8 | DMSO | 80 °C, 96 h | 86 | 66 |
| 9 | CF3CH2OH | 80 °C, 96 h | 73 | 60 |
| 10e | MeCN | 80 °C, 40 h | 95 | 7 |
| 11f | MeCN | 80 °C, 40 h | 70 | 26 |
| 12g | MeCN | 80 °C, 40 h | 71 | 51 |
| 13h | MeCN | 100 °C, 5 h | 10 | 9 |
At room temperature (MeOH), the reaction led only to the recovery of the starting materials, the target product 3a was not detected (entry 1). After 48 h at 60 °C in MeOH, the conversion of initial indole 1a and acetylene 2a was approximately 20%, and the yield of compound 3a was 18% (entry 2). In MeCN, under similar conditions, the reaction proceeded more efficiently: the product yield was 42% and the conversion of the starting pyrrolylacetylene 2a was 45% (entry 3). At a longer reaction time (72 h) in the same solvent, the yield increased up to 52% (entry 4). A further temperature increase to 80 °C (96 h) led to a nearly quantitative conversion of pyrrolylacetylene 2a and an 85% (1H NMR) yield of 3a (entry 6). In an attempt to attain better yields, we tried some other solvents, including, DMSO, MeNO2, and CF3CH2OH, but higher yields were not reached (60–78%, entries 7–9). All attempts to optimize the reaction conditions using a catalyst or activator were not successful. The addition of 10 mol% of tert-BuOK had a detrimental effect on the product yield, furnishing just 7% of 3a in MeCN at 80 °C for 40 h (entry 10). The same reaction in the presence of 10 mol% DABCO or PdCl2(MeCN)2 gave no improvement over the catalyst-free reaction (cf. entries 11, 12, and 5), while MW irradiation was ineffective (entry 13). Thus, MeCN, 80 °C, 96 h were found to be the best conditions for the assembly of acylethenylpyrroloimidazoindoles at this stage of the study.
Next, we evaluated the scope and limitations of this cycloaddition (MeCN, 80 °C, 96 h) using a variety of 3H-indoles 1 and acylpyrrolylacetylenes 2 (Scheme 1). The results showed that the yields of pyrroloimidazoindoles 3 were in the range of 9–80% (mostly 40–76%), and were very sensitive to the nature of the substituents in 3H-indole 1 and pyrrolylacetylene 2. In all the cases, the E-configuration of the acylethenyl substituents was observed. The reaction tolerated 3H-indoles bearing different substituents at the C5 position. Indole 1b containing the electron-donating Me-group was more reactive than those with electron-withdrawing groups to give pyrroloimidazoindole 3b in good yield (58%). The moderately electron-withdrawing substituents (F, Cl) in 3H-indoles diminished the yields of 3c (36%) and 3d (35%). The presence of a strong acceptor at position 5 of indole (cyano group in 1e and nitro group in 1f) did not prevent, but significantly reduced, the effectivity of the cycloaddition reaction; the yields of adducts 3e and 3f were from low to modest (9–15%). Apparently, the introduction of the electron-accepting substituents partially withdraws the electron density from the nitrogen atom and makes it less basic/nucleophilic.
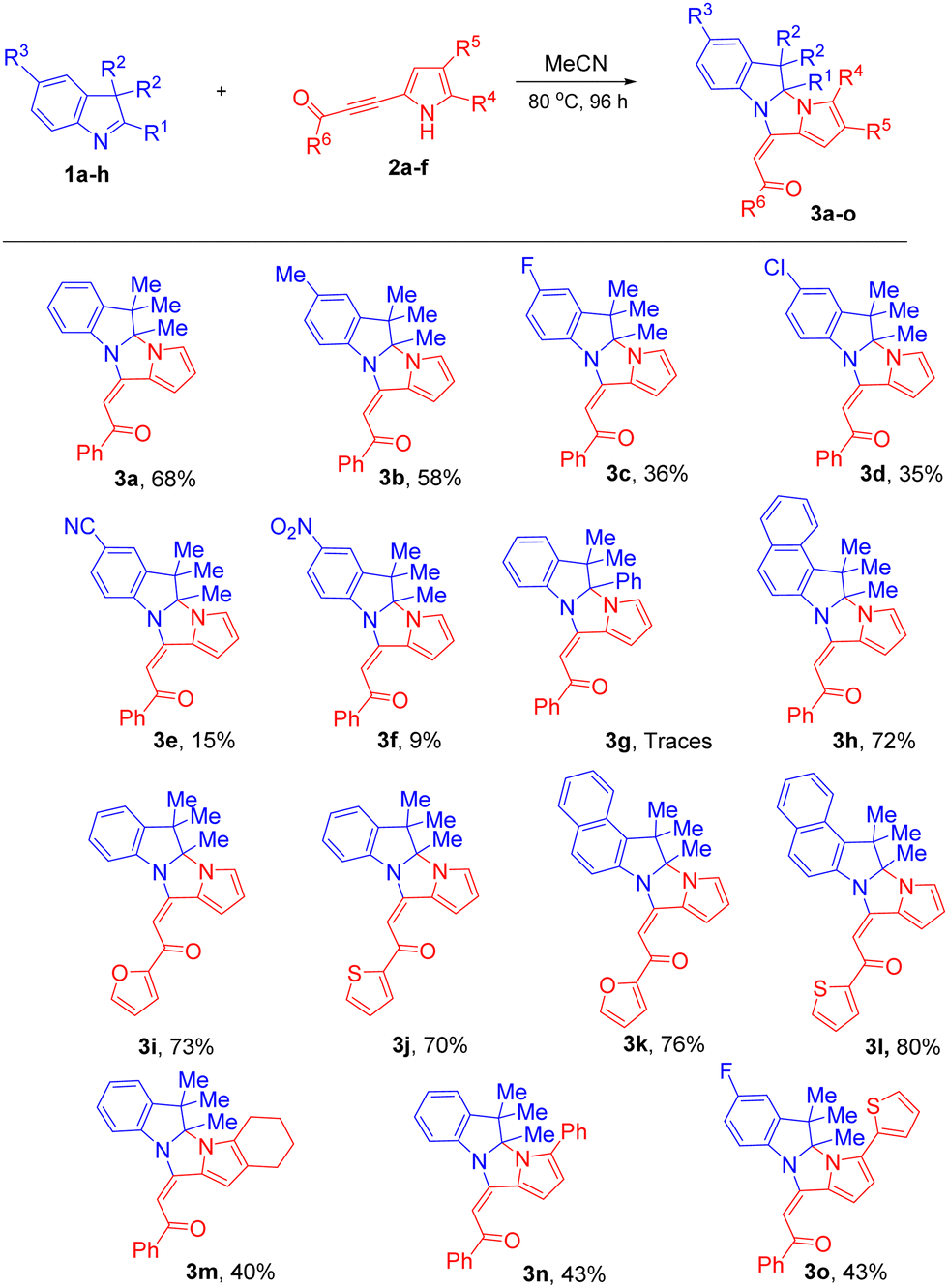 | ||
| Scheme 1 Synthesis of dihydropyrrolo[1′,2′:3,4]imidazo[1,2-a]indoles 3 in MeCN. Reaction conditions: 1 (0.5 mmol) and 2 (0.5 mmol) in MeCN (2 mL) at 80 °C for 96 h. Isolated yields after the purification by column chromatography are given. | ||
A limitation of the method was observed in the case when the Me-group at the C-2 position of 3H-indole was replaced by the phenyl group, possibly due to its electron-accepting nature and steric screening of the position 2 by its ortho-hydrogen. Therefore, the phenyl substituent in 1g impeded the reaction, and no conversion of this substrate was observed and the expected cycloaddition product was detected in only trace amounts.
Benzo[e]indole 1h more easily reacted with pyrrolylacetylene 2a, forming cycloadduct 3h in 72% yield. The effect of the extra benzene ring fused with the indole molecule likely consists of the conjugation extension over the whole system that improves the electron communication, thereby enhancing the basicity/nucleophilicity of the indole nitrogen.
We, then estimated the reaction scope relative to the acetylene substrates. The experiments showed that pyrrolylacetylenes 2b,c containing 2-furoyl and 2-thenoyl groups instead of the benzoyl group proved to be good substrates for this reaction; the yields of the corresponding cycloadducts 3i–l were 70–80%.
The substituents in the pyrrole ring of the starting acetylenes 2, both alkyl and aryl, slowed down the reaction and decreased the yields of the product 3m–o to 40–43%. This can be due to the steric screening of the reacting pyrrole nitrogen by bulky substituents.
Taking into account the growing interest in green approaches in modern organic chemistry and understanding that much more attractive in many regards would be the realization of the reaction under study in water (cheaper, safest, non-toxic, and the most environmentally friendly solvent), we performed the process in the aqueous medium. To our surprise, the results exceeded all our expectations. When we heated a heterogeneous mixture of 3H-indole 1a and pyrrolylacetylene 2a in H2O at 100 °C, soon after the reaction mixture looked pseudo-homogeneous, the insoluble stuff started to precipitate out and after 8 h, almost quantitative conversion of the initial reactants was reached, and the expected cycloadduct 3a was isolated (chromatography, silica gel) in 83% yield.
This approach was further examined and a wide range of acylpyrrolylacetylenes 2 was found to successfully undergo the cycloaddition with 3H-indoles in water under the above conditions (Scheme 2).
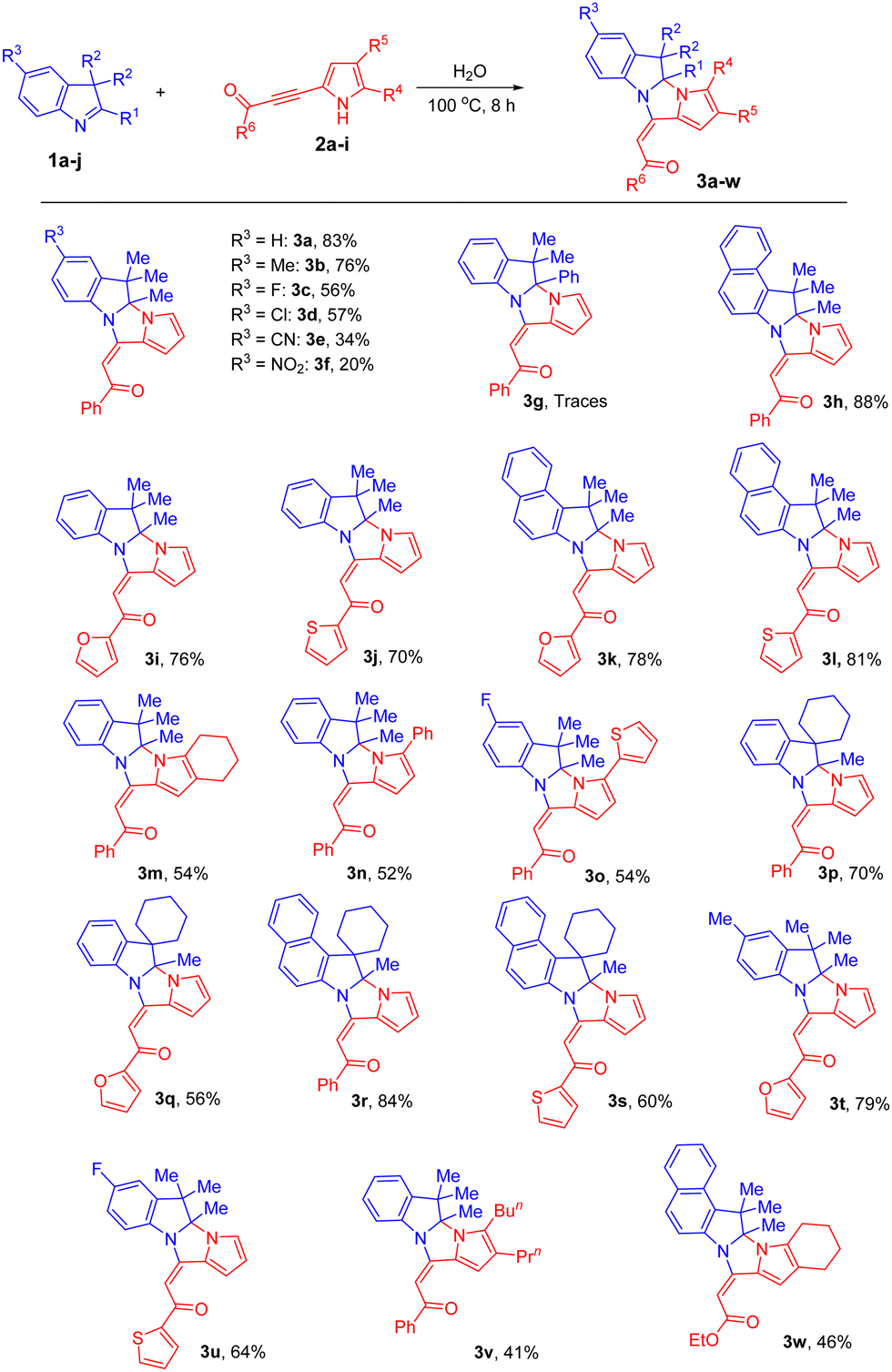 | ||
| Scheme 2 Synthesis of dihydropyrrolo[1′,2′:3,4]imidazo[1,2-a]indoles 3 in water. Reaction conditions: 1 (0.5 mmol) and 2 (0.5 mmol) in H2O (1 mL) at 100 °C for 8 h. Isolated yields after purification using column chromatography are given. | ||
Generally, various 3H-indoles and acylpyrrolylacetylenes participate in the reaction to exclusively form E-adducts in higher yields (20–88%) and much faster (8 h instead of 96 h) compared to those observed in MeCN (Table 1 and Scheme 1). Although the same regularities in the substituent effect occurred, in all the cases, the water-based approach provided better yields. For example, 5-cyano- 1e and 5-nitro- 1f 3H-indoles gave products 3e and 3f in 34% and 20% yields (Scheme 2), while in acetonitrile the yields were 15% and 9% (Scheme 1), respectively. Spirocyclic 3H-indoles 1i and 1j with the 3,3-attachment of the cyclohexane ring afforded polycyclic dihydropyrroloimidazoindoles of the original spirocyclic structure 3p–s, in reasonable to good yields (56–84%), which are first representatives of a novel family of fused polyheterocyclic systems. However, similar to the reaction in MeCN, with 2-phenyl-3H-indole 1g in water, only trace amounts of pyrroloimidazoindole 3g were detected in the reaction mixture. The same as in MeCN, retarding influence of the substitution in the pyrrole ring on the cyclization appears in the aqueous medium. So, the bulky alkyl groups (n-Bu and n-Pr) in the pyrrole ring of benzoylpyrrolylacetylene 2g expectedly retarded the reaction, and the yield of the corresponding cycloadduct 3v was only 41%. No side products were detected in the reaction mixtures. Additionally, we established that ethyl 3-(4,5,6,7-tetrahydroindol-2-yl)-2-propynoate (2h) complies well with the aqueous version of the reaction, delivering cycloadduct 3w in 46% yield under the above non-optimized (for this type of electron-deficient acetylenes) conditions. Generally, it is evident that the reaction with every reactant pair 1/2 needs to be separately optimized to achieve the best yield of the target cycloadducts for each case.
The structures of dihydropyrrolo[1′,2′:3,4]imidazo[1,2-a]indoles 3a–w were proven by NMR (1H, 13C, 15N, including 2D correlations, Fig. 2), IR spectroscopy, and mass spectrometry.
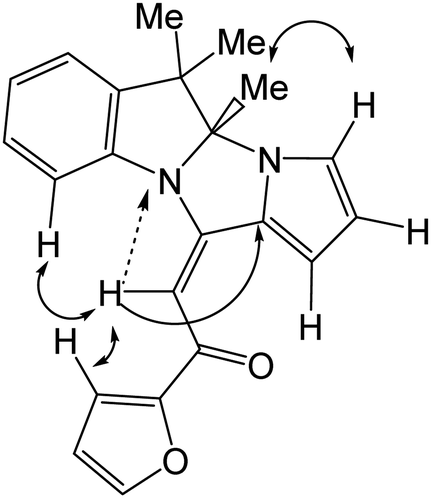 | ||
Fig. 2 Main NOESY (↔), 1H–13C (→), an d 1H–15N ( ) HMBC correlations for 3i. ) HMBC correlations for 3i. | ||
In the 2D NOESY spectra, cross-peaks between the signal of the olefin proton (δ 6.70 ppm) and the protons of the C-9 atom of indole moiety and the C-3 atom of furan ring were observed. The vicinal 3JC11a,H coupling constant 8.1 Hz corresponds to a trans position of the olefinic proton with respect to C-11a, that is, the E configuration of the adduct 3i (Fig. 2). Similar NOE effects in the NMR spectra of other pyrrolo[1′,2′:3,4]imidazo[1,2-a]indoles 3 were also observed.
To gain insights into the mechanism of pyrroloimidazoindole 3 formation, we conducted DFT calculations of [2+3] cycloaddition 3H-indole 1a with pyrrolylacetylene 2a. The level of the theory used was B2PLYP-D3BJ(PCM)/Def2-TZVPPD//wB97XD(PCM)/Def2-TZVPP. The results are depicted in Scheme 3.
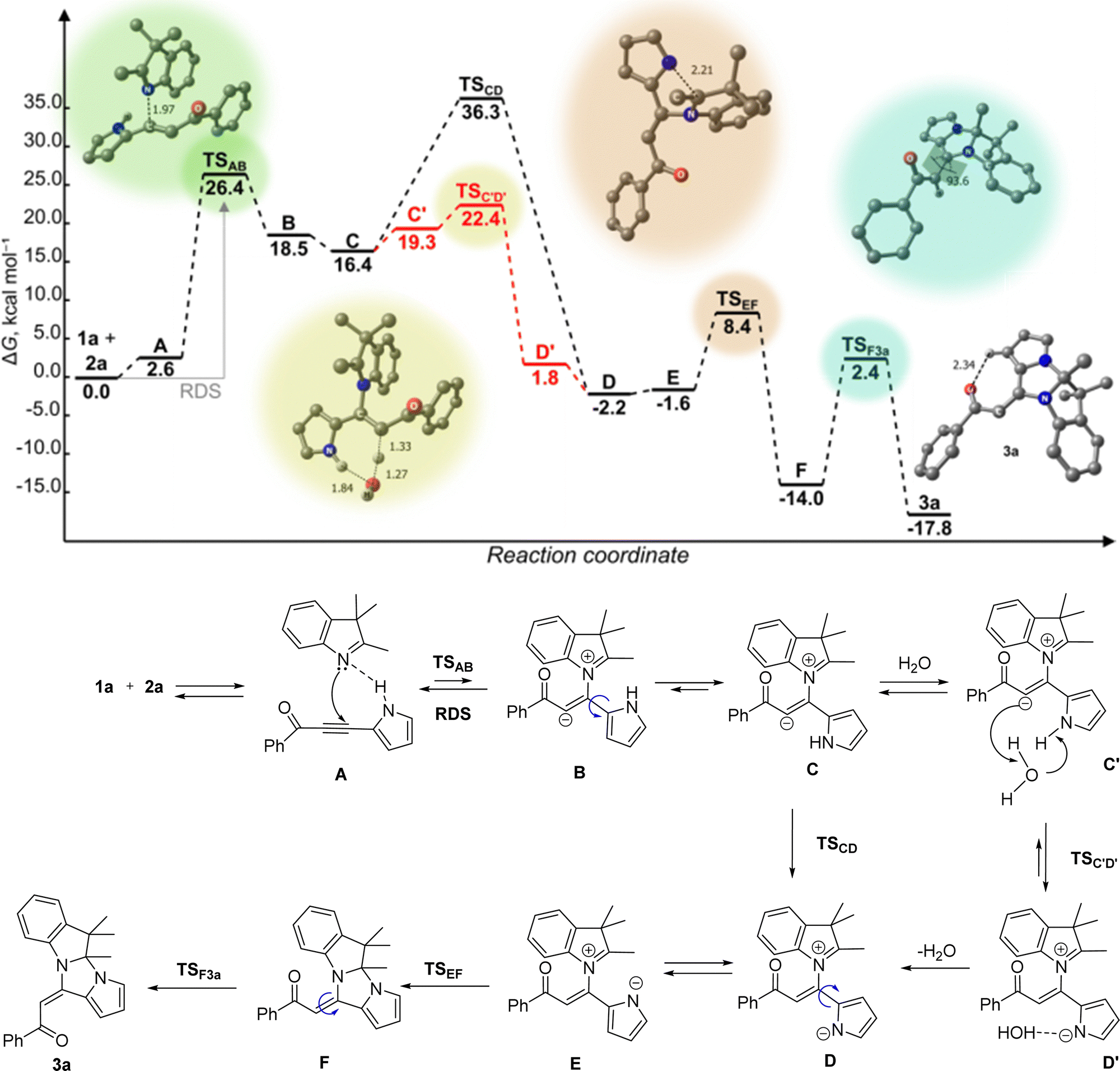 | ||
| Scheme 3 Free energy profile of (E)-1-phenyl-2-(4a,5,5-trimethyl-4a,5-dihydro-11H-pyrrolo[1′,2′:3,4]imidazo[1,2-a]indol-11-ylidene)ethan-1-one (3a) formation. The DFT calculations were carried out using the B2PLYP-D3BJ(PCM)/Def2-TZVPPD//wB97XD(PCM)/Def2-TZVPP level of theory. | ||
DFT analysis was already applied by us24,25 to the initial step of the interaction between N-nucleophilic center and electron-deficient acetylenes. According to this approach, the interaction of 1a and 2a was initiated by the formation of H-bonded pre-reaction complex A (Scheme 3). The subsequent attack of the nitrogen lone electron pair of 1a on Cβ atom of the triple bond in acetylene 2a produces the intermediate Bvia a high-lying transition state (TSAB, ΔΔG‡ = 23.8 kcal mol−1) that makes this interaction the rate-determining step of the whole process. The rotation of the pyrrole ring around the C2–C bond in intermediate B results in intermediate C (a conformational isomer of intermediate B). The latter may undergo an intramolecular H-shift to afford a quasi-stable intermediate D through the high-lying transition state TSCD, ΔΔG‡ = 19.9 kcal mol−1. However, a more favorable transformation C → D was initiated by water molecule precoordination so that the oxygen atom was H-bonded with the NH-pyrrole hydrogen, while one of the hydrogen atoms was coordinated with the C-nucleophilic center, intermediate C′ (′ – denotes the associated H2O molecule). This slightly increases the free energy of the system due to the entropy lowering (ΔΔG = 2.9 kcal mol−1). However, the further transformation C′ → D′ (→ D) proceeded significantly easily via the concerted low-lying transition state (TSC′D′, ΔΔG‡ 1.1 kcal mol−1). After 180° reorientation of the pyrrole anionic part (D⇄E), the N-anionic center of E attacks the carbocationic center of the indole moiety to give the adduct F through the corresponding TSEF (ΔΔG‡ = 10.0 kcal mol−1). The most interesting aspect here is how the reorientation of the benzoyl substituent (Z → E isomerization) occurs. The possible candidates for this role could be intermediates E or F. In the former case, predicted activation free energy is substantially high (ΔG‡ = 21.1 kcal mol−1 relative to the energy of the non-reacting system [1a + 2a]) compared with the latter. Therefore, the most probable and thermodynamically favorable transformation is F → 3a. The activation free energy ΔG‡ of the latter is only 2.4 kcal mol−1 with respect to the energy of the nonreacting [1a + 2a] system or 16.4 kcal mol−1 compared with the free energy of the intermediate F.
We also considered the relative probability of the two alternative processes, which may occur when the intermediate E′ attacks the carbocation position 2 of the indole (Fig. S1, ESI†). The first one is the oxygen center for the water attack on the carbocationic center (intermediate F′). The second one is the attacks of this position by the pyrrole N-anionic center (intermediate G′). DFT predicted the former one to be 4.8 kcal mol−1 less favorable compared with the latter option. This is well explained in terms of the HSAB concept, the softer base, a pyrrolate anion, should attack C2 carbon atom more easily than in the case of the harder oxygen base.
The quantum-chemical calculations are in agreement with the experimentally observed decisive effect of the water medium in the synthesis of pyrroloimidazoindole 3. The mechanism of [2+3]-cycloaddition in the aqueous medium could be rationalized as a micellar-like autocatalytic process. In the two-phase water/reactant mixture, the reacting indoles 1 and acetylenes 2 are self-organized in cross-aggregates of 1 (Lewis base)/2 (Lewis acid) types, which should be thermodynamically preferable compared to the homo-aggregates (1/1, 2/2). Consequently, in the cross-aggregates, the effective concentration of the reactants is much higher than that in a solution, and the reactant molecules accept the disposition corresponding to their dipole moments and inner electron distribution to produce the pre-reaction complexes A. All the intermediates represent typical surfactants consisting of hydrophobic (indole) and hydrophilic (acylpyrrolylacetylene) counterparts, both of non-ionic (A) and ionic (B–E) nature and hence accordingly26,27 can catalyze the micelle formation.
Conclusions
In summary, a green, in-water, catalyst-free implementation of stereoselective [2+3] 3H-indoles/acylpyrrolylacetylenes cycloaddition process to afford dihydropyrrolo[1′,2′:3,4]imidazo[1,2-a]indoles functionalized by acylethenyl groups of the E-configuration at up to 88% yield was achieved. Mechanistically, this reaction is assumed to represent a kind of micellar catalysis, when cross-aggregates of the starting compounds behave as surfactants and self-organized into micro-reactors, wherein, a favorable (relative to the charge and electron density distribution) mutual orientation of the reactants is realized. Performed DFT calculations of the mechanism of the major reaction occurring inside the micelle confirmed a key role of the water molecules within the whole cascade process. This water-based approach, except for being ecologically friendly, surpasses organic solvent-based protocols in terms of a much higher reaction rate (by more than 10 times), better yields, and simpler isolation procedures. All this together with the availability of the starting 3H-indoles and acylpyrrolylacetylenes and a suitable substrate scope allow this approach to be considered as a new general straightforward route to so far inaccessible but pharmacologically prospective fused polyheterocyclic structures.Author contributions
Ludmila A. Oparina: investigation, methodology, resources, writing–original draft. Kseniya V. Belyaeva: conceptualization, funding acquisition, methodology. Nikita A. Kolyvanov: investigation, visualization. Igor A. Ushakov: formal analysis, investigation. Denis N. Tomilin: resources, investigation. Lyubov N. Sobenina: methodology, supervision. Anton V. Kuzmin: formal analysis, software, data curation. Boris A. Trofimov: project administration, validation, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by a grant from the Russian Science Foundation (project no. 21-73-10134). The authors thank the Baikal Analytical Centre of Collective Use and Shared Research Facilities for Physical and Chemical Ultramicroanalysis, Limnological Institute, and SB RAS (HRMS-TOF Spectra) for the equipment.Notes and references
- C. L. Sun and Z. J. Shi, Transition-Metal-Free Coupling Reactions, Chem. Rev., 2014, 114, 9219–9280 CrossRef CAS.
- W. Zhang and W.-B. Yi, Pot, Atom, and Step Economy (Pase) Synthesis, Springer, 2019, pp. 49 Search PubMed.
- M. N. Elinson, A. N. Vereshchagin, Y. E. Anisina, S. K. Krymov, A. N. Fakhrutdinov, A. S. Goloveshkin and M. P. Egorov, Pot, atom and step economic (PASE) assembly of salicylaldehydes, malononitrile dimer and 4-hydroxypyridine-2(1H)-ones into medicinally relevant 5H-chromeno[2,3-b]pyridine scaffold, Mol. Diversity, 2020, 24, 617–626 CrossRef CAS.
- L. A. Oparina, K. V. Belyaeva, N. A. Kolyvanov, I. A. Ushakov, M. D. Gotsko, L. N. Sobenina, A. V. Vashchenko and B. A. Trofimov, Catalyst-Free Annulation of Acylethynylpyrroles with 1-Pyrrolines: a Straightforward Access to Tetrahydrodipyrrolo[1,2-a:1′,2′-c]imidazoles, J. Org. Chem., 2022, 87, 9518–9531 CrossRef CAS PubMed.
- L. A. Oparina, N. A. Kolyvanov, I. A. Ushakov, L. P. Nikitina, O. V. Petrova, L. N. Sobenina, K. B. Petrushenko and B. A. Trofimov, Contributing to Biochemistry and Optoelectronics: Pyrrolo[1′,2′:2,3]imidazo[1,5-a]indoles and Cyclohepta[4,5]pyrrolo[1,2-c]pyrrolo[1,2-a]imidazoles via [3+2] Annulation of Acylethynylcycloalka[b]pyrroles with Δ1-Pyrroline, Int. J. Mol. Sci., 2023, 24, 3404 CrossRef CAS.
- B. A. Trofimov, Z. V. Stepanova, L. N. Sobenina, A. I. Mikhaleva and I. A. Ushakov, Ethynylation of Pyrroles with 1-Acyl-2-Bromoacetylenes on Alumina: a Formally “Inverse Sonogashira Coupling”, Tetrahedron Lett., 2004, 45, 6513–6516 CrossRef CAS.
- L. N. Sobenina and B. A. Trofimov, Recent Strides in the Transition Metal-Free Cross-Coupling of Haloacetylenes with Electron-Rich Heterocycles in Solid Media, Molecules, 2020, 25, 2490 CrossRef CAS , and references cites therein.
- N. Koyama, Y. Inoue, M. Sekine, Y. Hayakawa, H. Homma, S. Ōmura and H. Tomoda, Relative and Absolute Stereochemistry of Quinadoline B, an Inhibitor of Lipid Droplet Synthesis in Macrophages, Org. Lett., 2008, 10, 5273–5276 CrossRef CAS.
- M. Wu and D. Ma, Total Syntheses of (±)-Spiroquinazoline, (−)-Alantryphenone, (+)-Lapatin A, and (−)-Quinadoline B, Angew. Chem., Int. Ed., 2013, 52, 9759–9762 CrossRef CAS PubMed.
- J. Peng, T. Lin, W. Wang, Z. Xin, T. Zhu, Q. Gu and D. Li, Antiviral Alkaloids Produced by the Mangrove-Derived Fungus Cladosporium sp. PJX-41, J. Nat. Prod., 2013, 76, 1133–1140 CrossRef CAS.
- M. T. J. Quimque, K. I. R. Notarte, R. A. T. Fernandez, M. A. O. Mendoza, R. A. D. Liman, J. A. K. Lim, L. A. E. Pilapil, J. K. H. Ong, A. M. Pastrana, A. Khan, D.-Q. Wei and A. P. G. Macabeo, Virtual screening-driven drug discovery of SARS-CoV2 enzyme inhibitors targeting viral attachment, replication, post-translational modification and host immunity evasion infection mechanisms, J. Biomol. Struct. Dyn., 2021, 39, 4316–4333 CrossRef CAS PubMed.
- S. Brogi, M. T. Quimque, K. I. Notarte, J. G. Africa, J. B. Hernandez, S. M. Tan, V. Calderone and A. P. Macabeo, Virtual Combinatorial Library Screening of Quinadoline B Derivatives against SARS-CoV-2 RNA-Dependent RNA Polymerase, Computation, 2022, 10, 7 CrossRef CAS.
- R. S. Chang, V. J. Lotti, R. L. Monaghan, J. Birnbaum, E. O. Stapley, M. A. Goetz, G. Albers-Schonberg, A. A. Patchett, J. M. Liesch, O. D. Hensens and J. P. Springer, A Potent Nonpeptide Cholecystokinin Antagonist Selective for Peripheral Tissues Isolated from Aspergillus alliaceus, Science, 1985, 230, 177–179 CrossRef CAS.
- S. M. Wong, L. L. Musza, G. C. Kydd, R. Kullnig, A. M. Gillum and R. Cooper, Fiscalins: New substance P inhibitors produced by the fungus Neosartorya fischeri. Taxonomy, fermentation, structures, and biological properties, J. Antibiot., 1993, 46, 545–553 CrossRef CAS PubMed.
- G. N. Belofsky, M. Anguera, P. R. Jensen, W. Fenical and M. Kock, Oxepinamides A-C and Fumiquinazolines H-I: Bioactive Metabolites from a Marine Isolate of a Fungus of the Genus Acremonium, Chem. – Eur. J., 2000, 6, 1355–1360 CrossRef CAS.
- H. Fujimoto, E. Negishi, K. Yamaguchi, N. Nishi and M. Yamazaki, Isolation of New Tremorgenic Metabolites from an Ascomycete, Corynascus setosus, Chem. Pharm. Bull., 1996, 44, 1843–1848 CrossRef CAS.
- K. H. Ahammad Uz Zaman, Z. Hu, X. Wu and S. Cao, Tryptoquivalines W and X, two new compounds from a Hawaiian fungal strain and their biological activities, Tetrahedron Lett., 2020, 61, 151730 CrossRef CAS PubMed.
- R. H. Jiao, S. Xu, J. Y. Liu, H. M. Ge, H. Ding, C. Xu, H. L. Zhu and R. X. Tan, Org. Lett., 2006, 8, 5709–5712 CrossRef CAS.
- I. Yamawaki, Y. Matsushita, N. Asaka, K. Ohmori, N. Nomura and K. Ogawa, Synthesis and aldose reductase inhibitory activity of acetic acid derivatives of pyrrolo[1,2-c]imidazole, Eur. J. Med. Chem., 1993, 28, 481–498 CrossRef CAS.
- D. Dal Ben, I. Antonini, M. Buccioni, C. Lambertucci, G. Marucci, A. Thomas, R. Volpini and G. Cristalli, Neuropeptide S receptor: recent updates on nonpeptide antagonist discovery, ChemMedChem, 2011, 6, 1163–1171 CrossRef CAS PubMed.
- M. Yamaoka and T. Hara, Pyrrolo[1,2-c]imidazole derivatives for use in the prophylaxis or treatment of cancer which is refractory to known cancer therapies, WO2009/1057795, 2009 Search PubMed.
- M. Zhang, Y. Ding, H.-X. Qin, Z.-G. Xu, H.-T. Lan, D.-L. Yang and C. Yi, One-pot synthesis of substituted pyrrole–imidazole derivatives with anticancer activity, Mol. Diversity, 2020, 24, 1177–1184 CrossRef CAS.
- D. Erşen, M. Ülger, S. Mangelinckx, M. Gemili, E. Şahin and Y. Nural, Synthesis and anti(myco)bacterial activity of novel 5,5-diphenylpyrrolidine N-aroylthiourea derivatives and a functionalized hexahydro-1H-pyrrolo[1,2-c]imidazole, Med. Chem. Res., 2017, 26, 2152–2160 CrossRef.
- K. V. Belyaeva, L. P. Nikitina, V. S. Gen', I. V. Saliy, L. N. Sobenina, A. V. Afonin, A. V. Kuzmin, A. V. Vashchenko and B. A. Trofimov, Benzo[1,4]diazocinone/pyrrole ensembles via the catalyst-free insertion of pyrrolylacetylenic ketones into benzimidazoles, ChemistrySelect, 2022, 7, e202204482 CrossRef CAS.
- K. V. Belyaeva, L. P. Nikitina, V. S. Gen', A. V. Kuzmin, A. V. Afonin and B. A. Trofimov, A straightforward access to 2-hydroxyoxazino[3,2-f]phenanthridines from phenanthridine, oxalylacetylenes and water, Mendeleev Commun., 2022, 32, 439 CrossRef CAS.
- N. Compagno, R. Profeta and A. Scarso, Recent advances in the synthesis of active pharmaceutical and agrochemical ingredients in micellar media, Curr. Opin. Green Sustainable Chem., 2023, 39, 100729 CrossRef CAS.
- T. Shen, S. Zhou, J. Ruan, X. Chen, X. Liu, X. Ge and C. Qian, Recent advances on micellar catalysis in water, Adv. Colloid Interface Sci., 2021, 287, 102299 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nj05049a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |