
Optimal synthesis conditions for NBF-modified 8,13-dihydroberberine derivatives†
A. D.
Zagrebaev
 *a,
V. V.
Butova
b,
A. A.
Guda
*a,
S. V.
Chapek
a,
O. N.
Burov
c,
S. V.
Kurbatov
c,
E. Yu.
Vinyukova
d,
M. E.
Neganova
d,
Yu. R.
Aleksandrova
d,
N. S.
Nikolaeva
d,
O. P.
Demidov
e and
A. V.
Soldatov
a
*a,
V. V.
Butova
b,
A. A.
Guda
*a,
S. V.
Chapek
a,
O. N.
Burov
c,
S. V.
Kurbatov
c,
E. Yu.
Vinyukova
d,
M. E.
Neganova
d,
Yu. R.
Aleksandrova
d,
N. S.
Nikolaeva
d,
O. P.
Demidov
e and
A. V.
Soldatov
a
aThe Smart Materials Research Institute, Southern Federal University, 344090 Rostov-on-Don, Russia. E-mail: guda@sfedu.ru
bInstitute of General and Inorganic Chemistry Bulgarian Academy of Sciences, Sofia, Bulgaria
cFaculty of Chemistry, Southern Federal University, 344090 Rostov-on-Don, Russia
dInstitute of Physiologically Active Compounds at Federal Research Center of Problems of Chemical Physics and Medicinal Chemistry, Russian Academy of Sciences, Severnij Pr. 1, Chernogolovka 142432, Russia
eDepartment of Chemistry, North Caucasus Federal University, Pushkin st. 1, Stavropol, 355009, Russia
First published on 5th December 2023
Abstract
Berberine is a natural alkaloid with a broad spectrum of biological activity. Derivatives of berberine with reduced structural stability are particularly promising for pharmacological applications. However, synthesizing disubstituted berberines poses a challenging task. The order and rate of reagent addition, mixing rate and trace amounts of acids in the atmosphere affect the purity and degradation. In this study, we developed a flow synthesis of 8,13-disubstituted berberines, which are particularly difficult to obtain in the batch. The reaction was performed in a 3D printed microfluidic chip, incorporating modules for rapid reagent mixing, optical diagnostics and a delay line to regulate the overall reaction time. The microfluidic flow system allowed us to synthesize hybrid berberine derivatives with an NBF fragment at the C-13 position, resulting in up to a 30% increase in product yields compared to classical batch synthesis. These substances exhibited high antioxidant activity without toxic effects on cell cultures, thus being potential candidates for novel pharmacological agents. The microfluidic approach, coupled with UV-Vis diagnostics, is a promising tool for optimizing and screening synthesis conditions of alkaloid derivatives that are challenging to obtain using conventional methods.
1. Introduction
Protoberberines represent an extensive class of tetracyclic natural alkaloids, that are often found in the roots of the plant families Rutaceae, Ranunculaceae, Papaveraceae, and Berberidaceae.1 Numerous studies have demonstrated the effectiveness of berberine (1) against metabolic syndrome,2 including diabetes,3–6 hypercholesterolemia,7 obesity,8 and hepatic steatosis.6 It has been shown to be effective against bacterial,9–11 viral,12–15 and fungal16 infections. Berberine and its derivatives exhibit a notable affinity for interacting with canonical and non-canonical DNA structures, such as G-quadruplexes.17–21 Berberine derivatives demonstrate significant tropism towards mitochondrial membranes, making them promising anti-tumor agents.22,23 Several recent reviews address the applications and functionalization methods of berberine.24–29Despite the diverse biological activity of berberine (1), low toxicity,11,30 and minimal side effects, protoberberine alkaloids alone have not been widely adopted in medicine. However, the combination of its unique properties make berberine a promising scaffold for versatile modifications and the development of new pharmaceutical compounds.31
Nucleophilic addition at the C-ring or nucleophilic substitution of the methoxy group in the D-ring are commonly employed for structural modification of berberine. Successful approaches, including those developed by our group,32–36 involve umpolung strategies.37 These approaches enable the reduction of the C-ring, followed by the use of electrophilic reagents to attack the C and D rings10,29,38–41 (Scheme 1).
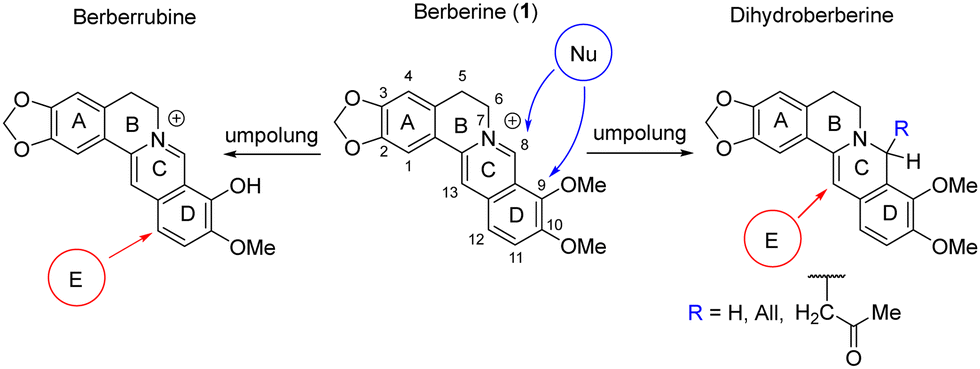 | ||
| Scheme 1 Umpolung approach for the structural modification of berberine. | ||
We have demonstrated the possibility of obtaining 13-substituted acetonyl- and dihydroberberine derivatives of types 2a–3a through their interaction with corresponding chloronitroaryls (including NBF-Cl).33,36 NMR spectroscopy indicated that these deeply green-colored compounds undergo intramolecular charge transfer, as reflected in the resonance structures 2b–3b (Scheme 2). According to quantum chemical calculations, the charge transfer from the berberine fragment to the nitroaryl moiety was found to be 0.30ē for dihydroberberine 2a and 0.27ē for 8-acetonyl-dihydroberberine 3a.33,36
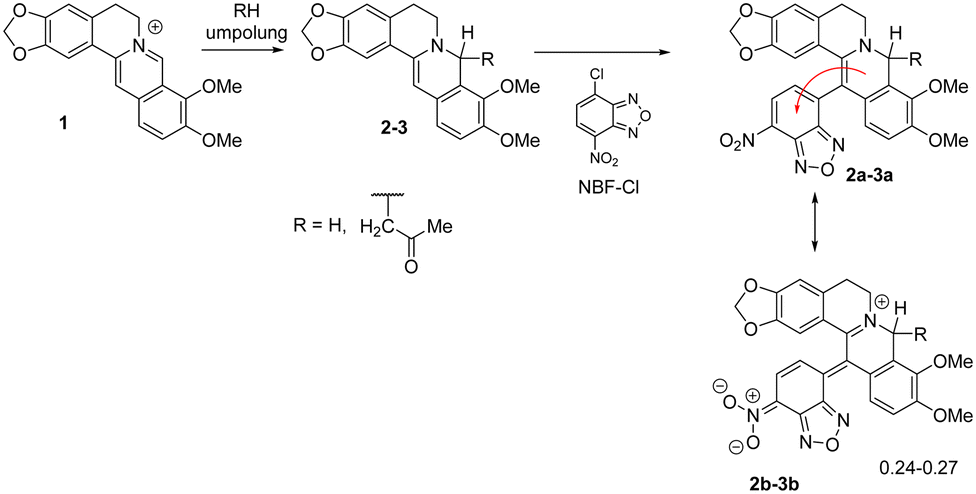 | ||
| Scheme 2 Zwitterionic disubstituted berberine derivatives. | ||
The excess electronic density on the NBF fragment, combined with the known propensity of 8,13-dihydroberberines to undergo oxidation,24–29,38 suggests the antioxidant properties of such compounds. However, their stable and reproducible synthesis was hindered by two main challenges. First, our earlier studies revealed that the yield and purity of the formed products dramatically depend on the order and rate of reagent addition to the reaction mixture, and their mixing efficiency. Hence, a key issue might be in the hindered mass transfer of reagents within the reaction mass, leading to a significant increase in the side reaction formation of products. Second, due to the intense coloration of reaction products, it is difficult to quantify the optimal reaction time. Both reagents and reaction products degrade rapidly on common chromatographic plates. A suboptimal reaction time can result in hydrolysis and oxidation of both reagents and reaction products.
Microfluidic flow systems are known to offer a promising alternative to traditional bulk synthesis,42,43 and they address the challenges described above. Thin capillaries are optimal for the rapid changes in reaction environment upon synthesis,44–47 along with effective heat and mass transfer and online monitoring at various stages of the reaction.48–51 This study aims to develop a microfluidic approach for synthesizing and online UV-Vis monitoring 8,13-disubstituted berberine derivatives and to investigate their antioxidant activity in vitro.
Current methods to produce microfluidic devices involve micromachining, hot embossing, etching and molding.52 However, these processes can be time consuming, imprecise, expensive or challenging to produce complex channel topologies. Furthermore, some of these processes require facilities such as clean rooms to ensure defect-free devices, and these are often expensive to build and maintain. Additive manufacturing offers superior accessibility and reliability. One of the most robust and versatile 3D printing techniques is digital light processing (DLP).53–55 The benefits of using 3D printed microfluidic devices for synthesis have been demonstrated in recent reviews.56,57 Van der Linden et al. demonstrated that DLP technology can produce complex 3D microchannel topologies that are difficult or nearly impossible using conventional fabrication methods.58 Bressan et al. reported a simple, inexpensive and efficient microfluidic device fabricated by a hybrid PDMS and 3D printing technique. The device is suitable for the flow synthesis of Ag and Au nanoparticles.59 Camarillo-Escobedo et al. developed a 3D-printed microfluidic device with integrated spectrophotometer and wireless connectivity and demonstrated its application for the analysis of fluoride content in ground waters.60 Guda et al. demonstrated 3D printed module devices for gold nanoparticle synthesis.61
Microfluidic technologies have gained significant popularity in the field of organic synthesis and catalysis.62 The production of various o-disubstituted benzenes in a continuous flow regime,62 screening applications of diazomethane,63 and anaerobic catalytic oxidation of stilbenes64 are examples. Small volumes of reagents inside microfluidic channels are promising for optimization tasks. In 2010, vitamin D3 was synthesized in a continuous-flow microfluidic reactor,65 and in 2020, Santana et al. provided optimized methods for the synthesis of several pharmaceutical compounds.66 To enhance the anticancer activity, Farahani et al. optimized flows and reagent concentrations in a microfluidic system to obtain chitosan nanoparticles loaded with berberine (1).67 The authors achieved a nanoparticle loading efficiency of up to 55% and demonstrated that chitosan nanoparticles loaded with berberine (1) exhibited an antiproliferative effect compared to the original berberine (1).
In this study, we optimized conditions for the microfluidic synthesis of 8,13-disubstituted berberine derivatives. A microfluidic reactor integrated a passive pearl mixer for the stoichiometric combination of reagents, along with the diagnostic ports for connecting optical fibers. The transparent surface of the chip enabled reaction kinetics observation through sharp color changes while quantitative analysis was based on UV-Vis spectra measured at various distances from the mixing point.
The combination of microfluidic techniques with previously developed batch approaches for synthesizing 8,13-disubstituted berberine derivatives allowed for a quantitative investigation of the reaction arylation kinetics and significantly increased the yield of the desired products. These results can further be applied to the synthesis of various 8,13-disubstituted berberine derivatives.
2. Results and discussion
2.1 Synthesis optimization for 8,13-disubstituted berberine derivatives
We chose the nitrobenzofurazanyl moiety as the main pharmacophore fragment for the synthesis of new 8,13-disubstituted berberines. Scheme 3 shows two stages of the reaction for obtaining products 4a–11a, involving nucleophilic addition of acetophenone to form intermediates 4–11 and subsequent electrophilic substitution of hydrogen at the C-13 position with the pharmacophore to form 4a–11a. The first phase of the reaction was carried out using a methodology previously developed by us without modifications,68 while the second stage of the process was the key focus of this study. It is worth noting that prior to our research, only 8-acetonyl, 8-alkyl, and 8-allyl-dihydroberberines (Scheme 1) were used for berberine modification. The use of phenacyls enhances the stability and preparative isolability of both activated intermediates 4–11 and the target products 4a–11a.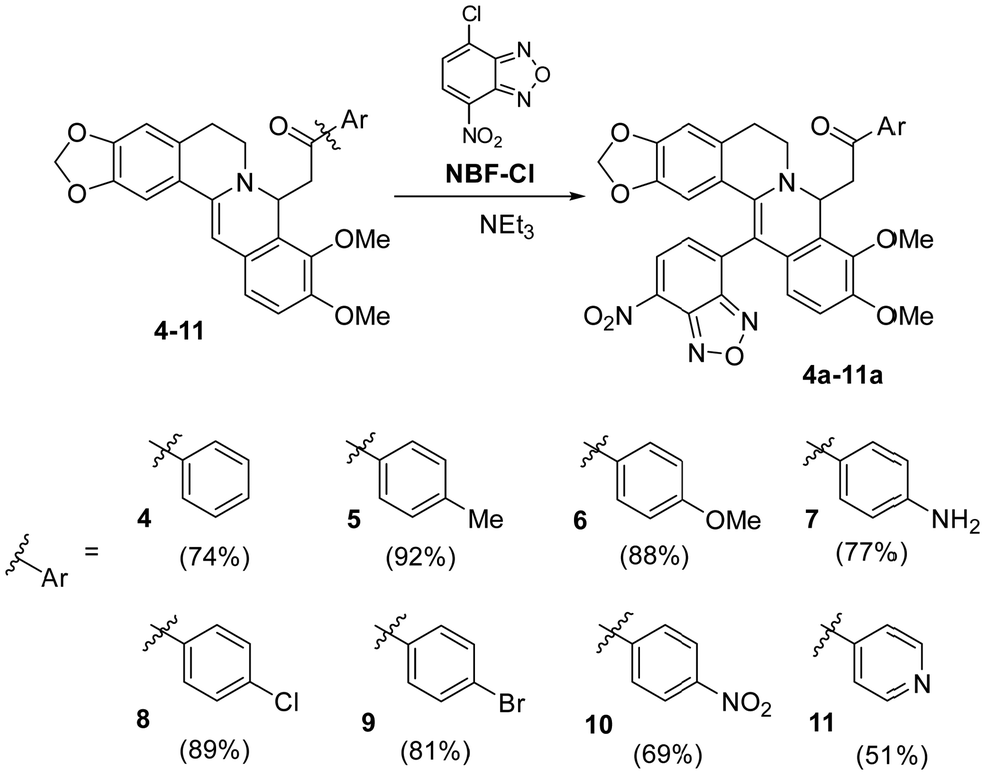 | ||
| Scheme 3 General scheme for the synthesis of derivatives 4a–11a. | ||
We observed that the yield of the target product depends on the rate and sequence of adding reagents to the reaction medium. If the NBF-Cl solution is added to the solution of substituted dihydroberberines 4–11, the product is formed with moderate yield. However, in the reverse approach when the berberine derivatives 4–11 were added to the NBF-Cl solution, the initial excess of NBF-Cl leads to the destruction of 8-substituted dihydroberberines 4–11, reverting them to the starting berberine (1). Optimal yields are achieved when the solutions of compounds 4–11 and NBF-Cl are uniformly mixed in a reaction vessel.
To achieve a stoichiometric ratio of reactants during the reaction and reduce contact with the surrounding environment, the synthesis method was integrated into a microfluidic flow system. Solutions from syringes no. 1, no. 2, and no. 3 were delivered by respective pumps and entered into a pearl mixer with 400 μm capillaries for complete mixing. The rate of delivery for each reagent was set at 1 μL s−1. After 8 min in the flow system the solution was kept in a vial for an additional 10 minutes before isolation of the target reaction products (see the Methods section for more details). It is interesting to note that, unlike batch synthesis, chip-based synthesis allowed pure target compounds to be obtained without the need for chromatographic purification methods. Table 1 presents the results of synthesis under conventional and microfluidic conditions (the reactions were carried out under the optimal conditions determined during the study. For more details, please refer to Table S1 in the appendix, ESI†). The reaction yields under microfluidic conditions exceed those in batch synthesis.
| Compound | 4a | 5a | 6a | 7a | 8a | 9a | 10a | 11a |
|---|---|---|---|---|---|---|---|---|
| a The experiment was conducted at ambient temperature, with reagent feed rates in each channel 1 μL s−1, and the total residence time of 11 minutes adjusted by delay line. The Reynolds number (Re) of liquid flow in the chip was ca 0.01 and was determined by a liquid density of 0.95 kg m−3; flow speed of 15 mm s−1; dynamic viscosity of 0.92 Pa s; characteristic scale of 0.45 mm. | ||||||||
| Yield, % (batch) | 54 | 59 | 72 | 38 | 62 | 66 | 65 | 40 |
| Yielda, % (microfluidic) | 74 | 92 | 88 | 77 | 89 | 81 | 69 | 51 |
2.2 In situ investigation of the hydrogen substitution reaction at C-13 of dihydroberberines
The common architecture of the pearl mixer was further extended with ports for an optical fiber as shown in Fig. 1. The transparent cured resin allowed direct spectral registration through the microfluidic chip walls without direct contact of the fiber with the reaction medium. Moreover, the fully printed UV cell enabled rational optimization of the optical path and decreased the volume of the transmission cells to only 0.62 μL for online spectral monitoring. Details of the geometrical parameters of the integrated flow cells are shown in Fig. 1.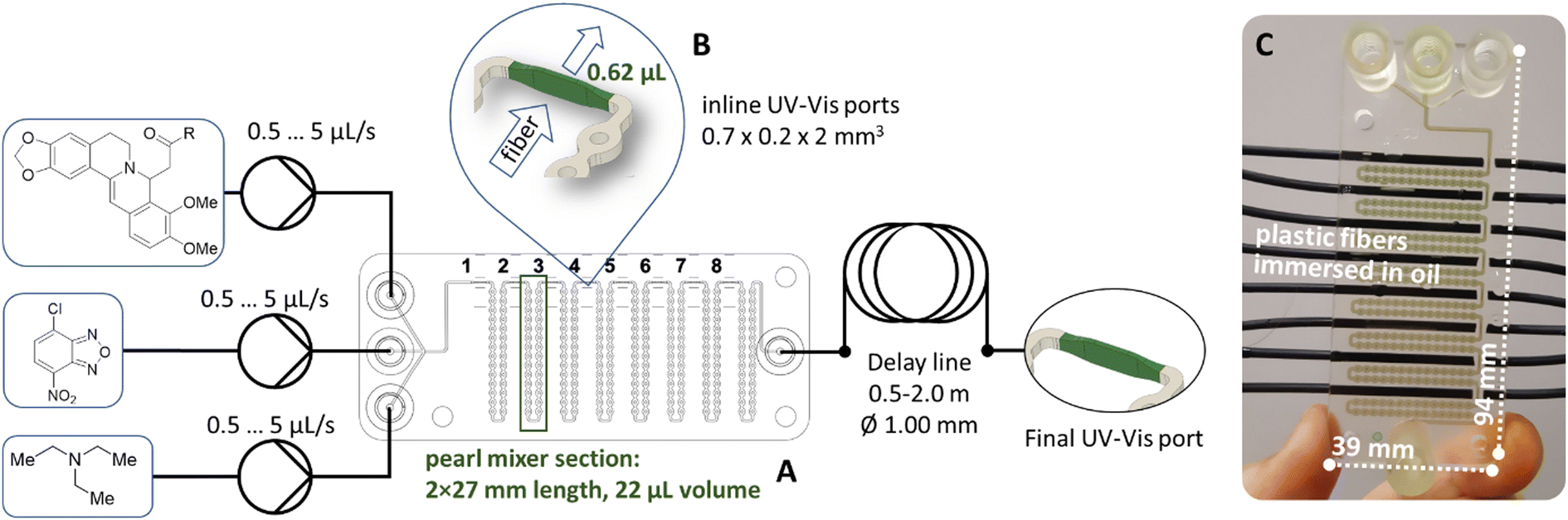 | ||
| Fig. 1 Scheme of the reaction setup. (A) Scheme of the 3D printed pearl mixer chip 94 × 39 × 4.5 mm with integrated ports with a 0.7 × 2 mm2 cross section and 0.2 mm optical pass for UV-Vis control. (B) Scheme of the UV-Vis flow cell integrated inside the pearl mixer. The height of the window for transmission is 850 μm, and optical path is 200 μm. (C) Photo of the microfluidic chip with installed optical fibers inside ports 1–8. | ||
The initial stages of the reaction were detected at ports 1–8 (Fig. 1), and for longer reaction times, a delay line with a length of 0.5 to 2 meters was installed after the chip. This delay line allowed us to extend the reaction time up to 10 minutes before recording the spectrum.
The use of UV-Vis detection was favored due to the dramatic color change of the reaction mixture during the first seconds after mixing. As shown in Fig. 2(a), the reagents alone are almost transparent in the range of 550–850 nm. Therefore, the optimal feature for detecting the product is the absorbance at 720 nm. The substituted berberine exhibits high optical density, which necessitated the use of a short optical path length for measurements. An optimal channel width of 200 μm was chosen in the point of spectrum registration in the 3D printed channel, enabling the registration of signals from both the reactants and products for a series of tested compounds in the range of optical density values A = 0.5…1.5.
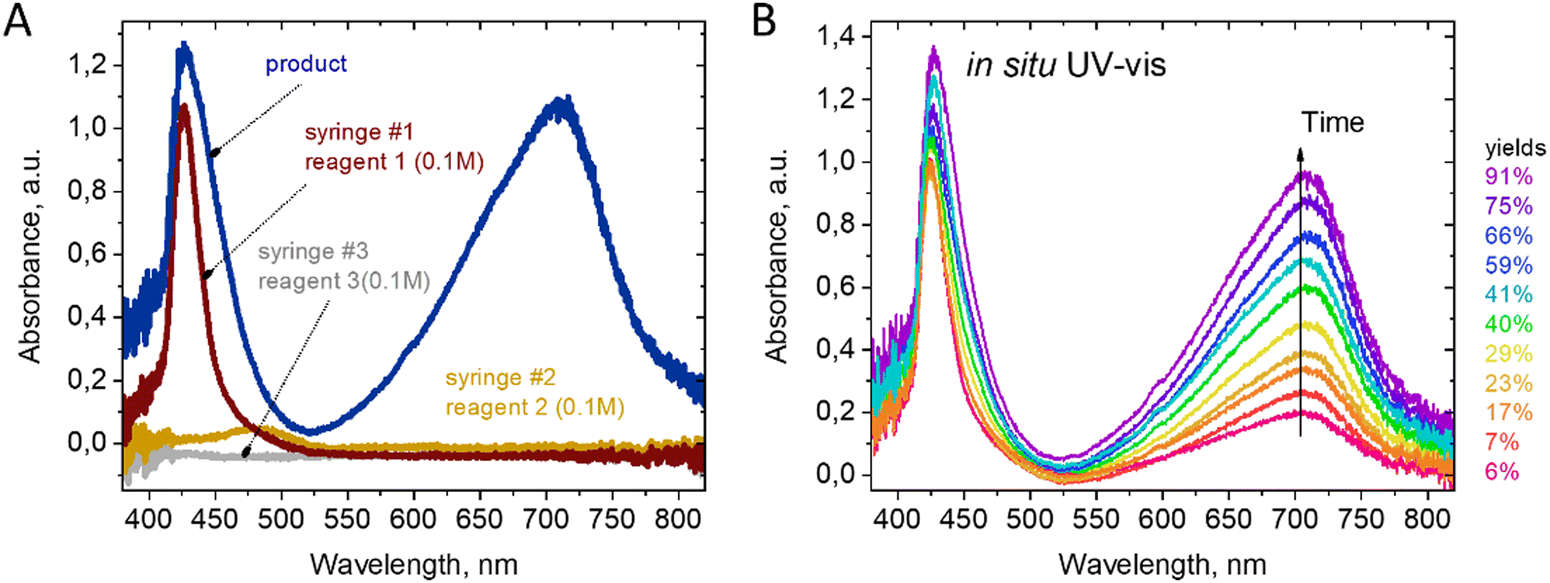 | ||
| Fig. 2 UV-Vis monitoring of the synthesis process of derivative 5a. (A) Spectra of the pure solution reactants and the product 5a. (B) Changes in optical absorption during the synthesis, measured in the microfluidic chip at different time delays after the moment of reagent mixing. | ||
Reaction kinetics studies were performed for three disubstituted berberines: 4a, 5a, and 10a, as these structures contain both donor and acceptor substituents in the p-position of the acetylphenone substituent. A series of spectra from ports 1–8 (Fig. 1) and remote port 9 after the delay line were obtained for each compound. Before each new synthesis the chip was cleaned and the UV-Vis cells by flushing with pure DMF solution (50 times the volume of chip) to remove traces of product and reagents from the channel surface. The reaction time was evaluated from the known flow speed and capillary volumes between ports. A Teflon tube with lengths of 0.5, 1.0, 1.5, and 2.0 m and an internal diameter of 1.0 mm was used as the external delay line to achieve longer reaction times in a flow regime. Variation of the reaction time was achieved by varying the rate of reagent delivery in the range of 0.5–5 μL s−1. The use of a delay line instead of further decreasing the flow speed maintained good mixing conditions inside the pearl mixer. This approach allowed us to achieve a range of time delays from 0.5 to 600 s. The kinetics study results of product formation are demonstrated in Fig. 3.
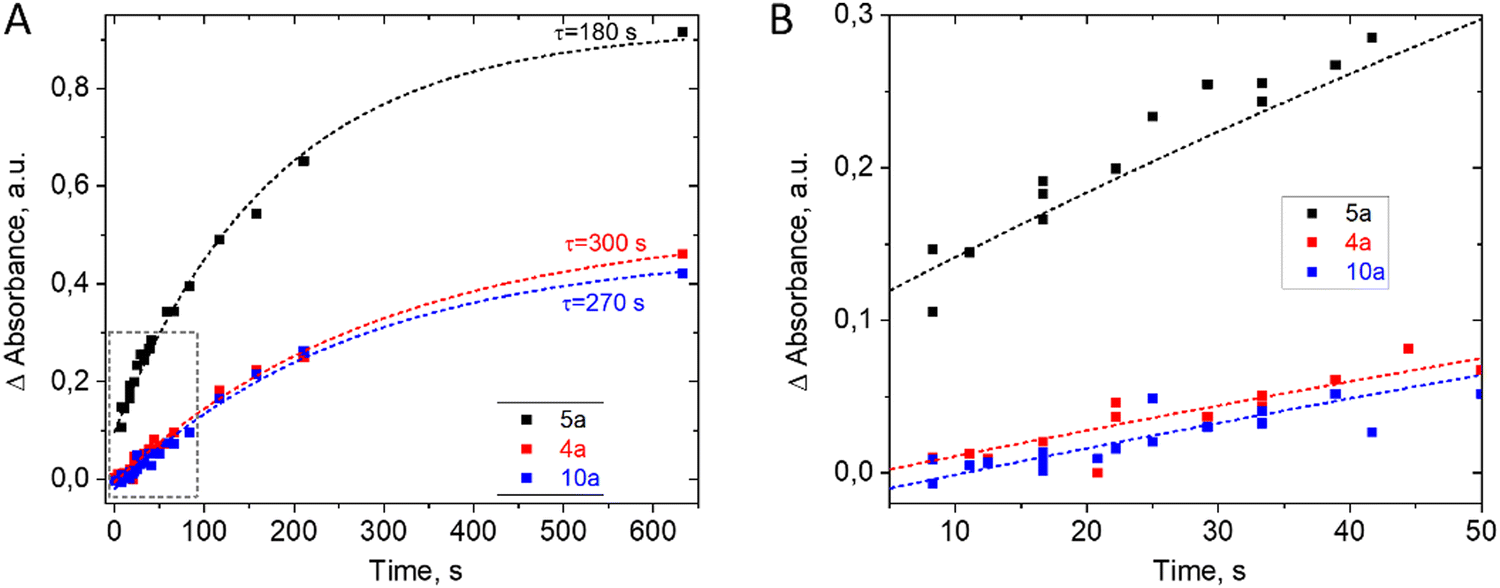 | ||
| Fig. 3 Changes in optical density of the reaction mixtures at 720 nm during the synthesis of derivatives 4a, 5a, and 10a, obtained for different time delays from the moment of reagent mixing in the microfluidic chip. Dashed lines show the fitting of the data with an exponential function of the form y = y0 + A·exp(−t/τ). The time delay was adjusted via syringe pumps and the length of delay line. The variation of Reynolds numbers in all experiments was in the range Re = 0.07…0.35. | ||
The kinetics study revealed that the time to reach equilibrium in the system exceeds 10 minutes. The presence of a donating group in the aryl substituent of dihydroberberine (4) should promote the preferential electrophilic substitution process at the C-13 position. This hypothesis is confirmed experimentally, and the highest reaction rate was observed for compound 5a, while compounds 4a and 10a exhibited similar values of the time constant. The slower reaction rate in derivatives 4a and 10a may be explained by the existence of electron-accepting fragments in the methyl–phenyl ketone group but further studies are needed for quantitative explanation.
The presence of a base in the reaction medium plays a crucial role in the berberine derivatives synthesis. Previous studies33,36 have shown that adding a base to the reaction mixture is a necessary condition for increasing the yield of pure compounds during isolation and crystallization. To investigate the influence of triethylamine (TEA) on the reaction process, we conducted a screening study by varying the reagents flow rates and TEA at the mixer inlet. We fixed the reaction time by maintaining a total flow rate of 4 μL s−1, resulting in a decrease in the flow rate of berberine derivative (5a) with NBF-Cl while simultaneously increasing the TEA flow rate. The intensity of the peak at 720 nm was chosen as an indicator of the yield of product 5a (similar to Fig. 3). We considered only short reaction times where the linear dependence of concentration of product 5a on time corresponds to the forward process only.
Fig. 4 demonstrates that the concentration of TEA has little impact on the rate of product formation. Varying TEA molar stoichiometry more than 2 times with respect to the berberine derivative affects the product yield by 10–15% only. The situation was opposite in batch synthesis. We have previously shown33,36 that the presence of bases in the reaction medium was absolutely necessary to observe product formation. The observed differences can be explained from SEAr–SNAr reactions, investigated previously for super-electrophilic nitrobenzofuroxanes.69,70 Existing kinetic studies of the interaction of π-excessive heterocycles and nitrochlorobenzodiazoles using deuterated substrates revealed that the reaction rate-limiting step involves the formation of a double Sigma-complex of Meisenheimer–Wheland type rather than proton detachment from it.71
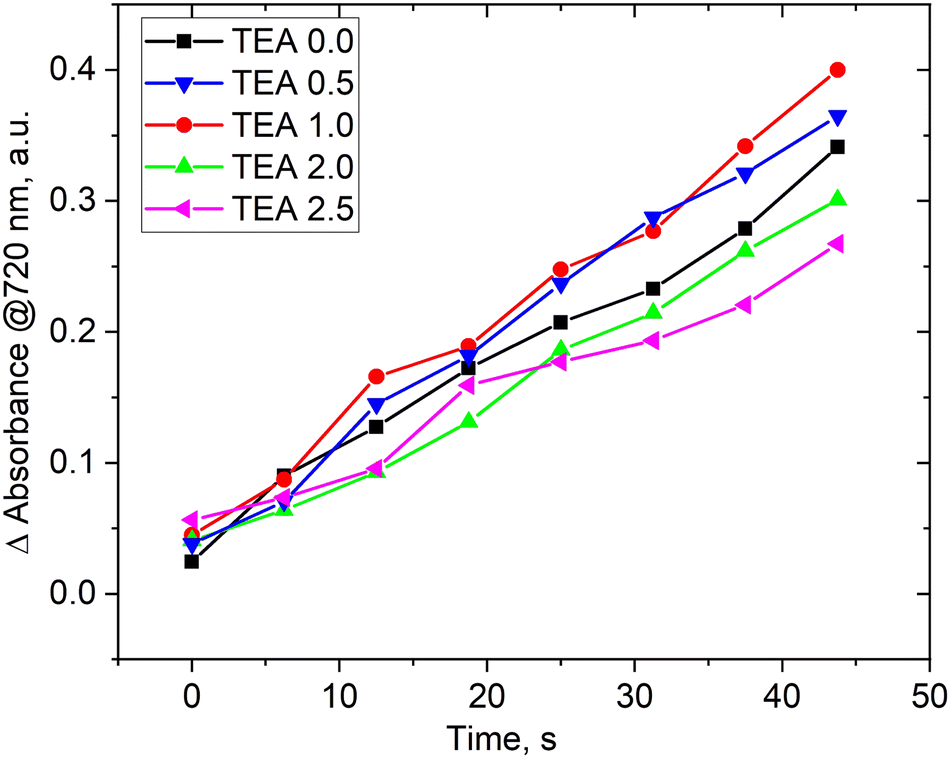 | ||
| Fig. 4 Change in the absorbance at 720 nm of product 5a with varying triethylamine (TEA) content in the reaction mixture. The TEA flow rate is indicated in its molar stoichiometry to berberine derivative (5a) while maintaining a total flow rate of 4 μL s−1. The values of changes in optical density at the maximum absorption of product at 720 nm were normalized to the total NBF-Cl and berberine derivatives flow rate, which were delivered in a stoichiometric ratio. | ||
The proposed scheme of the SEAr–SNAr reaction for products 4a–10a is depicted in Scheme 4. TEA was used as a base and may split protons from the Meisenheimer–Wheland complex (the transition state model in square brackets), thus shifting the equilibrium to the final product. But we don’t observe such effect. Therefore, the rate limiting step is the formation of this transition state complex itself but not its decomposition into a product accelerated by TEA and we observe little effect of TEA on the overall reaction rate. However, the presence of base is important on the stage of purification. According to Scheme 4 the final reaction mixture contains HCl molecules which may change the final product back into the initial berberine (1). TEA binds HCl and ensures structural stability of the product preventing its degradation. The role of TEA is thus not a catalyst for this reaction, as might have been expected a priori.
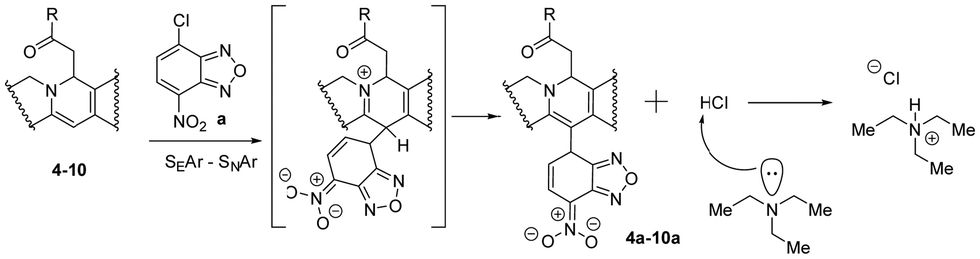 | ||
| Scheme 4 SEAr–SNAr reaction mechanism for the formation of berberine derivatives. | ||
2.3 Biological activity study
Early stages in neurodegenerative, cardiovascular, metabolic, and other diseases involve mitochondrial dysfunction and oxidative stress.72–74 These pathological processes are strongly interconnected, and creating therapies for these conditions has significant potential. We have evaluated the antioxidant and mitoprotective characteristics of the synthesized compounds, as well as their intrinsic cytotoxic effect.It is well known that berberines, being bioactive alkaloids, exhibit a wide range of pharmacological effects, with significant contributions from their antioxidant properties.75–77 Considering that oxidative stress plays a critical role in the pathogenesis of a wide spectrum of human diseases,78–82 the ability of compounds to modulate this process is a promising property of potential therapeutic agents.83–85
An investigation of the antioxidant properties was conducted for compounds that contain p-acetophenones as an additional (auxiliary) group at the position C-8 of the berberine skeleton. Previously, our work demonstrated that the substituent at the p-position exerts the maximal effect on the final biological activity of target derivatives.16 For this reason, we selected 4-substituted acetophenones with donor groups for further analysis.
The antioxidant activity of the compounds was investigated based on their ability to inhibit lipid peroxidation in rat brain homogenate, which can lead to membrane structure damage. To accelerate the oxidation process, 0.5 mM Fe(II) was used, promoting the generation of free radicals in the Fenton reaction.
All compounds effectively suppressed the lipid peroxidation process at a concentration of 0.1 mM (Fig. 5(a)). Moreover, a concentration-dependent “dose–response” relationship was observed, and IC50 values were calculated, representing the concentrations of the compounds causing 50% inhibition of the process, not exceeding 9 μM (Fig. 5(b)). As a reference substance, the well-known antioxidant trolox was used at a concentration of 100 μM, showing a similar level of activity to the tested compounds (Fig. 5(a)). This indicates the high antioxidant potential of berberine derivatives.
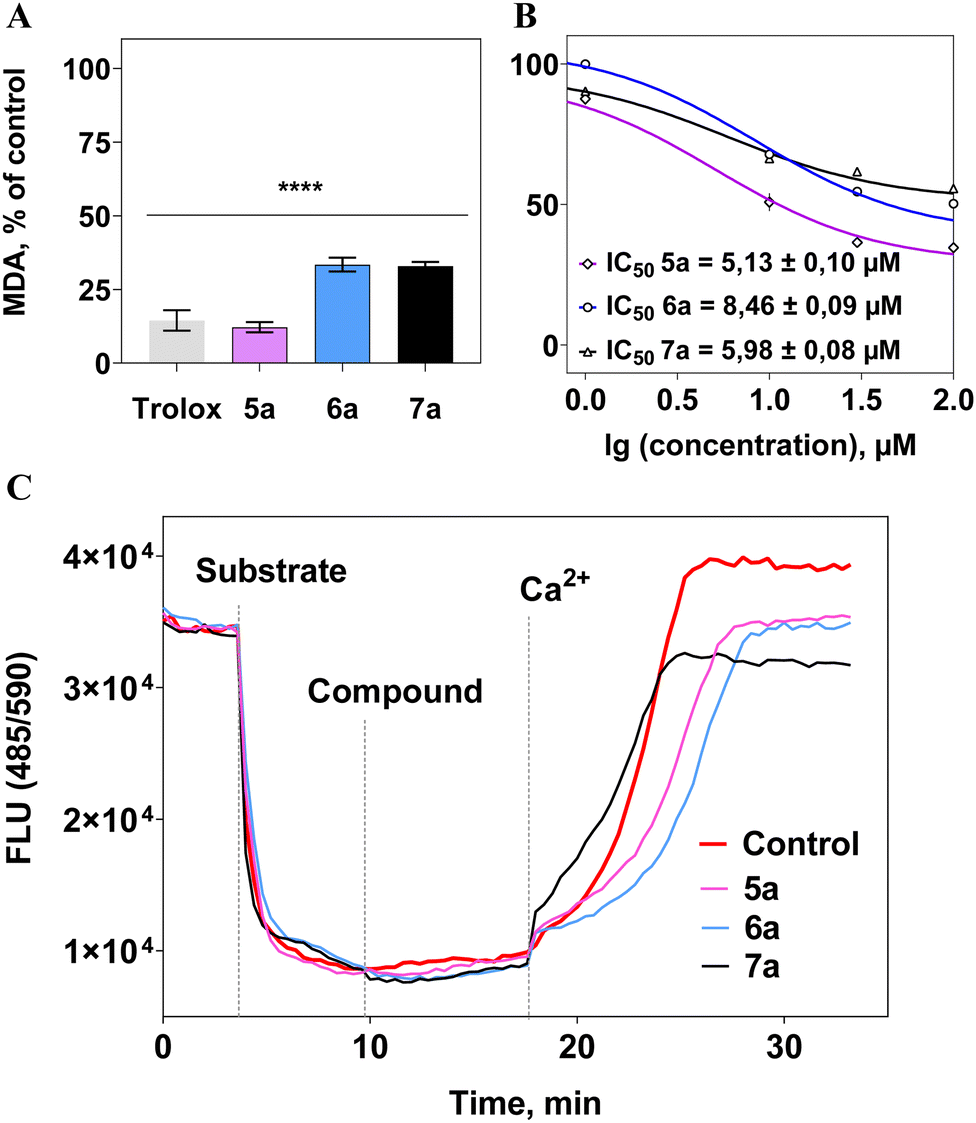 | ||
| Fig. 5 Investigation of the biological activity of the synthesized disubstituted berberine derivatives. (A) The effect of compounds at a concentration of 100 μM on lipid peroxidation in rat brain homogenate (2.5 mg mL−1), initiated by 0.5 mM Fe(II). Data are presented as the level of malondialdehyde (% relative to the control). **** – p ≤ 0.0001 (one-way ANOVA with Dunn's correction). (B) Concentration curves of the antioxidant effect of substances (1–300 μM) and the IC50 values of the inhibitory effect. (C) Influence of the tested compounds at a concentration of 30 μM on the membrane potential of isolated rat liver mitochondria (0.5 mg mL−1), energized with 5 mM succinate in the presence of 1 μM rotenone. The concentration of the depolarization inducer Ca2+ was 25 μM. | ||
It was found that the synthesized berberines did not affect the mitochondrial membrane potential (Ψm) in isolated rat liver mitochondria, suggesting no negative impact on the functioning of organelles crucial to cell viability.86–88 Furthermore, these investigated compounds did not influence the survival of both normal (HEK-293) and tumor-derived SH-SY5Y cells (IC50 > 0.1 mM).
In conclusion, the synthesized disubstituted berberine derivatives exhibit high antioxidant activity, without affecting mitochondrial potential or exerting any intrinsic toxicity on healthy and tumor cell lines. The discovered antioxidant activity of these synthesized berberine derivatives shows promise for the development of pharmaceutical drugs against neurodegenerative disorders,89,90 cardiovascular diseases,91–93 and metabolic disorders.94,95 An essential characteristic of such therapeutic agents is their lack of toxic effects. These findings recommend further in-depth investigations of these compounds as potential antioxidant agents.
3. Conclusion
We have successfully synthesized novel disubstituted derivatives of berberine using a microfluidic approach. The use of microfluidic systems allowed us to access previously unattainable reaction products and perform online reaction monitoring. The resulting quinone–imine compounds exhibited vibrant coloration and strong absorbance in the visible region, requiring use of the short optical path length of 0.2 mm integrated directly inside the microfluidic chip. The reaction starts directly after contact of the reagents with a linear increase in the product concentration during the first seconds of the reaction and subsequently follows an exponential asymptotic behavior. We found that the addition of trimethylamine (TEA), which was necessary during batch synthesis, did not significantly affect the reaction rate, and we concluded that its role lies in stabilizing the reaction product during extraction. The time needed for full conversion of the reagents was in the order of 10 minutes and we applied an external delay tube to the chip for equilibration.In vitro studies conducted on the three obtained products confirmed their potential as antioxidants. These investigations demonstrated the capability of these compounds against oxidative stress and its detrimental effects on the organism. The results indicate the promise of utilizing these structures in the development of future antioxidant drugs and therapeutics aimed at addressing various pathological conditions and diseases associated with oxidative stress. This opens new prospects for further research and advancements in the field of antioxidant therapy.
4. Experimental section
4.1 Batch synthesis
The classical synthesis method was employed. A weighed amount of reduced form berberine (DHB) (1 mmol) was dissolved in 15 mL of DMF while stirring. To this solution, 1 mmol of triethylamine (TEA) was added. Separately, 1 mmol of chloronitrobenzofurazane (NBF-Cl) was dissolved in 15 mL of chilled DMF and slowly added to the solution of activated form berberine (DHB) while continuously stirring. The reaction mixture was stirred in the dark for 2 hours. Subsequently, the mixture was poured into 250 mL of chilled distilled water (the addition of the solution to water leads to significant heating up to 50 °C). The resulting precipitate was filtered through a Büchner funnel, washed with 2 portions of chilled water, and then with 2 portions of hot water (90 °C). The dry residue was subjected to chromatography (Rf = 0.7) on SiO2 using a mixture of benzene and tetrachlorethylene (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as the eluent. The purified product was then recrystallized from a mixture of tetrachlorethylene/octane (in a ratio of 1:2). Yields of the products ranged from 38% to 72%.
:1) as the eluent. The purified product was then recrystallized from a mixture of tetrachlorethylene/octane (in a ratio of 1:2). Yields of the products ranged from 38% to 72%.
4.2 Microfluidic synthesis
The microfluidic synthesis is carried out by preparing three solutions in syringes as follows:Syringe #1: a solution containing 1 mmol of 8-substituted dihydroberberine (DHB) in 10 mL of DMF.
Syringe #2: a solution containing 1 mmol of 4-chloro-7-nitrobenzofurazane (NBF-Cl) in 10 mL of DMF.
Syringe #3: a solution containing 1 mmol of triethylamine (TEA) in 10 mL of DMF.
The microfluidic setup consists of a Pearl mixer chip (with a square channel cross section 0.45 × 0.45 m2 and 0.6 mm diameter of round obstacles) with 3 ports for the introduction of reagents and a delay line (2 m commercially available PFE pipe with a cross section of 0.79 mm2). The synthesis is conducted using custom software developed by our group. The microfluidic reactor was operated at room temperature, which was 24 °C. Each of the reagents was delivered into the microfluidic reactor at a rate of 1 μL per second, resulting in a total residence time of the reaction mixture in the reactor of 11 minutes. The mass yield of the reaction product was approximately 20–24 mg s−1 (excluding impurities, depending on the molecular weight of the synthesized product). The obtained solution was additionally kept in a closed vessel for 20 minutes immediately before purification.
To isolate the target products, the reaction mixture is poured into 250 mL of chilled distilled water. The resulting precipitate is filtered through a Büchner funnel, washed with two portions of chilled water and two portions of hot water (80 °C), each portion being 50 mL. The dry residue is recrystallized from a mixture of tetrachlorethylene and octane (in a ratio of 1:2). Yields of the products range from 69% to 92%.
4.3 NMR, FTIR, and UV-Vis characterization
The 1H and 13C NMR spectra were recorded on a Bruker DPX-250 spectrometer operating at 250 MHz and 63 MHz, respectively, using CDCl3 or DMSO-d6 as solvents, and TMS (tetramethylsilane) as an internal standard. Signal assignments in the 1H NMR spectra were made based on the data obtained from two-dimensional COSY spectra with mixing times of 0.6–1.3 seconds. The 13C NMR spectra exhibit signal overlap, resulting in fewer distinct signals compared to the theoretical number.High-resolution mass spectra were recorded on a Bruker micrOTOF II instrument using electrospray ionization in positive ion mode with a capillary voltage of 4500 V. The mass scanning range was from 50 to 3000 Da.
Melting points were determined using glass capillaries on a melting point apparatus (PTP).
These analytical techniques were used to characterize the synthesized compounds and obtain information about their molecular structure, functional groups, and purity.
FTIR spectra were measured using a Bruker Vertex 70 spectrometer in ATR (Attenuated Total Reflectance) geometry with an MCT detector and the Bruker Platinum ATR accessory. The spectra were recorded in the range of 5000 to 500 cm−1 with a resolution of 1 cm−1 and 64 scans. Additionally, a DTGS detector was used, and the spectra were recorded in the range of 5000 to 30 cm−1 with a resolution of 1 cm−1 and 64 scans. The reference sample was air.
UV-Visible (UV-Vis) spectroscopy was performed using a Shimadzu UV-2600 spectrophotometer.
4.4 Microfluidic devices and in situ UV-Vis diagnostics
The microfluidic devices (MFDs) used in the study were designed using computer-aided design (CAD) software (Fusion 360, Autodesk, USA) and fabricated using a FunToDo nano clear resin on a DLP 3D printer (ASIGA MAX UV, Sydney, Australia). The layer height for printing the MFDs was set at 10 μm, and the printing time for a 6 mm thick MFD was approximately 45 minutes. After printing, the device was removed from the build plate and subjected to ultrasonic cleaning in isopropyl alcohol (IPA) for 60 seconds at 35 kHz and 50W power, followed by manual flushing of the channels with IPA. The MFDs were then dried with compressed air, following an existing post-processing protocol.96 Additional UV light was applied for 2 minutes to improve the surface hydrophobicity, and the channels were filled with vaseline oil and left overnight.A custom-made dosing syringe pump system with borosilicate glass syringes was used to precisely control the flow of reactants and the carrier phase in the microfluidic system. Standard 1/4′′-28 UNF flangeless PTFE connectors and PFA tubes (1/32′′ ID) were used to connect the different parts of the microfluidic system. The rates of the reactants were varied in the range of 0.5 to 5 μm s−1 and delay line 0.5 to 2 m.
Spectra measurements were carried out using an optical fiber spectrometer (OceanFX fiber optic UV-Vis spectrometer, Ocean Insight, Orlando, USA) with an integration time of 35 ms and an acquisition interval of 100 ms. An Xe arc lamp setup served as the light source. During measurements, dark and background signals were automatically subtracted. In situ absorption signals were recorded directly through the transparent walls of the 3D printed microfluidic channels. For this purpose, the background signal from pure DMF (dimethylformamide) solvent was measured from the same microfluidic channel.
4.5 Biological activity
All animal work was conducted in accordance with the principles of Good Laboratory Practice in the Russian Federation (2016). The Bioethics Committee of IPAC RAS granted full approval for this research (approval no. 63, dated October 10, 2022).In the experiments, male non-inbred rats weighing 200–220 g were used. The animals were kept under standard conditions in a vivarium with a 12 hour light cycle and free access to water and food. All animal experiments were conducted following international principles and norms in accordance with the decisions of the Commission on Biological Ethics of the Institute of Advanced Research (IFAV RAS) (protocol no. 63, dated 10.10.2022).
The brain homogenate of rats was obtained by centrifugation at 1500 g and 4 °C, using a buffer containing 120 mM KCl and 20 mM HEPES at pH 7.4. Liver mitochondria of rats were obtained by differential centrifugation using buffers of various compositions.97 The quantitative determination of protein was carried out using the microburet method.
The impact of the compounds on the process of lipid peroxidation was evaluated using the Thiobarbituric Acid Reactive Substances (TBARS) test with modifications for a plate format.98
The mitochondrial membrane potential of rat liver mitochondria was measured using a plate reader analyzer, Victor 3 (PerkinElmer, USA), with the voltage-sensitive indicator safranin A.98 The mitochondrial suspension contained 0.5 mg of protein in 1 mL of buffer. The mitochondrial membrane energization was performed using 5 mM potassium succinate in the presence of 1 μM rotenone, and the maximum depolarization was induced by adding 25 μM CaCl2.
HEK-293 cells (human embryonic kidney cells) and SH-SY5Y cells (neuroblastoma) were cultured in DMEM medium (PanEco, Moscow, Russia) supplemented with 10% fetal bovine serum (HyClone®, Thermo Scientific), 2 mM L-glutamine (PanEco, Moscow, Russia), and 1% streptomycin (PanEco, Moscow, Russia) as an antibiotic, at 37 °C in a CO2 atmosphere (5%). The cell viability analysis was performed using the MTT assay.99
Author contributions
A. D. Zagrebaev – evaluation of all chemical experiments, writing – original draft. V. V. Butova – acquisition and interpretation of NMR, FTIR and UV-Vis spectra. A. A. Guda – methodology of in situ diagnostics, data processing, writing – review & editing. S. V. Chapek – design and manufacture of 3D printed microfluidic chips. O. N. Burov – acquisition and interpretation of NMR, FTIR and UV-Vis spectra. S. V. Kurbatov conceptualization of the chemical part. M. E. Neganova – conceptualization of biological part. E. Yu. Vinyukova, Yu. R. Aleksandrova, N. S. Nikolaeva – work with animals, evaluation of all biochemical experiments. O. P. Demidov – HRMS experiment and evaluation. A. V. Soldatov – funding acquisition, supervision.Conflicts of interest
There are no conflicts to declareAcknowledgements
This research was supported by the Strategic Academic Leadership Program of the Southern Federal University (“Priority 2030”). The authors are grateful to the “Centre for Collective Use of IPAC RAS” (IPAC research topic FFSN-2021-0013) for providing the opportunity to perform the experiments using laboratory animals.References
- M. Imanshahidi and H. Hosseinzadeh, Pharmacological and therapeutic effects of Berberis vulgaris and its active constituent, berberine, Phytother. Res., 2008, 22, 999–1012 CrossRef CAS.
- J. Yang, J. Yin, H. Gao, L. Xu, Y. Wang, L. Xu and M. Li, Berberine improves insulin sensitivity by inhibiting fat store and adjusting adipokines profile in human preadipocytes and metabolic syndrome patients, Endocr., Metab. Immune Disord.: Drug Targets, 2012, 2012, 363845 Search PubMed.
- J. Yin, H. Zhang and J. Ye, Traditional chinese medicine in treatment of metabolic syndrome, Endocr., Metab. Immune Disord.: Drug Targets, 2008, 8, 99–111 CrossRef CAS PubMed.
- H. Hui, G. Tang and V. L. Go, Hypoglycemic herbs and their action mechanisms, Chin. Med., 2009, 4, 11 CrossRef.
- J. Yin, H. Xing and J. Ye, Efficacy of berberine in patients with type 2 diabetes mellitus, Metabolism, 2008, 57, 712–717 CrossRef CAS.
- X. Xia, J. Yan, Y. Shen, K. Tang, J. Yin, Y. Zhang, D. Yang, H. Liang, J. Ye and J. Weng, Berberine improves glucose metabolism in diabetic rats by inhibition of hepatic gluconeogenesis, PLoS One, 2011, 6, e16556 CrossRef CAS.
- A. Bagade, V. Tumbigeremutt and G. Pallavi, Cardiovascular effects of berberine: a review of the literature, J. Restor. Med., 2017, 6, 37 CrossRef.
- W. Kong, J. Wei, P. Abidi, M. Lin, S. Inaba, C. Li, Y. Wang, Z. Wang, S. Si, H. Pan, S. Wang, J. Wu, Y. Wang, Z. Li, J. Liu and J. D. Jiang, Berberine is a novel cholesterol-lowering drug working through a unique mechanism distinct from statins, Nat. Med., 2004, 10, 1344–1351 CrossRef CAS.
- T. Patil, S. Patil, S. Patil and A. Patil, Antimicrobial Profile of Antidiabetic Drug: Berberine, Int. J. Pharm. Pharm. Res., 2015, 7, 45–50 Search PubMed.
- S. Samosorn, Development of berberine-based derivatives as novel antimicrobial agents, Bioorg. Med. Chem. Lett., 2016, 26(12), 2768–2773 CrossRef PubMed.
- A. H. Amin, T. V. Subbaiah and K. M. Abbasi, Berberine sulfate: antimicrobial activity, bioassay, and mode of action, Can. J. Microbiol., 1969, 15, 1067–1076 CrossRef CAS PubMed.
- W. Zha, G. Liang, J. Xiao, E. J. Studer, P. B. Hylemon, W. M. Pandak Jr., G. Wang, X. Li and H. Zhou, Berberine inhibits HIV protease inhibitor-induced inflammatory response by modulating ER stress signaling pathways in murine macrophages, PLoS One, 2010, 5, e9069 CrossRef PubMed.
- S. Mahata, A. C. Bharti, S. Shukla, A. Tyagi, S. A. Husain and B. C. Das, Berberine modulates AP-1 activity to suppress HPV transcription and downstream signaling to induce growth arrest and apoptosis in cervical cancer cells, Mol. Cancer, 2011, 10, 39 CrossRef CAS PubMed.
- L. W. Chin, Y. W. Cheng, S. S. Lin, Y. Y. Lai, L. Y. Lin, M. Y. Chou, M. C. Chou and C. C. Yang, Anti-herpes simplex virus effects of berberine from Coptidis rhizoma, a major component of a Chinese herbal medicine, Ching-Wei-San, Arch. Virol., 2010, 155, 1933–1941 CrossRef CAS PubMed.
- C. Q. Nguyen, T. H. M. Nguyen, T. T. T. Nguyen, T. B. H. Bui, T. T. Nguyen, N. T. Huynh, T. D. Le, T. M. P. Nguyen, D. T. Nguyen, M. T. Nguyen, M. Q. Pham, Q. D. Tran, H. P. Nguyen and A. K. Singh, Designs, Synthesis, Docking Studies, and Biological Evaluation of Novel Berberine Derivatives Targeting Zika Virus, J. Chem., 2021, 2021, 1–10 Search PubMed.
- K. D. Park, J. H. Lee, S. H. Kim, T. H. Kang, J. S. Moon and S. U. Kim, Synthesis of 13-(substituted benzyl) berberine and berberrubine derivatives as antifungal agents, Bioorg. Med. Chem. Lett., 2006, 16, 3913–3916 CrossRef CAS PubMed.
- M. Franceschin, L. Rossetti, A. D’Ambrosio, S. Schirripa, A. Bianco, G. Ortaggi, M. Savino, C. Schultes and S. Neidle, Natural and synthetic G-quadruplex interactive berberine derivatives, Bioorg. Med. Chem. Lett., 2006, 16, 1707–1711 CrossRef CAS.
- C. Bazzicalupi, M. Ferraroni, A. R. Bilia, F. Scheggi and P. Gratteri, The crystal structure of human telomeric DNA complexed with berberine: an interesting case of stacked ligand to G-tetrad ratio higher than 1:1, Nucleic Acids Res., 2013, 41, 632–638 CrossRef CAS PubMed.
- F. Wang, C. Wang, Y. Liu, W. Lan, H. Han, R. Wang, S. Huang and C. Cao, Colchicine selective interaction with oncogene RET G-quadruplex revealed by NMR, Chem. Commun., 2020, 56, 2099–2102 RSC.
- S. Soundarapandian, A. Alexander, A. S. Pillai, I. Enoch and S. Yousuf, G-Quadruplex binding of cavity-containing anthraquinonesulfonyl-beta-cyclodextrin conjugate. Effect of encapsulation of ethidium bromide and berberine, J. Biomol. Struct. Dyn., 2022, 40, 8301–8311 CrossRef CAS PubMed.
- K.-B. Wang, J. Dickerhoff and D. Yang, Solution structure of ternary complex of berberine bound to a dGMP–fill-in vacancy G-quadruplex formed in the PDGFR-β promoter, J. Am. Chem. Soc., 2021, 143, 16549–16555 CrossRef CAS PubMed.
- K. G. Lyamzaev, A. V. Pustovidko, R. A. Simonyan, T. I. Rokitskaya, L. V. Domnina, O. Y. Ivanova, I. I. Severina, N. V. Sumbatyan, G. A. Korshunova, V. N. Tashlitsky, V. A. Roginsky, Y. N. Antonenko, M. V. Skulachev, B. V. Chernyak and V. P. Skulachev, Novel mitochondria-targeted antioxidants: plastoquinone conjugated with cationic plant alkaloids berberine and palmatine, Pharm. Res., 2011, 28, 2883–2895 CrossRef CAS PubMed.
- X. Shi, T. Zhang, H. Lou, H. Song, C. Li and P. Fan, Anticancer Effects of Honokiol via Mitochondrial Dysfunction Are Strongly Enhanced by the Mitochondria-Targeting Carrier Berberine, J. Med. Chem., 2020, 63, 11786–11800 CrossRef CAS PubMed.
- L. Grycova, J. Dostal and R. Marek, Quaternary protoberberine alkaloids, Phytochemistry, 2007, 68(2), 150–175 CrossRef CAS PubMed.
- Y. Jin, D. B. Khadka and W. J. Cho, Pharmacological effects of berberine and its derivatives: a patent update, Expert Opin. Ther. Pat., 2016, 26(2), 229–243 CrossRef CAS PubMed.
- S. Singh, N. Pathak, E. Fatima and A. S. Negi, Plant isoquinoline alkaloids: Advances in the chemistry and biology of berberine, Eur. J. Med. Chem., 2021, 226, 113839 CrossRef CAS PubMed.
- M. Tillhon, L. M. Guaman Ortiz, P. Lombardi and A. I. Scovassi, Berberine: new perspectives for old remedies, Biochem. Pharmacol., 2012, 84, 1260–1267 CrossRef CAS PubMed.
- S. Gaba, A. Saini, G. Singh and V. Monga, An insight into the medicinal attributes of berberine derivatives: A review, Bioorg. Med. Chem., 2021, 38, 116143 CrossRef CAS.
- I. Nechepurenko, N. Salakhutdinov and G. Tolstikov, Berberine: chemistry and biological activity, Chem. Sustainable Dev., 2010, 18, 1–23 CAS.
- T. Schmeller, B. Latz-Bruning and M. Wink, Biochemical activities of berberine, palmatine and sanguinarine mediating chemical defence against microorganisms and herbivores, Phytochemistry, 1997, 44, 257–266 CrossRef CAS PubMed.
- G. A. Cordell, M. L. Quinn-Beattie and N. R. Farnsworth, The potential of alkaloids in drug discovery, Phytother. Res., 2001, 15, 183–205 CrossRef CAS PubMed.
- O. D. Demekhin, A. D. Zagrebaev, O. N. Burov, M. E. Kletskii, N. Y. V. Pavlovich, E. A. Bereznyak, M. V. Tsimbalistova and S. V. Kurbatov, The first 13-vinyl derivatives of berberine: synthesis and antimicrobial activity, Chem. Heterocycl. Compd., 2019, 55, 1128–1130 CrossRef CAS.
- O. N. Burov, S. V. Kurbatov, M. E. Kletskii, A. D. Zagrebaev and I. E. Mikhailov, Synthesis and structure of dihydroberberine nitroaryl derivatives – potential ligands for G-quadruplexes, Chem. Heterocycl. Compd., 2017, 53, 335–340 CrossRef CAS.
- O. D. Demekhin, O. N. Burov, M. Kletskii, S. V. Kurbatov, E. Bereznyak and A. V. Trishina, A Structural Modification of Berberine Using CH Acids and Ethoxyethylenes Based on Them, Chem. Heterocycl. Compd., 2022, 58, 621–627 CrossRef CAS.
- O. D. Demekhin, O. N. Burov, M. E. Kletskii, A. V. Lisovin, S. V. Kurbatov, E. A. Bereznyak and A. V. Trishina, New 13-vinyl derivatives of berberine: synthesis and characterization, Chem. Heterocycl. Compd., 2022, 58, 144–152 CrossRef CAS.
- O. N. Burov, S. V. Kurbatov, P. G. Morozov, M. E. Kletskii and A. V. Tatarov, Synthesis of the first 13-nitroaryl derivatives of 8-acetonylberberine, Chem. Heterocycl. Compd., 2015, 51, 772–774 CrossRef CAS.
- D. Seebach and D. Enders, Umpolung of Amine Reactivity. Nucleophilic?-(Secondary Amino)-alkylationvia Metalated Nitrosamines, Angew. Chem., Int. Ed. Engl., 1975, 14, 15–32 CrossRef.
- J. Wang, T. Yang, H. Chen, Y. N. Xu, L. F. Yu, T. Liu, J. Tang, Z. Yi, C. G. Yang, W. Xue and F. Yang, The synthesis and antistaphylococcal activity of 9, 13-disubstituted berberine derivatives, Eur. J. Med. Chem., 2017, 127, 424–433 CrossRef CAS.
- N. K. Dolla, C. Chen, J. Larkins-Ford, R. Rajamuthiah, S. Jagadeesan, A. L. Conery, F. M. Ausubel, E. Mylonakis, J. B. Bremner, K. Lewis and M. J. Kelso, On the Mechanism of Berberine-INF55 (5-Nitro-2-phenylindole) Hybrid Antibacterials, Aust. J. Chem., 2015, 67, 1471–1480 CrossRef.
- H. Sun, M. F. Ansari, B. Fang and C. H. Zhou, Natural Berberine-Hybridized Benzimidazoles as Novel Unique Bactericides against Staphylococcus aureus, J. Agric. Food Chem., 2021, 69, 7831–7840 CrossRef CAS.
- S. Ruan, Y. Zhang, S. Wu, Y. Gao, L. Yang, M. Li, Y. Yang, Z. Wang and S. Wang, A novel berberine-based colorimetric and fluorometric probe for Hg2+ detection and its applications in water samples, Inorg. Chem. Commun., 2021, 132, 108847 CrossRef CAS.
- M. Guidi, P. H. Seeberger and K. Gilmore, How to approach flow chemistry, Chem. Soc. Rev., 2020, 49, 8910–8932 RSC.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, The Hitchhiker's Guide to Flow Chemistry parallel, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed.
- V. Sans and L. Cronin, Towards dial-a-molecule by integrating continuous flow, analytics and self-optimisation, Chem. Soc. Rev., 2016, 45, 2032–2043 RSC.
- P. Jankowski, R. Kutaszewicz, D. Ogończyk and P. Garstecki, A microfluidic platform for screening and optimization of organic reactions in droplets, J. Flow Chem., 2020, 10, 397–408 CrossRef CAS.
- J. P. McMullen and K. F. Jensen, An Automated Microfluidic System for Online Optimization in Chemical Synthesis, Org. Process Res. Dev., 2010, 14, 1169–1176 CrossRef CAS.
- C. J. Taylor, A. Pomberger, K. C. Felton, R. Grainger, M. Barecka, T. W. Chamberlain, R. A. Bourne, C. N. Johnson, A. A. Lapkin and A. Brief, Introduction to Chemical Reaction Optimization, Chem. Rev., 2023, 123, 3089–3126 CrossRef CAS PubMed.
- J. Yue, J. C. Schouten and T. A. Nijhuis, Integration of Microreactors with Spectroscopic Detection for Online Reaction Monitoring and Catalyst Characterization, Ind. Eng. Chem. Res., 2012, 51, 14583–14609 CrossRef CAS.
- D. C. Fabry, E. Sugiono and M. Rueping, Online monitoring and analysis for autonomous continuous flow self-optimizing reactor systems, React. Chem. Eng., 2016, 1, 129–133 RSC.
- G. L. Nelson, A. M. Lines, J. M. Bello and S. A. Bryan, Online Monitoring of Solutions Within Microfluidic Chips: Simultaneous Raman and UV-Vis Absorption Spectroscopies, ACS Sens., 2019, 4, 2288–2295 CrossRef CAS.
- G. Tofighi, A. Gaur, D. E. Doronkin, H. Lichtenberg, W. Wang, D. Wang, G. Rinke, A. Ewinger, R. Dittmeyer and J.-D. Grunwaldt, Microfluidic Synthesis of Ultrasmall AuPd Nanoparticles with a Homogeneously Mixed Alloy Structure in Fast Continuous Flow for Catalytic Applications, J. Phys. Chem. C, 2018, 122, 1721–1731 CrossRef CAS.
- B. W. Pearre, C. Michas, J.-M. Tsang, T. J. Gardner and T. M. Otchy, Fast micron-scale 3D printing with a resonant-scanning two-photon microscope, Addit. Manuf., 2019, 30, 100887 CAS.
- S. V. Chapek, I. A. Pankin, D. V. Khodakova, A. A. Guda, A. S. Goncharova and A. V. Soldatov, Study of the Surface Morphology of Microfluidic-Chip Channels via X-Ray Tomography and Scanning Electron Microscopy, J. Surf. Invest.: X-Ray, Synchrotron Neutron Tech., 2023, 17, 392–396 CrossRef CAS.
- E. V. Sadyrin, A. L. Nikolaev, S. V. Chapek, D. V. Nazarenko, S. M. Aizikovich and Y.-C. Wang, Manufacturing Quality Evaluation of Photopolymer Resin 3D-Printed Scaffolds Using Microtomography, Springer International Publishing, 2023, pp. 619–630 Search PubMed.
- M. A. Marchenkova, S. V. Chapek, P. V. Konarev, K. B. Ilina, G. S. Peters, Y. V. Pisarevsky, V. A. Shishkov, A. V. Soldatov and M. V. Kovalchuk, 3D Printed Microfluidic Cell for SAXS Time-Resolved Measurements of the Structure of Protein Crystallization Solutions, Crystals, 2023, 13, 938 CrossRef CAS.
- A.-G. Niculescu, C. Chircov, A. C. Bîrcă and A. M. Grumezescu, Nanomaterials Synthesis through Microfluidic Methods: An Updated Overview, Nanomaterials, 2021, 11, 864 CrossRef CAS PubMed.
- A.-G. Niculescu, D. E. Mihaiescu and A. M. Grumezescu, A review of microfluidic experimental designs for nanoparticle synthesis, Int. J. Mol. Sci., 2022, 23, 8293 CrossRef CAS PubMed.
- P. J. E. M. Van Der Linden, A. M. Popov and D. Pontoni, Accurate and rapid 3D printing of microfluidic devices using wavelength selection on a DLP printer, Lab Chip, 2020, 20, 4128–4140 RSC.
- L. P. Bressan, J. Robles-Najar, C. B. Adamo, R. F. Quero, B. M. Costa, D. P. de Jesus and J. A. da Silva, 3D-printed microfluidic device for the synthesis of silver and gold nanoparticles, Microchem. J., 2019, 146, 1083–1089 CrossRef CAS.
- R. Camarillo-Escobedo, J. L. Flores-Nuñez, G. Garcia-Torales, E. Hernandez-Campos and J. M. Camarillo-Escobedo, 3D printed opto-microfluidic autonomous analyzer for photometric applications, Sens. Actuators, A, 2022, 337, 113425 CrossRef CAS.
- A. A. Guda, M. V. Kirichkov, V. V. Shapovalov, A. I. Muravlev, D. M. Pashkov, S. A. Guda, A. P. Bagliy, S. A. Soldatov, S. V. Chapek and A. V. Soldatov, Machine Learning Analysis of Reaction Parameters in UV-Mediated Synthesis of Gold Nanoparticles. The, J. Phys. Chem. C, 2023, 127, 1097–1108 CrossRef CAS.
- H. Usutani, Y. Tomida, A. Nagaki, H. Okamoto, T. Nokami and J. Yoshida, Generation and reactions of o-bromophenyllithium without benzyne formation using a microreactor, J. Am. Chem. Soc., 2007, 129, 3046–3047 CrossRef CAS PubMed.
- F. Mastronardi, B. Gutmann and C. O. Kappe, Continuous flow generation and reactions of anhydrous diazomethane using a Teflon AF-2400 tube-in-tube reactor, Org. Lett., 2013, 15, 5590–5593 CrossRef CAS PubMed.
- S. L. Bourne, S. V. Ley and A. Continuous, Flow Solution to Achieving Efficient Aerobic Anti-Markovnikov Wacker Oxidation, Adv. Synth. Catal., 2013, 355, 1905–1910 CrossRef CAS.
- S. Fuse, N. Tanabe, M. Yoshida, H. Yoshida, T. Doi and T. Takahashi, Continuous-flow synthesis of vitamin D3, Chem. Commun., 2010, 46, 8722 RSC.
- H. S. Santana, M. S. A. Palma, M. G. M. Lopes, J. Souza, G. A. S. Lima, O. P. Taranto and J. L. Silva, Microfluidic Devices and 3D Printing for Synthesis and Screening of Drugs and Tissue Engineering, Ind. Eng. Chem. Res., 2020, 59, 3794–3810 CrossRef CAS.
- M. Farahani, F. Moradikhah, I. Shabani, R. K. Soflou and E. Seyedjafari, Microfluidic fabrication of berberine-loaded nanoparticles for cancer treatment applications, J. Drug Delivery Sci. Technol., 2021, 61, 102134 CrossRef CAS.
- A. D. Zagrebaev, O. N. Burov, M. Kletskii, A. V. Lisovin, S. V. Kurbatov and O. D. Demekhin, The Synthesis and Investigation of New Electroneutral Berberine Derivatives, Chem. Heterocycl. Compd., 2022, 58, 45–57 CrossRef CAS.
- P. Rodriguez-Dafonte, F. Terrier, S. Lakhdar, S. Kurbatov and R. Goumont, Carbon Nucleophilicities of Indoles in SNAr Substitutions of Superelectrophilic 7-Chloro-4,6-dinitrobenzofuroxan and -benzofurazan, J. Org. Chem., 2009, 74, 3305–3315 CrossRef CAS PubMed.
- S. Kurbatov, P. Rodriguez-Dafonte, R. Goumont and F. Terrier, Superelectrophilic heterocycles: facile SNAr–SEAr couplings involving very weak carbon nucleophiles, Chem. Commun., 2003, 2150–2151 RSC.
- F. Terrier, Rate and equilibrium studies in Jackson-Meisenheimer complexes, Chem. Rev., 1982, 82, 77–152 CrossRef CAS.
- J. S. Bhatti, G. K. Bhatti and P. H. Reddy, Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies, Biochim. Biophys. Acta, Mol. Basis Dis., 2017, 1863, 1066–1077 CrossRef CAS PubMed.
- J. N. Peoples, A. Saraf, N. Ghazal, T. T. Pham and J. Q. Kwong, Mitochondrial dysfunction and oxidative stress in heart disease, Exp. Mol. Med., 2019, 51, 1–13 CrossRef CAS PubMed.
- G. C. Kujoth, A. Hiona, T. D. Pugh, S. Someya, K. Panzer, S. E. Wohlgemuth, T. Hofer, A. Y. Seo, R. Sullivan, W. A. Jobling, J. D. Morrow, H. Van Remmen, J. M. Sedivy, T. Yamasoba, M. Tanokura, R. Weindruch, C. Leeuwenburgh and T. A. Prolla, Mitochondrial DNA Mutations, Oxidative Stress, and Apoptosis in Mammalian Aging, Science, 2005, 309, 481–484 CrossRef CAS PubMed.
- A. K. Behera, M. M. Swamy, N. Natesh and T. K. Kundu, Garcinol and Its Role in Chronic Diseases, Springer International Publishing, 2016, pp. 435–452 Search PubMed.
- D. Song, J. Hao and D. Fan, Biological properties and clinical applications of berberine, Front. Med., 2020, 14, 564–582 CrossRef PubMed.
- E. Poopedi, M. Marimani, S. Y. Alomar, B. Aldahmash and A. Ahmad, Modulation of antioxidant defence system in response to berberine in Candida albicans, Yeast, 2021, 38, 157–169 CrossRef CAS PubMed.
- Z. Chen and C. Zhong, Oxidative stress in Alzheimer's disease, Neurosci. Bull., 2014, 30, 271–281 CrossRef CAS PubMed.
- H. Sies, C. Berndt and D. P. Jones, Oxidative Stress, Annu. Rev. Biochem., 2017, 86, 715–748 CrossRef CAS PubMed.
- J. S. Kimball, J. P. Johnson and D. A. Carlson, Oxidative Stress and Osteoporosis, J. Bone Jt. Surg., Am. Vol., 2021, 103, 1451–1461 CrossRef PubMed.
- C. Cabello-Verrugio, F. Simon, C. Trollet and J. F. Santibañez, Oxidative Stress in Disease and Aging: Mechanisms and Therapies 2016, Oxid. Med. Cell. Longevity, 2017, 2017, 1–2 CrossRef PubMed.
- T. Senoner and W. Dichtl, Oxidative Stress in Cardiovascular Diseases: Still a Therapeutic Target?, Nutrients, 2019, 11, 2090 CrossRef CAS PubMed.
- K. A. Ahmad, D. Yuan Yuan, W. Nawaz, H. Ze, C. X. Zhuo, B. Talal, A. Taleb, E. Mais and D. Qilong, Antioxidant therapy for management of oxidative stress induced hypertension, Free Radical Res., 2017, 51, 428–438 CrossRef CAS PubMed.
- M. A. Dkhil, D. Delic, H. A. El Enshasy and A. E. Abdel Moneim, Medicinal Plants in Therapy: Antioxidant Activities, Oxid. Med. Cell. Longevity, 2016, 2016, 1 CrossRef PubMed.
- L. Deng, C. Du, P. Song, T. Chen, S. Rui, D. G. Armstrong and W. Deng, The Role of Oxidative Stress and Antioxidants in Diabetic Wound Healing, Oxid. Med. Cell. Longevity, 2021, 2021, 1–11 Search PubMed.
- J. Nunnari and A. Suomalainen, Mitochondria: In Sickness and in Health, Cell, 2012, 148, 1145–1159 CrossRef CAS PubMed.
- E. Vringer and S. W. G. Tait, Mitochondria and cell death-associated inflammation, Cell Death Differ., 2023, 30, 304–312 CrossRef CAS PubMed.
- D. R. Green, L. Galluzzi and G. Kroemer, Mitochondria and the Autophagy – Inflammation – Cell Death Axis in Organismal Aging, Science, 2011, 333, 1109–1112 CrossRef CAS PubMed.
- Y. Feng and X. Wang, Antioxidant Therapies for Alzheimer's Disease, Oxid. Med. Cell. Longevity, 2012, 2012, 1–17 CrossRef PubMed.
- H. Khan, H. Ullah, M. Aschner, W. S. Cheang and E. K. Akkol, Neuroprotective Effects of Quercetin in Alzheimer's Disease, Biomolecules, 2019, 10, 59 CrossRef PubMed.
- N. Xia, A. Daiber, U. Förstermann and H. Li, Antioxidant effects of resveratrol in the cardiovascular system, Br. J. Pharmacol., 2017, 174, 1633–1646 CrossRef CAS PubMed.
- J. G. Gormaz and R. Carrasco, Antioxidant Supplementation in Cardiovascular Prevention, J. Am. Coll. Cardiol., 2022, 80, 2286–2288 CrossRef CAS PubMed.
- D.-D. Zhou, M. Luo, A. Shang, Q.-Q. Mao, B.-Y. Li, R.-Y. Gan and H.-B. Li, Antioxidant Food Components for the Prevention and Treatment of Cardiovascular Diseases: Effects, Mechanisms, and Clinical Studies, Oxid. Med. Cell. Longevity, 2021, 2021, 1–17 Search PubMed.
- J. Ahamad, I. Toufeeq, M. A. Khan, M. S. M. Ameen, E. T. Anwer, S. Uthirapathy, S. R. Mir and J. Ahmad, Oleuropein: A natural antioxidant molecule in the treatment of metabolic syndrome, Phytother. Res., 2019, 33, 3112–3128 CrossRef CAS PubMed.
- M. A. Alam, N. Subhan, M. M. Rahman, S. J. Uddin, H. M. Reza and S. D. Sarker, Effect of Citrus Flavonoids, Naringin and Naringenin, on Metabolic Syndrome and Their Mechanisms of Action, Adv. Nutr., 2014, 5, 404–417 CrossRef CAS PubMed.
- H. Gong, A. T. Woolley and G. P. Nordin, High density 3D printed microfluidic valves, pumps, and multiplexers, Lab Chip, 2016, 16, 2450–2458 RSC.
- O. A. Zalevskaya, Y. A. Gur’eva, A. V. Kutchin, Y. R. Aleksandrova, E. Y. Yandulova, N. S. Nikolaeva and M. E. Neganova, Palladium complexes with terpene derivatives of ethylenediamine and benzylamine: Synthesis and study of antitumor properties, Inorg. Chim. Acta, 2021, 527, 120593 CrossRef CAS.
- M. Neganova, A. Semakov, Y. Aleksandrova, E. Yandulova, S. Pukhov, L. Anikina and S. Klochkov, N-Alkylation of Anthracycline Antibiotics by Natural Sesquiterpene Lactones as a Way to Obtain Antitumor Agents with Reduced Side Effects, Biomedicines, 2021, 9, 547 CrossRef CAS PubMed.
- S. R. Chowdhury, J. Djordjevic, B. C. Albensi, C. Benedict and P. Fernyhough, Simultaneous evaluation of substrate-dependent oxygen consumption rates and mitochondrial membrane potential by TMRM and safranin in cortical mitochondria, Biosci. Rep., 2016, 36, e00286 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed NMR, UV-Vis and FTIR characterization of synthesized complexes. See DOI: https://doi.org/10.1039/d3nj04562e |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |