
Influence of neutral auxiliary ligands on crystal structure and magnetic behaviour of new [MnII2MnIII2] clusters supported by p-adamantylcalix[4]arene†
Alexander S.
Ovsyannikov
 *a,
Iuliia V.
Strelnikova
ab,
Aida I.
Samigullina
c,
Daut R.
Islamov
d,
Mikhail A.
Cherosov
a,
Ruslan G.
Batulin
a,
Airat G.
Kiiamov
a,
Aidar T.
Gubaidullin
b,
Pavel V.
Dorovatovskii
e,
Svetlana E.
Solovieva
a and
Igor S.
Antipin
a
*a,
Iuliia V.
Strelnikova
ab,
Aida I.
Samigullina
c,
Daut R.
Islamov
d,
Mikhail A.
Cherosov
a,
Ruslan G.
Batulin
a,
Airat G.
Kiiamov
a,
Aidar T.
Gubaidullin
b,
Pavel V.
Dorovatovskii
e,
Svetlana E.
Solovieva
a and
Igor S.
Antipin
a
aKazan Federal University, Kremlevskaya 18 str., 420008 Kazan, Russian Federation. E-mail: osaalex2007@rambler.ru
bA. E. Arbuzov Institute of Organic and Physical Chemistry, FRC Kazan Scientific Center, Russian Academy of Sciences, Arbuzova 8 str., 420088 Kazan, Russian Federation
cN. D. Zelinsky Institute of Organic Chemistry, Russian Academy of Sciences, Leninsky avenue 47, 119991 Moscow, Russian Federation
dLaboratory for structural analysis of biomacromolecules, FRC Kazan Scientific Center, Russian Academy of Sciences, Lobachevskogo 2/31 str., 420111 Kazan, Russian Federation
eNational Research Center “Kurchatov Institute”, Acad. Kurchatov 1 Sq., 123182 Moscow, Russian Federation
First published on 20th November 2023
Abstract
A series of new manganese complexes of general formula 32-Mn4Xn, displaying a highly-reproducible [MnII2MnIII2] cluster core formation, was observed when tetra-p-adamantylcalix[4]arene 3 was used in combination with N,N′-chelating auxiliary ligands, such as bipy (32-Mn4(bipy)2, bipy = 2,2′-bipyridyl) and phen (32-Mn4(phen)2, phen = 1,10-phenanthroline), as well as with DMF/H2O solvent molecules (32-Mn4(dmf)4, 32-Mn4(dmf)2(H2O)2), acting as O-donor coligands, in the crystalline phase. Single-crystal X-ray diffraction revealed that the involvement of auxiliary ligands in coordination with Mn-ions results in the distortion of their coordination sphere and leads to the controlled compression of the {Mn4}-cluster core of complexes. Moreover, the appearance of adamantyl groups at the upper rim of the macrocyclic backbone of 3 influences the crystalline self-assembly of obtained clusters, providing a preferable “side-to-head” orientation of the cluster molecules within the crystal, leading to the observation of relatively higher {Mn4}⋯{Mn4} distances than those revealed for the earlier reported calix[4]arene supported {Mn4} clusters. All 32-Mn4Xn clusters demonstrated ferromagnetic interactions between the metal atoms within the cluster core independent of the nature of the O-donor or N-donor auxiliary ligands.
Introduction
In recent decades, there has been a growing interest in the synthesis of new organic polydentate ligands capable of promoting a relatively strong magnetic response of paramagnetic metal ions, which can be achieved by controlling their coordination sphere distortion, offering a wide range of potential applications as smart materials.1–10 In particular, the design of new single-molecule magnetic compounds,11 which can retain their magnetization without a magnetic field and exhibit hysteresis below a certain temperature, through organic synthesis is a highly challenging scientific area. The focus has been on the development of new computing technologies,12 exploiting the quantum principles of data storage and operation.13 There are two main parameters to determine the effectiveness of single-molecule magnets. These are TB, the blocking temperature, and an anisotropic energy barrier, Ueff, which blocks the remagnetisation. A higher value of Ueff implies a higher TB. In this respect, the general trends in the rational design of single-molecule magnets focus on the formation of new metal–organic complexes displaying relatively high Ueff values and mechanistic studies of magnetic relaxation occurring in such systems. One of the feasible strategies that can be applied in order to achieve both these goals is to use suitable organic ligands, providing the appropriate coordination sites.Calix[4]arenes (see Scheme 1) belong to a family of macrocyclic ligands and have been intensively involved in the formation of magnetically active cluster complexes when coordinating with 3d- and 4f-metal ions, displaying various nuclearities and shapes.14–18 The possibility of functionalization of the upper and/or lower rims and CH2–methylene bridges makes them especially suitable molecular platforms for modulating the magnetic properties of such coordination compounds. Offering the preorganized tetradentate coordination pocket, composed of four closely disposed O-phenolate atoms, calix[4]arene 1 was found to be a particular complement for binding with such paramagnetic ions, such as Mn(III), Fe(III), Cu(II) or Ln(III) cations (Ln = Dy, Tb, Gd), forming in situ generated metal–organic sub-building block [1-M]−. The combination of the last one with other metal ions present in the reaction mixture led to the formation of homometallic {Mn2},19 {Mn4},20–25 {Cu9},26 {Ln6}27,28 (Ln = Dy, Tb, Gd) as well as heterometallic {Mn3Gd},29 {Mn2M2}29 (M = Tb, Gd, Ho, Y), {Mn4Ln4}30 (Ln = Dy, Tb, Gd), {Fe2Ln2}31 clusters. Generally, the described cluster complexes display sandwiched structures when the metallic cluster core is held between two or four calix[4]arene units (see Fig. 1a). Most of them, especially those based on the Mn-ions, have demonstrated attractive magnetic properties, including single-molecule magnetic behavior or magnetocaloric effect.
 | ||
| Scheme 1 Calix[4]arene family of macrocyclic ligands and their schematic representation. The upper and lower rims of the macrocyclic backbone are denoted by blue and red colours, respectively. | ||
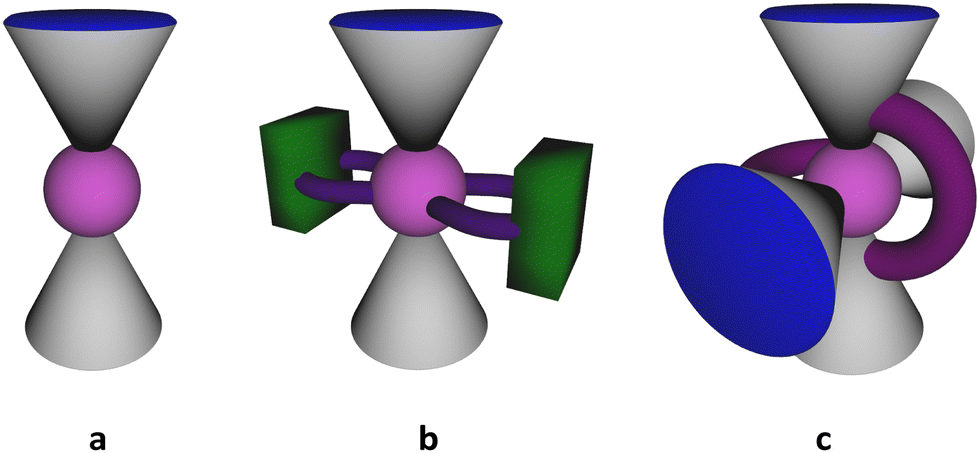 | ||
| Fig. 1 Schematic representation of sandwiched structures of manganese clusters formed by (a) 1 or 2, (b) 1 or 2 in combination with chelating auxiliary ligands, (c) CH2-tethered 1. | ||
One may distinguish a series of {MnII2MnIII2} clusters, generally prepared by fusing the in situ generated [1-MnIII]− or [2-MnIII]− sub-building blocks with MnII ions via μ3-OH bridges, and displayed the butterfly-like motif.20,21,24,32,33 It was found that the magnetic properties of these tetranuclear complexes can be fine-tuned by varying the N-/O-donor auxiliary ligands. In particular, the simple involvement of N-donor pyridine molecules instead of the O-donor DMF in the construction of the {MnII2MnIII2} cluster core afforded the Ueff parameter, equal to 29.4 K,32 exceeding that reported for the initial DMF-containing complex (16.5 K).21 Such a drastic change in the magnetic behavior was accompanied just by a subtle structural change in the coordination sphere of Mn(II) ions caused by substituting coordinated DMF molecules with pyridine molecules. Furthermore, the use of 2,2′-bipyridyl, containing two fused pyridyl moieties, as a neutral N,N′-donor chelating co-ligand, led to the formation of a similar {MnII2MnIII2} cluster core, exhibiting antiferromagnetic interactions between the metal ions (see Fig. 1b). In addition, another type of tetranuclear {MnII2MnIII2} cluster was synthesized when two O-donor phenyl phosphinate ligands were involved in the linkage of two [1-MnIIIMnII] species.22 A slight switching of the structural motif, in this case, resulted in the observation of a more complex magnetic behavior, demonstrating the ferromagnetic as well as antiferromagnetic interactions between metal ions.
An elegant strategy of structural modification of calix[4]arene-supported {MnII2MnIII2} clusters, consisting in the sequential replacement of Mn(II) ions by Ln(III) ions (Ln = Gd, Tb, Dy), was applied, leading to the formation of two types of heteronuclear clusters: {MnIII2MnII2LnIII} and {MnIII2LnIII2}.29 The thorough study of magnetic properties of these coordination compounds evidenced the diminishing magnitude of ferromagnetic interactions between MnIII and GdIII ions, compared to strong MnIII–MnII ferromagnetic exchange present in the {MnII2MnIII2} clusters.
In order to improve the magnetic properties, attempts to increase the nuclearity of such fascinating polynuclear Mn-based magnetic species were also undertaken. For example, the use of CH2-tethered bis-calix[4]arene led to the formation of a cluster core comprising 10 mixed-valent manganese(II/III) atoms34 (see Fig. 1c). However, the magnetic properties of the expanded core were found to be quite similar to the above-mentioned {MnII2MnIII2} systems, based on the non-tethered calix[4]arene ligand 1.
The reported examples clearly show that the control over magnetic properties of Mn-based calix[4]arene supported complexes can be successfully achieved due to rather small changes in the coordination environment of Mn ions, composing the ({MnII2MnIII2}) cluster core. Moreover, although many examples of Mn-based calixarene-supported clusters of various nuclearities are encountered in the literature, the influence of the functionalization of the upper rim of the calix[4]arene platform on the magnetic properties of these attractive coordination compounds, which can demonstrate single-molecule magnet behavior, is still unexplored.
In this work, we aimed to explore the influence of the upper rim modification of calix[4]arene ligand on the magnetic properties of the corresponding {Mn4} clusters by applying two general approaches: (a) the use of p-adamanthylcalix[4]arene 3, which, due to steric effect, can potentially prevent the inclusion of solvent molecules into the calix[4]arene cavity, allowing to control the coordination sphere of Mn(III) atoms; (b) the use of a series of neutral N,N′-donor chelating auxiliary ligands, displaying different distances between N-coordinating atoms, able to induce the distortions in the coordination sphere of Mn(II) atoms, by replacing the labile coordination positions occupied by solvent molecules.
Herein, we report the synthesis, structural characterization in the crystalline phase, and preliminary magnetic properties of a new family of {MnII2MnIII2} clusters, involving p-adamantylcalix[4]arene 3 as the main cluster forming ligand, from one side, and 1,10-phenanthroline and 2,2′-bipyridyl, as auxiliary ligands, from another side.
Experimental section
General
All chemicals were purchased from commercial suppliers and used without additional purification. Solvents were purified according to standard protocols. Calix[4]arene 3 was synthesised according to earlier reported procedures, and its purity was confirmed by 1H NMR spectroscopy.35Infrared spectra (IR) of powdered crystalline samples in KBr were recorded on a Tensor 27 Fourier-transform spectrometer in the range of 4000–400 cm−1 with an optical resolution of 1 cm−1 and an accumulation of 32 scans. Elemental analysis was performed on a EuroEA 3028-HT-OM Eurovector S.p.A. Powder X-ray diffraction (PXRD) data were collected on a Bruker D8 diffractometer using monochromatic Cu-Kα radiation with a scanning range between 3–40° using a scan step size of 8° nm−1.
The TGA/DSC measurements were performed on an STA 449C Jupiter synchronous microthermoanalyzer (Netzsch, Germany) at a heating rate of 10° min−1 under an argon atmosphere.
Crystallization conditions
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, s), 1459 (CC, s) 1254 (C–O, m). Elemental analysis found: C, 70.2%; H, 7.6%; N, 2.2%, calcd for C146H190O18Mn4N4: C, 69.90%; H, 7.63%; N, 2.23%.
O, s), 1599 (CC, w), 1459 (CC, s) 1248 (C–O, m). Elemental analysis found: C, 74.1%; H, 7.1%; N, 3.2%, calcd for C168H198O14Mn4N8: C, 72.76%; H, 7.20%; N, 4.04%.
O, s), 1517 (CC, w), 1459 (CC, s) 1252 (C–O, m). Elemental analysis found: C, 71.7%; H, 7.2%; N, 4.7%, calcd for C160H168O10Mn4N4: C, 76.05%; H, 6.70%; N, 2.22%.
O, s), 1459 (CC, s) 1254 (C–O, m). Elemental analysis found: C, 70.2%; H, 7.6%; N, 2.2%, calcd for C146H190O18Mn4N4: C, 69.90%; H, 7.63%; N, 2.23%.
O, s), 1599 (CC, w), 1459 (CC, s) 1248 (C–O, m). Elemental analysis found: C, 74.1%; H, 7.1%; N, 3.2%, calcd for C168H198O14Mn4N8: C, 72.76%; H, 7.20%; N, 4.04%.
O, s), 1517 (CC, w), 1459 (CC, s) 1252 (C–O, m). Elemental analysis found: C, 71.7%; H, 7.2%; N, 4.7%, calcd for C160H168O10Mn4N4: C, 76.05%; H, 6.70%; N, 2.22%.
Single crystal X-ray analysis
Diffraction data for single-crystals of 32-Mn4(dmf)4, 32-Mn4(dmf)2(H2O)2, 32-Mn4(bipy)2 and 32-Mn4(phen)2 were collected at 100 K on the ‘Belok/XSA’ beamline (λ = 0.7527 Å for 32-Mn4(dmf)4; λ = 0.7931 Å for 32-Mn4(dmf)4(H2O)2 and 32-Mn4(bipy)2, λ = 0.7927 Å for 32-Mn4(phen)2, φ-scans) of the Kurchatov Synchrotron Radiation Source (Moscow, Russian Federation).36,37 Diffraction patterns were collected using a Mardtb goniometer equipped with a Rayonix SX165 2D positional sensitive CCD detector at 100 K. In total, 300 frames were collected with an oscillation range of 1°. The data were indexed, integrated, and scaled; absorption correction was applied using the XDS program package.38 The structures were solved by direct methods using the software SHELXT.39 The structural model was investigated and refined using Olex2 software40 by a full-matrix least-squares method on F2 with anisotropic displacement. Non-hydrogen atoms were refined anisotropically. All hydrogen atoms were placed in calculated positions and included in the refinement within the Riding model with fixed isotropic displacement parameters Uiso(H) = 1.5Ueq(O), 1.2Ueq(N), and 1.2Ueq(C). All structures were found to be highly disordered. The quality of all crystals required moderate modelling using a range of restraints and constraints to account for the geometric and thermal parameters within parts of the structure.41The crystallographic data for 32-Mn4(dmf)4, 32-Mn4(dmf)2(H2O)2, 32-Mn4(bipy)2 and 32-Mn4(phen)2 are gathered in Table 1.
| Compound name | 32-Mn4(dmf)4 | 32-Mn4(dmf)2(H2O)2 | 32-Mn4(bipy)2 | 32-Mn4(phen)2 |
|---|---|---|---|---|
| Empirical formula | C148H182Mn4N4O14, C3H7NO, CH4O, 2(C2H3N) | C140H172Mn4N2O14, 2(C3H7NO), 2(H2O) | C156H170Mn4N4O10, 4(C3H7NO) | C160H168Mn4N4O10 |
| Formula weight, g mol−1 | 2647.97 | 2508.77 | 2773.09 | 2526.73 |
| Wavelength, Å | 0.7527 | 0.79312 | 0.79313 | 0.7927 |
| Temperature | 100(1) | 100(2) | 100(1) | 100(2) |
| Crystal system | Triclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c | P21/n | P21/n |
| a, Å | 16.780(3) | 14.071(3) | 16.357(3) | 13.162(3) |
| b, Å | 18.450(4) | 23.504(5) | 25.859(5) | 27.089(5) |
| c, Å | 23.540(5) | 21.891(4) | 18.043(4) | 23.389(5) |
| α, ° | 99.17(3) | 90 | 90 | 90 |
| β, ° | 101.29(3) | 96.28(3) | 95.27(3) | 96.43(3) |
| γ, ° | 96.07(3) | 90 | 90 | 90 |
| V, Å3 | 6985(3) | 7196(3) | 7600(3) | 8287(3) |
| Z/Z′ | 2/1 | 2/0.5 | 2/0.5 | 2/0.5 |
| ρ calc, g cm3 | 1.259 | 1.158 | 1.212 | 1.013 |
| μ, cm−1 | 0.486 | 0.544 | 0.522 | 0.467 |
| F(000) | 2824.0 | 2676.0 | 2948.0 | 2672.0 |
| Crystal size, mm3 | 0.3 × 0.12 × 0.08 | 0.15 × 0.03 × 0.02 | 0.2 × 0.2 × 0.1 | 0.12 × 0.05 × 0.03 |
| 2θ range for data collection, ° | 2.392 ≤ 2θ ≤ 58 | 2.846 ≤ 2θ ≤ 55.646 | 3.998 ≤ 2θ ≤ 61.872 | 4.144 ≤ 2θ ≤ 61.944 |
| Index ranges | 21 ≤ h ≤ 21, | −15 ≤ h ≤ 16, | 21 ≤ h ≤ 17, | −17 ≤ h ≤ 12, |
| −23 ≤ k ≤ 23, | −27 ≤ k ≤ 27, | −33 ≤ k ≤ 33, | −35 ≤ k ≤ 35, | |
| −30 ≤ l ≤ 30 | −25 ≤ l ≤ 25 | −14 ≤ l ≤ 23 | −29 ≤ l ≤ 30 | |
| Reflections collected/independ. | 126![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 005/31238 005/31238 |
45690/12198 |
41227/16429 |
55344/18416 |
| R int | 0.0957 | 0.1697 | 0.0480 | 0.0731 |
| Completeness to θmax/% | 99.8 | 99.3 | 94.8 | 97.0 |
| Data/restraints/parameters | 31238/42/1660 |
12198/65/690 |
16429/40/964 |
18416/524/416 |
| Goodness-of-fit on F2 | 1.014 | 1.036 | 1.044 | 1.284 |
| Final R indexes [I > 2σ(I)] R1/wR2 | 0.0636/0.1516 | 0.1091/0.2776 | 0.0633/0.1628 | 0.1336/0.3694 |
| Final R indexes [all data] R1/wR2 | 0.1178/0.1831 | 0.1987/0.3385 | 0.0927/0.1820 | 0.1960/0.4144 |
| ρ max/ρmin, e Å−3 | 0.63/−0.50 | 2.12/−0.60 | 1.44/−0.97 | 2.07/−0.67 |
For 32-Mn4(dmf)4, 32-Mn4(dmf)2(H2O)2, 32-Mn4(bipy)2 and 32-Mn4(phen)2, solvent mask command, generated by the BYPASS module within Olex2 software40 was applied to calculate 61, 257, 172, and 536 electrons per unit cell within the solvent accessible voids of 32 Å3, 253 Å3, 64 Å3, and 2148 Å3, respectively, which may correspond to the presence of undefined combinations of DMF, CH3CN, MeOH or H2O disordered solvent molecules, respectively.
CCDC Numbers: 2299403 (32-Mn4(dmf)4), 2299404 (32-Mn4(dmf)2(H2O)2), 2299407 (32-Mn4(bipy)2), 2299405 (32-Mn4(phen)2).1
Magnetic measurements
Magnetization of the prepared polycrystalline materials as a function of magnetic field and temperature was measured using the Physical Property Measurement System (PPMS-9, Quantum Design) with a vibrating sample magnetometry (VSM) option. The powdered samples were weighted (within an experimental error of 5%), pressed in gelatin capsules and mounted on the copper (gold plated) rod of PPMS-9 (with a typical background signal of 5 × 10−6 emu for the magnetic field applied).Results and discussion
The crystals of the obtained complexes, 32-Mn4(dmf)4, 32-Mn4(dmf)4(H2O)2, 32-Mn4bipy2 and 32-Mn4phen2, suitable for single crystal X-ray diffraction analysis, were grown at mild self-assembly conditions, using slow vapour diffusion of acetonitrile into the DMF/MeOH mixture, containing dissolved compound 3, MnCl2 salt and NEt3 in 10-fold excess with respect to 3. For 32-Mn4bipy2 and 32-Mn4phen2, a one-pot synthesis was performed; compound 3 was added to the reaction mixture simultaneously with appropriate N,N′-donor auxiliary ligands (colig) in a molar ratio 3/Mn/colig = 1/4/6 (colig = bipy and phen). All presented compounds are solvates, demonstrating different ratios of co-crystallised DMF, MeOH, H2O, and/or acetonitrile molecules, positions of which were established upon the refinement procedure. Whereas, the coordination of 3 with Mn(II) cations without adding the N,N′-donor co-ligands led to the formation of two complexes, displaying different crystalline phases and the nature of coordinated solvent molecules (32-Mn4(dmf)4 and 32-Mn4(dmf)4(H2O)2), for 32-Mn4(bipy)2 and 32-Mn4(phen)2, only one crystalline phase was observed.X-ray diffraction study
All prepared clusters (32-Mn4(dmf)4, 32-Mn4(dmf)4(H2O)2), 32-Mn4(bipy)2 and (32-Mn4(phen)2) are found to be centrosymmetric (see Fig. S1a–d, ESI†). Complex 32-Mn4(dmf)4 crystallizes in a triclinic P space group (see Fig. 2 and Table 1). There are two types of complex species in the crystal, differing by the calix[4]arene macrocyclic platform conformation, leading to small changes in the coordination environment of Mn atoms (see Scheme S1 and Tables S1–S4, ESI†). Both complex molecules of A-type (32-Mn4(dmf)4-A) and B-type (32-Mn4(dmf)4-B) are composed of two calix[4]arene molecules, four Mn atoms, and four coordinated DMF molecules. The macrocyclic platform of 3 in 32-Mn4(dmf)4-A presents a conformation close to the symmetrical cone, whereas, for 32-Mn4(dmf)4-B, calix[4]arene is found in a relatively more distorted conformation of a pinched cone, which was confirmed by analyzing the corresponding dihedral angles between the opposite aryl units of the macrocycle platform (see Table S5, ESI†). Two adamantyl groups of the opposite aryl units of 3 for 32-Mn4(dmf)4-A and one adamantyl group of 3 for 32-Mn4(dmf)4-B are found to be disordered over the two positions (see Experimental part). For 32-Mn4(dmf)4-A and 32-Mn4(dmf)4-B, the Mn(1) atom, located within the calix[4]arene cavity, is pentacoordinated due to the interaction with four O-phenolate atoms from the tetradeprotonated compound 34− and one O-atom, belonging to μ3-OH group, leading to a distorted vacant octahedral (C4v symmetry) coordination sphere, which was established by the SHAPE analysis42 (see Tables S6 and S7, ESI†). The Mn(2) atom demonstrates the coordination environment of six O-atoms, forming the distorted octahedral coordination sphere: two O-atoms belong to the phenolate groups and occupy the trans positions, two adjacent calix[4]arene molecules, two O-atoms of two cis μ3-OH groups and two DMF-molecules, located at side positions of the {Mn4}-cluster units. The BSV analysis (see Table S8, ESI†) confirmed that Mn(1) and Mn(2) atoms are in oxidation states (III) and (II), respectively, corresponding to the earlier reported familiar {Mn4} compounds.21 Two Mn(1) and two Mn(2) atoms form the {Mn4} cluster core, displaying an equilateral rhomboid geometry, with the side and opposite angle values practically identical for 32-Mn4(dmf)4-A and 32-Mn4(dmf)4-B (see Table S9, ESI†).
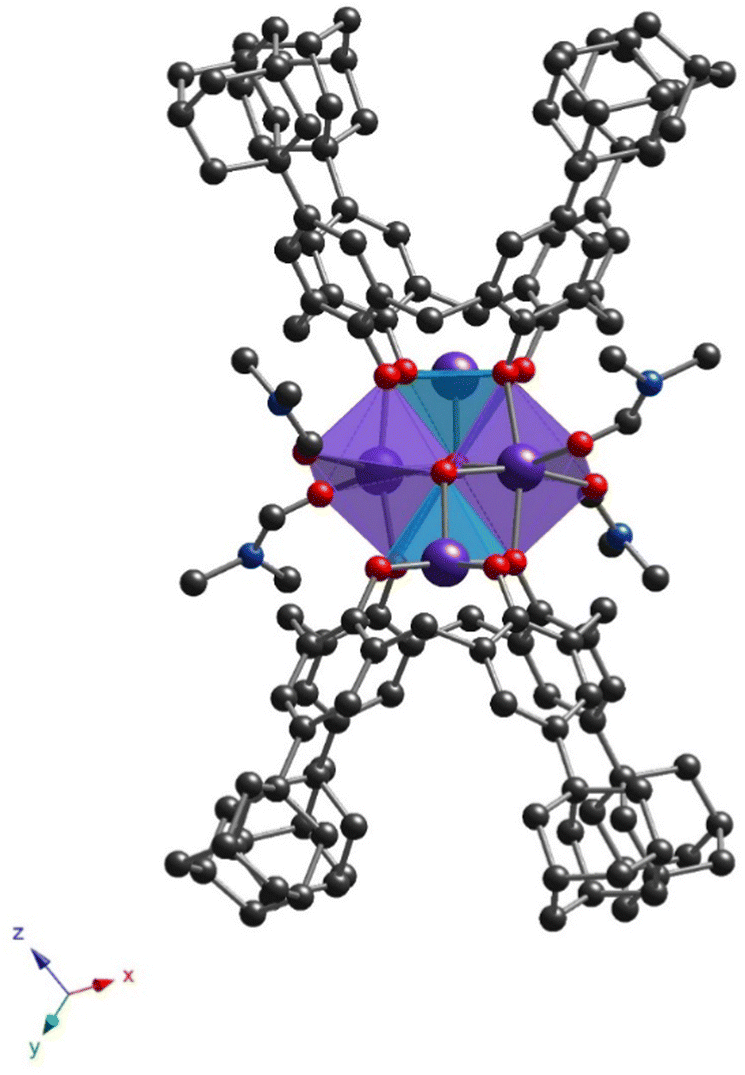 | ||
| Fig. 2 The structure of 32-Mn4(dmf)4 (only one independent molecule of A-type is presented) obtained by X-ray diffraction on single crystals. The C-, N-, O-, and Mn-atoms are represented by black, blue, red, and violet spheres, respectively. The Mn(1) and Mn(2) are denoted as light blue or violet polyhedra, respectively. Hydrogen atoms, non-coordinated solvent molecules, 32-Mn4(dmf)4-B, and disordered fragments are omitted for clarity. | ||
Both 32-Mn4(dmf)4-A and 32-Mn4(dmf)4-B molecules in the crystal form CH/π bonds with CH3CN solvent molecules, accommodated within the calix[4]arene hydrophobic cavity (dC(87)⋯C6centroid = 3.626(5)–3.703(4) Å and dC(90)⋯C6centroid = 3.437(3)–3.741(4) Å, respectively), and intermolecular H-bonds with DMF and MeOH molecules (d(O5A)⋯O(80) = 2.855(4) Å and d(O5B)⋯O(91A) = 2.817(5) Å, respectively) (see Fig. S2, ESI†).
In the crystal, 32-Mn4(dmf)4-A and 32-Mn4(dmf)4-B molecules are stacked in parallel rows, running along the 0y axis (see Fig. 3). The complex species, belonging to adjacent rows, display the perpendicular orientation, leading to the shortest {Mn4}⋯{Mn4} distance equal to 14.296(6) Å (see Fig. S5 and Table S10, ESI†).
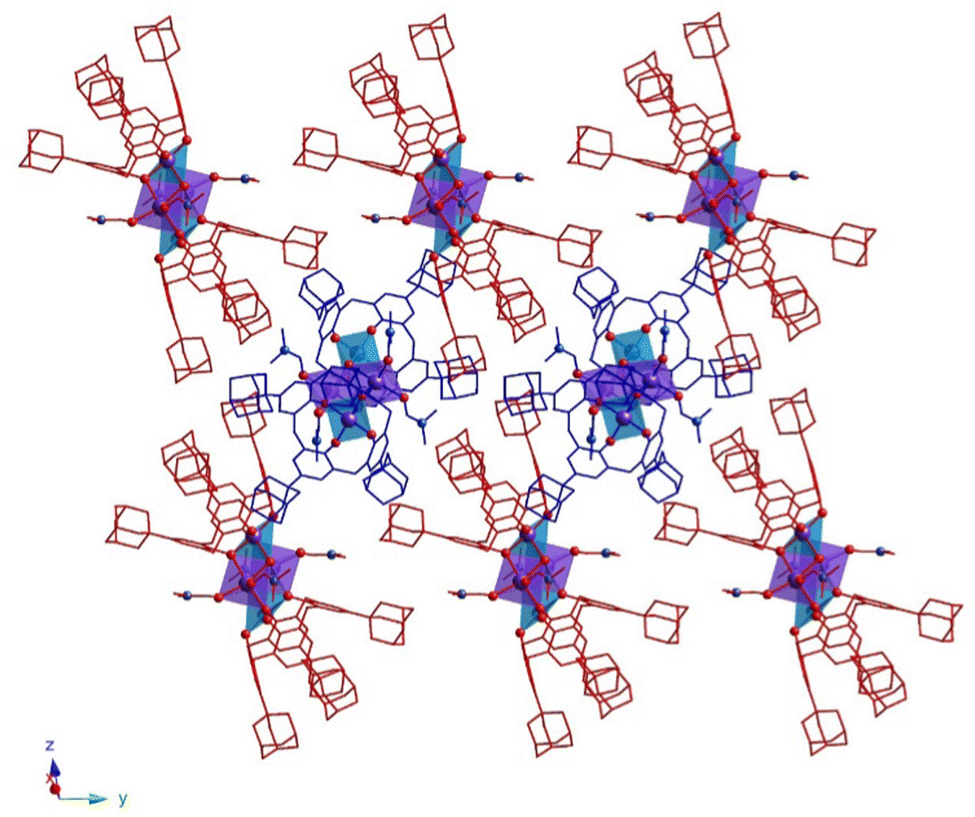 | ||
| Fig. 3 For 32-Mn4(dmf)4, a portion of the crystal packing along the 0y axis, showing perpendicular orientation of 32-Mn4(dmf)4-A (red colored) and 32-Mn4(dmf)4-B (blue colored) molecules in the crystal. The N-, O-, and Mn-atoms are represented by blue, red and violet spheres, respectively. The Mn(1) and Mn(2) are denoted as light blue or violet polyhedra, respectively. Hydrogen atoms, non-coordinated solvent molecules, and disordered fragments are omitted for clarity. | ||
The powder X-ray diffraction analysis for 32-Mn4(dmf)4 evidences a low matching between the simulated and experimental PXRD patterns, indicating the presence of another crystalline phase in the studied sample (see Fig. 4). In order to establish the structure of the unknown phase, another crystal from the same crystallization vial was analyzed using single crystal XRD, affording a new crystal structure 32-Mn4(dmf)2(H2O)2.
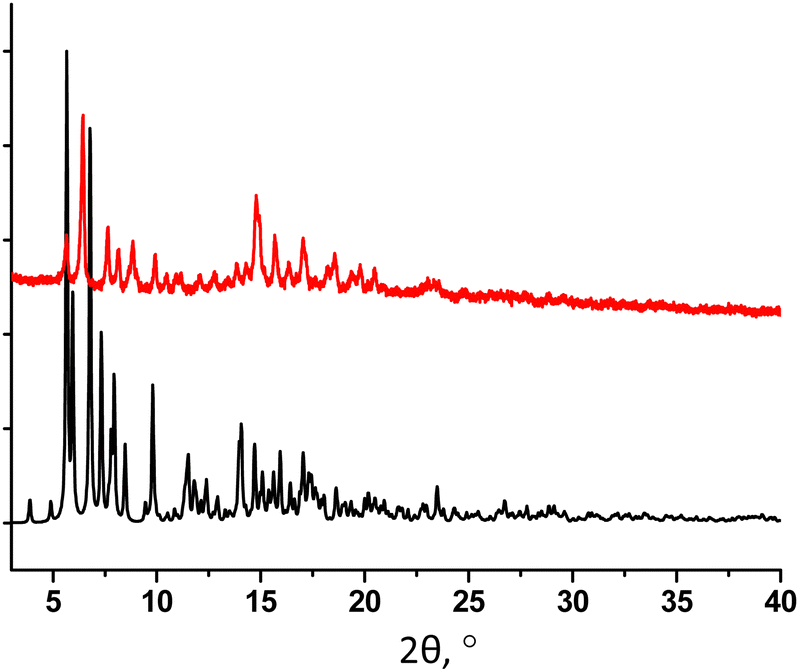 | ||
| Fig. 4 For 32-Mn4(dmf)4, a comparison of simulated (black) and experimental (red) PXRD patterns. | ||
Complex 32-Mn4(dmf)2(H2O)2 crystallizes in a monoclinic system (P21/c space group, see Table 1) and demonstrates a similar molecular structure, which differs from 32-Mn4(dmf)4 by the nature of coordination of Mn atoms to solvent molecules (two DMF molecules are replaced by H2O molecules) (see Fig. 5a).
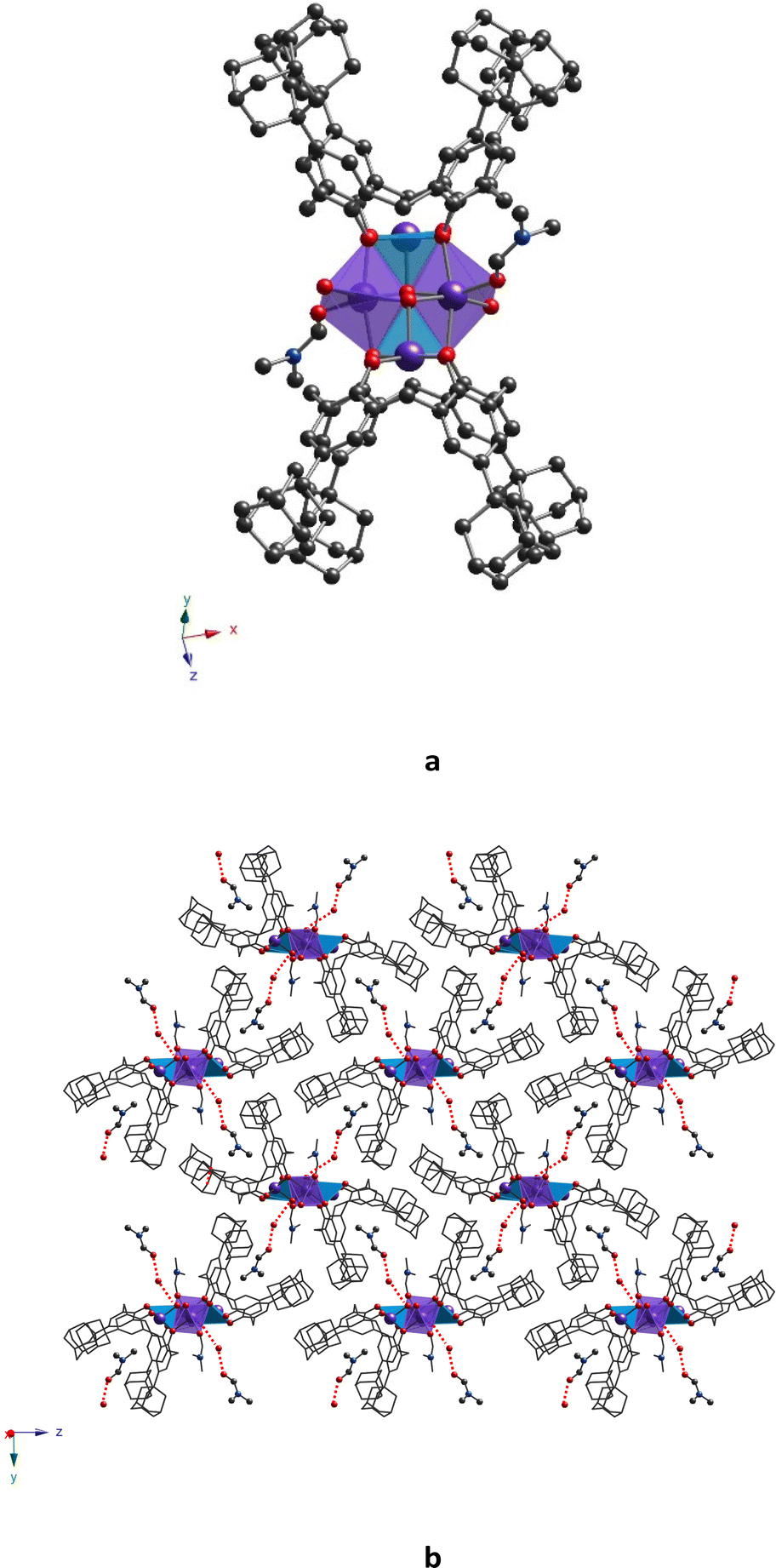 | ||
| Fig. 5 For 32-Mn4(dmf)2(H2O)2, (a) crystal structure of the complex and (b) a portion of the crystal packing, showing the formation of a non-covalent 2D molecular assembly (view along the y0z plane), resulting from intermolecular CH/π and H-bonding, involving complex species, co-crystallized DMF, and H2O molecules. The C-, N-, O-, and Mn-atoms are represented by black, blue, red, and violet spheres, respectively. Mn(1) and Mn(2) are denoted as light blue or violet polyhedra, respectively. Hydrogen atoms, non-coordinated solvent molecules, and disordered fragments are omitted for clarity. | ||
In contrast to 32-Mn4(dmf)4, only one independent molecule is found in the unit cell. As for 32-Mn4(dmf)4, two types of Mn atoms are present within the cluster unit: Mn(1), with oxidation state (III), coordinated with four O-phenolate atoms of the calix[4]arene ligand and one O-atom of μ3-OH, and Mn(2), with oxidation state (II), displaying an O6-coordination environment due to coordination with two O-phenolate atoms of 3, two O-atoms of cis μ3-OH groups, and two O-atoms, coming from one DMF and one H2O molecules. The substitution of DMF molecules by water molecules practically did not affect the coordination spheres of Mn atoms, which was attested by comparison of the coordination bonds and angles (see Tables S1–S4, ESI†) as well as by SHAPE analysis42 (see Tables S6 and S7, ESI†). As a result, the formation of a similar geometry for the rhomboid {Mn4} cluster core is observed (see Table S9, ESI†). The macrocyclic platform of 3 undergoes a relatively higher distortion from the ideal cone conformation compared to that of 32-Mn4(dmf)4, which is confirmed by the analysis of dihedral angles between the oppositely disposed aryl moieties (see Table S5, ESI†). One solvate DMF molecule is found to be accommodated in each hydrophobic cavity of each calix[4]arene molecule due to the CH/π interaction (dC(74)⋯C6centroid = 3.45(1)–3.49(1) Å and dC(75)⋯C6centroid = 3.663(1) Å, respectively, see Fig. S3, ESI†). Moreover, the formation of H-bonding between the O(5)-atoms of complex species belonging to μ3-OH groups and the O(7)-atom of co-crystallized H2O molecules occurs with a distance equal to 2.919(4) Å (see Fig. S3, ESI†). Additionally, the intermolecular H-bonding was also established between the co-crystallized H2O and DMF molecules (dO(7)⋯O(73) = 2.715(4) Å) (see Fig. S3, ESI†). As a result, the combination of CH/π and H-bonding interactions in the crystal of 32-Mn4(dmf)4 resulted in the 2D molecular network formation, located parallel to the y0z plane (see Fig. 5b) within the 2D layer. The dihedral angle between complex molecules, calculated as the angle between the planes, built on CH2 bridging moieties of macrocyclic platforms, belonging to calix[4]arene molecules of adjacent complex species, was found to be slightly decreased to 84.7(1)°, compared to that observed for 32-Mn4(dmf)4 (see Table S10, ESI†). In the 32-Mn4(dmf)2(H2O)2 crystal, the obtained 2D layers are stacked in a parallel fashion along the 0x axis, completing the crystal packing and leading to the shortest {Mn4}⋯{Mn4} distance, equal to 14.071(3) Å (see Fig. S6, ESI†).
The PXRD study of 32-Mn4(dmf)2(H2O)2 revealed a good match of the simulated and experimental diffraction patterns, evidencing that the sample is dominantly composed of the 32-Mn4(dmf)2(H2O)2 phase (see Fig. 6).
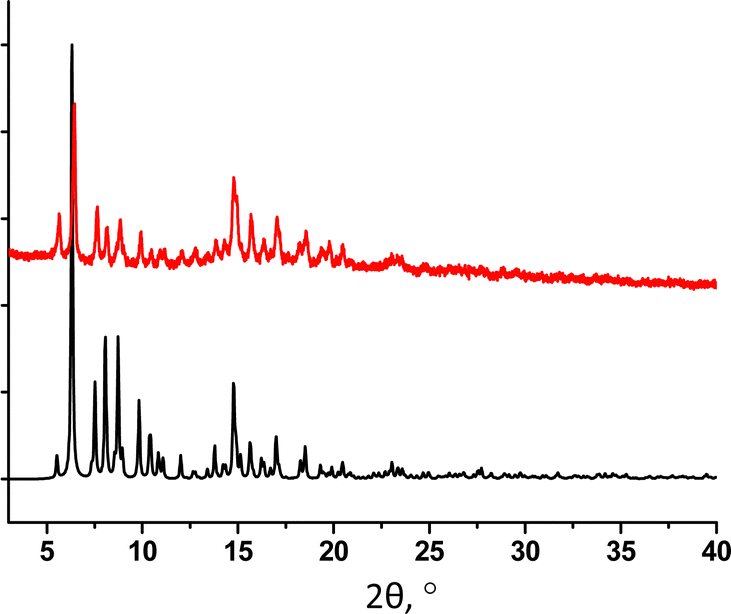 | ||
| Fig. 6 For 32-Mn4(dmf)2(H2O)2, a comparison of simulated (black) and experimental (red) PXRD patterns. | ||
Complex 32-Mn4(bipy)2, which crystallizes in the monoclinic system P21/n space group (see Table 1), displays a molecular structure similar to that observed for other {Mn4} clusters described above, 32-Mn4(dmf)4 and 32-Mn4(dmf)2(H2O)2. Here, the 2,2′-bipyridyl ligands replace the solvate molecules (DMF and/or H2O), cis located in the coordination sphere of the Mn(2) atom (see Fig. 7a). The macrocyclic platform of 3 is adopted in a cone conformation, displaying high symmetry: both oppositely placed aryl units of the macrocyclic platform form almost identical dihedral angles (see Table S5, ESI†), which does not significantly affect the Mn(1) atom coordination sphere, demonstrating similar coordination bond lengths and angles, compared to those revealed for 32-Mn4(dmf)4 and 32-Mn4(dmf)2(H2O)2. In contrast, the appearance of bipy ligands instead of DMF molecules has remarkably influenced the coordination sphere of the Mn(2) atom, resulting in a drastic deformation of the octahedral geometry of its coordination sphere (see Tables S3, S4 and S7, ESI†). In particular, unlike the practically symmetrical Mn(2)⋯O bonds observed for 32-Mn4(dmf)4 and 32-Mn4(dmf)2(H2O)2, in 32-Mn4(bipy)2, the Mn(2) atoms form non-symmetrical coordination bonds with N-atoms of the bipy ligand, leading to Mn(2)⋯N(1) and Mn(2)⋯N(2) distances of 2.175(3) Å and 2.272(3) Å, respectively. In addition, it should be noted the decrease of the angle between the O–Mn(2)–O coordination bonds from 91.3(1)°, 89.5(1)°, and 89.1(4)°, found for 32-Mn4(dmf)4 and 32-Mn4(dmf)2(H2O)2, respectively, to 74.1(1)° (N–Mn(2)–N angle) for 32-Mn4(bipy)2. The pyridyl moieties of the bipy ligand are not parallel and form a dihedral angle of 19.8(1)°. Comparing with 32-Mn4(dmf)4 and 32-Mn4(dmf)2(H2O)2, the rhomboid geometry of the {Mn4} cluster core of 32-Mn4(bipy)2 undergoes compression along Mn(1)–Mn(2) and Mn(2)⋯Mn(2) bonds, which is attested by decreased bond distances (see Table S9, ESI†).
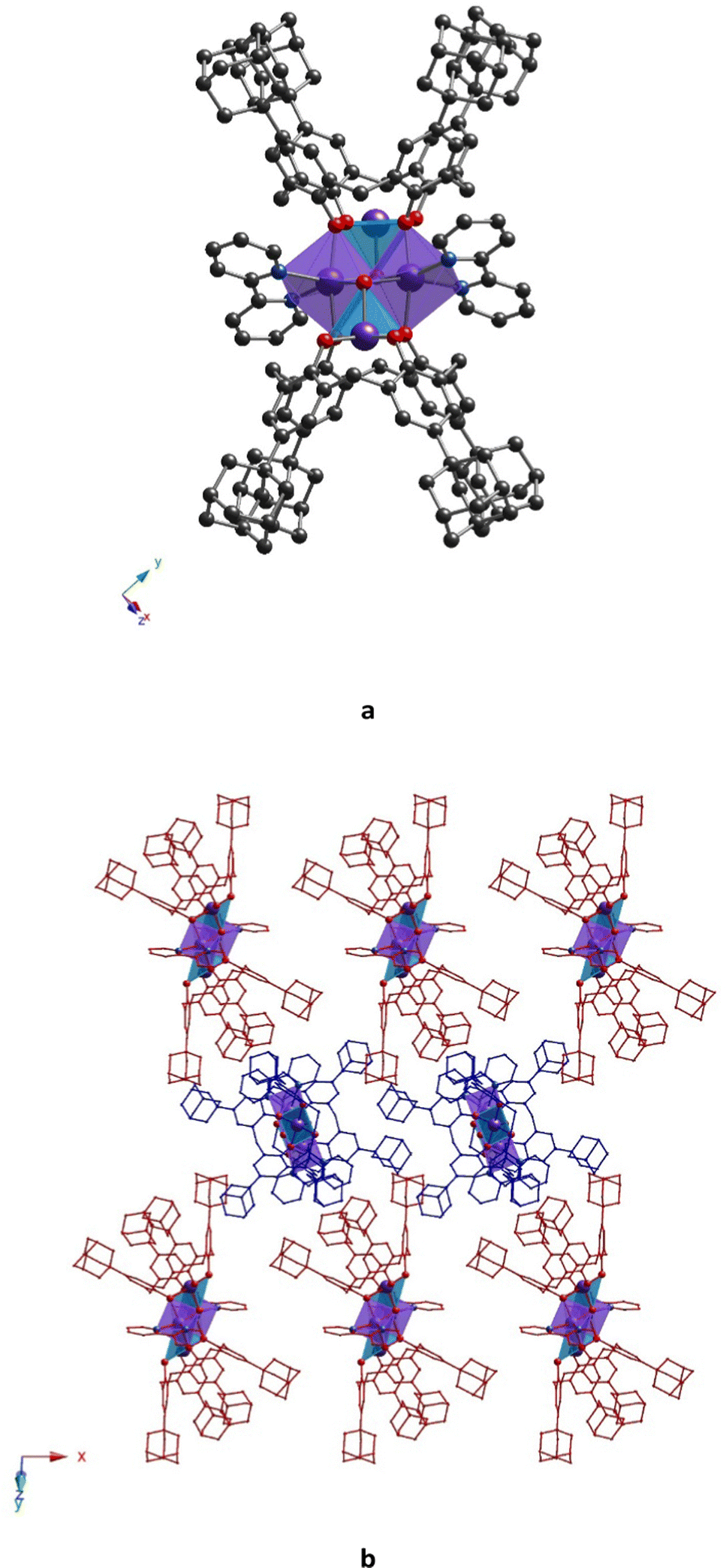 | ||
| Fig. 7 For 32-Mn4(bipy)2, (a) crystal structure of the complex and (b) a portion of crystal packing along the 0y axis, showing a near perpendicular orientation of complex species in the crystal. The C-, N-, O-, and Mn-atoms are represented by black, blue, red, and violet spheres, respectively. Mn(1) and Mn(2) are denoted as light blue or violet polyhedra, respectively. Hydrogen atoms, non-coordinated solvent molecules, and disordered fragments are omitted for clarity. | ||
One DMF molecule is found to be accommodated within the hydrophobic cavity of each calix[4]arene molecule of 32-Mn4(bipy)2 due to CH/π interactions between the C(88)-atom of methyl group and π-aromatic systems of aryl units of the macrocycle platform (dC(88)⋯C6cemtroid = 3.569(4)–3.742(4) Å) (see Fig. S4a, ESI†). Another DMF molecule also interacts with the calix[4]arene backbone via the exterior CH/π-bonding by involving the C(93) carbonyl atom (dC(93)⋯C6centroid = 3.542(5) Å). In addition, some short contacts between the O(6)- and O(7)-carbonyl atoms of DMF molecules with C(81), C(84), and C(85) atoms belonging to bipy molecules, with distances in the range 3.089(7)–3.361(5) Å, are present in the crystal (see Fig. S4b, ESI†).
In terms of crystal packing, the molecules of 32-Mn4(bipy)2 are stacked into parallel rows, running along the 0x axis, displaying the dihedral angle of 78.62(5)° between the planes, passing through the CH2-bridging moieties of calix[4]arene platforms, belonging to adjacent complex species (see Fig. 7b). Such crystalline arrangement allows to reach the shortest {Mn4}⋯{Mn4} distance, equal to 16.357(3) Å (see Fig. S7 and Table S10, ESI†).
The PXRD study for powdered sample of 32-Mn4(bipy)2 revealed a good match of simulated and experimental diffraction patterns, demonstrating high purity of the crystalline phase (see Fig. 8).
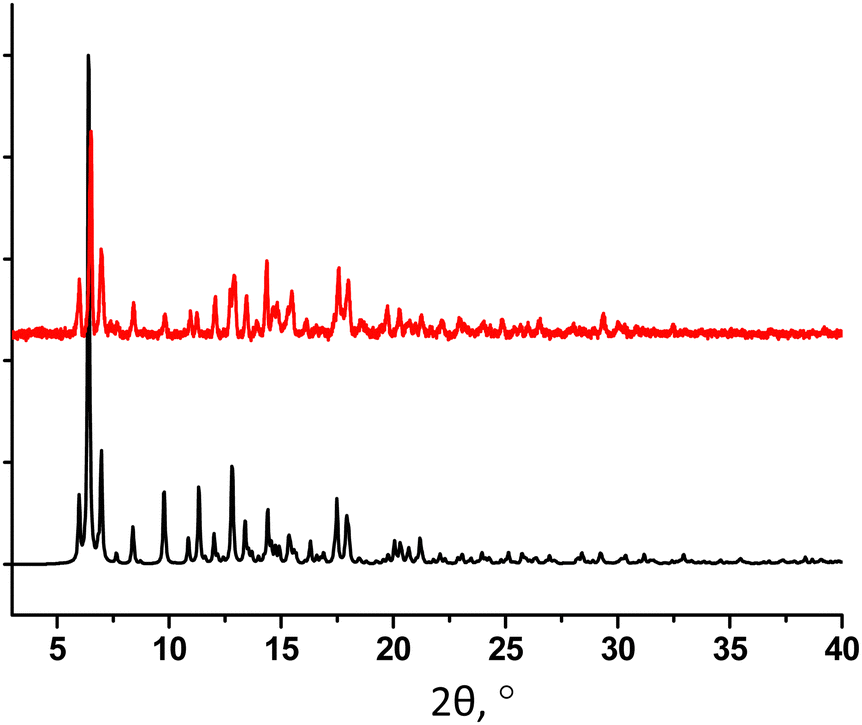 | ||
| Fig. 8 For 32-Mn4(bipy)2, a comparison of simulated (black) and experimental (red) PXRD patterns. | ||
The poor quality of the crystal did not allow us to obtain a high-resolution crystal structure for 32-Mn4(phen)2, even using the synchrotron radiation source (see Experimental part). However, the data obtained was quite good enough to establish the molecular structure of the complex. Similar to 32-Mn4(bipy)2, 32-Mn4(phen)2 crystallizes in a monoclinic P21/n space group and exhibits a very similar coordination pattern (see Fig. 9a and Table 1). The complex molecule is found to be disordered over two positions, leading to two kinds of clusters, 32-Mn4(phen)2-A* and 32-Mn4(phen)2-B*, in the crystal (see Fig. S4, ESI†). The phenanthroline molecules are found to be involved in coordination with Mn(2) atoms, which adopt an N2O4-distorted octahedral coordination sphere (see Table S7, ESI†). Due to the flat structure, phenanthroline ligands form two almost symmetrical coordination bonds with Mn(2) atoms (see Table S3, ESI†). Similar to 32-Mn4(bipy)2, the Mn(1) atoms are pentacoordinated with four O-phenolate atoms of the calix[4]arene platform and one O-atom of the μ3-OH group. Surprisingly, the involvement of phenanthroline ligands in coordination with Mn(2) atoms led to a distortion of the {Mn4} cluster core, resulting in the transformation of its rhomboid geometry into a parallelogram, which was attested by comparing the Mn(1)⋯Mn(2) bond lengths (3.271(6) Å, 3.108(8) Å and 3.119(8) Å, 3.37(1) Å for 32-Mn4(phen)2-A* and 32-Mn4(phen)2-B*, respectively, see Table S9, ESI†).
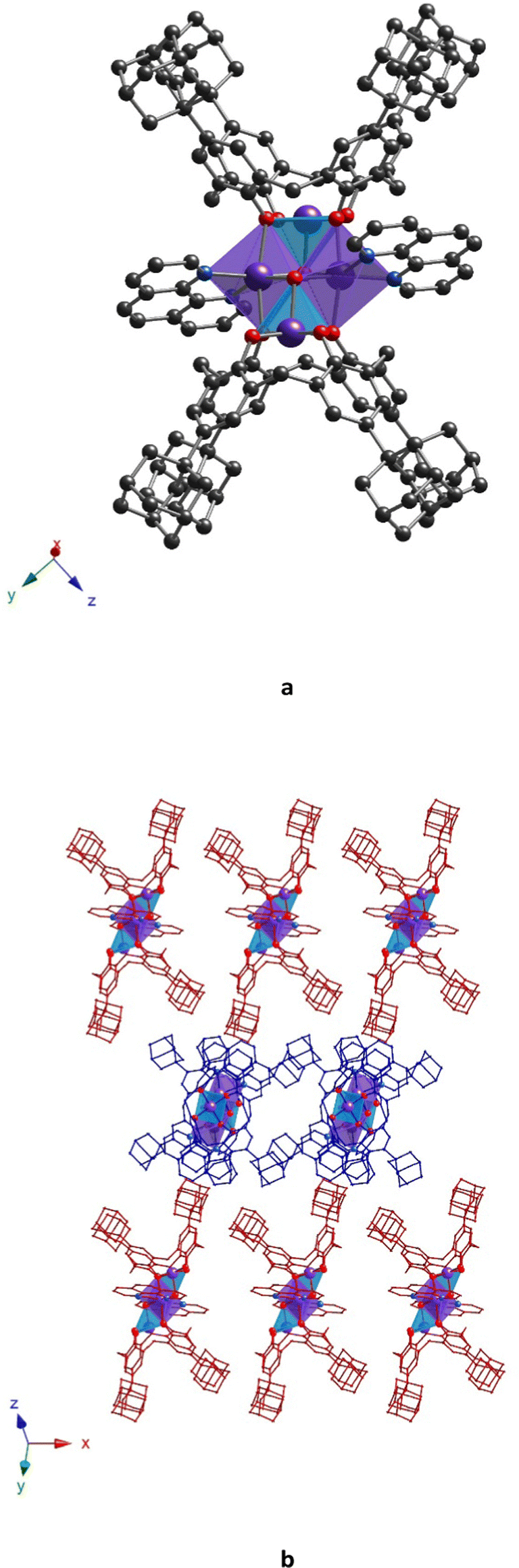 | ||
| Fig. 9 For 32-Mn4(phen)2, the crystal structure of the complex (a) and a portion of the crystal packing along the 0y axis, showing a near perpendicular orientation of the complex species within the crystal (b). The C-, N-, O-, and Mn-atoms are represented by black, blue, red, and violet spheres, respectively. Mn(1) and Mn(2) are denoted as light blue or violet polyhedra, respectively. Hydrogen atoms, non-coordinated solvent molecules, and disordered fragments are omitted for clarity. | ||
In the crystal, similar to 32-Mn4(bipy)2, the complex species of 32-Mn4(phen)2 are stacked in rows, running along the 0x axis (see Fig. 9b). The dihedral angle between the planes, passing through the CH2 bridging moieties of macrocycle platforms belonging to the calix[4]arenes lying in adjacent rows, is equal to 66.4(3)° (see Table S10, ESI†). The shortest {Mn4⋯Mn4} distance is found to be 13.162(3) Å, which is decreased compared to that found for 32-Mn4(bipy)2 (see Table S10 and Fig. S8, ESI†).
In contrast to 32-Mn4(dmf)2(H2O)2 and 32-Mn4(bipy)2, the crystal packing of 32-Mn4(phen)2 leads to the formation of large pores, filled by the disordered solvent molecules (see Fig. S9, ESI†). The solvent accessible volume, calculated by PLATON,43 was found to be 23.8% (1972 Å3 per unit cell).
For 32-Mn4(phen)2, the purity of the crystalline phase was confirmed using PXRD analysis. Although a partial decomposition of the crystalline sample is observed after separation from the mother liquor, the experimental PXRD pattern of the powdered sample demonstrates a good matching with the simulated one (see Fig. 10).
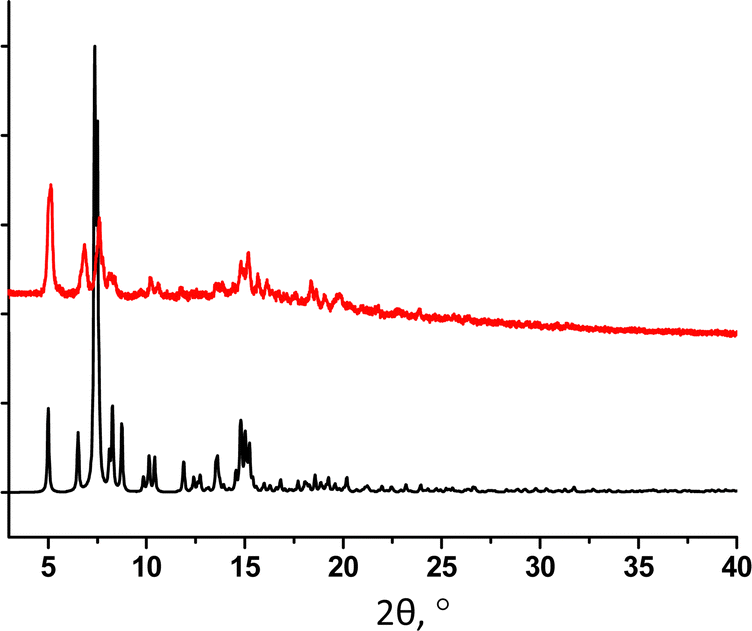 | ||
| Fig. 10 For 32-Mn4(phen)2, a comparison of simulated (black) and experimental (red) PXRD patterns. | ||
TGA study
Whereas the TG/DSC analysis of the powdered sample of Mn4(dmf)2(H2O)2 revealed good accordance of the quantity of solvate molecules with the X-ray diffraction data; for 32-Mn4(bipy)2, the TGA data confirmed the release of two instead of four refined DMF molecules (see Fig. S11 and S12, ESI†). For 32-Mn4(phen)2, presenting a porous crystal packing, it was established that the crystal unit additionally contains one CH3CN, two MeOH, and five DMF molecules located within the pores (see Fig. S13, ESI†). These results were also found to be in agreement with elemental analysis (see Experimental part).Magnetic properties
For 32-Mn4(dmf)2(H2O)2, 32-Mn4(bipy)2, and 32-Mn4(phen)2 samples, magnetization was measured (in ZFC and FC regimes) within the 5–300 K temperature range in the applied field of H = 100 Oe. All complexes displayed the temperature dependence of magnetic susceptibility χ(T), indicating the domination of ferromagnetic exchange between metal ions within the cluster core at low temperatures (see Fig. 11, left part). The approximation of obtained χ(T) curves was performed using the Curie–Weiss law χ = C/(T − Θ) with high R-squared values (99.7–99.9%), which allowed calculation of the material-specific Curie constant, C, and Curie temperature, Θ, for the studied paramagnetic complexes. For the 32-Mn4(dmf)2(H2O)2 sample, it was found that C = 0.0048 emu g−1 Oe−1(or cm3 g−1) and Θ = 8.22 K. For clarity, the inverse magnetic susceptibility 1/χ(T) is also shown (see Fig. 11, right part). For 32-Mn4(bipy)2, the Curie–Weiss law approximation gives a slightly different Curie constant, C = 0.00434 emu g−1 Oe−1(or cm3 g−1), while the Curie temperature decreased, Θ = 1.12 K. For 32-Mn4(phen)2, the χ(T) curve analysis showed an increased Curie constant, C = 0.00617 emu g−1 Oe−1(or cm3 g−1) with further decrease in Curie temperature, Θ = 0.13 K.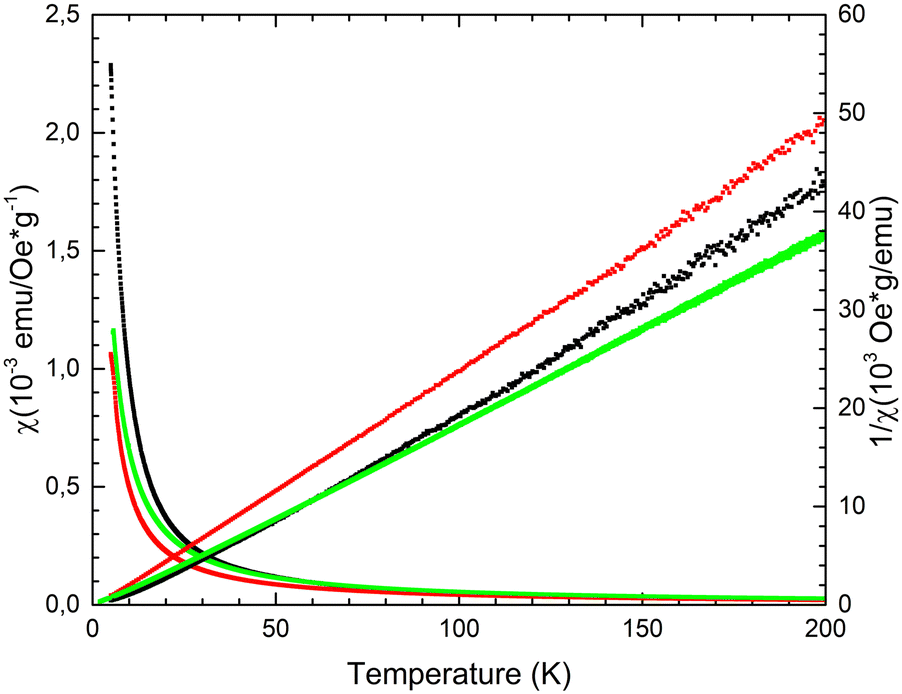 | ||
| Fig. 11 For 32-Mn4(dmf)2(H2O)2 (black dotted line), 32-Mn4(bipy)2 (red dotted line), and 32-Mn4(phen)2 (green dotted line), the χ(T) and 1/χ(T) dependences measured in the ZFC regime at H = 100 Oe. | ||
As reported for [Mn4]-complexes,21,22,32 the spin-only value, expected for an uncoupled [MnIII2MnII2] unit, is equal to 14.75 cm3 K mol−1 with g-factor 2.0 at an external field of 0.1 T. The high-temperature xMT values of samples 32-Mn4(dmf)2(H2O)2, 32-Mn4(phen)2, and 32-Mn4(bipy)2 are equal to 12.4, 13.4, and 19.7 cm3 K mol−1, respectively. For all samples, the xMT(T) plots showed that as the temperature decreases, xMT remains constant till 50 K (see Fig. 12). Below 50 K, the xMT value increases slowly, reaching a maximum of 27.1 cm3 K mol−1 and 16.9 cm3 K mol−1 at 6 K for 32-Mn4(bipy)2 and 32-Mn4(phen)2, respectively. Below 6 K, the corresponding xMT value decreases, reaching 23.1 cm3 K mol−1 and 12.6 cm3 K mol−1 at 5 K. For the sample 32-Mn4(dmf)2(H2O)2, the xMT value increases upon the temperature decrease, reaching a maximum of 30.3 cm3 K mol−1 at 5 K.
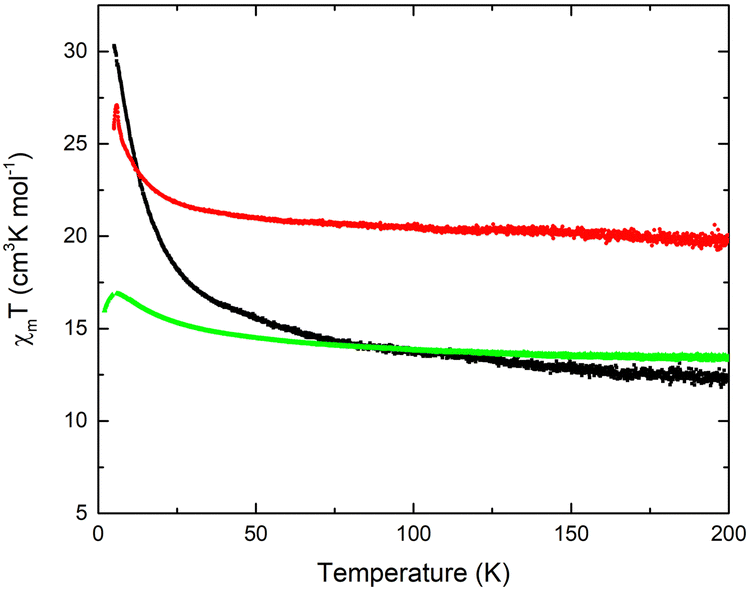 | ||
| Fig. 12 Temperature dependence of the xMT value for 32-Mn4(dmf)2(H2O)2 (black line), 32-Mn4(bipy)2 (red line), and 32-Mn4(phen)2 (green line). | ||
In order to confirm the ferromagnetic behavior, the field-dependent magnetization measurements for the obtained {Mn4} clusters were performed within the −9T⋯ +9T range of an externally applied magnetic field at 5 K. It was established that 32-Mn4(dmf)2(H2O)2 reaches saturation at 9T with the value of 16.8 μB per molecule, whereas for 32-Mn4(bipy)2 with Θ = 1.12 K, the saturation was not observed at 5 K within the studied range of the magnetic field, which is evidence of the contribution of paramagnetic ordering at high field (see Fig. 13). The field-dependent magnetization study for 32-Mn4(phen)2 almost saturates at 9T and T = 2 K with the value of 14.6μB per molecule (see Fig. S10, ESI†).
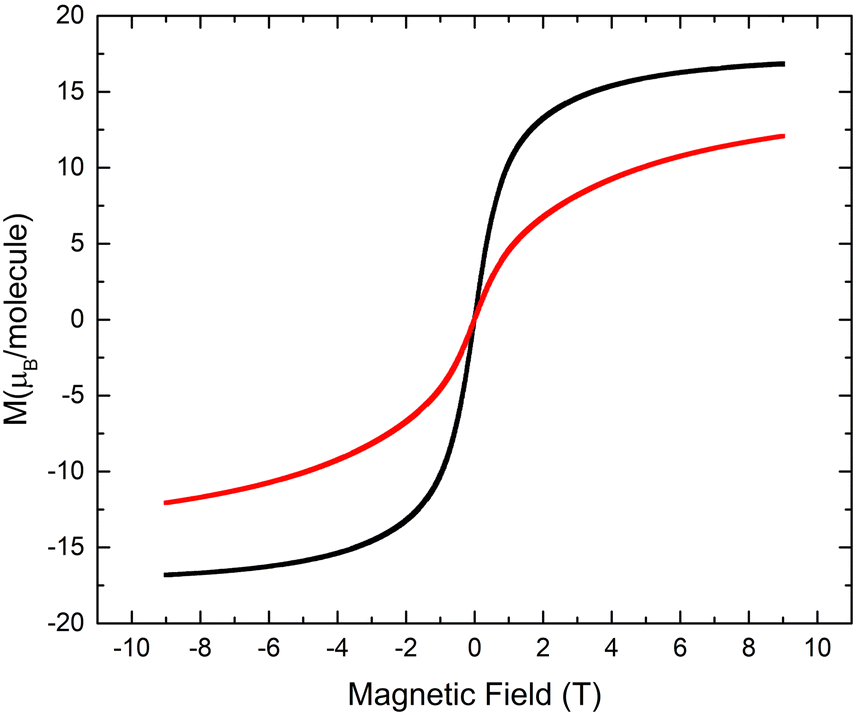 | ||
| Fig. 13 For 32-Mn4(dmf)2(H2O)2 (black line) and 32-Mn4(bipy)2, (red line), the field-dependent magnetization measured at T = 5 K. | ||
Discussion
The SCXRD studies revealed the formation of a well-reproducible {MnII2MnIII2} cluster core when calix[4]arene 3, bearing four bulky adamantyl substituents on the upper rim, was combined with manganese(II) chloride in a DMF/MeOH solution either with or without adding the N,N′-chelating auxiliary ligands. Due to similarities in the motif of complexes, one can thoroughly pursue the structural changes occurring within the coordination spheres of Mn(1) and Mn(2) atoms, both with distortion of the rhomboid {Mn4} cluster core, caused by the involvement of different auxiliary ligands in coordination with metal ions, and attempt to find the correlations with observed magnetic properties. In contrast to previously reported complexes 12-Mn4(dmf)6,2112-Mn4(dmf)4(H2O)221 and 22-Mn4(bipy)2(MeOH)2,33 all displaying an octahedral coordination environment for both Mn(1) and Mn(2) ions, it was found that the appearance of adamantyl groups at the upper rim of the calix[4]arene platform of 3 leads to the transformation of the octahedral coordination sphere (Oh symmetry) of Mn(1) atoms into the vacant octahedral coordination sphere (C4v symmetry) (see Table S5, ESI†), resulting from the prevention of interaction with O-donor coligands, such as MeOH or DMF solvent molecules, usually presenting in the reaction media upon cluster synthesis. That leads to enhancing the interaction between O5-pentacoordinated Mn(1) and μ3-OH groups, decreasing the Mn(1)⋯Mn(1) distances from 5.7556(6) Å and 5.68(1) Å, as reported for 12-Mn4(dmf)6,2112-Mn4(dmf)4(H2O)2,21 respectively, to 5.581(2), Å 5.616(2) Å and 5.577(2) Å, as established for 32-Mn4(dmf)4 and 32-Mn4(dmf)2(H2O)2, respectively. A similar trend was also observed when comparing the appropriate Mn(1)⋯Mn(2) distances (see Fig. 14a and Table S9, ESI†).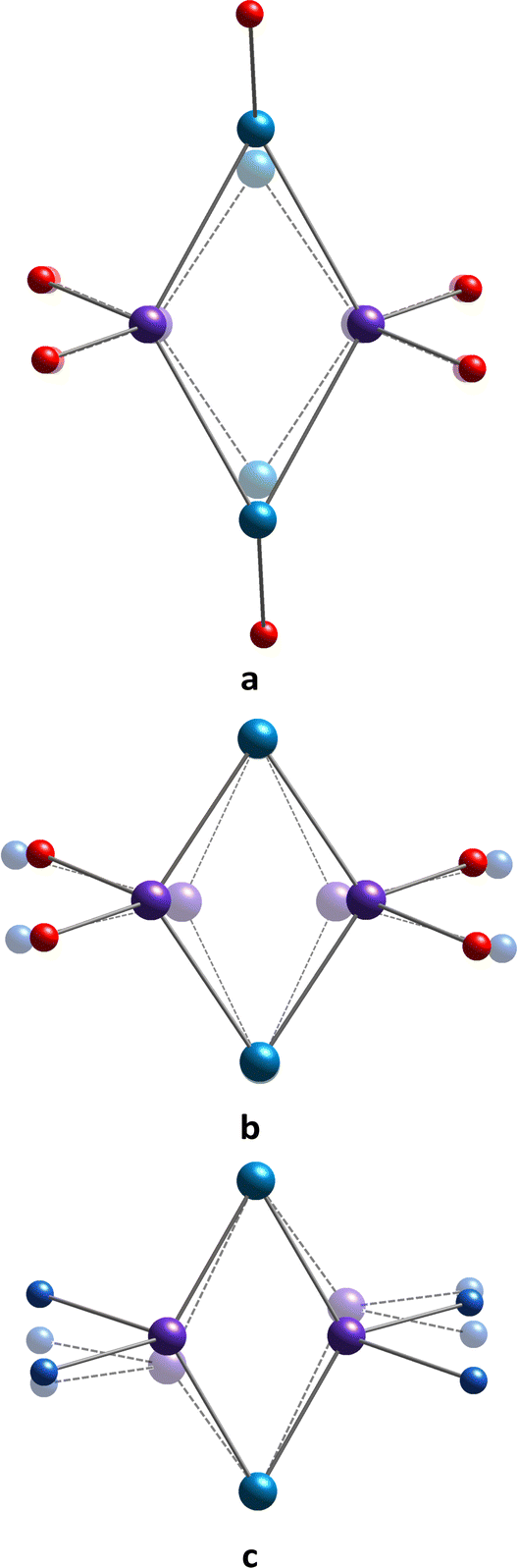 | ||
| Fig. 14 The schematic representation of {Mn4}-cluster core deformation (translucent mode) observed upon the elimination of the coordinated DMF molecule from the coordination sphere of Mn(1) atoms (a), the substitution of the coordinated solvent molecules by N,N′-chelating coligands (b), and the decreasing dihedral angle between the heterocyclic moieties of N,N′-chelating coligands (c). The N- and O-atoms are represented by blue and red spheres, respectively. Mn(1) and Mn(2) are coloured in light blue and violet, respectively. The O-atoms belonging to 3 and μ3-OH groups are omitted for clarity. | ||
Whereas the coordination sphere parameters for Mn(1) atoms are dependent mostly on the coordination of only DMF or MeOH solvent molecules, the environment of Mn(2) atoms can be controlled as well by varying the N,N-chelating auxiliary ligands. For 32-Mn4(bipy)2 and 32-Mn4(phen)2, it was established that the use of bipy or phen, differing by the intrinsic dihedral angle between the pyridyl moieties within the heterocycle structure, influences the Mn(2) atom coordination sphere, promoting elongation of the two corresponding coordination bonds, compared to those observed for H2O or DMF substituted complexes.21 As a result, the enhancement of the interaction between Mn(2) atoms and μ3-OH groups, disposed in the center of the metallic cluster, occurs, leading to a compression of the cluster core and decreasing the Mn(2)⋯Mn(2) distances to 3.1554(9) Å and 3.144(8) Å for 32-Mn4(bipy)2 and 32-Mn4(phen)2, respectively. It should be moted that for 32-Mn4(dmf)4 and 32-Mn4(dmf)2(H2O)2, the Mn(2)⋯Mn(2) distance was found to be 3.224(1) Å, 3.292(1) Å, and 3.276(2) Å, respectively (see Fig. 14b and Table S9, ESI†). A subsequent decrease in Mn(1)⋯Mn(2) distances was also detected.
When comparing a series of complexes involving bipy and phen coligands, one can notice the influence of the dihedral angle between the pyridyl moieties on the structure of the rhomboid {Mn4} cluster core. For 32-Mn4(bipy)2, it was found that the dihedral angle between the pyridyl moieties, resulting from the repulsion between ortho C–H bonds in the bipy molecule, causes the difference between the side lengths of polygons, built by four Mn-atoms (see Fig. 14c). The maximal deviation from the rhomboid geometry of the {Mn4} cluster core was revealed for 32-Mn4(phen)2 (see Table S9, ESI†). No more significant changes in the complex structures caused by variation of the nature of used N,N-chelating coliagnds were observed. The corresponding Mn(1)⋯Mn(1) and Mn(2)⋯Mn(2) distances were found to be similar for 32-Mn4(bipy)2 and 32-Mn4(phen)2.
Moreover, it is worth noting that the bulky adamantyl groups, presenting at the upper rim of the macrocycle platform of 3, play an important role in the crystalline self-assembly of these complexes. Whereas for earlier reported analogues, 12-Mn4(dmf)6,2112-Mn4(dmf)4(H2O)221 and 22-Mn4(bipy)2(MeOH)2,33 the “side-by-side” or “head-to-head” orientation of the complex species was underlined; all studied {Mn4} complexes, based on ligand 3, have demonstrated “side-to-head” orientation of the molecules, leading to the shortest inter-cluster distances, increasing in the order 32-Mn4(phen)2 < 32-Mn4(dmf)2(H2O)2 ≈ 32-Mn4(dmf)4, ≪ 32-Mn4(bipy)2 and reaching a value of 16.357(3) Å (for 32-Mn4(bipy)2), which significantly exceeds 12.5350(8) Å, the maximal one, found for the earlier described complex 12-Mn4(dmf)6.21
The magnetic property studies, performed for all prepared {Mn4} clusters, revealed a good agreement of χ(T) dependence with the Curie–Weiss law, indicating dominant ferromagnetic interactions between the metal ions within the cluster cores at low temperatures. Their magnetic behaviour was found to be dependent on the distortion of the rhomboid {Mn4} cluster core, which was controlled by the involvement of N,N′-chelating coligands in coordination with Mn(2) atoms. Whereas the involvement of bipy in coordination with Mn(2) atoms results in decreasing C and Θ constants as well as maximal field imposed magnetization parameters, which are apparently related to the compression of the cluster core and the decreasing Mn(2)⋯Mn(2) distance. The substitution of bipy by phen resulted in increasing C and maximal field imposed magnetization parameters, which probably can be associated with decreasing Mn(1)⋯Mn(2) distances. Thus, one may conclude that compression of the cluster core, imposed by N,N-chelating coligand coordination, dramatically influences the magnetic exchange between the Mn-atoms but, nevertheless, retains the dominant ferromagnetic interactions between the metallic centres. The last one was not observed for the earlier reported familiar cluster 22-Mn4(bipy)2(MeOH)2.33
The xMT(T) plots, obtained for 32-Mn4(dmf)2(H2O)2, 32-Mn4(phen)2, and 32-Mn4(bipy)2, demonstrated quite similar trends compared to those reported earlier for calix[4]arene-supported [Mn4] clusters and one calix[4]arene free analogue {MnIII4},44 also confirming the domination of ferromagnetic interactions within such systems (see Table 2). However, the comparison of the observed absolute xMT values indicates that the magnetic behavior of the {MnIII2MnII2} cluster core is notably susceptible to even small distortions in the coordination sphere of both Mn(III) and Mn(II) atoms, which makes these clusters an attractive platform for further investigations of magnetic coupling between the metallic centers.
| x M T (300 K), cm3 K mol−1 | x M T (max), cm3 K mol−1 | T (xMT. max), K | |
|---|---|---|---|
| phenPO2 = phenyl phosphinate. | |||
| 12-Mn4(dmf)6 21 | 15.5 | 24.5 | 5 |
| 12-Mn4(phenPO2)2(MeOH)4 22 | 14.7 | 17.7 | 17 |
| 22-Mn4(py4)(MeOH)4 32 | 14.2 | 50.5 | 6 |
| 32-Mn4(dmf)2(H2O)2 | 12.4 | 30.1 | 5 |
| 32-Mn4(bipy)2 | 19.7 | 27.1 | 6 |
| 32-Mn4(phen)2 | 13.4 | 16.9 | 6 |
| {MnIII4}44 | 12 | 28 | 20 |
Conclusions
The use of calix[4]arene 3 led to the generation of a series of new tetranuclear complexes, displaying the formation of the highly reproducible {MnIII2MnII2} cluster core, independent of the nature of the involved neutral O-donor (DMF, water) or N,N-chelating auxiliary ligands (2,2′-bipyridyl or 1,10-phenanthroline). Due to steric effects of bulky adamantyl substituents, located at the upper rim of 3, able to prevent the coordination with O-donor ligands, such as DMF or methanol molecules, the geometry of the coordination sphere of Mn(III) atoms was switched from octahedral to vacant octahedral, providing the {Mn4} core compression and decreasing Mn⋯Mn distances. Contrary to Mn(III) atoms, located within the calix[4]arene cavity, the coordination environment of Mn(II) atoms, disposed at the side positions of clusters, was controlled by the substitution of labile solvent DMF or H2O molecules by N,N′-chelating coligands, such as bipy or phen, providing the appropriate deformation of the octahedral geometry of their coordination sphere. Thus, the involvement of N,N-chelating coligands in coordination with Mn(II) atoms resulted in enhanced compression of the {Mn4}-cluster core, leading to partial degradation of the ferromagnetic exchange between the Mn-atoms, which was confirmed by the preliminary magnetic property measurements. To the best of our knowledge, the described fine-tuning of the distortion of rhomboid geometry of {Mn4} clusters, supported by calix[4]arenes and induced by the involvement of N,N-chelating coligands, was evidenced for the first time.The presence of four bulky substituents, grafted to the calix[4]arene backbone, drastically influenced the crystalline self-assembly of the studied {Mn4} clusters, compared to previously reported analogues and provided a “side-to-head” orientation of the complex species upon the crystal packing. Surprisingly for 32-Mn4(dmf)2(H2O)2, the formation of the 2D molecular network, supported by intermolecular H-bonding both with weak CH/π interactions, was revealed.
In order to get more insight into the magneto-structural correlations and single-molecule magnetic behaviour of the obtained {Mn4} clusters, the frequency dependence of magnetic susceptibility measurements, both with the quantum chemical calculation, using ab initio methods, have to be performed. The synthesis of new Mn-based clusters of mixed valences, using upper rim functionalized calix[4]arene derivatives, displaying controllable magnetic properties, is currently under investigation.
Author contributions
A. S. O., S. E. S., I. S. A. designed project, conception and methodology, D. R. I., A. I. S., A. T. G., P. V. D. performed X-ray diffraction analysis, I. V. S. carried out the synthesis of studied coordination compounds and acquired data, M. A. C., R. G. B. and A. G. K. performed magnetic properties studies, A. S. O., I. V. S., R. G. B. contributed in a draft version of the article writing. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Russian Science Foundation (grant number 19-73-20035) for financial support in the synthesis and crystal structure determination of Mn-complexes (A. S. O., I. V. S., S. E. S., I. S. A.). Magnetometry studies of prepared clusters have been supported by the Kazan Federal University Strategic Academic Leadership Program (PRIORITY-2030). The authors are grateful to the Kurchatov Complex For Synchrotron And Neutron Investigations for performing X-ray diffraction on single crystals study (P. V. D.), to the Spectral-Analytical Center of FRC Kazan Scientific Center of RAS for their help and support in IR-, EA, and PXRD-studies. Dr Mariia V. Kniazeva is warmly acknowledged for her help in the preparation of single crystals of obtained Mn-based clusters.References
-
B. Sieklucka and D. Pinkowic, Molecular Magnetic Materials: Concepts and Applications, Wiley-VCH Verlag GmbH & Co. KGaA, 2017 Search PubMed
.
- J. Kobylarczyk, E. Kuzniak, M. Liberka, S. Chorazy, B. Sieklucka and R. Podgajny, Coord. Chem. Rev., 2020, 419, 213394 CrossRef CAS
- S. Chorazy, J. J. Stanek, J. Kobylarczyk, S.-I. Ohkoshi, B. Sieklucka and R. Podgajny, Dalton Trans., 2017, 46, 8027–8036 RSC
- E. V. Eames, T. D. Harris and T. A. Betley, Chem. Sci., 2012, 3, 407–415 RSC
- V. V. Novikov, A. A. Pavlov, Y. V. Nelyubina, M.-E. Boulon, O. A. Varzatskii, Y. Z. Voloshin and R. E. P. Winpenny, J. Am. Chem. Soc., 2015, 137(31), 9792–9795 CrossRef CAS PubMed
- J. Nehrkorn, I. A. Valuev, M. A. Kiskin, A. S. Bogomyakov, E. A. Suturina, A. M. Sheveleva, V. I. Ovcharenko, K. Holldack, C. Herrmann, M. V. Fedin, A. Schnegg and S. L. Veber, J. Mater. Chem. C, 2021, 9, 9446–9452 RSC
- T. M. Muzioł, N. Tereba, R. Podgajny, R. Pełka, D. Czernia, M. Wiśniewski, S. Koter and G. Wrzeszcz, Int. J. Mol. Sci., 2022, 23, 1556 CrossRef
- S. Zhang, H. Wu, L. Sun, H. Ke, S. Chen, B. Yin, Q. Wei, D. Yang and S. Gao, J. Mater. Chem. C, 2017, 5, 1369 RSC
- M. Barwiolek, D. Jankowska, A. Kaczmarek-Kędziera, I. Lakomska, J. Kobylarczyk, R. Podgajny, P. Popielarski, J. Masternak, M. Witwicki and T. M. Muzioł, Int. J. Mol. Sci., 2023, 24, 3017 CrossRef CAS
- S. M. Hossain, S. Kamilya, S. Ghosh, R. Herchel, M. A. Kiskin, S. Mehta and A. Mondal, Cryst. Growth Des., 2023, 23(3), 1656–1667 CrossRef CAS
- A. Zabala-Lekuona, J. Manuel Seco and E. Colacio, Coord. Chem. Rev., 2021, 441, 213984 CrossRef CAS
- J. M. Zadrozny, J. Niklas, O. G. Poluektov and D. E. Freedman, ACS Cent. Sci., 2015, 1, 488–492 CrossRef CAS PubMed
- E. Moreno-Pineda and W. Wernsdorfer, Nat. Rev. Phys., 2021, 3, 645–659 CrossRef CAS
- R. O. Fuller, G. A. Koutsantonis and M. I. Ogden, Coord. Chem. Rev., 2020, 402, 213066 CrossRef CAS
- L. R. B. Wilson, M. Coletta, M. Evangelisti, S. Piligkos, S. J. Dalgarno and E. K. Brechin, Dalton Trans., 2022, 51, 4560 RSC
- A. S. Ovsyannikov, I. V. Strelnikova, I. D. Shutilov, D. R. Islamov, P. V. Dorovatovskii, A. T. Gubaidullin, A. S. Agarkov, S. E. Solovieva and I. S. Antipin, Crystals, 2023, 13, 1017 CrossRef CAS
- C. Desroches, G. Pilet, S. A. Borshch, S. Parola and D. Luneau, Inorg. Chem., 2005, 44, 9112–9120 CrossRef CAS
- M. Lamouchi, E. Jeanneau, G. Novitchi, D. Luneau, A. Brioude and C. Desroches, Inorg. Chem., 2014, 53, 63–72 CrossRef CAS
- S. M. Taylor, J. M. Frost, R. McLellan, R. D. McIntosh, E. K. Brechin and S. J. Dalgarno, CrystEngComm, 2014, 16, 8098 RSC
- G. Karotsis, S. J. Teat, W. Wernsdorfer, S. Piligkos, S. J. Dalgarno and E. K. Brechin, Angew. Chem., Int. Ed., 2009, 48, 8285–8288 CrossRef CAS
- S. M. Taylor, G. Karotsis, R. D. McIntosh, S. Kennedy, S. J. Teat, C. M. Beavers, W. Wernsdorfer, S. Piligkos, S. J. Dalgarno and E. K. Brechin, Chem. – Eur. J., 2011, 17, 7521 CrossRef CAS
- S. M. Taylor, R. D. McIntosh, C. M. Beavers, S. J. Teat, S. Piligkos, S. J. Dalgarno and E. K. Brechin, Chem. Commun., 2011, 47, 1440–1442 RSC
- L. R. B. Wilson, M. Coletta, M. Evangelisiti, S. Piligkos, S. J. Dalgarno and E. K. Brechin, Dalton Trans., 2022, 51, 4213–4226 RSC
- A. Fong, L. McCormick, S. J. Teat, E. K. Brechin and S. J. Dalgarno, Supramol. Chem., 2018, 30(5–6), 504–509 CrossRef CAS
- A. S. Ovsyannikov, I. V. Strelnikova, A. A. Iova, A. S. Agarkov, D. R. Islamov, P. V. Dorovatovskii, S. E. Solovieva and I. S. Antipin, J. Struct. Chem., 2024, 65(2) DOI:10.26902/JSC_id122224
- S. Sanz, K. Ferreira, S. J. Dalgarno and E. K. Brechin, Chem. Commun., 2011, 47, 9042–9044 RSC
- S. Sanz, R. D. McIntosh, C. M. Beavers, S. J. Teat, M. Evangelisti, E. K. Brechin and S. J. Dalgarno, Chem. Commun., 2012, 48, 1449–1451 RSC
- Y. Bi, G. Xu, W. Liao, S. C. Du, R. P. Deng and B. W. Wang, Sci. China: Chem., 2012, 55, 967–972 CrossRef CAS
- M. A. Palacios, R. McLellan, C. M. Beavers, S. J. Teat, H. Weihe, S. Piligkos, S. J. Dalgarno and E. K. Brechin, Chem. – Eur. J., 2015, 21, 11212 CrossRef CAS PubMed
- G. Karotsis, S. Kennedy, S. J. Teat, C. M. Beavers, D. A. Fowler, J. J. Morales, M. Evangelisti, S. J. Dalgarno and E. K. Brechin, J. Am. Chem. Soc., 2010, 132(37), 12983–12990 CrossRef CAS PubMed
- S. Sanz, K. Ferreira, R. D. McIntosh, S. J. Dalgarno and E. K. Brechin, Chem. Commun., 2011, 47, 9042–9044 RSC
- S. M. Aldoshin, I. S. Antipin, V. I. Ovcharenko, S. E. Solov’eva, A. S. Bogomyakov, D. V. Korchagin, G. V. Shilov, E. A. Yur’eva, F. B. Mushenok, K. V. Bozhenko and A. N. Utenyshev, Russ. Chem. Bull., 2013, 62, 536–542 CrossRef CAS
- S. M. Aldoshin, I. S. Antipin, S. E. Solov’eva, N. A. Sanina, D. V. Korchagin, G. V. Shilov, F. B. Mushenok, A. N. Utenyshev and K. V. Bozhenko, J. Mol. Struct., 2015, 1081, 217–223 CrossRef CAS
- M. Coletta, R. McLellan, A. Waddington, S. Sanz, K. J. Gagnon, S. J. Teat, E. K. Brechin and S. J. Dalgarno, Chem. Commun., 2016, 52, 14246–14249 RSC
- A. N. Khomich, E. A. Shokova and V. V. Kovalev, Synlett, 1994, 1027–1028 CrossRef CAS
- V. A. Lazarenko, P. V. Dorovatovskii, Y. V. Zubavichus, A. S. Burlov, Y. V. Koshchienko, V. G. Vlasenko and V. N. Khrustalev, Crystals, 2017, 7(11), 325 CrossRef
- R. D. Svetogorov, P. V. Dorovatovskii and V. A. Lazarenko, Cryst. Res. Technol., 2020, 55(5), 1900184 CrossRef CAS
- W. Kabsch, Acta Crystallogr., 2010, D66, 125–132 CrossRef
- G. M. Sheldrick, Acta Crystallogr., 2015, A71, 3–8 CrossRef
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. J. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS
- I. A. Guzei, J. Appl. Crystallogr., 2014, 47, 806–809 CrossRef CAS
-
M. Llunell, D. Casanova, J. Cirera, J. M. Bofill, P. Alemany, S. Alvarez, M. Pinsky and D. Avnir, SHAPE, version 2.3, University of Barcelona, Barcelona, Spain, and Hebrew University of Jerusalem, Jerusalem, Israel, 2013 Search PubMed
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS
- R. McLellan, M. A. Palacios, E. K. Brechin and S. J. Dalgarno, ChemPlusChem, 2014, 79, 667 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Contains ORTEP views, details of the refinement procedure, tables with characteristics of coordination bonds, ligand conformations, SHAPE-/BVS- analysis, {Mn4} cluster units parameters, crystal packing details, TGA/DSC traces, IR data for 32-Mn4(dmf)4, 32-Mn4(dmf)2(H2O)2, 32-Mn4(bipy)2, 32-Mn4(phen)2. CCDC 2299403–2299405 and 2299407. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3nj04809h |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |