
Guanidinium iodide salts as single component catalysts for CO2 to epoxide fixation†
Ángela
Mesías-Salazar
 ad,
René S.
Rojas
*a,
Fernando
Carrillo-Hermosilla
*c,
Javier
Martínez
b,
Antonio
Antiñolo
c,
Oleksandra S.
Trofymchuk
d,
Fabiane M.
Nachtigall
e,
Leonardo S.
Santos
f and
Constantin G.
Daniliuc
g
ad,
René S.
Rojas
*a,
Fernando
Carrillo-Hermosilla
*c,
Javier
Martínez
b,
Antonio
Antiñolo
c,
Oleksandra S.
Trofymchuk
d,
Fabiane M.
Nachtigall
e,
Leonardo S.
Santos
f and
Constantin G.
Daniliuc
g
aLaboratorio de Química Inorgánica, Facultad de Química Universidad Católica de Chile, Casilla 306, Santiago-22 6094411, Chile. E-mail: rrojasg@uc.cl
bInstituto de Ciencias Químicas, Facultad de Ciencias, Isla Teja, Universidad Austral de Chile, Valdivia 5090000, Chile
cDepartamento de Química Inorgánica, Orgánica y Bioquímica-Centro de Innovación de Química Avanzada ORFEO-CINQA, Universidad de Castilla-La Mancha, Avda. Camilo José Cela, 10, Ciudad Real, E-13071, Spain. E-mail: Fernando.Carrillo@uclm.es
dDepartamento de Química Orgánica y Fisicoquímica, Universidad de Chile, Facultad de Ciencias Químicas y Farmacéuticas, Sergio Livingstone 1007, Casilla 233, Santiago 8380492, Chile
eInstituto de Ciencias Químicas Aplicadas – Universidad Autónoma de Chile, Talca 3467987, Chile
fLaboratory of Asymmetric Synthesis, Chemistry Institute of Natural Resources, Universidad de Talca, P.O. Box 747, Talca, Chile
gOrganisch-Chemisches Institut der Universität Münster, Corrensstrasse 40, Münster 48149, Germany
First published on 17th November 2023
Abstract
In this study, we present the synthesis, characterization and catalytic reactions of a new family of one-component catalysts based on guanidinium salts. The synthesis of cyclic carbonates from epoxides and CO2 at 70–80 °C under 1–5 bar of CO2 pressure is demonstrated using the new one-component guanidinium salts with various substituents. The 1e catalytic system, which has isopropyl-type substituents in its structure, has the best conversion rates; the TON for the synthesis of styrene carbonate was 92. The production of cyclic carbonates from the internal epoxides is another application for these catalytic systems with the conversion of cyclohexane oxide to the respective cyclic carbonate at 86% with 1e catalyst. Finally, ESI-MS was used to examine the interaction of the new organocatalysts with CO2 and epichlorohydrin. These findings led to catalysts made of organic molecules that are suitable for organocatalysis since they do not require a co-catalyst and can operate effectively at moderate pressures and temperatures.
Introduction
The chemical conversion of carbon dioxide (CO2) by its fixation into high-value-added products has been one of the principal focuses and challenges for the scientific community to contribute to the decrease in CO2 emissions in the atmosphere.1–4 Related to this, the attention to the catalytic transformation of this molecule has increased enormously in recent years5–8 for the preparation of a great variety of different products, such as methanol,9,10 carbamates,11–13 formic acid,14,15 and amines.16 It is important to note that according to a recent study, only 0.36% of CO2 emissions were employed as feedstock for the industrial preparation of value-added chemicals.17 It would therefore be desirable to develop new CO2 technologies (capture, separation, transport, and use) that would help to make these processes more viable.18–20In that sense, an outstanding example that implies the utilization of CO2 is its cycloaddition into epoxides to afford cyclic organic carbonates.21–23 These compounds have shown their ability to be used as polar aprotic solvents in chemical processes24,25 and as electrolyte solvents in lithium-ion batteries.26,27 It is relevant to point out that a large number of catalyst systems, such as metal-salen complexes,28–38 metal organic frameworks39–42 and ionic liquids,43–45 have been recently prepared to carry out the synthesis of cyclic carbonates with excellent yields and selectivities. However, the multistep preparation, meticulous purification, and the elevated price associated with the synthesis of most metal-based catalysts could be the bottlenecks of their large-scale production, which make it desirable to develop metal-free catalysts or organocatalysts to perform the cyclic carbonate production. In fact, numerous compounds of this type based on N-heterocyclic carbenes,46,47 phosphonium,48–51 or ammonium salts49,52–55 have reported exceptional results for this process.
Among the organocatalysts that have achieved brilliant results for this catalytic transformation, the hydrogen-bond donor (Hcat) catalysts have aroused the interest of several research groups as Hcat assists the epoxy ring-opening,56 which is the rate-determining step for the CO2 fixation into cyclic carbonates,57 by hydrogen bonding with the oxygen atom of the epoxide in an analogous way to metal Lewis acid-based catalysts. In this way, it facilitates the nucleophilic attack from the halide moiety on the epoxide (Scheme 1). Afterwards, the insertion of the CO2 molecule follows with the corresponding formation of the alkyl carbonate intermediate, which finally undergoes an intramolecular ring-closing to yield the cyclic carbonate product (Scheme 1).
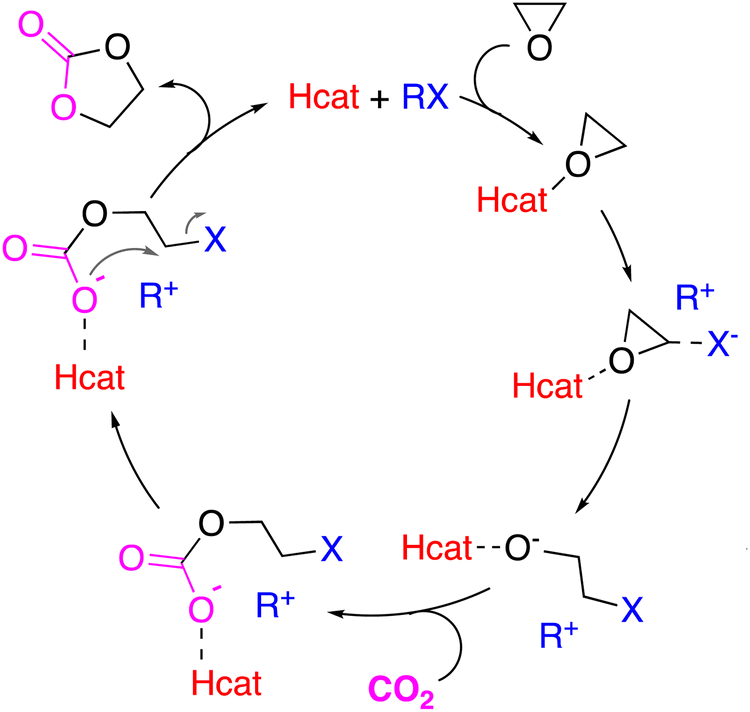 | ||
| Scheme 1 Proposed mechanism for the cycloaddition reaction of CO2 to epoxides co-catalyzed by an organic halide (RX) with the cooperation of an Hcat. | ||
The number of Hcat organocatalysts has grown lately and they are mainly based on fluorinated alcohols,58 silanediols,59 phenol-derived compounds,60–64 phosphonium alcohols,65,66 and organic acids.67,68 These compounds catalyzed the cycloaddition of epoxides and CO2 into cyclic carbonates in excellent yields under relatively medium reaction conditions. On the other hand, guanidines, which are a potential type of Hcat catalysts, have rarely been investigated since these compounds typically catalyze this reaction through the activation of the CO2 molecule, which often requires elevated temperatures (>100 °C) and high CO2 pressures (>10 bar).69–76
In contrast, a mechanism based on the interaction between hydrogen-bond donor (Hcat) and epoxide has been less reported. Relative to this, it is worth highlighting that recently we were able to prepare a set of aromatic mono- and bis(guanidines), which acted as efficient catalysts for this catalytic process at low temperatures and CO2 pressure, as a result of the hydrogen-bond formation between the oxygen atom of the epoxide and the N–H group of the guanidine, although the use of the co-catalyst (Bu4NI) remained necessary.77 It is relevant to note that recently bifunctional organocatalysts based on guanidinium salts for the CO2 fixation into cyclic carbonates have been reported (Fig. 1), however, the catalytic cycle of this process has hardly been studied.78–81
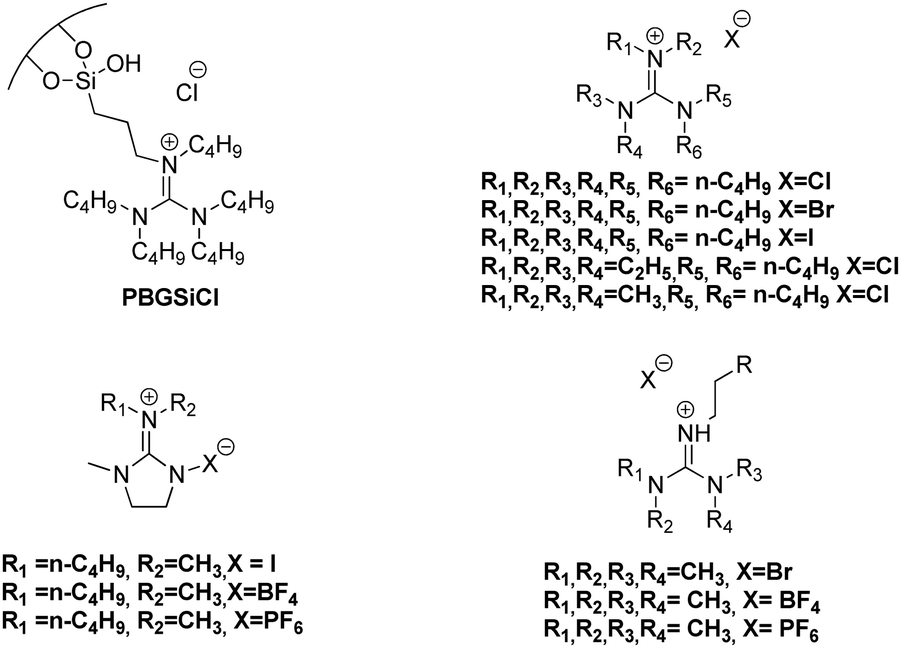 | ||
| Fig. 1 Bifunctional organocatalysts based on guanidinium salts for the CO2 fixation into cyclic carbonates. | ||
For this reason, and with the principal challenge to continue contributing to this area, herein, we describe the synthesis and characterization of a set of novel and effective aromatic mono- and bis(guanidinium)iodides, which are excellent hydrogen-bond catalysts for the synthesis of cyclic carbonates without the utilization of a co-catalyst. Furthermore, research on the reaction intermediate formation has been carried out through electrospray ionization (ESI) experiments.
Results and discussion
Synthesis and catalytic activity of guanidinium salts
The synthesis of the guanidinium iodide salts 1a–d and (bis)guanidinium diiodide 1e–j was carried out employing an acid–base reaction between mono-and (bis)guanidines77,82,83 and 1 or 2 equivalents of hydroiodic acid, obtaining the corresponding salts in quantitative yields (Scheme 2, See ESI† for details).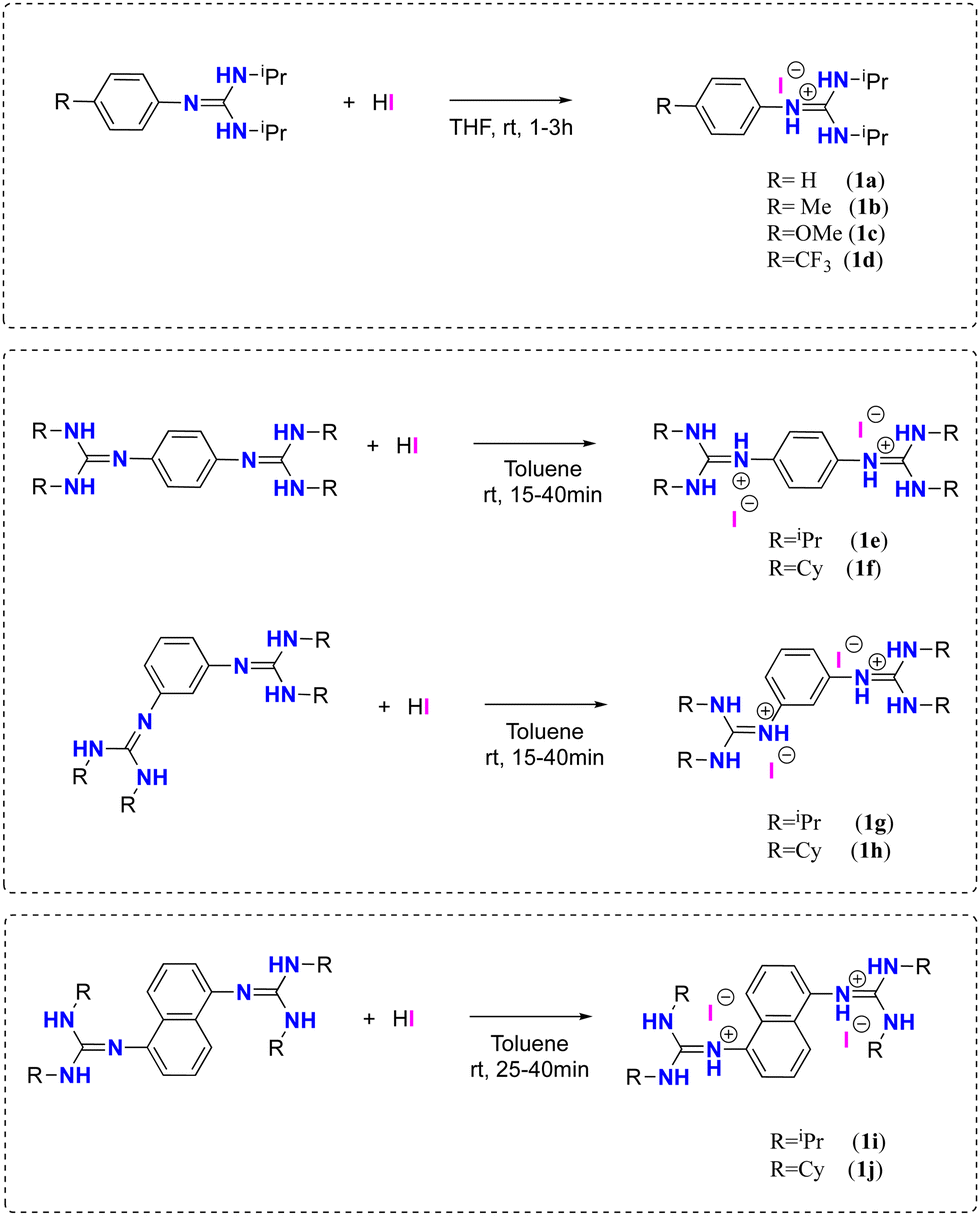 | ||
| Scheme 2 Synthesis of the guanidinium iodide salts 1a–d and (bis)guanidinium diiodide 1e–j. | ||
Through slow crystallization from acetonitrile, it was possible to obtain two single crystals suitable for X-ray crystal structural analysis. The XP drawings for compounds 1e and 1j are shown in Fig. 2. The structures obtained were consistent with the performed spectroscopic and elemental analyses. The structures confirmed the presence of iodine bound by an ionic bond whose average distances is around 3.73 Å for 1e and 3.72 Å for 1j. For compound 1e in the solid state, the formation of a complex 3D network was observed, with the iodine anions involved in three N–H hydrogen bonds (two N–H groups from the same guanidine unit and one N–H group from a neighboring molecule) (see the ESI,† Fig. S50 and Table S2, Page S36 and S37).
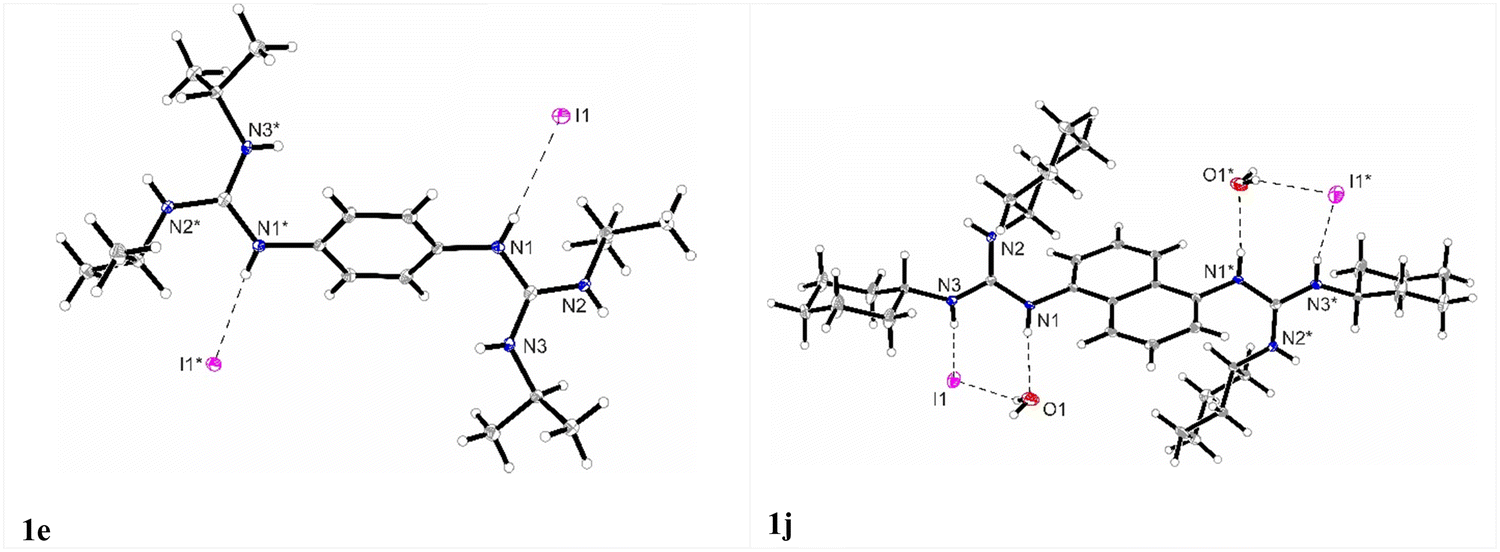 | ||
| Fig. 2 XP drawings of 1e and 1j at 100 K. Displacement ellipsoids are drawn at the 50% probability level. | ||
For compound 1j an additional water molecule co-crystallized in the asymmetric unit. A dimeric-type structure was formed involving hydrogen bonds between the two N–H groups, two iodine anions, and two water molecules. The two iodine anions are bridged through the water molecules and interact each with two neighbouring N–H groups (see the ESI,† Fig. S52, Table S3, Page S38). While the two N–H groups of the guanidine unit are involved in hydrogen bonds with two different iodine anions, being responsible for the building of a 3D network, the third N–H group stabilized the water molecule.
The N–H protons were identified in the 1H-NMR spectra as broad signals and showed a noticeable shift towards low fields, indicating the presence of a proton with a higher degree of acidity than those corresponding to starting guanidines (See ESI†).
Having prepared the different 1a–j guanidinium salts and with the evidence that mono and (bis)guanidines possess catalytic activity under mild reaction conditions of pressure and temperature,74 we decided to explore the catalytic activity of these new salts as one-component systems for the preparation of cyclic carbonates from CO2 and a range of epoxides. First, the ability of these salts 1a–j to transform styrene oxide (2a) into styrene carbonate (3a) was chosen to optimize the reaction conditions. The reactions were carried out at 70 °C and 1 bar of constant pressure of CO2 in the absence of the solvent for 18 h, using 2 mol% of guanidinium iodide salts 1a–d or 1 mol% of diiodide salts of (bis)guanidinium 1e–j as catalysts, keeping the number of N–H bonds constant, considering these bonds as the active centers for the synthesis of cyclic carbonates. It is worth mentioning that the studied guanidinium salts have shown a high solubility in styrene oxide (2a) at 70 °C. The reaction conversions were determined by 1H-NMR and the respective results are shown in Table 1.
|
|
||||
|---|---|---|---|---|
| Entry | Guanidinium salt | Conversion (%)b | TONc | TOF (h−1)d |
| a Reactions were carried out at 70 °C and 1 bar CO2 pressure for 18 h using 2 mol% of guanidinium iodide salts 1a–d or 1 mol% (bis)guanidinium iodide salts under solvent-free conditions unless otherwise specified. b Determined by 1H NMR spectroscopy of the crude reaction mixture. c TON = moles of product/moles of catalyst. d TOF = TON/time (h). | ||||
| 1 | 1a | 45 | 23 | 1.27 |
| 2 | 1b | 40 | 20 | 1.11 |
| 3 | 1c | 37 | 19 | 1.06 |
| 4 | 1d | 0 | 0 | 0 |
| 5 | 1e | 92 | 92 | 5.11 |
| 6 | 1f | 48 | 48 | 2.67 |
| 7 | 1g | 62 | 62 | 3.44 |
| 8 | 1h | 53 | 53 | 2.94 |
| 9 | 1i | 79 | 79 | 4.39 |
| 10 | 1j | 54 | 54 | 3.00 |
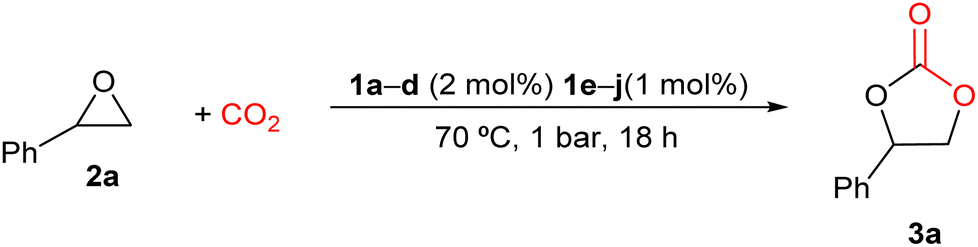
Meanwhile, compounds 1a–c showed lower reaction conversions (37–41%), and the guanidinium salt 1d was not active for the synthesis of styrene carbonate 3a (see Table 1, entries 1–4).
On the other hand, (bis)guanidinium diiodide salts 1e–j were found to be more active than guanidinium iodide salts in this catalytic process (Table 1, entries 5–10). In general, the salts with substituted iPr groups (1e, 1g, and 1i) displayed higher catalytic activity than the salts with the Cy groups. This was attributed to the different steric bulkiness of the involved groups, which would affect the bond with the epoxide. Among all the salts that were tested, the (bis)guanidinium diiodide salt 1e was selected for further catalytic studies, since it presented the highest conversion towards the formation of styrene carbonate with a 92% conversion and a TOF of 5.11 h−1 (Table 1, entry 5).
Therefore, 1e was used to obtain different cyclic carbonates from CO2 and different epoxides under mild conditions of pressure (1 bar CO2) and temperature (70 °C), for 18 h. As shown in Fig. 3, this catalytic system under study has an evident synthetic potential as an organocatalyst, being highly active toward the formation of cyclic carbonates starting from terminal epoxides with different functional groups.
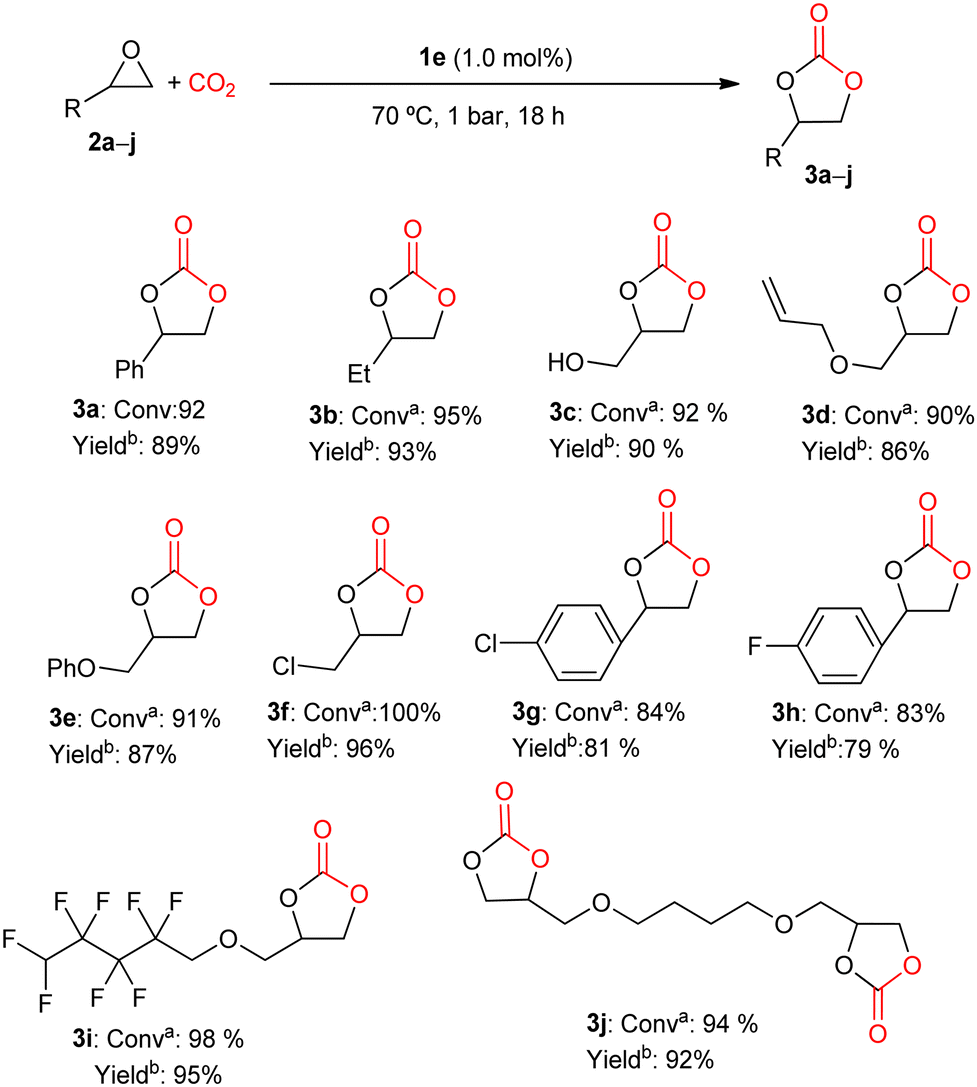 | ||
Fig. 3 Cyclic carbonates synthesis. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Conversion was determined by 1H NMR spectroscopy of the crude reaction mixture. bIsolated yield obtained from purified cyclic carbonate. Conversion was determined by 1H NMR spectroscopy of the crude reaction mixture. bIsolated yield obtained from purified cyclic carbonate. | ||
Following that, we decided to consider this one-component system's behavior as a more difficult issue while looking into its catalytic activity toward internal epoxides. It is important to highlight that organocatalyst 1e was able to prepare cis-1,2-cyclohexene carbonate (5a) and cis-1,2-cyclopentene carbonate (5b) from their corresponding epoxides 4a and 4b in exceptional yields, 82% and 80%, respectively (Fig. 4) using 2 mol% of catalyst loading at 85 °C and 10 bar of CO2 pressure for 24 h under solvent-free conditions.
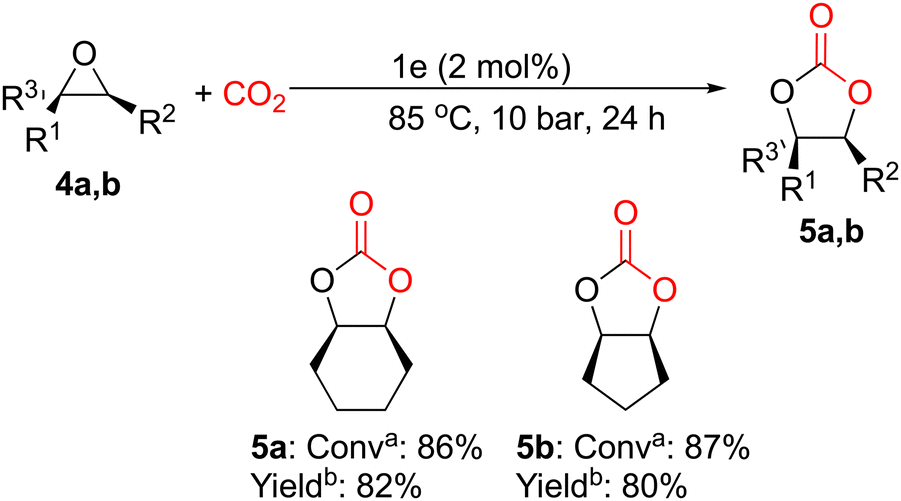 | ||
| Fig. 4
a
Conversion was determined by 1H NMR spectroscopy of the crude reaction mixture. bIsolated yield obtained from the purified cyclic carbonate. | ||
Electrospray ionization mass spectrometry (ESI-MS) reaction studies
Electrospray ionization mass spectrometry (ESI-MS) was applied to identify the active species,84–86 which could be involved in the model catalytic formation of 4-(chloromethyl)-1,3-dioxolan-2-one using bisguanidinium 1e as the catalyst (Scheme 3). The employed catalytic setup is depicted in Fig. 5.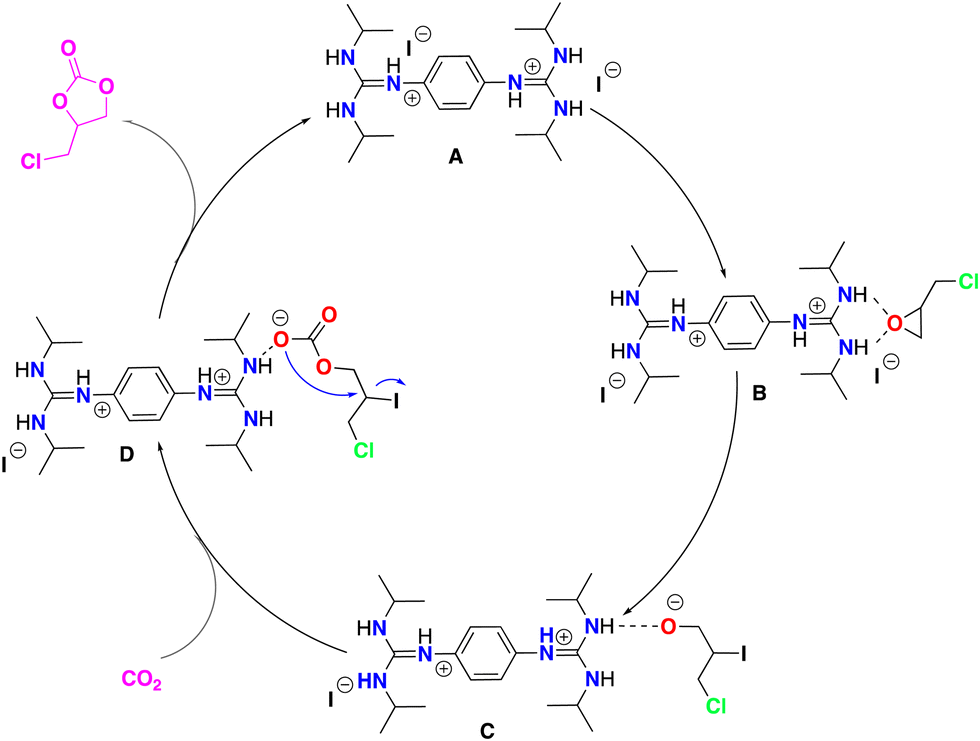 | ||
| Scheme 3 Plausible mechanism for the synthesis of 1,3-dioxolan-2-one catalyzed by 1e. | ||
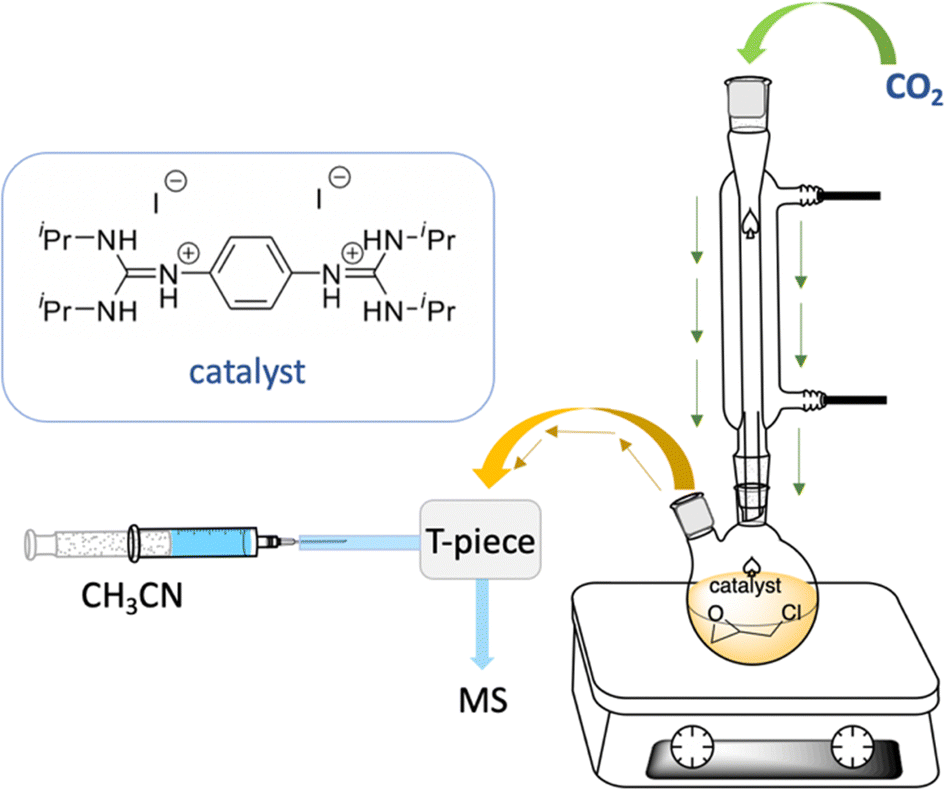 | ||
| Fig. 5 ESI-MS experimental setup. | ||
In short, 4.6 mg of the bisguanidinium catalyst (7.9 mmol) was mixed in 20.0 mL of commercial epichlorohydrin in a reactional flask at 70–80 °C, as shown in Fig. 5. CO2 gas (1–3 psi) was purged using a Peek tubing, and a second Peek tubing allowed the transfer of the reaction mixture directly to the ESI-MS ion source. The reaction solution exceeded the concentration range permissible for ESI-MS analyses. Therefore, it was diluted with acetonitrile prior to conducting the experiments. On-line dilution of the solution with acetonitrile using a microreactor resulted in a clean spectrum, which afforded cationic species that were easily identified and allowed the online continuous flow analysis of the reaction mixture. Fig. 6 displays clean MS spectra of the cationic species identified during the monitoring of the first 3 h of the reaction.
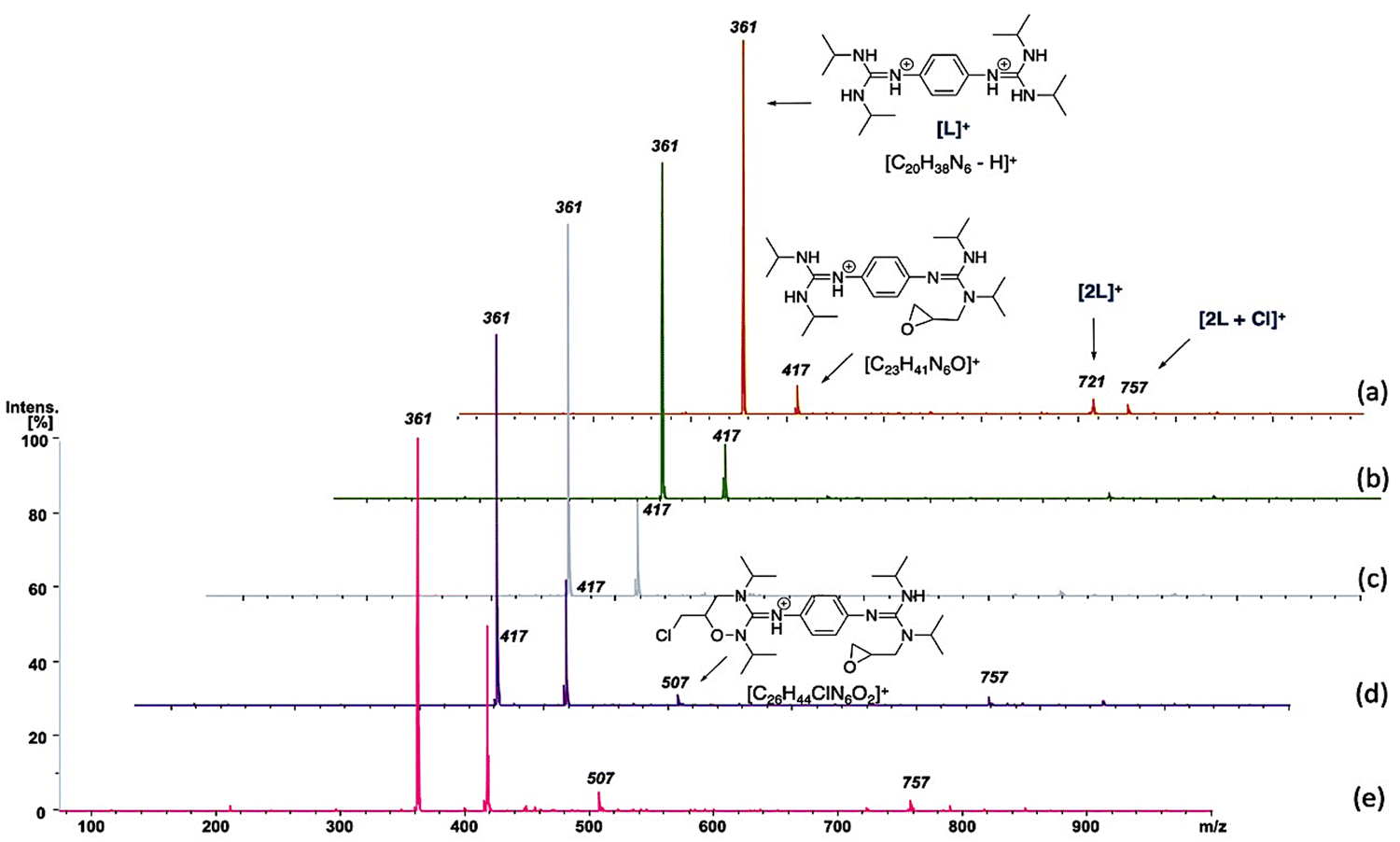 | ||
| Fig. 6 In situ ESI-MS monitoring. (a), (b), (c), (d), and (e) represent 3, 57, 99, 118 and 147 min, respectively, after the onset of the reaction. | ||
At the beginning of the reaction (3 min), we observed the presence of the catalyst ([C20H38N6-H]+) of m/z 361, which was present throughout the course of the reaction. In addition, we detected a dimer of cations [2L]+ of m/z 721, which corresponded to two catalyst units, as well as the species [2L+Cl]+ of m/z 757. Interestingly, from the first minutes of the reaction, the cation [C23H41N6O]+ of m/z 417 was observed, which probably corresponded to the species B (see Scheme 3), where the catalyst interacts with epichlorohydrin and, surprisingly, as the reaction time passes, the concentration of this species increases. Similar interactions of amines and their derivatives have been previously reported.87–89
Finally, at 118 and 147 minutes of the reaction, the formation of the cationic species [C26H44ClN6O2]+ of m/z 507, responsible for the epichlorohydrin opening (from the B to C step in Scheme 3) was observed. It is worth mentioning that no species with inserted CO2 were detected, probably because this step is very fast. However, while the rate-limiting step of the reaction is probably the opening of epichlorohydrin with nucleophilic attack of I-, the alkoxide intermediate species of m/z 507 was undoubtedly observed after 118 min. The alkoxide intermediate is postulated as the main species that suffers from CO2 insertion to achieve the final product in this class of organocatalyzed reactions. Although the catalytic species shown in Scheme 3 are not directly visible during the monitoring of the reaction by ESI-MS, we believe that the derivatives that we can see by monitoring are analogous to the coordination and opening species of the epoxide.
Conclusions
In this study, we reported new bisguanidium salt-based one-component catalysts that exhibited good activity in the synthesis of the cyclic carbonates from various epoxides and CO2 under conditions of moderate temperature (70–85 °C) and CO2 pressure (1–10 bar). It is important to note that of the new organocatalysts, the one with isopropyl groups in its structure exhibits the best performance. Since this structure has less steric restriction than bisguanidines with a cyclohexyl-type substituent, it allowed for more effective coordination of the epoxide during its coordination, which is the first step of the catalytic cycle. The reaction was studied by ESI-MS, and although the catalytic species that we believe were formed during the catalytic cycle, they were not directly observed; some derivatives such as [C26H44ClN6O2]+ of m/z 507, responsible for the epichlorohydrin opening and the cation [C23H41N6O]+ of m/z 417, which probably corresponds to the species B where the catalyst is interacting with epichlorohydrin could be observed. Our further studies will concentrate on various uses of these novel organocatalysts.Author contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. M.-S. is grateful for FONDECYT Postdoctoral fellowship 3230173. O. S. T. is grateful for FONDECYT Regular fellowship 1220241. J. M. is grateful for FONDECYT Iniciación fellowship 11230124. F. M. N. thanks FONDECYT Regular fellowship 1210576. L.S.S. thanks FONDECYT Regular fellowship 1220618. R. S. R. thanks FONDECYT Regular fellowship 1200748 and 1230537. F. C.-H. and A. A. acknowledge financial support from MCIN/AEI/10.13039/501100011033 (Project numbers PID2020-117353GB-I00 and RED2022-134287-T) and the Universidad Castilla-La Mancha (Project number 2022-GRIN-34031).References
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS.
- J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2018, 118, 434–504 CrossRef CAS.
- P. Tyagi, D. Singh, N. Malik, S. Kumar and R. Singh Malik, Mater. Today, 2023, 65, 133–165 CrossRef CAS.
- T. Yan, H. Liu, Z. X. Zeng and W. G. Pan, J. CO2 Util., 2023, 68, 102355 CrossRef CAS.
- L. F. Bobadilla, L. Azancot, L. A. Luque-Álvarez, G. Torres-Sempere, M. González-Castaño, L. Pastor-Pérez, J. Yu, T. Ramírez-Reina, S. Ivanova, M. A. Centeno and J. A. Odriozola, Chemistry, 2022, 4, 1250–1280 CrossRef CAS.
- M. Andérez-Fernández, E. Pérez, S. Ferrero, C. M. Álvarez, J. Gumiel, Á. Martín and M. D. Bermejo, Chem. Eng. J., 2023, 453, 139741 CrossRef.
- D. M. Fernandes, A. F. Peixoto and C. Freire, Dalton Trans., 2019, 48, 13508–13528 RSC.
- L.-Q. Qiu, X. Yao, Y.-K. Zhang, H.-R. Li and L.-N. He, J. Org. Chem., 2023, 88, 4942–4964 CrossRef CAS.
- Y. A. Alli, P. O. Oladoye, O. Ejeromedoghene, O. M. Bankole, O. A. Alimi, E. O. Omotola, C. A. Olanrewaju, K. Philippot, A. S. Adeleye and A. S. Ogunlaja, Sci. Total Environ., 2023, 868, 161547 CrossRef CAS.
- K. Wang, M. Cheng, F. Xia, N. Cao, F. Zhang, W. Ni, X. Yue, K. Yan, Y. He, Y. Shi, W. Dai and P. Xie, Small, 2023, 19, 2207581 CrossRef CAS.
- X. Li, J. Benet-Buchholz, E. C. Escudero-Adán and A. W. Kleij, Angew. Chem., Int. Ed., 2023, 62, e202217803 CrossRef CAS.
- K. Takeuchi, K. Matsumoto, N. Fukaya, K. Osakada, K. Sato and J.-C. Choi, Dalton Trans., 2022, 51, 15631–15643 RSC.
- J. Zhou, S. Yang, W. Wan, L. Chen and J. Chen, J. Catal., 2023, 418, 178–189 CrossRef CAS.
- T. Miah, P. Demoro, I. Nduka, F. De Luca, S. Abate and R. Arrigo, Chem. Phys. Chem., 2023, 24, e202200589 CrossRef CAS PubMed.
- N. Yusuf, F. Almomani and H. Qiblawey, Fuel, 2023, 345, 128178 CrossRef CAS.
- H. Niu, L. Lu, R. Shi, C.-W. Chiang and A. Lei, Chem. Commun., 2017, 53, 1148–1151 RSC.
- A. Otto, T. Grube, S. Schiebahn and D. Stolten, Energy Environ. Sci., 2015, 8, 3283–3297 RSC.
- Y. Wang, Z. Yan, J. Deng and G. Luo, Sep. Purif. Technol., 2023, 317, 123872 CrossRef CAS.
- B. Dziejarski, R. Krzyżyńska and K. Andersson, Fuel, 2023, 342, 127776 CrossRef CAS.
- J. Boualavong, K. G. Papakonstantinou and C. A. Gorski, Chem. Eng. Sci., 2023, 274, 118673 CrossRef CAS.
- A. J. Kamphuis, F. Picchioni and P. P. Pescarmona, Green Chem., 2019, 21, 406–448 RSC.
- S. Bhattacharjee, S. Chongdar, A. Modak, P. Bhanja, B. K. Jena and A. Bhaumik, Green Chem., 2022, 24, 8853–8862 RSC.
- A. Chowdhury, S. Bhattacharjee, R. Chatterjee and A. Bhaumik, J. CO2 Util., 2022, 65, 102236 CrossRef CAS.
- A. Rehman, F. Saleem, F. Javed, A. Ikhlaq, S. W. Ahmad and A. Harvey, J. Environ. Chem. Eng., 2021, 9, 105113 CrossRef CAS.
- A. Kesküla, A.-L. Peikolainen, P. A. Kilmartin and R. Kiefer, Polymers, 2021, 13, 3466 CrossRef.
- A. V. Parfeneva, A. M. Rumyantsev, D. A. Lozhkina, M. Y. Maximov and E. V. Astrova, Batteries, 2022, 8, 91 CrossRef CAS.
- Y. Fernandes, A. Bry and S. De Persis, J. Power Sources, 2019, 414, 250–261 CrossRef CAS.
- V. Aomchad, S. Del Gobbo, P. Yingcharoen, A. Poater and V. D’Elia, Catal. Today, 2021, 375, 324–334 CrossRef CAS.
- J. Kiriratnikom, N. Laiwattanapaisarn, K. Vongnam, N. Thavornsin, P. Sae-Ung, S. Kaeothip, A. Euapermkiati, S. Namuangruk and K. Phomphrai, Inorg. Chem., 2021, 60, 6147–6151 CrossRef CAS.
- A. Vidal-López, S. Posada-Pérez, M. Solà, V. D’Elia and A. Poater, Green Chem. Eng., 2022, 3, 180–187 CrossRef.
- H. Fish, S. Hart, K. J. Lamb, M. North, S. C. Z. Quek, A. C. Whitwood, B. Woods and X. Wu, Dalton Trans., 2021, 50, 587–598 RSC.
- J. Liu, G. Yang, Y. Liu, D. Zhang, X. Hu and Z. Zhang, Green Chem., 2020, 22, 4509–4515 RSC.
- F. Della Monica, B. Maity, T. Pehl, A. Buonerba, A. De Nisi, M. Monari, A. Grassi, B. Rieger, L. Cavallo and C. Capacchione, ACS Catal., 2018, 8, 6882–6893 CrossRef CAS.
- Y. Xu, D. Yuan, Y. Wang and Y. Yao, Dalton Trans., 2017, 46, 5848–5855 RSC.
- H. Ullah, B. Mousavi, H. A. Younus, Z. A. K. Khattak, S. Suleman, M. T. Jan, B. Yu, S. Chaemchuen and F. Verpoort, J. Catal., 2019, 377, 190–198 CrossRef CAS.
- R. Luo, W. Zhang, Z. Yang, X. Zhou and H. Ji, J. CO2 Util., 2017, 19, 257–265 CrossRef CAS.
- R. E. Barker, L. Guo, C. J. A. Mota, M. North, L. P. Ozorio, W. Pointer, S. Walberton and X. Wu, J. Org. Chem., 2022, 87, 16410–16423 CrossRef CAS.
- Y. Kang, B. Wang, R. Nan, Y. Li, Z. Zhu and X.-Q. Xiao, Inorg. Chem., 2022, 61, 8806–8814 CrossRef CAS PubMed.
- L.-N. Ma, L. Zhang, W.-F. Zhang, Z.-H. Wang, L. Hou and Y.-Y. Wang, Inorg. Chem., 2022, 61, 2679–2685 CrossRef CAS PubMed.
- Y.-H. Zou, Y.-B. Huang, D.-H. Si, Q. Yin, Q.-J. Wu, Z. Weng and R. Cao, Angew. Chem., Int. Ed., 2021, 60, 20915–20920 CrossRef CAS.
- Q.-J. Wu, J. Liang, Y.-B. Huang and R. Cao, Acc. Chem. Res., 2022, 55, 2978–2997 CrossRef CAS.
- T. K. Pal, D. De and P. K. Bharadwaj, Fuel, 2022, 320, 123904 CrossRef CAS.
- Y. Qu, Y. Chen and J. Sun, J. CO2 Util., 2022, 56, 101840 CrossRef CAS.
- Y. Fu, Y. Xu, Z. Zeng, A.-R. Ibrahim, J. Yang, S. Yang, Y. Xie, Y. Hong, Y. Su, H. Wang, Y. Wang, L. Peng, J. Li and W. L. Queen, Green Energy Environ., 2023, 8, 478–486 CrossRef CAS.
- Á. F. Arruda da Mata, N. Glanzmann, P. H. Fazza Stroppa, F. Terra Martins, R. P. das Chagas, A. D. da Silva and J. L. S. Milani, New J. Chem., 2022, 46, 12237–12243 RSC.
- N. Liu, Y.-F. Xie, C. Wang, S.-J. Li, D. Wei, M. Li and B. Dai, ACS Catal., 2018, 8, 9945–9957 CrossRef CAS.
- K. Zhang, Z. Liu and N. Liu, Appl. Organomet. Chem., 2021, 35, e6347 CrossRef CAS.
- Y. Hu, Z. Wei, A. Frey, C. Kubis, C.-Y. Ren, A. Spannenberg, H. Jiao and T. Werner, ChemSusChem, 2021, 14, 363–372 CrossRef CAS.
- A. F. Eftaiha, A. K. Qaroush, A. K. Hasan, W. Helal and F. M. Al-Qaisi, React. Chem. Eng., 2022, 7, 1807–1817 RSC.
- J. Steinbauer, C. Kubis, R. Ludwig and T. Werner, ACS Sustainable Chem. Eng., 2018, 6, 10778–10788 CrossRef CAS.
- C. Ren, A. Spannenberg and T. Werner, Asian J. Org. Chem., 2022, 11, e202200156 CrossRef CAS.
- S. E. Lyubimov, P. V. Cherkasova and B. Chowdhury, Russ. Chem. Bull., 2022, 71, 404–407 CrossRef CAS.
- Q. Yi, T. Liu, X. Wang, Y. Shan, X. Li, M. Ding, L. Shi, H. Zeng and Y. Wu, Appl. Catal., B, 2021, 283, 119620 CrossRef CAS.
- D. Kim, S. Subramanian, D. Thirion, Y. Song, A. Jamal, M. S. Otaibi and C. T. Yavuz, Catal. Today, 2020, 356, 527–534 CrossRef CAS.
- W. Ding, J. Qiao, L. Zeng, H. Sun and T. Peng, Int. J. Greenhouse Gas Control, 2023, 123, 103843 CrossRef CAS.
- M. Liu, X. Wang, Y. Jiang, J. Sun and M. Arai, Catal. Rev., 2019, 61, 214–269 CrossRef CAS.
- W. Natongchai, S. Pornpraprom and V. D’Elia, Asian J. Org. Chem., 2020, 9, 801–810 CrossRef CAS.
- M. Alves, B. Grignard, A. Boyaval, R. Méreau, J. De Winter, P. Gerbaux, C. Detrembleur, T. Tassaing and C. Jérôme, ChemSusChem, 2017, 10, 1128–1138 CrossRef CAS.
- A. M. Hardman-Baldwin and A. E. Mattson, ChemSusChem, 2014, 7, 3275–3278 CrossRef CAS PubMed.
- A. Rostami, M. Mahmoodabadi, A. Hossein Ebrahimi, H. Khosravi and A. Al-Harrasi, ChemSusChem, 2018, 11, 4262–4268 CrossRef CAS.
- J. Martínez, F. De La Cruz-Martínez, M. M. De Sarasa Buchaca, M. P. Caballero, R. M. Ojeda-Amador, M. D. Salvador, G. Fregapane, J. Tejeda, J. A. Castro-Osma and A. Lara-Sánchez, J. Environ. Chem. Eng., 2021, 9, 105464 CrossRef.
- J. A. Castro-Osma, J. Martínez, F. de la Cruz-Martínez, M. P. Caballero, J. Fernández-Baeza, J. Rodríguez-López, A. Otero, A. Lara-Sánchez and J. Tejeda, Catal. Sci. Technol., 2018, 8, 1981–1987 RSC.
- J. Catalá, M. P. Caballero, F. De La Cruz-Martínez, J. Tejeda, J. A. Castro-Osma, A. Lara-Sánchez, J. M. García-Vargas, M. T. García, M. J. Ramos, I. Gracia and J. F. Rodríguez, J. CO2 Util., 2022, 61, 102060 CrossRef.
- P. Yingcharoen, C. Kongtes, S. Arayachukiat, K. Suvarnapunya, S. V. C. Vummaleti, S. Wannakao, L. Cavallo, A. Poater and V. D’Elia, Adv. Synth. Catal., 2019, 361, 366–373 CrossRef CAS.
- T. Werner and H. Büttner, ChemSusChem, 2014, 7, 3268–3271 CrossRef CAS.
- H. Büttner, J. Steinbauer and T. Werner, ChemSusChem, 2015, 8, 2655–2669 CrossRef.
- Y. Yang, Y. Guo, C. Gao, M. North, J. Yuan, H. Xie and Q. Zheng, ACS Sustainable Chem. Eng., 2023, 11, 2634–2646 CrossRef CAS.
- S. Arayachukiat, C. Kongtes, A. Barthel, S. V. C. Vummaleti, A. Poater, S. Wannakao, L. Cavallo and V. D’Elia, ACS Sustainable Chem. Eng., 2017, 5, 6392–6397 CrossRef CAS.
- X. Zhang, Y.-B. Jia, X.-B. Lu, B. Li, H. Wang and L.-C. Sun, Tetrahedron Lett., 2008, 49, 6589–6592 CrossRef CAS.
- X. Zhang, N. Zhao, W. Wei and Y. Sun, Catal. Today, 2006, 115, 102–106 CrossRef CAS.
- E. M. Maya, E. Rangel-Rangel, U. Díaz and M. Iglesias, J. CO2 Util., 2018, 25, 170–179 CrossRef CAS.
- D. Wei-Li, J. Bi, L. Sheng-Lian, L. Xu-Biao, T. Xin-Man and A. Chak-Tong, J. Mol. Catal. A: Chem., 2013, 378, 326–332 CrossRef.
- A. Chen, C. Chen, Y. Xiu, X. Liu, J. Chen, L. Guo, R. Zhang and Z. Hou, Green Chem., 2015, 17, 1842–1852 RSC.
- B. Liu, M. Liu, L. Liang and J. Sun, Catalysts, 2015, 5, 119–130 CrossRef.
- K. M. K. Yu, I. Curcic, J. Gabriel, H. Morganstewart and S. C. Tsang, J. Phys. Chem. A, 2010, 114, 3863–3872 CrossRef CAS.
- R. D. do E. Santo, R. M. Capitao and E. R. P. González, Guanidines as Reagents Catal. II, 2017, pp. 27–74 Search PubMed.
- Á. Mesías-Salazar, J. Martínez, R. S. Rojas, F. Carrillo-Hermosilla, A. Ramos, R. Fernández-Galán and A. Antiñolo, Catal. Sci. Technol., 2019, 9, 3879–3886 RSC.
- N. Aoyagi, Y. Furusho and T. Endo, Synthesis, 2020, 150–158 CAS.
- H. Seo and H. Kim, Bull. Korean Chem. Soc., 2019, 40, 169–172 CrossRef CAS.
- H. Xie, H. Duan, S. Li and S. Zhang, New J. Chem., 2005, 29, 1199–1203 RSC.
- X.-Y. Dou, J.-Q. Wang, Y. Du, E. Wang and L.-N. He, Synlett, 2007, 3058–3062 CAS.
- F. Werlinger, R. Caprile, V. Cárdenas-Toledo, B. Tarraff, Á. Mesías-Salazar, R. S. Rojas, J. Martínez, O. S. Trofymchuk and M. E. Flores, ACS Omega, 2023, 8, 21540–21548 CrossRef CAS PubMed.
- A. Mesias-Salazar, O. S. Trofymchuk, C. G. Daniliuc, A. Antiñolo, F. Carrillo-Hermosilla, F. M. Nachtigall, L. S. Santos and R. S. Rojas, J. Catal., 2020, 382, 150–154 CrossRef CAS.
- L. S. Santos and J. O. Metzger, Rapid Commun. Mass Spectrom., 2008, 22, 898–904 CrossRef CAS PubMed.
- R. Tokala, D. Bora, S. Sana, F. M. Nachtigall, L. S. Santos and N. Shankaraiah, J. Org. Chem., 2019, 84, 5504–5513 CrossRef CAS.
- L. S. Santos and J. O. Metzger, Angew. Chem., Int. Ed., 2006, 45, 977–981 CrossRef.
- D. B. G. Williams and A. Cullen, J. Org. Chem., 2009, 74, 9509–9512 CrossRef CAS.
- A. Simaioforidou, Y. Georgiou, A. Bourlinos and M. Louloudi, Polyhedron, 2018, 153, 41–50 CrossRef CAS.
- X. Wang, J. Yin and X. Wang, Macromolecules, 2011, 44, 6856–6867 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2267508 and 2267509. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3nj03959e |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |