
Synthesis of functionalized disiloxanes with nonconventional fluorescence via oxa-Michael addition reaction†
Rui
Wang
a,
Shengyu
Feng
 a,
Hailong
Liu
ab,
Gang
Yi
b and
Dengxu
Wang
*a
a,
Hailong
Liu
ab,
Gang
Yi
b and
Dengxu
Wang
*a
aNational Engineering Research Center for Colloidal Materials & Key Laboratory of Special Functional Aggregated Materials, Ministry of Education, Shandong Key Laboratory of Advanced Organosilicon Materials and Technologies, School of Chemistry and Chemical Engineering, Shandong University, Jinan 250100, P. R. China. E-mail: dxwang@sdu.edu.cn
bShandong Dongyue Organosilicon Materials Co., Ltd., Zibo 256401, P. R. China
First published on 23rd November 2023
Abstract
Developing a new organosilicon synthetic methodology is significant because it enables the design and synthesis of organosilicon materials with novel structures and functionality, thereby expanding their potential applications. Herein, we introduce the classical oxa-Michael addition reaction to prepare functionalized disiloxanes by reacting 1,3-bis(3-hydroxypropyl)-1,1,3,3-tetramethyldisiloxane with various commercial vinyl compounds containing electron-withdrawing groups, while employing a phosphazene base as the catalyst. The impact of various factors, including catalytic systems, reaction solvent, and reaction temperature, on the efficiency of the process was investigated. It was found that functionalized disiloxanes can be obtained under mild conditions with moderate-to-high yields. Remarkably, these compounds exhibit nonconventional fluorescence due to the cluster-triggered emission of chromophores, such as cyano and sulfone. Moreover, the fluorescence performance of these compounds surpasses that of the analogs lacking Si–O–Si units. This observation is distinct from previous reports suggesting that the enhancement is commonly achieved by the formation of coordination bonds between the heteroatoms and the Si–O units. This implies that the introduction of a Si–O unit can efficiently enhance the fluorescence emission by weak interactions between a Si atom and an O atom from ether groups. These findings demonstrate the efficiency of the oxa-Michael addition reaction as an organosilicon synthetic methodology, thus opening up the possibilities for the development of more organosilicon materials with unique structures and enhanced functionality.
Introduction
Intense efforts have been devoted to developing new organosilicon synthetic methodologies because the species and applications of organosilicon materials are far from comparable to conventional carbon-based analogous materials, despite the fact that silicon (26.3%) is much more abundant in nature than carbon (0.026%).1–4 Compared to numerous strategies employed in synthesizing carbon-based materials, the number of strategies available for generating organosilicon materials is limited. Traditional synthetic methodologies, such as direct synthesis, hydrolysis and polycondensation, and hydrosilylation reactions, have been known for more than 80 years and are still the most important and widely used techniques in the laboratory and industry.5,6 Given the increasing requirements for organosilicon materials in emerging areas (e.g., biomedicine and sensors), developing novel and efficient synthetic methodologies is highly desirable.By introducing classical or emerging organic or polymerization methodologies into the preparation of organosilicon molecules, polymers, and materials,7 some new synthetic methodologies have been developed. Click reactions have emerged as a prominent approach due to their remarkable advantages, such as mild reaction conditions, fast speed, high yields, and tolerance of functional groups.8 Two notable examples, the thiol–ene reaction9 and aza-Michael addition reaction10 have been extensively employed to synthesize diverse functional silanes,11 polysiloxanes,12 and silicone elastomers,13 reported by us and other researchers, and the generated materials have demonstrated novel applications in self-healing materials, ultraviolet (UV) light-emitting diodes, and sensors.12,14 From a mechanistic perspective, the thiol–ene and aza-Michael addition reactions are classified as classical Michael addition reactions when initiated by vinyl groups substituted with electron-withdrawing moieties, and the thiol–ene reaction is also called the thiol-Michael addition reaction.15 Similarly, when the thiol or amine group is replaced by a hydroxyl group, this reaction is known as the oxa-Michael addition reaction. However, this reaction has lagged behind the other two Michael reactions due to the relatively low reactivity of the hydroxyl group, despite its wide application in organic synthesis16,17 and polymerization.18 Nevertheless, by utilizing an environmentally friendly and efficient catalyst known as a phosphazene base, the oxa-Michael addition reaction can also exhibit click-like characteristics. This catalyst has been successfully employed in the preparation of copolymers and hyperbranched polymers.19,20 Surprisingly, there have been no reports on the utilization of this reaction for the synthesis of organosilicon materials.
Herein, we present the oxa-Michael addition reaction as a new synthetic methodology for the preparation of functionalized disiloxanes from 1,3-bis(3-hydroxypropyl)-1,1,3,3-tetramethyl-disiloxane (BHTDS) and various vinyl-containing monomers. The effects of various factors on the efficiency of the reaction were investigated, and the mechanism was discussed. It was found that the use of a phosphazene base as a catalyst renders the reaction mild, affording moderate-to-high yields of the disiloxanes. These findings indicate that the oxa-Michael addition reaction is promising as an efficient strategy for the development of various organosilicon materials. Moreover, the afforded disiloxanes exhibit remarkable nonconventional fluorescence, which is superior to analogs without Si–O linkers.
Results and discussion
Optimization of reaction conditions
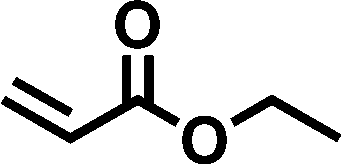
Although the oxa-Michael addition reaction has been widely employed in organic synthesis and multiple reaction conditions have been devised,16,17 it is still necessary to optimize the reaction conditions when applied to the preparation of functionalized disiloxanes because of the sensitivity of Si–O and Si–C bonds in the presence of bases, which commonly act as catalysts in this reaction. Therefore, several factors, such as the catalytic system, reaction temperature, and solvent, were meticulously optimized to achieve the successful synthesis of disiloxanes.To evaluate the catalytic activity of previously reported catalysts, a model reaction involving ethyl acrylate with moderate electron-withdrawing capacity and ethanol was chosen (Scheme 1). Various catalysts, as outlined in previous literature, were tested in this reaction, including sodium carbonate (Na2CO3),21 cesium carbonate (Cs2CO3),22 Cs2CO3/CuCl2,23 4-dimethylaminopyridine (DMAP),24,25 triphenyl-phosphine (PPh3),26–28 sodium tert-butoxide (t-BuONa),29 a phosphazene base,30 and binaphthol phosphate.31,32 The results revealed that most catalysts exhibited unsatisfactory catalytic activity in this case. For instance, when Na2CO3, Cs2CO3, Cs2CO3/CuCl2, and PPh3 were used as catalysts, no significant products were found after 6 h at room temperature (Table 1, entries 1–3 and 5). The conversion rate was only 7% when DMAP was utilized as a catalyst (Table 1, entry 4). Interestingly, the reaction could be completed within 3 h when a phosphazene base t-BuP2 was employed as the catalyst (Table 1, entry 7). While t-BuONa also displayed catalytic activity, its effectiveness was comparatively lower than that of t-BuP2, yielding a conversion rate of 44% (Table 1, entry 6), likely due to its limited solubility in organic solvents. When a chiral phosphoric acid was tested as the catalyst, no significant product generation was observed (Table 1, entry 8). These findings indicate that the phosphazene base (t-BuP2) is an effective catalyst, consistent with its high efficiency in polymerization processes.20
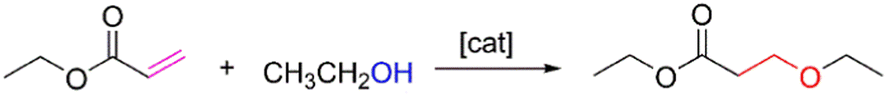 | ||
| Scheme 1 Synthetic route of ethyl acrylate and ethanol. | ||
| Entry | Catalyst | Solvent | T (°C) | Time (h) | Conv.b (%) |
|---|---|---|---|---|---|
| a Reaction conditions: ethyl acrylate (1 mmol), ethanol (1 mmol), catalyst (10 mol%), N2. b NMR calculation yield. c nr = no reaction. d nd = not detected. | |||||
| 1 | Na2CO3 | THF | rt | 6 h | nrc |
| 2 | Cs2CO3 | THF | rt | 6 h | nrc |
| 3 | CsCO3/CuCl2 | THF | 39 | 6 h | ndd |
| 4 | DMAP | THF | rt | 6 h | 7% |
| 5 | PPh3 | THF | rt | 6 h | nrc |
| 6 | t-BuONa | THF | rt | 6 h | 44 |
| 7 | t-BuP2 | THF | rt | 3 h | 98 |
| 8 | Binaphthyl phosphate | THF | rt | 6 h | ndd |
Considering the high catalytic activity of t-BuP2, it was utilized to prepare functionalized disiloxanes. To evaluate its applicability and optimize the reaction conditions, a model reaction involving the reaction between BHTDS and ethyl acrylate was investigated (Scheme 2). The results are summarized in Table 2. Similar to the model reaction of ethyl acrylate and ethanol, t-BuP2 also demonstrated good catalytic effect in this model reaction, yielding approximately 75% conversion at room temperature for 6 h (Table 2, entry 1). Furthermore, t-BuP2 maintained a comparable level of catalytic efficiency even when the reaction time was reduced to 3 h (Table 2, entry 4).
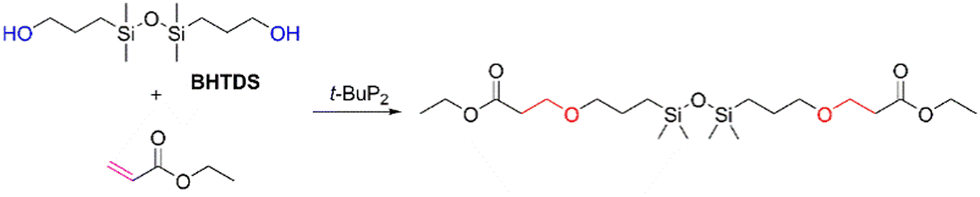 | ||
| Scheme 2 Oxa-Michael reaction of BHTDS and ethyl acrylate. | ||
| Entry | Catalyst (mol%) | Solvent | T (°C) | Time (h) | Conv.b (%) |
|---|---|---|---|---|---|
| a Reaction conditions: BHTDS (1 mmol), ethyl acrylate (1 mmol), N2. b Determined by 1H NMR. | |||||
| 1 | t-BuP2 (10) | CH2Cl2 | rt | 6 | 75 |
| 2 | t-BuP2 (5) | CH2Cl2 | rt | 6 | 52 |
| 3 | t-BuP2 (5) | CH2Cl2 | rt | 3 | 40 |
| 4 | t-BuP2 (10) | CH2Cl2 | rt | 3 | 72 |
| 5 | t-BuP2 (10) | Toluene | rt | 3 | 60 |
| 6 | t-BuP2 (10) | MeCN | rt | 3 | 50 |
| 7 | t-BuP2 (10) | THF | rt | 3 | 70 |
| 8 | t-BuP2 (10) | THF | 60 | 3 | 72 |
| 9 | t-BuP2 (10) | THF | 60 | 6 | 70 |
| 10 | t-BuP2 (10) | THF | 60 | 9 | 75 |
| 11 | t-BuP2 (10) | CH2Cl2 | rt | 12 | 70 |
| 12 | t-BuP2 (10) | CH2Cl2 | rt | 24 | 72 |
| 13 | t-BuP2 (10) | CH2Cl2 | rt | 36 | 73 |
| 14 | t-BuP2 (10) | CH2Cl2 | rt | 48 | 75 |
The effect of several solvents on the catalytic activity was examined. Given that protonic solvents have a negative effect on the nucleophilic ability of reagents, four nonprotonic solvents with low boiling points, dichloromethane, tetrahydrofuran (THF), toluene, and acetonitrile, which are easily removable during the subsequent post-treatment process, were selected (Table 2, entries 4–7). The results revealed that the use of dichloromethane and THF as solvents resulted in nearly identical conversion, both hovering around 70%. When toluene and acetonitrile were employed as solvents, a slightly lower conversion rate was observed.
The effects of catalyst dosage, reaction time, and reaction temperature were further investigated. It was found that the reaction conversion could be effectively improved from 52% to 75% by increasing the catalyst dosage from 5 mol% to 10 mol% and maintaining the reaction time of 6 h (Table 2, entries 1 and 2). Moreover, even when the reaction time was reduced to 3 h, a conversion of 72% could still be achieved with a catalyst dosage of 10 mol% (Table 2, entry 4), in comparison to the conversion of 40% with a catalyst dosage of 5 mol% (Table 2, entry 3). However, prolonging the reaction time to 12 h, 24 h, 36 h, and 48 h did not afford any significant changes in conversion, as it consistently remained around 70% (Table 2, entries 11–14). To provide additional evidence, the reactions were monitored by proton nuclear magnetic resonance (1H NMR) spectroscopy, as illustrated in Fig. S1 (ESI†). The integrations of the protons from double bonds at δ 5.8–6.4 ppm (Hd, He, and Hf) exhibited similarities, indicating comparable consumption of the double bonds. Additionally, the reaction temperature also does not obviously influence the conversion. Even when the temperature was raised to 60 °C, the conversion remained around 70% (Table 2, entries 7–10).
Moreover, transesterification also occurred between BHTDS and ethyl acrylate during the process of oxa-Michael addition. As shown in Scheme S1 (ESI†), the transesterification reaction of BHTDS and ethyl acrylate afforded the formation of acrylate-containing disiloxanes (C1 and C2) along with ethanol. Subsequently, the oxa-Michael reaction of these aforementioned compounds yielded by-products A and B. This mechanism can be verified by the appearance of new peaks at 3.45 ppm (Hc) and 4.21 ppm (Hi) in Fig. S1 (ESI†), which are consistent with previous reports.33
Based on these results, it can be concluded that t-BuP2 demonstrates good catalytic activity in the reaction, and dichloromethane as the solvent has less effect on efficiency. To further optimize the reaction conditions, orthogonal experiments were conducted using dichloromethane as the reaction solvent, with t-BuP2 serving as the catalyst. The factors considered in these experiments were the amount of catalyst (2.5 mol%, 5 mol%, and 10 mol%), reaction temperature (room temperature, 30 °C, and 40 °C), and reaction time (3 h, 6 h, and 9 h). The experimental data are summarized in Table 3 and the corresponding NMR spectra are shown in Fig. S2 (ESI†). The results revealed that higher conversion was achieved with a catalyst dosage of 10 mol% (Table 3, entries 1–3) compared to catalyst dosages of 5 mol% and 2.5 mol% (Table 3, entries 4–9). However, when the catalyst dosage was 10 mol%, increasing the reaction temperature and prolonging the reaction time did not significantly improve the conversion rate, as the conversion rate remained around 72%. Therefore, the optimal conditions for the synthesis of functionalized disiloxanes involve using 10 mol% of t-BuP2 as the catalyst, dichloromethane as the solvent, and conducting the reaction at room temperature.
Synthesis of functionalized disiloxanes via the oxa-Michael addition reaction
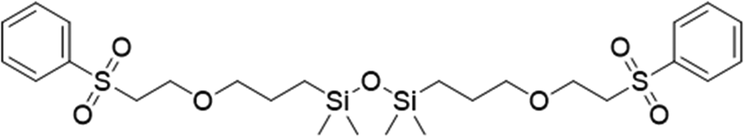
To assess the applicability of the optimized reaction conditions, they were employed in the preparation of various functionalized disiloxanes, as depicted in Scheme 3. As expected, the conversion rates were significantly influenced by the electron-withdrawing ability of the substituents attached to the double bonds because this reaction follows a nucleophilic addition mechanism, and compounds bearing strong electron-withdrawing groups (EWG) display enhanced reactivity. For instance, when acrylonitrile, methyl vinyl sulfone, and phenyl vinyl sulfone were used as starting materials, the desired products were obtained in high conversion rates of 85%–95% (Table 4, entries 1–3), even when the reaction time was shortened to 1 h or 2 h (Table 4, entries 10–13). Conversely, as the electron-withdrawing ability of the substituents decreased, the conversion rates diminished to10%–75% (Table 4, entries 4–9). Additionally, the conversion of FDSi-6 (25%, Table 4, entry 6) was much lower than that of FDSi-4 (65%, Table 4, entry 4). This discrepancy can be attributed to the increased steric hindrance imposed by the methyl methacrylate moiety in comparison to methyl acrylate. The structures of the products were fully determined using Fourier-transform infrared, 1H NMR, 13C NMR, 29Si NMR, and high-resolution mass spectrometries (Fig. S7–S43, ESI†) with the exception of FDSi-8 and FDSi-9 due to challenges in purifying the crude products. | ||
| Scheme 3 Synthetic route of various functionalized disiloxanes via the oxa-Michael reaction. | ||
| Entry | Substrates | Products | Time (h) | Conv.b (%) | Yieldc (%) | |
|---|---|---|---|---|---|---|
| a Reaction condition: BHTDS (1 mmol), substrate (1 mmol), room temperature, t-BuP2 (10 mol%), N2. b Determined by 1H NMR. c Isolated yields. | ||||||

|
FDSi-1 |
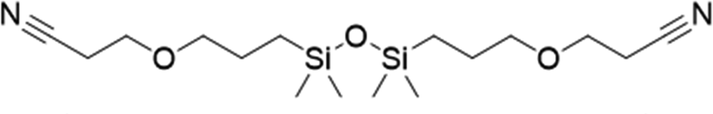
|
3 | 95 | 93.5 | |
| 2 |
|
FDSi-2 |
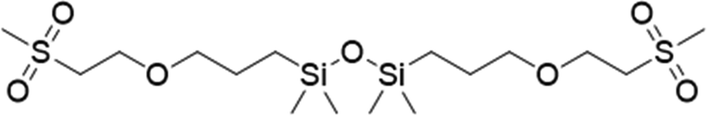
|
3 | 90 | 87.6 |
| 3 |
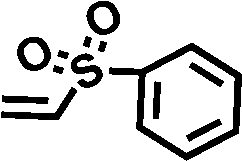
|
FDSi-3 |
|
3 | 85 | 83.5 |
| 4 |
|
FDSi-4 |
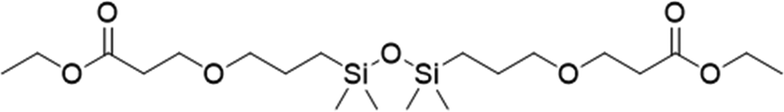
|
3 | 70 | 70.2 |
| 5 |

|
FDSi-5 |
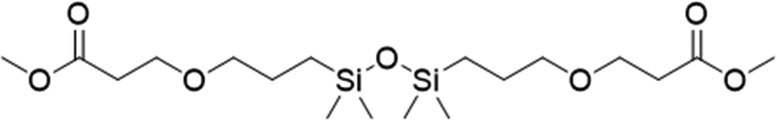
|
3 | 65 | 40.2 |
| 6 |
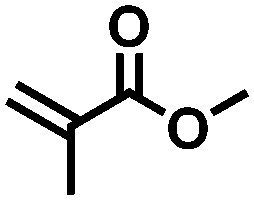
|
FDSi-6 |

|
3 | 25 | 10.0 |
| 7 |

|
FDSi-7 |
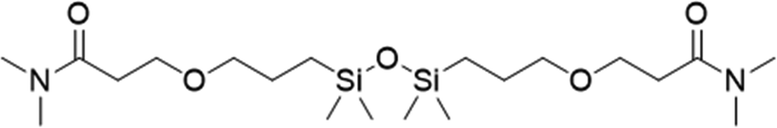
|
3 | 10 | 8.5 |
| 8 |
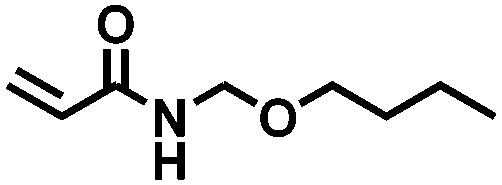
|
FDSi-8 |
|
3 | 20 | — |
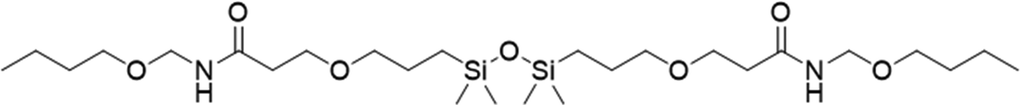
| 9 |
|
FDSi-9 |
|
3 | 40 | — |
| 10 |

|
FDSi-1 | 2 | 93 | — | |
| 11 |
|
FDSi-1 | 1 | 90 | — | |
| 12 |
|
FDSi-2 | 2 | 85 | — | |
| 13 |
|
FDSi-3 | 2 | 81 | — | |
Based on these results, it can be concluded that t-BuP2 is an efficient catalyst for the synthesis of functionalized disiloxanes via the oxa-Michael addition reaction due to its super-strong alkalinity and weak nucleophilicity.34 The reaction can be conveniently performed at ambient temperature, thereby mitigating the occurrence of self-polymerization of vinyl monomers at elevated temperatures. Nevertheless, the efficiency of the current reaction is slightly lower compared to other oxa-Michael addition reactions catalyzed by t-BuP2. For instance, the conversion rate in the model reaction involving ethyl acrylate and ethanol reaches an impressive 98%, significantly surpassing the 75% conversion observed for the reaction between BHTDS and ethyl acrylate. Moreover, the completion of the present reaction time requires hours and is much longer than a few minutes in the reaction between the acrylate double bonds and a primary alcohol reported by Jiang's group.20 This difference can be attributed to the presence of competing factors in the reaction. Previous studies have demonstrated that a phosphazene base can act as a catalyst for the ring-opening polymerization of cyclosiloxanes due to its strong basicity.35 Thus, t-BuP2 exhibits dual reactivity by not only catalyzing the oxa-Michael addition reaction but also engaging in the cleavage of the Si–O–Si bond. Consequently, this competitive process can diminish the catalytic efficiency of t-BuP2. The reaction mechanism is illustrated in Scheme 4.
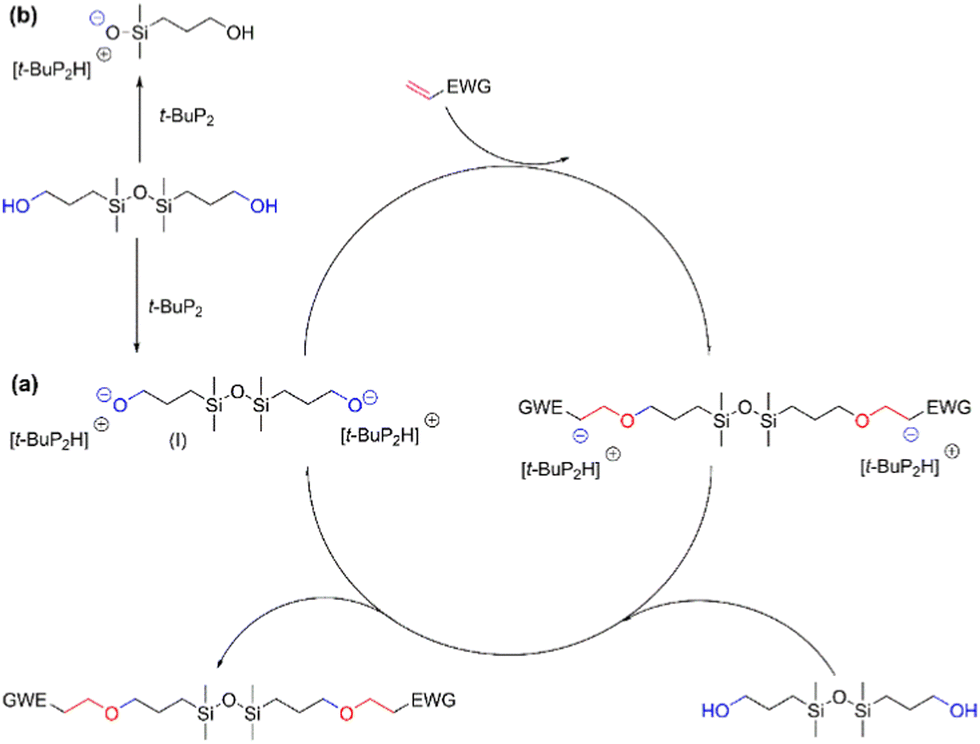 | ||
| Scheme 4 Plausible mechanism of the reaction between BHTDS and various substrates. | ||
Initially, t-BuP2 deprotonates the hydroxyl groups in BHTDS, yielding the formation of an intermediate (I) harboring a negatively charged oxygen ion (or alkoxide). This intermediate subsequently engages in a nucleophilic addition reaction with the electron-deficient Michael acceptor in the β-position to the EWG in the substrates. Thereby, a carbanion in α-position to the EWG is formed, which is further pronated by another BHTDS closing the cycle, while the final functionalized disiloxane is formed (Scheme 4a).18 Concurrently, t-BuP2 also attacks the Si–O–Si unit during the reaction, generating a silanolate product (Scheme 4b).35,36 These two forms of competition mean completion of the reaction requires a longer time.
Photophysical properties of functionalized disiloxanes
Based on previous reports, compounds or polymers containing specific chromophores, such as sulfone, ester, and cyano groups, can display intriguing nonconventional fluorescence in the absence of substantial conjugation.37–40 In the case of functionalized disiloxanes, the presence of these chromophores could render them capable of nonconventional fluorescence characteristics. To elucidate this, the fluorescent spectra of FDSi-1, FDSi-2, FDSi-4, and FDSi-7 as representative examples were measured in CH2Cl2 solution. As anticipated, these compounds exhibit similar emission spectra and blue fluorescence, with maximum emission wavelengths (λmax) of 421 nm, 445 nm, 426 nm, and 428 nm for FDSi-1, FDSi-2, FDSi-4, and FDSi-7, respectively, when excited at 365 nm (Fig. 1). Moreover, this phenomenon can be visualized under 365 nm UV light (Fig. S3, ESI†). This finding can be explained by the cluster-triggered emission mechanism.37–40 In these compounds, when the chromophores cluster in close proximity to each other, through-space electronic communication is achieved by various interactions, including electron overlap between lone pairs and π electrons, dipole–dipole interactions, and n–π interactions. Consequently, extended electronic conjugation is established, giving rise to nonconventional fluorescence.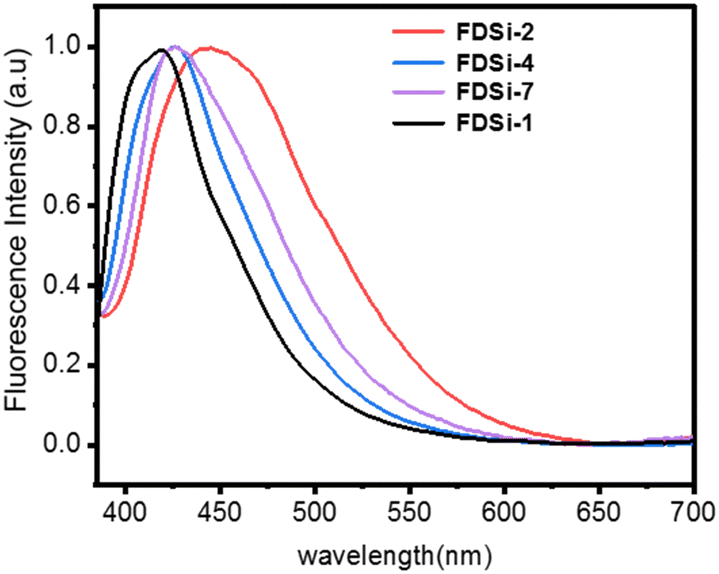 | ||
| Fig. 1 Fluorescence spectra of functionalized disiloxanes (FDSi) in CH2Cl2 solution (M = 10−7 mol L−1, excitation wavelength (λex) is 365 nm). | ||
Considering the similar emission behavior observed for these compounds, FDSi-1 and FDSi-2 were selected as representative examples for further investigation into their fluorescence properties. As shown in Fig. 2a and b, the fluorescent spectra of FDSi-1 and FDSi-2 were examined at different concentrations. It was found that both of them in CH2Cl2 solutions are almost nonfluorescent when the concentrations are lower than 0.1 mg mL−1. This finding can be explained by the limited opportunities for aggregation of individual chromophores in dilute solution, impeding the formation of clusters; even if the clusters are formed, the molecular motion is pronounced and active in dilute solution, affording negligible or weak fluorescence. In contrast, as the concentration increases, the fluorescence intensities are significantly enhanced. For instance, in the case of the FDSi-1 solution, the intensity at a concentration of 10 mg mL−1 is approximately six times higher than that at 1 mg mL−1. This pronounced enhancement can be ascribed to the effective aggregation of the chromophores at higher concentration,41 along with the formation of clusters comprising cyano and sulfone groups in the case of FDSi-1 and FDSi-2, respectively. Additionally, fluorescence enhancement with an increase in concentration can be clearly observed under 365 nm UV light, as shown in Fig. S4 (ESI†).
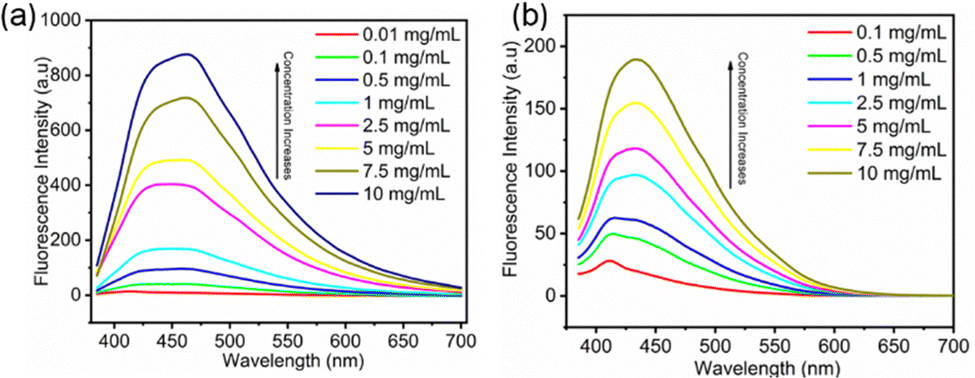 | ||
| Fig. 2 Fluorescence spectra of functionalized disiloxanes FDSi-1 (a) and FDSi-2 (b) at different concentrations in CH2Cl2 solution (λex = 365 nm). | ||
To prove the contribution of aggregation to nonconventional fluorescence, the fluorescence spectra of FDSi-1 and FDSi-2 were examined in dichloromethane/petroleum ether (CH2Cl2/PE) solutions. As illustrated in Fig. 3a, the fluorescence intensity of FDSi-1 gradually improved by changing the volume ratio of CH2Cl2/PE from 100%![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0 to 60%:40%. However, the fluorescence intensity decreased as the proportion of PE was further increased. It was found that when the ratio of CH2Cl2/PE was 20%:80%, the fluorescence intensity greatly decreased. The difference can be also observed in photographs of these solutions under 365 nm UV light (Fig. S5a, ESI†). Unlike FDSi-1, the fluorescence intensity of FDSi-2 progressively increases as the volume ratio of CH2Cl2/PE is tuned from 100%:0 to 20%:80% (Fig. 3b), and Fig. S5b (ESI†) shows photographs of these solutions under 365 nm UV light. It is known that CH2Cl2 serves as a good solvent for disiloxanes, whereas PE is considered a poor solvent. Thus, as the content of PE in the mixed solvents increases, the solubility of the samples progressively decreases, thereby facilitating chromophore aggregation and yielding higher fluorescence intensities. These findings underscore the pivotal role played by chromophore aggregation in the context of nonconventional fluorescence. The unexpected decrease in intensity for FDSi-1 when the PE content in the mixed solvents is high may be attributed to poor dispersibility of the sample. Furthermore, when FDSi-2 (as an example) was dissolved in various solvents, including THF, dimethylformamide, CH2Cl2, ethanol, and acetone, similar emission profiles were observed. However, the fluorescence intensity exhibited significant variation depending on the solvent employed (Fig. S6, ESI†), indicating the presence of distinct emissive clusters in different solvents. The intensities of FDSi-2 in THF and CH2Cl2 were notably higher compared to those that in other solvents, suggesting that nonprotonic solvents favor fluorescence enhancement. This phenomenon can be attributed to interactions between lone-pair electrons in the nonprotonic solvent molecules and the chromophores, enhancing their aggregation and degree of conjugation.42
:0 to 60%:40%. However, the fluorescence intensity decreased as the proportion of PE was further increased. It was found that when the ratio of CH2Cl2/PE was 20%:80%, the fluorescence intensity greatly decreased. The difference can be also observed in photographs of these solutions under 365 nm UV light (Fig. S5a, ESI†). Unlike FDSi-1, the fluorescence intensity of FDSi-2 progressively increases as the volume ratio of CH2Cl2/PE is tuned from 100%:0 to 20%:80% (Fig. 3b), and Fig. S5b (ESI†) shows photographs of these solutions under 365 nm UV light. It is known that CH2Cl2 serves as a good solvent for disiloxanes, whereas PE is considered a poor solvent. Thus, as the content of PE in the mixed solvents increases, the solubility of the samples progressively decreases, thereby facilitating chromophore aggregation and yielding higher fluorescence intensities. These findings underscore the pivotal role played by chromophore aggregation in the context of nonconventional fluorescence. The unexpected decrease in intensity for FDSi-1 when the PE content in the mixed solvents is high may be attributed to poor dispersibility of the sample. Furthermore, when FDSi-2 (as an example) was dissolved in various solvents, including THF, dimethylformamide, CH2Cl2, ethanol, and acetone, similar emission profiles were observed. However, the fluorescence intensity exhibited significant variation depending on the solvent employed (Fig. S6, ESI†), indicating the presence of distinct emissive clusters in different solvents. The intensities of FDSi-2 in THF and CH2Cl2 were notably higher compared to those that in other solvents, suggesting that nonprotonic solvents favor fluorescence enhancement. This phenomenon can be attributed to interactions between lone-pair electrons in the nonprotonic solvent molecules and the chromophores, enhancing their aggregation and degree of conjugation.42
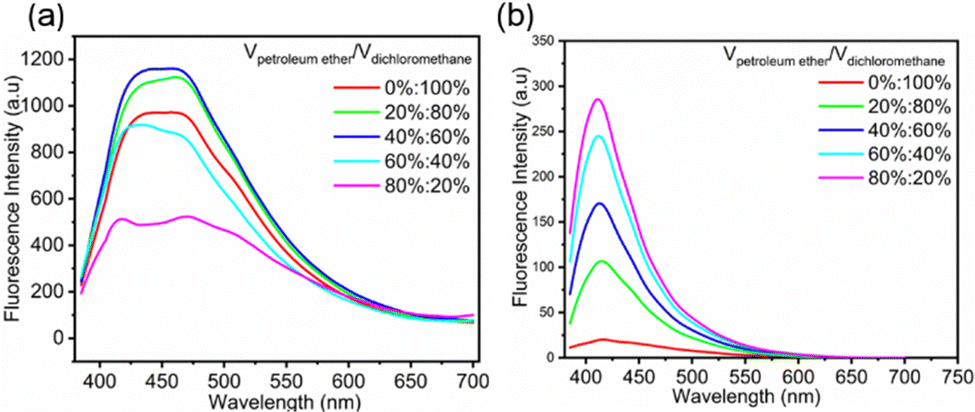 | ||
| Fig. 3 Fluorescence spectra of FDSi-1 (a) and FDSi-2 (b) in CH2Cl2/PE solvent mixture (0.01 mol L−1, λex = 365 nm). | ||
Si–O–Si effect on the nonconventional fluorescence
To investigate the impact of the Si–O–Si unit on nonconventional fluorescence, the silicon-free compound FD-J was synthesized via the oxa-Michael addition reaction of 1,6-hexanediol and acrylonitrile (Scheme S2, ESI†). Previously, we found that Si–O bonds can enhance the nonconventional fluorescence in cyano-containing organosilicon molecules due to the formation of N → Si coordination. This coordination results in shorter distances between cyano clusters, facilitating aggregation and the development of extended electronic conjugation. Consequently, this reduces nonradiative decay rates and promotes fluorescence emission.42 Interestingly, FDSi-1 exhibits an obvious red-shift and much higher fluorescence intensity than FD-J at the same concentration (Fig. 4a), even though no N → Si coordination occurs. This finding can be attributed to the introduction of Si–O–Si units, which play a crucial role in enhancing fluorescence. To confirm this hypothesis, the fluorescence emission spectra of these compounds and a mixture of FD-J and hexamethyldisiloxane (MM) were tested using different excitation wavelengths. The results demonstrated that the mixture displays higher fluorescence intensity than FD-J but lower intensity than FDSi-1 (Fig. 4a and b). Based on these findings, it can be inferred that Si–O–Si units have a beneficial effect on chromophore aggregation and fluorescence enhancement, even when not directly incorporated into the structure. This contribution is speculated to result from the weak interaction between Si and O in the ether group, yielding the formation of O → Si coordination, similar to N → Si coordination.41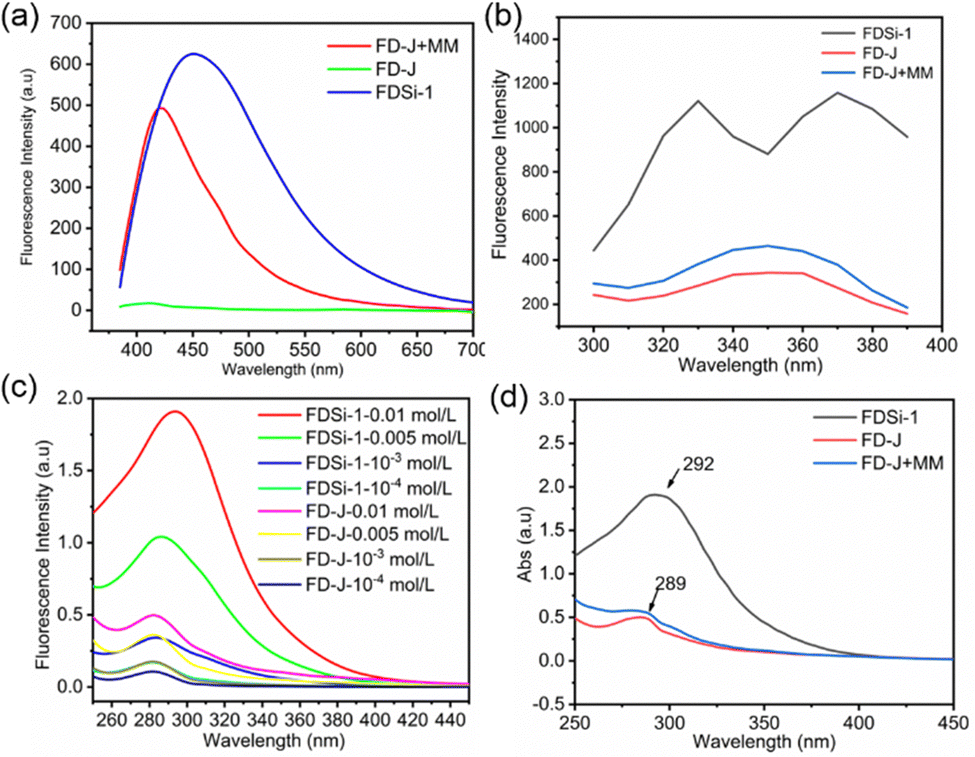 | ||
| Fig. 4 (a) and (b) Comparison of fluorescence emission intensity of FDSi-1, FD-J and the mixture in CH2Cl2 solution (0.01 mol L−1, λex = 365 nm) (a), and in CH2Cl2 solution (0.01 mol L−1) under different excitation wavelengths (b); (c) UV/Vis absorption spectra of FDSi-1 and FD-J at various concentrations in CH2Cl2 solution; (d) UV/Vis absorption spectra of FDSi-1, FD-J and the mixture (0.01 mol L−1). | ||
Additionally, the presence of silicon-induced aggregation can also be proved by UV-visible absorption spectra. As depicted in Fig. 4c, the absorption is progressively enhanced for both FDSi-1 and FD-J with increasing concentration, and the maximum absorption peaks experience a slight red-shift. Notably, the UV absorption of FDSi-1 is significantly stronger than that of FD-J at the same concentration. Moreover, the addition of MM improves the absorption of FD-J, although the absorption intensity remains lower than that of FDSi-1 (Fig. 4d). Therefore, the UV results provide further evidence that the introduction of Si–O–Si units promotes the aggregation of unconventional chromophores. This effect is observed both intermolecularly and intramolecularly, consistent with the fluorescent results.
Conclusions
We synthesized a series of functionalized disiloxanes via the oxa-Michael addition reaction from 1,3-bis(3-hydroxypropyl)-1,1,3,3-tetramethyldisiloxane and various vinyl monomers containing cyano, sulfone, ester, and amide groups. The influence of catalyst species, reaction temperature, reaction time, and solvents on the reaction efficiency was investigated. The results demonstrated that the reaction can be conducted under mild conditions, with moderate-to-high product yields, with the utilization of 10 mol% of phosphazene base t-BuP2 as the catalyst and dichloromethane as the solvent at room temperature. These disiloxanes exhibit intriguing nonconventional fluorescence, outperforming analogs lacking Si–O–Si units, thereby providing evidence for the presence of an Si–O induced aggregation effect and fluorescence enhancement. Thus, the oxa-Michael addition reaction proves to be an effective methodology for the preparation of disiloxanes with unique properties. Unlike traditional organosilicon synthetic methods, such as hydrosilylation reactions, this methodology obviates the need for precious metal catalysts, illustrating its potential for applications in the development of novel organosilicon functional materials with diverse uses, such as sensing and wearable devices. Further optimization of the reaction conditions may be pursued in future investigations, considering the strong basicity of t-BuP2. Currently, ongoing efforts involve the application of this innovative strategy in the design and synthesis of new organosilicon materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by Fluorine Silicone Materials Collaborative Fund of Shandong Provincial Natural Science Foundation (ZR2020LFG011 and ZR2021LFG001), National Natural Science Foundation of China (22271175 and 52173102), and Young Scholars Program of Shandong University.Notes and references
- J. Liu, Y. Yao, X. Li and Z. Zhang, Chem. Eng. J., 2020, 408, 127262 CrossRef.
- C. Rücker and K. Kümmerer, Chem. Rev., 2015, 115, 466–524 CrossRef.
- E. Yilgör and I. Yilgör, Prog. Polym. Sci., 2014, 39, 1165–1195 CrossRef.
- M. P. Wolf, G. B. Salieb-Beugelaar and P. Hunziker, Prog. Polym. Sci., 2018, 83, 97–134 CrossRef CAS.
- D. Wang, J. Klein and E. Mejía, Chem. – Asian J., 2017, 12, 1180–1197 CrossRef CAS.
- L. D. de Almeida, H. Wang, K. Junge, X. Cui and M. Beller, Angew. Chem., Int. Ed., 2021, 60, 550–565 CrossRef CAS PubMed.
- D. Wang, J. Cao, D. Hang, W. Li and S. Feng, Prog. Chem., 2019, 31, 110–120 CAS.
- C. O. Kappe and E. Van der Eycken, Chem. Soc. Rev., 2010, 39, 1280–1290 RSC.
- Y. Zuo, J. Cao and S. Feng, Adv. Funct. Mater., 2015, 25, 2754–2762 CrossRef CAS.
- H. Lu, L. Feng, S. Li, J. Zhang, H. Lu and S. Feng, Macromolecules, 2015, 48, 476–482 CrossRef CAS.
- A. K. Tucker-Schwartz, R. A. Farrell and R. L. Garrell, J. Am. Chem. Soc., 2011, 133, 11026–11029 CrossRef CAS PubMed.
- Y. Zuo, X. Liang, J. Yin, Z. Gou and W. Lin, Coord. Chem. Rev., 2021, 447, 214166 CrossRef CAS.
- Y. J. Zuo, Z. M. Gou, C. Q. Zhang and S. Y. Feng, Macromol. Rapid Commun., 2016, 37, 1052–1059 CrossRef CAS.
- R. Sun, S. Feng, D. Wang and H. Liu, Chem. Mater., 2018, 30, 6370–6376 CrossRef CAS.
- D. P. Nair, M. Podgórski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2014, 26, 724–744 CrossRef CAS.
- Y. Wang and D. M. Du, Org. Chem. Front., 2020, 7, 3266–3283 RSC.
- C. F. Nising and S. Brase, Chem. Soc. Rev., 2012, 41, 988–999 RSC.
- K. Ratzenböck, S. M. Fischer and C. Slugovc, Monatsh. Chem., 2023, 154, 443–458 CrossRef.
- Q. Jiang, Y. Zhang, Y. Du, M. Tang, L. Jiang, W. Huang, H. Yang, X. Xue and B. Jiang, Polym. Chem., 2020, 11, 1298–1306 RSC.
- H. J. Yang, Y. K. Zuo, J. D. Zhang, Y. Y. Song, W. Y. Huang, X. Q. Xue, Q. M. Jiang, A. B. Sun and B. B. Jiang, Polym. Chem., 2018, 9, 4716–4723 RSC.
- S.-H. Guo, S.-Z. Xing, S. Mao, Y.-R. Gao, W.-L. Chen and Y.-Q. Wang, Tetrahedron Lett., 2014, 55, 6718–6720 CrossRef CAS.
- V. B. Gudise, P. C. Settipalli, E. K. Reddy and S. Anwar, Eur. J. Org. Chem., 2019, 2234–2242 CrossRef CAS.
- S.-i Matsuoka, Y. Hoshiyama, K. Tsuchimoto and M. Suzuki, Chem. Lett., 2017, 46, 1718–1720 CrossRef CAS.
- H. Wang, F. Cheng, W. He, J. Zhu, G. Cheng and J. Qu, Biointerphases, 2017, 12, 02C414 CrossRef PubMed.
- S. Strasser, C. Wappl and C. Slugovc, Polym. Chem., 2017, 8, 1797–1804 RSC.
- N. Ziegenbalg, R. Lohwasser, G. D'Andola, T. Adermann and J. C. Brendel, Polym. Chem., 2021, 12, 4337–4346 RSC.
- S. Strasser and C. Slugovc, Catal. Sci. Technol., 2015, 5, 5091–5094 RSC.
- S. M. Fischer, S. Renner, A. D. Boese and C. Slugovc, Beilstein J. Org. Chem., 2021, 17, 1689–1697 CrossRef CAS PubMed.
- Y. Murata and J. i Uenishi, Tetrahedron, 2016, 72, 4962–4967 CrossRef CAS.
- Q. Jiang, Y. Du, Y. Zhang, L. Zhao, L. Jiang, W. Huang, H. Yang, X. Xue and B. Jiang, J. Polym. Sci., 2020, 58, 2718–2727 CrossRef CAS.
- W. Wu, X. Li, H. Huang, X. Yuan, J. Lu, K. Zhu and J. Ye, Angew. Chem., Int. Ed., 2013, 52, 1743–1747 CrossRef CAS PubMed.
- Q. Gu, Z.-Q. Rong, C. Zheng and S.-L. You, J. Am. Chem. Soc., 2010, 132, 4056–4057 CrossRef CAS PubMed.
- H. Yang, Y. Zuo, J. Zhang, Y. Song, W. Huang, X. Xue, Q. Jiang, A. Sun and B. Jiang, Polym. Chem., 2018, 9, 4716–4723 RSC.
- S. Boileau and N. Illy, Prog. Polym. Sci., 2011, 36, 1132–1151 CrossRef CAS.
- J. Shi, N. Zhao, S. Xia, S. Liu and Z. Li, Polym. Chem., 2019, 10, 2126–2133 RSC.
- P. C. Hupfield and R. G. Taylor, J. Inorg. Organomet. Polym., 1999, 9, 17–34 CrossRef CAS.
- M. Sun, C.-Y. Hong and C.-Y. Pan, J. Am. Chem. Soc., 2012, 134, 20581–20584 CrossRef CAS.
- X. Miao, T. Liu, C. Zhang, X. Geng, Y. Meng and X. Li, Phys. Chem. Chem. Phys., 2016, 18, 4295–4299 RSC.
- Q. Zhou, B. Cao, C. Zhu, S. Xu, Y. Gong, W. Z. Yuan and Y. Zhang, Small, 2016, 12, 6586–6592 CrossRef CAS PubMed.
- W. I. Lee, Y. Bae and A. J. Bard, J. Am. Chem. Soc., 2004, 126, 8358–8359 CrossRef CAS.
- Y. Zuo, Z. Gou, W. Quan and W. Lin, Coord. Chem. Rev., 2021, 438, 213887 CrossRef CAS.
- H. Lu, Z. Hu and S. Feng, Chem. – Asian J., 2017, 12, 1213–1217 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Complete experimental procedures and characterization data for the prepared compounds. See DOI: https://doi.org/10.1039/d3nj04004f |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |