
Photoelectrochemical properties of p-type CuBi2O4 prepared by spray pyrolysis of carbon-free precursor aqueous solution combined with post-annealing treatment†
Kaisei
Wakishima
a,
Tomohiro
Higashi
 *b,
Akira
Nagaoka
ac and
Kenji
Yoshino
*a
*b,
Akira
Nagaoka
ac and
Kenji
Yoshino
*a
aElectrical and Electronic Engineering Program, Faculty of Engineering, University of Miyazak, 1-1 Gakuen-kibanadai-nishi, Miyazaki 889-2192, Japan. E-mail: t0b114u@cc.miyazaki-u.ac.jp
bInstitute for Tenure Track Promotion, University of Miyazaki, 1-1 Gakuen-kibanadai-nishi, Miyazaki 889-2192, Japan. E-mail: t_higashi@cc.miyazaki-u.ac.jp
cResearch Center for Sustainable Energy & Environmental Engineering, University of Miyazaki, 1-1 Gakuen-kibanadai-nishi, Miyazaki 889-2192, Japan
First published on 22nd November 2023
Abstract
Energy conversion using semiconductor-based photoelectrodes for photoelectrochemical (PEC) water splitting is a promising technology that contributes to an environmentally friendly society. CuBi2O4 is a p-type semiconductor material that can be used in visible-light-responsive photocathodes. To date, the fabrication of CuBi2O4 has been examined using wet processes such as spray pyrolysis of precursor solutions containing organic solvents and organic additives. Spray pyrolysis causes problems such as CO2 emission during the decomposition of the precursor solution. In this study, we established a carbon-free method for the fabrication of CuBi2O4 using a precursor solution comprising Cu(NO3)2 and Bi(NO3)3 dissolved in a dilute nitric acid aqueous solution. The aqueous precursor solution was sprayed onto F-doped tin oxide-coated glass substrates, followed by post-annealing treatment in the temperature range of 400–700 °C for 1 h in air. An annealing temperature over 500 °C provides CuBi2O4 as revealed by the results of X-ray diffraction, scanning electron microscopy, and X-ray photoelectron spectroscopy characterization. The PEC properties of the CuBi2O4 photoelectrodes were investigated to determine the optimal post-annealing conditions for maximizing the photocurrent density under AM 1.5G solar illumination. The optimized CuBi2O4 photoelectrodes generated a photocurrent density of −0.94 mA cm−2 at 0.6 V vs. a reversible hydrogen electrode in a potassium phosphate aqueous solution with the H2O2 sacrificial reagent. This study provides rational guidelines for visible-light-absorbing p-type metal oxide-based photoelectrodes utilizing a carbon-free preparation process.
1. Introduction
Solar hydrogen produced from water is one of the most promising renewable fuels to tackle the problems of environmental pollution and energy crisis.1–5 Among the processes of solar hydrogen production, photoelectrochemical (PEC) water splitting to generate hydrogen and oxygen has been recognized as the key technology, using abundant solar energy and water resources.4–6 One approach to realizing efficient solar water splitting is by creating a PEC cell comprising a photocathode for the hydrogen evolution reaction (HER) and a photoanode for the oxygen evolution reaction (OER).6–8 PEC cells can exhibit overall water splitting to produce hydrogen and oxygen without the application of an external bias voltage under simulated sunlight illumination. The PEC cell can relax the thermodynamic requirements for water splitting, such as the positions of the conduction band minimum (CBM) of the photoanode and the valence band maximum (VBM) of the photocathode, as shown in Fig. S1 (ESI†),8,9 through a two-step light excitation process similar to Z-scheme photocatalytic water splitting. Typically, photogenerated electrons in the CBM of the photocathode and the photogenerated holes in the VBM of the photoanode drive the reduction and oxidation reactions of water, respectively. With the synergistic effect of two driving forces of both photoelectrodes,8,9 the photogenerated electrons of the photoanode (in the CBM) are transported to the photocathode (in the VBM) through the external circuit, generating a photocurrent attributable to the spontaneous overall water splitting.Various semiconductor materials have been designed as photoelectrodes to realize efficient PEC water splitting.10–12 Narrow-bandgap semiconductors such as chalcogenides,13–15 silicon,16,17 and perovskite solar cells (PSCs)18–21 have been employed as photocathodes or photovoltaic (PV) cells in the PEC HER. For the case of the PEC OER, wide-bandgap semiconductors such as metal oxides,22–24 metal (oxy)nitrides,25–29 and bimetallic oxide composites30,31 have been extensively investigated. Further, overall water splitting by the PEC cell based on the photocathode/photoanode combination has been examined to realize a high level of solar-to-hydrogen (STH) energy conversion efficiency. An STH energy conversion efficiency of more than 10% for commercial solar hydrogen production is required to be cost-competitive with a steam reforming process,3,9 and it is still challenging. For instance, Huang et al. demonstrated a solar-to-hydrogen (STH) energy conversion efficiency of 3.17% using a PEC cell consisting of a Cu2ZnSnS4 chalcogenide-based photocathode for the HER and a BiVO4 metal oxide-based photoanode for the OER.32 Kobayashi et al. reported an STH efficiency of 3.7% using a PEC cell composed of a CuIn0.5Ga0.5Se2-based photocathode and a BiVO4-based photoanode.13 In addition, PV-assisted PEC (PV-PEC) cells have been examined to achieve a high level of STH energy conversion efficiency for overall water splitting. Wang et al. demonstrated an STH efficiency of 6.5% from a PV-PEC cell composed of PSC-based PV for the HER and a dual-BiVO4 photoanode for the OER.19 A PV-PEC cell composed of CuInSe2-based PV for the HER and a Ta3N5-based semitransparent photoanode for the OER has achieved an STH efficiency of 10%.33 Recently, semitransparent photoanodes based on metal oxynitride materials such as LaTiO2N, SrTaO2N, and CaTaO2N have been developed.27–29 The onset potential for the OER delivered from the oxynitride-based semitransparent photoanodes has been reported to be below 0.0 V vs. a reversible hydrogen electrode (VRHE),27–29 which is expected to further improve the STH efficiency on the PV-PEC device. At the present stage, the PV-PEC cell achieved a higher STH efficiency than the PEC cell with a photocathode/photoanode combination. The development of efficient photocathodes for the PEC HER applicable to a PEC cell is required to further increase the STH efficiency.
The PEC HER on Cu-based metal oxide semiconducting materials such as CuO, Cu2O, CuFeO2, and CuBi2O4 has been extensively investigated because of their abundant resources and low toxicity.34,35 Among them, the CuBi2O4 (CBO) material can be recognized as one of the promising candidates for the photoelectrode material for the PEC HER involving CO2 reduction.36–48 CBO has a tetragonal crystal structure and consists of stacks of square planar Cu(II)O4 groups linked to distorted trigonal Bi(III)O6 polyhedral groups.34 The bandgap (Eg) energy of the CBO material has been reported to be 1.5–1.8 eV (adsorption edge wavelength up to 830 nm),37,38,42,44 which leads to a theoretical STH energy conversion efficiency limit of ∼24% at an external quantum efficiency (EQE) of 100%.34 The STH limit of 24% can lead to a theoretical maximum cathodic photocurrent density of 19.7 mA cm−2 under AM 1.5G solar illumination (1 Sun) when an EQE of 100% and an Eg of 1.8 eV were assumed. Moreover, the flat band potential of CBO in contact with an aqueous electrolyte solution is located at ∼1.2 VRHE,37,41,42 enabling the application of a photovoltage to drive the PEC reduction reaction. These light-absorption features, theoretically expected PEC performances, and an energy level alignment with the standard redox potential of the HER have promoted advanced research for CBO-based photocathodes involving a PEC cell for non-biased overall water splitting.36,48–51 Wang et al. have reported that the CBO-based photoelectrode prepared through a spray pyrolysis method produces a photocurrent density of −2.0 mA cm−2 at 0.6 VRHE in the presence of an H2O2 sacrificial reagent.41 In addition, −2.66 mA cm−2 at 0.6 VRHE generated by the optimized CBO photoelectrodes has been established by Xu et al.47 Non-biased overall water splitting by a PEC cell with a combination of photoanodes for the OER such as TiO2, BiVO4, and ZnIn2S4 has been investigated, but typically exhibits an STH energy conversion efficiency of less than 1.0%.36,48–51 Further development of efficient CBO-based photoelectrodes applicable to PEC cells is in demand.
The CBO-based photoelectrode is typically processed by heating the CBO precursor compounds such as a Cu/Bi complex deposited on the conductive substrates (e.g., F-doped tin oxide coated glass (FTO) and Au) in air.35–39 CBO precursor compounds have been prepared on the substrates by deposition of the precursor solution using wet processes such as electrodeposition,36–39,49,52 drop-casting methods,36,44,45,48,50 spin-coating methods,46,47 and spray pyrolysis methods.40–43,51,53 Concerning the wet process to prepare CBO-based photocathodes, most of the precursor solution is based on non-aqueous organic solvents (e.g., ethanol and dimethyl sulfoxide) and acetic acid with organic additives (e.g., polyethylene glycol (PEG) and triethyl orthoformate (TEOF)) to dissolve and stabilize the Cu and Bi complex in the solvent.38,41,46,47 The role of the organic additives in the precursor solution is to mitigate the hydrolyzed precipitation of Bi subnitrate species as time advances in the precursor solution.38,41,46 Very recently, Joos et al. have established an impressive synthesis route using citrate-ligand-based aqueous-sol–gel methods to fabricate the CBO photoelectrode prepared by a combination of spin-coating and thermal annealing in air.46 However, CO2 emissions associated with the combustion and decomposition of carbon species present in the precursor compounds (ligands of the metal complex) and the solvents in which they are dissolved are considered issues. To overcome this, it is preferable to establish a method for the preparation of precursor compounds via a carbon-free wet process, leading to a large-scale process with a low environmental impact.
In this study, we report a carbon-free synthesis method for preparing CBO-based photoelectrodes using spray pyrolysis of an aqueous precursor solution. The aqueous precursor solution was composed of Cu nitrate (Cu(NO3)2) and Bi nitrate (Bi(NO3)3) dissolved in the dilute aqueous nitric acid medium as a solvent. In this precursor aqueous solution, precipitation due to the hydrolysis of Bi and Cu ions was not observed, indicating the stability of Cu and Bi ions in the solvent. The aqueous precursor solution was sprayed onto the FTO substrates which kept the temperature at 400 °C. The resulting specimens were further annealed at several temperatures in the range of 400–700 °C in air. This post-annealing treatment at temperatures over 500 °C provided a CBO polycrystalline film on the FTO substrate. The impact of the post-annealing treatment was evaluated using X-ray photoelectron spectroscopy (XPS) and PEC measurements under simulated sunlight illumination. In the presence of an H2O2 sacrificial reagent as an electron acceptor, the optimized CBO-based photoelectrodes showed a photocurrent density of −0.94 mA cm−2 at 0.6 VRHE with an onset potential of 1.05 VRHE. The investigation of the fabrication procedure and characterization of the CBO photocathode in this study will contribute to the establishment of a preparation protocol for visible-light-responsive metal oxide-based photoelectrode materials through a carbon-free wet process.
2. Results and discussion
2.1. Structural and morphological properties of CuBi2O4
CuBi2O4 (CBO) thin films were prepared via spray pyrolysis using a precursor solution consisting of Cu nitrate and Bi nitrate ([Cu]/[Bi] = 0.50) dissolved in a dilute nitric acid aqueous solution, followed by an additional annealing treatment at a specific temperature. Initially, the FTO used as a substrate was heated to and kept at 400 °C when the precursor solution was sprayed to form a precursor film on FTO. The resulting film on FTO was subsequently annealed in air at temperatures ranging from 400 to 700 °C for 1 h. We defined the additional annealing process as the post-annealing treatment. Details of the fabrication process, including the preparation of the precursor solution and the spray pyrolysis protocol, are provided in the Experimental section and ESI,† (Fig. S2).Fig. 1a presents the X-ray diffraction (XRD) patterns of the specimens processed with the post-annealing treatment (500, 600, and 700 °C for 1 h), the as-deposited film (without post-annealing treatment), and CBO powder as a reference (COD-1006028). The peaks at 26.6°, 33.9°, 37.9°, and 51.7° are attributed to the FTO substrates (marked with an asterisk in Fig. 1a). The XRD pattern of the as-deposited sample (without post-annealing treatment) shows a broad peak at approximately 28°, which overlaps with the XRD signals delivered from the FTO substrate, indicating the low crystallinity of the as-deposited sample. The samples after post-annealing treatment at temperatures over 500 °C generated XRD patterns consistent with the CBO reference with a tetragonal structure and in good agreement with reported literature.39,41,47 Regardless of the temperatures for post-annealing treatment (ranging from 500 to 700 °C), XRD peaks were observed at 21.1°, 28.2°, and 46.9°, which correspond to the CBO crystalline planes of (200), (211), and (411), respectively. The increase in the post-annealing temperature increases the intensities of the peaks at 21.1°, 28.2°, and 46.9°, indicating the enhanced crystallinity of the CBO. Specifically, the intensity of the peak at 28.2°, assignable to the (211) crystal plane of CBO, increased significantly. The impurities from the residual precursors (Cu and Bi nitrates) were absent in the specimens after post-annealing treatment at a temperature over 500 °C (Fig. S3, ESI†).
 | ||
| Fig. 1 Structural characterization of CuBi2O4 (CBO) on the FTO substrate prepared by spray pyrolysis of Cu nitrate and Bi nitrate ([Cu2+]/[Bi3+] = 0.50) containing precursor aqueous solution at 400 °C together with post-annealing treatment in air. (a) X-ray diffraction (XRD) patterns of samples processed with post-annealing treatment at 500 °C (blue), 600 °C (green), and 700 °C (black), and without post-annealing treatment of the as-deposited (As-depo) sample (red). The XRD peak derived from the FTO substrate is indicated by an asterisk (*). The XRD patterns (COD-1006028, colored in pink) correspond to the crystalline planes of CBO as a reference. (b) Top-view scanning electron microscopy (SEM) image of bare FTO. (c)–(g) Top-view and cross-sectional SEM images of the as-deposited sample at 400 °C (c) and samples processed at 500 °C (d) and (g), 600 °C (e), and 700 °C (f). Scale bars in SEM images indicate 500 nm. | ||
To eliminate the effect of chemical interference from the FTO layer during CBO preparation, the CBO thin film deposited on a bare-glass substrate was prepared in the same manner for the CBO on FTO. The XRD pattern of the sample deposited on the glass substrate processed with post-annealing treatment was in good agreement with the reference patterns of CBO (Fig. S4, ESI†), indicating that a processing temperature over 500 °C is required to produce CBO from the precursor solution containing Cu and Bi nitrates. The weak XRD signals at 35.5° and 38.7° observed in the as-deposited sample are assignable to the monoclinic CuO of (002) and (111) crystal planes, respectively.41,42 As shown in Fig. S5 (ESI†), thermogravimetry-differential thermal analysis (TG-DTA) in air revealed that the precursor compounds (mixed with Cu(NO3)2 and Bi(NO3)3, [Cu]/[Bi] = 0.50) are fully decomposed at a temperature exceeding 500 °C and generate Cu–Bi oxide, which is in good agreement with the results of the XRD patterns of the resulting films processed with the post-annealing treatment. The thermal decomposition temperature of the precursor compound (mixed with Cu(NO3)2 and Bi(NO3)3) is lower than that of Bi(NO3)3. Cu(NO3)2 showed the thermal decomposition into Cu oxide (CuO) at a temperature over 270 °C, whereas Bi(NO3)3 exhibits two-step thermal decomposition: BiONO3 is formed above 350 °C and pyrolyzed completely at a temperature over 520 °C to produce Bi oxide (Bi2O3).54–56 These results indicate that the post-annealing treatment for the as-deposited film provides the CBO on the FTO substrate through the thermal decomposition of the precursor compound.
Top-view and cross-sectional scanning electron microscopy (SEM) images obtained from the bare FTO substrate, as-deposited film (without post-annealing treatment), and the CBO samples with post-annealing treatment (500, 600, and 700 °C for 1 h) are presented in Fig. 1b–g. The bare FTO substrate had a densely packed structure with a rough surface morphology of 200–500 nm (Fig. 1b). The as-deposited film covered the surface of the FTO and was found to have a protrusion of approximately 500 nm in size (Fig. 1c). The SEM image obtained from the as-deposited film indicated separate dark and bright regions corresponding to pores of approximately 200 nm size and the deposited precursor, respectively. The formation of pores in the as-deposited film is likely attributed to the nonuniform nucleation–growth process during the thermal decomposition of the precursor solution containing Cu(NO3)2 and Bi(NO3)3 and the thermal evaporation of the solvent (dilute nitric aqueous solution). The reason for the formation of pores in the as-deposited film can be interpreted by the basis of homogeneous and heterogeneous nucleation processes similar to the metal oxide composite-based thin films prepared using an aerosol-assisted chemical vapor deposition technique.57–59 In the present study, the deposition temperature (substrate temperature) was kept at 400 °C, producing the mixture of Cu oxide and Bi subnitrate that have different pyrolysis temperatures (see Fig. S5, ESI†). Thus, the formation of pores with an agglomerated film is likely due to the homogeneous nucleation simultaneously with the heterogenous nucleation-growth reaction.
In sharp contrast to the as-deposited film, the SEM images obtained from the CBO samples processed with the post-annealing treatment indicate significant surface morphological changes (Fig. 1d–f). After the post-annealing treatment of the samples, no pores or protrusions were observed. The grain size grew significantly when the post-annealing temperature was raised from 500 to 700 °C. The SEM images of the samples with post-annealing treatment at 500 °C, 600 °C, and 700 °C indicated particle sizes of ∼200 nm (Fig. 1d), ∼400 nm (Fig. 1e), and ∼500 nm (Fig. 1f), respectively. The increased particle size associated with an increase in the crystallite size of CBO is in reasonable agreement with the XRD patterns of the CBO samples. Even when the post-annealing temperature was increased, no correlation was observed with the surface morphology of the underlying FTO. From the cross-sectional SEM image (Fig. 1g), the film thicknesses of CBO and FTO were 330 and 600 nm, respectively. The thickness of the CBO produced herein was found to be larger than that of previously reported CBO (approx. 270 nm) on FTO fabricated through a spray pyrolysis method using acetic acid/ethanol solvent containing organic additives of PEG and TEOF.41–43 While the CBO grains grew in the direction normal to the FTO surface, a continuous layer consisting of CBO grains with a thickness of approximately 20 nm was observed. No cracks or pores were observed between the bottom CBO layer and the FTO surface. Surface protrusions and gaps were observed in the as-deposited sample (Fig. S6, ESI†), and the thickness from the bottom layer of the film to the top surface was approximately 1100 nm. It can be interpreted that the post-annealing treatment produced a densely packed film of CBO on the FTO substrate, whereas the film thickness decreased via annealing.
To evaluate the impact of the post-annealing treatment on the light-absorption characteristics of the CBO samples, diffuse reflectance spectroscopy (DRS) was performed (Fig. 2). Irrespective of the post-annealing temperature, the CBO samples absorbed visible and near-infrared light at wavelengths of up to 830 nm, which is in good agreement with previously reported CBO materials.41,44,47 The as-deposited sample exhibited further light absorption beyond 830 nm, whereas the CBO samples after post-annealing treatment showed reduced light absorption at wavelengths longer than 830 nm. Specifically, the CBO samples with post-annealing temperatures of 600 and 700 °C showed reduced light absorption at wavelengths longer than 830 nm. Fig. S7 (ESI†) shows the DRS spectra of the samples prepared by spray pyrolysis at 400 °C using aqueous precursor solutions consisting only of Cu(NO3)2 and Bi(NO3)3. The Bi oxide (BiOx) sample delivered from Bi(NO3)3 showed light absorption at wavelengths longer than 520 nm, which gradually increased with increasing wavelength, whereas Cu oxide (CuOx) exhibited gradual attenuation of light absorption at wavelengths longer than approximately 650 nm. The increase in light absorption at longer wavelengths observed in the as-deposited sample of CBO is most likely attributable to residual BiOx and subnitrate of BiONO3. Gottesman et al. reported that the light-absorption features related to the optical bandgap energy of CBO vary with the concentration of the defects and impurities inside CBO.60 In addition, cation doping such as Co2+ into CBO induces a shift of the absorption edge wavelength toward a longer wavelength.53 The bandgap energy related to the absorption edge wavelength of the CBO needs to be determined more accurately. The light response of CBO to an actual PEC process will be discussed based on the results of the wavelength-dependent PEC characterization described later.
 | ||
| Fig. 2 Diffuse reflectance spectra (DRS) obtained from the samples prepared on the FTO substrate via spray pyrolysis at 400 °C (As-depo, red) combined with post-annealing treatment in air at 500 °C (blue), 600 °C (green), and 700 °C (black). | ||
2.2. Analysis of the surface chemical composition of CuBi2O4
Polycrystalline CBO thin films were successfully developed via spray pyrolysis using a carbon-free aqueous precursor solution and post-annealing in air. As discussed above, the temperature of the post-annealing treatment significantly affected the morphology and light-absorption properties of the CBO thin film. The surface chemical states and surface atomic ratios of Cu and Bi are crucial factors limiting the semiconducting properties related to the PEC performance of CBO materials. To gain an in-depth understanding of the CBO thin films prepared herein, especially regarding the surface chemical states of the samples, XPS was conducted.The wide-scan XPS spectrum (Fig. S8, ESI†) clearly shows the presence of Cu, Bi, and O in the materials, regardless of the post-annealing treatment. The as-deposited samples showed the N 1s peak centered at a binding energy (BE) of 406.6 eV, which is assignable to the signals derived from the nitrates (BiONO3) produced during the spray pyrolysis at 400 °C. In contrast, the N 1s signal on the samples is fully diminished by the post-annealing treatment at temperatures over 500 °C (Fig. S9a, ESI†). The Sn 3d peak originating from the FTO substrates was observed in all the samples, and no drastic difference in the Sn 3d peak was observed after the post-annealing treatment (Fig. S9b, ESI†).
Fig. 3 displays the high-resolution XPS (HR-XPS) spectra of the as-deposited film (abbreviated to As-depo in the figure) and CBO samples processed with post-annealing treatment at 500 °C and 700 °C for 1 h. The HR-XPS spectra contained Cu 2p peaks centered at BEs of 934.0 eV for Cu 2p3/2 and 953.8 eV for Cu 2p1/2 with a spin–orbit separation (ΔBE) of 19.8 eV (Fig. 3a). These peaks were consistent with the literature and can be assigned to the valence number of Cu2+.37,38,44,61 On the other hand, the CBO processed at 700 °C showed two different peak pairs: 934.0 eV coupled with 953.8 eV (attributable to Cu2+) and 932.4 eV (Cu 2p3/2) coupled with 952.2 eV (Cu 2p1/2), both with ΔBEs of 19.8 eV. The Cu 2p peak pair observed at lower BEs indicates the emergence of reduced Cu species at the surface of the CBO. Based on the peak fitting (Table S1, ESI†), the surface concentration proportions of Cu2+ and reduced Cu were calculated to be 76.6% and 23.4%, respectively. The peaks centered at BEs of 941.5, 944.0, and 962.3 eV correspond to the satellite peaks of Cu 2p, which is in good agreement with the literature.44,61 In the case of the sample processed at 700 °C, the intensity ratio of the satellite peaks at 941.5 and 944.0 eV was inverted in comparison with the other samples. This inversion of the peak intensity ratio may be caused by the presence of reduced Cu species on the CBO surface, providing a satellite peak at approximately 944 eV, which is a characteristic feature of reduced Cu (Cu1+ (Cu2O) and Cu0 (metal)).61
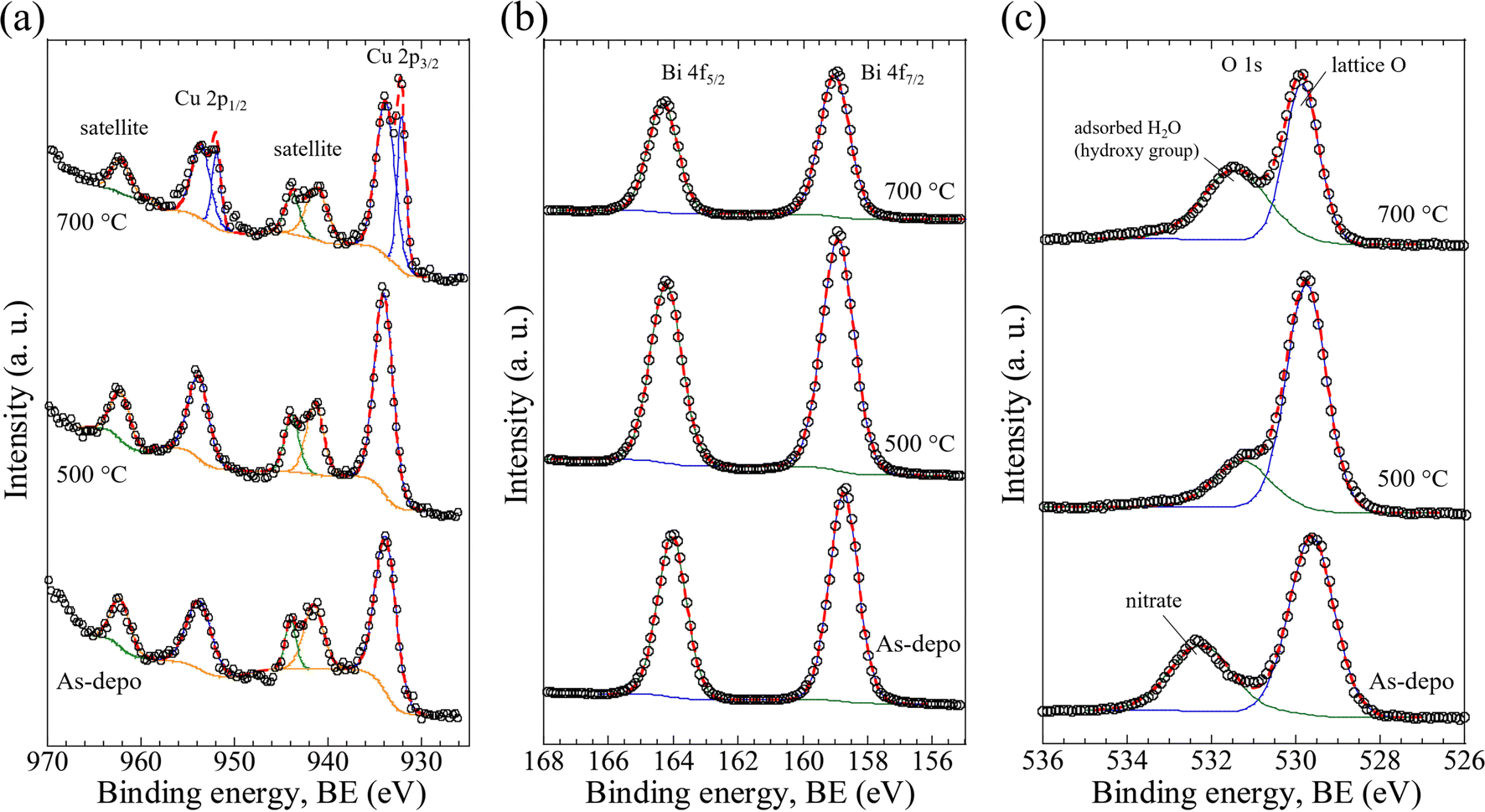 | ||
| Fig. 3 X-ray photoelectron spectroscopy (XPS) data for CBO on the FTO substrate prepared by spray pyrolysis of Cu nitrate and Bi nitrate ([Cu2+]/[Bi3+] = 0.50) containing precursor aqueous solution at 400 °C (As-depo) together with post-annealing treatment in air at temperatures of 500 °C and 700 °C. High-resolution XPS spectra of Cu 2p (a), Bi 4f (b), and O 1s (c). Empty circles, solid lines (blue, green, and orange), and dashed lines (red) indicate the experimental data, fitted curves, and composite curves used in peak fitting, respectively. | ||
Fig. 3b presents the HR-XPS spectra of Bi 4f. The Bi 4f peaks centered at BEs of 158.8 eV for Bi 4f7/2 and 164.1 eV for Bi 4f5/2 with a ΔBE of 5.3 eV. These peaks are consistent with those reported in the literature and can be attributed to Bi3+.44,62 Even though the as-deposited sample contained BiONO3, no significant difference was observed in the Bi 4f HR-XPS spectra compared to the samples after post-annealing treatment. Quantitative XPS analysis based on peak fitting enabled estimation of the surface atomic concentration ratios of Cu and Bi, [Cu/(Cu + Bi)] and [Bi/(Cu + Bi)], assuming that the peak areas were derived from Cu2+ and Bi3+, respectively. The peak fitting revealed that the surface atomic concentration ratios of Cu and Bi for as-deposited samples and samples annealed at 500 and 700 °C were 1.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6.4, 1.0:4.2, and 1.0:4.0, respectively (Table S2, ESI†). The decreased Bi ratio with the post-annealing treatment was probably due to the formation and growth of CBO associated with the decomposition of BiONO3 in the as-deposited sample.
:6.4, 1.0:4.2, and 1.0:4.0, respectively (Table S2, ESI†). The decreased Bi ratio with the post-annealing treatment was probably due to the formation and growth of CBO associated with the decomposition of BiONO3 in the as-deposited sample.
The O 1s HR-XPS spectra of the as-deposited film and the sample after post-annealing treatment are shown in Fig. 3c. In the samples processed by the post-annealing treatment (500 and 700 °C), the peaks centered at BEs of 529.8 and 531.4 eV were clearly observed. These peaks can be attributed to the lattice oxygen (O2−) in CBO and the adsorbed H2O (or hydroxy groups, OH−) at the surface, respectively.37 For the as-deposited sample, the peak centered at 532.3 eV with a higher BE shift of 0.9 eV was observed. The peak that emerged at a higher BE was most likely ascribed to the contribution of the nitrate species (BiONO3) in the as-deposited film, which is consistent with the results of the N 1s HR-XPS analysis (Fig. S9a, ESI†). The decreased peak intensity at 529.8 eV was probably due to the formation of reduced Cu species associated with the elimination of lattice oxygen from the CBO when the post-annealing temperature increased, which is in reasonable agreement with the results of the Cu 2p HR-XPS analysis (Fig. 3a). Quantitative XPS analysis revealed that the surface chemical state of the specimen depended on the post-annealing temperature. Thus, the surface quality correlated with the PEC properties of the CBO-based photoelectrode, as discussed in the next section.
2.3. Photoelectrochemical properties of CuBi2O4-based photocathodes
The PEC properties of the CBO-based photoelectrodes, including the photocurrent density (J) and onset potential (Eon), were assessed under simulated AM 1.5G solar illumination (100 mW cm−2). The measurements were performed using a three-electrode configuration and 0.5 M potassium phosphate buffered aqueous solution (KPi, pH = 7.0) or 0.5 M KPi containing 0.1 M H2O2 sacrificial reagent. The details of the PEC measurements are described in the Experimental section and Fig. S10 (ESI†).Fig. 4a–c presents the typical photocurrent density–photoelectrode potential (J–E) curves for the CBO-based photoelectrodes with and without post-annealing treatment, measured under the illumination of chopped AM 1.5G solar light. Irrespective of the electrolyte composition (with and without H2O2), all the photoelectrodes showed cathodic photocurrent in response to simulated sunlight illumination. The cathodic photocurrent is derived from the p-type semiconducting properties of the CBO material, which means that the photogenerated electrons are transferred into the solid/liquid interface to drive the photoreduction reaction.5 According to the literature,34,43,52 the bare CBO photoelectrodes without insertion of a surface protection layer or cocatalyst-loading for the HER exhibit self-photoreduction most probably due to the reduction of Cu elements inside the CBO material, namely photocorrosion. However, the addition of sacrificial reagents (such as H2O2) as electron acceptors to the aqueous electrolyte solution promotes the electron transfer process of photogenerated carriers to the electron acceptors at the solid/liquid interface, thereby mitigating the self-photoreduction reaction of CBO. As shown in Fig. 4a–c, in the presence of an H2O2 electron acceptor, the CBO-based photoelectrodes generated a larger cathodic J value over the measured photoelectrode potential (E) in the range of 0.2–1.1 VRHE compared to those in electrolyte without H2O2. The cathodic J at 0.6 VRHE of CBO-based photoelectrodes is summarized in Table 1.
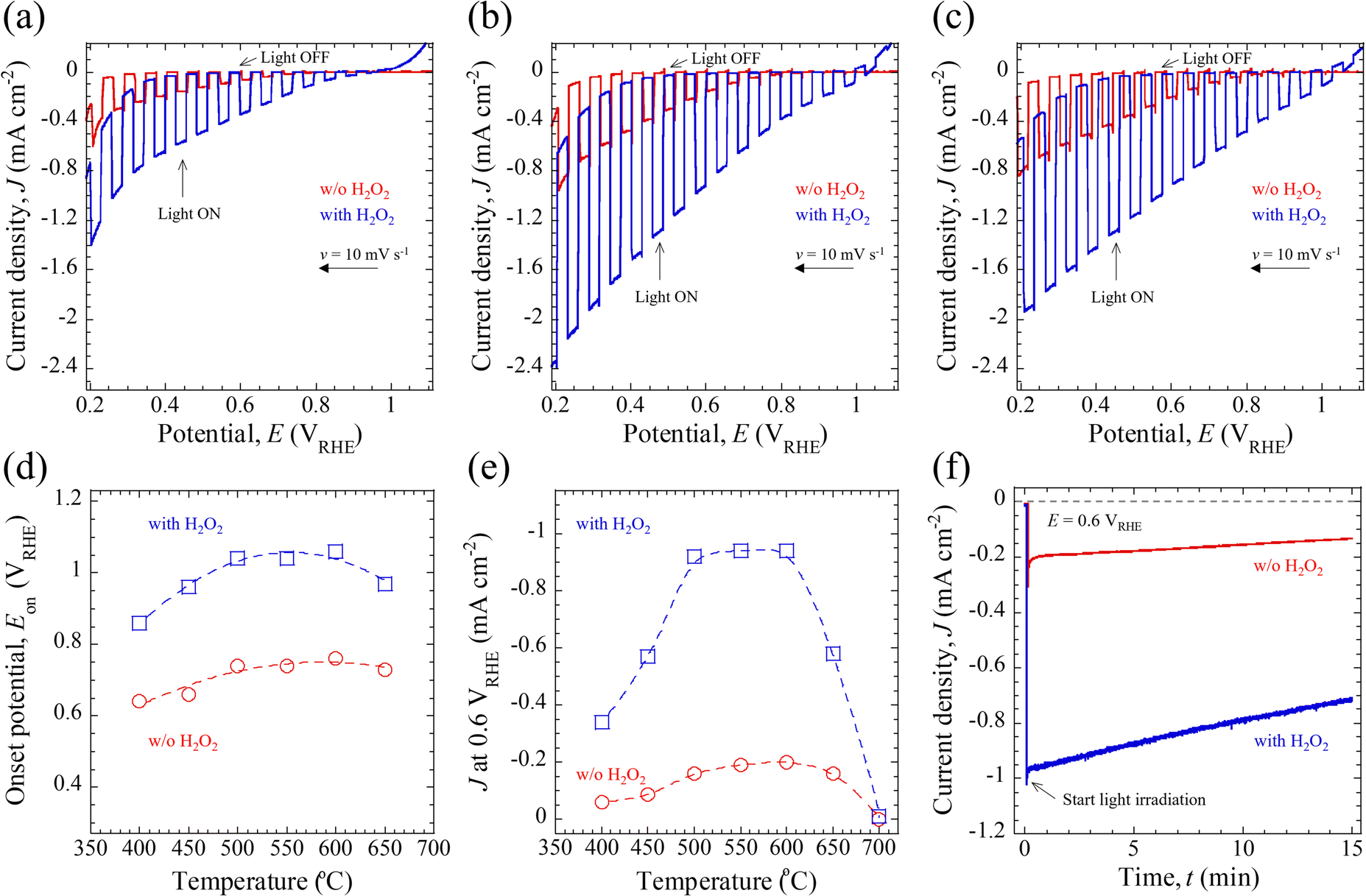 | ||
| Fig. 4 Photoelectrochemical properties of CBO-based photoelectrodes prepared via spray pyrolysis with post-annealing treatment. Photocurrent density vs. photoelectrode potential (J–E) curves for the as-deposited sample (a) and CBO processed with post-annealing treatment at 500 °C (b) and 700 °C (c) acquired under chopped simulated AM 1.5G solar illumination with an intermittent period of 6 s and a potential sweep rate (v) of 10 mV s−1 in 0.5 M potassium phosphate buffered aqueous solution (KPi) at pH 7.0 with 0.1 M H2O2. (d) and (e) Plots of onset potential (Eon) and photocurrent density (J) at 0.6 VRHE for the CBO-based photoelectrodes as a function of annealing temperature. (f) Photocurrent density vs. time (J–t) curves for the CBO-based photoelectrode annealed at 500 °C for 1 h measured at a constant E of 0.6 VRHE under continuous simulated AM 1.5G solar illumination. The J–t curves were obtained in the 0.5 M KPi aqueous solution with (blue) and without (w/o, red) 0.1 M H2O2 sacrificial reagent. | ||
| Photocurrent density, J, at 0.6 VRHE (mA cm−2) | ||||
|---|---|---|---|---|
| As-deposited | 400 °C | 500 °C | 600 °C | |
| With H2O2 | −0.34 | −0.44 | −0.92 | −0.94 |
| Without H2O2 | −0.06 | −0.07 | −0.16 | −0.20 |
The increase in J over the measured E in the presence of H2O2 is attributable to the reduction reaction of H2O2 to produce H2O together with successfully mitigating the self-photoreduction reaction of the CBO photoelectrode. Regardless of the samples and electrolyte compositions, J increased when E was reduced toward the negative photoelectrode potential. This increase in J with more negative E is attributed to the greater band bending at the CBO/electrolyte interface and likely also the CBO/H2O2 solid/liquid interface, which facilitates the electron transfer process. When the post-annealing treatment was conducted at 700 °C in air, the CBO-based photoelectrode exhibited a significantly reduced photocurrent (J less than −0.05 mA cm−2) at E more negative than 0.95 VRHE (Fig. S11a, ESI†). One plausible explanation for the inferior J–E property of the CBO processed at 700 °C is due to the carrier recombination at the reduced Cu species (Fig. 3a) and increased film resistivity (ρ) of the FTO-coated glass substrate (Fig. S11b, ESI†). The increase in the ρ of the FTO prevents an efficient carrier transfer toward the external circuit from inside CBO during the light excitation process, leading to the reduced photocurrent. While the crystallinity of the CBO films increased with rising annealing temperature (Fig. 1a), the PEC property was limited by the electrical conductivity of the back-contact carrier collection substrate of FTO glass. This problem could be solved by using a noble metal layer or substrate (such as Au),39 whose electrical properties do not deteriorate even when high-temperature post-annealing treatment is conducted.
Mott–Schottky (MS) analysis was performed using the CBO-based photoelectrodes processed with and without (w/o) post-annealing treatment to prove the p-type semiconducting properties at the CBO/electrolyte solid/liquid interface (Fig. S12, ESI†). When the direct current (DC) potential was changed to positive under the application of an alternating current (AC) potential modulation of 10 mVrms at a frequency of 1000 Hz, each sample showed a decrease in the reciprocal of the square of the surface differential capacitance (1/C2), and the plots of 1/C2vs. DC potential showed a negative slope of 1/C2. This result demonstrated that the CBO-based photoelectrodes showed p-type semiconductor properties at the electrified solid/liquid interface and are consistent with the J–E characteristics exhibiting a cathodic photocurrent in response to light illumination. From the MS analysis in Fig. S12 (ESI†), the flat band potential (EFB) of CBO-based photoelectrodes processed with the post-annealing treatment was found to be 1.20 VRHE. The EFB of CBO-based photoelectrodes prepared in this study is close to the previously reported EFB of CBO photoelectrodes evaluated by the MS analysis.41,42
Fig. 4d shows the annealing temperature-dependent Eon, where a cathodic photocurrent was generated in response to AM 1.5G solar illumination. The Eon in this study was defined as the E required to generate a J of −0.05 mA cm−2 based on the J–E characteristics. Regardless of the electrolyte composition (with and without H2O2), the Eon for the CBO-based photoelectrodes showed a potential shift toward a positive E with an increase in annealing temperature up to 600 °C. In the presence of the H2O2 electron acceptor, the Eon for the CBO photoelectrode was located at 0.22–0.30 V more positive than those observed in the KPi electrolyte. Specifically, in the case of H2O2 addition, the Eon for the as-deposited sample (400 °C) and the specimens annealed at 500 °C and 600 °C were 0.86 VRHE, 1.04 VRHE, and 1.06 VRHE, respectively. In comparison with the previously reported CBO-based photoelectrode prepared through spray pyrolysis, in which photoelectrodes were also evaluated in the presence of the H2O2 sacrificial reagent, the Eon of the sample was observed to be around 1.0 VRHE.34 The Eon observed in the CBO-based photoelectrodes developed herein is comparable to those previously reported, whereas this study employs a non-organic-solvent-based precursor solution for the fabrication of CBO thin films.
Since the operating potential in non-biased overall water splitting of the PEC cell with a photocathode/photoanode combination is typically 0.6 VRHE,5,6,14 we discuss the J values recorded at 0.6 VRHE as a function of the annealing temperature (Fig. 4e). The values of J at 0.6 VRHE generated by the photoelectrodes in the electrolyte containing H2O2 were approximately five times higher than those in the electrolyte without H2O2. Regardless of the electrolyte composition (with and without H2O2), the CBO-based photoelectrodes showed enhanced J values with the increase in the annealing temperature up to 600 °C. When the annealing temperature was further increased to 650 °C and 700 °C, the J values decreased with the annealing temperature. Table 1 and Table S3 in the ESI,† summarize the PEC properties of the CBO-based photoelectrodes, including Eon and J at 0.6 VRHE. Specifically, in the presence of a sacrificial reagent, the values of J generated by the samples processed at 400, 500, and 600 °C were found to be −0.44 mA cm−2, −0.92 mA cm−2, and −0.94 mA cm−2, respectively. A photocurrent gain of about −0.5 mA cm−2 was achieved when the temperature of the post-annealing treatment was raised from 400 °C to 600 °C. Comparing the J–E characteristics with the previously reported CBO-based photoelectrodes prepared through the wet process involving a spray pyrolysis of organic solution and thermal annealing aimed at enhancing the PEC performance (Tables S4 and S5, ESI†), the J at 0.6 VRHE generated by CBO prepared in this work (aqueous precursor solution) was found to be almost half that of the CBO photoelectrode prepared using ethanol/acetic acid-based precursor solution with PEG and TEOF.41,42,51 In contrast, the J at 0.6 VRHE generated by CBO prepared in this work is comparable to those reported CBO photoelectrodes prepared using ethanol/acetic acid-based precursor solution without organic additives together with post-annealing treatment in air (Table S5, ESI†).40,53
Fig. 4f shows the photocurrent density–time (J–t) curves of CBO-based photoelectrodes in the electrolyte with and without H2O2 recorded at 0.6 VRHE under the illumination of continuous AM 1.5G solar light. The J–t curves were acquired with the CBO photoelectrodes processed with post-annealing treatment at 500 °C. At the beginning of light illumination (t = 0 min), a spike-like cathodic photocurrent was observed. This spike-like photocurrent mainly involves a double-layer charging current as a non-faradaic process. To exclude the contribution of the non-faradaic current, we defined the initial photocurrent density (Jin) as J observed at t = 1 min. The Jin of the photoelectrodes acquired in the electrolyte with and without the H2O2 sacrificial reagent was found to be −0.94 mA cm−2 and −0.19 mA cm−2, respectively. The values of Jin herein are consistent with the observations from the J–E curves shown in Fig. 4b. The CBO photoelectrode continued to produce a photocurrent at 0.6 VRHE, and the J decreased gradually with time. During the first 15 minutes of the PEC operation, the J values reduced to −0.72 mA cm−2 (with H2O2) and −0.13 mA cm−2 (w/o H2O2), respectively. The ratio of photocurrent density after 15 min (Jt=15 min) and Jin (Jt=15 min/Jin) is 0.77 (with H2O2) and 0.68 (w/o H2O2), respectively. Although the CBO-based photoelectrode in the presence of the sacrificial reagent exhibited an improved and stabilized photocurrent, degradation due to the self-photoreduction reaction inside the CBO was not fully mitigated.
To examine the surface chemical states of the CBO-based photoelectrodes after the stability test, XPS analysis was performed for the samples that were tested in the electrolyte with and without H2O2 (Fig. S13, ESI†). The HR-XPS spectrum of Cu 2p can be fitted to the two different oxidation states of Cu (Cu2+ and reduced Cu), indicating the self-reduction reaction (photocorrosion) of CBO that occurred after the stability test. The peak intensity at a BE of 932.4 eV, attributable to the reduced Cu species after the stability test without H2O2, was larger than that of the sample after the stability test with H2O2 (Fig. S13a, ESI†). In sharp contrast, the HR-XPS spectrum of Bi 4f obtained in the stability test without H2O2 was deconvoluted into two different Bi oxidation states (Fig. S13b, ESI†). The major XPS peaks at BEs of 159.8 eV (Bi 4f7/2) and 165.1 eV (Bi 4f5/2) with a ΔBE of 5.3 eV overlapped with the shoulder XPS peaks at BEs of 158.8 eV (Bi 4f7/2) and 164.1 eV (Bi 4f5/2). The major XPS peaks of Bi 4f observed at higher BEs imply that the surface chemical state of Bi3+ in the CBO changed to a further oxidized state during the PEC operation. The emergence of the further oxidized state of Bi in CBO after the PEC operation has previously been reported by Berglund et al.44 According to the literature,62 the Bi 4f peaks appearing at higher BEs were determined to be Bi5+. As shown in Fig. S13c (ESI†), the O 1s HR-XPS spectrum obtained after the stability test showed a significant increase in the peak intensity centered at 531.4 eV, which was derived from the adsorbed H2O and/or hydroxyl groups at the surface.
The PEC characteristics of the BiOx- and CuOx-based photoelectrodes prepared in the same manner using a dilute aqueous nitric acid precursor solution containing Bi(NO3)3 or Cu(NO3)2 are shown in Fig. S14 (ESI†). While the BiOx-based photoelectrode generated an anodic photocurrent in response to simulated AM 1.5G solar illumination, the CuOx-based photoelectrode exhibited a cathodic photocurrent (Fig. S14a and b, ESI†). The contribution of BiOx to the cathodic photoresponse of CBO is negligible because the anodic photocurrent can be generated at E more positive than 0.6 VRHE. The evaluation of CuOx under the same conditions as the durability test of the CBO photoelectrode showed that CuOx exhibited a more rapid decay in photocurrent than CBO (Fig. S14c and d, ESI†). The ratio of photocurrent density after 15 min (Jt=15 min) and Jin (Jt=15 min/Jin) is 0.53 (with H2O2) and 0.22 (w/o H2O2), respectively. Compared to the stability of the J–t characteristics of CBO-based photoelectrodes (Table S6, ESI†), the durability of CBO is superior to that of CuOx (CuO and Cu2O).60,63
Finally, the wavelength-dependent incident photon-to-current density conversion efficiency (IPCE) was probed to evaluate the impact of post-annealing treatment for the CBO-based photoelectrodes. The IPCE values for the samples processed by the post-annealing treatment (400, 500, and 600 °C for 1 h) were acquired at an E of 0.6 VRHE in KPi electrolyte with and without the H2O2 sacrificial reagent (Fig. 5a–c). These data indicate that CBO-based photoelectrodes showed appreciable cathodic photocurrent in response to light absorption at wavelengths below ∼740 nm, irrespective of the electrolyte composition (with and w/o H2O2). The self-photoreduction reaction (photocorrosion) and H2O2 reduction reaction derived from CBO-based photoelectrodes are driven by light absorption at wavelengths of up to ∼740 nm. The CBO photoelectrode that came into contact with the electrolyte containing H2O2 had about three times higher IPCEs over the measured wavelengths of monochromatic light than the sample measured in the KPi electrolyte (w/o H2O2). In addition, the IPCE values of the CBO photoelectrode processed at 500 °C and 600 °C are larger than that of the sample processed at 400 °C over the measured wavelengths of incident light. The tendency of the IPCE data supports the validity of the J–E and J–t characteristics obtained under the illumination of simulated sunlight (Fig. 4, Table 1 and Table S3, ESI†).
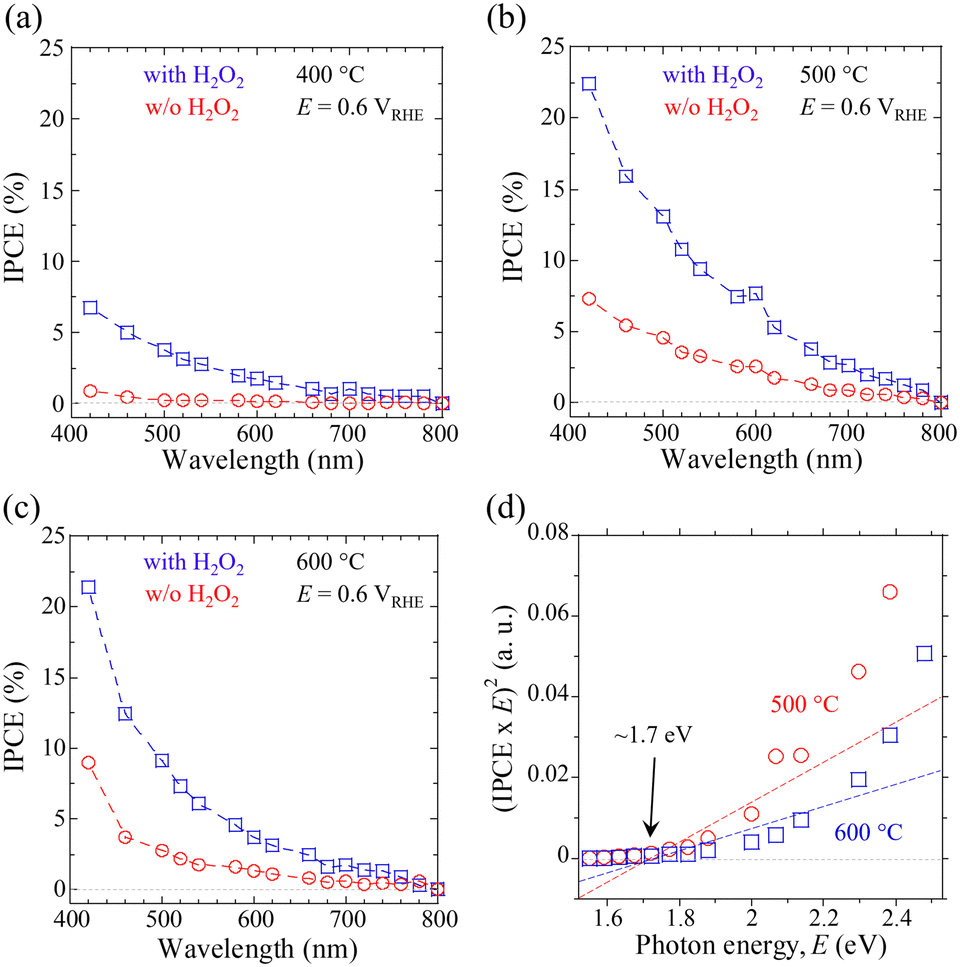 | ||
| Fig. 5 Incident-photon-to-current conversion efficiency (IPCE) spectra for CBO-based photoelectrodes with post-annealing treatment at (a) 400 °C, (b) 500 °C, and (c) 600 °C. The IPCE data were acquired in 0.5 M KPi buffered aqueous solution (pH = 7.0) with (square) and without (w/o, circle) the H2O2 sacrificial reagent. (d) Tauc plots derived from the IPCE spectra in panels (b) and (c). | ||
The IPCEs decreased with increasing wavelength of incident monochromatic light. Specifically, the CBO photoelectrode processed at 500 °C showed IPCE values (in the presence of H2O2) at 420 nm and 700 nm of 22.4% and 2.6%, respectively. Light absorption at a short wavelength (such as 420 nm) produces photogenerated carriers that move a short distance toward the reaction site at the solid/liquid interface, compared to photogenerated carriers generated by light of a long wavelength. On the other hand, the carriers generated by the incident light with a long wavelength (such as 700 nm) need to travel a longer distance compared to the photogenerated electrons produced by short wavelengths. To generate a photocurrent related to the IPCE values, the photogenerated carriers must achieve a solid/liquid interface against the charge recombination process in the CBO. According to the previously reported absorption coefficient (a) of 103–105 cm−1 (in the range of 300–800 nm) for the CBO material,41,44 the optical penetration depth (α−1) of the incident light of 420 nm is expected to be within 10–100 nm, which is close to the surface region of the CBO photoelectrode. In contrast, the α−1 of the incident light at 700 nm reaches the order of a micrometer, which is significantly longer than the film thickness (∼330 nm) of the CBO photoelectrode produced in this study.
The onset of IPCE in response to monochromatic light is related to the absorption edge wavelength of the material, which contributes to the actual light response in the PEC process. Tauc plots derived from the IPCE spectra of CBO photoelectrodes processed at 500 and 600 °C enabled us to estimate a bandgap energy of 1.7 eV (Fig. 5d), which is in good agreement with the value reported in the literature.38,40 There was no significant difference in the band gap energy of CBO photoelectrodes estimated from the Tauc plots between the samples processed at 500 °C and 600 °C. Fig. S15 (ESI†) presents the energy level diagram of CBO based on the Eg of 1.7 eV and the EFB of 1.20 VRHE estimated from the MS analysis (Fig. S12, ESI†). The CBM of the CBO is located at a more negative value than the standard redox potential of the HER (0.0 VRHE), enabling the electron transfer process (PEC reduction process) at the solid/liquid interface under light illumination.
To realize high IPCE values at long wavelengths near the absorption edge wavelength of the CBO, the photogenerated carriers (electrons and holes) within the CBO must move greater distances to reach the solid/liquid interface. The impact of film thickness on the PEC properties of CBO-based photoelectrodes is crucial for utilizing photons at longer wavelengths to improve the overall efficiency. Surface modifications, such as the addition of a protective layer and a cocatalyst, serve as advanced strategies to prevent surface photocorrosion and promote the electron transfer process (such as the HER and CO2-reduction); these also mitigate photocorrosion during the PEC operation.43,51,52,64 To further clarify the structure–activity and carrier transfer processes of CBO under light excitation, spectroscopic techniques such as photoluminescence (PL) emission spectroscopy,40 pump–probe spectroscopy,42 and AC impedance spectroscopy65 should be employed. These comprehensive investigations will be performed in a future study aiming to provide an in-depth insight into the physicochemical properties of CBO and enhance its PEC energy conversion efficiency.
3. Conclusions
CuBi2O4 (CBO)-based photoelectrodes with a visible-light response were fabricated by spray pyrolysis of a carbon-free Cu and Bi nitrate-containing dilute nitric acid aqueous solution, followed by post-annealing treatment in air. The as-deposited sample prepared on the FTO was found to have a low-crystallinity CuO phase and BiONO3 produced by the thermal decomposition of Bi(NO3)3. The effect of the post-annealing treatment on the as-deposited sample was examined to produce a single-phase CBO thin film on FTO. Various characterization techniques including XRD, SEM, and XPS analysis enabled us to determine a threshold annealing temperature of 500 °C, which provides a polycrystalline CBO thin film on the FTO substrate. Quantitative XPS analysis demonstrated that the ratios of the surface Cu and Bi concentrations on the CBO were far from stoichiometric in all samples, regardless of the post-annealing temperature. However, the increase in post-annealing temperature was found to achieve a closer-to-stoichiometric surface composition than that of the sample processed at a lower temperature. The CBO-based photoelectrodes processed by post-annealing treatment at 500 °C and 600 °C showed an almost equivalent photocurrent density of −0.94 mA cm−2 at 0.6 VRHE under simulated AM 1.5G solar illumination in the presence of the H2O2 sacrificial reagent, while the as-deposited sample and the sample processed at 400 °C containing the BiONO3 impurity exhibited a photocurrent at 0.6 VRHE of −0.34 mA cm−2 and −0.44 mA cm−2, respectively. When the post-annealing temperature exceeds 650 °C, the photocurrent density generated by the CBO photoelectrodes at 0.6 VRHE reduced significantly. This reduced photocurrent is most likely attributable to the thermal degradation of the electrical conductivity of the FTO back contact due to high-temperature annealing. The optimized CBO-based photoelectrodes (processed at 500 and 600 °C) generated appreciable photocurrents in response to incident light at wavelengths up to 740 nm. Based on the Tauc plot derived from the wavelength-dependent IPCE spectra, the CBO material demonstrates a bandgap energy of ∼1.7 eV.Further investigation of the impact of deposition temperature during the spray pyrolysis of the precursor aqueous solution involving the protocol of post-annealing treatment is beneficial to gain an in-depth understanding of the correlation with the CBO film structure and the PEC properties. Moreover, the surface modification with a thin functional layer and the loading of the cocatalyst onto the CBO are future prospects for this work to improve the PEC performance as well as to mitigate the self-photoreduction (photocorrosion) of CBO. These investigations to enhance the PEC water splitting efficiency of CBO will be further performed in a future study. We believe that our work provides a carbon-free synthesis method to fabricate Cu-based ternary oxide-based semiconducting materials and contributes to the future development of strategies for designing efficient CBO-based photocathodes for the HER, applicable to PEC cells for non-biased solar water splitting.
4. Experimental
4.1. Preparation of CuBi2O4 by spray pyrolysis and post-annealing treatment
All chemicals were used as received. Cu(NO3)2·3H2O (≥99.9%, Fujifilm Wako Pure Chemical Corp.) and Bi(NO3)3·5H2O (99.9%, Fujifilm Wako Pure Chemical Corp.) were dissolved in dilute nitric acid (10 vol%, Fujifilm Wako Pure Chemical Corp.) to prepare a precursor aqueous solution for spray pyrolysis. The molar concentrations of Cu(NO3)2 and Bi(NO3)3 in the precursor solution were adjusted to 0.05 M and 0.10 M ([Cu2+]/[Bi3+] = 0.50), respectively. The F-doped tin oxide-coated glass substrates (FTO; AGC, 10 mm × 5 mm × 1.1 mm) were sequentially cleaned by sonication in ultrapure water, 2-propanol, acetone, and ultrapure water, and the substrates were then dried in a constant temperature dryer at 60 °C for 2 h. Subsequently, the FTO substrates were irradiated with UV light for 20 min to eliminate surface contaminants. The FTO substrates were placed on a hot plate and kept at a temperature of 400 °C. The distance between the FTO surface and the spray nozzle of the atomizer was set to 15 cm. The aqueous precursor solution was sprayed onto the FTO surface at about 0.2 mL for 0.5 s in one cycle. The precursor solution was sprayed for 100 cycles at an intermittent period of 1.0 s. The area sprayed in one cycle was approximately 314 cm2, which was sufficient to cover the entire surface of the FTO substrate. The obtained samples were further annealed in air at 400, 450, 500, 550, 600, 650, and 700 °C for 1 h using a muffle furnace.4.2. Characterization of the samples
The structural and optical properties of the samples prepared on the FTO were characterized using X-ray diffraction (XRD; X’Pert-Pro MRD, PANalytical and MiniFlex 600-C, Rigaku), scanning electron microscopy (SEM; S-5500, Hitachi), and diffuse reflectance spectroscopy (DRS; V-670, JASCO). Thermogravimetry-differential thermal analysis (TG-DTA, Thermo Plus EVO2 TG-DTA8122, Rigaku) was performed to obtain the TG and DTA curves. An alumina pan was used as the container, and an empty container was used as the reference material. The surface chemical state of the samples was analyzed by X-ray photoelectron spectroscopy (XPS, ULTRA2, KRATOS) equipped with a charge neutralizer and employing Al Kα radiation. The XPS C 1s peak (285.0 eV) was used to calibrate the binding energy (BE) of the XPS spectrum.4.3. Photoelectrochemical measurements
Indium metal was soldered onto the FTO surface to form an ohmic contact. A lead wire was connected to the indium metal with an additional indium solder, and the unused region was covered with epoxy resin for insulation. The typical geometric area (light-receiving area) of the prepared photoelectrodes was 0.4 cm2. PEC data were acquired in 0.5 M potassium phosphate buffered aqueous solution (KPi) with the pH adjusted to 7.0 by mixing 0.5 M K2HPO4 (Fujifilm Wako Pure Chemical Corp.) and 0.5 M KH2HPO4 (Fujifilm Wako Pure Chemical Corp.) aqueous solutions. The photoelectrode potential was controlled using a potentiostat (HSV-110, Hokuto Denko) with a three-electrode configuration comprising a coiled Pt wire as the counter electrode, Ag/AgCl/saturated KCl as the reference electrode, and the CBO-based photoelectrodes as the working electrode. Before the measurements, the 0.5 M KPi electrolyte was degassed by Ar gas bubbling (purity >99.9999 vol%) for 30 min to remove the oxygen dissolved in the electrolyte. As an electron acceptor and a sacrificial reagent, 0.1 M H2O2 (Fujifilm Wako Pure Chemical Corp.) was added to the 0.5 M KPi aqueous solution. A solar simulator (XES-40S2, SAN-EI Electrical. Co. Ltd) applying AM1.5G irradiation (1 sun, 100 mW cm−2) was employed as the light source. The photoelectrode potential was converted to a reversible hydrogen electrode potential (ERHE) using eqn (1):| ERHE = E(Ag/AgCl/sat’d KCl) + 0.197 V + 0.059 × pH | (1) |
In the Mott–Schottky (MS) analysis via AC impedance measurements, a 0.5 M KPi buffered aqueous solution with the pH adjusted to 7.0 was used as the electrolyte. Prior to applying the AC potential modulation, the electrolyte was degassed using Ar gas for 30 min, and cyclic voltammetry (CV) of the CBO-based photoelectrodes was performed in the photoelectrode potential range of 0.60–1.20 VRHE with a v of 10 mV s−1 in the dark. Then the electrolyte was degassed again for 5 min. The photoelectrode potential was controlled using a potentiostat (VersaSTAT 3, AMETEK). The data in the MS plot were acquired at an AC potential modulation (ΔEac) of 10 mVrms and a potential modulation frequency (f) of 1000 Hz. The flat band potential (EFB) of the CBO-based photoelectrode was estimated from the MS plot using eqn (2):66
 | (2) |
Wavelength-dependent incident photon-to-current density conversion efficiency (IPCE) spectra were obtained using a MAX-303 Xe light source (Asahi Spectra) equipped with bandpass filters to generate monochromatic light. The IPCE was calculated using eqn (3):
 | (3) |
Author contributions
K. W. and T. H. designed the research and performed the experimental work related to the preparation of photoelectrodes, PEC measurements, and XPS analyses, and also analyzed the data. A. N. and K. Y. performed the materials characterization and were also involved in data analysis. T. H. and K. Y. planned and supervised the project. K. W. and T. H. wrote the manuscript. All authors contributed to manuscript revisions.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was funded in part by a Grant-in-Aid for Scientific Research (C) (no. 23K04917) from JSPS. The authors wish to thank Prof. K. Sakai at the University of Miyazaki for providing technical support during the XPS and TG-DTA analyses.References
- H. Song, S. Luo, H. Huang, B. Deng and J. Ye, ACS Energy Lett., 2022, 7, 1043–1065 CrossRef CAS.
- Y. Wang, A. Sharma, T. Duong, H. Arandiyan, T. Zhao, D. Zhang, Z. Su, M. Garbrecht, F. J. Beck, S. Karuturi, C. Zhao and K. Catchpole, Adv. Energy Mater., 2021, 11, 2101053 CrossRef CAS.
- B. A. Pinaud, J. D. Benck, L. C. Seitz, A. J. Forman, Z. Chen, T. G. Deutsch, B. D. James, K. N. Baum, G. N. Baum, S. Ardo, H. Wang, E. Miller and T. F. Jaramillo, Energy Environ. Sci., 2013, 6, 1983–2002 RSC.
- J. R. McKone, N. S. Lewis and H. B. Gray, Chem. Mater., 2014, 26, 407–414 CrossRef CAS.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- Y. Kawase, T. Higashi, K. Domen and K. Takanabe, Adv. Energy Sustainability Res., 2021, 2, 2100023 CrossRef CAS.
- T. Higashi, H. Nishiyama, V. Nandal, Y. Pihosh, Y. Kawase, R. Shoji, M. Nakabayashi, Y. Sasaki, N. Shibata, H. Matsuzaki, K. Seki, K. Takanabe and K. Domen, Energy Environ. Sci., 2022, 15, 4761–4775 RSC.
- T. Higashi, Y. Sasaki, Y. Kawase, H. Nishiyama, M. Katayama, K. Takanabe and K. Domen, Catalysts, 2021, 11, 584 CrossRef CAS.
- T. Higashi, Y. Shinohara, A. Ohnishi, J. Liu, K. Ueda, S. Okamura, T. Hisatomi, M. Katayama, H. Nishiyama, T. Yamada, T. Minegishi and K. Domen, ChemPhotoChem, 2017, 1, 167–172 CrossRef CAS.
- M. I. Díez-García and R. Gómez, Solar RRL, 2022, 6, 2100871 CrossRef.
- D. K. Lee, D. Lee, M. A. Lumley and K.-S. Choi, Chem. Soc. Rev., 2019, 48, 2126–2157 RSC.
- K. T. Fountaine, H. J. Lewerenz and H. A. Atwater, Nat. Commun., 2016, 7, 13706 CrossRef CAS.
- H. Kobayashi, N. Sato, M. Orita, Y. Kuang, H. Kaneko, T. Minegishi, T. Yamada and K. Domen, Energy Environ. Sci., 2018, 11, 3003–3009 RSC.
- H. Kaneko, T. Minegishi, M. Nakabayashi, N. Shibata, Y. Kuang, T. Yamada and K. Domen, Adv. Funct. Mater., 2016, 26, T4570–T4577 CrossRef.
- B. Koo, S.-W. Nam, R. Haight, S. Kim, S. Oh, M. Cho, J. Oh, J. Y. Lee, B. T. Ahn and B. Shin, ACS Appl. Mater. Interfaces, 2017, 9, 5279–5287 CrossRef CAS.
- S. K. Karuturi, H. Shen, A. Sharma, F. J. Beck, P. Varadhan, T. Duong, P. R. Narangari, D. Zhang, Y. Wan, J.-H. He, H. H. Tan, C. Jagadish and K. Catchpole, Adv. Energy Mater., 2020, 10, 2000772 CrossRef CAS.
- D. Xue, M. Kan, X. Qian and Y. Zhao, ACS Sustainable Chem. Eng., 2018, 6, 16228–16234 CrossRef CAS.
- J. Park, K.-Y. Yoon, T. Kim, H. Jang, M.-J. Kwak, J. Y. Kim and J.-H. Jang, Nano Energy, 2020, 76, 105089 CrossRef CAS.
- S. Wang, P. Chen, Y. Bai, J.-H. Yun, G. Liu and L. Wang, Adv. Mater., 2018, 30, 1800486 CrossRef.
- Gurudayal, D. Sabba, M. H. Kumar, L. H. Wong, J. Barber, M. Grätzel and N. Mathews, Nano Lett., 2015, 15, 3833–3839 CrossRef CAS PubMed.
- J. Luo, Z. Li, S. Nishiwaki, M. Schreier, M. T. Mayer, P. Cendula, Y. H. Lee, K. Fu, A. Cao, M. K. Nazeeruddin, Y. E. Romanyuk, S. Buecheler, S. D. Tilley, L. H. Wong, A. N. Tiwari and M. Grätzel, Adv. Energy Mater., 2015, 5, 1501520 CrossRef.
- Y. Pihosh, I. Turkevych, K. Mawatari, J. Uemura, Y. Kazoe, S. Kosar, K. Makita, T. Sugaya, T. Matsui, D. Fujita, M. Tosa, M. Kondo and T. Kitamori, Sci. Rep., 2015, 5, 11141 CrossRef.
- Z. Pan, R. Yanagi, T. Higashi, Y. Pihosh, S. Hu and K. Katayama, Sustain. Energy Fuels, 2022, 6, 2067–2074 RSC.
- M. Kim, B. Lee, H. Ju, J. Y. Kim, J. Kim and S. W. Lee, Adv. Mater., 2019, 31, 1903316 CrossRef.
- Y. Asakura, T. Higashi, H. Nishiyama, H. Kobayashi, M. Nakabayashi, N. Shibata, T. Minegishi, T. Hisatomi, M. Katayama, T. Yamada and K. Domen, Sustain. Energy Fuels, 2018, 2, 73–78 RSC.
- T. Higashi, H. Nishiyama, Y. Otsuka, Y. Kawase, Y. Sasaki, M. Nakabayashi, M. Katayama, T. Minegishi, N. Shibata, K. Takanabe, T. Yamada and K. Domen, ChemSusChem, 2020, 13, 1974–1978 CrossRef CAS.
- Z. Ma, K. Piętak, J. Piątek, J. Reed DeMoulpied, A. Rokicińska, P. Kuśtrowski, R. Dronskowski, S. Zlotnik, R. H. Coridan and A. Slabon, Chem. Commun., 2020, 56, 13193–13196 RSC.
- C. Lu, J. Chen, K. Piętak, A. Rokicińska, P. Kuśtrowski, R. Dronskowski, J. Yuan, S. Budnyk, S. Złotnik, R. H. Coridan and A. Slabon, Chem. Mater., 2022, 34, 6902–6911 CrossRef CAS.
- C. Lu, N. J. O'Brien, P. Rouf, R. Dronskowski, H. Pedersen and A. Slabon, Green Chem. Lett. Rev., 2022, 15, 658–670 CrossRef CAS.
- M. A. Mansoor, K. Munawar, S. P. Lim, N. M. Huang, M. Mazhar, M. J. Akhtar and M. Siddique, New J. Chem., 2017, 41, 7322–7330 RSC.
- M. A. Mansoor, M. Mazhar, V. McKee and Z. Arifin, Polyhedron, 2014, 75, 135–140 CrossRef CAS.
- D. Huang, K. Wang, L. Li, K. Feng, N. An, S. Ikeda, Y. Kuang, Y. Ng and F. Jiang, Energy Environ. Sci., 2021, 14, 1480–1489 RSC.
- Y. Pihosh, V. Nandal, T. Higashi, R. Shoji, R. Bekarevich, H. Nishiyama, T. Yamada, V. Nicolosi, T. Hisatomi, H. Matsuzaki, K. Seki and K. Domen, Adv. Energy Mater., 2023, 13, 2301327 CrossRef CAS.
- C. Li, J. He, Y. Xiao, Y. Li and J.-J. Delaunay, Energy Environ. Sci., 2020, 13, 3269–3306 RSC.
- S. A. Monny, Z. Wang, M. Konarova and L. Wang, J. Energy Chem., 2021, 61, 517–530 CrossRef CAS.
- B. Meena, M. Kumar, R. K. Hocking, S. Juodkazis, V. Biju, P. Subramanyam and C. Subrahmanyam, Energy Fuels, 2023, 37, 14280–14289 CrossRef.
- J. Jin, J. Hu, J. Qu, G. Cao, Y. Lei, Z. Zheng, X. Yang and C. M. Li, ACS Appl. Mater. Interfaces, 2022, 14, 17509–17519 CrossRef CAS.
- D. Kang, J. C. Hill, Y. Park and K.-S. Choi, Chem. Mater., 2016, 28, 4331–4340 CrossRef CAS.
- D. Cao, N. Nasori, Z. Wang, Y. Mi, L. Wen, Y. Yang, S. Qu, Z. Wang and Y. Lei, J. Mater. Chem. A, 2016, 4, 8995–9001 RSC.
- Y. Wang, J. Hu, S. Liu, D. Zhu, Z. Li and A. Song, Int. J. Hydrogen Energy, 2022, 47, 37774–37782 CrossRef CAS.
- F. Wang, A. Chemseddine, F. F. Abdi, R. van de Krol and S. P. Berglund, J. Mater. Chem. A, 2017, 5, 12838–12847 RSC.
- F. Wang, W. Septina, A. Chemseddine, F. F. Abdi, D. Friedrich, P. Bogdanoff, R. van de Krol, S. D. Tilley and S. P. Berglund, J. Am. Chem. Soc., 2017, 139, 15094–15103 CrossRef CAS.
- A. Song, I. Levine, R. van de Krol, T. Dittrich and S. P. Berglund, Chem. Sci., 2020, 11, 11195–11204 RSC.
- S. P. Berglund, F. F. Abdi, P. Bogdanoff, A. Chemseddine, D. Friedrich and R. van de Krol, Chem. Mater., 2016, 28, 4231–4242 CrossRef CAS.
- G. Liu, R. Cai, Z. Lv, G. Ma, J. Li, J. Jin, X. Zhong and F. Li, J. Catal., 2023, 424, 130–139 CrossRef CAS.
- B. Joos, K. Elen, J. van den Ham, N. Meulendijks, P. Buskens, A. Paulus, K. Wouters, J. Manca, J. D'Haen, S. Shukla, B. Vermang, M. Van Bael and A. Hardy, Adv. Sustain. Syst, 2023, 7, 2300083 CrossRef CAS.
- Y. Xu, J. Jian, F. Li, W. Liu, L. Jia and H. Wang, J. Mater. Chem. A, 2019, 7, 21997–22004 RSC.
- Z. Jiang, H. Geng, X. Cai, L. Mao, Y. Zhao and X. Gu, Mater. Sci. Semicond. Process., 2021, 134, 105989 CrossRef CAS.
- S. A. Monny, L. Zhang, Z. Wang, B. Luo, M. Konarova, A. Du and L. Wang, J. Mater. Chem. A, 2020, 8, 2498–2504 RSC.
- J. Li, M. Griep, Y. Choi and D. Chu, Chem. Commun., 2018, 54, 3331–3334 RSC.
- A. Song, P. Bogdanoff, A. Esau, I. Y. Ahmet, I. Levine, T. Dittrich, T. Unold, R. van de Krol and S. P. Berglund, ACS Appl. Mater. Interfaces, 2020, 12, 13959–13970 CrossRef CAS PubMed.
- Q. Zhang, B. Zhai, Z. Lin, X. Zhao and P. Diao, J. Phys. Chem. C, 2021, 125, 1890–1901 CrossRef CAS.
- C. G. O. Bruziquesi, M. C. P. Stolzemburg, R. R. de Souza, M. Rodriguez, M. L. Rocco, P. E. A. Salomão, A. E. Nogueira, Z. E. López-Cabaña, M. C. Pereira and A. C. Silva, Int. J. Hydrogen Energy, 2023, 48, 3456–3465 CrossRef CAS.
- P. Hao, Z. Zhao, J. Tian, Y. Sang, G. Yu, H. Liu, S. Chen and W. Zhou, Acta Mater., 2014, 62, 258–266 CrossRef CAS.
- N. Henry, M. Evain, P. Deniard, S. Jobic, F. Abraham and O. Mentré, Z. Naturforsch. B, 2005, 60, 322–327 CrossRef CAS.
- H. Tagawa, Bull. Inst. Environ. Sci. Technol., Yokohama Natl. Univ., 1987, 14, 41–57 CAS.
- M. A. Mansoor, M. A. Ehsan, V. McKee, N.-M. Huang, M. Ebadi, Z. Arifin, W. J. Basirun and M. Mazhar, J. Mater. Chem. A, 2013, 1, 5284–5292 RSC.
- M. A. Mansoor, M. Mazhar, M. Ebadi, H. N. Ming, M. A. Mat Teridi and L. Kong Mun, New J. Chem., 2016, 40, 5177–5184 RSC.
- M. A. Mansoor, M. Mazhar, A. Pandikumar, H. Khaledi, H. Nay Ming and Z. Arifin, Int. J. Hydrogen Energy, 2016, 41, 9267–9275 CrossRef CAS.
- R. Gottesman, A. Song, I. Levine, M. Krause, A. T. M. N. Islam, D. Abou-Ras, T. Dittrich, R. van de Krol and A. Chemseddine, Adv. Funct. Mater., 2020, 30, 1910832 CrossRef CAS.
- T. Ghodselahi, M. A. Vesaghi, A. Shafiekhani, A. Baghizadeh and M. Lameii, Appl. Surf. Sci., 2008, 255, 2730–2734 CrossRef CAS.
- H. Fan, G. Wang and L. Hu, Solid State Sci., 2009, 11, 2065–2070 CrossRef CAS.
- N. H. Lam, N. T. N. Truong, N. Le, K.-S. Ahn, Y. Jo, C.-D. Kim and J. H. Jung, Sci. Rep., 2023, 13, 5776 CrossRef CAS PubMed.
- A. Song, Z. Li, B. Wulan, S. Liu, B. Zhao, D. Zhu and J. Hu, J. Alloys Compd., 2023, 960, 170769 CrossRef CAS.
- Z. Zhang, B. Zhu and X. Guan, J. Phys. Chem. Lett., 2022, 13, 2356–2364 CrossRef CAS PubMed.
- W. P. Gomes and D. Vanmaekelbergh, Electrochim. Acta, 1996, 41, 967–973 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental data, Fig. S1–S15, and Tables S1–S6. See DOI: https://doi.org/10.1039/d3nj04878k |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |