
Aroyl-isothiocyanates/isoselenocyanates as precursors to obtain novel cis-3-aroyl-thiourea/urea-β-lactams: design, synthesis, docking and biological evaluation†
Pankaj
Kumar‡
 a,
Jaswinder
Kaur‡
a,
Sumeeta
Kumari
b,
Sakshi
Paliwal
b,
Shiwani
Berry
c,
Anil Kumar
Pinnaka
*b and
Aman
Bhalla
*a
a,
Jaswinder
Kaur‡
a,
Sumeeta
Kumari
b,
Sakshi
Paliwal
b,
Shiwani
Berry
c,
Anil Kumar
Pinnaka
*b and
Aman
Bhalla
*a
aDepartment of Chemistry and Centre of Advanced Studies in Chemistry, Panjab University, Chandigarh, 160014, India. E-mail: amanbhalla@pu.ac.in
bMicrobial Type Culture Collection and Gene Bank, CSIR-Institute of Microbial Technology, Chandigarh, 160036, India. E-mail: apinnaka@imtech.res.in
cDepartment of Chemistry and Chemical Sciences, Central University of Himachal Pradesh, Shahpur Campus, Shahpur, Kangra-176206, HP, India
First published on 16th November 2023
Abstract
Herein, a novel synthetic methodology was devised to synthesize cis-3-aroyl-urea-β-lactams by reacting aroyl isoselenocyanates with cis-3-amino-β-lactams in a THF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O mixture (9:1). This synthetic approach eliminates the previous requirement for the challenging synthesis of unstable aroyl isocyanates and the low-yield acylation of urea to obtain aroyl urea derivatives. Further optimized conditions for nucleophilic reaction of aroyl isothiocyanates with cis-3-amino-β-lactams to obtain cis-3-aroyl-thiourea-β-lactams were developed. The stereochemistry of cis-3-aroyl-thiourea/urea-β-lactams (5a–f/8a–f) was elucidated by analyzing the J values of C3–H and C4–H of β-lactams. Subsequently, we evaluated the antibacterial activity of these compounds against both Gram-positive bacteria (S. aureus and B. cereus) and Gram-negative bacteria (P. aeruginosa and E. coli). Remarkably, among all the synthesized compounds, 8e exhibited potent activity against all bacterial strains, with a notable MIC value of 6.25 μg mL−1 against B. cereus, surpassing ampicillin (MIC = 25 μg mL−1) by four-fold. Additionally, compounds 8d and 8e demonstrated promising antifungal properties, with an MIC value of 1.5 μg mL−1 against C. tropicalis, comparable to fluconazole (MIC = 1 μg mL−1). Furthermore, compounds 8d and 8e exhibited 80% and 75% cell viability against THLE-2 cells at a concentration of 25 μg mL−1. Molecular docking analysis provided additional support for the observed in vitro antibacterial and antifungal activities of compound 8e.
:H2O mixture (9:1). This synthetic approach eliminates the previous requirement for the challenging synthesis of unstable aroyl isocyanates and the low-yield acylation of urea to obtain aroyl urea derivatives. Further optimized conditions for nucleophilic reaction of aroyl isothiocyanates with cis-3-amino-β-lactams to obtain cis-3-aroyl-thiourea-β-lactams were developed. The stereochemistry of cis-3-aroyl-thiourea/urea-β-lactams (5a–f/8a–f) was elucidated by analyzing the J values of C3–H and C4–H of β-lactams. Subsequently, we evaluated the antibacterial activity of these compounds against both Gram-positive bacteria (S. aureus and B. cereus) and Gram-negative bacteria (P. aeruginosa and E. coli). Remarkably, among all the synthesized compounds, 8e exhibited potent activity against all bacterial strains, with a notable MIC value of 6.25 μg mL−1 against B. cereus, surpassing ampicillin (MIC = 25 μg mL−1) by four-fold. Additionally, compounds 8d and 8e demonstrated promising antifungal properties, with an MIC value of 1.5 μg mL−1 against C. tropicalis, comparable to fluconazole (MIC = 1 μg mL−1). Furthermore, compounds 8d and 8e exhibited 80% and 75% cell viability against THLE-2 cells at a concentration of 25 μg mL−1. Molecular docking analysis provided additional support for the observed in vitro antibacterial and antifungal activities of compound 8e.
1. Introduction
β-Lactam drugs are harnessed as formidable tools for combating bacterial infections.1 Their mode of action involves the inactivation of penicillin-binding proteins (PBPs) by acylating the catalytic serine residue, which is pivotal for synthesizing the bacterial cell wall.2 Paradoxically, the overabundance of production and imprudent application of these drugs have expedited the emergence of antibacterial resistance.3 This resistance arises through a range of mechanisms, encompassing alterations in PBPs and modifications in the outer membrane, which hold particular importance within Gram-negative bacterial strains.4,5 Additionally, the synthesis of β-lactamases further exacerbates this resistance by enzymatically hydrolyzing β-lactam drugs, rendering them ineffective.6 Analogous challenges pertaining to resistance have been observed in the application and efficacy of antifungal agents.7Ergosterol, a pivotal constituent of fungi, assumes a vital role in modulating membrane permeability and fluidity. The focal point of antifungal agents involves targeting and inhibition of essential genes such as CYP51, CYP51B, and ERG11, responsible for the biosynthesis of ergosterol.8 Furthermore, these drugs also act upon fungal cell walls and interact with ergosterols directly.9 The main driving force behind antifungal resistance can be attributed to genetic modifications and the heightened expression of these critical genes.10 These adaptive genetic shifts collectively undermine the effectiveness of antifungal interventions, underscoring a substantial hurdle in the cure of fungal infections.
Aroyl thiourea and urea represent important structural motifs found in a wide array of bioactive compounds, including those with antimicrobial, antioxidant, antifungal, anticancer, and insecticidal properties (Fig. 1I–V).11–15 Even these moieties are found in various commercially available drugs and bioactive molecules known for their therapeutic properties including β-lactams (Fig. 1VI–VIII).16–18 These motifs play a vital role as linkers during the synthesis of pharmacophoric units, enabling the connection of two functional groups into a single hybrid structure through the coupling of isothiocyanates or isocyanates with amines.19 However, the exploration of β-lactam containing aroyl-thiourea/urea compounds in terms of their synthesis and biological activities remains unknown.
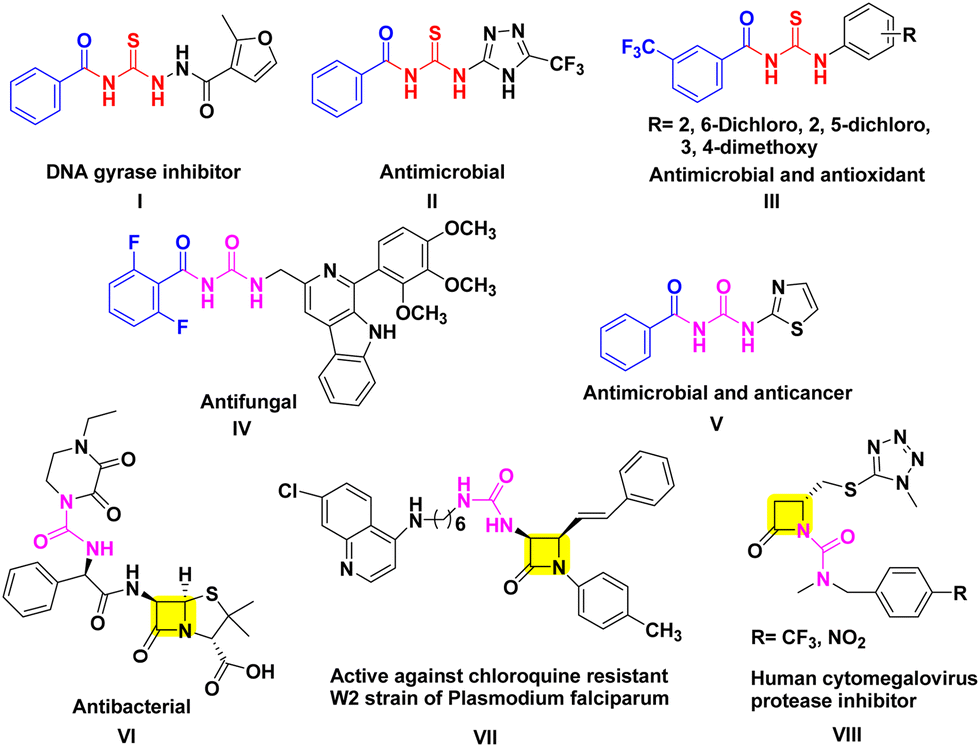 | ||
| Fig. 1 Biologically active aroyl thiourea/urea derivatives. | ||
Although the synthesis of aroyl isothiocyanates is relatively straightforward, obtaining aroyl isocyanates presents significant hurdles due to their demanding synthetic procedures, challenges in isolation, instability, and lower yields.20,21 In addition, other traditional methods involving acylation of ureas and carbodiimides also suffer from similar limitations.22 These factors collectively pose substantial difficulties in the synthesis of aroyl urea derivatives (Scheme 1a). To address these issues, Liptrot and colleagues introduced a modification to the traditional method by employing palladium-catalyzed carbonylation of aryl halides with urea nucleophiles in the presence of carbon monoxide (Scheme 1b).21 Over the past decade, there have been notable advancements in the field of novel approaches for synthesizing urea compounds, either through in situ methods or by isolating isocyanates.23 Despite these advancements, the challenges in obtaining aroyl urea derivatives persist when utilizing these methods.
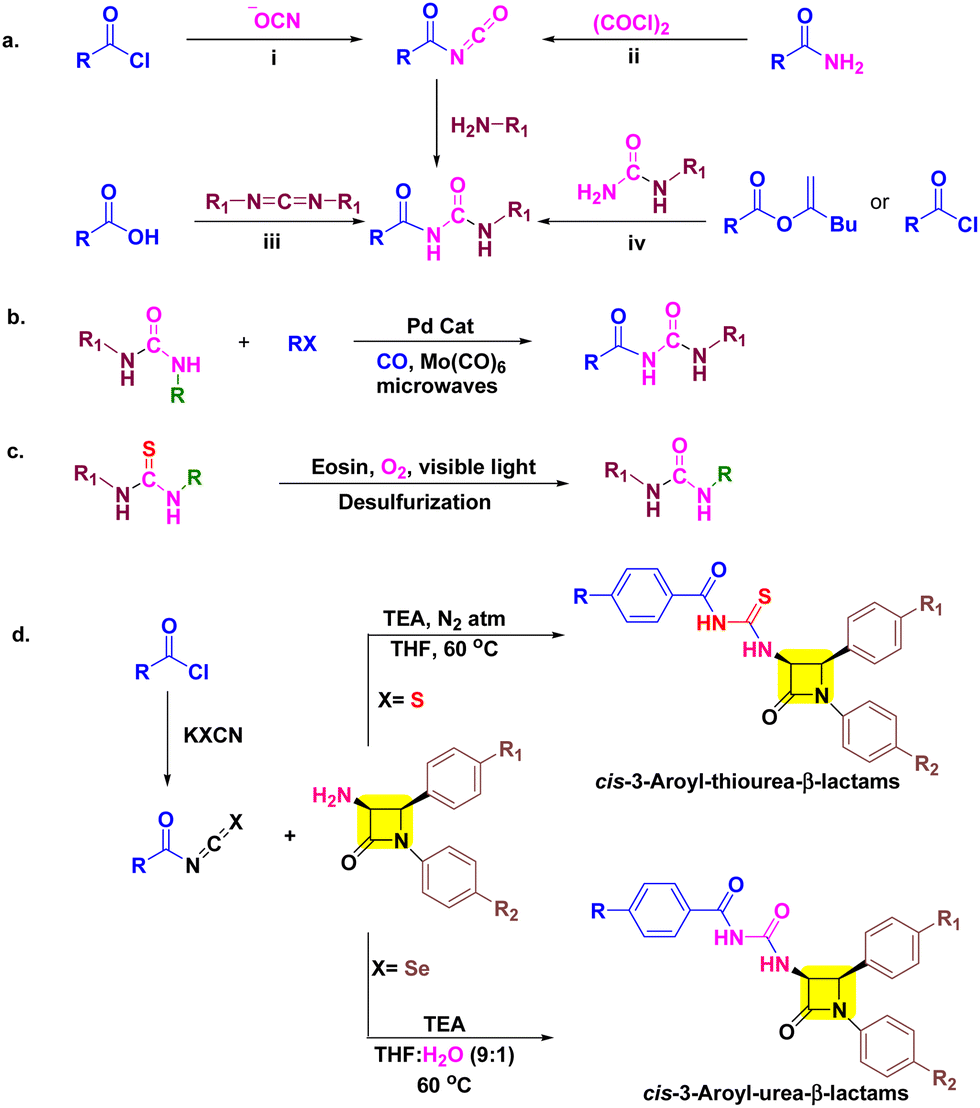 | ||
| Scheme 1 (a) Traditional approaches to aroyl urea (b) aroyl urea synthesis by Liptrot's group, (c) the desulfurization method developed by Gan's group for thiourea to urea conversion and (d) our work. | ||
The need for novel bioactive derivatives, molecular diversity, and aforementioned biological activity of aroyl thioureas/ureas and β-lactams stimulated us to club these moieties into a single pharmacophoric entity. The designed target molecules are almost identical, differing solely by the presence of oxygen (in urea) and sulfur (in thiourea) (Scheme 1d). This subtle variance enables us to predict the impact of this elemental alteration on interactions with biological targets of β-lactams (PBPs) and thiourea/urea (ergosterol biosynthesis)8–10,24,25 based on antimicrobial and cytotoxic assay. Furthermore, in silico studies including docking simulations and ADME parameters were performed for better interpretation of results.
As our research involves the synthesis of chalcogen containing bioactive molecules (O, S and Se), C-3 functionalization of β-lactams, and based on structure–activity relationship we have identified some target 3-aroyl-thiourea/urea-β-lactams 5a–f/8a–f as antimicrobial agents (Fig. 2).26–32 We initially considered a strategy for synthesizing 3-aroyl-thiourea derivatives by coupling aroyl isothiocyanates with 3-amino-β-lactams. However, the extended reaction times and additional synthetic steps required to obtain 3-urea-β-lactams using the Liptrot method prompted us to explore alternative approaches. Recently, the Gan research group introduced a method that caught our attention, allowing for the direct synthesis of urea derivatives from isothiocyanates through visible-light-promoted desulfurization (Scheme 1c).33 This innovative method has inspired us to develop an approach for efficiently obtaining 3-aroyl-urea-β-lactams in high yields and shorter reaction times by utilizing easily accessible aroyl isoselenocyanates (Scheme 1d).
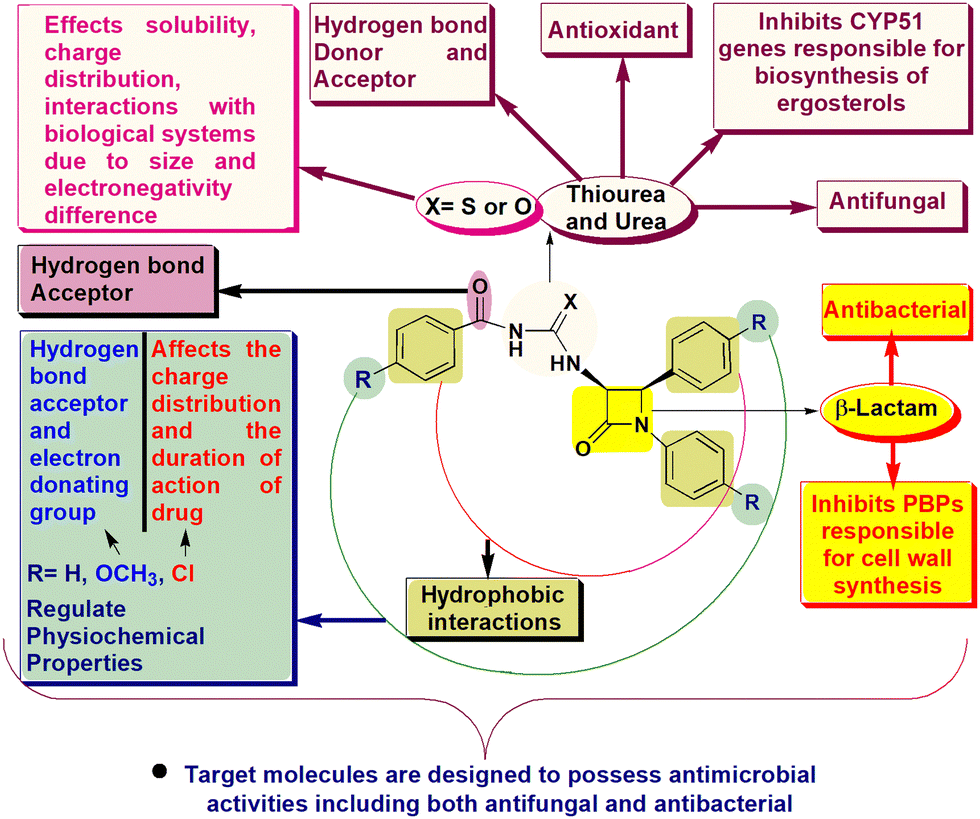 | ||
| Fig. 2 Designing of 3-aroyl-thiourea/urea-β-lactams as antimicrobial agents. | ||
2. Results and discussion
2.1 Chemistry
Initially aroyl isothiocyanate 3 was synthesized following the earlier literature and utilized in situ for nucleophilic-trapping with 3-amino-β-lactam 4a (Table 1, entries 1 and 2) in the presence of TEA at 60 °C. However, only 42% yield of target cis-3-aroyl-thiourea-β-lactam 5a was achieved in acetone solvent, while trace amounts were obtained in acetonitrile. Furthermore, 3 was isolated and reacted with 4a in acetone which resulted in 72% yield of target 5a (Table 1, entry 3). Also, performing the reaction in the absence of TEA has resulted in diminished yield (Table 1, entry 4). Interestingly, in THF solvent the yield of target 5a was reduced to 44% along with the synthesis of 40% cis-3-aroyl-urea-β-lactam 8a as a co-product (Table 1, entry 5). This suggested the potential involvement of water in a process substituting sulfur with oxygen. This was further confirmed by doing the reaction in a nitrogen atmosphere in dry THF resulting in the formation of 5a in 92% yield in 3 h (Table 1, entry 8). Also, decreasing the temperature by 5 °C results in decreased yield (Table 1, entry 9), while increasing the temperature by 5 °C has a negligible effect (Table 1, entry 10). However, the formation of 3-aroyl-urea-β-lactam 8a led us to utilize aroyl isoselenocyante 7 as a precursor. Gratefully, 7 results in target 8a in THF with 43% yield at 60 °C (Table 1, entry 11). Subsequently, altering the solvent does not show any gradual increase in the yield (Table 1, entries 12–14). To our delight, at room temperature (RT) the yield increased to 63% (Table 1, entry 15). However, a trace amount of 8a was detected in acetone solvent at RT (Table 1, entry 16). Notably, taking THF solvent in combination with H2O in a 9:1 ratio has allowed us to obtain 8a in 86% yield (Table 1, entry 17).
|
|
|||||
|---|---|---|---|---|---|
| Sr No. | 2 or 6 | Solvent | Temp (°C) | %Yield (5a) | %Yield (8a) |
| 1 (1 mmol, 1 equiv.), 2 or 6 (1.2 equiv.), 3 (1 equiv.), 4a (1.2 equiv.), 7 (1 equiv.), TEA (2 equiv.).a In situ2.b TEA not added.c N2 atm. Reaction time = 5 h for 5a and 7 h for 8a. | |||||
| 1a | 2 | Acetone | 60 | 42 | — |
| 2a | 2 | Acetonitrile | 60 | Trace | — |
| 3 | 2 | Acetone | 60 | 72 | — |
| 4b | 2 | Acetone | 60 | 23 | — |
| 5 | 2 | THF | 60 | 44 | 40 |
| 6 | 2 | 1,2-DCE | 60 | 15 | — |
| 7 | 2 | Toluene | 60 | Trace | — |
| 8c | 2 | Dry THF | 60 | 92 | — |
| 9c | 2 | Dry THF | 55 | 84 | — |
| 10c | 2 | Dry THF | 65 | 91 | — |
| 11 | 6 | THF | 60 | — | 43 |
| 12 | 6 | Acetonitrile | 60 | — | Trace |
| 13 | 6 | Toluene | 60 | — | 47 |
| 14 | 6 | 1,2-DCE | 60 | — | 36 |
| 15 | 6 | THF | RT | — | 63 |
| 16 | 6 | Acetone | RT | — | Trace |
| 17 | 6 | THF:H2O (9:1) |
RT | — | 86 |
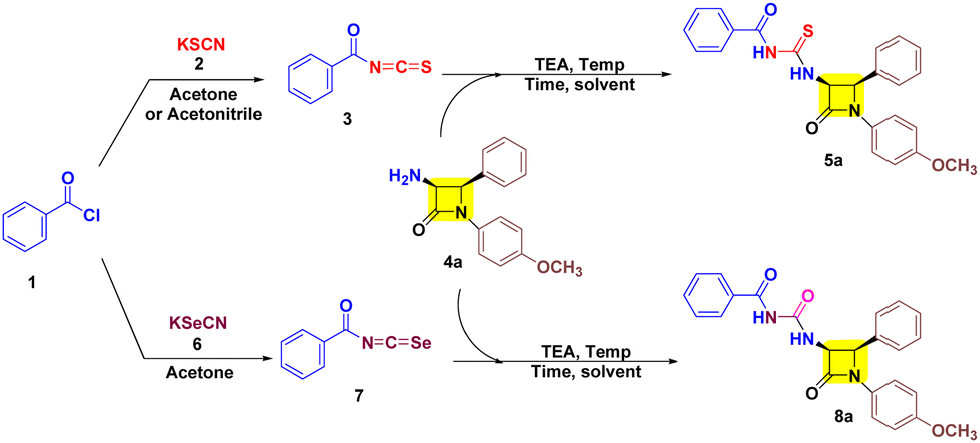
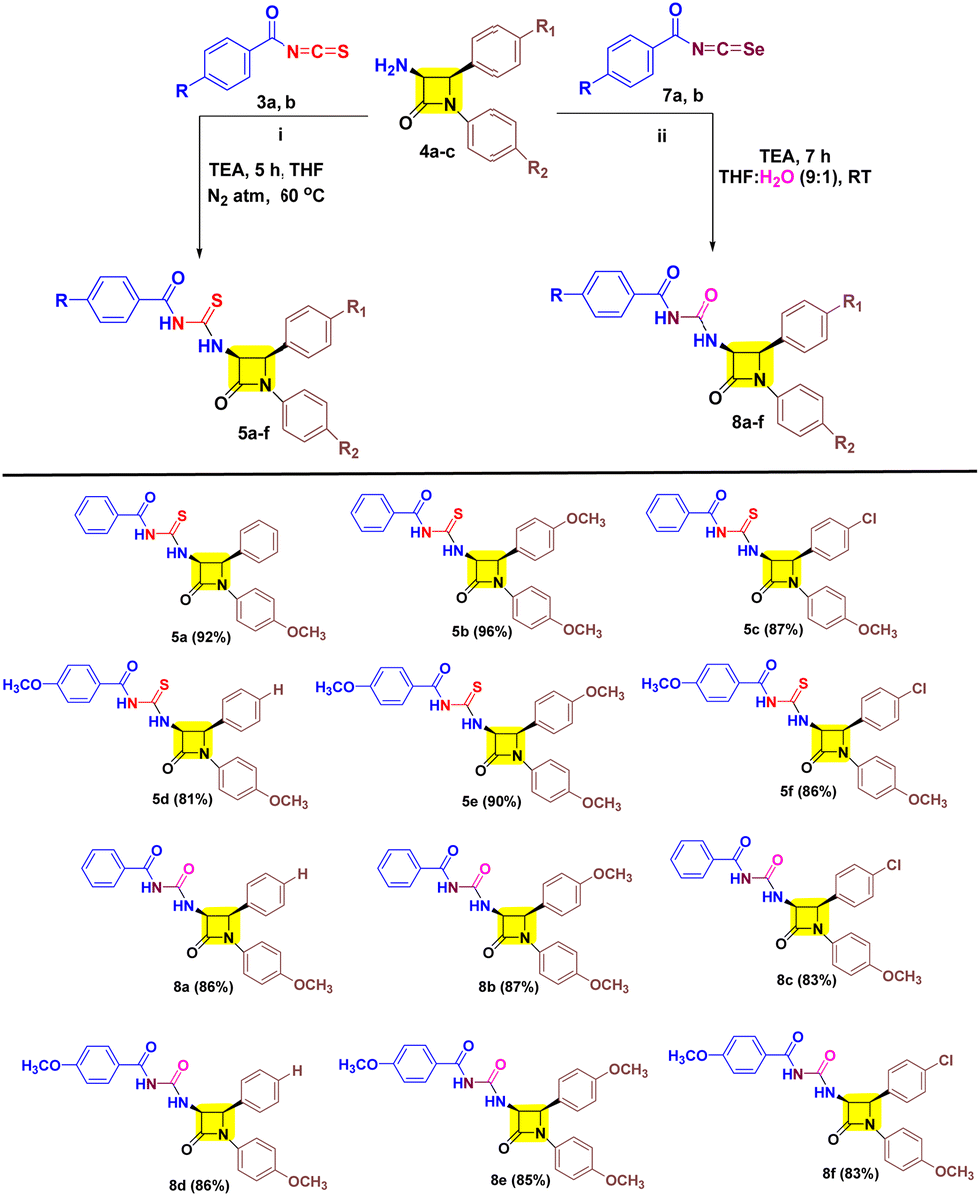
After successfully developing the conditions for 5a and 8a we proceeded to investigate the nucleophilic reactivities of other cis-3-amino-β-lactam derivatives 4 with aroyl isothiocyanates 3 and aroyl isoselenocyanates 7 to obtain target 5a–f/8a–f (Table 2). We found that inclusion of substituents on aryl rings with varying electronic properties (the electron donating group (OCH3), the electron withdrawing group (Cl), or an electron neutral group) is well tolerated and afforded target products 5a–f in excellent yield. It was noted that compound 5b exhibited the highest yield (96%) with the aroyl group having no substituents and the p-OCH3-aryl group present at both N-1 and C-4 positions of β-lactam. Furthermore, a decreased yield is obtained for compound 5a (92%) with one methoxy group at the N-1 aryl ring followed by compound 5e (90%) with a methoxy substituent on each aryl group of β-lactam. Also, a decreased yield was witnessed for compounds 5c (87%) and 5f (86%) with one p-chloro on the C-4 aryl ring of β-lactam. Unsurprisingly, lower yield was obtained for compound 5d (81%) with an electron rich group on aroyl and an electron poor group on the C-4 aryl of β-lactam. After successfully investigating the scope of cis-3-aroyl-thiourea-β-lactams 5a–f we analysed the nucleophilic trapping of aroyl isoselenocyanates with 3-amino-β-lactams 4 to obtain target cis-3-aroyl-thiourea-β-lactams 8a–f. However, a lower yield of target compounds 8a–f (83–87%) was observed with respect to 5a–f (81–96%). In addition, a similar yield was also witnessed for every substituent of 8a–f.
| 3 (1 equiv.), 4 (1.2 equiv.), 7 (1 equiv.), TEA (2 equiv.). |
|---|
|
Furthermore, we established the stereochemistry of the target cis-3-aroyl-thiourea/urea-β-lactams 5a–f/8a–f by analyzing the coupling constant (J) values of C3–H and C4–H, with J values greater than 3 serving as criteria.
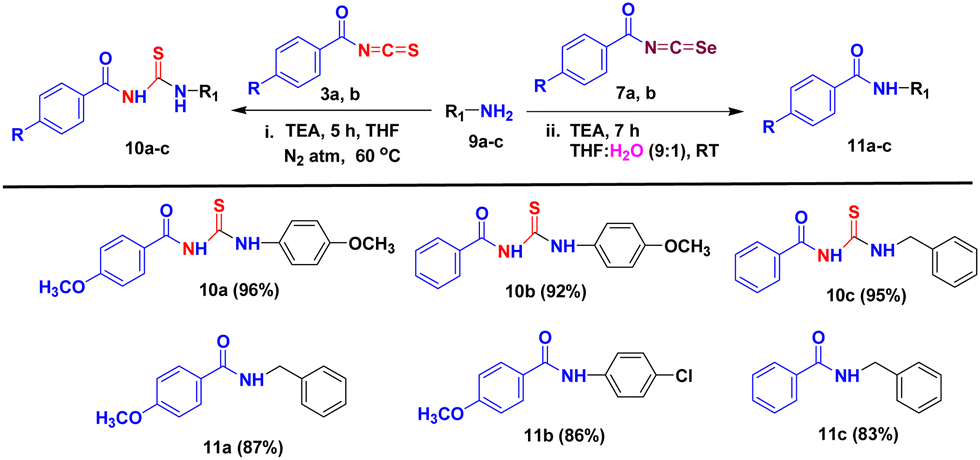
We conducted experiments to assess the practicality of applying our developed methods to produce various aroyl thiourea/urea derivatives. This involved testing a range of aromatic and aliphatic amines (9a–c) to react with aroyl isothiocyanates/isoselenocyanates 3a, b/7a, b under our optimized reaction conditions, as outlined in Table 3. We were pleased to discover that our approach displayed a broad substrate compatibility, resulting in excellent yields of the desired aroyl thiourea derivatives 10a–c when treated with benzoyl isothiocyanates, while amides 11a–c were formed when benzoyl isoselenocyanates were used.
| 3 (1 equiv.), 9 (1.2 equiv.), 7 (1 equiv.), TEA (2 equiv.). |
|---|
|
We conducted the reaction under dark conditions to gain deeper insights into the impact of visible light on the reaction mechanism. However, this did not hinder the synthesis of target 8a as outlined in Scheme 2a. With this result in mind and previous literature we outlined a probable mechanism as shown in Scheme 2b.21,29 The reaction begins with the abstraction of protons from 3-amino-β-lactam 4a to afford the corresponding 3-amino-β-lactam anion I. I then attacks the electrophilic aroyl isoselenocyanate 7a carbon and furnishes intermediate II. Furthermore, nucleophilic attack by water on II results in intermediate III whose proton is abstracted by TEA to give target derivative 8a with the removal of selenium.
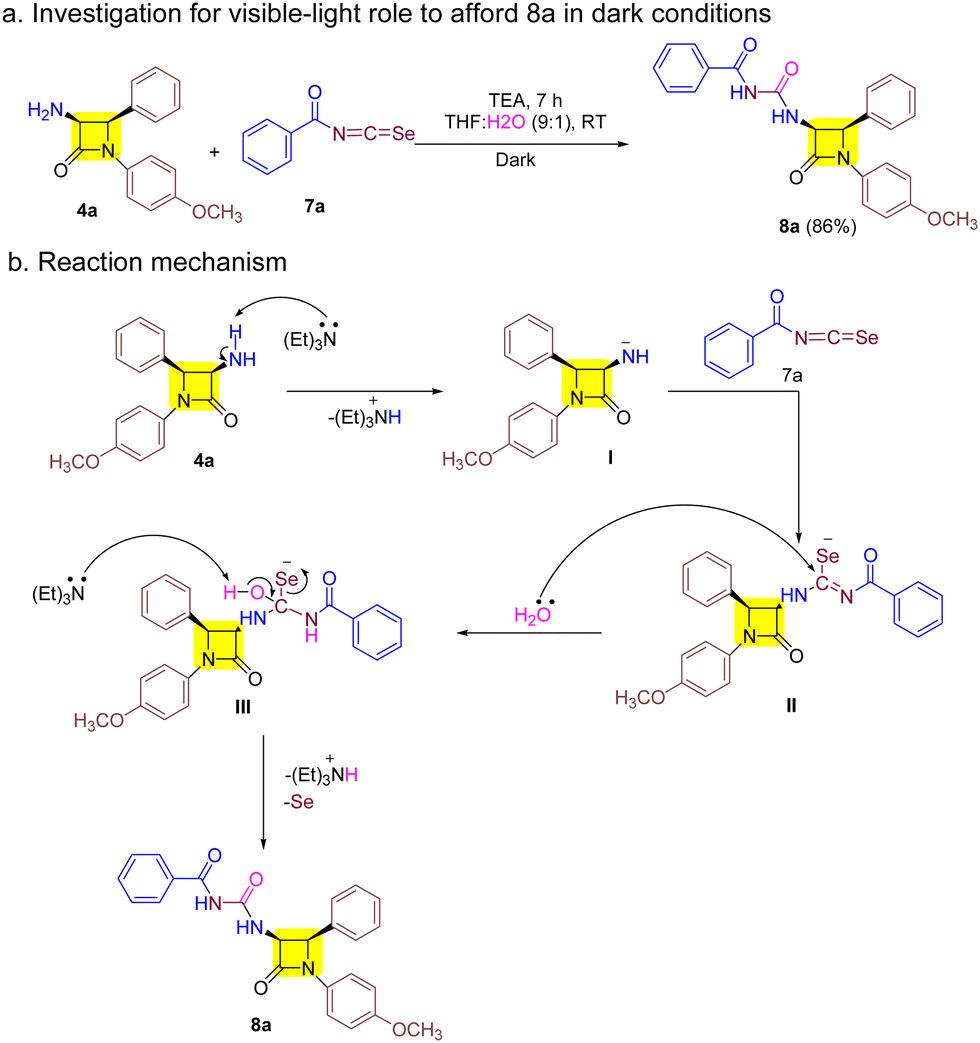 | ||
| Scheme 2 (a) Reaction under dark conditions and (b) the probable mechanism of synthesis of 8. | ||
2.2 Biological studies
| Compounds | Gram-positive bacteriaa | Gram-negative bacteriab | Fungusc | |||
|---|---|---|---|---|---|---|
| S. A. | B. C. | P. A. | E. C. | C. A. | C. T. | |
| a S. A, S. aureus MTCC 1430; B. C, B. cereus MTCC 9490. b P. A, P. aeruginosa MTCC 1934; E. C, E. coli MTCC 1610. c C. A, C. albicans MTCC 1637; C. T, C. tropicalis MTCC 184. | ||||||
| 5a | — | — | 50 | — | — | — |
| 5b | 25 | 12.5 | 25 | 25 | — | — |
| 5c | — | — | — | — | — | — |
| 5d | — | — | — | — | 12.5 | 6.25 |
| 5e | 50 | 25 | 50 | — | — | — |
| 5f | — | — | — | — | 25 | 12.5 |
| 8a | — | — | — | — | 50 | 25 |
| 8b | — | — | — | — | 25 | 25 |
| 8c | — | 50 | — | — | 25 | 12.5 |
| 8d | 50 | 12.5 | 50 | 25 | 1.5 | 1.5 |
| 8e | 12.5 | 6.25 | 25 | 25 | 1.5 | 1.5 |
| 8f | — | — | — | — | 12.5 | 12.5 |
| Ampicillin | 3.12 | 25 | 25 | 12.5 | — | — |
| Vancomycin | 6.25 | 1.5 | — | — | — | — |
| Penicillin G | 50 | 3.12 | — | — | — | — |
| Fluconazole | — | — | — | — | 0.5 | 1 |
2.2.1.1 In vitro antibacterial activity. Out of the twelve target compounds, three compounds including 5b, 8d and 8e have shown activity against all the tested bacterial strains.
This is followed by compound 5e which has shown significant activity against both Gram positive strains and one Gram negative bacterium. Furthermore, compounds 5a and 8c were selectively active against P. aeruginosa and B. cereus respectively. Some of the target molecules surpass the reference drug ampicillin, indicating their potency to combat bacterial infections. Among cis-3-aroylthiourea-β-lactams, 5b was found to inhibit all strains of bacteria with MIC values ranging from 12.5–25 μg mL−1. The incorporation of EDG methoxy at the para position of the phenyl ring on N-1 and C-4 positions of the β-lactam ring in both 5b and 5e has shown positive effects. However, introduction of one more EDG methoxy on the para position of the benzoyl ring in 5e lowers the activity as compared to 5b by two-fold, although no inhibitory activity of 5e was observed against E. coli. Also, the presence of only one methoxy group on the phenyl ring attached to N-1 and the absence of substituents on other aryl rings in 5a enabled this molecule to fight P. aeruginosa (MIC = 50 μg mL−1). The rest of the other derivatives 5c, 5d, and 5f showed no inhibitory activity, indicating that the absence of substituents and the presence of the EWG chloro on the phenyl ring at the C-4 position make these molecules inactive towards the tested strains showing their detrimental effects. However, the presence of the methoxy group on the para positions of all the phenyl rings of compound 8e makes it more potent to inhibit the bacterial growth with MIC values ranging from 6.25–25 μg mL−1, while removing one methoxy group from the phenyl ring at the C-4 position of β-lactam results in 2–4 times decrease in the activity of 8d. Furthermore, compound 8c having a chloro group in place of methoxy attached to the phenyl ring at the C-4 position of β-lactam and no substituents on the benzoyl group shows a negative effect on the antibacterial activity and is found to inhibit the growth of the B. cereus bacterial strain only having an MIC value of 50 μg mL−1. Furthermore, no activity was obtained in the case of 8a, 8b and 8f indicating a downregulated effect of the combination of substituents and their placement. Concluding the results, aroylurea-β-lactam 8e, bearing three methoxy substituents, exhibited favorable antimicrobial properties, while the removal of one methoxy group from the aroyl moiety completely inactivates compound 8b towards all tested bacterial strains. In contrast, the presence of a methoxy group negatively impacted the bioactivity of the thiourea moiety in 5e, rendering it less active compared to 5b, which lacked a methoxy group on the aroyl group (Fig. 3).
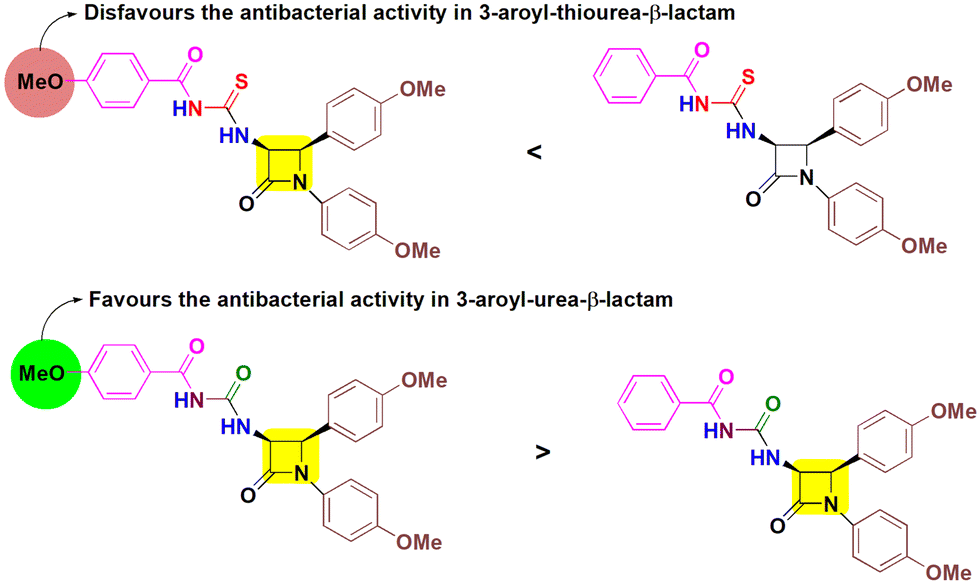 | ||
| Fig. 3 Comparable effect of the methoxy group substituted at the aroyl moiety on the antibacterial activity of cis-3-aroylthiourea-β-lactams (5b and 5e) and cis-3-aroylurea-β-lactams (8b and 8e). | ||
Comparing the antibacterial activity results of cis-3-aroyl-thiourea/urea-β-lactam derivatives 5a–f/8a–f, cis-3-aroylurea-β-lactams showed better activity against Gram-positive bacteria. With reference to ampicillin activity against the B. cereus bacterial strain with an MIC value of 25 μg mL−1, both compounds 5b and 8e exhibit better antibacterial activity with MIC values of 12.5 μg mL−1 and 6.25 μg mL−1, but still 8e is more potent than 5b indicating a remarkable inhibition effect of the benzoylurea moiety. Comparing the results with penicillin G having an MIC value of 3.12 against B. cereus, compound 8e has shown two-fold less activity and compound 5b has shown four-fold less activity. For Gram-negative bacterial strains, 5b and 8e gave equivalent results, while compounds 5a, 5e and 8d showed two-fold activity with respect to ampicillin against P. aeruginosa. Furthermore, two-fold activity was observed for compounds 5b, 8d and 8e in contrast to ampicillin with an MIC value of 12.5 against E. coli.
2.2.1.2 Antifungal activity of target compounds. The antifungal activity data for the target cis-3-aroyl-thiourea/urea-β-lactam derivatives 5a–f/8a–f with fluconazole as the reference drug revealed most of the compounds to exhibit inhibitory properties. Among the six cis-3-aroylthiourea-β-lactam derivatives (5a–f), only 5d, featuring two methoxy groups at the para position of the benzoyl group and a phenyl ring at the N-1 position of the β-lactam ring, exhibited superior antifungal activity compared to 5f. Although 5f has a similar molecular diversity to 5d, the presence of the chloro group at the phenyl ring incorporated at the C-4 position of the β-lactam moiety results in decreased activity, indicating the opposing effect of the EWG. Furthermore, the rest of the other derivatives 5a–c and 5e are completely devoid of antifungal activity, indicating inadequate alterations of the molecular heterogeneity to make the molecules biologically active. To our delight, all the cis-3-aroylurea-β-lactam derivatives 8a–f were found to be active against both the tested fungi. Both 8d and 8e remarkably inhibited the fungus growth with an MIC value of 1.5 μg mL−1 for both tested fungal strains. This can be attributed to the commendatory effect of the presence of three methoxy groups at the para position of each aryl ring in 8e and two methoxy groups at the para position of the benzoyl group and the phenyl ring at the N-1 position of the β-lactam ring in 8d. Remaining other molecules were also observed to have antifungal activity with MIC values ranging from 12.5–50 μg mL−1, which are 8–34 fold lower than those of 8d and 8e.
Comparing the antifungal activity results of derivatives 5a–f and 8a–f, again cis-3-aroylurea-β-lactam derivatives 8a–f were found to be superior in inhibiting the growth of fungus. Moreover, the best-found active derivatives 8d and 8e are 8-fold better against C. albicans (1.5 μg mL−1) and 4 times against C. tropicalis (1.5 μg mL−1) compared to 5d with MIC values of 12.5 μg mL−1 and 6.25 μg mL−1. Comparing the results of antifungal activity of 8d and 8e against the reference drug fluconazole revealed that they have three-fold decreased activity against C. albicans and 0.5-fold less activity against C. tropicalis.
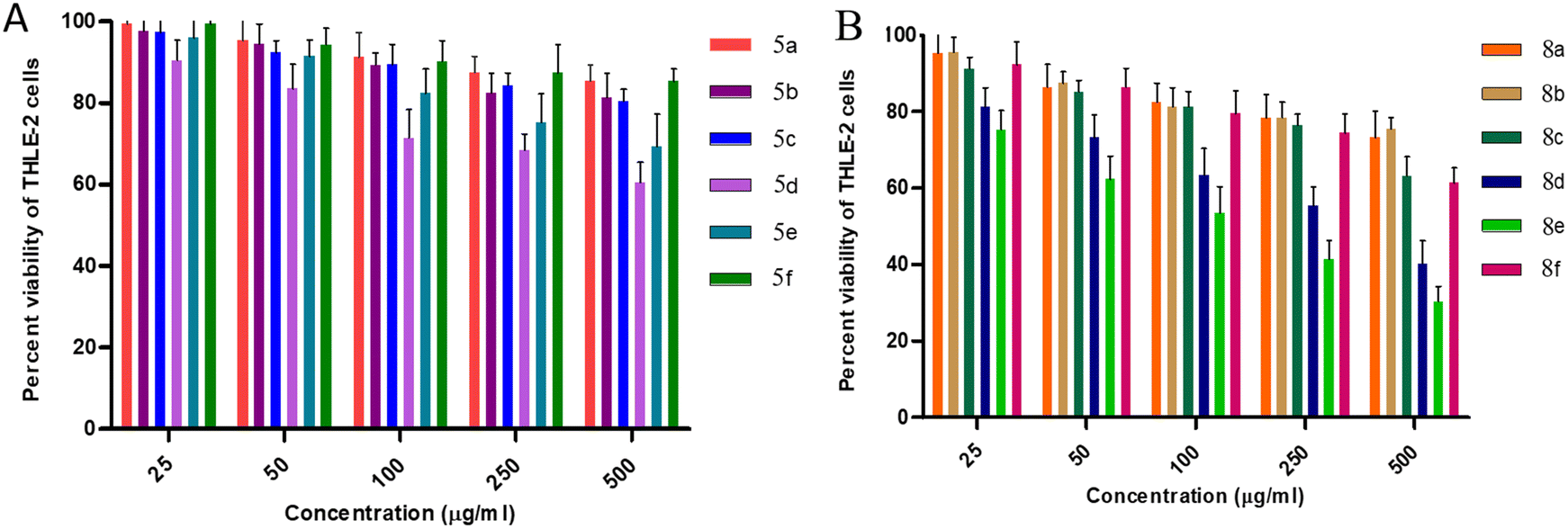 | ||
| Fig. 4 Cytotoxic effects of intended compounds (A) 5a–f and (B) 8a–f on THLE-2 cells. | ||
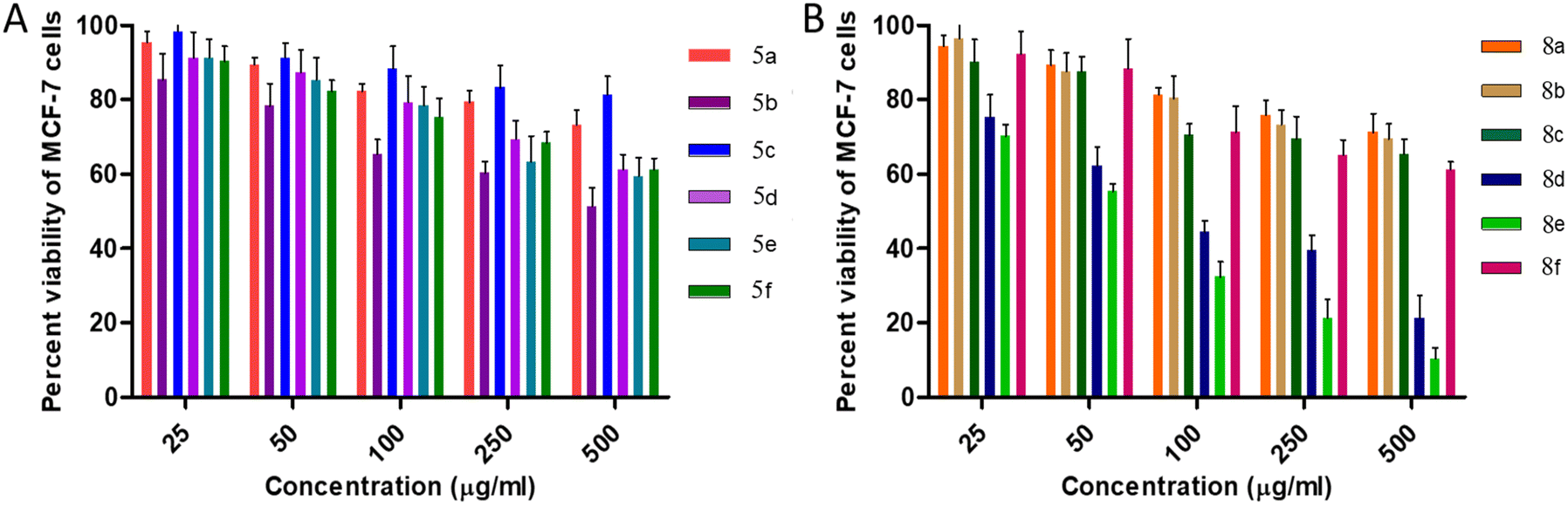 | ||
| Fig. 5 Cytotoxic effects of intended compounds (A) 5a–f and (B) 8a–f on MCF-7 cells. | ||
For the THLE-2 cell line, it was observed that at 25 μg mL−1 concentration of 5a–f, approx. 95–99% of the cells were viable except in the case of 5d, where 90% of the cells were viable. Furthermore, upon increasing the concentration to 50 μg mL−1, 90–95% of the cells were still viable, while 80% cell viability is observed for 5d. Cytotoxic evaluation of 8a–f derivatives at 25 μg mL−1 concentration revealed more than 90% cell viability except in the case of 8e and 8d with 75% and 80% cell viability, respectively. Furthermore, 85–90% of viable cells were observed excluding 60% for 8e and 70% for 8d upon increasing the concentration to 50 μg mL−1. These results favour the potential of synthesized molecules as antimicrobial agents with MIC values ranging from 1.5–50 μg mL−1. However, comparing the effect of thiourea and urea moieties, cis-3-aroylthiourea-β-lactam derivatives 8a–f were observed to possess less cytotoxicity against THLE-2 cell lines.
Initial screening performed against human breast adenocarcinoma cells (MCF-7) reveals that 80% of the cells remain viable for all 5a–f derivatives at 50 μg mL−1 concentration. Furthermore, at 50 μg mL−1 concentration of cis-3-aroylurea-β-lactams 8a–f, again more than 80% viable cells were found except for 8e and 8d. Moreover, urea containing derivatives 8a–f are found to be more toxic to the MCF-7 cell line in comparison to thiourea containing derivatives 5a–f.
2.3 In silico studies
To predict the binding affinities of the designed molecules with their biological targets and their toxicity, molecular docking simulations and ADME parameters were estimated using computational methods.| Compounds | PDB 1VQQ/5TZ1 | ||
|---|---|---|---|
| Binding energy (kcal mol−1) | Vdw_hb_desolv_energy (kcal mol−1) | Total internal energy (kcal mol−1) | |
| 5a | −6.62/−4.84 | −8.43/−6.75 | −2.36/−2.87 |
| 5b | −6.72/−7,56 | −8.87/−9.98 | −1.75/−1.92 |
| 5c | −6.63/−6.61 | −8.63/−8.65 | −2.18/−2.60 |
| 5d | −6.18/−4.19 | −8.46/−6.73 | −2.08/−1.94 |
| 5e | −8.24/−4.65 | −10.63/−7.32 | −2.85/−2.54 |
| 5f | −5.99/−5.43 | −8.28/−7.75 | −2.30/−1.87 |
| 8a | −5.93/−6.51 | −7.34/−7.96 | −2.28/−2.35 |
| 8b | −5.91/−6.38 | −7.65/−8.14 | −2.32/−2.42 |
| 8c | −6.76/−6.41 | −8.21/−7.78 | −2.26/−2.15 |
| 8d | −6.27/−6.92 | −7.95/−8.39 | −2.35/−2.45 |
| 8e | −6.09/−8.23 | −7.96/−10.18 | −2.30/−2.32 |
| 8f | −5.63/−6.43 | −7.19/−8.08 | −2.39/−2.31 |
Among the synthesized derivatives 5a–f/8a–f, compounds 5b and 8e demonstrated promising antibacterial activity. We generated 3D and 2D interaction models for these derivatives within the active site of the target protein using the Discovery Studio Visualizer, as illustrated in Fig. 6 and 7. Upon observing these visual representations, an intriguing finding emerged regarding the binding pattern of thiourea and urea derivatives with the protein residues. Notably, the resonance form of thiourea with a negatively charged sulfur and a nitrogen atom attached to the C-4 carbon, carrying a positive charge, engaged in interactions with various amino acids (Fig. 6). This significant alteration led the sulfide group to partake in a conventional hydrogen bond interaction with LYS 273 in 5b. Moreover, the NH group linked to the aroyl moiety engaged in hydrogen bond interactions with GLN 292 and HIS 293. The methoxy group located at the para position of the N-phenyl and C4-phenyl rings also formed hydrogen bond interactions with LYS 148 and ASP 295. Additionally, interactions such as π–π/cationic/alkyl interactions with TYR 272, HIS 293, LYS 148, and LYS 273 further contributed to stabilizing the ligand-receptor complex. The interaction pattern of compound 8e with the protein revealed hydrogen bond formation between the NH group and ASP 275, as well as between the methoxy group and GLU 294 and ASP 295, both connected to the aroyl group (Fig. 7). Furthermore, a π-donor hydrogen bond was observed between the C4-phenyl ring and VAL 277. Additionally, π-alkyl interactions involving LYS 273, HIS 293, TYR 297, and LYS 316 played a role in allowing the ligand to adopt an active conformation. Literature also identified ASN146, SER149, LYS148, LYS273, VAL277, GLN292, HIS293, GLU294, ASP295, TYR297, ARG298, VAL299, THR300, ILE314, GLU315, and LYS316 within the protein (1VQQ) as crucial residues for interactions with ligands to facilitate the active conformation of the ligands. This finding further reveals that the observed amino acids undergo interactions with molecules 5b and 8e.
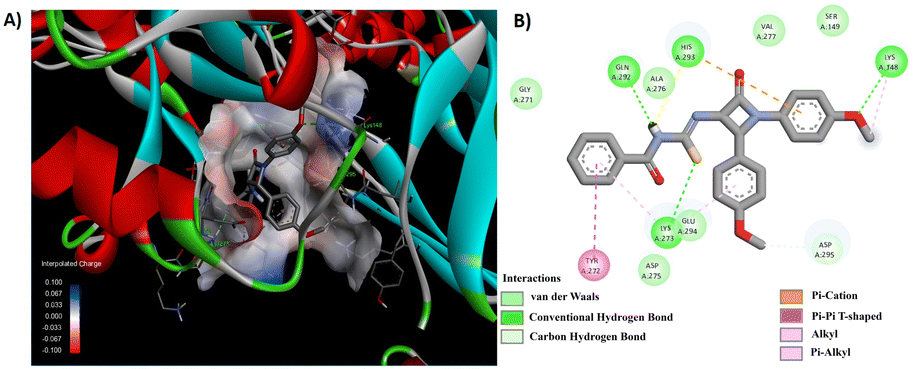 | ||
| Fig. 6 (A) 3D conformation and (B) 2D conformation of 5b docked to PBP (PDB ID: 1VQQ). | ||
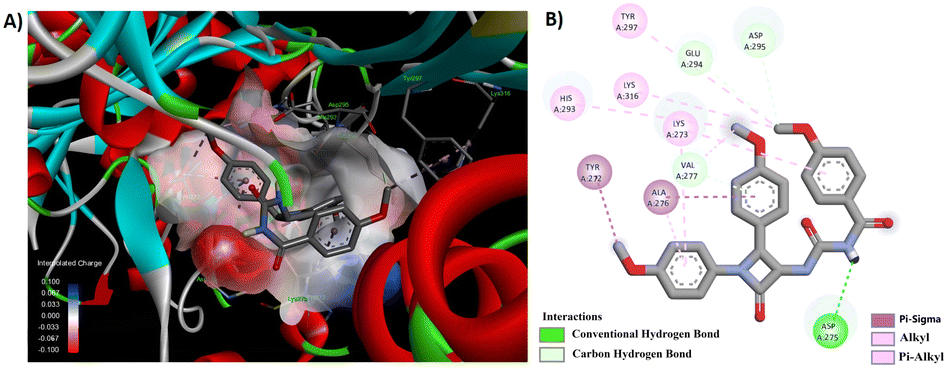 | ||
| Fig. 7 (A) 3D conformation and (B) 2D conformation of 8e docked to PBP (PDB ID: 1VQQ). | ||
Through a meticulous examination of the binding behavior of the most effective antifungal agents, namely 8d and 8e, it was ascertained that both compounds were engaged in interactions with similar amino acids (Fig. 8 and 9). Hydrogen bond interactions with TRP 54, established by the methoxy group present in the aroyl ring of 8d and the aroyl carbonyl in 8e, were observed. Furthermore, LYS 78 formed conventional hydrogen bonds with the methoxy group at the N1-phenyl ring in 8d and the C4-phenyl ring in 8e. Additionally hydrogen bonds were identified between the aroyl carbonyl group and ALA 62/SER 63, as well as between the methoxy group at the N1-phenyl ring and ALA 62, for 8d. Other interactions, such as π–π and alkyl interactions with TRP 54 and PHE 52 in 8d and ALA 62 and PHE 58 in 8e, contributed to the stability of the ligand–receptor complex.
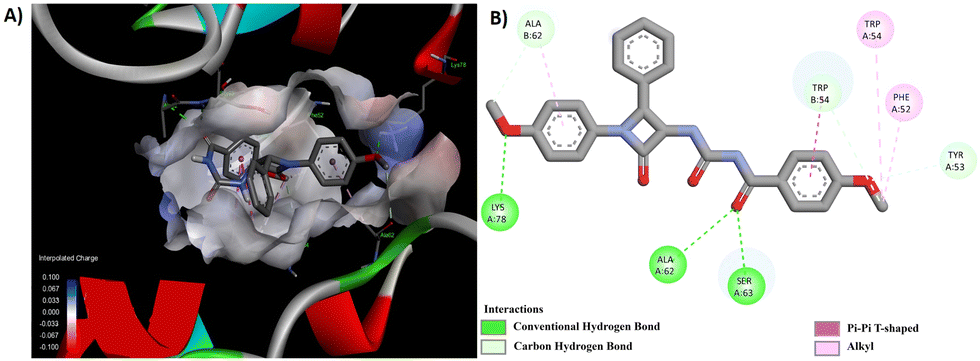 | ||
| Fig. 8 (A) 3D conformation and (B) 2D conformation of 8d docked to CYP51 (PDB ID: 5TZ1). | ||
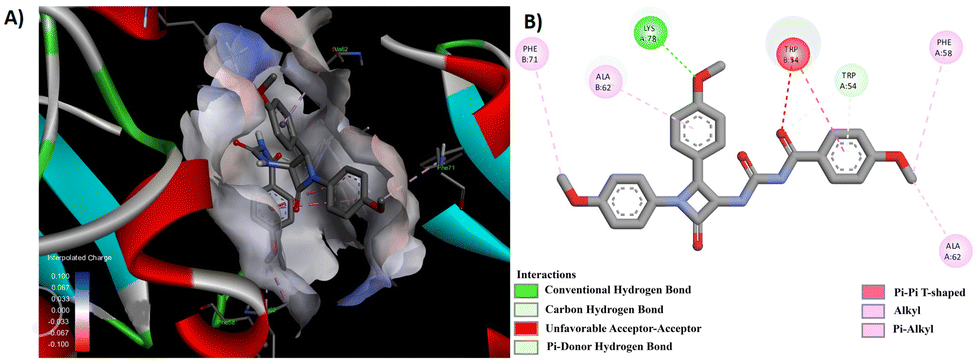 | ||
| Fig. 9 (A) 3D conformation and (B) 2D conformation of 8e docked to CYP51 (PDB ID: 5TZ1). | ||
3. Experimental
3.1 Synthesis
Column chromatography was performed using Merck Silica Gel (60–120 mesh) as the stationary phase, and elution was performed using a mixture of ethyl acetate (EtOAc) and n-hexane as the mobile phase. Analytical thin-layer chromatography (TLC) was carried out on aluminium plates pre-coated with silica gel F254, and the spots were visualized under UV light. Unless specifically mentioned, all reagents used in this study were acquired from commercial suppliers and were employed without additional purification. Benzoylchloride (Avra), potassium selenocyanate (Alfa Aesar), aniline (Merck), anisaldehyde (Fluka), phosphorus oxychloride (Merck), triethylamine (Qualigen) and all other commercially available compounds/reagents/solvents were used directly without any additional purification steps. Prior to use, dichloromethane was subjected to a thorough drying process and distilled over anhydrous phosphorus pentoxide (P2O5). Furthermore, it underwent a subsequent distillation over CaH2 to ensure the complete elimination of any residual water content before application in the experiment.
1-Benzoyl-3-[1-(4-methoxy-phenyl)-2-oxo-4-phenyl-azetidin-3-yl]-thiourea (5a). Yield: 92%; 1H NMR (500 MHz, DMSO) δ 11.53 (s, 1H), 11.14 (d, J = 8.3 Hz, 1H), 7.87 (dd, J = 5.2, 3.3 Hz, 1H), 7.79 (dd, J = 8.3, 1.1 Hz, 2H), 7.63–7.59 (m, 1H), 7.49–7.43 (m, 3H), 7.38–7.34 (m, 2H), 7.31 (d, J = 7.8 Hz, 1H), 7.28–7.25 (m, 2H), 6.95–6.92 (m, 2H), 6.19 (dd, J = 8.3, 5.3 Hz, 1H), 5.70 (d, J = 5.3 Hz, 1H), 3.71 (s, 3H). 13C NMR (126 MHz, DMSO) δ 181.50, 168.26, 161.90, 155.84, 133.50, 133.09, 131.46, 131.07, 130.10, 129.25, 128.50, 128.42, 128.27, 127.32, 118.45, 114.42, 63.73, 60.41, 59.85, 55.16. HRMS (ESI+): m/z calcd for [M + Na]+ C24H21N3NaO3S 454.1201, found 454.1165.
1-Benzoyl-3-[1,2-bis-(4-methoxy-phenyl)-4-oxo-azetidin-3-yl]-thiourea (5b). Yield: 96%; 1H NMR (500 MHz, DMSO) δ 11.53 (s, 1H), 11.18 (d, J = 8.1 Hz, 1H), 7.81 (dd, J = 8.4, 1.1 Hz, 2H), 7.64–7.60 (m, 1H), 7.48 (dd, J = 8.1, 7.6 Hz, 2H), 7.27–7.22 (m, 4H), 6.95–6.90 (m, 4H), 6.12 (dd, J = 8.1, 5.3 Hz, 1H), 5.63 (d, J = 5.2 Hz, 1H), 3.71 (s, 3H), 3.67 (s, 3H). 13C NMR (126 MHz, DMSO) δ 181.46, 168.28, 161.96, 159.13, 155.79, 133.09, 131.53, 130.12, 128.60, 128.45, 128.28, 125.06, 118.47, 114.38, 113.96, 63.79, 59.56, 55.15, 54.93. HRMS (ESI+): m/z calcd for [M + Na]+ C25H23N3NaO4S 484.1307, found 484.1283.
1-Benzoyl-3-[2-(4-chloro-phenyl)-1-(4-methoxy-phenyl)-4-oxo-azetidin-3-yl]-thiourea (5c). Yield: 87%; 1H NMR (500 MHz, DMSO) δ 11.55 (s, 1H), 11.16 (d, J = 7.9 Hz, 1H), 7.82–7.80 (m, 2H), 7.65–7.60 (m, 1H), 7.50–7.46 (m, 2H), 7.43–7.40 (m, 2H), 7.36–7.34 (m, 2H), 7.27–7.24 (m, 2H), 6.94–6.92 (m, 2H) 6.13 (dd, J = 7.8, 5.3 Hz, 1H), 5.72 (d, J = 5.2 Hz, 1H), 3.71 (s, 3H). 13C NMR (126 MHz, DMSO) δ 181.51, 168.28, 161.65, 155.92, 133.13, 132.89, 132.61, 131.48, 129.94, 129.31, 128.44, 128.43, 128.31, 118.45, 114.45, 63.83, 59.34, 55.17. HRMS (ESI+): m/z calcd for [M + Na]+ C24H20ClN3NaO3S 488.0812, found 488.0835.
1-(4-Methoxy-benzoyl)-3-[1-(4-methoxy-phenyl)-2-oxo-4-phenyl-azetidin-3-yl]-thiourea (5d). Yield: 81%; 1H NMR (500 MHz, DMSO) δ 11.53 (s, 1H), 11.18 (d, J = 8.1 Hz, 1H), 7.81 (dd, J = 8.3, 1.1 Hz, 2H), 7.65–7.60 (m, 1H), 7.48 (dd, J = 10.7, 5.0 Hz, 2H), 7.28–7.23 (m, 4H), 6.94–6.90 (m, 4H), 6.12 (dd, J = 8.1, 5.3 Hz, 1H), 5.63 (d, J = 5.2 Hz, 1H), 3.71 (s, 3H), 3.67 (s, 3H). 13C NMR (126 MHz, DMSO) δ 181.45, 168.27, 161.95, 159.12, 155.79, 133.08, 131.52, 130.11, 128.59, 128.44, 128.27, 125.06, 118.46, 114.37, 113.95, 63.78, 59.55, 55.14, 54.92.
1-[1,2-Bis-(4-methoxy-phenyl)-4-oxo-azetidin-3-yl]-3-(4-methoxy-benzoyl)-thiourea (5e). Yield: 90%; 1H NMR (500 MHz, DMSO) δ 11.33 (s, 1H), 11.29 (d, J = 8.0 Hz, 1H), 7.90–7.82 (m, 2H), 7.27–7.21 (m, 4H), 6.99 (dd, J = 9.5, 2.3 Hz, 2H), 6.91 (dt, J = 8.7, 4.7 Hz, 4H), 6.12 (dd, J = 8.0, 5.3 Hz, 1H), 5.62 (d, J = 5.2 Hz, 1H), 3.82 (s, 3H), 3.70 (s, 3H), 3.66 (s, 3H). 13C NMR (126 MHz, DMSO) δ 181.62, 167.46, 163.18, 162.05, 159.12, 155.80, 130.80, 130.13, 128.60, 125.11, 123.24, 118.47, 114.37, 113.95, 113.65, 63.80, 59.64, 55.47, 55.14, 54.92. HRMS (ESI+): m/z calcd for [M + Na]+ C26H25N3NaO5S 514.1413, found 514.1419.
1-[2-(4-Chloro-phenyl)-1-(4-methoxy-phenyl)-4-oxo-azetidin-3-yl]-3-(4-methoxy-benzoyl)-thiourea (5f). Yield: 86%; 1H NMR (500 MHz, DMSO) δ 11.53 (s, 1H), 11.18 (d, J = 8.1 Hz, 1H), 7.81 (dd, J = 8.3, 1.1 Hz, 2H), 7.62 (dt, J = 8.5, 1.1 Hz, 1H), 7.48 (dd, J = 10.8, 4.9 Hz, 2H), 7.27–7.23 (m, 3H), 6.94–6.90 (m, 4H), 6.12 (dd, J = 8.1, 5.3 Hz, 1H), 5.63 (d, J = 5.2 Hz, 1H), 3.71 (s, 3H), 3.67 (s, 3H). 13C NMR (126 MHz, DMSO) δ 181.45, 168.28, 161.95, 159.12, 155.79, 133.08, 131.52, 130.11, 128.59, 128.44, 128.27, 125.06, 118.46, 114.37, 113.95, 63.78, 59.55, 55.15, 54.92.
:1). The advancement of the reaction was monitored via TLC using a mobile phase consisting of 40% ethyl acetate and n-hexane. Once the reaction reached completion, the crude product was subjected to a series of washes using water (3 × 10 mL) and brine (3 × 10 mL). The organic layer was subsequently isolated, dried utilizing anhydrous sodium sulfate, and evaporated under reduced pressure to obtain 1-benzoyl-3-[1-(4-methoxy-phenyl)-2-oxo-4-phenyl-azetidin-3-yl]-urea 8a as a solid material with a white color.
1-Benzoyl-3-[1-(4-methoxy-phenyl)-2-oxo-4-phenyl-azetidin-3-yl]-urea (8a). Yield: 86%; 1H NMR (500 MHz, DMSO) δ 10.78 (s, 1H), 8.86 (d, J = 9.1 Hz, 1H), 7.85–7.79 (m, 2H), 7.63–7.57 (m, 1H), 7.46 (t, J = 7.8 Hz, 2H), 7.38 (t, J = 7.5 Hz, 2H), 7.33–7.24 (m, 5H), 6.97–6.90 (m, 2H), 5.69 (dd, J = 9.1, 5.5 Hz, 1H), 5.60 (d, J = 5.4 Hz, 1H), 3.71 (s, 3H). 13C NMR (126 MHz, DMSO) δ 168.12, 163.27, 155.72, 152.89, 133.95, 132.83, 128.67, 128.35, 128.20, 127.97, 126.87, 118.28, 114.41, 60.06, 59.71, 55.14. HRMS (ESI+): m/z calcd for [M + Na]+ C24H21N3NaO4 438.1430, found 438.1410.
1-Benzoyl-3-[1,2-bis-(4-methoxy-phenyl)-4-oxo-azetidin-3-yl]-urea (8b). Yield: 87%; 1H NMR (500 MHz, DMSO) δ 10.80 (s, 1H), 8.90 (d, J = 8.5 Hz, 1H), 7.84 (d, J = 7.1 Hz, 2H), 7.60–7.59 (m, 1H), 7.48–7.46 (m, 2H), 7.26–7.18 (m, 4H), 6.93 (t, J = 7.7 Hz, 4H), 5.63 (d, J = 7.8 Hz, 1H), 5.54 (d, J = 4.0 Hz, 1H), 3.71 (s, 3H), 3.69 (s, 3H). 13C NMR (126 MHz, DMSO) δ 168.19, 163.36, 159.09, 155.68, 152.93, 132.84, 131.89, 130.33, 128.35, 128.29, 128.00, 125.51, 118.31, 114.37, 114.16, 59.72, 55.14, 54.93. HRMS (ESI+): m/z calcd for [M + Na]+ C25H23N3NaO5 468.1535, found 468.1556.
1-Benzoyl-3-[2-(4-chloro-phenyl)-1-(4-methoxy-phenyl)-4-oxo-azetidin-3-yl]-urea (8c). Yield: 83%; 1H NMR (500 MHz, DMSO) δ 10.73 (s, 1H), 8.81 (d, J = 8.7 Hz, 1H), 7.79–7.73 (m, 2H), 7.56–7.50 (m, 1H), 7.39 (dd, J = 17.1, 8.3 Hz, 4H), 7.24 (d, J = 8.5 Hz, 2H), 7.21–7.16 (m, 2H), 6.86 (d, J = 9.1 Hz, 2H), 5.58 (dd, J = 8.7, 5.5 Hz, 1H), 5.54 (d, J = 5.4 Hz, 1H), 3.64 (s, 3H). 13C NMR (126 MHz, DMSO) δ 168.26, 163.06, 155.81, 152.91, 133.13, 132.89, 132.78, 131.86, 130.22, 128.94, 128.65, 128.40, 128.01, 118.29, 114.47, 59.90, 59.54, 55.18. HRMS (ESI+): m/z calcd for [M + Na]+ C24H20ClN3NaO4 472.1040, found 472.1025.
1-(4-Methoxy-benzoyl)-3-[1-(4-methoxy-phenyl)-2-oxo-4-phenyl-azetidin-3-yl]-urea (8d). Yield: 86%; 1H NMR (500 MHz, DMSO) δ 10.80 (s, 1H), 8.90 (d, J = 9.1 Hz, 1H), 7.84 (dd, J = 8.4, 1.2 Hz, 2H), 7.63–7.57 (m, 1H), 7.47 (dd, J = 10.7, 4.9 Hz, 2H), 7.28–7.19 (m, 4H), 6.97–6.90 (m, 4H), 5.63 (dd, J = 9.1, 5.4 Hz, 1H), 5.54 (d, J = 5.4 Hz, 1H), 3.71 (s, 3H), 3.69 (s, 3H). 13C NMR (126 MHz, DMSO) δ 168.19, 163.35, 159.08, 155.67, 152.92, 132.83, 131.88, 130.33, 128.36, 128.28, 127.99, 125.51, 118.30, 114.37, 114.16, 59.71, 55.14, 54.93. HRMS (ESI+): m/z calcd for [M + Na]+ C25H23N3NaO5 468.1535, found 468.1507.
1-[1,2-Bis-(4-methoxy-phenyl)-4-oxo-azetidin-3-yl]-3-(4-methoxy-benzoyl)-urea (8e). Yield: 85%; 1H NMR (500 MHz, CDCl3) δ 9.03 (d, J = 9.0 Hz, 1H), 8.58 (s, 1H), 7.73 (d, 2H), 7.30 (d, 2H), 7.21 (d, 2H), 6.94–6.91 (m, 2H), 6.90–6.87 (m, 2H), 6.83–6.79 (m, 2H), 5.64 (dd, J = 9.0, 5.3 Hz, 1H), 5.31 (d, J = 5.3 Hz, 1H), 3.85 (s, 3H), 3.76 (s, 3H), 3.74 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 166.65, 163.62, 163.06, 159.88, 156.40, 153.67, 130.72, 129.68, 128.34, 125.18, 124.05, 118.80, 114.54, 114.40, 114.11, 60.91, 60.38, 55.56, 55.46, 55.26. HRMS (ESI+): m/z calcd for [M + Na]+ C26H25N3NaO6 498.1641, found 498.1615.
1-[2-(4-Chloro-phenyl)-1-(4-methoxy-phenyl)-4-oxo-azetidin-3-yl]-3-(4-methoxy-benzoyl)-urea (8f). Yield: 83%; 1H NMR (500 MHz, CDCl3) δ 9.03 (d, J = 8.8 Hz, 1H), 8.46 (s, 1H), 7.76–7.68 (m, 2H), 7.36–7.32 (m, 2H), 7.31–7.27 (m, 2H), 7.25–7.21 (m, 2H), 6.96–6.91 (m, 2H), 6.85–6.80 (m, 2H), 5.66 (dd, J = 8.8, 5.4 Hz, 1H), 5.33 (d, J = 5.3 Hz, 1H), 3.85 (s, 3H), 3.77 (s, 3H).13C NMR (126 MHz, CDCl3) δ 166.70, 163.72, 162.58, 156.58, 153.53, 134.65, 132.11, 130.44, 129.67, 129.30, 128.48, 123.86, 118.69, 114.50, 114.18, 60.77, 60.47, 55.57, 55.48. HRMS (ESI+): m/z calcd for [M + Na]+ C25H22ClN3NaO5 502.1146, found 502.1145.
1-(4-Methoxy-benzoyl)-3-(4-methoxy-phenyl)-thiourea (10a). Yield: 96%; 1H NMR (500 MHz, CDCl3) δ 12.48 (s, 1H), 9.09 (s, 1H), 7.85–7.83 (m, 2H), 7.57–7.54 (m, 2H), 6.99–6.96 (m, 2H), 6.93–6.90 (m, 2H), 3.86 (s, 3H), 3.81 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 178.82, 166.39, 164.00, 158.20, 130.71, 129.74, 125.84, 123.55, 114.46, 114.07, 55.64, 55.48.
1-Benzoyl-3-(4-methoxy-phenyl)-thiourea (10b). Yield: 92%; 1H NMR (500 MHz, CDCl3) δ 12.40 (s, 1H), 9.14 (s, 1H), 7.87 (dt, J = 8.5, 1.6 Hz, 2H), 7.65–7.61 (m, 1H), 7.59–7.54 (m, 2H), 7.54–7.49 (m, 2H), 6.96–6.90 (m, 2H), 3.81 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 178.63, 166.98, 158.28, 133.71, 131.71, 130.60, 129.21, 127.53, 125.85, 114.12, 55.50.
1-Benzoyl-3-benzyl-thiourea (10c). Yield: 95%; 1H NMR (500 MHz, CDCl3) δ 11.02 (s, 1H), 9.13 (s, 1H), 7.82 (dt, J = 8.5, 1.6 Hz, 2H), 7.63–7.57 (m, 1H), 7.52–7.46 (m, 2H), 7.41–7.28 (m, 5H), 4.91 (d, J = 5.4 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 180.10, 166.89, 136.19, 133.60, 131.75, 129.15, 128.91, 127.97, 127.95, 127.50, 49.83.
N-Benzyl-4-methoxy-benzamide (11a). Yield: 87%; 1H NMR (500 MHz, CDCl3) δ 7.75 (dd, J = 6.9, 4.9 Hz, 2H), 7.37–7.33 (m, 3H), 7.29 (ddd, J = 9.6, 7.5, 3.3 Hz, 2H), 6.94–6.89 (m, 2H), 6.33 (s, 1H), 4.63 (d, J = 5.7 Hz, 2H), 3.84 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 166.87, 162.26, 138.39, 129.43, 128.78, 128.76, 128.61, 127.93, 127.59, 127.47, 126.65, 113.80, 55.42, 44.11.
N-Benzyl-benzamide (11b). Yield: 86%; 1H NMR (500 MHz, CDCl3) δ 8.30 (s, 1H), 7.90–7.78 (m, 2H), 7.57 (t, J = 15.7 Hz, 2H), 7.39–7.28 (m, 2H), 7.04–6.91 (m, 2H), 3.88 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 162.89, 137.14, 129.06, 128.95, 122.42, 121.39, 115.48, 114.04, 55.49.
N-(4-Chloro-phenyl)-4-methoxy-benzamide (11c). Yield: 83%; 1H NMR (500 MHz, CDCl3) δ 7.79 (dt, J = 8.5, 1.7 Hz, 2H), 7.52–7.47 (m, 1H), 7.42 (ddd, J = 6.8, 4.5, 1.2 Hz, 2H), 7.37–7.33 (m, 4H), 7.32–7.27 (m, 1H), 6.45 (s, 1H), 4.65 (d, J = 5.7 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 167.37, 138.19, 134.41, 131.57, 129.48, 128.81, 128.61, 127.94, 127.65, 126.97, 44.16.
3.2 In vitro antimicrobial evaluation
The compounds were evaluated using microtiter plate dilution assay against pathogenic strains Staphylococcus aureus MTCC 1430, Bacillus cereus MTCC 9490, Pseudomonas aeruginosa MTCC 1934, and Escherichia coli MTCC 1610 grown in nutrient broth (NB). Additionally, antifungal activity was determined against Candida albicans MTCC 1637 and Candida tropicalis MTCC 184. A two-fold serial dilution technique was used to examine the test compounds against the above-mentioned strains, and known antibiotics and DMSO were used as positive and vehicle controls respectively. The antimicrobial efficacy is reported in terms of MIC values, i.e. the lowest concentrations that inhibited microorganism growth. The MIC value was determined by repeating each experiment three times (Table 2).3.3 Cytotoxicity assay
Normal human hepatic (THLE-2) and human breast adenocarcinoma (MCF-7) cells were maintained at 37 °C in a humidified incubator containing 5% CO2 in DMEM and RPMI-1640, respectively, 10% fetal bovine serum, 100 μg mL−1 streptomycin and 100 U mL−1 penicillin. 1 × 104 cells per well were inoculated in a 96-well plate. After culturing for 24 h, media were carefully aspirated from the wells and the cells were further treated with different compounds which were diluted in the respective media followed by incubation for 24 h. MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) was added at 5 mg mL−1 concentration in each well and incubated for 4 h. Furthermore, the residual medium was removed and 100 μL of DMSO was added to each well to dissolve formazan. Absorbance was recorded at 540 nm using a microplate reader. The cell viability was calculated from the three independent experiments (Fig. 4 and 5).3.4 Molecular docking
The crystal structures of Methicillin-resistant Staphylococcus aureus (MRSA) (PDB ID: 1VQQ) and CYP51 (5TZ1) were downloaded from the protein data bank (https://www.rcsb.org/). The Autodock 4.2.6 tool was employed for molecular docking investigations and the Biovia Discovery Studio visualizer was utilized for exploring the protein–ligand binding interactions.4. Conclusions
In the present study, we designed and synthesized cis-3-aroyl-thiourea/urea-β-lactams with comparable structural variability. In this work, while traditional routes were optimized for nucleophilic capturing of aroyl isothiocyanates with cis-3-amino-β-lactams to obtain cis-3-aroyl-thiourea-β-lactams, a new synthetic methodology was developed to attain cis-3-aroyl-urea-β-lactams. To achieve this, aroyl isoselenocyanates were synthesized and then reacted with cis-3-amino-β-lactams in THF:H2O (9:1). This synthetic strategy bypasses the earlier need for harsh synthesis of unstable aroyl isocyanates and low yielding acylation of urea to obtain aroyl urea derivatives. Furthermore, we assessed the antibacterial activity of these compounds against Gram-positive bacteria (S. aureus and B. cereus) and Gram-negative bacteria (P. aeruginosa and E. coli). Notably, 3-aroyl-thiourea-β-lactams 5b displayed the highest antibacterial potency against B. cereus (MIC: 12.5 μg mL−1), while maintaining over 90% viability of THLE-2 cells at concentrations of up to 50 μg mL−1. Among the 3-aroyl-urea-β-lactams, 8e displayed activity against all bacterial strains with a notable MIC value of 6.25 μg mL−1 against B. cereus (four-fold better than ampicillin). In addition, 8d and 8e were identified as promising antifungal agents with an MIC of 1.5 μg mL−1 against both Candida species (comparable to fluconazole with an MIC of 0.5 μg mL−1). Also, 8d and 8e show 80% and 75% cell viability against THLE-2 cells at a concentration of 25 μg mL−1. Molecular docking analysis also predicted interactions between the compounds and key amino acid residues in target PBPs (PDB ID: 1VQQ) and CYP51 (PDB ID: 5TZ1), supporting the in vitro activities of 5b and 8e. Furthermore, the computed ADME parameters suggest favorable drug-like characteristics for all molecules, as they adhered to Lipinski's rule of five, Veber's rule, and Muegge's rule.
Author contributions
Pankaj Kumar: conceptualization, methodology, synthesis, data collection and interpretation, molecular docking, original draft preparation; Jaswinder Kaur: conceptualization, methodology, synthesis, data collection and interpretation, molecular docking, original draft preparation; Sumeeta Kumari: in vitro biological assay (antibacterial and antifungal); Sakshi Paliwal: supplementary data collection, IC50 curves and IC50 value calculations; Shiwani Berry: reviewing and editing; Anil Kumar Pinnaka: conceptualization, validation, formal analysis, resources, supervision, reviewing and editing; and Aman Bhalla: conceptualization, methodology, validation, formal analysis, resources, supervision, reviewing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Science and Engineering Research Board (SERB-DST), New Delhi, Government of India, Project No. SR/FT/CS-037/2010 dated 28-10-2010, Council of Scientific and Industrial Research (CSIR) New Delhi, India vide Award No. – F. No. 09/135(0823)/2018-EMR-I, University Grant Commission (UGC), New Delhi, India vide Award No. – F. No. 21/(CSIR-UGC NET JUNE 2019) and No. F.30-581/2021 (BSR), and Department of Biotechnology, Ministry of Science and Technology, New Delhi, India vide Award No. DBTHRDPMU/JRF/BET-23/I/2023-24/118.Notes and references
- R. Gaynes, Emerg. Infect. Dis., 2017, 23, 849–853 CrossRef.
- L. Decuyper, K. Magdalenić, M. Verstraete, M. Jukič, I. Sosič, E. Sauvage, A. M. Amoroso, O. Verlaine, B. Joris, S. Gobec and M. D'hooghe, Chem. – Eur. J., 2019, 25, 16128 CrossRef CAS PubMed.
- F. Prestinaci, P. Pezzotti and A. Pantosti, Pathog Glob Health., 2015, 109, 309 CrossRef PubMed.
- A. Fedarovich, K. A. Djordjevic, S. M. Swanson, Y. K. Peterson, R. A. Nicholas and C. Davies, PLoS One, 2012, 7, e44918 CrossRef CAS.
- N. C. Rosas and T. Lithgow, Trends Microbiol., 2022, 30, 544 CrossRef CAS PubMed.
- T. Idowu, D. Ammeter, M. Brizuela, G. Jackson, S. Alam and F. Schweizer, Bioorg. Med. Chem. Lett., 2020, 30, 127575 CrossRef CAS PubMed.
- S. Nami, A. Aghebati-Maleki, H. Morovati and L. Aghebati-Maleki, Biomed. Pharmacother., 2019, 110, 857 CrossRef CAS PubMed.
- N. M. Revie, K. R. Iyer, N. Robbins and L. E. Cowen, Curr. Opin. Microbiol., 2018, 45, 70 CrossRef CAS.
- M. J. Torres-Ossandón, A. Vega-Gálvez, C. E. Salas, J. Rubio, E. Silva-Moreno and L. Castillo, Int. J. Food Microbiol., 2019, 289, 7 CrossRef.
- M. C. Fisher, A. Alastruey-Izquierdo, J. Berman, T. Bicanic, E. M. Bignell, P. Bowyer, M. Bromley, R. Brüggemann, G. Garber, O. A. Cornely and S. J. Gurr, Nat. Rev. Microbiol., 2022, 20, 557 CrossRef CAS PubMed.
- H. E. Hashem, A. E. G. E. Amr, E. S. Nossier, E. A. Elsayed and E. M. Azmy, Molecules, 2020, 25, 2766 CrossRef CAS.
- L. C. S. Pinheiro, L. V. B. Hoelz, M. L. G. Ferreira, L. G. Oliveira, R. F. A. Pereira, A. M. do Valle, L. S. P. André, J. Scaffo, F. R. Pinheiro, T. A. N. Ribeiro, D. Sachs, A. C. R. F. Pascoal, N. Boechat and F. Aguiar-Alves, Lett. Appl. Microbiol., 2020, 71, 645 CrossRef CAS PubMed.
- A. Maalik, H. Rahim, M. Saleem, N. Fatima, A. Rauf, A. Wadood, M. I. Malik, A. Ahmed, H. Rafique, M. N. Zafar, M. Riaz, L. Rasheed and A. Mumtaz, Bioorg. Chem., 2019, 88, 102946 CrossRef CAS.
- Z. J. Zhang, Y. Zeng, Z. Y. Jiang, B. S. Shu, V. Sethuraman and G. H. Zhong, Pest. Manag. Sci., 2018, 74, 1736 CrossRef CAS.
- F. M. Sroor, A. M. Othman, M. A. Tantawy, K. F. Mahrous and M. E. El-Naggar, Bioorg. Chem., 2021, 112, 104953 CrossRef CAS PubMed.
- M. Blair, J. M. Côté, A. Cotter, B. Lynch, L. Redahan and P. T. Murray, Am. J. Nephrol., 2021, 52, 85 CrossRef CAS PubMed.
- P. Singh, R. Raj, P. Singh, J. Gut, P. J. Rosenthal and V. Kumar, Eur. J. Med. Chem., 2014, 71, 128 CrossRef CAS PubMed.
- C. Yoakim, W. W. Ogilvie, D. R. Cameron, C. Chabot, I. Guse, B. Haché, J. Naud, J. A. O’meara, R. Plante and R. Déziel, J. Med. Chem., 1998, 41, 2882 CrossRef CAS.
- A. Puglisi, M. Benaglia, L. Raimondi, L. Lay and L. Polettia, Org. Biomol. Chem., 2011, 9, 3295 RSC.
- M. Z. Deng, P. Caubere, J. P. Senet and S. Lecolier, Tetrahedron, 1988, 44, 6079 CrossRef CAS.
- D. Liptrot, L. Alcaraz and B. Roberts, Adv. Synth. Catal., 2010, 352, 2183 CrossRef CAS.
- Z. Cui, Y. Ling, B. Li, Y. Li, C. Rui, J. Cui, Y. Shi and X. Yang, Molecules, 2010, 15, 4267 CrossRef CAS.
- J. Yang, L. Chen, Y. Dong, J. Zhang and Y. Wu, Synth. Commun., 2022, 52, 63 CrossRef CAS.
- I. A. Hampe, J. Friedman, M. Edgerton and J. Morschhäuser, PLoS Pathog., 2017, 13, 1006655 CrossRef PubMed.
- H. Harikrishnan, B. Arayambath, V. K. Jayaraman, K. Ekambaram, E. A. Ahmed, P. Senthilkumar, H. I. M. Ibrahim, A. Sundaresan and K. Thirugnanasambantham, World J. Microbiol. Biotechnol., 2022, 38, 224 CrossRef.
- R. Kaur, D. Desai, S. Amin, K. Raza, A. Bhalla, P. Yadav and N. Kaushal, Mol. Cell. Biochem., 2023, 478, 621 CrossRef CAS.
- A. Garg, D. Desai, A. Bhalla, S. Thakur, P. Rastogi and N. Kaushal, Sci. Rep., 2023, 13, 9301 CrossRef CAS.
- P. Ghanghas, M. Sharma, D. Desai, K. Raza, A. Bhalla, P. Kumar, D. Narula, S. Amin, S. N. Sanyal and N. Kaushal, Biol. Trace Elem. Res., 2021, 200, 635 CrossRef.
- A. Bhalla, G. Modi, P. Yadav, P. Kumar, S. S. Bari and G. Hundal, Tetrahedron Lett., 2020, 61, 152098 CrossRef CAS.
- D. Narula, S. S. Bari, G. Singh, R. Sharma, A. Garg and A. Bhalla, J. Sulfur Chem., 2023, 1 Search PubMed.
- A. Vijeata, G. R. Chaudhary, A. Bhalla and S. Chaudhary, ACS Appl. Bio Mater., 2023, 6, 1849 CrossRef CAS.
- R. Ronchetti, G. Moroni, A. Carotti, A. Gioiello and E. Camaioni, RSC Med. Chem., 2021, 12, 1046 RSC.
- Z. Gan, G. Li, Q. Yan, W. Deng, Y. Y. Jiang and D. Yang, Green Chem., 2020, 22, 2956 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Copies of the 1H NMR, 13C{1H} NMR and HRMS spectra of new compounds (pdf). See DOI: https://doi.org/10.1039/d3nj04485h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |