
Revealing charge-shift bonds in H3+ and their metallic analogs M3+ (M = Li, Na, K) through electron density topology†
Ricardo
Pino-Rios
 *ab
*ab
aInstituto de Estudios de la Salud, Universidad Arturo Prat., Iquique, 1100000, Chile. E-mail: rpinorios@unap.cl
bQuímica y Farmacia, Facultad de Ciencias de la Salud, Universidad Arturo Prat., Casilla 121, Iquique 1100000, Chile
First published on 29th August 2023
Abstract
The bonding pattern of H3+ and its metallic isomers M3+ (M = Li, Na, and K) have been studied through the topological criterion by the quantum theory of atoms in molecules (QTAIM), interacting quantum atoms (IQA) and valence bond (VB) approach at the Coupled Cluster level. The results show that for the studied compounds, it is possible to state that they are not ring-type structures but structures in which the atoms are bound through electrostatic and covalent interactions (in that order of priority) to a non-nuclear attractor (NNA) located in the geometric center of the atomic positions. A series of resonance structures have been proposed following the VB model, including those of the covalent and ionic type with the participation of the NNA, which would explain the computed results. Additionally, a simple way of calculating the percentages of the contribution of each resonance structure using the delocalization and localization indexes is proposed. Calculated values show that the ionic resonance structures have a high contribution in the four studied systems, so it is possible to conclude that these compounds have charge-shift bonds.
Introduction
The H3+ cluster is the lightest of all atomic clusters. It was detected first by Sir J. J. Thomson 1911 in his report on experiments regarding positive rays of electricity.1 This small cluster is among the most abundant in the interstellar medium due to the ideal temperature and pressure conditions. However, it was not detected until 1989 by Drossart et al. while investigating Jupiter's ionosphere. Since then, it has been detected in different celestial environments.2–6 Another fascinating feature of this compound is its chemical bonding;7 according to previous studies, this compound is the simplest example of a molecule with a delocalized bond that also possesses aromaticity. The two electrons owned by H3+ indicate the existence of a three-center-two-electron bond (3c–2e) enabling compliance with Hückel's 4n + 2 rule,8 although this rule was strictly given for the π electron count. Havenith et al. have shown the existence of a diatropic ring current,9 a pattern present in aromatic rings. Negative values of NICS also support the ring current results.From the electron density topology view, the picture is very different. Some authors indicate that this compound is not even a ring.10,11 Reports of the topology of the electron density of H3+ show a maximum at the geometric center of hydrogen atom positions. This maximum is known as a non-nuclear attractor (NNA) in the context of Bader's Quantum Theory of Atoms in Molecules (QTAIM).12 Additionally, the absence of bond paths linking the hydrogen atoms and those connecting the NNA and hydrogen atoms supports the idea that there is no cyclic structure. Although it has been shown on several occasions that a bond path does not unequivocally express the existence of a chemical bond as traditionally known, it does indicate that there are specific attractive interactions or electronic exchanges between two electron density maxima.13
Some factors may contradict both explanations. First, the charges on H3+ are equally distributed on the hydrogen atoms since it is an equilateral triangle, so each one should have a charge of +0.33. It is plausible that the electrostatic repulsion is greater than the covalent contribution, so the ring becomes very unstable. Additionally, diatropic currents are also observed in atomic nuclei, which could explain the presence of a ring current in a cluster as small as H3+, and even the presence of a non-nuclear attractor could also account for the existence of the diatropic current shown by Havenith et al.9
The isovalent Li3+ cluster also presents differences concerning the nature of its bonding pattern. Since it has two valence electrons, this bond is expected to be delocalized and therefore present an aromatic character. However, magnetically induced current density calculations show that it does not exhibit diatropic ring current, typical of aromatic systems. Havenith described Li3+ as a non-aromatic σ-compound.9 Foroutan-Nejad et al. analyzing the topology of the electron density in Li3+ showed that there is no presence of covalent bonds but rather establish that this cluster is the smallest that presents metallic bonding. Regarding the analysis of the chemical bonding in heavier isovalent analogs Na3+ and K3+, no studies have been reported. However, the fact that H3+ and Li3+ are so similar but apparently present great differences in their bonding pattern makes the analysis of heavier isovalent clusters appealing.
This work aims to give new information regarding the chemical bonding pattern of these small but exciting systems. To achieve this, the quantum theory of atoms in molecules has been applied, and additionally, an energy decomposition analysis between the atoms that form the molecule has been carried out in order to gain insights into their ionic and covalent contributions by applying the interacting quantum atoms (IQA) method. This method allows a decomposition of the interaction energies between the atoms with the advantage that no arbitrary fragment selection is required. IQA has been recently reviewed by Pendás and coworkers showing the ability of this method to explain various chemical phenomena related to bonding.14
Computational methods
Geometry optimization of H3+ and heavier analogues has been carried out at the CCSD(T)15/def2-TZVP16 level using Gaussian 16 (G16) software.17 Vibrational frequencies indicate a minimum in the potential energy surface (NImag = 0). Due to the limitation of G16 to obtain the wfx file at the CCSD(T) level, single-point calculations were performed to obtain the wave function (wfx) file by using the density=current command at the CCSD/def2-TZVP level. Topological analysis of the electron density and interacting quantum atoms calculations have been performed using the AIMAll program.18The gradient, in conjunction with the Laplacian of the electron density, allows to identify and characterize critical points (CPs) that permit an analysis of the chemical bond pattern through its topology. First of all, it is possible to identify density maxima of the (3, −3) which are typically located at the atomic positions. These points are known as nuclear attractors (NACP). However, the maxima can also be found in areas that do not coincide with the positions of the atoms; these critical points are known as non-nuclear attractors (NNACP). Additionally, it is possible to identify first-order saddle points in the electron density. These (3, −1) critical points are known as bond critical points (BCPs) and provide information about the interaction between two maxima (either a nuclear or non-nuclear attractor). It is possible to determine the binding between two maxima through bond paths, which link BCPs and NACPs (or NNACPs) through the maximum electron density. Finally, the second-order saddle points and minima of the electron density are known as ring (RCP) and cage critical points (CCP), which appear in the center when electron density maxima are linked through the ring- or three-dimensional cage-shaped bond paths. In addition, delocalization and localization indexes are computed. The first provides information of electrons shared between two atoms (or NNAs), and the second indicates the electrons located in a given atom (or NNA).
The interacting quantum atoms method allows a non-arbitrary decomposition of the interaction energy between two atoms based on the electron density topology. It allows the acquisition of coulombic (related to classical electrostatic interactions) and exchange contributions (related to quantum and covalent effects). More technical details about QTAIM and IQA can be found in the famous Bader book12 and the recent review by Pendás and coworkers,14 respectively.
Results and discussion
Scheme 1 shows the molecular graph obtained at the CCSD/def2-TZVP level. As previously reported. Four maxima (3, −3) of the electron density (red dots) are found, three located at the positions of the hydrogen atoms and one at the geometrical center. There are also saddle points between the NNA and the hydrogen atoms. These points, known as BCPs (blue dots) according to QTAIM, indicate interaction between two maxima. Finally, the black lines indicate the bond paths detected for H3+.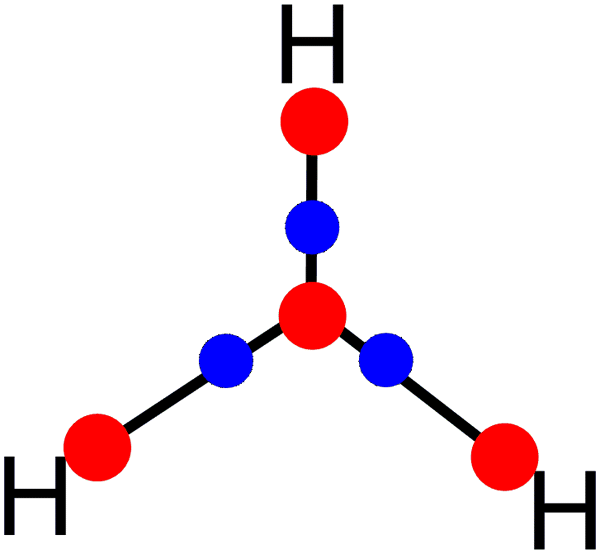 | ||
| Scheme 1 The QTAIM molecular graph of H3+ obtained at the CCSD/def2-TZVP level. | ||
The results shown are similar to others reported previously.11 Additionally, the delocalization/localization (DI/LI) indexes and charges for H3+ have been computed. Table 1 shows that the charges on the hydrogen atoms are positive. However, they are higher than the expected value of +0.33 because the NNA has a negative charge (−0.1230), which (from an electrostatic point of view) would hold the atoms together. Additionally, the LI values indicate a much lower electron localization in the NNA than for the hydrogen atoms. Complementarily, the DI values indicate how the electrons are distributed between two atoms (or NNA, in this case). It is possible to observe a delocalization of 0.3377 a.u. for H–H interactions, while for H–NNA, it is 0.0767 a.u.
| Basin | Charge | LI | DI | |
|---|---|---|---|---|
| H1 | 0.3743 | 0.2496 | H1–H2 | 0.3377 |
| H2 | 0.3743 | 0.2496 | H1–H3 | 0.3377 |
| H3 | 0.3743 | 0.2496 | H2–H3 | 0.3377 |
| NNA | −0.1230 | 0.0079 | H1–NNA | 0.0767 |
| — | H2–NNA | 0.0767 | ||
| — | H3–NNA | 0.0767 | ||
According to the calculated charges, the appropriate bonding pattern is the one observed in Scheme 2a, while the notoriously higher DI values and lower localization values indicate that the best pattern is described by Scheme 2b. So, what is the most appropriate description for this compound?
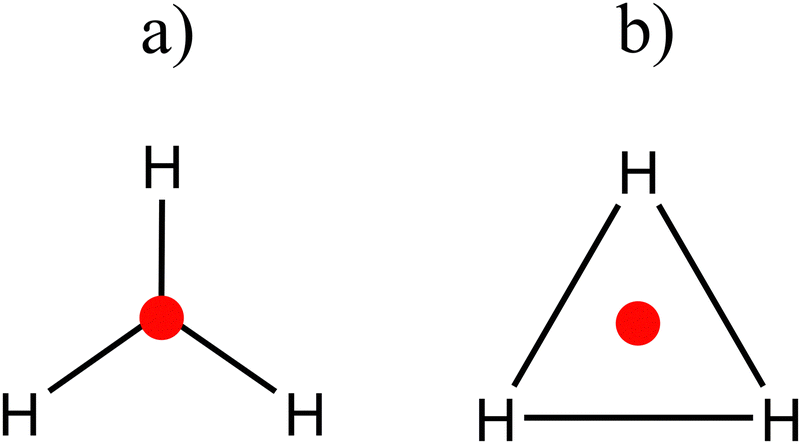 | ||
| Scheme 2 Description of the possible chemical bonding patterns for H3+ according to the results of Table 1. | ||
To have more detail regarding the contributions in the interactions of two atoms, energy decomposition calculations were carried out under the IQA approach. Table 2 shows that the interaction energy between hydrogen atoms is completely repulsive (2.16 kcal mol−1). The absolute value of the exchange contribution (covalent) is lower than the repulsive (positive) coulombic (ionic) contribution. On the other hand, the H–NNA interactions are negative; even the ionic and covalent contributions contribute to the total interaction by 60% and 40%, respectively, showing that the coulombic contributions are notoriously more important.
| V IQA,Inter(A,B) | V C,IQA,Inter(A,B) | V X,IQA,Inter(A,B) | |
|---|---|---|---|
| H1–H2 | 2.16 | 63.72 | −61.56 |
| H1–H3 | 2.16 | 63.72 | −61.56 |
| H2–H3 | 2.16 | 63.72 | −61.56 |
| H1–NNA | −45.25 | −25.81 | −19.45 |
| H2–NNA | −45.25 | −25.81 | −19.45 |
| H3–NNA | −45.25 | −25.81 | −19.45 |
Results indicate that in order to define the bonding pattern in H3+, electrostatic and covalent interactions must be considered. This can be described using classical chemical criteria under the valence bond approach through resonance structures based on the delocalization/localization indicators19 shown in Scheme 3.
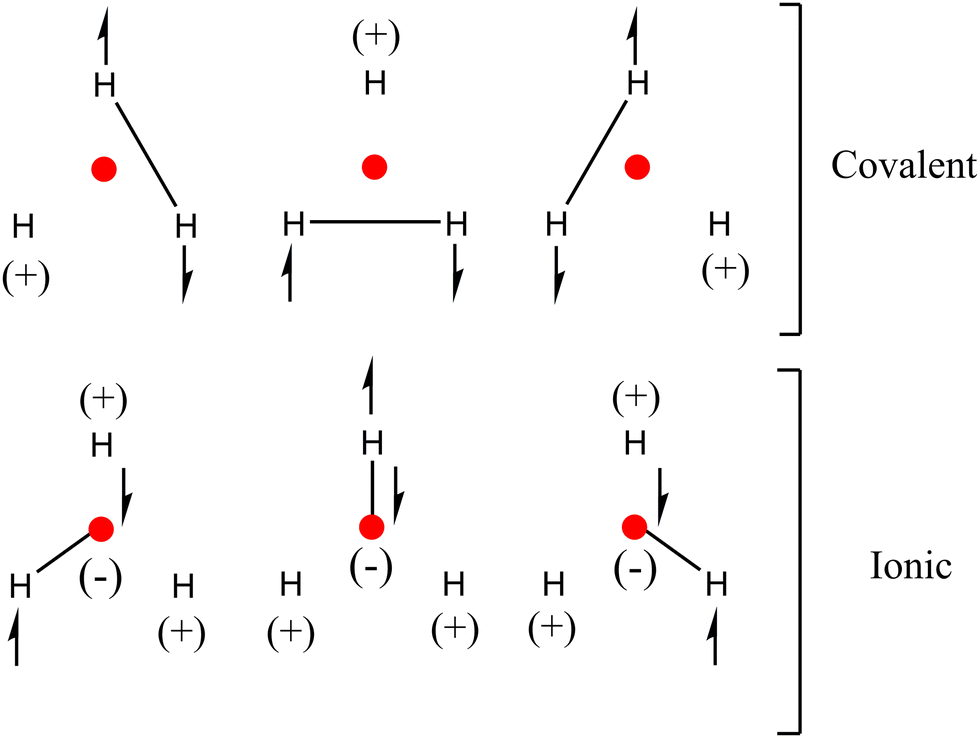 | ||
| Scheme 3 Proposed resonance structures for H3+ according to the presented QTAIM and IQA results. | ||
The covalent resonance structures describe the chemical bond between the H–H atoms (DI = 0.3377 a.u.), which are delocalized around the triangle formed by the hydrogens according to the DI values. However, the importance of the covalent H–NNA (DI = 0.0767 a.u. VIQA,Inter(A,B) = −19.45 kcal mol−1) interactions must also be considered, which, although they contribute to a lesser degree, are not negligible. The proposed structures also allow us to explain the charges in the atomic positions (including the NNA).
The delocalization and localization values must be considered for a complete bond analysis since DI + LI = # of total electrons. Scheme 4 shows fully ionic resonance structures (LIH = 0.2496; LINNA = 0.0079, in a.u.) with no covalent bonding but multiple ionic interactions. The contributions of these resonance structures would be small, the least contributing being the one where the electron pair is located in the NNA. The proposed structures not only allow us to understand the chemical bonding but also explain the diatropic current in the center of the triangle, which would be due to the localization of at least one electron. It is possible to verify this by observing the diatropic current of the hydrogen atom shown in Fig. S1 in the ESI.†
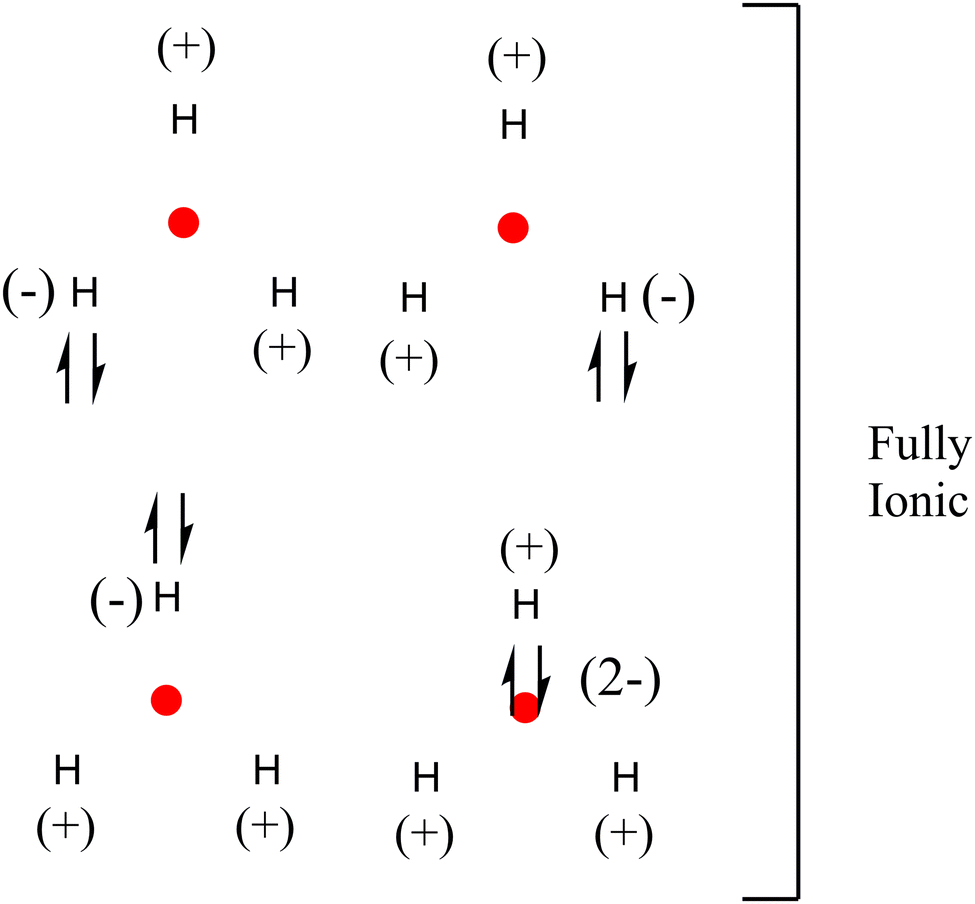 | ||
| Scheme 4 Proposed fully ionic resonance structures for H3+ according to the results obtained by QTAIM and IQA. | ||
When ionic resonance structures have important contributions for the description of chemical bonding, we have charge-shift bonds.20,21 This type of bond has been widely documented and allows us to explain the bonds in different molecules, for example, F2 and XeF2,22 among others.23 For this case, the contributions of all the resonance structures mentioned can be quantified using the DI and LI values. The DI and LI values can be divided by two electrons and multiplied by 100 to have a percentage value: covalent structures contribute 51% (17% each), while ionic structures contribute 12% (4% each). Finally, fully ionic resonance structures contribute a total of 37% (12.3% for those where the electron pair is located on each atom and 0.4% for those where the pair is located in the center of the triangle). The total sum of the ionic contributions is 49% which would confirm the charge-shift nature of the chemical bond.
The same analysis has been carried out for the metal analogs, obtaining remarkable results. First, the topology of the M3+ is identical to that of H3+ (see Fig. S2–S5, ESI†) with an NNA in the center of the triangle formed by the atoms bonded through bond paths. Additionally, the coulombic and exchange contributions show the same tendency as H3+, indicating that we are dealing with compounds that could possess charge-shift bonds. For this reason, we have calculated the contributions using the DI and LI values. However, in the case of heavier molecules, only the two valence electrons of each cluster have been used.
In the case of Li3+, the resonance structures that contribute the most are the ionic ones (see Table 3). It is very striking that the structure with three Li+ cations surrounding the electron pair located at the center of the triangle (fully ionic) is the structure that contributes the most (40.6%). Additionally, the ionic structure, where there are “covalent” bonds between the metal and the NNA, is in second place with 45.8%. As can be seen, the covalent contributions are meager. These results agree with those presented by Foroutan-Nejad et al., who indicated that covalent bonding in Li3+ does not exist. The best way to describe the chemical bonding pattern in this structure is through ionic resonance forms. Based on the calculation of ionic contributions, it is feasible to say that Li3+ consists of an electron pair surrounded by Li+ cations and not a ring structure.
| M3+ | Contributions (%) | |||
|---|---|---|---|---|
| Computed using delocalization index | Computed using localization index | |||
| Covalent | Ionic | Fully ionic | ||
| (M–M) × 3 | (NNA–M) × 3 | M+ M+ M− NNA | M+ M+ M+ NNA− | |
| H3+ | 50.7 | 11.5 | 37.4 | 0.4 |
| Li3+ | 6.2 | 45.8 | 7.5 | 40.6 |
| Na3+ | 16.6 | 47.3 | 18.0 | 18.3 |
| K3+ | 23.3 | 41.5 | 24.0 | 10.7 |
For Na3+ and K3+, the picture changes slightly. The fully ionic structures contribute up to 36%, while the contribution of the covalent structures is higher for K3+. In both cases, the structures in which the NNA is covalently bonded to the metal atoms are the major contributing structures. Due to the high contributions of the ionic and fully ionic structures, both compounds present charge-shift bonds and would be better described as non-cyclic structures.
Conclusions
The chemical bonding of the small H3+ cluster and its metallic analogous Li3+, Na3+, and K3+ has been described through topological analysis of the electron density, energy decomposition, and valence bond theory in the form of resonance structures proposed based on the numerical values obtained. The results show the necessity to consider covalent and ionic descriptions on these small systems. These contributions are 51% and 49% for H3+ according to the simple model using the delocalization and localization values obtained from the topological analysis. Fully ionic structures contribute 38% of the total, so the binding pattern in H3+ possesses a charge-shift nature. Additionally, the description of a charge-shift bond considers all the features presented in the description of the delocalized ring at 3c–2e and the one where the NNA holds the hydrogen nuclei together through electrostatic interactions proposed in the past.For heavier analogs, the description is somewhat different: in the case of Li3+, the ionic and fully ionic resonance structures are the major contributors, it being the case that the best way to describe the Li3+ bonding pattern is through an electron pair surrounded by Li+ cations. While for Na3+ and K3+, the results are very similar to each other. Covalent structures have become more relevant, but ionic structures are still the most important, including the fully ionic structures, which contribute up to 35% in both cases.
Although in the four isovalent systems studied, different descriptions of the bonding pattern have been obtained, in all cases, it is concluded that the ionic and fully ionic contributions are important (the latter being around 35% for Na3+ and K3+ and 48% for Li3+) leading to the conclusion that these compounds possess charge-shift bonds. Finally, to the best of the author's knowledge, this is the first time that NNAs have been used for the proposition of resonance structures.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The author acknowledges the financial support of the National Agency for Research and Development (ANID) through FONDECYT projects 1230571.References
- J. J. Thomson, London, Edinburgh Dublin Philos. Mag. J. Sci., 1911, 21, 225–249 CrossRef CAS.
- J.-P. Maillard, P. Drossart, J. K. G. Watson, S. J. Kim and J. Caldwell, Astrophys. J., 1990, 363, L37 CrossRef CAS.
- T. R. Geballe, M. F. Jagod and T. Oka, Astrophys. J., 1993, 408, L109 CrossRef CAS.
- L. M. Trafton, T. R. Geballe, S. Miller, J. Tennyson and G. E. Ballester, Astrophys. J., 1993, 405, 761 CrossRef CAS.
- T. R. Geballe and T. Oka, Nature, 1996, 384, 334–335 CrossRef CAS.
- B. J. McCall, T. R. Geballe, K. H. Hinkle and T. Oka, Science, 1998, 279, 1910–1913 CrossRef CAS PubMed.
- L. Pauling, The Nature of the Chemical Bond and the Structure of Molecules and Crystals: An Introduction to Modern Structural Chemistry, Cornell University Press, 1960 Search PubMed.
- E. Hückel, Z. Phys., 1931, 70, 204–286 CrossRef.
- R. W. A. Havenith, F. De Proft, P. W. Fowler and P. Geerlings, Chem. Phys. Lett., 2005, 407, 391–396 CrossRef CAS.
- A. Sadjadi and M. Abdzadeh, J. Chem. Res., 2004, 2004, 797–801 CrossRef.
- C. Foroutan-Nejad and P. Rashidi-Ranjbar, THEOCHEM, 2009, 901, 243–248 CrossRef CAS.
- R. F. W. Bader and R. F. Bader, Atoms in Molecules: A Quantum Theory, Clarendon Press, 1990 Search PubMed.
- R. F. W. Bader, J. Phys. Chem. A, 2009, 113, 10391–10396 CrossRef CAS.
- J. M. Guevara-Vela, E. Francisco, T. Rocha-Rinza and Á. Martín Pendás, Molecules, 2020, 25, 4028 CrossRef CAS PubMed.
- R. J. Bartlett and M. Musiał, Rev. Mod. Phys., 2007, 79, 291–352 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- T. A. Keith, AIMAll (Version 19.02.13), Overland Park KS, USA, 2019, https://aim.tkgristmill.com Search PubMed.
- R. Pino-Rios, D. Inostroza and W. Tiznado, Angew. Chem., Int. Ed., 2021, 60, 12747–12753 CrossRef CAS.
- S. Shaik, D. Danovich, W. Wu and P. C. Hiberty, Nat. Chem., 2009, 1, 443–449 CrossRef CAS.
- S. Shaik, D. Danovich, J. M. Galbraith, B. Braïda, W. Wu and P. C. Hiberty, Angew. Chem., Int. Ed., 2020, 59, 984–1001 CrossRef CAS PubMed.
- B. Braïda and P. C. Hiberty, Nat. Chem., 2013, 5, 417–422 CrossRef.
- S. Shaik, D. Danovich, W. Wu and P. C. Hiberty, The Chemical Bond, 2014, pp. 159–198 DOI:10.1002/9783527664696.ch5.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nj03674j |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |