
High stereoselectivity synthesis of Z-3-methyleneisoindolin-1-ones on a Cu/ETS-10 catalyst via domino coupling–cyclization without the use of protective groups and ligands†
Huiling
Hu
,
Changjun
Liu
*,
Chaojie
Zhu
,
Chenghong
Liu
and
Tiandi
Tang
*
Jiangsu Key Laboratory of Advanced Catalytic Materials and Technology, School of Petrochemical Engineering, Changzhou University, Changzhou, Jiangsu 213164, P. R. China. E-mail: 20070303032@smail.cczu.edu.cn; tangtiandi@cczu.edu.cn
First published on 6th December 2023
Abstract
Controlling the spatial selectivity of the coupling sites in two-molecule reactions is critical for stereoselective product synthesis. Herein, we report a novel strategy that uses basic ETS-10 zeolite-supported copper (Cu/ETS-10) as a catalyst for the highly selective synthesis of Z-3-methyleneisoindolin-1-ones in the absence of ligands. It was found that the rigidity of the zeolite skeleton was conducive to the high configurational selectivity of the product. The Cu/ETS-10 catalyst was easily recovered by simple filtration and could be reused five times without loss of its initial activity.
Introduction
Developing a simple and efficient catalytic system with high regioselectivity and stereoselectivity is critical for the synthesis of high-value fine chemicals.1 The 3-methyleneisoindolin-1-ones are very important structural moieties in natural extracts and bioactive molecules. In particular, Z-3-methyleneisoindolin-1-ones with good anaesthetic and sedative properties are significant blocks for synthesizing high-value compounds, such as natural products, functional materials and bioactive heterocyclic molecules.2A series of feasible strategies have been developed for the synthesis of Z-3-methyleneisoindolin-1-one compounds owing to their wide applications (Scheme 1), such as Wittig reaction (Scheme 1(a)),3 Aza–Heck reaction (Scheme 1(b)),4 Aza–Henry reaction (Scheme 1(c)),5 the cyanation-hydrolysis of 2-haloacetophenones (Scheme 1(d)),6 acid-catalyzed dehydration (Scheme 1(e))7 and phosphorylate condensation (Scheme 1(f)).8 However, these strategies suffer from various drawbacks, such as harsh reaction conditions, poor stereoselectivity, the pre-synthesis of starting materials and the complexity of multicomponent transformations, which are not conducive to practical applications.
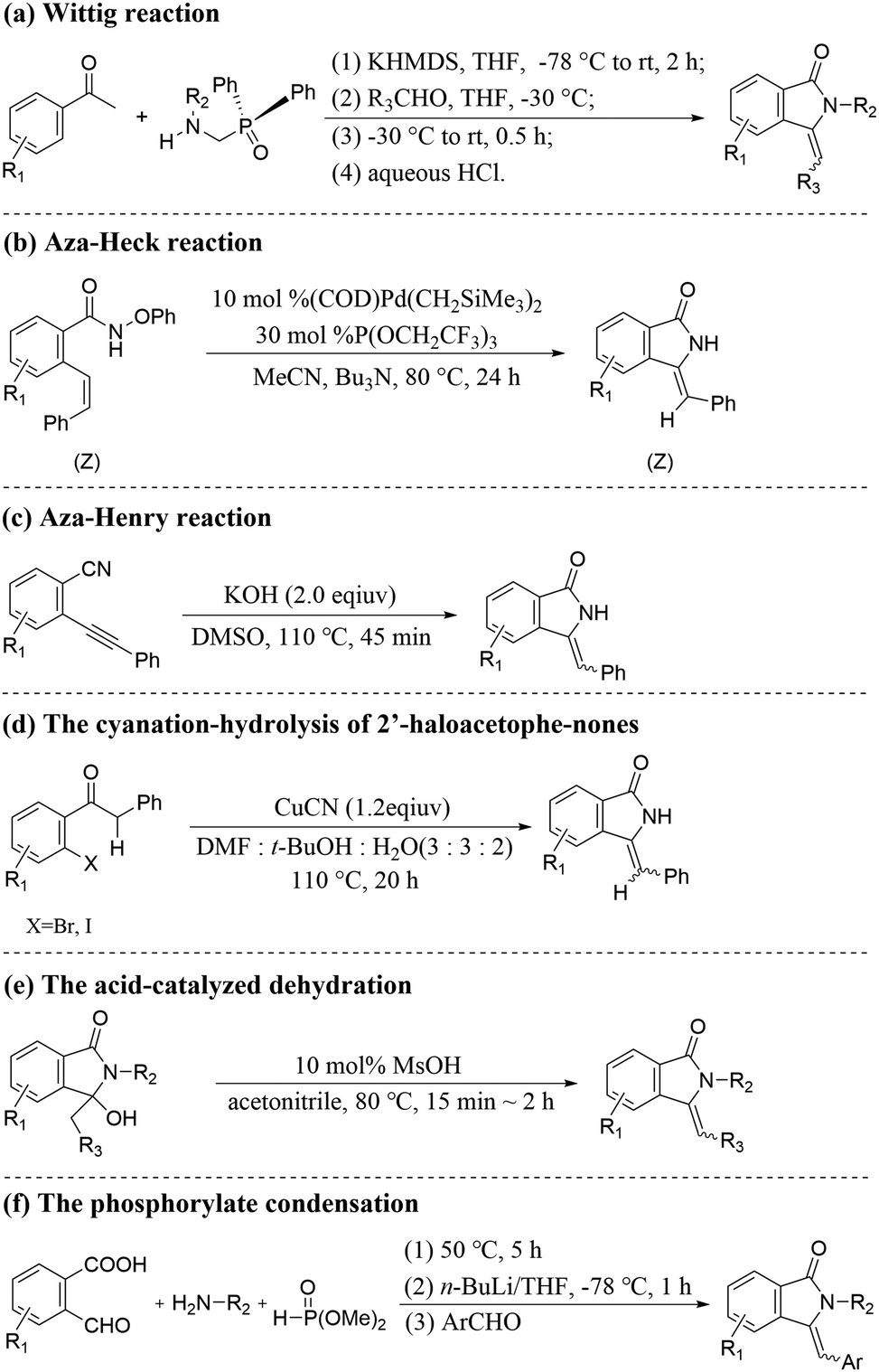 | ||
| Scheme 1 Some strategies for the synthesis of 3-methyleneisoindolin-1-ones. | ||
In contrast, the use of cheap and readily available 2-halobenzamides and aromatic terminal alkynes through domino coupling–cyclization to synthesize Z-3-methyleneisoindolin-1-ones is regarded as a direct and powerful method. In this method, various metal-catalyzed9 reaction systems have been developed, among which the cheap and easily available copper-based catalysts9c–f,h,i,k have captured wide attention. However, solving the problem of poor regio- and stereoselectivity is still one of the ongoing challenges in the synthesis processes.
In the process of modern organic synthesis, selective functional group protection and the introduction of ligands into metal-catalyzed systems are important strategies to synthesize complex organic compounds and achieve the required regioisomer.10 In order to synthesize Z-3-methyleneisoindolin-1-ones with high selectivity, the protected 2-halobenzamides were chosen as the starting substrate, such as 2-halo-N-phenylbenzamide9d and N-(quinolin-8-yl)benzamides9e,f (Scheme 2(a)). Although Z-3-methyleneisoindolin-1-ones can be highly stereoselectively obtained from these protected 2-halobenzamide compounds with directing groups, long reaction times and ligands are normally needed for improving the reaction activity. Additionally, an extra step of deprotection of the amine functionality of the final isoindoline is required, which can lead to the formation of by-products and complicate the reaction system. Considering these drawbacks, a series of ligands, such as L-proline,9c 2,2′-biimidazole,9d and D-glucosamine,9k have been reported to achieve target products with high stereoselectivity and avoid the use of protected 2-halobenzamides (Scheme 2(b)). The ligands can not only improve the catalytic activity, but also increase the steric hindrance of the formed Cu-L structure, which can generate efficiently Z-3-methyleneisoindolin-1-ones. However, the use of soluble metal salts as catalysts, along with the addition of costly organic ligands, could increase production costs and bring an environmental burden. Consequently, from the viewpoint of environmentally friendly processes and atomic economic efficiencies, it is highly desirable to develop a straightforward and efficient protocol to stereoselectively construct Z-3-methyleneisoindolin-1-ones.
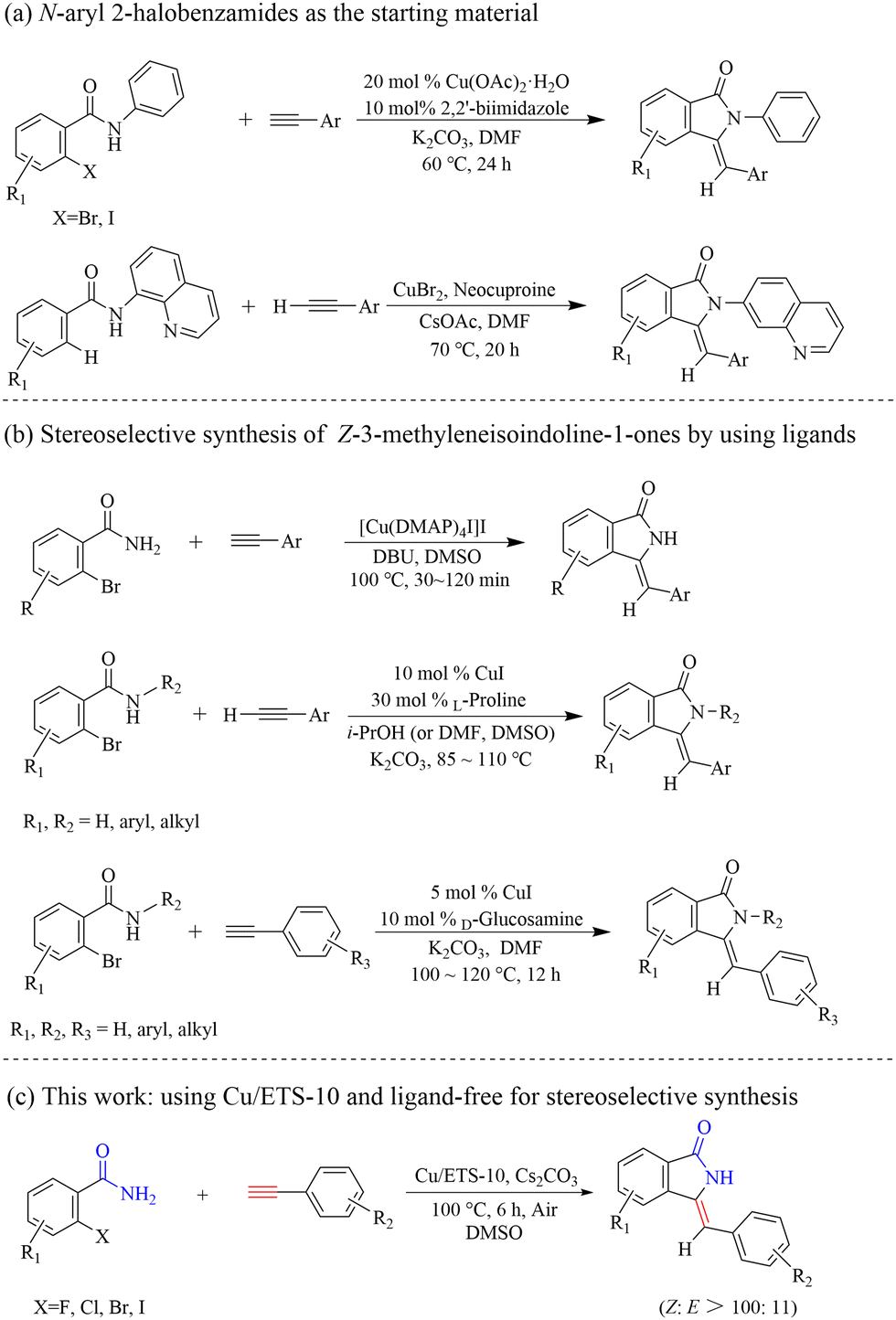 | ||
| Scheme 2 High stereoselective synthesis of Z-3-methyleneisoindolin-1-ones. | ||
It is well-known that zeolites with ordered microporous systems, distinct framework topologies, good spatial nanoconfinement effects, and superior thermal/chemical stabilities are ideal supports for metal catalysts in industry.11 The metal species can be easily introduced into the zeolite crystals by a variety of simple methods for constructing functional metal catalysts with strong metal–support interaction.11b,12 In particular, the unique framework structure and the surface acidic/basic sites of zeolites could decorate the electronic properties of the assembled metal species, which can improve their catalytic performance.11b,12b,13 In addition, the active sites in zeolite catalysts usually can afford the target products with high efficiency and selectivity due to their unique local chemical environment, such as coordination, pore confinement, structural rigidity, steric effects, etc., which can affect the spatial state of adsorbed molecules on its surface, and thus determine the direction of subsequent guest molecules to attack adsorbed molecules.14 The crystalline titanosilicate ETS-10 zeolite consists of corner-sharing tetrahedral silicon (SiO4) and octahedral titanium (TiO6)2− linked through bridging oxygen atoms, composing a three dimensional 12-ring pore system. The presence of each tetravalent Ti atom in an octahedron generates two negative charges, which are balanced by extra-framework Na+ and K+ cations.15 This framework structure of ETS-10 with K+, Na+, can prevent the formation of coke and therefore drastically increase the catalyst lifetime.16 The unique skeleton structure of ETS-10 endows it with strong basicity, which is suitable for application as a solid catalyst or support in some base-catalyzed reactions.17
Herein, we skillfully utilized the basic characteristics, steric effect and host–guest interaction with adsorbed molecules of ETS-10 to prepare a supported Cu catalyst (Cu/ETS-10) for the domino coupling–cyclization of 2-halobenzamides with alkynes (Scheme 2(c)). Under the condition that no protected 2-halobenzamides or ligands were used, the catalytic system can effectively produce Z-3-methyleneisoindolin-1-ones with high stereoselectivity.
Experimental section
Material synthesis
The zeolite ETS-10 was synthesized from a gel with the molar composition of 1.0TiO2:5.7SiO2:4.7Na2O:1.3K2O:163H2O. In a typical synthesis, 10 mL NaOH (26.9 wt%) solution and 11.8 mL KOH (17.5 wt%) solution were added to a 15.2 mL water glass with stirring for 60 min at room temperature. Then, 13 g TiCl3 (17 wt%) was slowly added to the above mixture. The obtained gel was stirred for 2 h and transferred into a 50 mL autoclave to crystallize at 230 °C for 3 days. After filtrating, drying, and calcining at 475 °C for 5 h, the solid sample of ETS-10 was obtained. The zeolite Beta, ZSM-5 and Silicalite-1 were synthesized according to the procedure described in our previous work.18The zeolite supported Cu catalyst (Cu/ETS-10) with Cu loading of 3.0 wt% was prepared by the incipient wetness impregnation method using the required amount of copper nitrate aqueous solution. After impregnation, the sample was placed in an air atmosphere for 8 h, further dried at 120 °C for 12 h, and calcined in air at 450 °C for 4 h. The corresponding catalyst samples were designated as Cu/ETS-10, Cu/Beta, Cu/ZSM-5 and Cu/Silicalite-1, respectively.
Characterization
The X-ray diffraction (XRD) patterns of the supports and calcined catalysts were obtained on a D/MAX 2500/PC powder (Rigaku) using Cu Kα radiation.Nitrogen physisorption measurements were carried out using a Micromeritics TriStar II 3020 apparatus. Before the measurement, the sample was degassed for 4 h at 200 °C. Pore size distribution was calculated using the Barrett–Joyner–Halenda (BJH) model.
The X-ray photoelectron spectroscopic (XPS) experiment of the catalyst was performed on an ESCALAB MK II system to obtain the chemical valence of the metal species.
Scanning electron microscopy (SEM) images of the sample were carried out on a SUPRA 5 field emission electron microscope with an accelerating voltage of 5 kV to observe the morphology of the samples. A transmission electron microscopy (TEM) experiment was performed on a JEM-2100F microscope with a limited line resolution capacity of 1.4 Å, under a voltage of 200 kV. Before characterization by TEM, the sample was dispersed in ethanol and dropped on a Cu-grid coated with carbon membrane.
Infrared (IR) spectra of the substrate chemisorbed catalyst sample were obtained on a Bruker TENSOR 27 infrared spectroscope equipped with an in situ reactor cell.
Thermogravimetric (TG) analysis was performed at a ramp of 10 °C min−1 under flowing N2 (20 mL min−1) on an STA 6000 instrument. The TG sample was approximately 10 mg.
Catalytic activity measurements
Unless otherwise stated, all reagents were obtained from commercial sources (purity >99%) and were used without further purification. A typical procedure for the coupling–cyclization of 2-halobenzamides with terminal alkynes was as follows. A mixture of 2-iodobenzamide (0.7 mmol), phenylacetylene (0.5 mmol), 1,3,5-trimethylbenzene (as internal standard, 0.5 mmol), catalysts (30 mg), Cs2CO3 (0.1 mmol) and DMSO (1.5 mL) was added in a 10 mL reaction tube. The reaction mixture was heated at 100 °C under vigorous stirring (500 rpm) for the required time (6 h). After reaction, the catalyst was removed by filtration. The liquid product was collected and analyzed by an Agilent 7890B GC (gas chromatography). The phenylacetylene conversion and product selectivity were calculated as follows.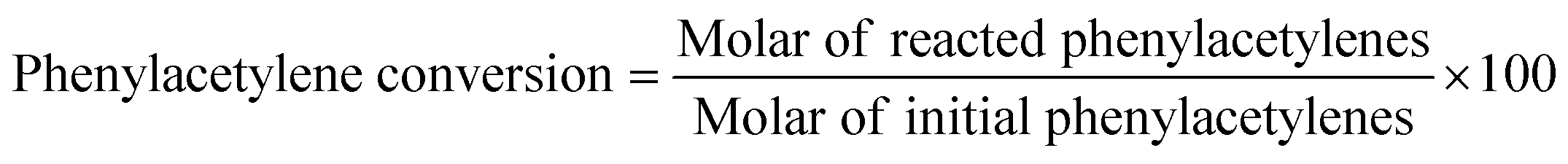
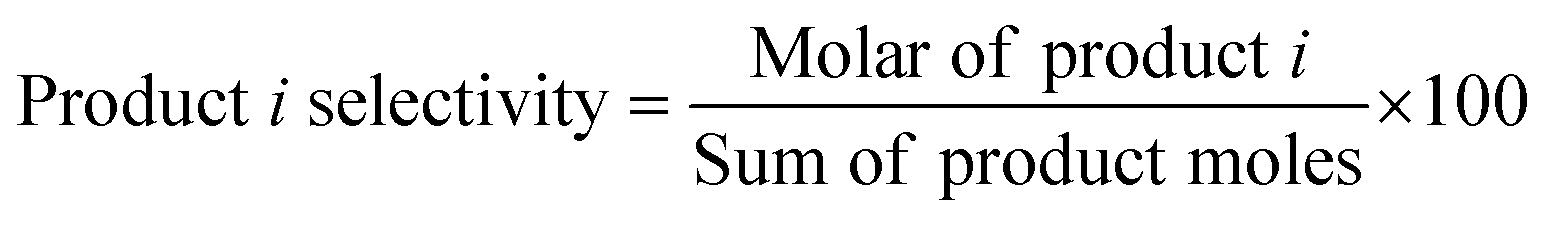
| Product i yield = Phenylacetylene conversion × Product i selectivity × 100 |
Results and discussion
The XRD pattern in Fig. 1(a) shows that the Cu/ETS-10 has characteristic peaks in the range of 5–50°, associated with the ETS-10 zeolite structure. After loading Cu and calcination, Cu/ETS-10 exhibited a slightly weaker diffraction peak intensity compared to ETS-10, suggesting that the crystallinity of the zeolite was marginally reduced. Notably, no obvious characteristic peaks associated with Cu species were observed in the XRD pattern of the Cu/ETS-10 sample, indicating that the Cu species with small particle sizes were well-dispersed in the Cu/ETS-10.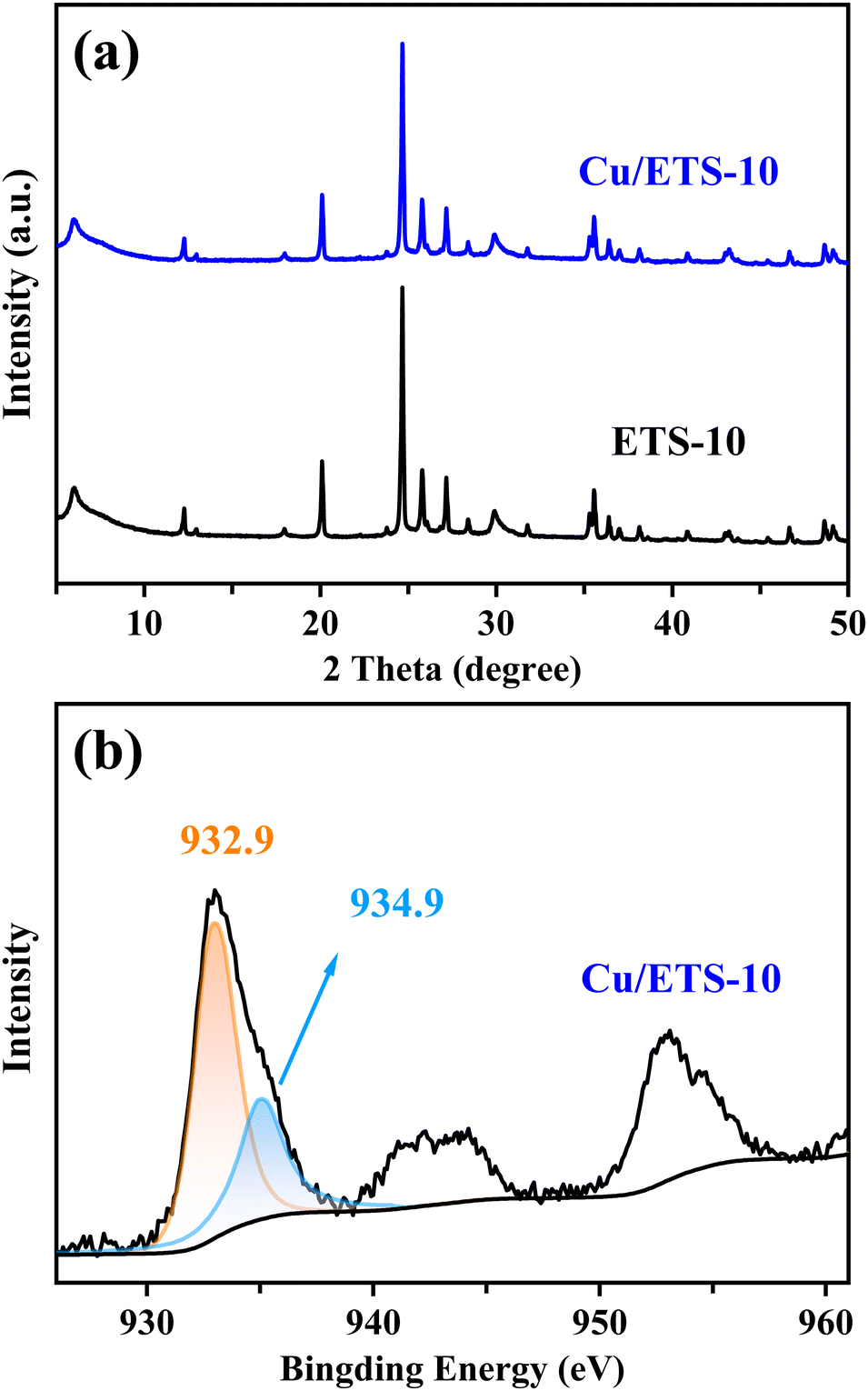 | ||
| Fig. 1 (a) XRD patterns of the ETS-10 and Cu/ETS-10 samples. (b) XPS spectrum of the Cu/ETS-10 sample. | ||
To investigate the electron state of the Cu species in the Cu/ETS-10 catalyst, XPS analysis was performed. For the Cu/ETS-10 catalyst, the XPS spectrum of Cu2p3/2 showed two binding energies at approximately 932.9 and 934.9 eV (Fig. 1(b)), which were characteristic of the Cu+ and Cu2+ species, respectively.19 The XPS spectra revealed that the higher content of Cu+ in the Cu/ETS-10 catalyst compared to Cu2+ was attributed to the presence of a strong metal–support interaction on the catalyst, leading to the transfer of electrons from the framework oxygen to the Cu species.20 Dynamic transformation, such as valence states of the metal caused by metal–support interaction, are ubiquitous for supported metal catalysts during catalytic processes.11b
The SEM image in Fig. 2(a) shows that the ETS-10 zeolite exhibited a quadrate-tapered morphology with large particles with a size of 8–10 μm, which could be conducive to the formation of good specific surface area, external surface area (375 and 104 m2 g−1, Table 1) and structural rigidity. After loading Cu and calcination, these corresponding texture parameters (367 and 98 m2 g−1) did not change obviously. Additionally, it was evident from Fig. 2(b) that most of the Cu nanoparticles with a size of 2–5 nm were well dispersed on the surface of the ETS-10 zeolite.
 | ||
| Fig. 2 (a) SEM images of ETS-10. (b) TEM images of Cu/ETS-10. | ||
We initiated our investigations with the use of 2-iodobenzamide (1a) and phenylacetylene (2a) as the model substrate for the optimization of the reaction conditions (Table 2). The reaction did not occur in the absence of the catalyst or just introducing ETS-10 as a catalyst (entries 1 and 2). In addition, the bases played an important role in this reaction because only 3% of 3a was generated without them (entry 3). Commonly used copper salts, such as CuO2, Cu(NO3)2, Cu2O, and CuI, were proven to be active, but still suffered from low yields of 3a (12–29%, entries 4–7) and low Z-isomer selectivity of 3a (43–62%). Moreover, the large number of by-product 4a of phenylacetylene coupling (27–57%) reduced the atom economy of the coupling–cyclization reaction. These results demonstrated that the use of Cu salt alone as a catalyst did not efficiently catalyze this reaction.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Base | Solvent | Yieldb (%) | TONd | |
3a (Z![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :E)c :E)c |
4a | |||||
| a Reaction conditions: 1a (0.7 mmol), 2a (0.5 mmol), catalyst (30 mg), base (20 mol%, 0.1 mmol), solvent (1.5 mL), at 100 °C for 6 h. b Obtained by GC. The internal standard method was used with 1,3,5-trimethylbenzene as the internal standard. c The ratio of Z and E was determined by 1H NMR analysis of the product 3a mixture. d TON, the conversion number of reactant molecules was based on the unit mole number of metals. e N.d. = not detected. f The Cu content was equivalent to that of 30 mg of Cu/ETS-10. | ||||||
| 1 | None | Cs2CO3 | DMSO | N.d.e | N.d. | — |
| 2 | ETS-10 | Cs2CO3 | DMSO | N.d. | N.d. | — |
| 3 | Cu/ETS-10 | None | DMSO | 3 | 2 | 1.7 |
| 4 | CuOf | Cs2CO3 | DMSO | 29 (59:41) |
29 | 20.4 |
| 5 | Cu(NO3)2f | Cs2CO3 | DMSO | 12 (62:38) |
27 | 13.7 |
| 6 | Cu2Of | Cs2CO3 | DMSO | 25 (50:50) |
35 | 21.1 |
| 7 | CuIf | Cs2CO3 | DMSO | 14 (43:57) |
57 | 25.0 |
| 8 | Cu/ETS-10 | Cs2CO3 | DMSO | 97 (100:0) |
3 | 35.3 |
| 9 | Cu/ETS-10 | K2CO3 | DMSO | 81 (100:0) |
11 | 32.4 |
| 10 | Cu/ETS-10 | Na2CO3 | DMSO | 58 (100:0) |
8 | 23.3 |
| 11 | Cu/ETS-10 | KOH | DMSO | 71 (100:0) |
7 | 27.5 |
| 12 | Cu/ETS-10 | NaOH | DMSO | 56 (100:0) |
16 | 25.4 |
| 13 | Cu/ETS-10 | NEt3 | DMSO | 21 (100:0) |
13 | 12.0 |
| 14 | Cu/ETS-10 | Cs2CO3 | Toluene | 19 (100:0) |
21 | 14.1 |
| 15 | Cu/ETS-10 | Cs2CO3 | Dioxane | 24 (100:0) |
10 | 12.0 |
| 16 | Cu/ETS-10 | Cs2CO3 | THF | 38 (100:0) |
11 | 17.2 |
| 17 | Cu/ETS-10 | Cs2CO3 | DCE | 27 (100:0) |
34 | 21.5 |
| 18 | Cu/ETS-10 | Cs2CO3 | DMF | 67 (100:0) |
19 | 30.3 |
| 19 | Cu/ETS-10 | Cs2CO3 | MeCN | 53 (100:0) |
17 | 24.7 |
| 20 | Cu/Beta | Cs2CO3 | DMSO | 31 (89:11) |
2 | 11.6 |
| 21 | Cu/ZSM-5 | Cs2CO3 | DMSO | 44 (85:15) |
9 | 18.7 |
| 22 | Cu/Silicalite-1 | Cs2CO3 | DMSO | 53 (83:17) |
5 | 20.4 |
In contrast, Cu/ETS-10 can boost this reaction in high yield (97%, entry 8). In addition, the good performance of Cu/ETS-10 catalyst depended on the employed bases and solvents. Using different bases, such as Cs2CO3, K2CO3, Na2CO3, KOH, NaOH and NEt3 (entries 8–13) or different solvents, such as dimethyl sulfoxide (DMSO), toluene, dioxane, tetrahydrofuran (THF), dichloroethane (DCE), N,N-dimethylformamide (DMF), and acetonitrile (MeCN) (entries 8, 14–19), the yield of product 3a was significantly different and the base Cs2CO3 and solvent DMSO were beneficial to the coupling–cyclization reaction. When using other zeolites with a large number of acid sites, such as Beta and ZSM-5, and Silicalite-1 (it has almost no acidity) as supports (entries 20–22), relatively low yields of 3a (31–53%) were obtained.
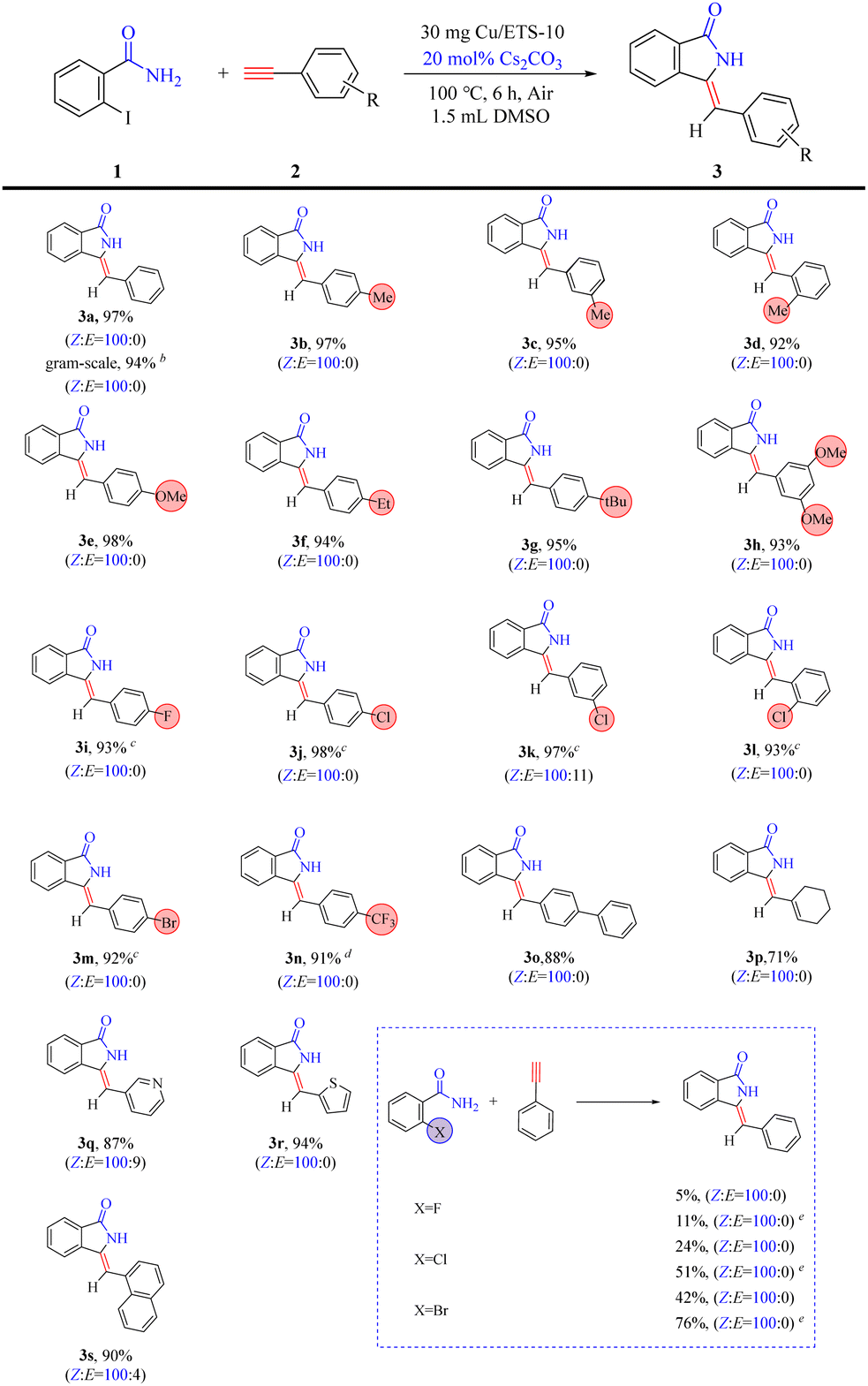
Having the optimized reaction conditions in hand, the scope of the reaction was explored. The protocol catalyzed by Cu/ETS-10 was proven to be general (Table 3). The reaction was tolerant to the electronics of alkynes, as electron-neutral (3a), electron-rich (3b–3h), and electron-deficient (3i–3n) alkynes reacted to yield the corresponding products in good yield (>91%) and high Z-isomer selectivity (>90%). However, phenylacetylene compounds containing electron-deficient groups required a longer reaction time. The heterocyclic and polycyclic alkynes (3o–3s) also exhibited superior coupling–cyclization ability with good product yields (>87%) and high ratios of Z:E. Moreover, this protocol can be used in a gram-scale experiment without a significant decrease in yield and stereoselectivity (entry 3a).
| a Reaction conditions: 1 (0.7 mmol), 2 (0.5 mmol), Cu/ETS-10 (30 mg), and Cs2CO3 (0.1 mmol) in 1.5 mL DMSO at 100 °C for 6 h; the yields were calculated by GC. The internal standard method was used with 1,3,5-trimethylbenzene as the internal standard. The ratio of Z and E was determined by 1H NMR analysis of the product 3 mixture. b 1 (70 mmol), 2 (50 mmol), Cu/ETS-10 (3.0 g), Cs2CO3 (20 mol%, 10 mmol) in 150 mL DMSO at 100 °C for 24 h. c 8 h. d 10 h. e 12 h. |
|---|
|
Furthermore, as shown in Table 3, the reaction conditions described above were found to be less efficient for the coupling–cyclization of fluorine, chlorine, and bromine-substituted benzamide. This was likely attributed to the strong electronegativity of these elements, which hindered the cyclization process in the domino reaction. Surprisingly, a high selectivity of the Z-isomer was still achieved from these low-activity substrates, suggesting that the rigidity of the catalyst crystal favored the formation of the Z-isomer.
The results in Table 3 showed that the substrates with electron-rich groups had higher reactivity relative to the ones with electron-deficient groups. Thus, the competition reactions between alkynes containing different electronic properties with 1a were further carried out (Scheme 3). With the electron-deficient trifluoromethyl-derived alkyne (2n) and electron-rich methoxy-derived alkyne (2e) as mixed substrates, the yields of 3n and 3e were 29% and 56%, respectively (Scheme 3(a)).
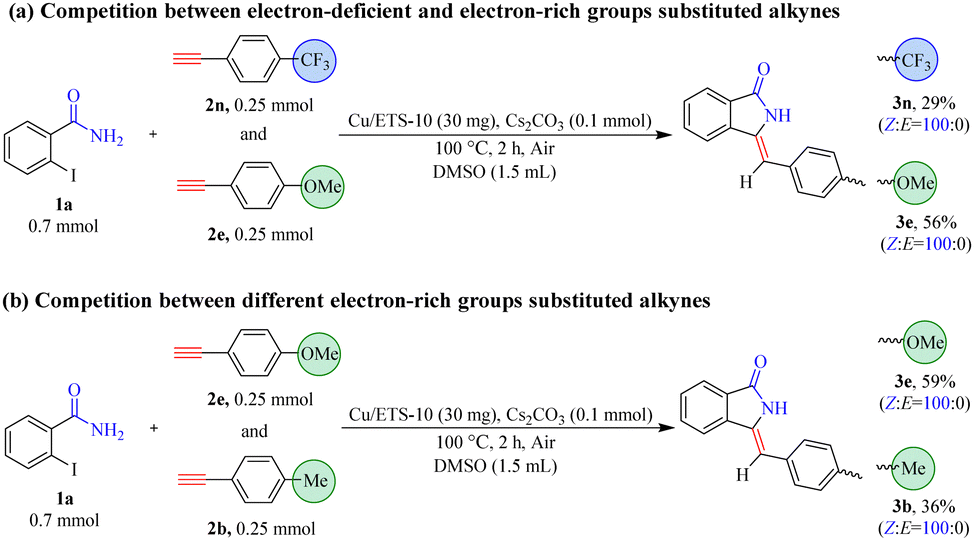 | ||
| Scheme 3 (a) Competitive reaction between substrates containing an electron-deficient group (2n) and an electron-rich group (2e). (b) Competitive reaction between substrates containing different electron-rich groups (2b and 2e). | ||
When the strong electron-rich methoxy-derived alkyne (2e) and relatively mild electron-rich methyl-derived alkyne (2b) were mixed as mixed substrates (Scheme 3(b)), the yield of 3e (59%) was higher than that of 3b (36%). These results demonstrated that the electronic nature of the phenylacetylene compounds was an important factor that can affect the subsequent coupling and/or cyclization reaction, and the reactivity of substrates with electron-rich groups was higher than the ones with electron-deficient groups. This was most likely due to alkynes with high electron density facilitating coordination with metals to form metal–π-complex intermediates, which can promote subsequent coupling reactions with 2-iodobenzamide compounds.21
In order to explore the influence of the rigid crystal structure of the catalyst on the configurational selectivity during the reaction, the Z- and E-isomer distributions of the coupling–cyclization reaction of phenylacetylene with 2-iodobenzamide were investigated at different reaction temperatures (Fig. 3, the detailed experimental procedure see the ESI†). When CuO was employed as the catalyst, the selectivity of the Z-isomer was strongly affected by temperature, increasing from 37% to 59% as the temperature increased (Fig. 3 up). As expected, when using Cu/ETS-10 with structural rigidity as the catalyst, the stereoselectivity of the Z-isomer was always 100% regardless of the temperature (Fig. 3 down).
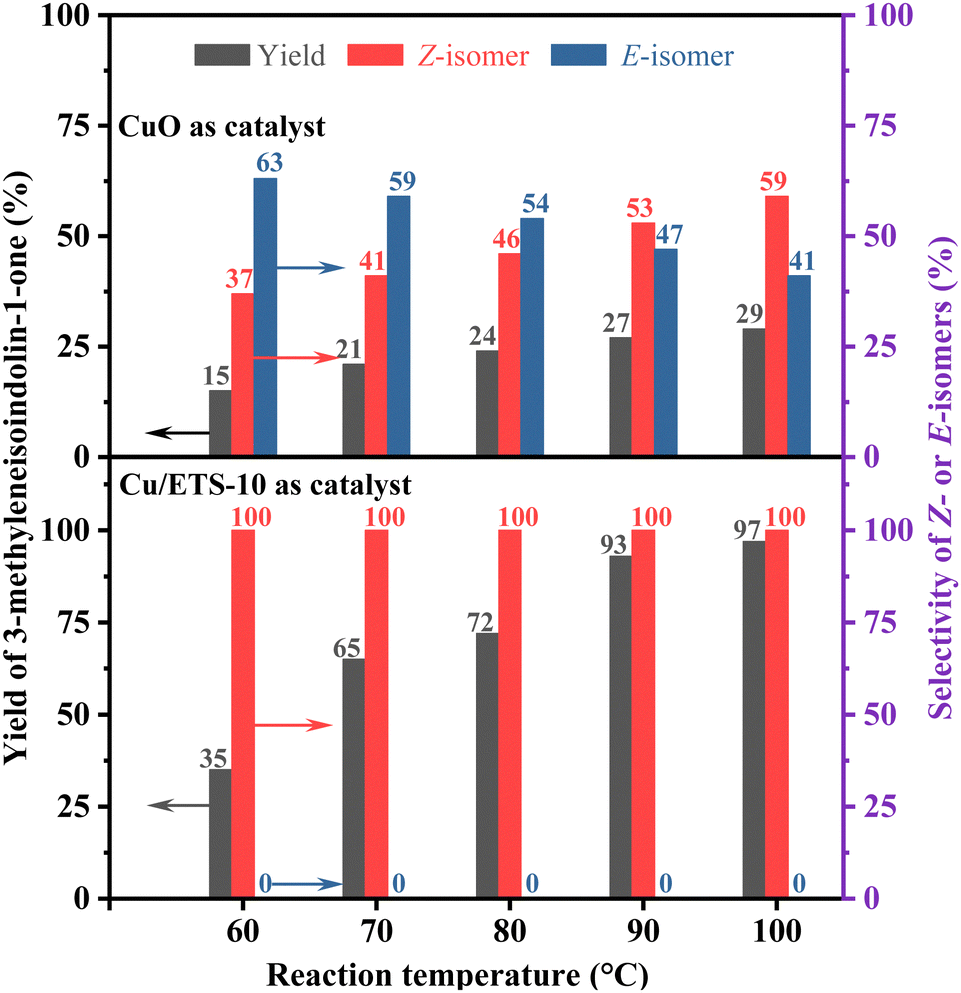 | ||
| Fig. 3 The effect of reaction temperature on the product yield and selectivity over the CuO (up) and Cu/ETS-10 (down) catalysts. | ||
To gain a preliminary understanding of the elementary reaction steps for the coupling–cyclization reaction, several control experiments were performed to explore the initial activation step (Scheme 4). The pre-activation of 2a by Cu/ETS-10 at 100 °C for 1 h can greatly promote the coupling–cyclization reaction and obtain a higher yield of 3a (66%, Scheme 4(c)) than the standard reaction (37%, Scheme 4(a)) and pre-activation of 1a (41%, Scheme 4(b)). These results suggested that the intermediate generated through the activation of 2a by Cu/ETS-10 effectively promoted the progress of the reaction. The activation of 2a by Cu/ETS-10 was also confirmed by IR spectroscopy (Fig. 4). At room temperature, the absorption band at 2109 cm−1 was assigned to the stretching vibration of the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond in the phenylacetylene molecule. When the sample was heated to 100 °C at 10 °C min−1 and kept at 100 °C, this stretching vibration gradually shifted to 2101 cm−1.
C bond in the phenylacetylene molecule. When the sample was heated to 100 °C at 10 °C min−1 and kept at 100 °C, this stretching vibration gradually shifted to 2101 cm−1.
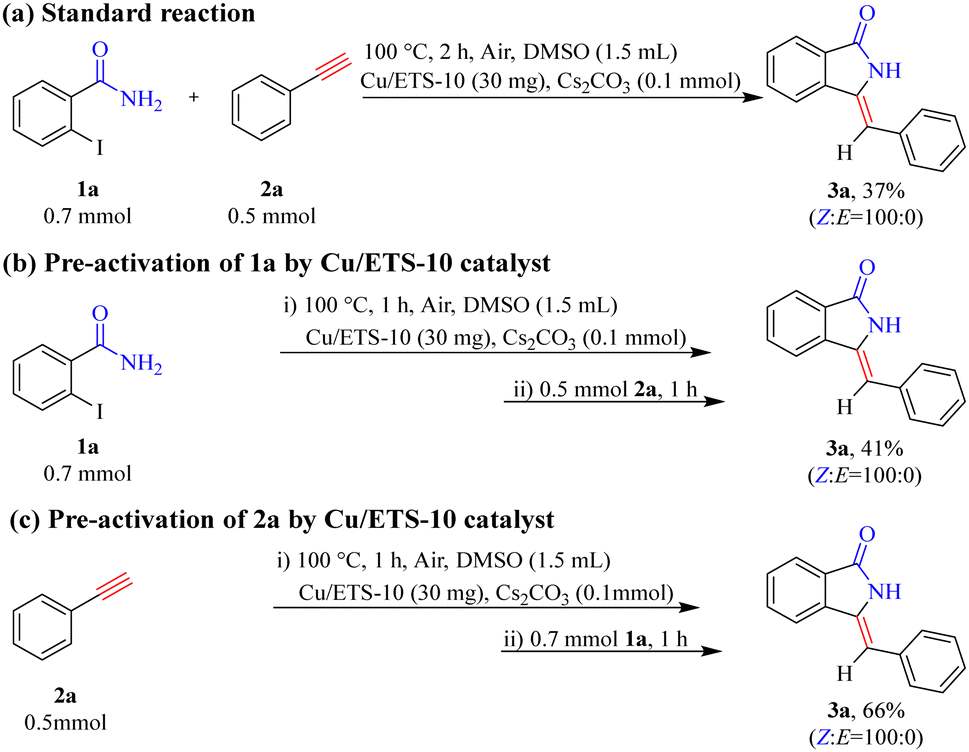 | ||
| Scheme 4 The control experiments. | ||
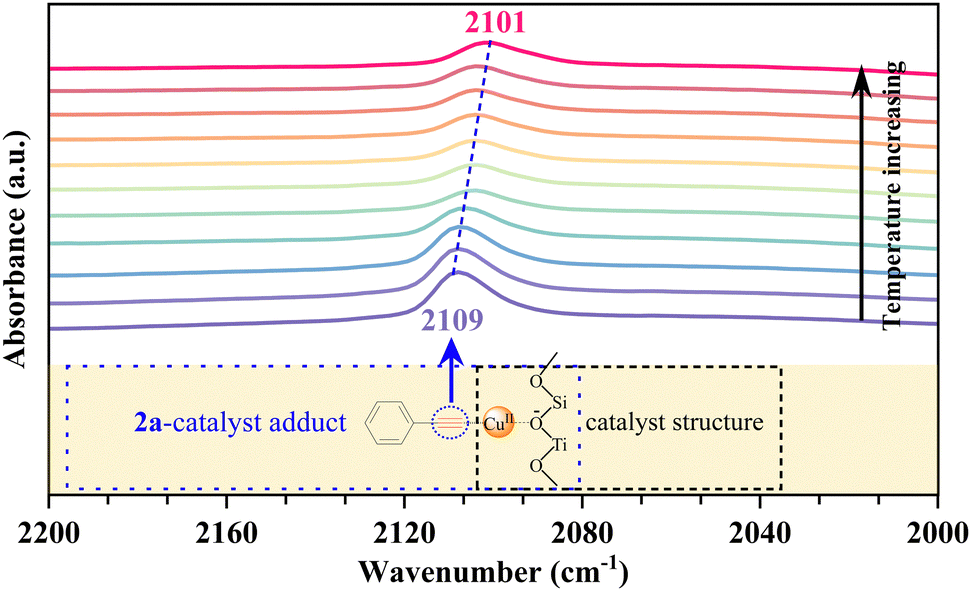 | ||
| Fig. 4 The IR spectroscopy of the Cu/ETS-10 catalyst absorbed 2a. | ||
Combining the discussions above and literature reports,9c,22 a proposed mechanism of the Z-3-methyleneisoindolin-1-ones synthesis catalyzed by Cu/ETS-10 was depicted in Scheme 5. Firstly, the CuI species on Cu/ETS-10 can convert dynamically (CD) into CuII species, then phenylacetylene was dehydrogenated by the base and coordinated with CuII sites of Cu/ETS-10 to form a metal–π-complex intermediate A. The intermediate A underwent a Sonogashira type coupling with the C–I bond of 2-iodobenzamide and then coordinated with CuII sites again to form the intermediate B. It is worth noting that the electron-rich groups can effectively enhance the electron cloud density of the alkynyl groups, which can increase the stability of the metal–π-complex intermediates A and B. In this case, the electron pairs transferred from the CC bond of intermediates B to CuII sites accompanying the deprotonation of the amide moiety, and an intermediate C was formed. This amino anion triggers the cyclization reaction by nucleophilic attack on the proximate carbocation to generate a Cu0-stabilized cyclized intermediate D that has high Z-isomer selectivity due to the structural rigidity and steric resistance of the Cu/ETS-10 catalyst. Finally, with the transference of electron pairs from Cu0 to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of intermediate D and the reduction of the Z-product by base-H, the Z-3-methyleneisoindolin-1-one was formed accompanying the regeneration of the catalyst.
C bond of intermediate D and the reduction of the Z-product by base-H, the Z-3-methyleneisoindolin-1-one was formed accompanying the regeneration of the catalyst.
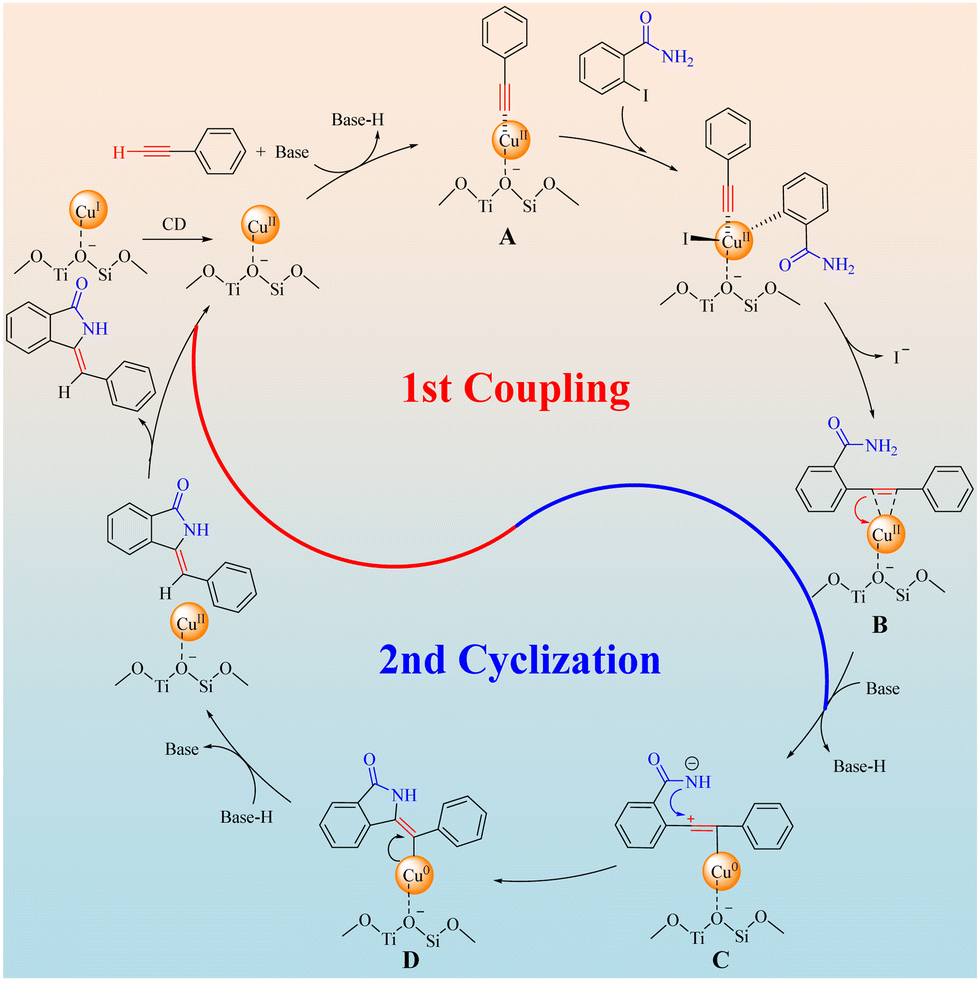 | ||
| Scheme 5 The proposed mechanism. | ||
The reusability of the catalyst was also surveyed (Fig. 5). At the end of the reaction, the catalyst was filtered and separated, then subsequently washed with ethyl acetate. The obtained solid catalyst was dried at 120 °C for 12 h before the next cycle. The results demonstrated that the catalyst can be reused at least five times without significant activity loss (>89% yield of Z-3a).
 | ||
| Fig. 5 The reusability of the Cu/ETS-10 catalyst. | ||
The spent catalyst was analyzed by XRD and TG. The characteristic diffraction peaks assigned to the ETS-10 crystal structure in the range of 5–50° were well maintained for the spent Cu/ETS-10 catalyst (Fig. 6(a)), indicating that the zeolite structure was not affected by the reaction. The zeolite framework in the Cu/ETS-10 catalyst played an important role in its high catalytic stability. In addition, the spent and fresh catalysts exhibited similar weight loss by the TG analyses (Fig. 6(b)), indicating that a large amount of coke was not formed during the coupling–cyclization reaction over Cu/ETS-10. In addition, compared with fresh Cu/ETS-10, the binding energy of Cu2p3/2 for the spent Cu/ETS-10 sample did not change, which indicated that the surface structure of the catalyst was maintained after the reaction (Fig. S1, p. S4, in Section 5 of ESI†). These results all indicated that Cu/ETS-10 has good practical application value.
 | ||
| Fig. 6 (a) XRD patterns of the spent and fresh Cu/ETS-10 catalysts. (b) TG curves of the spent and fresh Cu/ETS-10 catalysts. | ||
Conclusions
In summary, we have developed a practical and highly efficient method for synthesizing Z-3-methyleneisoindolin-1-ones through the cyclization coupling of 2-iodobenzamide with phenylacetylene. The catalytic system composed of a basic ETS-10 zeolite-supported copper catalyst with strong metal–support interaction exhibited excellent reactivity and stereoselectivities. Preliminary studies found that the structural rigidity of the zeolite crystal can determine the stereo configuration of the intermediate. This method has good catalyst stability and wide substrate scope. Moreover, the gram-scale synthesis also highlighted the synthetic practicality of this method.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21972011 and 22178029). The authors thank the anonymous reviewers for their invaluable advice.Notes and references
- (a) M. J. Climent, A. Corma and S. Iborra, Chem. Rev., 2011, 111, 1072–1133 CrossRef CAS PubMed; (b) Y. Li, L. Li and J. Yu, Chem, 2017, 3, 928–949 CrossRef CAS.
- (a) E. De Clercq, J. Med. Chem., 1995, 38, 2491–2517 CrossRef CAS PubMed; (b) H. M. Botero Cid, C. Tränkle, K. Baumann, R. Pick, E. Mies-Klomfass, E. Kostenis, K. Mohr and U. Holzgrabe, J. Med. Chem., 2000, 43, 2155–2164 CrossRef CAS; (c) Y. Chia, F. Chang, C. Teng and Y. Wu, J. Nat. Prod., 2000, 63, 1160–1163 CrossRef CAS.
- A. Couture, E. Deniau and P. Grandclaudon, Tetrahedron, 1997, 53, 10313–10330 CrossRef CAS.
- S. A. Shuler, G. Yin, S. B. Krause, C. M. Vesper and D. A. Watson, J. Am. Chem. Soc., 2016, 138, 13830–13833 CrossRef CAS PubMed.
- S. Verma, M. Kumar and A. K. Verma, Org. Lett., 2020, 22, 130–134 CrossRef CAS PubMed.
- T. Banik and K. P. Kaliappan, Chem. – Eur. J., 2021, 27, 628–633 CrossRef CAS PubMed.
- N. Topolovčan, F. Duplić and M. Gredičak, Eur. J. Org. Chem., 2021, 3920–3924 CrossRef.
- M. A. Reyes-González, A. Zamudio-Medina and M. Ordóñez, Tetrahedron Lett., 2012, 53, 5756–5758 CrossRef.
- (a) N. G. Kundu and M. W. Khan, Tetrahedron, 2000, 56, 4777–4792 CrossRef CAS; (b) S. Couty, B. Liegault, C. Meyer and J. Cossy, Tetrahedron, 2006, 62, 3882–3895 CrossRef CAS; (c) L. Li, M. Wang, X. Zhang, Y. Jiang and D. Ma, Org. Lett., 2009, 11, 1309–1312 CrossRef CAS; (d) J. Pan, Z. Xu, R. Zeng and J. Zou, Chin. J. Chem., 2013, 31, 1022–1026 CrossRef CAS; (e) J. Dong, F. Wang and J. You, Org. Lett., 2014, 16, 2884–2887 CrossRef CAS PubMed; (f) Y. Zhang, Q. Wang, H. Yu and Y. Huang, Org. Biomol. Chem., 2014, 12, 8844–8850 RSC; (g) L. Zhang, X. Hao, Z. Liu, X. Zheng, S. Zhang, J. Niu and M. Song, Angew. Chem., Int. Ed., 2015, 54, 10012–10015 CrossRef CAS; (h) S. B. Munoz, A. N. Aloia, A. K. Moore, A. Papp, T. Mathew, S. Fustero, G. A. Olah and G. K. Surya Prakash, Org. Biomol. Chem., 2016, 14, 85–92 RSC; (i) D. Hazarika and P. Phukan, Chemistryselect, 2018, 3, 2474–2478 CrossRef CAS; (j) B. C. Fillmore, J. Price, R. Dean, A. A. Brown, A. Decken and S. Eisler, Acs Omega, 2020, 5, 22914–22925 CrossRef CAS; (k) S. K. Singh, M. S. Yadav, A. S. Singh, A. K. Agrahari, N. Mishra, S. Kumar and V. K. Tiwari, Acs Omega, 2021, 6, 21125–21138 CrossRef CAS.
- (a) G. Sartori, R. Ballini, F. Bigi, G. Bosica, R. Maggi and P. Righi, Chem. Rev., 2004, 104, 199–250 CrossRef CAS; (b) L. Ping, D. S. Chung, J. Bouffard and S. Lee, Chem. Soc. Rev., 2017, 46, 4299–4328 RSC; (c) D. Wang, A. B. Weinstein, P. B. White and S. S. Stahl, Chem. Rev., 2018, 118, 2636–2679 CrossRef CAS PubMed; (d) R. Connon, B. Roche, B. V. Rokade and P. J. Guiry, Chem. Rev., 2021, 121, 6373–6521 CrossRef CAS.
- (a) X. Wang, Y. Ma, Q. Wu, Y. Wen and F. Xiao, Chem. Soc. Rev., 2022, 51, 2431–2443 RSC; (b) Q. Zhang, S. Gao and J. Yu, Chem. Rev., 2023, 123, 6039–6106 CrossRef CAS PubMed.
- (a) D. Kubička, N. Kumar, T. Venäläinen, H. Karhu, I. Kubičková, H. Österholm and D. Y. Murzin, J. Phys. Chem. B, 2006, 110, 4937–4946 CrossRef PubMed; (b) Q. Sun, N. Wang and J. Yu, Adv. Mater., 2021, 33, 2104442 CrossRef CAS.
- Z. Hu, J. Han, Y. Wei and Z. Liu, ACS Catal., 2022, 12, 5060–5076 CrossRef CAS.
- (a) J. Sivaguru, T. Poon, R. Franz, S. Jockusch, W. Adam and N. J. Turro, J. Am. Chem. Soc., 2004, 126, 10816–10817 CrossRef CAS; (b) S. Saravanamurugan, A. Riisager, E. Taarning and S. Meier, Chem. Commun., 2016, 52, 12773–12776 RSC; (c) Q. Zhang, J. Yu and A. Corma, Adv. Mater., 2020, 32, 2002927 CrossRef; (d) L. Liu, M. Lopez-Haro, D. M. Meira, P. Concepcion, J. J. Calvino and A. Corma., Angew. Chem., Int. Ed., 2020, 59, 15695–15702 CrossRef CAS.
- (a) M. W. Anderson, O. Terasaki, T. Ohsuna, A. Philippou, S. P. MacKay, A. Ferreira, J. Rocha and S. Lidin, Nature, 1994, 367, 347–351 CrossRef CAS; (b) N. C. Jeong, Y. J. Lee, J. Park, H. Lim, C. Shin, H. Cheong and K. B. Yoon, J. Am. Chem. Soc., 2009, 131, 13080–13092 CrossRef CAS.
- I. Yarulina, K. De Wispelaere, S. Bailleul, J. Goetze, M. Radersma, E. Abou-Hamad, I. Vollmer, M. Goesten, B. Mezari, E. J. M. Hensen, J. S. Martínez-Espín, M. Morten, S. Mitchell, J. Perez-Ramirez, U. Olsbye, B. M. Weckhuysen, V. Van Speybroeck, F. Kapteijn and J. Gascon, Nat. Chem., 2018, 10, 804–812 CrossRef CAS PubMed.
- (a) A. Philippou, J. Rocha and M. W. Anderson, Catal. Lett., 1999, 57, 151–153 CrossRef CAS; (b) Y. Goa, P. Wu and T. Tatsumi, J. Catal., 2004, 224, 107–114 CrossRef CAS.
- (a) T. Tang, C. Yin, L. Wang, Y. Ji and F. Xiao, J. Catal., 2007, 249, 111–115 CrossRef CAS; (b) W. Fu, L. Zhang, D. Wu, M. Xiang, Q. Zhuo, K. Huang, Z. Tao and T. Tang, J. Catal., 2015, 330, 423–433 CrossRef CAS; (c) W. Fu, L. Zhang, T. Tao and T. Tang, J. Ind. Eng. Chem., 2021, 95, 376–387 CrossRef CAS.
- (a) F. Bin, X. Wei, B. Li and K. S. Hui, Appl. Catal., B, 2015, 162, 282–288 CrossRef CAS; (b) S. Chen, Z. Shao, Z. Fang, Q. Chen, T. Tang, W. Fu, L. Zhang and T. Tang, J. Catal., 2016, 338, 38–46 CrossRef CAS.
- (a) P. Vanelderen, J. Vancauwenbergh, B. F. Sels and R. A. Schoonheydt, Coord. Chem. Rev., 2013, 257, 483–494 CrossRef CAS; (b) W. Fu, C. Yin, Y. Feng, L. Zhang, F. Cheng, Z. Fang, C. Zhu and T. Tang, Appl. Catal., A, 2021, 609, 117907 CrossRef CAS.
- (a) R. Chinchilla and C. Nájera., Chem. Rev., 2007, 107, 874–922 CrossRef CAS PubMed; (b) S. Yamada, Chem. Rev., 2018, 118, 11353–11432 CrossRef CAS PubMed.
- (a) S. Dhara, R. Singha, M. Ghosh, A. Ahmed, Y. Nuree, A. Das and J. K. Ray, RSC Adv., 2014, 4, 42604–42607 RSC; (b) K. V. Arundhathi, P. Vaishnavi, T. Aneeja and G. Anilkumar, RSC Adv., 2023, 13, 4823–4834 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, characterization and copies of NMR spectra. See DOI: https://doi.org/10.1039/d3nj04522f |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |