
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrothermal conversion of cerium oxalate to CeO2: a parade of oxalate and water coordination modes†
Navid
Assi
a,
Christophe
Lahoud
b,
Petr
Brázda
c,
Dominika
Zákutná
a,
Daniel N.
Rainer
d,
Jakub
Hraníček
e,
Jan
Rohlíček
c and
Václav
Tyrpekl
 *a
*a
aDepartment of Inorganic Chemistry, Faculty of Science, Charles University, Hlavova, 2030/8, Prague 12800, Czech Republic. E-mail: vaclav.tyrpekl@natur.cuni.cz
bUniversité Aix-Marseille, Faculty of Science, Site St. Jérôme, 52 Avenue Escadrille Normandie Niemen, Marseille 13013, France
cDepartment of Structure Analysis, Institute of Physics of the Czech Academy of Sciences, Prague 18221, Czech Republic
dDepartment of Physical and Macromolecular Chemistry, Faculty of Science, Charles University, Hlavova, 2030/8, Prague 12800, Czech Republic
eDepartment of Analytical Chemistry, Faculty of Science, Charles University, Hlavova, 2030/8, Prague 12800, Czech Republic
First published on 11th December 2023
Abstract
Hydrothemal conversion of lanthanide/actinide oxalate salts to nanocrystalline oxide has gained technological importance in the chemistry of f-elements. Herein, we present a study describing the mechanism behind the hydrothermal recrystallization and decomposition of cerium (and Ce-Gd) oxalate to oxide. Structural determination of all intermediate nanocrystalline products was possible thanks to three-dimensional electron diffraction. This work reveals the enormous structural abilities of simple oxalate ligands.
Oxalates are low-cost, simple, but essential species in the chemistry of transition metals, lanthanides,1 and actinides.2 Thanks to the low solubility of oxalate salts, oxalic acid is used: as a separation agent of metal ions from aqueous solutions; as a ligand in coordination compounds; in metal–organic frameworks. These salts can be converted readily to nano- and microcrystalline oxides by an appropriate calcination cycle, typically beyond 600 °C.3,4 Recently, Walter et al.5 reported the conversion of uranium(IV) oxalates to oxide under hydrothermal conditions at mild temperatures (160–250 °C). The authors extended their work on the hydrothermal decomposition of various actinide oxalates (Th, U, Np, Pu)6 and their solid solutions.7,8 Typically, an oxalate dispersion in pure water (pH adjusted or in the presence of hydrazine) is loaded into an autoclave and treated at 100–250 °C. This procedure results in agglomerates of AnO2 nanoparticles of approximate size 3 to 10 nm. Later, comprehensive studies were conducted by Manaud et al. on Th(IV)9 and U(IV)10 while changing the main reaction parameters (pH, temperature, and time). There was a clear link between the treatment temperature, pH of the solution, and the crystallite size and morphology of the final product. Recently, Baumann et al.11 compared the hydrothermal and thermal conversion of Pu(IV) oxalate to nanocrystalline PuO2 mentioning most of the positive aspects of the hydrothermal route: lower temperature, shorter reaction time, and lower content of residual carbon. All the above-mentioned works described a drastic change in the morphology of the starting powder during the process. However, tracking of the reaction mechanism is complicated due to the hydrothermal conditions, so determination of the structure of intermediates is complicated due to nanocrystallinity. Therefore, the detailed reaction mechanisms are not known. Few studies have described the hydrothermal treatment of cerium oxalate. Flower-like CeO2 microcrystals were hydrothermally synthesized from an oxalate precursor in an alkaline environment and hydrogen peroxide.11 A similar procedure yielding hierarchical mesoporous ceria was obtained in the presence of some amino acids previously.12 Recently, mixed U1−xCexO2+δ·nH2O (0.1 ≤ x ≤ 0.7) solid solutions were prepared using this method.13
Herein, we focused on the hydrothermal conversion of pure cerium oxalate hydrate to oxide. Ceria has been in the spotlight for many decades, mainly due to its applicability in fuel cells,14 catalysis,15 and Pu surrogates.16 The mechanism of the decomposition and recrystallisation of oxalate (which is responsible for such a drastic change in the morphology of the agglomerates) was studied in detail. This was possible thanks to the newly developed technique of three-dimensional electron diffraction (3D ED), which enabled complete determination of the structure without the need for large crystals.17–22 In addition, we tested the ability of this route to convert mixed Ce1−xGdx(C2O4)3·10H2O solid solutions to Ce1−xGdxO2−x/2 (x = 0.1, 0.2, 0.3).
Pure and mixed oxalates were prepared by heterogeneous precipitation as detailed previously1,23 (a description is in the ESI†). Briefly, for the hydrothermal conversion, 250 mg (0.35 mmol) of Ce2(C2O4)3·10H2O or Ce1−xGdx(C2O4)3·10H2O (x = 0.1, 0.2, 0.3) was placed in a Teflon™-lined autoclave (capacity of 50 mL) together with distilled water (21 mL) or nitric acid solution of the desired pH. Solutions of HClO4 and HCl, as nonoxidizing acids, were also tested instead of HNO3. The mixture was treated at 180–220 °C for ≥ 24 h. The final product was purified by repeated centrifugation and dried at 50 °C under air. All prepared materials were analysed by powder X-ray diffraction (PXRD), scanning electron microscopy (SEM), transmission electron microscopy (TEM), and thermogravimetric analysis (TGA). The sample was treated in pure water (pH = 7, 220 °C, 48 h) and analysed using 3D ED. A detailed description of the analytical techniques is given in the ESI.†
Ce2(C2O4)3·10H2O can be converted completely to oxide in aqueous nitric acid solution (pH = 1) at >180 °C. Optimal conditions were found to be 24 h and 220 °C to yield a powder of grain size 200–500 nm (Fig. 1). The final size of CeO2 crystals increased with the applied temperature (see the micrographs in Fig. S1 and infrared spectra in Fig. S2, ESI†).
 | ||
| Fig. 1 Transmission (top) and scanning (bottom) electron micrographs of CeO2 powder obtained by the hydrothermal decomposition of Ce2(C2O4)3·10H2O in nitric acid solution (pH = 1) at 220 °C for 24 h. | ||
A short study of the hydrothermal conversion of Ce1−xGdx(C2O4)3·10H2O (x = 0.1, 0.2, 0.3) solid solutions showed that the conditions mentioned above (HNO3 solution, pH = 1) were valid for mixed oxides giving homogeneous nanocrystalline Ce1−xGdxO2−x/2 (see Fig. S3 and S4 and PXRD in Fig. S5, ESI†). However, the yield of Gd(III) incorporation in CeO2 was not quantitative and showed some fluctuations (see ICP-MS analyses in Table S1, ESI†). Further studies in this direction are beyond the scope of the current work.
The presence of an oxidizing acid (HNO3) solution was of paramount importance. It facilitated the oxidation of Ce(III) ions to Ce(IV) ions, leading to the oxide. Hydrothermal conversion in HCl or HClO4 (non-oxidizing acids) solutions of pH = 1 did not lead to CeO2 (see the XRD results of samples obtained after 24 h at 220 °C in Fig. S6, ESI†). Different concentrations of nitric acid (pH = 1 to 7) in the autoclave batch led to different degrees of conversion. Fig. 2 shows Ce2(C2O4)3·10H2O treated in solutions of pH 1 to 7 (HNO3). In pure water (pH = 7), recrystallisation and a drastic change in morphology occurred, but decomposition to CeO2 was not reached (Fig. 2, top and middle row). The original cerium oxalate with a flat needle morphology (Fig. S6, ESI†) changed to large rods (from 10 × 10 × 100 μm and larger) with well-developed crystal facets parallel to the elongation direction. Increasing the concentration of nitric acid led to the decomposition of the recrystallized material to oxide (Fig. 2, bottom row). This process was accompanied by full disintegration of the rods to nanocrystals, which sometimes remained assembled to the initial rod shape. A change in agglomerate morphology has been described for all hydrothermally converted oxlates.5–10 This is one of the main differences with thermal conversion, during which the initial oxalate shape is retained.1,3,23,24
 | ||
| Fig. 2 Scanning electron micrographs of cerium oxalate with different degrees of conversion to CeO2. Assemblies of the recrystallised phase with initial Ce2(C2O4)3·10H2O (top), recrystallisation and change of morphology to rods (middle), and disintegration of the rods to nanocrystalline CeO2 (bottom). | ||
Recrystallisation was also studied from the crystallographic viewpoint. The PXRD patterns of selected samples were plotted (Fig. 3). Unfortunately, structure matching from PXRD was possible only for the initial cerium oxalate and final cerium dioxide. Tracking the intermediate phases and resolving their structures was difficult due to the nanocrystallinity of the material and the coexistence of several phases in the samples. This was overcome thanks to 3D ED enabling structural determination of small, unstable, or problematic crystals unsuitable for single-crystal XRD. The structure of the initial oxalate is well known. It crystallizes in the P21/c space group with four Ce(III) ions in the elementary cell, each bonded to three oxalate groups and three water molecules in the primary coordination sphere.25 Two additional water molecules are present in each of the four cavities formed by the oxalate coordination polymer. Therefore, the molecular formula should be written as Ce2(C2O4)3(H2O)6·4H2O, as for some An(III) oxalates with a similar structure.26 There are two formula units within the elementary cell. The recrystallized sample at 220 °C (pH = 7, 24 and 48 h) was selected for structural analysis by 3D ED. This sample showed full recrystallization of the oxalate but low decomposition to oxide (Fig. 3). In total, 114 datasets were indexed. Except for the known initial decahydrate structure (42 indexed datasets), four other structures were observed (72 indexed datasets in total), as shown in Table 1 (full information is given in Table S2, ESI†). The structures were all solved ab initio by the Superflip program.27 The structure present in most of the transformed oxalate hydrates was the orthorhombic P212121 of Ce2(C2O4)3(H2O)4. Isostructural Pr and Nd oxalates have been described in the ICSD database.28,29 The other three structures have not been reported.
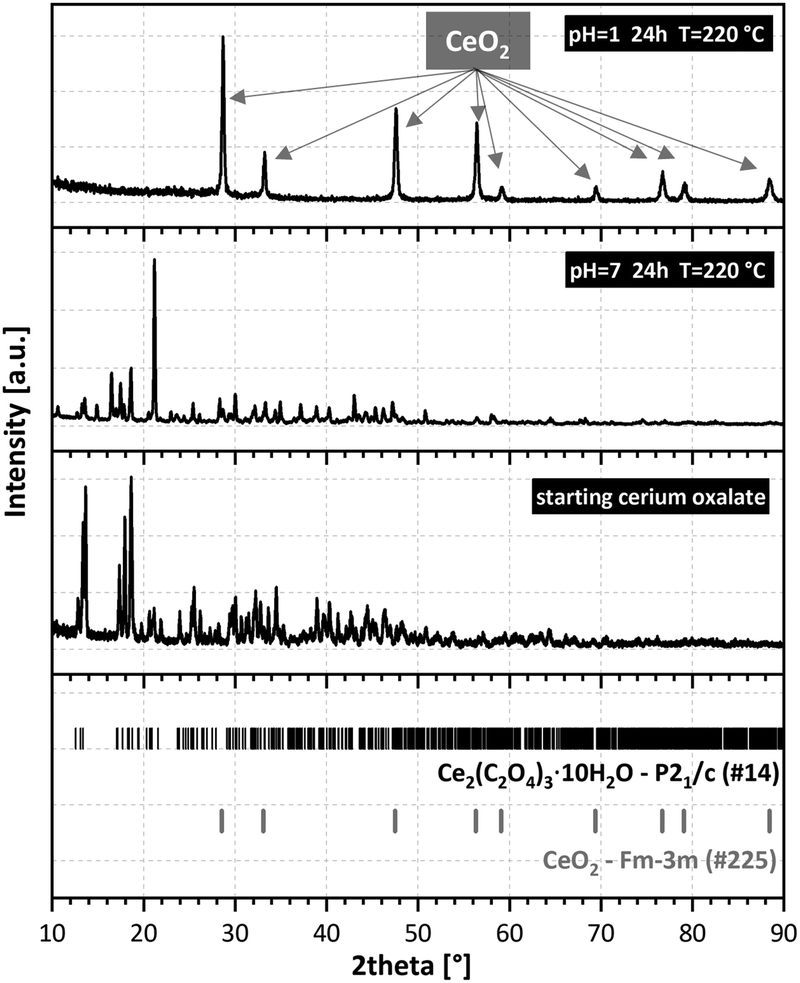 | ||
| Fig. 3 Powder X-ray diffraction of the initial Ce2(C2O4)3·10H2O (bottom), hydrothermally treated (middle), and final CeO2 (top). | ||
| Stoichiometric formula | Ce2(C2O4)3(H2O)4 | Ce2(C2O4)3(H2O)4·H2O | Ce2(C2O4)3(H2O)4·H2O | Ce2(C2O4)3(H2O)4·H2O |
| Occurrence (% of cases) | 40 | 39 | 14 | 7 |
| Crystal system | Orthorhombic | Triclinic | Orthorhombic | Monoclinic |
| Space group | P212121 |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
Pbcn | P21/n |
| a, b, c (Å) | 8.64(1), 9.59(1), 16.87(1) | 10.50(1), 11.77(1), 12.97(1) | 22.06(8), 12.96(1), 10.61(1) | 21.67(1), 10.51(1), 12.97(1) |
| α, β, γ (°) | 90, 90, 90 | 92.91(4), 90.58(4), 112.95(4) | 90, 90, 90 | 90, 93.58(4), 90 |
| Z | 4 | 4 | 8 | 8 |
The second most abundant phase was a structure with triclinic P symmetry. If all the water molecules were fully occupied, then the structure was pentahydrate Ce2(C2O4)3(H2O)4·H2O (see the detailed discussion of the level of hydration in the ESI†). Four water molecules in the formula unit were coordinated to cerium ions and the fifth one resided in a pore. Two other phases, orthorhombic Pbcn and monoclinic P21/n, were significantly less abundant and much more disordered judging from the observed diffuse scattering. Both phases were pentahydrate, with four water molecules coordinated to two independent cerium atoms. The fifth water molecule was in a cavity. Hydrolysis or formation of hydroxo-complexes were not observed until the final oxalate decomposition. Probably a higher pH would be required to observe such behavior, as has been observed with Nd.30 The results of 3D ED were supported by PXRD Rietveld analysis of the sample treated at 220 °C for 48 h (Fig. S7, ESI†). The best fit was obtained using the P212121 and P phases having the highest abundance in the sample.
Evidently, the four structures had common structural motives worth mentioning. In all structures, oxalate ions adopted bridging coordination modes (Fig. S8, ESI†) to connect cerium ions with coordination modes 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, 1:2 + 1 and 1:2 + 2 (Fig. S8, top from left, ESI†), extending the list of oxalate coordination modes for rare-earth ions.31 The oxalates in the P212121 structure formed infinite chains along two nearly perpendicular directions, which created a “wine rack” structural motif (Fig. 4, left). Higher coordination numbers in the Pbcn, P and P21/n structures were probably caused by a specific organization of the infinite chains of oxalates with the 1:2 coordination mode solely, all aligned in the same direction. Layers of oxalates with higher denticity were then perpendicular to these polymer chains (Fig. 4, right), here 1:2 and 1:2 + 2 modes were present.
:2, 1:2 + 1 and 1:2 + 2 (Fig. S8, top from left, ESI†), extending the list of oxalate coordination modes for rare-earth ions.31 The oxalates in the P212121 structure formed infinite chains along two nearly perpendicular directions, which created a “wine rack” structural motif (Fig. 4, left). Higher coordination numbers in the Pbcn, P and P21/n structures were probably caused by a specific organization of the infinite chains of oxalates with the 1:2 coordination mode solely, all aligned in the same direction. Layers of oxalates with higher denticity were then perpendicular to these polymer chains (Fig. 4, right), here 1:2 and 1:2 + 2 modes were present.
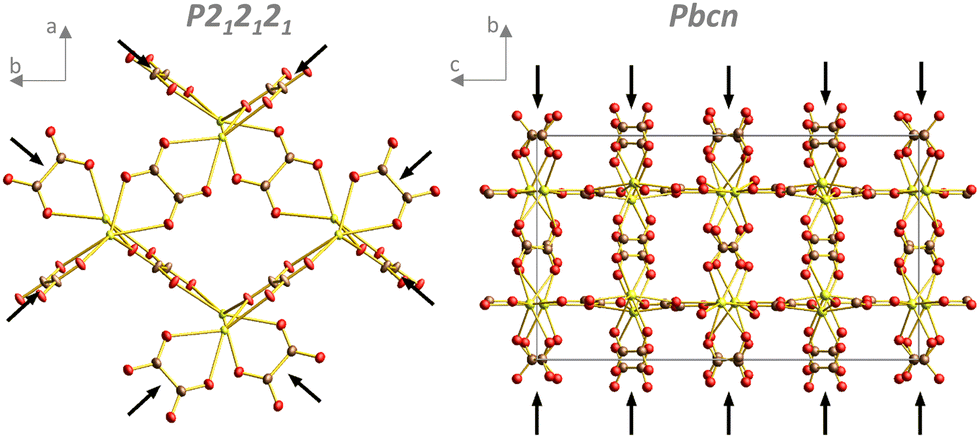 | ||
| Fig. 4 Oxalates in the P212121 structure form infinite chains running along two nearly perpendicular directions (black arrows; the other oxalate molecule and coordinated water molecules have been omitted for clarity). Pbcn structure with infinite oxalate chains running only along one direction marked by black arrows (coordinated water molecules and water in pores have been omitted for clarity). | ||
Six cerium coordination polyhedra were found in the four structures. Type 1 comprised the cerium ion coordinated by nine ligands. Two water molecules were in the cis conformation, with one monodentate oxalate and three bidentate oxalates (Fig. S9, ESI†). Types 2, 3, and 4 are shown in Fig. S10 (ESI†). Coordination was achieved by 10 ligands, two water molecules in trans positions, and two oxalates in the perpendicular direction forming infinite chains. There were two bidentate oxalates in the other perpendicular direction. Type 5 revealed sole coordination by five oxalates (nine oxalate oxygens (Fig. S11, ESI†)), four bidentates and one monodentate. Type 6 involved non-participation in the infinite chains and, except for three bidentate oxalates, it was coordinated to four water molecules (Fig. S12, ESI†). For more details, see the ESI.†
As mentioned above, in the Pbcn, P21/n, and P structures, there were oxalate layers perpendicular to the direction of the infinite oxalate chains. The structural motifs in these layers were almost identical for Pbcn and P21/n phases (Fig. 5, left and middle). The difference was that four water molecules were in the layer in the Pbcn structure, but an oxalate replaced it in the P21/n phase. The four coordinated molecules in Pbcn pointed outwards from the layer in the direction of the infinite oxalate chains and were coordinated to one cerium atom (type 4). In the P structure, the layer motif was slightly simpler (Fig. 5, right).
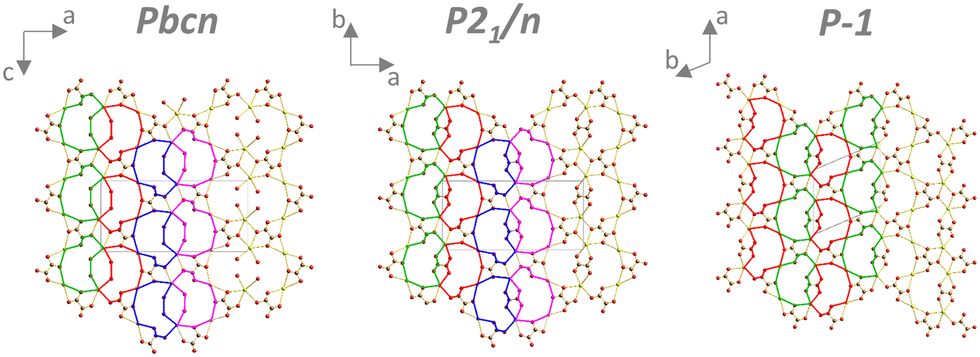 | ||
| Fig. 5 Structural motifs of oxalate layers perpendicular to the infinite coordination polymer chains. Coordinated water molecules (except those forming the oxalate layer and water in pores) have been omitted for clarity. | ||
The global water content in the sample treated at 220 °C for 24 h was measured by thermogravimetry, and Ce2(C2O4)3·3.5H2O is shown in Fig. S13 (ESI†). From the crystallographic analysis considering two main phases, the composition was estimated to be Ce2(C2O4)3·4.5H2O (i.e., one water molecule more than determined by thermogravimetry). This was probably caused by lower occupancy of water molecules coordinated to cerium (type 4) and those within the pores (deeper discussion is provided in the ESI†).
Similar to that in,32,33 hydrothermal crystallization was typically not a single-step process. Here, we reported the mechanism of hydrothermal conversion of Ce2(C2O4)3·10H2O to oxide. The whole mechanism is expressed graphically in Fig. 6.
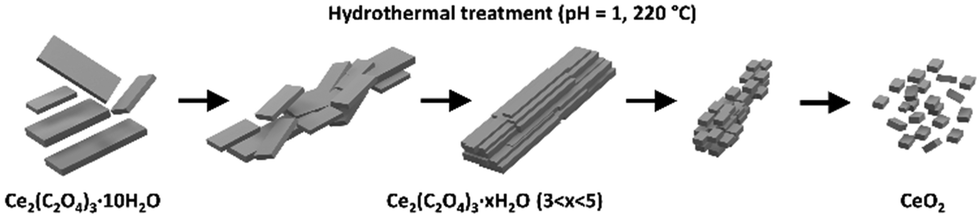 | ||
| Fig. 6 Graphical illustration of the mechanism of hydrothermal conversion of Ce2(C2O4)3·10H2O to CeO2. | ||
Hydrothermal dehydration and recrystallisation of oxalate take place prior to the conversion to oxide, which is responsible for the enormous change in morphology during this process. 3D ED was crucial for structural determination of nanocrystalline samples. The loss of water was linked with an increase in the denticity and coordination mode of the oxalate ligand, and elicited several structurally analogous phases. The change in the oxidation state of cerium, together with oxalate decomposition, seemed to be the final one-step process visible as a disintegration of the recrystallised phases to oxide. In addition to structural investigations, we tested the possibility of hydrothermal conversion of Ce-Gd oxalates to mixed oxides.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This study was funded by the Czech Science Foundation (GAČR) (project 20-20936Y). P.B. and J. R. acknowledge GAČR (project 21-05926X), the CzechNanoLab Research Infrastructure (MEYS CR, LM2018110) and the Operational Programme Research, Development and Education by the EU ESIF and MEYS CR (SOLID21 CZ.02.1.01/0.0/0.0/16_019/0000760).Notes and references
- A. Alemayehu, D. Zákutná, S. Kohúteková and V. Tyrpekl, J. Am. Ceram. Soc., 2022, 105, 4621 CrossRef CAS.
- F. Abraham, B. Arab-Chapelet, M. Rivenet, C. Tamain and S. Grandjean, Coord. Chem. Rev., 2014, 266–267, 28 CrossRef CAS.
- V. Tyrpekl, J.-F. Vigier, D. Manara, T. Wiss, O. Dieste Blanco and J. Somers, Low temperature decomposition of U(IV) and Th(IV) oxalates to nanograined oxide powders, J. Nucl. Mater, 2015, 460, 200 CrossRef CAS.
- L. De Almeida, S. Grandjean, N. Vigier and F. Patisson, Eur. J. Inorg. Chem., 2012, 4986 CrossRef CAS.
- O. Walter, K. Popa and O. Dieste Blanco, Open Chem., 2016, 14, 170 CAS.
- K. Popa, O. Walter, O. Dieste Blanco, A. Guiot, D. Bouëxière, J. Y. Colle, L. Martel, M. Naji and D. Manara, CrystEngComm, 2018, 20, 4614 RSC.
- L. Balice, D. Bouëxière, M. Cologna, A. Cambriani, J. F. Vigier, E. De Bona, G. D. Sorarù, C. Kübel, O. Walter and K. Popa, J. Nucl. Mater., 2018, 498, 307 CrossRef CAS.
- V. Baumann, K. Popa, O. Walter, M. Rivenet, G. Senentz, B. Morel and R. J. M. Konings, Nanomaterials, 2023, 13, 340 CrossRef CAS PubMed.
- J. Manaud, J. Maynadié, A. Mesbah, M. O. J. Y. Hunault, P. M. Martin, M. Zunino, N. Dacheux and N. Clavier, Inorg. Chem., 2020, 59, 14954 CrossRef CAS PubMed.
- J. Manaud, J. Maynadié, A. Mesbah, M. O. J. Y. Hunault, P. M. Martin, M. Zunino, D. Meyer, N. Dacheux and N. Clavier, Inorg. Chem., 2020, 59, 3260 CrossRef CAS PubMed.
- W. Liu, L. Feng, C. Zhang, H. Yang, J. Guo, X. Liu, X. Zhang and Y. Yang, J. Mater. Chem. A, 2013, 1, 6942 RSC.
- G. Zhang, Z. Shen, M. Liu, C. Guo, P. Sun., Z. Yuan, B. Li, D. Ding and T. Chen, J. Phys. Chem. B, 2006, 110, 25782 CrossRef CAS PubMed.
- S. Benarib, N. Dacheux, X. F. Le Goff, J. Lautru, L. Di Mascio and N. Clavier, Dalton Trans., 2023, 52, 10951–10968 RSC.
- H. Inaba and H. Tagawa, Solid State Ionics, 1996, 83, 1 CrossRef CAS.
- T. Montini, M. Melchionna, M. Monai and P. Fornasiero, Chem. Rev., 2016, 116, 5987 CrossRef CAS PubMed.
- K. Choi, W. Tong, R. D. Maiani, D. E. Burkes and Z. A. Munir, J. Nucl. Mater., 2010, 404, 210 CrossRef CAS.
- L. Palatinus, P. Brázda, M. Jelínek, J. Hrdá, G. Steciuk and M. Klementová, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2019, 75, 512 CrossRef CAS PubMed.
- P. Brázda, L. Palatinus and M. Babor, Science, 2019, 364, 667 CrossRef PubMed.
- P. Brázda, M. Klementová, Y. Krysiak and L. Palatinus, IUCrJ, 2022, 9, 735 CrossRef PubMed.
- P. B. Klar, Y. Krysiak, H. Xu, G. Steciuk, J. Cho, X. Zou and L. Palatinus, Nat. Chem., 2023, 15, 848–855 CrossRef CAS PubMed.
- L. Palatinus, V. Petříček and C. A. Correa, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2015, A71, 235 CrossRef PubMed.
- V. Petříček, L. Palatinus, J. Plášil and M. Dušek, Z. Kristallogr., 2023, 238, 271 Search PubMed.
- V. Tyrpekl, P. Markova, M. Dopita, P. Brázda and M. A. Vacca, Inorg. Chem., 2019, 58, 10111 CrossRef CAS PubMed.
- R. Podor, N. Clavier, J. Ravaux, L. Claperde and N. Dacheux, J. Amer. Ceram. Soc., 2012, 95, 3683 CrossRef CAS.
- Dicerium trioxalate decahydrate, ICSD: 109637, Inorganic Crystal Structure Database, https://www2.fiz-karlsruhe.de/icsd_home.html, January 2018.
- C. Tamain, B. Arab-Chapelet, M. Rivenet., X. F. Legoff, G. Loubert, S. Grandjean and F. Abraham, Inorg. Chem., 2016, 55, 51 CrossRef CAS PubMed.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786 CrossRef CAS.
- J. C. Trombe and J. Jaud, J. Chem. Crystallogr., 2003, 33, 19 CrossRef CAS.
- C. J. Hao and H. Xie, Acta Cryst., 2012, E68, m444 Search PubMed.
- A. Mer, M. Rivenet, L. De Almeida, S. Granjean and F. Abraham, Inorg. Chem. Commun., 2013, 31, 90 CrossRef CAS.
- M. Hernández-Molina, P. A. Lorenzo-Luis and C. Ruiz-Pérez, CrystEngComm, 2001, 16, 1 Search PubMed.
- L. Wang, R. Zhao, C. Wang, L. Yuan, Z. Gu., C. Xiao, S. Wang, X. Wang, Y. Zhao, Z. Chai and W. Shi, Chem. – Eur. J., 2014, 20, 12655 CrossRef CAS PubMed.
- L. Wang, R. Zhao, X. Wang, L. Mei, L. Yuan, S. Wang, Z. Chai and W. Shi, CrystEngComm, 2014, 16, 10469 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2296196–2296199. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3nj04635d |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |