
Quest for ultralong C–C bonds in [1.1.1]propellane derivatives: a theoretical study†
Nargish
Sultana
a,
Amlan J.
Kalita
 ab and
Ankur K.
Guha
*a
ab and
Ankur K.
Guha
*a
aAdvanced Computational Chemistry Centre, Cotton University, Panbazar, Guwahati, Assam 781001, India. E-mail: ankurkantiguha@gmail.com
bDepartment of Chemistry, University of Science and Technology, Meghalaya 793101, India
First published on 29th November 2023
Abstract
Constructing hydrocarbons with a long C–C bond has attracted chemists for a long time. Herein, we envision the formation of an ultra-long C–C bond in [1.1.1]propellane derivatives through quantum chemical calculations. Fluorination of the wing –CH2 group elongates the C–C distance by more than 0.1 Å compared to the parent molecule. Similarly, SiR2 (R = H, F) substituted derivatives have much longer C–C distances of greater than 1.9 Å. The longest C–C distance reported here is 2.234 Å in the beryllium substituted derivative. Interestingly, most of the molecules have intact C–C interaction (except BH substituted one) as confirmed by detailed electronic structure analysis. Due to the toxicity related issue for Be, SiR2 substituted [1.1.1]propellane derivatives may become viable and are expected to show an ultra-long C–C distance of greater than 1.9 Å.
One of the central themes of chemistry is to envision new molecules with predesigned properties.1 In addition to potential applications, they create new concepts in chemical bonding and reactivity. One such long-standing challenge for chemists has been the synthesis and isolation of molecules with long C–C single bonds.2–4 Suzuki et al. reported an ultralong C–C bond of 1.806(2) Å in highly strained dihydropyracylene and explained the stability on the basis of dispersion forces between the aromatic rings.5 Substituted ortho-carboranes and their derivatives have also been investigated extensively in this context. Oliva and co-workers reported the C–C bond 1,2-(SH)2-1,2-ortho-carborane as 1.803 Å based on calculations.6 Elongated C–C bonds in derivatives of closo-[3-Co(η5-NC4H4)-1,2-C2B9H11] have also been reported by Llop et al.7 They also determined the crystal structure of closo-1,2-(SPh)2-1,2-C2B10H10 and reported a long C–C bond of 1.798(3) Å.8 Oliva et al. have demonstrated that C–C bonds can be elongated to as much as 2.638 Å by tuning the donor ability of the substituents.9 Xie and co-workers reported the X-ray structure for 1,2-(NHMes)2-o-carborane (Mes = 2,4,5-trimethylphenyl) with a long C–C distance of 1.990(4) Å.10 Several derivatives of 1,2-(NHR)2-o-carborane have been reported with long C–C bonds.11,12 Dutta and co-workers demonstrated that the C–C distance in diamino-o-carborane can be elongated to as large as 2.001, 2.011 and 1.807 Å by tuning the N(lone pair) → σ*(C–C) donor–acceptor “negative hyperconjugation” interaction.12
In the quest for long C–C bonds, [1.1.1]propellane needs attention (I, Scheme 1). The central bond (C1–C2) in [1.1.1]propellane has attracted chemists since its prediction and realization after a decade.13,14 The unusually long C–C bond length of 1.59 (±0.01) Å determined from the X-ray structure remained unexplained for a long time.15–22 Indeed, calculations reveal that the central C–C bond has non-bonding or slightly antibonding character, which results in such elongation.13,23–26 Shaik and co-workers investigated the bonding within the valence bond framework, which made them ascertain this bond as “charge-shift” (CS).22,27–30 Moreover, an experimental charge density study has also revealed no charge accumulation at the middle of the bond.31 The inherently long central C–C bond has also attracted chemists to look for ways to shorten the length. Jemmis and co-workers showed that halogen bonding is an effective way to shorten the central bond.32 Substituting the wing –CH2 group by electron donors such as NH, O and S has also been shown to shorten and even re-establish the lost covalency of the central bond.33 This implies that most of the studies on [1.1.1]propellane have been focussed on shortening the central C–C bond. As the central C–C bond in the parent [1.1.1]propellane is already elongated,1,2 we envisioned that further elongation is possible with an appropriately chosen substituent. Before that the electronic structure of the parent structure is worthy of investigation to get some hint towards the nature of substituents to be utilized for this purpose. Natural bond orbital (NBO)34 reveals extensive hyperconjugation between the adjacent σ(Ci–Cj) bonds to σ*(C1–C2) bonds (Ci–Cj are adjacent C–C bonds to the central C1–C2 bonds where Ci–Cj ≠ C1–C2) and σ(C1–C2) bonds to adjacent σ*(Ci–Cj) bonds. The stabilization energy arising from the interaction σ(C1–C2) → σ*(Ci–Cj) amounts to 81 kcal mol−1 while σ(Ci–Cj) → σ*(C1–C2) amounts to 93.6 kcal mol−1. Both of these second order interactions result in the removal of electron density from the C1–C2 bonding orbital while accumulation in the C1–C2 antibonding orbital and thereby lengthening the C1–C2 bond. This implies that substituting the wing –CH2 by more electron withdrawing groups will draw more electron density from the C1–C2 bond, thereby making the central bond elongated.
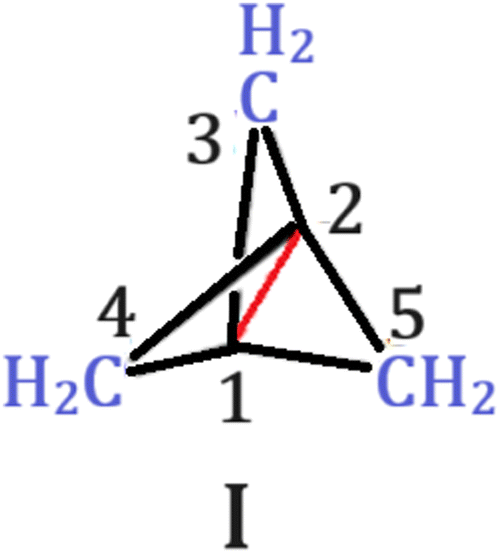 | ||
| Scheme 1 Structure of [1.1.1]propellane. The central C–C bond is shown in the red line. | ||
With that in mind, we tried to investigate the effect of different substitution patterns on the central C–C bond length (Scheme 2).
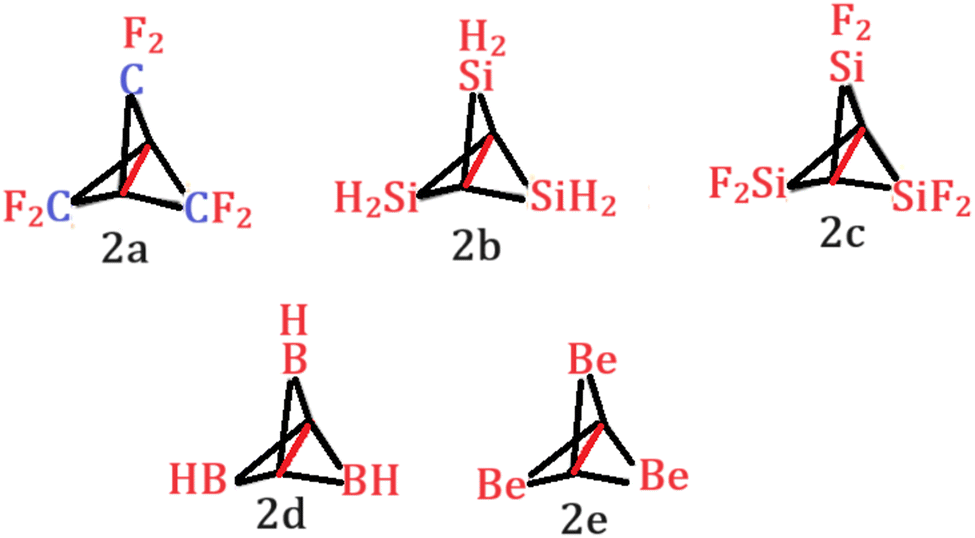 | ||
| Scheme 2 [1.1.1]Propellane derivatives considered in this study. | ||
All these molecules were fully optimized at the B2PLYP/6-311++G** and MP2(full)/6-311++G** levels.‡![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 35,36 Both these levels produced excellent agreement with the X-ray data for the parent [1.1.1]propellane. B2PLYP produces 1.589 Å, while MP2 produces 1.602 Å. Unless otherwise noted, all results will be discussed based on B2PLYP results. All these structures were characterized to be local minima on the potential energy surface. Bonding analyses were performed using the natural bonding orbital (NBO) scheme.34 All these calculations were performed using the Gaussian16 suite of programs.37 The singlet state of these molecules is the ground state and the diradical triplet states are higher in energy with a significant singlet–triplet gap (ΔEST, Table S1, ESI†), indicating that diradical character may be absent in these molecules. However, the ΔEST values may not adequately describe the biradical character. We therefore investigated the biradical character using complete-active space self-consistent field (CASSCF) theory using two electrons in two orbitals, i.e., CASSCF(2,2). Electron density analyses38 were also performed using the Multiwfn program code.39
35,36 Both these levels produced excellent agreement with the X-ray data for the parent [1.1.1]propellane. B2PLYP produces 1.589 Å, while MP2 produces 1.602 Å. Unless otherwise noted, all results will be discussed based on B2PLYP results. All these structures were characterized to be local minima on the potential energy surface. Bonding analyses were performed using the natural bonding orbital (NBO) scheme.34 All these calculations were performed using the Gaussian16 suite of programs.37 The singlet state of these molecules is the ground state and the diradical triplet states are higher in energy with a significant singlet–triplet gap (ΔEST, Table S1, ESI†), indicating that diradical character may be absent in these molecules. However, the ΔEST values may not adequately describe the biradical character. We therefore investigated the biradical character using complete-active space self-consistent field (CASSCF) theory using two electrons in two orbitals, i.e., CASSCF(2,2). Electron density analyses38 were also performed using the Multiwfn program code.39
Central C–C bond lengths and second-order donor–acceptor interactions are summarized in Table 1. It is evident from Table 1 that the central C–C bond has elongated to 1.706 Å in 2a when H atoms of the wing –CH2 groups are substituted with F atoms. Substitution by SiH2 results in significant elongation of the C–C bond to 1.975 Å in 2b. SiF2 substitution results in more elongation (2.083) in 2c. Electron deficient substituents like BH (2d) and Be (2e) also result in significant elongation. The longest C–C bond reported here is for Be substituted propellane 2e. Natural bond orbital analyses indicate significant second order interaction such as donation from the σ bonding orbital of C–C to the neighboring σ* antibonding orbital. These secondary interactions make the central C–C bond elongated. It is to be noted that for BH substituted propellane, NBO calculations were not performed as the central C–C bond is broken (discussed below). Similarly, Be substituted propellane is also not considered for NBO calculation as the 2p orbital of Be is not considered as valence in NBO calculation. Rather it is considered as the Rydberg state. It should be noted that the hyperconjugative effect alone may not dictate the lengthening of the C–C bond. Atomic sizes of the substituents also have a role to play. For example, although the amount of hyperconjugation in 2b is lower compared to 2a, still 2b has a longer C–C bond (Table 1). This is due to the bigger size of Si compared to C at the wing position. Similar effects are also seen for BH and Be substituted propellanes.
| Molecules | d C–C | d C–E | ∠E–C–E | E 2 σ(C–C) → σ*(C–E) | E 2 σ(C–E) → σ*(C–C) |
|---|---|---|---|---|---|
| a Central C–C bond is broken. b NBO is not performed as NBO does not consider the 2p orbital of Be as valence; rather it considers it to be the Rydberg state. | |||||
| I | 1.589 | 1.521 | 95.1 | 81.0 | 93.6 |
| 2a | 1.706 | 1.531 | 91.9 | 86.7 | 83.5 |
| 2b | 1.975 | 1.867 | 94.6 | 30.4 | 54.8 |
| 2c | 2.083 | 1.851 | 91.4 | 42.0 | 74.3 |
| 2d | 2.011 | 1.545 | 82.2 | ||
| 2e | 2.234 | 1.640 | 78.7 | ||
The elongation of the central C–C bond may also be gauged from other geometrical parameters such as C–E bond length and the angle around the central carbon atom, ∠E–C–E. All these are provided in Table 1. In general, lengthening of the C–E distance and decrease in the ∠E–C–E angle will lead to lengthening of the central C–C bond. Propellane I and 2a have similar C–E bonds as the substituents have similar linkages (CH2 in I and CF2 in 2a). The C–E bond in 2a is longer than that in I which makes the central C–C bond longer in 2a. Similarly, the angle around the central carbon atom, i.e., ∠E–C–E is more acute (less obtuse) in 2a compared to I. This geometrical constraint around the central C–C bond makes it more elongated. However, the case for SiR2 substituted propellane cannot be straightforwardly measured from these geometrical constraints. For example, geometrical constraints should result in more elongated bonds in 2b compared to 2c. However, the reverse is found. The actual trend in C–C bond elongation may be ascertained from both geometrical constraints as well as hyperconjugative effects. In 2c, despite the shorter C–E bond, the higher amount of hyperconjugation may lead to a longer C–C bond than that in 2b. 2d and 2e have different C–E linkages; however, their geometrical strain can be indicated by the ∠E–C–E angle. With a decrease in ∠E–C–E angle, the central C–C bond elongates.
Is the C–C bond really intact in these studied compounds? As most of the studied compounds have very elongated C–C bonds, we wanted to investigate whether the bonds are intact or not. We, therefore, performed electron density analysis38 of the studied compounds. Fig. 1 shows the molecular graph of these compounds. The presence of the bond path and its associated bond critical point (BCP) is a clear indication of bonding interaction.38 In all of the studied molecules, the formation of the bond critical path as well as (3, −1) BCP for the central C–C bond is observed except for 2d. In addition, three ring critical points (yellow dot) were also observed in 2a, 2b, 2c and 2e, indicating the formation of three rings which also hints at the presence of central C–C interaction. This indicates that the central C–C bond is intact in all these molecules whereas it has broken in 2d. Only a cage critical point (green dot) is observed in 2d. Surprisingly, 2e has a longer C–C bond length of 2.234 Å, but still it is bonded. This is one of the longest C–C bonds reported in the literature. Why is there no C–C interaction in 2d with a smaller C–C distance than that in 2e? Is there a biradical character involved in 2d? We therefore calculated the biradical character of 2d using CASSCF(2,2) calculation on the B2PLYP optimized geometry. We have explicitly considered C–C σ and C–C σ* orbitals as the active space. Higher natural occupancy of the C–C σ* orbital indicates higher biradical character. It is evident from Fig. 2 that the natural occupancy of the C–C σ* orbital of 2d is higher compared to that in 2e. Therefore, biradical character is more prominent in 2d which results in the absence of central C–C interaction. In contrast, 2e has lower biradical character as revealed by the lower occupancy of the C–C σ* orbital. We also investigated their electron density parameters (Table S2, ESI†). It has been well-established that the central C–C bond in [1.1.1]propellane has a charge-shift character22,27–30 with a positive value of Laplacian of electron density, ∇2ρBCP at the BCP, which indicates that excess kinetic energy dominates at the BCP. Moreover, the electron densities at the C–C bond critical points (0.09–0.18, Table S2, ESI†) are similar or smaller than the well-established charge shift F–F (0.25) and Cl–Cl (0.014) bonds.27 Does elongation of the C–C bond alter its character? Table S2 (ESI†) reveals that the values of ∇2ρBCP are all positive at the C–C bond critical point, indicating that elongation does not alter the charge-shift character.
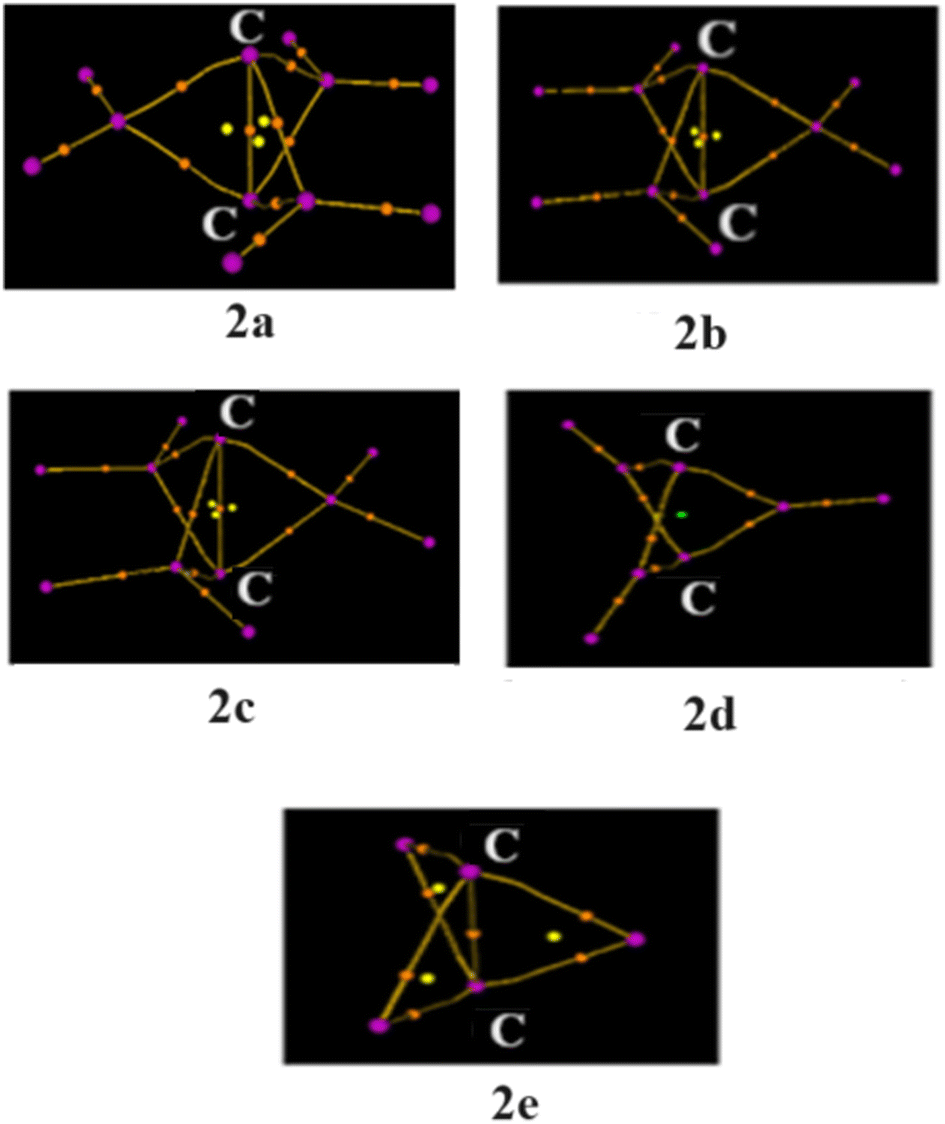 | ||
| Fig. 1 Molecular graph of 2a–2e showing the presence of C–C BCP and the bond critical point (orange dots) and absence of it in 2d. Yellow dots represent ring critical points and green dots represent cage critical points (in 2d). | ||
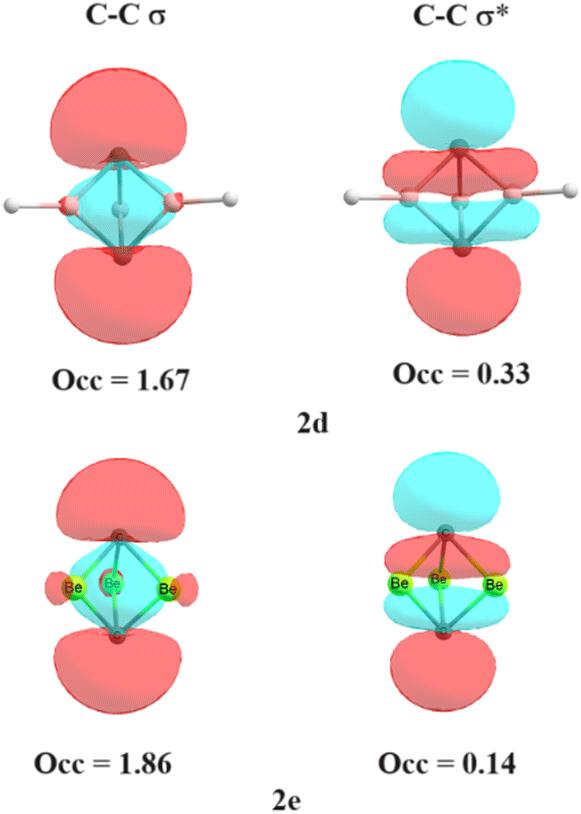 | ||
| Fig. 2 Natural occupancies (in e) of the C–C σ and σ* orbitals of 2d and 2e calculated at the CASSCF(2,2)/6-311++G** level. | ||
Conclusions
In summary, density functional calculations have been carried out to search for ultra-long C–C bonds in [1.1.1]propellane derivatives. All the studied derivatives have longer C–C bonds compared to the parent one. Fluorination at the wing –CH2 results in a longer C–C bond compared to the parent one. Similarly, the SiR2 (R = H, F) substituent also elongates the C–C bond. The longest C–C bond reported here is 2.234 Å for Be substituted [1.1.1]propellane, which retains the interaction even after significant elongation. This is one of the longest computed C–C bonds reported in the literature. With a caution regarding the toxicity of Be, we feel that SiR2 (R = H, F) substituted [1.1.1]propellane derivatives with C–C distance of greater than 1.9 Å may be experimentally realizable and will create a record for the ultra-long bonded C–C distance.5,40Author contributions
The project was conceived by A. K. G. N. S. and A. J. K. did the calculations. A. K. G. wrote the whole draft and all authors checked the manuscript.Conflicts of interest
Authors declare no conflict of interest.Acknowledgements
The computational facility of Cotton University is acknowledged.Notes and references
- S. J. Lippard, Nature, 2002, 416, 587 CrossRef CAS.
- S. P. Schreiner, L. V. Chernish, P. A. Gunchenko, E. Y. Tikhonchuk, H. Hausmann, M. Serafin, S. Schlecht, J. E. P. Dahl, R. M. K. Carlson and A. A. Fokin, Nature, 2011, 477, 308–311 CrossRef.
- M. Kaupp, B. Metz and H. Stoll, Angew. Chem., Int. Ed., 2000, 39, 4607–4609 CrossRef CAS.
- S. Kammermeier, P. G. Jones and R. Herges, Angew. Chem., Int. Ed. Engl., 1997, 36, 1757–1760 CrossRef CAS.
- Y. Ishigaki, T. Shimajiri, T. Takeda, R. Katoono and T. Suzuki, Chem, 2018, 4, 795–806 CAS.
- J. M. Oliva and C. Vinas, J. Mol. Struct., 2000, 556, 33–42 CrossRef CAS.
- J. Llop, C. Vinas, F. Teixidor, L. Victori, R. Kivekas and R. C. C. Sillanpaa, Organometallics, 2001, 20, 4024–4030 CrossRef CAS.
- J. Llop, C. Vinas, J. M. Oliva, F. Teixidor, M. A. Flores, R. Kivekas and R. Sillanpaa, J. Organomet. Chem., 2002, 657, 232–238 CrossRef CAS.
- J. M. Oliva, N. L. Allan, P. V. R. Schleyer, C. Vinas and F. Teixidor, J. Am. Chem. Soc., 2005,(127), 13538–13547 CrossRef CAS.
- Y. Wu, J. Zhang and Z. Xie, Chin. Chem. Lett., 2019, 30, 1530–1532 CrossRef CAS.
- J. Li, R. Pang, Z. Li, G. Lai, X.-Q. Xiao and T. Muller, Angew. Chem., Int. Ed., 2019, 58, 1397–1401 CrossRef CAS PubMed.
- N. Mondal, A. K. Pal, P. Gain, A. Zohaib and A. Dutta, J. Am. Chem. Soc., 2020, 142, 5331–5337 CrossRef.
- M. D. Newton and J. M. Schulman, J. Am. Chem. Soc., 1972, 94, 773 CrossRef CAS.
- K. B. Wiberg and F. H. Walker, J. Am. Chem. Soc., 1982, 104, 5239 CrossRef CAS.
- J. E. Jackson and L. C. Allen, J. Am. Chem. Soc., 1984, 106, 591 CrossRef CAS.
- K. B. Wiberg, W. P. Dailey, F. H. Walker, S. T. Waddell, L. S. Crocker and M. D. Newton, J. Am. Chem. Soc., 1985, 107, 7247 CrossRef CAS.
- D. Feller and E. R. Davidson, J. Am. Chem. Soc., 1987, 109, 4133 CrossRef CAS.
- (a) R. P. Messmer and P. A. Schultz, J. Am. Chem. Soc., 1986, 108, 7407 CrossRef CAS; (b) R. M. Jarret and L. Cucumano, Tetrahedron Lett., 1990, 31, 171 CrossRef CAS; (c) A. Ebrahimi, F. Deyhimi and H. Roohi, J. Mol. Struct., 2003, 626, 223 CrossRef CAS; (d) M. Pecul, H. Dodziuk, M. Jaszunski, O. Lukin and J. Leszczynski, Phys. Chem. Chem. Phys., 2001, 3, 1986 RSC; (e) L. B. Krivdin, Magn. Reson. Chem., 2004, 42, 1 CrossRef CAS.
- K. B. Wiberg, J. Am. Chem. Soc., 1983, 105, 1227 CrossRef CAS.
- K. B. Wiberg, R. F. W. Bader and C. D. H. Lau, J. Am. Chem. Soc., 1987, 109, 985 CrossRef CAS.
- K. B. Wiberg, Chem. Rev., 1989, 89, 975 CrossRef CAS.
- W. Wu, J. Gu, J. Song, S. Shaik and P. C. Hiberty, Angew. Chem., 2009, 121, 1435–1438 CrossRef.
- W. Stohrer and R. Hoffmann, J. Am. Chem. Soc., 1972, 94, 779 CrossRef CAS.
- E. Honegger, H. Huber and E. Heilbronner, J. Am. Chem. Soc., 1985, 107, 7172–7174 CrossRef CAS.
- O. Schafer, M. Allan, G. Szeimies and M. Sanktjohansert, J. Am. Chem. Soc., 1992, 114, 8180–8186 CrossRef CAS.
- R. Laplaza, J. Contreras-Garcia, F. Fuster, F. Volatron and P. Chaquin, Chem. – Eur. J., 2020, 26, 6839–6845 CrossRef CAS PubMed.
- S. Shaik, D. Danovich, W. Wu and P. C. Hiberty, Nat. Chem., 2009, 1, 443–449 CrossRef CAS.
- S. Shaik, D. Danovich, J. M. Galbraith, B. Braïda, W. Wu and P. C. Hiberty, Angew. Chem., Int. Ed., 2020, 132, 996–1013 CrossRef.
- S. Shaik, D. Danovich, B. Silvi, D. Lauvergnat and P. C. Hiberty, Chem. – Eur. J., 2005, 11, 6358–6371 CrossRef CAS PubMed.
- L. Zhang, F. Ying, W. Wu, P. C. Hiberty and S. Shaik, Chem. – Eur. J., 2009, 15, 2979–2989 CrossRef CAS PubMed.
- M. Messerschmidt, S. Scheins, L. Grubert, M. Pätzel, G. Szeimies, C. Paulmann and P. Luger, Angew. Chem., Int. Ed., 2005, 44, 3925–3928 CrossRef CAS.
- J. Joy, E. Akhil and E. D. Jemmis, Phys. Chem. Chem. Phys., 2018, 20, 25792–25798 RSC.
- N. Sultana and A. K. Guha, ChemistrySelect, 2023, 8, e202302714 CrossRef CAS.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899 CrossRef CAS.
- S. Grimme, J. Chem. Phys., 2006, 124, 03418 CrossRef.
- C. Møller and M. S. Plesset, Phys. Rev., 1934, 46, 618 CrossRef.
- M. J. Frisch, et al., Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- R. W. F. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, Oxford, 1990 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- J. L. Adcock, A. A. Gakh, J. L. Pollitte and C. Woods, J. Am. Chem. Soc., 1992, 114(10), 3980–3981 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nj04166b |
| ‡ Cartesian coordinates of all the optimized geometries at the B2PYP/6-311++G** and MP2(Full)/6-311++G** levels. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |