
Enhanced oxidative degradation of tetracycline by visible light-promoted g-C3N4 modified Cu3(OH)4SO4/Cu7S4 composites under an air atmosphere†
Yan
Wang
,
Haoran
Li
,
Daqing
Chen
,
Danhua
Ge
 * and
Xiaojun
Chen
*
* and
Xiaojun
Chen
*
College of Chemistry and Molecular Engineering, Nanjing Tech University, Nanjing, 211800, P. R. China. E-mail: gedanhua@njtech.edu.cn; chenxj@njtech.edu.cn
First published on 23rd November 2023
Abstract
Herein, a simple solvothermal method was used to prepare a CuSx/g-C3N4 (CSG) heterojunction as an efficient oxygen activator containing sulfur vacancies (SVs). The as-prepared catalyst can activate oxygen to effectively degrade tetracycline (TC). The degradation rate of TC (20 mg L−1) reached 98.9% in 90 min over CSG (0.02 g L−1) under an air atmosphere, which maintained good performance within a wide pH range of 6–10. It was almost unaffected by any light source and water quality, indicating a highly efficient oxygen activator and good stability. Moreover, the trapping experiments and EPR results revealed that the main reactive oxygen species (ROSs) were singlet oxygen (1O2) and hydroxyl radicals (˙OH). CSG (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) enabled the activation of oxygen-produced ROS for removing TC even in the dark, which was in accordance with the EPR results. In addition, the CSG catalyst exhibited a good removal performance in practical wastewater treatment. Our study demonstrated that the CSG catalyst had the potential to remove organic pollutants from wastewater.
:1) enabled the activation of oxygen-produced ROS for removing TC even in the dark, which was in accordance with the EPR results. In addition, the CSG catalyst exhibited a good removal performance in practical wastewater treatment. Our study demonstrated that the CSG catalyst had the potential to remove organic pollutants from wastewater.
Introduction
Tetracycline (TC), as a broad-spectrum antibiotic, has been applied in various fields including agriculture and livestock products and bacterial infections because of its low production cost and broad-spectrum antibacterial activity.1 However, it is reported that microbial resistance to antibiotics can result from the long-term enrichment of TC, which produces direct toxicity to native microorganisms at the same time.2 Thus, the excessive use of antibiotics causes significant risks to eco-environments and human health, and has become a global problem in environmental governance.3 Various international health organizations have set maximum residue levels of 100 ppb for TC in fish and milk.3 Unfortunately, the diversified receiving aquatic environments including drinking water can determine the residues of TC.4,5 Given these, it is urgent to develop efficient methods for the removal of TC from aquatic environments.Up to now, many techniques have been proposed to remove TC, such as adsorption, membrane filtration, ion exchange and advanced oxidation processes (AOPs).1,3,6,7 Among these methods, AOPs (such as electrochemical oxidation, ultrasonic degradation, photocatalysis, microwave irradiation, Fenton/Fenton-like oxidation and so on) have proven a good application for the treatment of refractory organic pollutants in the environment.8–15 Currently, the conventional peroxymonosulfate (PMS) and H2O2 activation processes based on AOPs by transition metals or UV irradiation are gaining researchers' attention.16,17 However, using dissolved oxygen (O2) as an environmentally safe oxidant to generate radical or anionic reactive oxygen species (ROS: ˙OH, ˙O2−, and 1O2) has been rarely reported to remove refractory organics in wastewater or groundwater.18
Owing to their similar redox properties to Fe, Cu-based Fenton-like catalysts also have excellent catalytic performance for the degradation of organic pollutants, such as Cu2(OH)PO4/g-C3N4, Cu2O@g-C3N4, Cu-modified alkalinized g-C3N4 and CoSx–CuSx/copper foam.19–22 Among them, copper sulfide (CuSx), a p-type indirect band-gap semiconductor material, has attracted extensive interest due to its nontoxicity, variable valence state, resource abundance, relative eco-friendliness and variable stoichiometries with different band-gap energies and electronic properties.23 Nevertheless, the insufficient active sites of CuS have resulted in unsatisfactory degradation efficiency. In addition, Cu3(OH)4SO4 has been proven to be photocatalytically active under NIR irradiation because of its unique structure.24 Two different Cu2+ environments in Cu3(OH)4SO4 forming CuOm and Cu′On polyhedra can be joined by Cu–O–Cu′ bridges. Moreover, SO4 as an acceptor ligand provides empty orbitals (namely, the σ* orbital of the S–O bonds) for extra electrons, so that the empty states of Cu3(OH)4SO4 around its gap band minimum appear as the orbital character of SO4. Accordingly, the “siphoning” of photogenerated electrons into SO4 would boost the separation of photogenerated electron–hole pairs in Cu3(OH)4SO4.24
Graphite-like carbon nitride (g-C3N4) is a kind of metal-free polymeric semiconductor, which is facilely prepared by thermal polycondensation. In recent years, g-C3N4 has exhibited good performance in environmental pollutant removal and photocatalytic hydrogen generation because of its excellent physical and chemical stability, low cost, unique electronic structure and narrow band gap (2.7 eV).25,26 For example, g-C3N4 as a visible light photocatalyst was employed in the degradation of rhodamine B (RhB) by activating H2O2.27 However, the inevitable shortcomings of low utilization efficiency and rapid recombination of photoinduced electron–hole pairs would limit the photocatalytic activity of g-C3N4.26 Surprisingly, the formation of a heterojunction by combining g-C3N4 has been proven to be an effective strategy to improve its performance. For example, Cai et al. designed g-C3N4/CuS heterojunctions for the degradation of RhB and MB.26 Fe and Cu doped g-C3N4 catalysts were employed to activate PMS for degrading ofloxacin with excellent catalytic activity.28
In this regard, a novel CuSx/g-C3N4 (CSG) heterojunction with sulfur vacancies (SVs) was successfully synthesized by the formation of Cu3(OH)4SO4 and Cu7S4 coupled with g-C3N4 for efficient antibiotic removal. The obtained CSG was characterized by SEM, TEM, XRD, BET and XPS measurements. The experimental results show that the CSG heterojunction exhibits a high performance in activating O2 for the removal of TC even in the dark, owing to sufficient SV contributions. In addition, it was found that structural stability and more metallic active sites were guaranteed by g-C3N4. This proposed study of CSG heterojunctions provides a new prospect for the exploitation of efficient materials to destroy organic pollutants.
Experimental section
Materials and reagents
All chemicals and solvents used in the experiments were of analytical grade and used directly without additional treatments. Tetracycline (TC), copper acetate monohydrate (Cu(OAc)2·H2O), thiourea (CH4N2S), cetyltrimethylammonium bromide (CTAB), ethanol, urea, cupric chloride (CuCl2) and ammonium hydroxide (NH3·H2O) were purchased from Sinopharm Chemical Reagent Co. Ltd, China. Deionized water was prepared in the laboratory and used throughout the experiments. Ethylenediamine was obtained from Shanghai Aladdin Biochemical Technology Co., Ltd. Copper(II) sulfate pentahydrate (CuSO4·5H2O) was purchased from Sigma-Aldrich (Finland).Preparation of the CSG heterojunction
In this study, the CSG heterojunction was synthesized by a solvothermal method according to a previously reported procedure with some modifications.29 In brief, 10 g of urea was placed in a muffle furnace and heated at 550 °C for 3 h with a heating rate of 10 °C min−1. After cooling to room temperature, a pale yellow solid powder was obtained (g-C3N4) for further use. Then, 0.1 g of the as-synthesized g-C3N4, 0.182 g of CTAB, 0.3433 g of Cu(OAc)2·H2O and 0.4933 g of CH4N2S were dispersed in 30 mL of ethanol with ultrasonication. After that, the solution was transferred to a three-neck round bottom flask and heated at 65 °C for 3 h. The cyan-grey solid product was collected by centrifugation, washed three times with ethanol and dried in an oven at 60 °C for 12 h. Finally, the material was ground and calcined in a tube furnace at 400 °C for 4 h, which was named as CSG (4:1) according to the mass ratio of Cu(OAc)2·H2O and g-C3N4.
For comparison, the samples were obtained under the same conditions mentioned above with different Cu amounts, and were marked as CSG (1:4), CSG (1:1) and CSG (9:1). In addition, the sample was prepared without g-C3N4 and named as CS. We synthesized Cu3(OH)4SO4, Cu7S4, Cu3(OH)4SO4/g-C3N4 and Cu7S4/g-C3N4 following the previous reports.24,30 For Cu3(OH)4SO4, 0.02 mol of CuSO4·5H2O was dissolved in 80 mL of deionized water containing 1 mL of NH3·H2O. After stirring for 30 min, the suspension was transferred to a 120 mL sealed PTFE-lined automated crucible and hydrothermally treated at 150 °C for 12 h. Finally, it was separated by centrifugation with ethanol and dried in an oven overnight. For the Cu3(OH)4SO4/g-C3N4 heterojunction, the other operations were the same as those for Cu3(OH)4SO4, except for the addition of 1.4 g of g-C3N4 powder. For Cu7S4, 0.618 g of CuCl2 was added to 50 mL of ethylenediamine with constant stirring. When the colour of the solution changed from clear to dark blue, 0.182 g of CH4N2S was added to the above solution. After stirring for 10 min, the solutions were transferred to a Teflon-lined stainless steel autoclave, sealed and kept at 150 °C for 12 h. For Cu7S4/g-C3N4, the other operations were the same as those for Cu7S4, except for the addition of g-C3N4 powder. In addition, Cu3(OH)4SO4/Cu7S4 (1:1) was prepared by mixing Cu3(OH)4SO4 and Cu7S4 in a mass ratio of 1:1.
Material characterization
The morphologies and surface structures of the as-prepared samples were determined by scanning electron microscopy (FESEM, Fei Talos F200x G2) and transmission electron microscopy (TEM, Fei Talos-F200s). The structures and elemental compositions of the samples were confirmed by powder X-ray diffraction (XRD, MiniFlex600), electron paramagnetic resonance spectroscopy (ESR, Bruker EMXplus-6/1) and X-ray photoelectron spectroscopy (XPS, Axis supra). Absorbance measurements were performed using a UV-Vis-NIR spectrophotometer (LAMBDA950, PerkinElmer). The Brunauer–Emmett–Teller (BET) surface area was calculated by monitoring the N2 adsorption/desorption using a NOVA 2000 surface area analyzer (Quantachrome) at 77 K.General procedure for the removal of TC
First, we chose 20 mg L−1 TC in the comparative photocatalytic experiment. 20 mg of the CSG catalyst was selected and added to a 20 mL TC aqueous solution. The process lasted for 1.5 h under visible light irradiation. A visible light source (780 nm ≥ λ ≥ 420 nm) was supplied by a daylight lamp (30 W). The absorbance of the solution was measured using a UV-vis spectrophotometer. In addition, to test the durability and recyclability of the material, recycling experiments were carried out. After each cycle, the solid catalyst was filtered, washed with deionized water to remove residual contaminants, dried overnight at 60 °C, and ready for reuse.Result and discussion
As shown in Fig. 1, XRD spectra are employed to verify the composite compositions and crystalline properties of the prepared pure g-C3N4, CSG (1:4), CSG (1:1), CSG (4:1), and CSG (9:1) heterojunctions, as well as Cu3(OH)4SO4, Cu7S4, Cu3(OH)4SO4/g-C3N4 and Cu7S4/g-C3N4. For g-C3N4, the characteristic peaks at 12.8° and 27.3° correspond to (100) and (002) facets, respectively, suggesting the successful preparation of g-C3N4. In addition, the (100) and (002) facets in the g-C3N4 phase are associated with the in-plane packing of the tri-s-triazine facet and the interlayer stacking of the conjugated aromatic rings, respectively.28 For CS, the characteristic peaks at 27.6°, 29.3°, 31.6°, 32.9°, 47.9°, 52.6° and 59.3° correspond to the (101), (102), (103), (006), (110), (108) and (116) planes of CuS (JCPDS NO. 06-0464), and other peaks at 27.6°, 31.6°, 46.1° and 54.6° correspond to the (111), (200), (220) and (311) planes of Cu7.2S4 (JCPDS NO. 24-0061). Moreover, six peaks of CSG (4:1) and CSG (9:1) heterojunctions at 14.4°, 16.1°, 17.9°, 24.3°, 32.9° and 34.6° were obviously observed, which match well with those of synthetic antlerite Cu3(OH)4SO4 (JCPDS card No. 07-0407).31 Meanwhile, another four peaks of CSG (4:1) and CSG (9:1) heterojunctions at 32.0°, 35.3°, 42.1° and 46.2° correspond to the diffraction plane of (220), (031), (124) and (224) of Cu7S4 (JCPDS card No. 33-0489), respectively. And the intensity of g-C3N4 peaks gradually becomes weaker. However, CSG (1:4) and CSG (1:1) heterojunctions exhibited only g-C3N4 peaks. This indicates that Cu-based materials (Cu3(OH)4SO4 and Cu7S4 combined as CuSx in this study) were successfully introduced into g-C3N4via our synthetic strategy when a certain amount of Cu(OAc)2·H2O was added. The XRD patterns of Cu3(OH)4SO4, Cu7S4, Cu3(OH)4SO4/g-C3N4 and Cu7S4/g-C3N4 are shown in Fig. 1B. The diffraction peaks matched well with those in Fig. 1A, suggesting the successful synthesis of the samples.
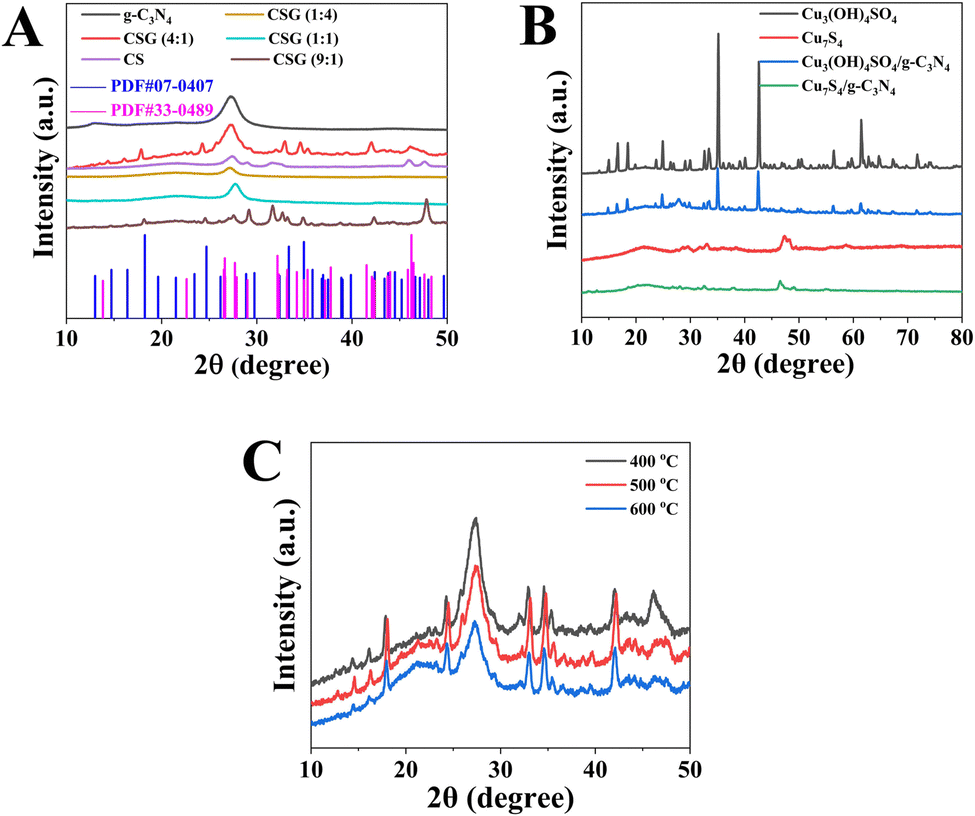 | ||
| Fig. 1 (A) XRD patterns of g-C3N4, CSG (1:4), CSG (1:1), CSG (4:1), CSG (9:1) heterojunctions and CS. (B) XRD patterns of Cu3(OH)4SO4, Cu7S4, Cu3(OH)4SO4/g-C3N4 and Cu7S4/g-C3N4. (C) XRD patterns of CSG (4:1) heterojunctions treated at different calcination temperatures. | ||
SEM and TEM were utilized to reveal the surface morphologies of the as-prepared samples. Fig. 2 shows the SEM and TEM images of pure g-C3N4 and CSG (4:1) heterojunctions. As shown in Fig. 2A, the obtained g-C3N4 consists of layered structures and irregular granules with many pores because ammonia and CO2 gases are released during urea pyrolysis.32 The SEM image of CS displayed nanoparticle morphology (Fig. S1, ESI†). The SEM and TEM images of the CSG (4:1) heterojunctions display similar porous-layered structures, suggesting that the introduction of Cu3(OH)4SO4 and Cu7S4 does not change the skeleton structure of g-C3N4 (Fig. 2B–E). In addition, the components and structure were assessed using TEM elemental mapping, which indicated a uniform distribution of Cu, S, C and N in the CSG (4:1) heterojunctions (Fig. 2F). As shown in Fig. S2A (ESI†), the lattice stripe spacing of g-C3N4 cannot be observed in the HRTEM image, which may be derived from a low degree of order in the layers of g-C3N4.33 As seen from the HRTEM image of CSG (4:1) in Fig. S2B (ESI†), the observed lattice fringe spacing of 0.280 nm indicated the (220) planes of Cu7S4, and a spacing of 0.267 nm corresponded to the (310) planes of Cu3(OH)4SO4.
 | ||
| Fig. 2 SEM images of (A) pure g-C3N4 and (B) and (C) CSG (4:1) heterojunctions. TEM images (D) and (E) and TEM elemental mapping (F) of CSG (4:1) heterojunctions. | ||
The N2 adsorption–desorption experiments were performed to investigate the BET specific surface areas (SBET) of pure CS, pure g-C3N4 and CSG (4:1) heterojunctions. Fig. S3 (ESI†) reveals that all samples exhibit type IV isotherms, which proves the existence of mesoporous structures. CSG (4:1) (29.24 m2 g−1) has a BET surface area between that of pure g-C3N4 (54.85 m2 g−1) and CS (14.33 m2 g−1). This phenomenon may be because the deposited CS nanoparticles obstructed some mesopores in g-C3N4. Thus, the CSG (4:1) heterojunctions with a large surface area can be expected to promote the activity in the adsorption and photocatalytic degradation because more reactive sites and stronger adsorption ability may derive from a high surface area.34
The valence states and bond configurations of the as-synthesized heterojunctions were evaluated in detail by XPS. As shown in Fig. 3A, the survey spectrum indicates the presence of C, N, S and Cu elements, which is consistent with the XRD analysis. The signal peak of C 1s at 284.8 eV is attributed to the sp2 C–C from carbon-containing contaminations, while the other peak at 288.0 eV is from the sp2 N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond in the graphitic structure (Fig. 3B).34 The spectrum of N 1s is fitted to four regions at around 398.7 eV, 399.2 eV, 400.7 eV and 404.7 eV, which is attributed to the bonding of C–NC, N(N–(C)3), N–H and π excitations, respectively (Fig. 3C).29,31 The high-resolution XPS spectrum of Cu 2p shows two binding energy peaks located at 932.4 eV and 952.5 eV, which are assigned to Cu+ 2p3/2 and Cu+ 2p1/2, respectively (Fig. 3D). And the other two peaks centered at 933.1 eV and 953.3 eV are ascribed to Cu2+ 2p3/2 and Cu2+ 2p1/2, respectively. For the S 2p spectra (Fig. 3E), the characteristic peaks of S 2p3/2 and S 2p1/2 appear at 161.8 eV and 164.1 eV, which represent metal–sulfur bonds and sulfur with low coordination on the surface, respectively. It has been previously reported that the S 2p1/2 core level is generally connected to the SVs in the structure.35 In addition, the XPS curves of pure g-C3N4 and CS are shown in Fig. 3. The chemical state of N 1s in CSG (4:1) was similar to that of g-C3N4, and a slight shift in the binding energy revealed that the heterojunction was formed between g-C3N4 and CS (Fig. 3C). The high-resolution Cu 2p spectra of CS contain shakeup satellites, indicating the presence of a d9 configuration in Cu2+ (Fig. 3D).36 However, the high-solution Cu 2p spectra of CSG (4:1) had no shakeup satellites, suggesting a decrease in the content of Cu2+. More valence electrons of CSG (4:1) were transferred to Cu2+ so that the binding energy was reduced.
N bond in the graphitic structure (Fig. 3B).34 The spectrum of N 1s is fitted to four regions at around 398.7 eV, 399.2 eV, 400.7 eV and 404.7 eV, which is attributed to the bonding of C–NC, N(N–(C)3), N–H and π excitations, respectively (Fig. 3C).29,31 The high-resolution XPS spectrum of Cu 2p shows two binding energy peaks located at 932.4 eV and 952.5 eV, which are assigned to Cu+ 2p3/2 and Cu+ 2p1/2, respectively (Fig. 3D). And the other two peaks centered at 933.1 eV and 953.3 eV are ascribed to Cu2+ 2p3/2 and Cu2+ 2p1/2, respectively. For the S 2p spectra (Fig. 3E), the characteristic peaks of S 2p3/2 and S 2p1/2 appear at 161.8 eV and 164.1 eV, which represent metal–sulfur bonds and sulfur with low coordination on the surface, respectively. It has been previously reported that the S 2p1/2 core level is generally connected to the SVs in the structure.35 In addition, the XPS curves of pure g-C3N4 and CS are shown in Fig. 3. The chemical state of N 1s in CSG (4:1) was similar to that of g-C3N4, and a slight shift in the binding energy revealed that the heterojunction was formed between g-C3N4 and CS (Fig. 3C). The high-resolution Cu 2p spectra of CS contain shakeup satellites, indicating the presence of a d9 configuration in Cu2+ (Fig. 3D).36 However, the high-solution Cu 2p spectra of CSG (4:1) had no shakeup satellites, suggesting a decrease in the content of Cu2+. More valence electrons of CSG (4:1) were transferred to Cu2+ so that the binding energy was reduced.
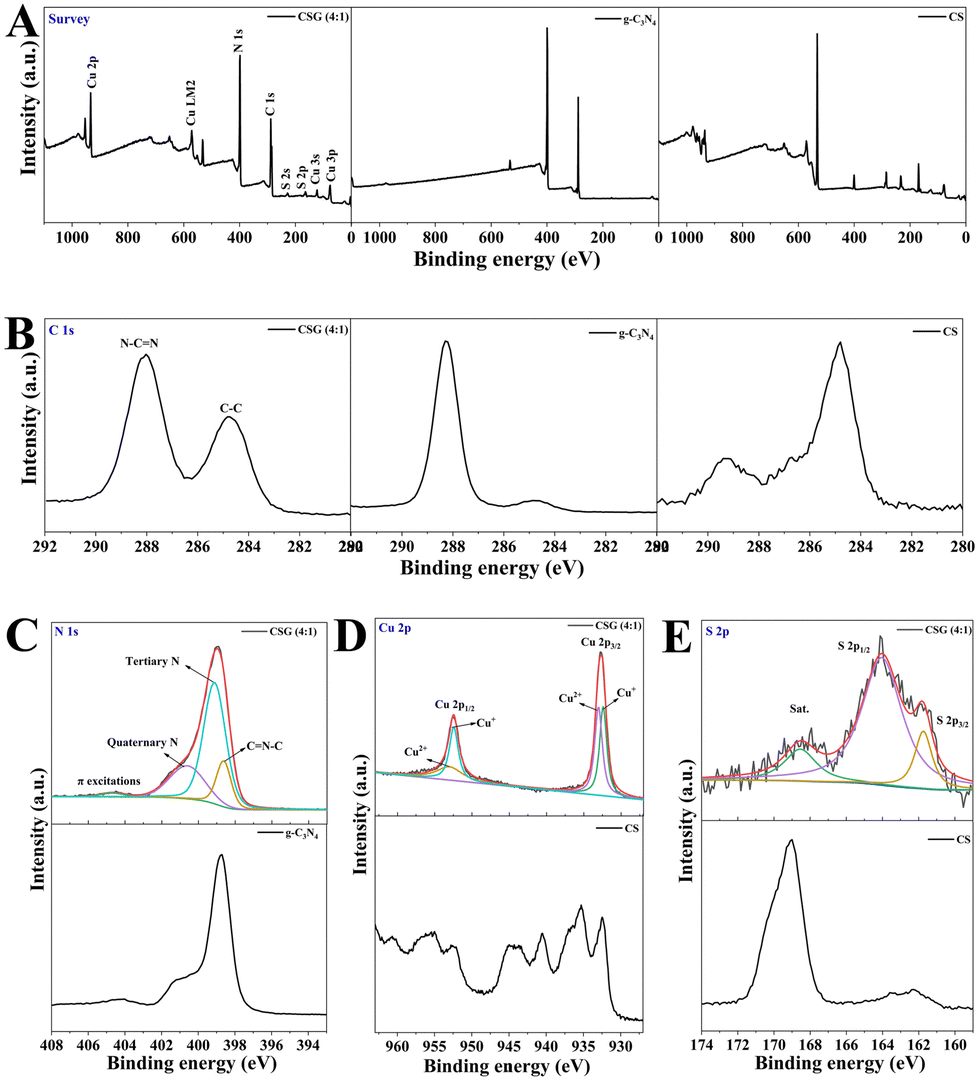 | ||
| Fig. 3 XPS of g-C3N4, CS and CSG (4:1) heterojunctions: (A) survey spectra, (B) C 1s, (C) N 1s, (D) Cu 2p and (E) S 2p high-resolution spectra. | ||
To investigate the catalytic effect of CSG (4:1) heterojunctions on TC abatement under an air atmosphere, the control experiments were performed. The effects of light irradiation and dark conditions on TC removal are shown in Fig. S4 (ESI†). The removal efficiency of TC was visibly lowered without being exposed to light, suggesting that CSG (4:1) enabled the activation of oxygen-produced ROS for removing TC even without light irradiation.37 The initial TC concentration and the catalyst amount were 20 mg L−1 and 1.0 mg mL−1, respectively. Fig. 4A shows the effect of Cu on TC removal efficiency over catalysts with different Cu loading amounts. For pure g-C3N4, TC decreased by only 8.9% after 90 min. In addition, the removal efficiency of TC over the CS sample without g-C3N4 was lower than that of CSG (1:1) and CSG (4:1), suggesting the importance of g-C3N4. Among the g-C3N4, CS, CSG (1:4), CSG (1:1), CSG (4:1) and CSG (9:1) catalysts, CSG (4:1) achieved the highest removal of TC, indicating that a moderate Cu amount may elevate the active site density on the surface of g-C3N4.38 Thereby, the CSG (4:1) heterojunctions were chosen for further investigation. In addition, the removal efficiencies of TC over CSG (4:1) samples obtained at different calcination temperatures (300–600 °C) are illustrated in Fig. 4B. Optimum catalytic activity of the CSG (4:1) was observed after calcination treatment at 400 °C, which brought about 96.3% of TC abatement at 90 min. As shown in Fig. 1C, the XRD patterns of the samples treated at 500 °C and 600 °C agreed with those treated at 400 °C, which suggested that the structures of the catalysts did not change with temperature. CSG (4:1) is considered as the product after the heat treatment at 400 °C. As shown in Fig. 4C, according to the Langmuir–Hinshelwood model, the degradation rate constant of TC over the CSG (4:1) was calculated following the equation ln(C0/C) = kt, where C0 and C represent the initial concentration and the concentration over time of the TC solution, respectively, and the slope k is the apparent reaction rate constant (min−1).34 As shown in Fig. 4D, the CSG (4:1) at 400 °C provides the highest slope k value (0.049 min−1) for TC degradation.
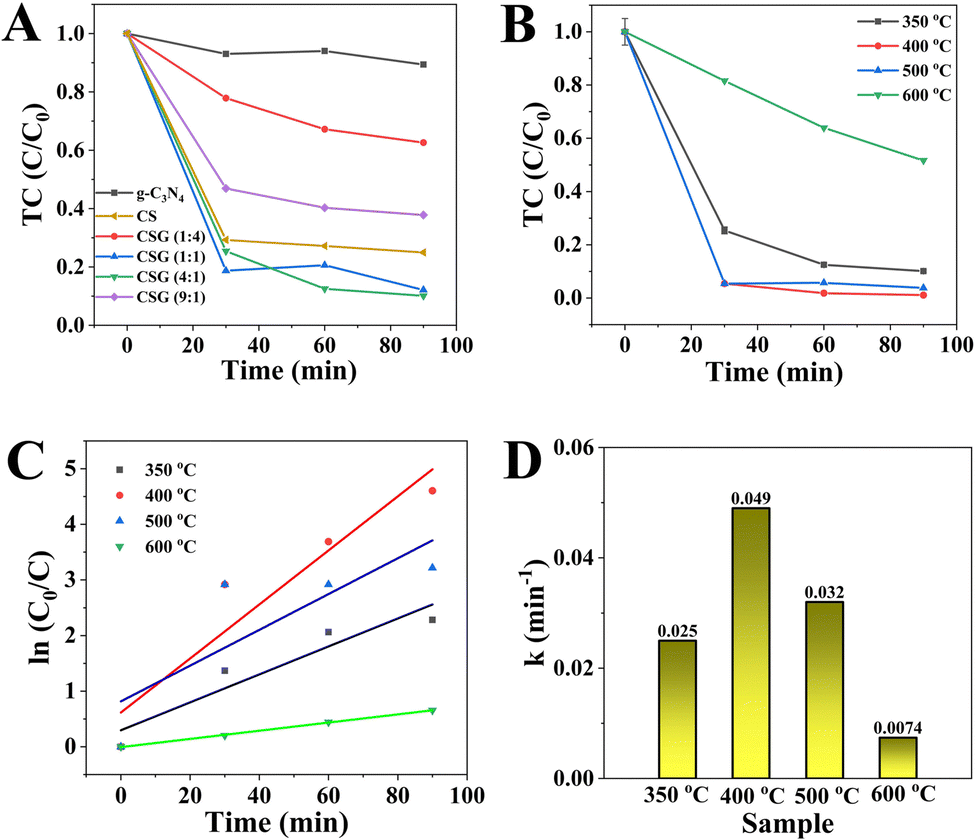 | ||
| Fig. 4 (A) Effect of the Cu(OAc)2 mass ratio on TC abatement over different catalysts. (B) Effect of calcination temperature on TC abatement over CSG (4:1) heterojunctions. (C) Pseudo first-order kinetic plots and (D) corresponding rate constants of TC abatement over CSG (4:1) heterojunctions at different calcination temperatures. | ||
UV-vis absorption spectra were utilized to characterize the optical absorption properties of g-C3N4, CS and CSG (4:1). As shown in Fig. 5A, g-C3N4 has a steep absorption edge at around 450 nm with an absorption band of less than 450 nm, which was ascribed to the reported optical band gap of ∼2.7 eV, suggesting a rather low photoconversion efficiency.26 With the formation of a heterojunction, the absorption intensity of CSG (4:1) increased, indicating enhanced visible light absorption. PL spectra and photoelectrochemical measurements were employed to study the separation and migration of photogenerated charges.8 The diagrams of (αhν)2vs. (hν) obtained using Tauc's method are shown in Fig. 5B. The Eg of pure g-C3N4 = 2.74 eV in accordance with the previous report.26 In addition, the Eg of CS was calculated to be 1.96 eV, while for CSG (4:1), the Eg was 1.93 eV. Within a certain range, the reduced Eg can facilitate the transfer of photoelectrons from the valence band to the conduction band.
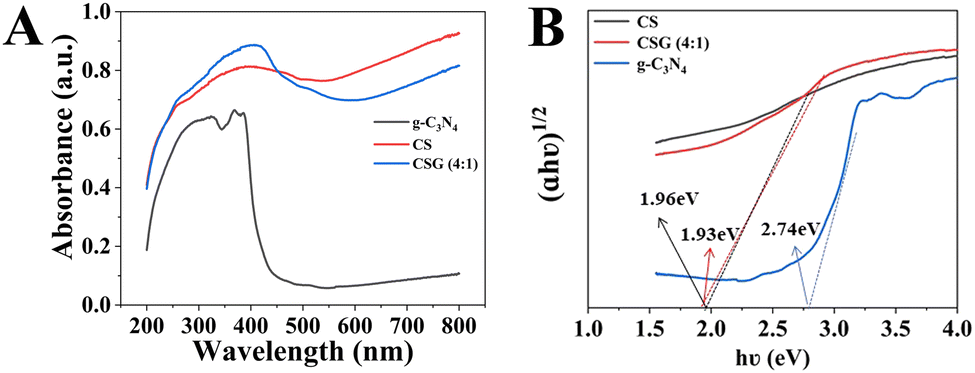 | ||
| Fig. 5 (A) UV-vis absorption spectra and (B) bandgap fitting by the Kubelka–Munk function of g-C3N4, CS and CSG (4:1). | ||
Compared with g-C3N4 and CS (Fig. 6A), the CSG heterojunction exhibited the highest photocurrent intensity, suggesting that the CSG heterojunction can enhance charge separation.39,40Fig. 6B shows representative Nyquist plots of the impedance response of the samples. As expected, the CSG heterojunction presented the lowest charge transport resistance, revealing that the formation of heterojunctions can boost the charge mobility and photogenic electron–hole separation efficiency.41–43 These results are consistent with the transient photocurrent performance. Besides, Fig. 6C depicts the PL spectra of g-C3N4, CS, CSG (4:1), Cu3(OH)4SO4/g-C3N4, Cu7S4/g-C3N4 and Cu3(OH)4SO4/Cu7S4, and g-C3N4 presented the highest PL emission intensity. However, the PL peak intensity of the CSG heterojunction was the lowest, demonstrating that the combination of g-C3N4 and CS can restrain the recombination of photogenerated electron–hole pairs.44 The time-resolved photoluminescence (TRPL) test was employed to characterize the fluorescence lifetime of the carrier.45 As shown in Fig. 6D, the average fluorescence lifetimes of pure g-C3N4, CS and CSG (4:1) were 5.62, 4.00 and 3.64 ns, respectively. Among the three, the shortest fluorescence lifetime of CSG (4:1) indicated the most rapid electron transfer rate through the interface.46,47
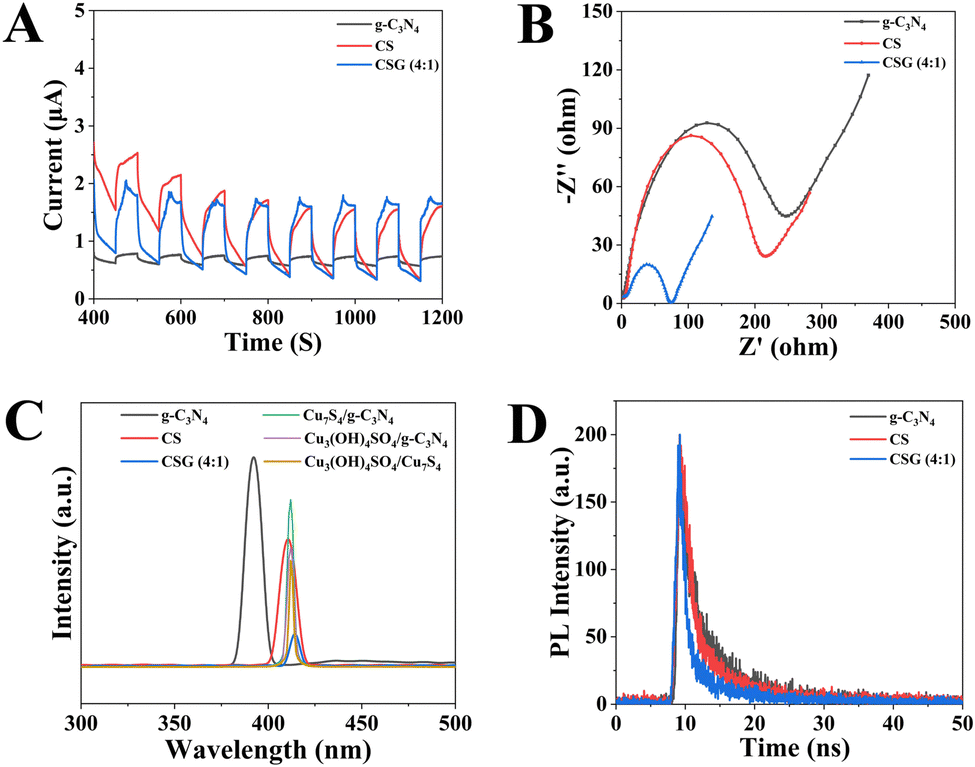 | ||
| Fig. 6 (A) Transient photocurrent performance, (B) electrochemical impedance spectra and (C) PL spectra of g-C3N4, CS, CSG (4:1), Cu3(OH)4SO4/g-C3N4, Cu7S4/g-C3N4 and Cu3(OH)4SO4/Cu7S4. (D) Time-resolved PL decays of g-C3N4, CS and CSG (4:1). | ||
In the next set of experiments, the effects of various parameters including the pH value, catalyst dosage, TC concentration and temperature in the removal process over CSG (4:1) heterojunctions were systematically investigated. Fig. 7A shows the influence of pH on the removal efficiency of TC. The highest TC removal efficiency was obtained at pH 6.0, and a profitable environment for TC abatement was achieved under weakly acidic, neutral and alkaline conditions. This result was consistent with that of the previous report, suggesting that the peracidity was bad for the degradation of TC molecules.48 Significantly, TC molecules can easily attract ˙OH radicals for self-oxidation due to their negative charges and high electric density in neutral alkaline conditions.49 The effect of CSG (4:1) dosage is illustrated in Fig. 7B. The removal efficiency of TC increased with the increase of the catalyst dosage from 0.5 to 1.0 mg mL−1, and the efficiency remained roughly the same as the CSG (4:1) dosage increased from 1.0 to 2.0 mg mL−1. Therefore, subsequent experiments in this study were performed with 1.0 mg mL−1 owing to the principle of saving dosage. As shown in Fig. 7C, it is apparent that the removal efficiency first improved as the TC concentration increased from 10 to 20 mg L−1. However, the removal efficiency decreased with a continuous increase of the TC concentration from 20 to 60 mg L−1. Therefore, the TC concentration of 20 mg L−1 was selected for further study. As shown in Fig. 7D, the temperature had almost no effect on TC removal efficiency after 90 min. Thus, 25 °C was chosen based on mild conditions. The optimal pH, catalyst dosage, TC concentration and temperature were 6.0, 1.0 mg mL−1, 20 mg L−1 and 25 °C, respectively.
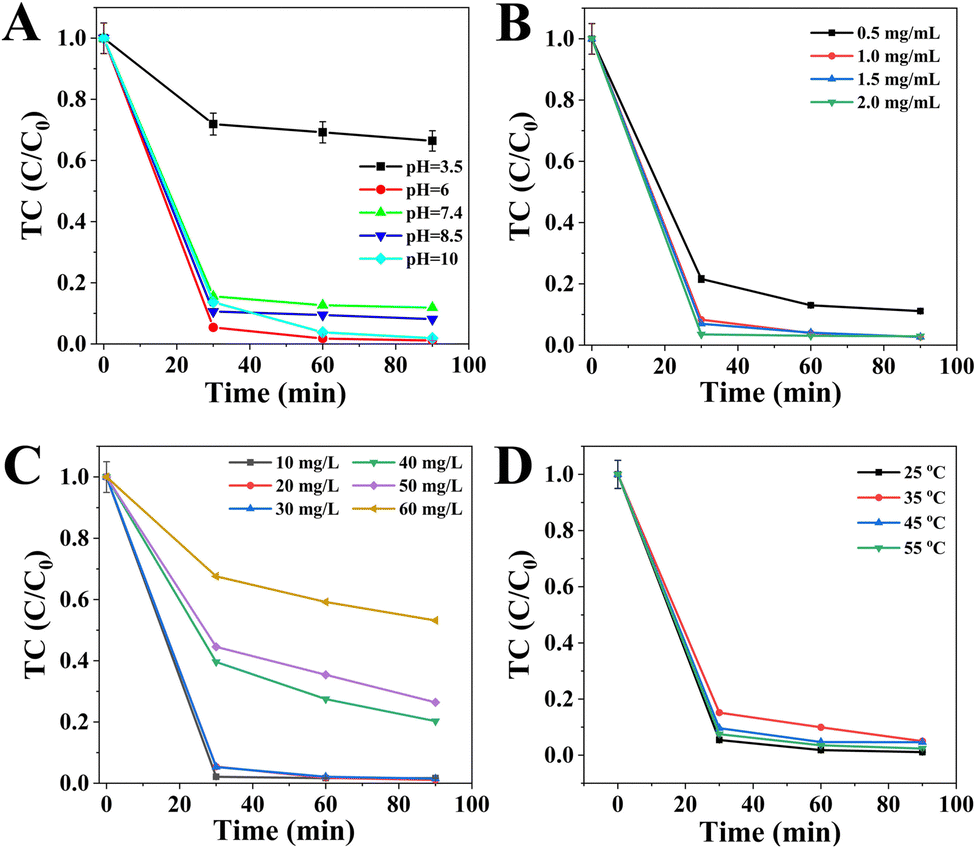 | ||
| Fig. 7 Effects of various parameters including pH (A), catalyst dosage (B), TC concentration (C) and temperature (D) on the removal efficiency of TC over CSG (4:1) heterojunctions. | ||
Then, we evaluated the photoactivity of the samples under optimal conditions. As shown in Fig. 8, the removal efficiencies of TC over Cu3(OH)4SO4, Cu7S4, Cu3(OH)4SO4/g-C3N4, Cu7S4/g-C3N4 and Cu3(OH)4SO4/Cu7S4 (1:1) after 90 min were 68.0%, 62.4%, 67.9%, 32.8% and 70.5%, respectively. It follows that whether it is a single material or a pairwise composite material, the performance was better than that of pure g-C3N4. However, as expected, all data showed that the synthesized CSG (4:1) heterojunction still performed the best (96.3%). The preparation processes of the obtained Cu7S4/Cu3(OH)4SO4, Cu3(OH)4SO4/g-C3N4, and Cu7S4/g-C3N4 were different from that of our CSG (4:1). As a result, the number of components in a material cannot be regulated in the same way. There was no synergistic effect in Cu7S4/Cu3(OH)4SO4, Cu3(OH)4SO4/g-C3N4, and Cu7S4/g-C3N4, possibly because excess g-C3N4 covered the material to reflect the synergistic effect, thus reducing the catalytic efficiency. The enhancement of the removal efficiency of the CSG (4:1) heterojunction may be due to the synergistic effect of Cu3(OH)4SO4, Cu7S4, and g-C3N4, which can produce effective heterojunctions and reduce the recombination efficiency of electron–hole pairs.50,51 However, the synergistic effect of Cu7S4/Cu3(OH)4SO4/g-C3N4 needs to be further explored. A synergy coefficient may be used for quantifying the synergistic effect of Cu7S4/Cu3(OH)4SO4/g-C3N4 on the reaction process.52 Also, density functional theory calculations may be utilized to understand the enhanced TC removal activity.53 Under the optimized conditions, the relative degradation activity of the CSG (4:1) heterojunction maintained 91.1% after 8 cycles and 68.2% after 10 cycles (Fig. S5, ESI†), indicating that the CSG (4:1) heterojunction was a durable and recyclable catalyst for TC removal. As shown in Fig. S6 (ESI†), the SEM and TEM images of CSG (4:1) after the reaction still showed porous-layered structures as before. In addition, we investigated the surface state of CSG (4:1) after the reaction by XPS. As shown in Fig. S7 (ESI†), compared to that before the reaction (Fig. 3), there was no obvious difference in the XPS spectra in the C 1s, N 1s, Cu 2p and S 2p regions. This indicated that CSG (4:1) after the reaction still maintained its composition and the chemical state of all elements.
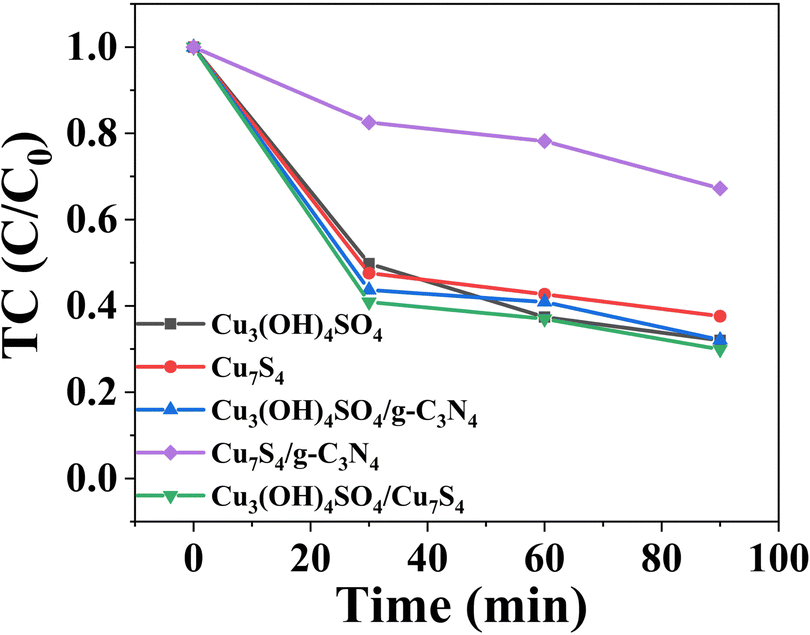 | ||
| Fig. 8 Removal efficiencies of TC over Cu3(OH)4SO4, Cu7S4, Cu3(OH)4SO4/g-C3N4, Cu7S4/g-C3N4 and Cu3(OH)4SO4/Cu7S4 (1:1). | ||
Free-radical quenching experiments were carried out by using specific free radical scavengers. As shown in Fig. 9A, the removal efficiency of TC is 98.6% without quenchers. The addition of tert-butanol (TBA) and ethylene diamine tetraacetic acid (EDTA) to the CSG (4:1) heterojunctions/TC/air system, except for p-benzoquinone (BQ), demonstrated that the main active species were ˙OH and vacancies in the removal process of TC. Additionally, electron spin resonance spectroscopy (ESR) was used to confirm the existence of reactive oxygen species (ROSs) in the removal process (Fig. 9).54,55 As expected, the ˙OH was found using a DMPO trapping agent with four characteristic peaks of DMPO-˙OH with a relative intensity of 1:2:2:1 (Fig. 9B). Meanwhile, 1O2 was monitored using a TEMP trapping agent with TEMP-1O2 signals of 1:1:1 triplet signal intensity (Fig. 9C). Besides, all signals intensities were basically same in the dark and light, which suggested that the CSG (4:1) heterojunctions generated reactive species ˙OH and 1O2 without an additional light source. As shown in Fig. 9A, the removal efficiency of TC decreased to 56.1% after the addition of L-histidine (L-His). When BQ was added, the removal efficiency of TC also decreased, suggesting the presence of ˙O2− during 1O2 generation. In addition, we have performed the elimination of TC under different atmospheres. Fig. S8 (ESI†) shows that the N2 atmosphere led to a reduction in the removal efficiency of TC, which demonstrated that 1O2 was derived from dissolved oxygen.56 Therefore, some ˙O2− could react with water to generate 1O2.22,57 As shown in Fig. 8D, the ESR spectrum of the CSG (4:1) heterojunctions displays a nearly isotropic signal at g of 2.005, which is ascribed to the free electrons trapped by the SVs in CSG (4:1) heterojunctions.58 Significantly, the SVs can serve as the active sites for oxygen atoms to interact with the catalyst surface, which is beneficial to lower adsorption energy and promote the generation of ROSs.
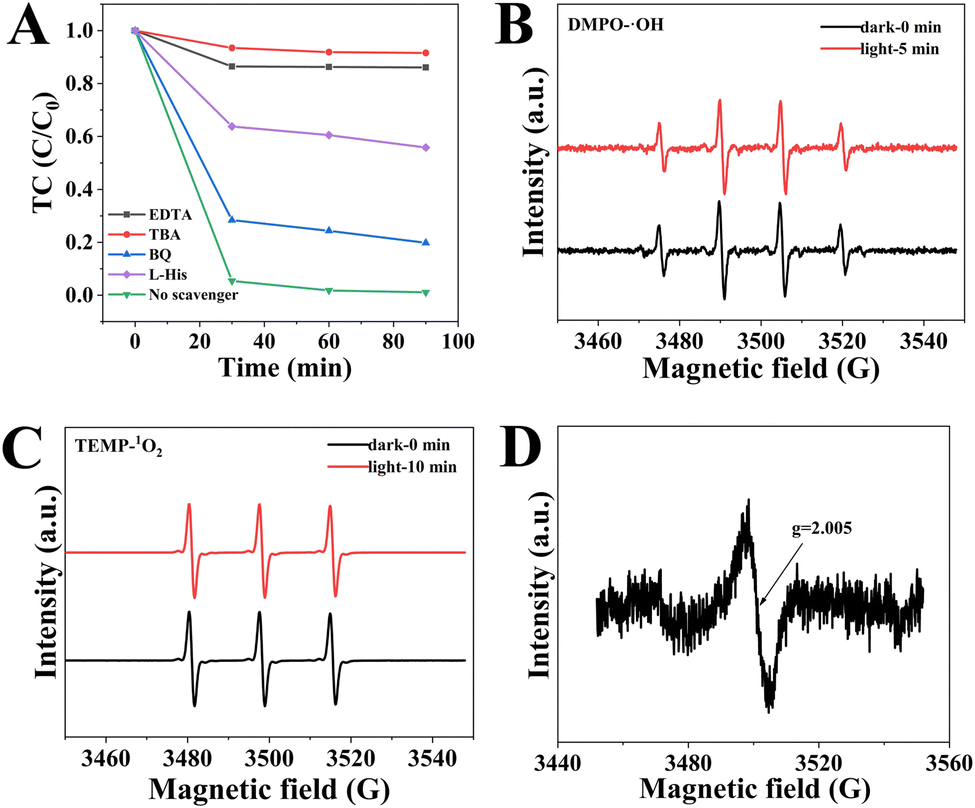 | ||
| Fig. 9 (A) Effect of scavenges on the removal activity of TC. ESR spectra of (B) DMPO-˙OH and (C) TEMP-1O2. (D) EPR spectra of the CSG (4:1) heterojunctions. | ||
Based on the above analysis, we proposed a possible mechanism for TC removal on a CSG (4:1) heterojunction. The ˙O2− intermediates were generated from O2 adsorbed to SVs by delocalized electrons and then transferred to 1O2.58,59 With respect to the active species of Cu+ in the CSG (4:1) heterojunction, it can catalyze H2O2 to ˙OH in the system.19,60Eqn (1)–(4) show the corresponding illustration.
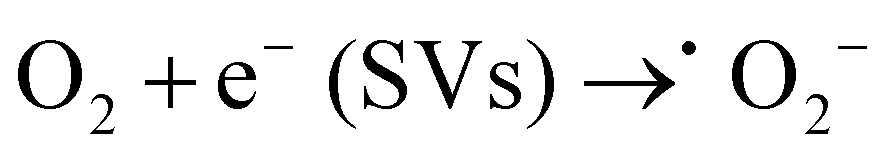 | (1) |
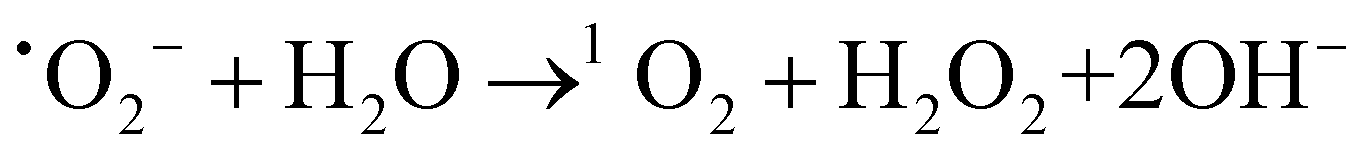 | (2) |
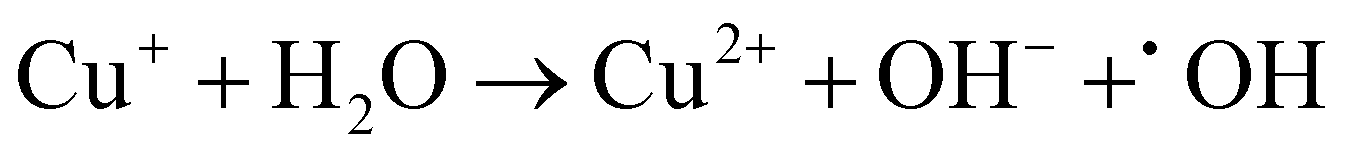 | (3) |
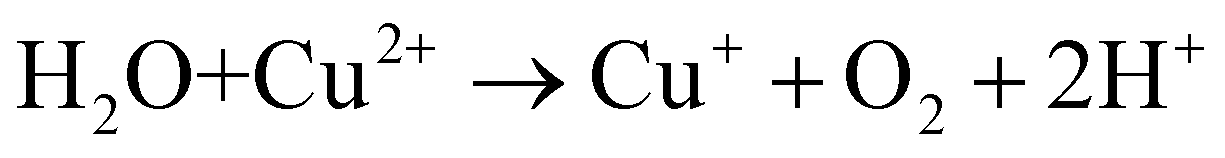 | (4) |
The mineralization efficiency of TC over g-C3N4 and CSG (4:1) was explored and the TOC values were measured to be 5.58% and 9.21%, respectively, which were lower than the literature data.61 The main reasons might be the following: (1) the main active species were ˙OH and 1O2 in this work, in which 1O2 is reported not to be a competent ROS for organic pollutant control.44 (2) TC would produce intermediates other than CO2 and H2O by breaking different bonds, and it is reported that high TC removal rates were normally accompanied by poor mineralization efficiencies.61,62 (3) The short reaction time made it inefficient for TC to be mineralized. To identify the intermediates produced after TC degradation, LC-MS and ESI-MS tests were performed, as shown in Fig. S9 (ESI†). Fig. S9A and B (ESI†) show the LC-MS analysis of the TC solution before and after degradation. It can be seen that the TC in the solution became very low after 90 min of degradation. The three intermediates with m/z values of 301.1403, 407.1340 and 423.1658 (Fig. S9C, ESI†) might correspond to the dehydroxylation and dehydration products of TC molecules, displaying a low mineralization efficiency.63 The degradation ability of CSG (4:1) on TC in different water environments (deionized water, tap water, and rainwater) was investigated using different water qualities. Fig. S10 (ESI†) shows that there was a 10% difference in the degradation efficiency between tap water and ultrapure water, and it may be due to some interfering substances in tap water. Thus, the degradation level in the actual water samples was maintained at a very high level, which indicated that the material was basically adapted to all aqueous environments.
Conclusions
In summary, we successfully synthesized a new type of CSG heterojunction with SVs for efficient removal of TC. The obtained CSG composites were characterized by SEM, TEM, XRD, BET and XPS. Such heterostructured structures of the CSG (4:1) composite can activate oxygen to the 1O2 radical, leading to considerable enhancement in TC removal under visible light irradiation. Experimental studies by radical trapping and EPR revealed that the main ROS are 1O2 and˙OH, and the presence of SVs was confirmed. Besides, CSG (4:1) enabled the activation of oxygen-produced ROS for removing TC even in the dark. 1O2 was generated by direct oxidation or conversion of oxygen by electrons provided by SVs in solution, while ˙OH was generated by the intermediate H2O2 produced in the experiment activated by Cu+ ions. Thus far, the proposed CSG composites can remove TC under mild conditions without additional chemical oxidants or energy inputs. Furthermore, the method is widely used, pH-adaptable, highly reusable and environmentally friendly.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the project fund of the National Natural Science Foundation of China (21575064) and the Natural Science Foundation of the Jiangsu Higher Education Institutions of China (22KJB150025).Notes and references
- D. Wang, F. Jia, H. Wang, F. Chen, Y. Fang, W. Dong, G. Zeng, X. Li, Q. Yang and X. Yuan, J. Colloid Interface Sci., 2018, 519, 273–284 CrossRef CAS.
- J. Jin, Z. Yang, W. Xiong, Z. Zhou, R. Xu, Y. Zhang, J. Cao, X. Li and C. Zhou, Sci. Total Environ., 2019, 650, 408–418 CrossRef CAS.
- W. Zhang, L. Wang, Y. Su, Z. Liu and C. Du, Appl. Surf. Sci., 2021, 566, 150708 CrossRef CAS.
- N. Kemper, Ecol. Indic., 2008, 8, 1–13 CrossRef CAS.
- Y. Valcarcel, S. Gonzalez Alonso, J. L. Rodriguez-Gil, A. Gil and M. Catala, Chemosphere, 2011, 84, 1336–1348 CrossRef CAS.
- N. Qi, P. Wang, C. Wang and Y. Ao, J. Hazard. Mater., 2017, 341, 187–197 CrossRef CAS.
- H. Li, J. Hu, Y. Meng, J. Su and X. Wang, Sci. Total Environ., 2017, 603–604, 39–48 CrossRef CAS.
- Y. Shi, C. Zhang, Z. Yang, X. Liu, X. Zhang, C. Ling, J. Cheng, C. Liang, C. Mao and L. Zhang, J. Phys. Chem. C, 2022, 126, 21847–21856 CrossRef CAS.
- R. Nazari, L. Rajic, A. Ciblak, S. Hernández, I. E. Mousa, W. Zhou, D. Bhattacharyya and A. N. Alshawabkeh, Chemosphere, 2019, 216, 556–563 CrossRef CAS.
- J. Dewulf, H. Van Langenhove, A. De Visscher and S. Sabbe, Ultrason. Sonochem., 2001, 8, 143–150 CrossRef CAS PubMed.
- X. Zhang, K. Yue, R. Rao, J. Chen, Q. Liu, Y. Yang, F. Bi, Y. Wang, J. Xu and N. Liu, Appl. Catal., B, 2022, 310, 121300 CrossRef CAS.
- C.-L. Lee, H.-Y. Lee, K.-H. Tseng, P. K. Andy Hong and C.-J. G. Jou, Environ. Chem. Lett., 2011, 9, 355–359 CrossRef CAS.
- R. Xie, J. Cao, X. Xie, D. Lei, K. Guo, H. Liu, Y. Zeng and H. Huang, Chem. Eng. J., 2020, 401, 126077 CrossRef CAS.
- H. Shen, X. Zhan, S. Hong, L. Xu, C. Yang, A. W. Robertson, L. Hao, F. Fu and Z. Sun, Nano Res., 2023, 16, 10713–10723 CrossRef CAS.
- H. Shen, C. Yang, W. Xue, L. Hao, D. Wang, F. Fu and Z. Sun, Chem. – Eur. J., 2023, 29, e202300748 CrossRef CAS.
- Y. Zhang, X. Ren, L. Yang and Z. Chen, J. Mater. Sci., 2021, 56, 17556–17567 CrossRef CAS.
- L. Li, C. Hu, L. Zhang and B. Shi, J. Hazard. Mater., 2021, 406, 124739 CrossRef CAS PubMed.
- D. Cheng, A. Neumann, S. Yuan, W. Liao and A. Qian, Environ. Sci. Technol., 2020, 54, 4091–4101 CrossRef CAS PubMed.
- Q. Dong, Y. Chen, L. Wang, S. Ai and H. Ding, Appl. Surf. Sci., 2017, 426, 1133–1140 CrossRef CAS.
- C. Chen, Y. Zhou, N. Wang, L. Cheng and H. Ding, RSC Adv., 2015, 5, 95523–95531 RSC.
- L. Liu, Y. Qi, J. Hu, Y. Liang and W. Cui, Appl. Surf. Sci., 2015, 351, 1146–1154 CrossRef CAS.
- A. Du, H. Fu, P. Wang, C. Zhao and C.-C. Wang, J. Hazard. Mater., 2022, 426, 128134 CrossRef CAS PubMed.
- M. P. Ravele, O. A. Oyewo, S. Ramaila, L. Mavuru and D. C. Onwudiwe, Catalysts, 2021, 11, 1238 CrossRef CAS.
- G. Wang, B. Huang, Z. Li, Z. Wang, X. Qin, X. Zhang, Y. Dai and M.-H. Whangbo, Chem. – Eur. J., 2015, 21, 13583–13587 CrossRef CAS.
- X. Liu, A. Jin, Y. Jia, T. Xia, C. Deng, M. Zhu, C. Chen and X. Chen, Appl. Surf. Sci., 2017, 405, 359–371 CrossRef CAS.
- Z. Cai, Y. Zhou, S. Ma, S. Li, H. Yang, S. Zhao, X. Zhong and W. Wu, J. Photochem. Photobiol., A, 2017, 348, 168–178 CrossRef CAS.
- Y. Cui, Z. Ding, P. Liu, M. Antonietti, X. Fu and X. Wang, Phys. Chem. Chem. Phys., 2012, 14, 1455–1462 RSC.
- Y. Tian, Q. Li, M. Zhang, Y. Nie, X. Tian, C. Yang and Y. Li, J. Cleaner Prod., 2021, 315, 128207 CrossRef CAS.
- X. Li, Y. Guo, L. Yan, T. Yan, W. Song, R. Feng and Y. Zhao, Chem. Eng. J., 2022, 429, 132234 CrossRef CAS.
- X. Cao, Q. Lu, X. Xu, J. Yan and H. Zeng, Mater. Lett., 2008, 62, 2567–2570 CrossRef CAS.
- N. Koga, A. Mako, T. Kimizu and Y. Tanaka, Thermochim. Acta, 2018, 467, 11–19 CrossRef.
- M. Arumugam, M. Tahir and P. Praserthdam, Chemosphere, 2022, 286, 131765 CrossRef CAS PubMed.
- M. T. Uddin, Y. Nicolas, C. Olivier, T. Toupance, L. Servant, M. M. Müller, H.-J. Kleebe, J. Ziegler and W. Jaegermann, Inorg. Chem., 2012, 51, 7764–7773 CrossRef CAS PubMed.
- P. Gao, X. Chen, M. Hao, F. Xiao and S. Yang, Chemosphere, 2019, 228, 521–527 CrossRef CAS PubMed.
- C. Han, T. Zhang, J. Li, B. Li and Z. Lin, Nano Energy, 2020, 77, 105165 CrossRef CAS.
- C. K. Wu, M. Yin, S. O’ Brien and T. Koberstein, Chem. Mater., 2006, 18, 6054–6058 CrossRef CAS.
- T. D. Trang, E. Kwon, J.-C. Wen, N. N. Huy, V. S. Munagapati, S. Ghotekar, K.-P. Yu and K.-Y. A. Lin, J. Mol. Liq., 2023, 389, 122832 CrossRef CAS.
- F. Guo, W. Shi, H. Wang, H. Han, H. Li, H. Huang, Y. Liu and Z. Kang, Catal. Sci. Technol., 2017, 7, 3325–3331 RSC.
- C. Zhao, X. Li, L. Yue, S. Yuan, X. Ren, Z. Zeng, X. Hu, Y. Wu and Y. He, J. Alloys Compd., 2023, 968, 171956 CrossRef CAS.
- Z. Feng, L. Zeng, Q. Zhang, S. Ge, X. Zhao, H. Lin and Y. He, J. Environ. Sci., 2020, 87, 149–162 CrossRef CAS.
- K. Wang, Z. Guan, X. Liang, S. Song, P. Lu, C. Zhao, L. Yue, Z. Zeng, Y. Wu and Y. He, Ultrason. Sonochem., 2023, 100, 106616 CrossRef CAS.
- C. Zhao, X. Li, L. Yue, X. Ren, S. Yuan, Z. Zeng, X. Hu, Y. Wu and Y. He, ACS Appl. Nano Mater., 2023, 6, 15709–15720 CrossRef CAS.
- Y. Shi, X. Wang, X. Liu, C. Ling, W. Shen and L. Zhang, Appl. Catal., B, 2020, 277, 119229 CrossRef CAS.
- Y. Shi, Z. Yang, L. Shi, H. Li, X. Liu, X. Zhang, J. Cheng, C. Liang, S. Cao, F. Guo, X. Liu, Z. Ai and L. Zhang, Environ. Sci. Technol., 2022, 56, 14478–14486 CrossRef CAS PubMed.
- R. Du, C. Wang, L. Guo, R. A. Soomro, B. Xu, C. Yang, F. Fu and D. Wang, Small, 2023, 19, 2302330 CrossRef CAS PubMed.
- Y.-L. Li, Q. Zhao, X.-J. Liu, Y. Liu, Y.-J. Hao, X.-J. Wang, X.-Y. Liu, D. Hildebrandt, F.-Y. Li and F.-T. Li, Small Struct., 2023, 2300177 CrossRef.
- H. Bhatt, T. Goswami, D. K. Yadav, N. Ghorai, A. Shukla, G. Kaur, A. Kaur and H. N. Ghosh, J. Phys. Chem. Lett., 2021, 12, 11865 CrossRef CAS PubMed.
- L. Wang, W. Zhang, Y. Su, Z. Liu and C. Du, Appl. Clay Sci., 2021, 213, 106238 CrossRef CAS.
- H. Dong, X. Guo, C. Yang and Z. Ouyang, Appl. Catal., B, 2018, 230, 65–76 CrossRef CAS.
- D. Masih, Y. Ma and S. Rohani, Appl. Catal., B, 2017, 206, 556–588 CrossRef CAS.
- F. Dai, Y. Wang, R. Zhao, X. Zhou, J. Han and L. Wang, Int. J. Hydrogen Energy, 2020, 45, 28783–28791 CrossRef CAS.
- S. Jing, X. Yan, Y. Xiong, T. Li, J. Xiong, T. Hu, Z. Wang, L. Lou and X. Ge, J. Energy Chem., 2023, 84, 34–40 CrossRef CAS.
- Z. Jiang, W. Wan, H. Li, S. Yuan, H. Zhao and P. K. Wong, Adv. Mater., 2018, 30, 1706108 CrossRef.
- T. Ma, C. Yang, L. Guo, R. A. Soomro, D. Wang, B. Xu and F. Fu, Appl. Catal., B, 2023, 330, 122643 CrossRef CAS.
- C. Yang, Y. Zhang, F. Yue, R. Du, T. Ma, Y. Bian, R. Li, L. Guo, D. Wang and F. Fu, Appl. Catal., B, 2023, 338, 123057 CrossRef CAS.
- C. Zhao, L. Meng, H. Chu, J.-F. Wang, T. Wang, Y. Ma and C.-C. Wang, Appl. Catal., B, 2023, 321, 122034 CrossRef CAS.
- H. Zeng, L. Deng, H. Zhang, C. Zhou and Z. Shi, J. Hazard. Mater., 2020, 400, 123297 CrossRef CAS.
- L. Wu, P. Guo, X. Wang, H. Li, X. Zhang, K. Chen and P. Zhou, Chem. Eng. J., 2022, 446, 136759 CrossRef CAS.
- S. Wang, S. Gao, J. Tian, Q. Wang, T. Wang, X. Hao and F. Cui, J. Hazard. Mater., 2020, 387, 121995 CrossRef CAS PubMed.
- J. Long, L. Xu, L. Zhao, H. Chu, Y. Mao and D. Wu, Chemosphere, 2020, 241, 125034 CrossRef CAS PubMed.
- Y. Ma, H. Xiong, Z. Zhao, Y. Yu, D. Zhou and S. Dong, Chem. Eng. J., 2018, 351, 967–975 CrossRef CAS.
- S. Dong, S. Dong, X. Tian, Z. Xu, D. Ma, B. Cui, N. Ren and B. E. Rittmann, J. Hazard. Mater., 2016, 302, 386–394 CrossRef CAS.
- T. Jiang, Z. Peng, M. Xie, X. Fang, Y. Hong, Z. Huang, W. Gao, Z. Zhou, L. Li and Z. Zhu, Anal. Methods, 2020, 12, 535–543 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nj04276f |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |