
Transition-metal-free synthesis and photophysical studies of highly functionalized (E)-stilbenes†
Ritu
Mamgain
,
Gana
R. J.
,
Abhrajeet
Malik
and
Fateh V.
Singh
 *
*
Chemistry Department, School of Advanced Sciences (SAS), Vellore Institute of Technology, Chennai, Vandalur-Kelambakkam Road, Chennai-600 127, Tamil Nadu, India. E-mail: fatehveer.singh@vit.ac.in
First published on 21st November 2023
Abstract
Metal-free synthesis of highly functionalized (E)-stilbenes cored with electronic donor and acceptor functionalities was achieved through a carbanion-induced ring transformation reaction between (E)-4-(4-(dimethylamino)phenyl)but-3-en-2-one 12 and 6-aryl-2H-pyran-2-ones 9 in the presence of a base. The procedure has several advantages, including mild reaction conditions, readily available precursors, metal-free synthesis, simple setup, and the synthesis of various substituted regioselective (E)-stilbenes in good yields. All the synthesized compounds 13a–j were found to be thermally stable and exhibited fluorescence in the range of 436–490 nm. A considerable positive solvatochromic behaviour in different solvents of varying polarity was observed, and the concentration study indicated the non-aggregating behaviour of all the synthesized compounds.
Introduction
Stilbenes can be distinguished by the presence of two aromatic rings connected through an ethylene bridge. Although stilbene (1,2-diphenylethene) does not exist in nature, its derivatives have been obtained from numerous plant species as phytoalexins.1–4 Stilbene is found in (E)- and (Z) conformations, each of which shows unique pharmaceutical properties as well as exhibits excellent photophysical and photochemical properties. The varied spectrum of pharmaceutical activities exhibited by stilbene and its analogues include antimicrobial,5–7 anticancer,8,9 antileukemic,10,11 antiproliferative,12 anti-inflammatory,13 antioxidants,14 anti-HIV,15,16 and anti-herpes simplex virus.17 Resveratrol, oxyresveratrol, pterostilbene, piceatannol, isorhapotigenin, schweinfurthins E, combretastatin A-4 (CA-4) and rumexoid are a few stilbene-cored isolated natural products known for their biological activities.18–28Apart from biological features, stilbenes also have promising prospects in materials science. The derivatives exhibit industrial applications in electrochemistry, dyes, organic light emitting diodes (LEDs), fluorescent lighting, and optical brighteners.29–32 In Fig. 1 we have included recently synthesised fluorescent stilbenes 1–4.33–35 Kanvah's group reported the synthesis of a pH-sensitive cyanostilbene-based fluorophore 1. By replacing the donor group dimethylaniline with carbazole, the compound 2 exhibited a noticeable blue shift in the absorption spectrum due to reduced electron-donating properties of the nitrogen involved in the aromatic structure. Jiang et al. reported a cyanostilbene derivative with a characteristic D–π–A structure. A strong intramolecular charge transfer (ICT) process was observed on modifying the electron donor to a methoxy group and the electron acceptor to a nitro group in molecule 3. Furthermore, this compound exhibited the capability to serve as a fluorescent sensor for detecting traces of water in both THF and dioxane.35 Synthesis and comprehensive photophysical study of E/Z-isomerization of fluorescent diphenylamine-linked stilbene derivatives 4 indicated their potential utility as materials for transporting holes in the production of electroluminescent devices.34 The literature review exhibits that the substitution groups, both electron-donating and electron-withdrawing groups, present on stilbene significantly affect it's photophysical properties.
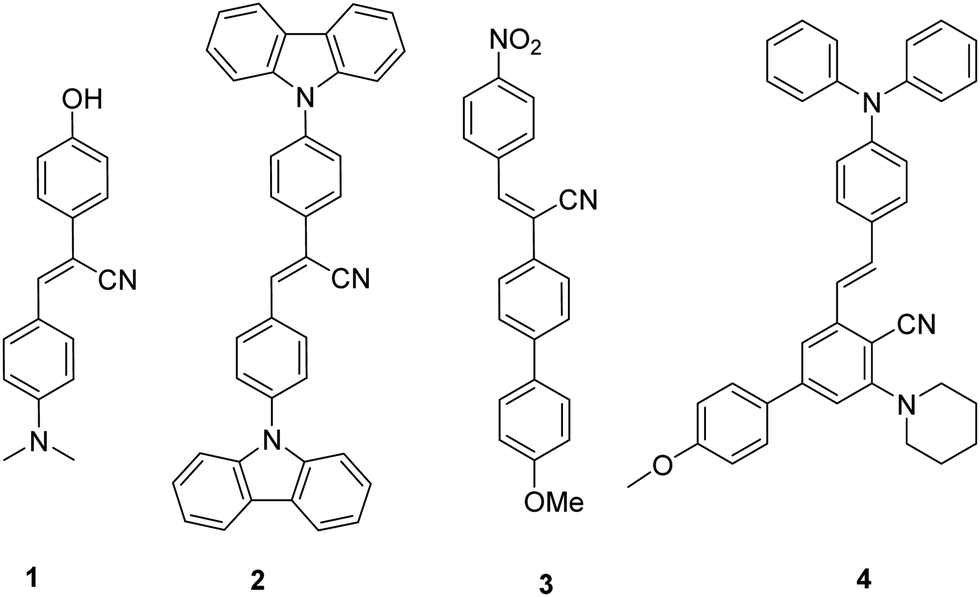 | ||
| Fig. 1 Stilbene-cored scaffolds with fluorescent properties 1–4. | ||
Due to the expanding applications of stilbene-based compounds in various research areas, several synthesis methods have been developed over the years. The well-known methods to synthesize stilbene derivatives include transition metal couplings, such as Sonogashira, Mizoroki–Heck, Negishi, Suzuki–Miyaura, Stille, Grubbs & McMurry, Knoevenagel–Doebner, Wittig, or Horner–Wadsworth–Emmons (HWE) olefination, Ramberg–Bucklund reactions, and Perkin aldol condensation.36–38
In the course of our continuous investigation into substituted aromatic systems involving donors and acceptors,39–46 we have devised an efficient approach to craft donor–acceptor π-conjugated (D–π–A) stilbenes. In this molecule, the secondary amine at the C4 position acts as the donor group and the CN group acts as the acceptor functionality. In this procedure of ring transformation reaction, carbanions are used at ambient temperature. Customized variations of stilbenes enable us to incorporate our preferred donor and acceptor components, enhancing the internal charge transfer (ICT) properties, as both donor and acceptor groups are within a single molecule.
In this piece of work, we explored the photophysical attributes of tailor-made stilbene in solution. To investigate how the introduction of dimethylamine, acting as an electronic donating group, on one aromatic ring, and the presence of a cyano group, an electron-withdrawing group, along with a secondary amine serving as an electron-donating group on another aromatic ring of stilbene, affects its photophysical properties, we plan to synthesize highly functionalized derivatives of (E)-stilbene via ring transformation of 2H-pyran-2-ones 9 using (E)-4-(4-(dimethylamino)phenyl)but-3-en-2-one 12 under mild reaction conditions. The push and pull system involved in the scaffold helps to enhance the photoluminescence of the moiety.
Results and discussion
Knowing the importance of stilbene in pharmaceuticals and various industrial applications, we attempted to synthesize highly functionalized stilbenes 13 under mild conditions, starting with 6-aryl-4-amino-2H-pyran-2-ones 9. The starting material 9 was prepared in two steps (Scheme 1).47–49 In the initial step, functionalized aryl ketones 6 reacted with ethyl 2-cyano-3,3-dimethylsulfanylacrylate 5 in the presence of KOH in DMSO to give 2-pyranones 7 in 57–73% yields. In the subsequent step, 6-aryl-4-amino-2H-pyran-2-ones 9 were produced in 71–85% by reacting the synthesized 2-pyranones 7 with secondary amines 8 in methanol at reflux temperature (Scheme 1).50,51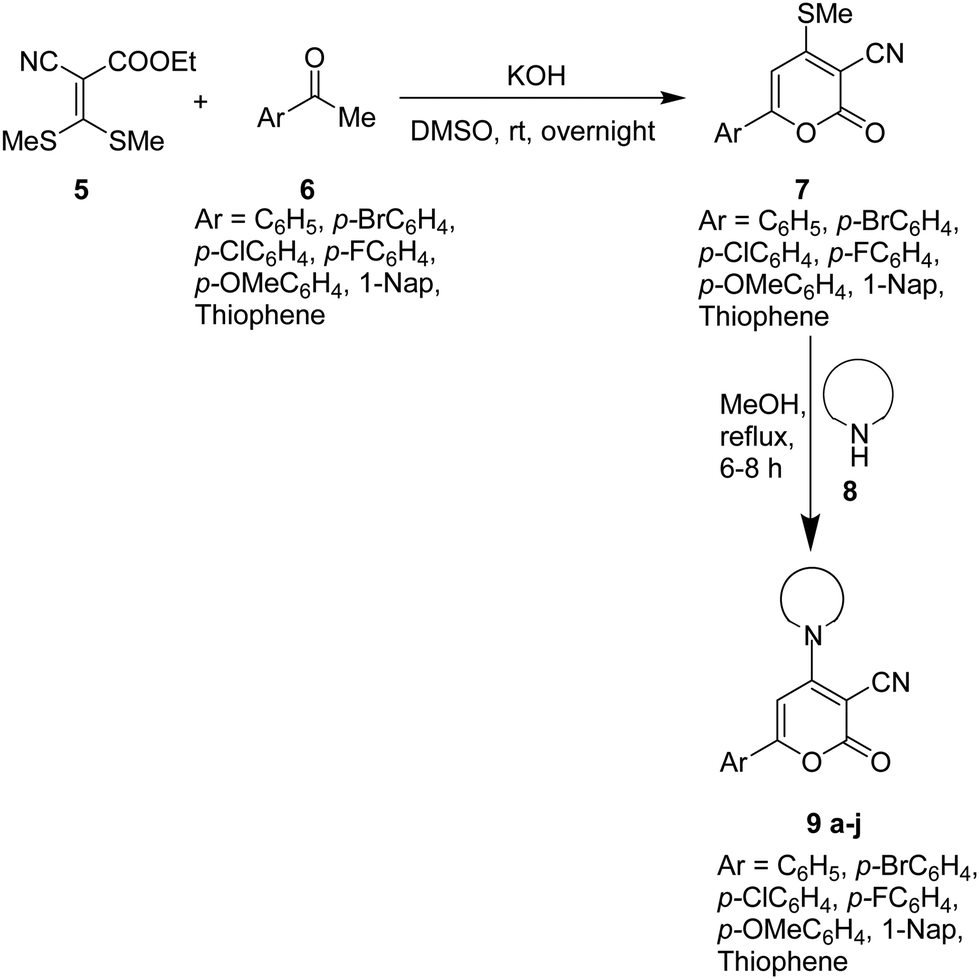 | ||
| Scheme 1 Synthesis of 6-aryl-4-amino-2-oxo-2H-pyran-3-carbonitriles 9a–j as a starting precursor. | ||
Furthermore, (E)-4-(4-(dimethylamino)phenyl)but-3-en-2-one 12 was synthesised by Aldol condensation starting with p-dimethylamino benzaldehyde 10 in an excess of acetone 11 in the presence of 10% NaOH solution (Scheme 2).
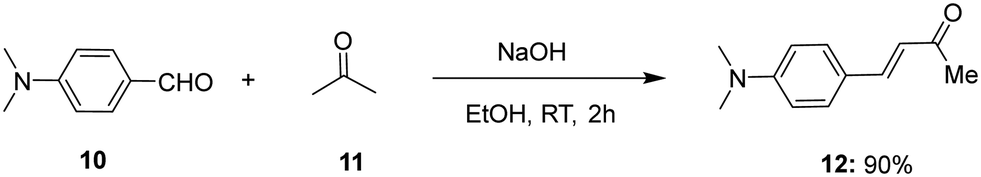 | ||
| Scheme 2 Synthesis of (E)-4-(4-(dimethylamino)phenyl)but-3-en-2-one 12via aldol condensation. | ||
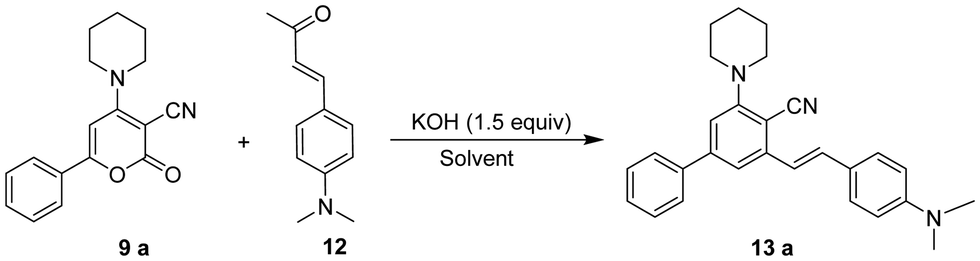
The focus of our research was initially to find the ideal reaction conditions for the ring transformation reaction. As a model reaction, we selected 2-oxo-6-phenyl-4-(piperidin-1-yl)-2H-pyran-3-carbonitrile 9a (1 mmol, 1 equiv.) and (E)-4-(4-(dimethylamino)phenyl)but-3-en-2-one 12 (1 mmol, 1 equiv.) as the substrate. At first, the reaction was performed at room temperature in DMF using KOH as a base. The reaction didn’t complete even after 16 h and the product isolated was obtained in trace amounts (Table 1, entry 1). The reaction was then carried out using a sonicator at ambient temperature for 5 h. On monitoring the reaction by TLC, we found that (E)-4-(4-(dimethylamino)phenyl)but-3-en-2-one 12 could not be fully consumed and the yield of isolated product was very low (Table 1 entry 2). When we increased the 2-oxo-6-phenyl-4-(piperidin-1-yl)-2H-pyran-3-carbonitrile 9a to 2 equiv., the product was obtained in 82% yield. The reaction time required to complete the reaction at room temperature was 16 h (Table 1, entry 3). When we increased the temperature to 50 °C, the reaction time decreased up to 6 h and the yield increased to 85% (Table 1, entry 4). However, a further increase in temperature did not show any increase in yield or decrease in reaction time.
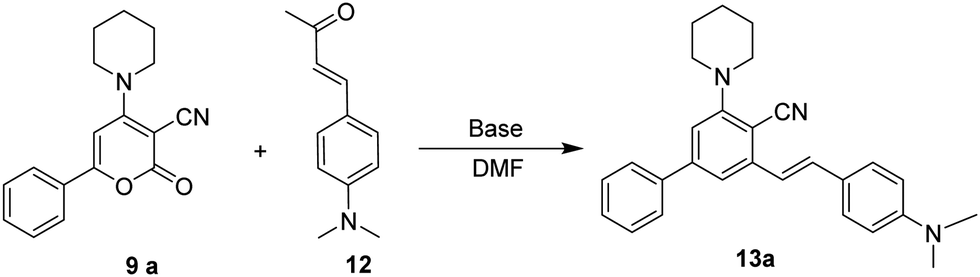
We then observed the effect of various bases on the yield of the required product 13. Reaction performed using NaOH gave a good yield of the product, but the reaction time was long (Table 1, entry 5). On the other hand, LiOH, K2CO3, and NaHCO3 gave a low yield even on prolonged reaction time (Table 1, entries 6–8). In the case of Et3N, no reaction was observed, and the starting material was recovered (Table 1, entry 9).
After the initial findings, which suggested that using 2 equiv. of 2-oxo-6-phenyl-4-(piperidin-1-yl)-2H-pyran-3-carbonitrile 9a, 1 equiv. of (E)-4-(4-(dimethylamino) phenyl)but-3-en-2-one 12 and KOH as the base at 50 °C for 5–8 h were the optimal reaction conditions for obtaining the required product 13a, we focused on selecting a suitable solvent for the ring transformation. In the beginning, various polar and non-polar solvents were used with KOH as a base under the above-mentioned conditions. Ring-transformed product 13a was produced in 5–8 hours in more polar aprotic solvents such as DMF and DMSO, with yields of 85% and 78%, respectively (Table 2, entries 1 and 2).
|
|
|||
|---|---|---|---|
| Entry | Solvent | Time (h) | 13a Yield (%) |
| 1 | DMF | 6 | 85 |
| 2 | DMSO | 8 | 78 |
| 3 | MeCN | 12 | 55 |
| 4 | EtOAc | 12 | 50 |
| 5 | Toluene | 14 | 62 |
| 6 | CHCl3 | 15 | 35 |
| 7 | DCM | 15 | 32 |
| 8 | EtOH | 20 | 38 |
| 9 | MeOH | 20 | 35 |
| 10 | 1,4-Dioxane | 20 | — |
| 11 | THF | 20 | — |
| 12 | Benzene | 20 | — |
However, when MeCN and EtOAc were utilised as the solvents, the yield of the transformation reaction was somewhat reduced (Table 2, entries 3 and 4). Similarly in toluene, the reaction under given conditions gave a moderate yield of 62% (Table 2 entry 5). On performing the reaction in CHCl3 and DCM the product was obtained in a low yield even after a long reaction time (Table 2, entries 6 and 7). Furthermore, on using polar protic solvents such as EtOH and MeOH the product obtained was in a reasonable yield (Table 2 entries 8 and 9). The reaction did not work in solvents like 1,4-dioxane, THF and benzene (Table 2, entries 10–12).
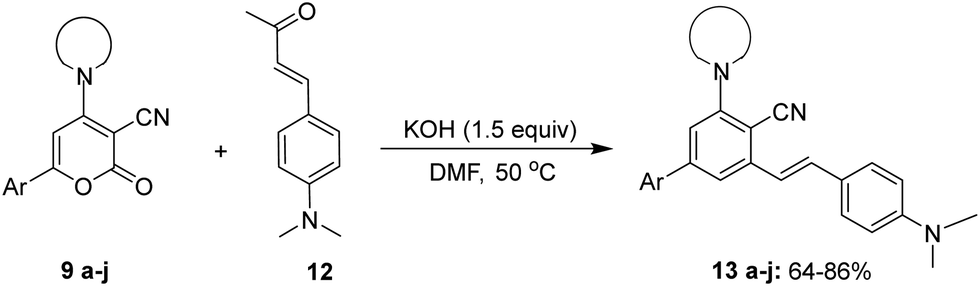
With the optimised reaction conditions obtained from the above screening of solvents, bases and temperature, we synthesised (E)-3-(4-(dimethylamino)styryl)-5-(piperidin-1-yl)-[1,1′-biphenyl]-4-carbonitrile 13avia ring transformation of 2-oxo-6-phenyl-4-(piperidin-1-yl)-2H-pyran-3-carbonitrile 9a (2 equiv.) in the presence of (E)-4-(4-(dimethylamino)phenyl)but-3-en-2-one 12 (1 equiv.) and KOH (1.5 equiv.) dissolved in 5 ml of DMF at 50 °C to give the product in 85% yield. Initially, 6-aryl-4-amino-2H-pyran-2-ones 9 with electron-donating and withdrawing substituents on the aryl at the C-6 position were analyzed.
On keeping piperidine as an amino group at position C-4 the 6-aryl-4-(piperidin-1-yl)-2H-pyran-2-ones bearing electron-withdrawing groups in the phenyl ring 9b–9d yielded the ring transformation product in excellent yield (Table 3, entries 2–4). In order to investigate the viability of the desired product in the presence of an electron-donating group, p-MeO-phenyl 9e was selected for ring transformation. Product 13e was found to have a reasonable yield, although the obtained yield was slightly lower when compared to electron-withdrawing groups (Table 3, entry 5). Similarly, on employing bulky groups like naphthalene and heterocyclic thiophene at the C-6 position instead of the phenyl group, the ring transformation products 13f and 13g resulted in moderate to good yields (Table 3, entries 6 and 7). Furthermore, to determine the effect of different cyclic secondary amines such as N-phenylpiperazine and morpholine, the ring transformation of 9h–9j was analyzed (Table 3, entries 8–10) and it was found that the reaction worked very well yielding product in 70–78% yield. Thus, it was observed that the precursor 6-aryl-4-amino-2H-pyran-2-ones 9 demonstrated adaptability towards the ring transformation reaction and successfully tolerated both electron-donating and withdrawing groups on the aryl at the C-6 position and different cyclic secondary amines at the C-4 position under the given reaction conditions.
|
|
|||||
|---|---|---|---|---|---|
| S. No. | Comp. No. | Ar | Amine | Time (h) | % Yield |
| 1 | 13a | C6H5 | Piperidine | 6 | 85 |
| 2 | 13b | p-BrC6H4 | Piperidine | 6 | 75 |
| 3 | 13c | p-FC6H4 | Piperidine | 6 | 86 |
| 4 | 13d | p-ClC6H4 | Piperidine | 6 | 78 |
| 5 | 13e | p-OMeC6H4 | Piperidine | 7 | 65 |
| 6 | 13f | 1-Naphthyl | Piperidine | 8 | 64 |
| 7 | 13g | Thiophene | Piperidine | 5 | 72 |
| 8 | 13h | Thiophene | N-Phenylpiperazine | 7 | 70 |
| 9 | 13i | p-FC6H4 | N-Phenylpiperazine | 6 | 74 |
| 10 | 13j | p-FC6H4 | Morpholine | 6 | 78 |
Based on previous studies, a plausible mechanism for the ring transformation of 2-oxo-6-phenyl-4-Amino-2H-pyran-3-carbonitrile to highly functionalized stilbene is illustrated in Scheme 3.52–54 The reaction mechanism starts with the in situ generation of enolate in the presence of a base, possibly by deprotonation of the active methylene group of 12. The enolate formed acts as a nucleophile and attacks the C-6 position of 6-aryl-2H-pyran-2-ones 9 through Michael addition to generate an intermediate [A]. Instantly, the intramolecular cyclization at the C-3 location of the pyran ring and the carbonyl functionality in intermediate [A] produces a bicyclic intermediate [B]. Furthermore, by decarboxylation and dehydration of intermediate [B], intermediate [C] is formed, which finally undergoes aromatization to give highly functionalized stilbene 13.
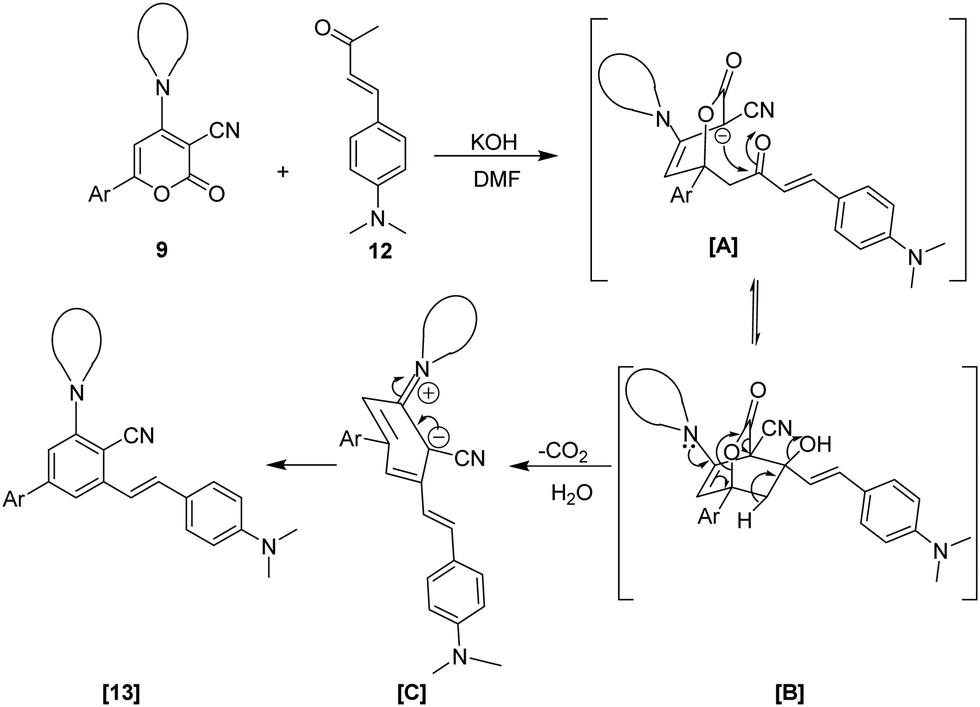 | ||
| Scheme 3 Plausible mechanism towards biaryl-cored stilbenes 13via ring transformation of 2-oxo-6-phenyl-4-(piperidin-1-yl)-2H-pyran-3-carbonitrile 9 with (E)-4-(4-(dimethylamino)phenyl)but-3-en-2-one 12. | ||
Photophysical properties of biaryl-cored stilbenes
The photophysical characteristics of each synthesized compound 13a–j were investigated by examining their ultraviolet-visible absorption spectra and photoluminescence (PL) spectra in CHCl3 at a concentration of 5 × 10−5 M. The donor–acceptor system present in the derivatives of stilbene exhibits intramolecular charge transfer (ICT). In addition to the electron-donating amino group and the electron-withdrawing cyano group, an additional donor group, p-dimethylamino styryl, was added to the system to explore the effect on the photophysical properties. The absorption spectra of 13a–j exhibit two major bands: a short-range maximum wavelength (λmax) between 261–295 nm, which could be associated with the π–π* electronic transition due to the conjugated stilbene backbone and a less intense band at the long-range maximum wavelength (λmax) in the range of 380–407 nm, due to the presence of the tertiary amino group (Fig. 2).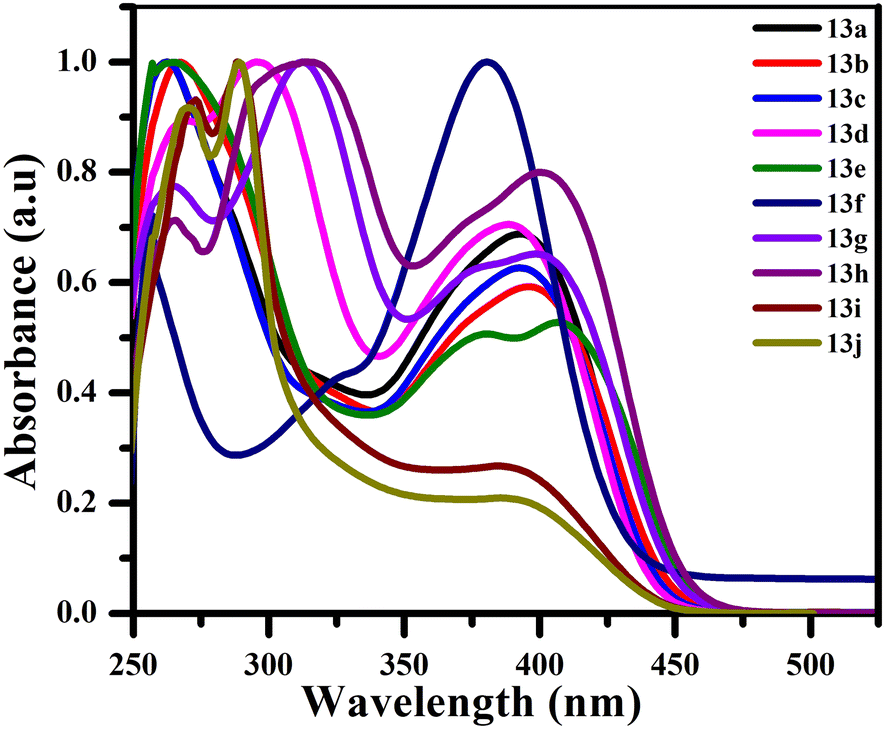 | ||
| Fig. 2 Absorbance spectra for biaryl stilbene 13a–j. | ||
On examining the emission spectra of all the functionalised stilbene compounds 13a–j the maximum emission wavelength appeared in the range of 436–490 nm (Fig. 3). A notable blue shift was observed from 481 nm to 436 nm when the simple phenyl is replaced with naphthyl. On comparing different para substituents on the phenyl group, it was found that the emission maxima in the presence of electron-withdrawing functionalities, such as F 13c and Cl 13d, decreased in comparison to the electron donating groups such as OMe 13f. On changing the phenyl ring with heterocyclic ring thiophene 13h the wavelength maxima increase to 490 nm.
 | ||
| Fig. 3 Emission spectra for biaryl-stilbene 13a–j. | ||
Additionally, by comparing the emission spectra of the compounds 13c, 13i, and 13j, it can be deduced that the ascending order of the emission maxima of the stilbene derivatives with various amino groups at para position is in the order piperidine < morpholine < N-phenylpiperazine.
Furthermore, using the widely used Tauc's approach,55,56 the optical band gaps of all the synthesised compounds, 13a–j, were determined from the absorption spectra. It is centred on the Tauc–Davis–Mott equation (eqn (1)), which explains the relation between the band gap Eg and the absorption coefficient (α).
| (αhν)n = A(hν − Eg) | (1) |
| Entry | Comp. | λ max,abs (nm) | λ max,em (nm) | Band gap (eV) Eg | Stoke's shift (Δ![[small upsilon, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0d5.gif) cm−1) cm−1) |
|---|---|---|---|---|---|
| 1 | 13a | 261, 393 | 481 | 4.0 | 4655 |
| 2 | 13b | 266, 395 | 487 | 3.86 | 4782 |
| 3 | 13c | 262, 393 | 477 | 4.07 | 4480 |
| 4 | 13d | 295, 388 | 474 | 3.72 | 4676 |
| 5 | 13e | 255, 380 | 436 | 2.96 | 3380 |
| 6 | 13f | 264, 407 | 482 | 3.93 | 3823 |
| 7 | 13g | 264, 311, 393 | 487 | 3.55 | 4911 |
| 8 | 13h | 265, 315, 400 | 490 | 3.45 | 4592 |
| 9 | 13i | 289, 388 | 488 | 4.04 | 5281 |
| 10 | 13j | 270, 288, 391 | 486 | 4.10 | 4999 |
To examine the solvatochromic effect and non-aggregating behaviour of the synthesized compounds, the highly fluorescent product 13h was selected for the study. To explore the effects of concentration on its photoluminescence, compound 13h in chloroform from 2.5 × 10−6 to 10−3 M concentration was studied (Fig. 4). It is evident from the photoluminescence spectrum of 13h that the fluorescence intensity of the molecule gradually increased until 10−4 M, but on increasing the concentration above it, the intensity starts dropping. At 10−3 M, no emission was observed due to concentration quenching. The emission band of 13h solely complemented the monomer's 491 nm band at all concentrations. It emerged from the study that there were no intermolecular π–π interactions or stacking, as no bathochromic shifts or extra peaks for excimer formation were observed.
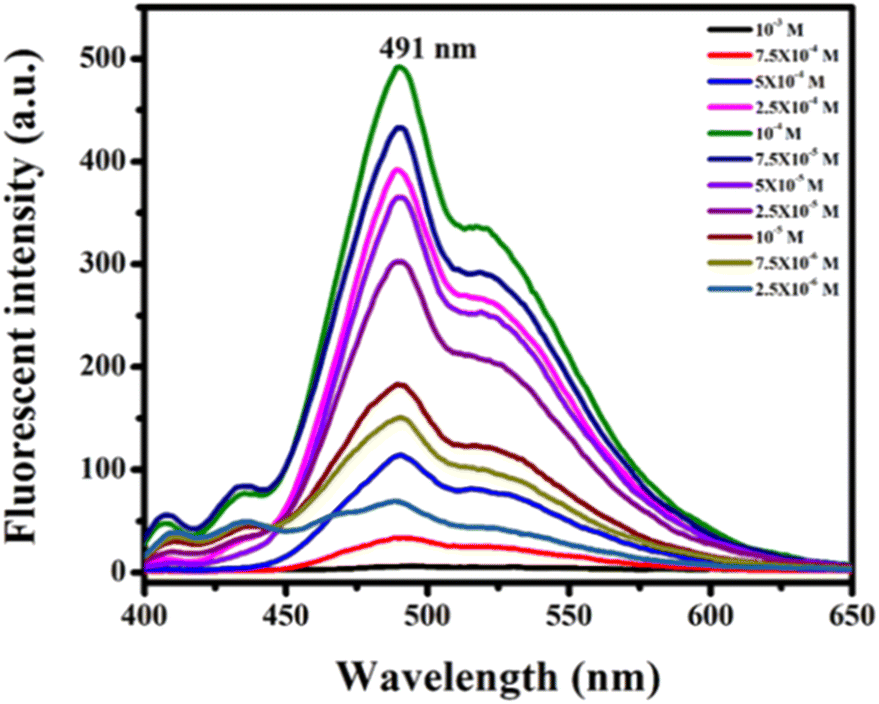 | ||
| Fig. 4 Effect of concentration on the fluorescence emission spectrum of 13h in CHCl3. | ||
To investigate the stabilization of the ground and excited states of the synthesized compound 13h the absorption and photoluminescence spectra in a series of solvents of differing polarity indices such as cyclohexane, toluene, chloroform, ethanol, DMF and DMSO were examined. It was observed that no significant changes appeared in the absorption spectra but in the photoluminescence, on changing the solvent polarity in increasing order, a substantial bathochromic shift was observed. The positive solvatochromism ranged in wavelength from 437 nm in nonpolar cyclohexane to 501 nm in moderately polar chloroform to 572 nm in highly polar DMSO (Fig. 5).
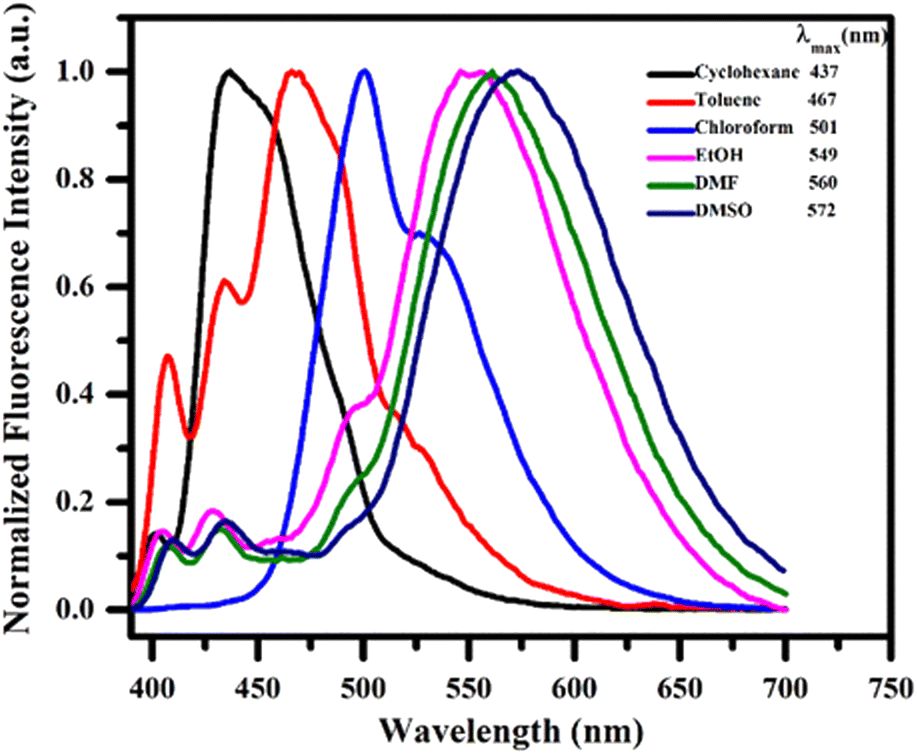 | ||
| Fig. 5 Fluorescence spectra of compound 13h in different solvents. | ||
Another piece of evidence for the solvent sensitivity of the emission spectra was provided by means of the Lippert−Mataga plot (Fig. 6).57 A linear correlation (r2 = 0.985) between the Stokes shift and the orientation polarizability (Δf) with a steeper slope indicates a larger fluorescence solvatochromism. The positive solvatochromism effect revealed that the polar excited state of the molecule is strongly stabilized by polar solvents in comparison to the ground state. Charge transfer from the donor amino group to the acceptor cyano group in compounds 13a–j may be considered a feasible route for ICT-induced fluorescence.58,59
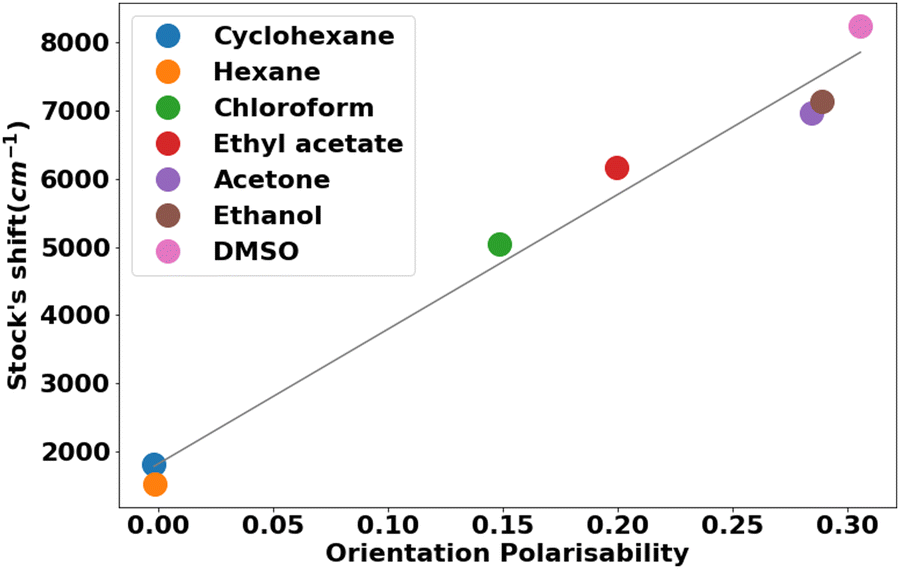 | ||
| Fig. 6 Lippert–Mataga plot of 13h in solvents of increasing polarity. | ||
Thermo-gravimetric analysis
In addition, thermogravimetric analysis (TGA) and differential thermal analysis (DTA) were performed to investigate the thermal stability of the highly functionalized stilbene 13a. The TGA thermograms (Fig. 7), show that 13a are thermally stable under the nitrogen atmosphere up to 333.5 °C after which mass loss was noted with subsequent decomposition in the range of 650–700 °C. Furthermore, the DTA curve showed melting of 13a through an endothermic peak at 173 °C.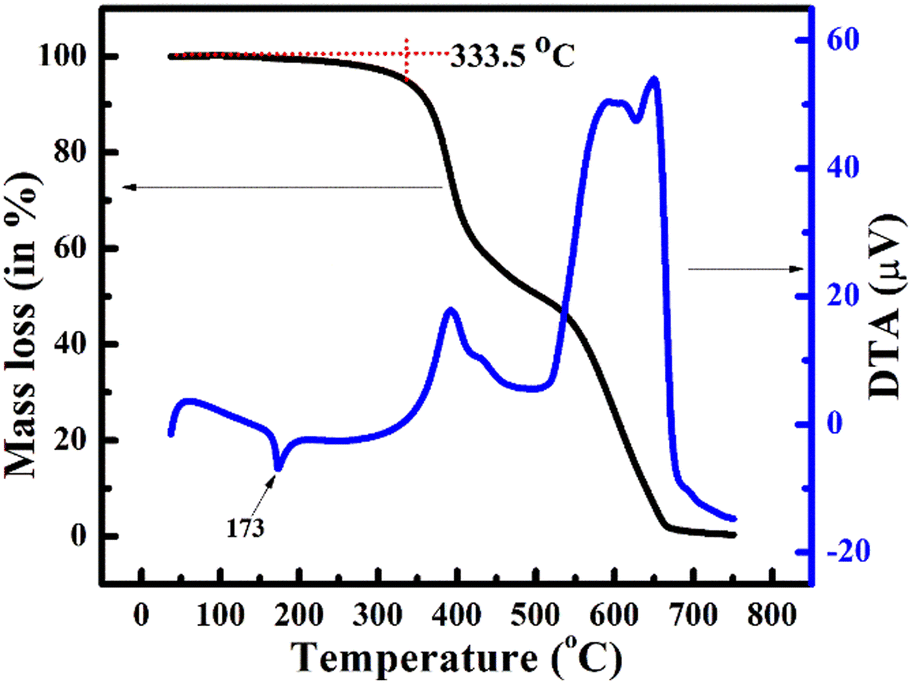 | ||
| Fig. 7 Thermogravimetric analysis (TGA) and differential thermal analysis (DTA) plot of 13a under a nitrogen atmosphere. | ||
Materials and methods
All chemicals were reagent grade and were purchased from Avra Synthesis Pvt. Ltd and Alfa Aesar, Thermo Fisher Scientific India Private Limited and were utilised without further purification. For monitoring reactions, thin-layer chromatography (TLC) was performed on Merck pre-coated sheets of silica gel 60 and column chromatography was performed with silica gel (60–120 mesh). The temperature of the water bath was maintained by the addition or removal of water. Melting points were measured in open capillary tubes using REMI DDMS 2545 melting point equipment. The 1H and 13C NMR spectra were acquired using deuterated chloroform (CDCl3, δ 77.00 ppm for 13C NMR) as the solvent and tetramethylsilane (Me4Si, δ 0 ppm) as the internal standard on a Bruker AV-400 at 400 and 100 MHz, respectively. The chemical shifts (δ) are given in ppm, whereas the coupling constants (J) are given in Hertz. Signal peaks are designated as s, singlet; d, doublet; t, triplet; q, quartet and m, multiplet. The molecular weights of the compounds (m/z) were determined using a Shimadzu GC-mass spectrometer-QP2020 and also on a WATERSXEVO G2-XS-QToF High-Resolution Mass Spectrometer. FT-IR spectral analysis was carried out on a PerkinElmer Spectrum Two FT-IR Spectrometer using ATR in the range of 4000–450 cm−1. Absorption spectra were recorded on a Cary100 UV-Visible spectrophotometer and emission spectra were obtained on a Varian Cary Eclipse Fluorescence Spectrophotometer.Experimental section
General procedure: for the synthesis of biaryl-cored stilbenes 13a–j34,60,61
To a mixture of 4-amino-3-cyano-6-aryl-2H-pyranones (2.0 mmol, 2.0 equiv.) and (E)-4-(4-(dimethylamino)phenyl)but-3-en-2-one (1.0 mmol, 1.0 equiv.) in dry DMF (6 ml), powdered KOH (1.5 mmol, 1.5 equiv.) was added. The reaction mixture was stirred at 50 °C for a certain period, as indicated in Table 3. The progress of the reaction was monitored by TLC. After the reaction was complete, the resultant mixture was poured into ice-cold water and neutralized with 10% HCl. The precipitate obtained was filtered and purified using column chromatography on silica using 2% ethyl acetate in hexane as the eluent to afford the product in good yield, which was characterized by spectroscopic analysis.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), IR (ATR): 2208 (CN) cm−1; 1H NMR (400 MHz, CDCl3): δ = 1.53–1.57 (m, 2H, CH2), 1.72–1.77 (m, 4H, 2CH2), 2.93 (s, 6H, 2CH3), 3.10 (t, J = 8 Hz, 4H, 2NCH2), 6.63 (d, J = 8.0 Hz, 2H, ArH), 6.92 (s, 1H, ArH), 7.13 (d, J = 16 Hz, 1H, CH), 7.23 (d, J = 16 Hz, 1H, CH), 7.31–7.43 (m, 6H, ArH), 7.53 (d, J = 8.0 Hz, 2H, Ar); 13C NMR (100 MHz, CDCl3): δ = 24.1, 26.2, 40.34, 53.63, 104.0, 112.2, 115.4, 116.3, 117.7, 120.3, 124.6, 127.3, 128.3, 128.9, 133.3, 140.5, 143.5, 145.8, 150.7, 158.3; HRMS (m/z): calcd for C28H29N3 407.2361, found 408.2442 [M++1]+.
:2), IR (ATR): 2204 (CN) cm−1; 1H NMR (400 MHz, CDCl3): δ = 1.52–1.58 (m, 2H, CH2), 1.70–1.77 (m, 4H, 2CH2), 2.93 (s, 6H, 2CH3), 3.11 (t, J = 4.8 Hz, 4H, 2NCH2), 6.62–6.64 (m, 2H, ArH), 6.86 (s, 1H, ArH), 7.15 (d, J = 16 Hz, 1H, CH), 7.21 (d, J = 16 Hz, 1H, CH), 7.36–7.41 (m, 5H, ArH), 7.51 (d, J = 8.0 Hz, 2H, Ar); 13C NMR (100 MHz, CDCl3): δ = 24.1, 26.2, 40.3, 53.5, 104.1, 112.1, 114.8, 115.8, 117.5, 119.9, 122.6, 124.3, 128.3, 128.8, 131.9, 133.4, 139.4, 143.5, 144.4, 150.7, 158.3; HRMS (m/z): calcd for C28H28BrN3 485.1467, found 486.1538 [M+ + 1]+, 488.1314 [M+ + 3]+.
:2), IR (ATR): 2203 (CN) cm−1; 1H NMR (400 MHz, CDCl3): δ = 1.53–1.57 (m, 2H, CH2), 1.71–1.77 (m, 4H, 2CH2), 2.93 (s, 6H, 2CH3), 3.11 (t, J = 4 Hz, 4H, 2NCH2), 6.62 (d, J = 8 Hz, 2H, ArH), 6.86 (s, 1H, ArH), 7.06–7.11 (m, 2H, CH & ArH), 7.15–7.25 (m, 2H, CH & ArH), 7.36–7.41 (m, 3H, ArH), 7.48–7.51 (m, 2H, Ar); 13C NMR (100 MHz, CDCl3): δ = 24.1, 26.2, 40.4, 53.6, 104.0, 112.2, 115.1, 115.7, 115.9, 116.0, 117.5, 120.1, 124.5, 128.4, 128.9, 129.0, 133.4, 136.65, 143.6, 144.6, 150.3, 158.1, 161.6, 164.0; HRMS (m/z): calcd for C28H28FN3 425.2267, found 426.2292 [M+ + 1]+.
:1), IR (ATR): 2207 (CN) cm−1; 1H NMR (400 MHz, CDCl3): δ = 1.52–1.54 (m, 2H, CH2), 1.93–1.96 (m, 4H, 2CH2), 2.93 (s, 6H, 2CH3), 3.57 (t, J = 4.0 Hz, 4H, 2NCH2), 6.55 (s, 1H, ArH), 6.62–6.64 (m, 2H, ArH), 7.08–7.12 (m, 2H, CH & ArH), 7.24 (d, J = 16 Hz, 1H, CH), 7.34–7.41 (m, 4H, ArH), 7.45–7.48 (m, 2H, ArH); 13C NMR (100 MHz, CDCl3): δ = 25.8, 40.3, 50.5, 93.8, 110.4, 111.7, 112.2, 119.7, 120.9, 124.8, 128.3, 128.5, 128.9, 133.1, 134.2, 139.4, 144.3, 150.6, 152.0. HRMS (m/z): calcd for C28H28ClN3 441.1972, found 442.1980 [M+ + 1]+, and 444.1806 [M+ + 3]+.
:2), IR (ATR): 2205 (CN) cm−1; 1H NMR (400 MHz, CDCl3): δ = 1.52–1.57 (m, 2H, CH2), 1.71–1.76 (m, 4H, 2CH2), 2.93 (s, 6H, 2CH3), 3.09 (t, J = 4.0 Hz, 4H, 2NCH2), 3.79 (s, 3H, OCH3) 6.60–6.65 (m, 2H, ArH), 6.89–6.94 (m, 3H, ArH), 7.16 (d, J = 16 Hz, 1H, CH), 7.22 (d, J = 16 Hz, 1H, CH), 7.39–7.42 (m, 3H, ArH), 7.46–7.49 (m, 2H, ArH); 13C NMR (100 MHz, CDCl3): δ = 23.1, 25.2, 39.3, 52.6, 54.4, 102.4, 111.2, 113.3, 113.9, 114.8, 119.4, 123.6, 127.4, 132.1, 142.3, 144.3, 149.7, 157.2, 158.9. HRMS (m/z): calcd for C29H31N3O 437.2467, found 438.2580 [M+ + 1]+.
:1), IR (ATR): 2210 (CN) cm−1; 1H NMR (400 MHz, CDCl3): δ = 1.18–1.34 (m, 2H, CH2), 1.73–1.96 (m, 4H, 2CH2), 2.90 (s, 6H, 2CH3), 3.00–3.11 (m, 4H, 2NCH2), 6.61 (m, 2H, ArH), 6.84 (s, 1H, ArH), 7.10 (d, J = 16 Hz, 1H, CH), 7.27 (d, J = 12 Hz, 1H, CH), 7.37–7.45 (m, 7H, ArH), 7.63–7.82 (m, 3H, ArH); 13C NMR (100 MHz, CDCl3): δ = 24.1, 26.2, 40.3, 53.6, 104.0, 112.2, 117.7, 118.2, 119.0, 120.1, 124.5, 125.3, 125.6, 126.0, 126.4, 128.3, 128.4, 131.2, 133.4, 133.7, 139.4, 143.1, 145.5, 150.7, 157.89 HRMS (m/z): calcd for HRMS (m/z): calcd for C32H31N3 457.2518, found 458.2593 [M+ + 1]+.
:1), IR (ATR): 2203 (CN) cm−1; 1H NMR (400 MHz, CDCl3): δ = 1.52–1.57 (m, 2H, CH2), 1.71–1.77 (m, 4H, 2CH2), 2.94 (s, 6H, 2CH3), 3.11 (t, J = 4.0 Hz, 4H, 2NCH2), 6.63 (d, J = 8.0 Hz, 2H, ArH), 6.95 (s, 1H, ArH), 7.04–7.06 (m, 1H, ArH), 7.12 (d, J = 16 Hz, 1H, CH), 7.19 (d, J = 16 Hz, 1H, CH), 7.29 (d, J = 8.0 Hz, 1H, ArH), 7.34 (d, J = 8.0, 1H, ArH), 7.41–7.45 (m, 3H, ArH); 13C NMR (100 MHz, CDCl3): δ = 24.2, 26.1, 40.3, 53.4, 103.6, 112.1, 113.8, 114.8, 117.6, 120.0, 124.5, 126.2, 128.2, 128.4, 133.5, 138.5, 143.4, 143.6, 150.7, 158.4; HRMS (m/z): calcd for C26H27N3S 413.1926, found 414.1988 [M+ + 1]+.
:1), IR (ATR): 2204 (CN) cm−1; 1H NMR (400 MHz, CDCl3): δ = 2.95 (s, 6H, 2CH3), 3.32–3.38 (m, 8H, 4NCH2), 6.49 (s, 1H, ArH), 6.62–6.70 (m, 2H, ArH), 6.82–7.07 (m, 6H, CH & ArH), 7.24–7.52 (m, 7H, CH & ArH) 13C NMR (100 MHz, CDCl3): δ 40.7, 48.7, 51.8, 103, 112.0, 115.1, 116.0, 116.2, 116.8, 120.1, 124.2, 128.9, 129.1, 133.8, 143.6, 145.8, 150.6, 151.2, 156.7; HRMS (m/z): calcd for C31H30N4S 490.2191, found 491.2209 [M+ + 1]+.
:1), IR (ATR): 2204 (CN) cm−1; 1H NMR (400 MHz, CDCl3): δ = 2.94 (s, 6H, 2CH3), 3.31–3.37 (m, 8H, 4CH2), 6.64 (d, J = 8Hz 2H, ArH), 6.83 (t, J = 8.0 Hz, 1H, CH), 6.92–6.94 (m, 3H, ArH), 7.09–7.13 (m, 2H, CH & ArH), 7.19–7.26 (m, 4H, CH & ArH), 7.41–7.45 (m, 3H, ArH), 7.50–7.54 (m, 2H, ArH); 13C NMR (100 MHz, CDCl3): δ = 40.3, 49.5, 51.9, 104.0, 112.2, 115.0, 115.8, 116.0, 116.4, 116.9, 117.4, 119.8, 120.1, 124.3, 128.5, 128.9, 129.0, 129.1, 133.8, 143.8, 145.0, 150.8, 151.2, 156.8; HRMS (m/z): calcd for C33H31FN4 502.2533 found 503.2610 [M+ + 1]+.
:1), IR (ATR): 2204 (CN) cm−1; 1H NMR (400 MHz, CDCl3): δ = 2.94 (s, 6H, 2CH3), 3.14 (t, J = 4.0 Hz, 4H, 2CH2), 3.86 (t, J = 4.0 Hz, 4H, 2CH2), 6.62 (d, J = 8.0 Hz, 2H, ArH), 6.86 (s, 1H, ArH), 7.07–7.20 (m, 4H, CH & ArH), 7.39–7.43 (m, 3H, ArH), 7.48–7.52 (m, 2H, ArH); 13C NMR (100 MHz, CDCl3): δ = 40.2, 52.2, 67.0, 103.9, 112.1, 114.9, 115.7, 116.0, 117.0, 117.4, 119.5, 124.3, 128.4, 128.9, 129.0, 133.8, 136.3 143.9, 145.0, 150.9, 156.8, 161.8, 164.3; HRMS (m/z): calcd for C27H26FN3O 427.2060 found 427.2101 [M + 1]+.
:1), IR (ATR): 2208 (CN) cm−1; 1H NMR (400 MHz, CDCl3): δ = 1.53–1.57 (m, 2H, CH2), 1.72–1.77 (m, 4H, 2CH2), 2.93 (s, 6H, 2CH3), 3.10 (t, J = 8 Hz, 4H, 2NCH2), 6.63 (d, J = 8.0 Hz, 2H, ArH), 6.92 (s, 1H, ArH), 7.13 (d, J = 16 Hz, 1H, CH), 7.23 (d, J = 16 Hz, 1H, CH), 7.31–7.43 (m, 6H, ArH), 7.53 (d, J = 8.0 Hz, 2H, Ar); 13C NMR (100 MHz, CDCl3): δ = 24.1, 26.2, 40.34, 53.63, 104.0, 112.2, 115.4, 116.3, 117.7, 120.3, 124.6, 127.3, 128.3, 128.9, 133.3, 140.5, 143.5, 145.8, 150.7, 158.3; HRMS (m/z): calcd for C28H29N3 407.2361, found 408.2442 [M++1]+.
:2), IR (ATR): 2204 (CN) cm−1; 1H NMR (400 MHz, CDCl3): δ = 1.52–1.58 (m, 2H, CH2), 1.70–1.77 (m, 4H, 2CH2), 2.93 (s, 6H, 2CH3), 3.11 (t, J = 4.8 Hz, 4H, 2NCH2), 6.62–6.64 (m, 2H, ArH), 6.86 (s, 1H, ArH), 7.15 (d, J = 16 Hz, 1H, CH), 7.21 (d, J = 16 Hz, 1H, CH), 7.36–7.41 (m, 5H, ArH), 7.51 (d, J = 8.0 Hz, 2H, Ar); 13C NMR (100 MHz, CDCl3): δ = 24.1, 26.2, 40.3, 53.5, 104.1, 112.1, 114.8, 115.8, 117.5, 119.9, 122.6, 124.3, 128.3, 128.8, 131.9, 133.4, 139.4, 143.5, 144.4, 150.7, 158.3; HRMS (m/z): calcd for C28H28BrN3 485.1467, found 486.1538 [M+ + 1]+, 488.1314 [M+ + 3]+.
:2), IR (ATR): 2203 (CN) cm−1; 1H NMR (400 MHz, CDCl3): δ = 1.53–1.57 (m, 2H, CH2), 1.71–1.77 (m, 4H, 2CH2), 2.93 (s, 6H, 2CH3), 3.11 (t, J = 4 Hz, 4H, 2NCH2), 6.62 (d, J = 8 Hz, 2H, ArH), 6.86 (s, 1H, ArH), 7.06–7.11 (m, 2H, CH & ArH), 7.15–7.25 (m, 2H, CH & ArH), 7.36–7.41 (m, 3H, ArH), 7.48–7.51 (m, 2H, Ar); 13C NMR (100 MHz, CDCl3): δ = 24.1, 26.2, 40.4, 53.6, 104.0, 112.2, 115.1, 115.7, 115.9, 116.0, 117.5, 120.1, 124.5, 128.4, 128.9, 129.0, 133.4, 136.65, 143.6, 144.6, 150.3, 158.1, 161.6, 164.0; HRMS (m/z): calcd for C28H28FN3 425.2267, found 426.2292 [M+ + 1]+.
:1), IR (ATR): 2207 (CN) cm−1; 1H NMR (400 MHz, CDCl3): δ = 1.52–1.54 (m, 2H, CH2), 1.93–1.96 (m, 4H, 2CH2), 2.93 (s, 6H, 2CH3), 3.57 (t, J = 4.0 Hz, 4H, 2NCH2), 6.55 (s, 1H, ArH), 6.62–6.64 (m, 2H, ArH), 7.08–7.12 (m, 2H, CH & ArH), 7.24 (d, J = 16 Hz, 1H, CH), 7.34–7.41 (m, 4H, ArH), 7.45–7.48 (m, 2H, ArH); 13C NMR (100 MHz, CDCl3): δ = 25.8, 40.3, 50.5, 93.8, 110.4, 111.7, 112.2, 119.7, 120.9, 124.8, 128.3, 128.5, 128.9, 133.1, 134.2, 139.4, 144.3, 150.6, 152.0. HRMS (m/z): calcd for C28H28ClN3 441.1972, found 442.1980 [M+ + 1]+, and 444.1806 [M+ + 3]+.
:2), IR (ATR): 2205 (CN) cm−1; 1H NMR (400 MHz, CDCl3): δ = 1.52–1.57 (m, 2H, CH2), 1.71–1.76 (m, 4H, 2CH2), 2.93 (s, 6H, 2CH3), 3.09 (t, J = 4.0 Hz, 4H, 2NCH2), 3.79 (s, 3H, OCH3) 6.60–6.65 (m, 2H, ArH), 6.89–6.94 (m, 3H, ArH), 7.16 (d, J = 16 Hz, 1H, CH), 7.22 (d, J = 16 Hz, 1H, CH), 7.39–7.42 (m, 3H, ArH), 7.46–7.49 (m, 2H, ArH); 13C NMR (100 MHz, CDCl3): δ = 23.1, 25.2, 39.3, 52.6, 54.4, 102.4, 111.2, 113.3, 113.9, 114.8, 119.4, 123.6, 127.4, 132.1, 142.3, 144.3, 149.7, 157.2, 158.9. HRMS (m/z): calcd for C29H31N3O 437.2467, found 438.2580 [M+ + 1]+.
:1), IR (ATR): 2210 (CN) cm−1; 1H NMR (400 MHz, CDCl3): δ = 1.18–1.34 (m, 2H, CH2), 1.73–1.96 (m, 4H, 2CH2), 2.90 (s, 6H, 2CH3), 3.00–3.11 (m, 4H, 2NCH2), 6.61 (m, 2H, ArH), 6.84 (s, 1H, ArH), 7.10 (d, J = 16 Hz, 1H, CH), 7.27 (d, J = 12 Hz, 1H, CH), 7.37–7.45 (m, 7H, ArH), 7.63–7.82 (m, 3H, ArH); 13C NMR (100 MHz, CDCl3): δ = 24.1, 26.2, 40.3, 53.6, 104.0, 112.2, 117.7, 118.2, 119.0, 120.1, 124.5, 125.3, 125.6, 126.0, 126.4, 128.3, 128.4, 131.2, 133.4, 133.7, 139.4, 143.1, 145.5, 150.7, 157.89 HRMS (m/z): calcd for HRMS (m/z): calcd for C32H31N3 457.2518, found 458.2593 [M+ + 1]+.
:1), IR (ATR): 2203 (CN) cm−1; 1H NMR (400 MHz, CDCl3): δ = 1.52–1.57 (m, 2H, CH2), 1.71–1.77 (m, 4H, 2CH2), 2.94 (s, 6H, 2CH3), 3.11 (t, J = 4.0 Hz, 4H, 2NCH2), 6.63 (d, J = 8.0 Hz, 2H, ArH), 6.95 (s, 1H, ArH), 7.04–7.06 (m, 1H, ArH), 7.12 (d, J = 16 Hz, 1H, CH), 7.19 (d, J = 16 Hz, 1H, CH), 7.29 (d, J = 8.0 Hz, 1H, ArH), 7.34 (d, J = 8.0, 1H, ArH), 7.41–7.45 (m, 3H, ArH); 13C NMR (100 MHz, CDCl3): δ = 24.2, 26.1, 40.3, 53.4, 103.6, 112.1, 113.8, 114.8, 117.6, 120.0, 124.5, 126.2, 128.2, 128.4, 133.5, 138.5, 143.4, 143.6, 150.7, 158.4; HRMS (m/z): calcd for C26H27N3S 413.1926, found 414.1988 [M+ + 1]+.
:1), IR (ATR): 2204 (CN) cm−1; 1H NMR (400 MHz, CDCl3): δ = 2.95 (s, 6H, 2CH3), 3.32–3.38 (m, 8H, 4NCH2), 6.49 (s, 1H, ArH), 6.62–6.70 (m, 2H, ArH), 6.82–7.07 (m, 6H, CH & ArH), 7.24–7.52 (m, 7H, CH & ArH) 13C NMR (100 MHz, CDCl3): δ 40.7, 48.7, 51.8, 103, 112.0, 115.1, 116.0, 116.2, 116.8, 120.1, 124.2, 128.9, 129.1, 133.8, 143.6, 145.8, 150.6, 151.2, 156.7; HRMS (m/z): calcd for C31H30N4S 490.2191, found 491.2209 [M+ + 1]+.
:1), IR (ATR): 2204 (CN) cm−1; 1H NMR (400 MHz, CDCl3): δ = 2.94 (s, 6H, 2CH3), 3.31–3.37 (m, 8H, 4CH2), 6.64 (d, J = 8Hz 2H, ArH), 6.83 (t, J = 8.0 Hz, 1H, CH), 6.92–6.94 (m, 3H, ArH), 7.09–7.13 (m, 2H, CH & ArH), 7.19–7.26 (m, 4H, CH & ArH), 7.41–7.45 (m, 3H, ArH), 7.50–7.54 (m, 2H, ArH); 13C NMR (100 MHz, CDCl3): δ = 40.3, 49.5, 51.9, 104.0, 112.2, 115.0, 115.8, 116.0, 116.4, 116.9, 117.4, 119.8, 120.1, 124.3, 128.5, 128.9, 129.0, 129.1, 133.8, 143.8, 145.0, 150.8, 151.2, 156.8; HRMS (m/z): calcd for C33H31FN4 502.2533 found 503.2610 [M+ + 1]+.
:1), IR (ATR): 2204 (CN) cm−1; 1H NMR (400 MHz, CDCl3): δ = 2.94 (s, 6H, 2CH3), 3.14 (t, J = 4.0 Hz, 4H, 2CH2), 3.86 (t, J = 4.0 Hz, 4H, 2CH2), 6.62 (d, J = 8.0 Hz, 2H, ArH), 6.86 (s, 1H, ArH), 7.07–7.20 (m, 4H, CH & ArH), 7.39–7.43 (m, 3H, ArH), 7.48–7.52 (m, 2H, ArH); 13C NMR (100 MHz, CDCl3): δ = 40.2, 52.2, 67.0, 103.9, 112.1, 114.9, 115.7, 116.0, 117.0, 117.4, 119.5, 124.3, 128.4, 128.9, 129.0, 133.8, 136.3 143.9, 145.0, 150.9, 156.8, 161.8, 164.3; HRMS (m/z): calcd for C27H26FN3O 427.2060 found 427.2101 [M + 1]+.
Conclusions
In conclusion, we have synthesized highly functionalized stilbenes with a donor–acceptor system. The products 13a–j were obtained in good to excellent yields via carbanion-induced ring transformation reaction of 6-aryl-2H-pyran-2-ones 9 with (E)-4-(4-(dimethylamino)phenyl)but-3-en-2-one 12 in the presence of a base. The advantages of our procedure are easily accessible precursors, mild reaction conditions, metal-free synthesis, easy workup, and flexibility in introducing electronic donating and withdrawing groups in the stilbene backbone. In addition, the photophysical characteristics of these thermally stable highly functionalized stilbene molecules were investigated using UV-Vis and fluorescence spectroscopy. All of the synthesized compounds 13a–j exhibited fluorescence in the range of 436–490 nm and showed positive solvatochromic behaviour with solvents of different polarities. Moreover, other photophysical parameters such as the optical band gap and Stokes shift were calculated. Based on the interesting photophysical results obtained in this study, further application of these compounds for material and biological imaging is underway.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful to SAIF department, VIT Vellore and Department of Physics and STIC, Kochi for TG-DTA data. Ritu Mamagin acknowledges the support from the Department of Science and Technology, New Delhi, India, for WOS-A project SR/WOS-A/CS-107/2018. Moreover, Fateh V. Singh is thankful to CSIR New Delhi [Grant No.: 02/(0330)/17-EMR-II] for the financial support.References
- S. Mochida, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2010, 12, 5776–5779 CrossRef CAS PubMed.
- C. G. Fraga, K. D. Croft, D. O. Kennedy and F. A. Tomás-Barberán, Food Funct., 2019, 10, 514–528 RSC.
- C. F. Xiao, L. Y. Tao, H. Y. Sun, W. Wei, Y. Chen, L. W. Fu and Y. Zou, Chin. Chem. Lett., 2010, 21, 1295–1298 CrossRef CAS.
- Y. Q. Li, Z. L. Li, W. J. Zhao, R. X. Wen, Q. W. Meng and Y. Zeng, Eur. J. Med. Chem., 2006, 4, 1084–1089 CrossRef.
- J. Gabaston, T. Richard, B. Biais, P. Waffo-Teguo, E. Pedrot, M. Jourdes, M. F. Corio-Costet and J. M. Mérillon, Ind. Crops Prod., 2017, 103, 267–273 CrossRef CAS.
- E. Giacomini, S. Rupiani, L. Guidotti, M. Recanatini and M. Roberti, Curr. Med. Chem., 2016, 23, 2439–2489 CrossRef CAS.
- M. Wan, L. Hua, Y. Zeng, P. Jiao, D. Xie, Z. Tong, G. Wu, Y. Zhou, Q. Tang and F. Mo, Cellulose, 2017, 24, 3209–3218 CrossRef CAS.
- M. Roberti, D. Pizzirani, D. Simoni, R. Rondanin, R. Baruchello, C. Bonora, F. Buscemi, S. Grimaudo and M. Tolomeo, J. Med. Chem., 2003, 46, 3546–3554 CrossRef CAS.
- A. Sharma, M. Kaur, J. K. Katnoria and A. K. Nagpal, Curr. Med. Chem., 2018, 25, 4740–4757 CrossRef CAS.
- M. Gao, M. Wang, K. D. Miller, G. W. Sledge, G. D. Hutchins and Q. H. Zheng, Bioorg. Med. Chem. Lett., 2006, 16, 5767–5772 CrossRef CAS PubMed.
- J. Zheng and V. D. Ramirez, Biochem. Biophys. Res. Commun., 1999, 261, 499–503 CrossRef CAS.
- P. A. Segun, O. O. Ogbole, F. M. Ismail, L. Nahar, A. R. Evans, E. O. Ajaiyeoba and S. D. Sarker, Fitoterapia, 2019, 134, 151–157 CrossRef CAS.
- A. Benayahoum, S. Bouakkaz, T. Bordjiba and M. Abdaoui, J. Mol. Liq., 2019, 275, 221–231 CrossRef CAS.
- Y. J. Cai, J. G. Fang, L. P. Ma, L. Yang and Z. L. Liu, Biochem. Biophys. Acta, 2003, 1637, 31–38 CAS.
- L. X. Wang, A. Heredia, H. Song, Z. Zhang, B. Yu, C. Davis and R. Redfield, J. Pharm. Sci., 2004, 93, 2448–2457 CrossRef CAS.
- K. Likhitwitayawuid, B. Sritularak, K. Benchanak, V. Lipipun, J. Mathew and R. F. Schinazi, Nat. Prod. Res., 2005, 19, 177–182 CrossRef CAS PubMed.
- J. J. Docherty, M. M. Fu, B. S. Stiffler, R. J. Limperos, C. M. Pokabla and A. L. De Lucia, Antivir. Res., 1999, 43, 145–155 CrossRef CAS PubMed.
- C. Faustino, A. P. Francisco, V. Isca and N. Duarte, Curr. Pharm. Des., 2018, 24, 4270–4311 CrossRef CAS PubMed.
- P. de Medina, R. Casper, J. F. Savouret and M. Poirot, J. Med. Chem., 2005, 48, 287–291 CrossRef CAS PubMed.
- P. Lorenz, S. Roychowdhury, M. Engelmann, G. Wolf and T. F. W. Horn, Nitric oxide, 2003, 9, 64–76 CrossRef CAS.
- S. A. Andrabi, M. G. Spina, P. Lorenz, U. Ebmeyer, G. Wolf and T. F. W. Horn, Brain Res., 2004, 1017, 98–107 CrossRef CAS.
- C. Breuer, G. Wolf, S. A. Andrabi, P. Lorenz and T. F. Horn, Neurosci. Lett., 2006, 393, 113–118 CrossRef CAS.
- D. McCormack and D. McFadden, J. Surg. Res., 2012, 173, 53–61 CrossRef.
- Y. L. Tang and S. W. Chan, Phytother. Res., 2014, 28, 1581–1588 CrossRef CAS.
- K. Banik, A. M. Ranaware, C. Harsha, T. Nitesh, S. Girisa, V. Deshpande and A. B. Kunnumakkara, Pharmacol. Res., 2020, 153, 104635 CrossRef CAS.
- Y. Kimura, H. Okuda and M. Kubo., J. Ethnopharmacol., 1995, 45, 131–139 CrossRef CAS.
- N. C. Ulrich, J. G. Kodet, N. R. Mente, C. H. Kuder, J. A. Beutler, R. J. Hohl and D. F. Wiemer, Bioorg. Med. Chem., 2010, 18, 1676–1683 CrossRef CAS PubMed.
- A. Chaudhary, S. N. Pandeya, P. Kumar, P. P. Sharma, S. Gupta, N. Soni, K. K. Verma and G. Bhardwaj, Mini Rev. Med. Chem., 2007, 12, 1186–1205 CrossRef.
- J. McNulty and D. McLeod, Tetrahedron Lett., 2011, 52, 5467–5470 CrossRef CAS.
- S. I. Um, Y. Kang and J. K. Lee, Dyes Pigm., 2007, 75, 681–686 CrossRef CAS.
- M. Grätzel, Nature, 2001, 414, 338–344 CrossRef.
- Y. C. Zhu, D. H. He and Z. R. Yang, Spectrochim. Acta, Part A, 2009, 72, 417–420 CrossRef PubMed.
- V. Palakollu and S. Kanvah, New J. Chem., 2014, 38, 5736–5746 RSC.
- S. Mishra, P. Awasthi, J. Singh, R. K. Gupta, V. Singh, R. Kant, R. Jeet, D. Goswami and A. Goel, J. Org. Chem., 2018, 83, 3669–3678 CrossRef CAS.
- Y. Zhang, C. Liang and S. A. Jiang, New J. Chem., 2017, 41, 8644–8649 RSC.
- A. K. Sinha, V. Kumar, A. Sharma, A. Sharma and R. Kumar, Tetrahedron, 2007, 63, 11070–11077 CrossRef CAS.
- J. E. Robinson and R. J. K. Taylor, Chem. Commun., 2007, 1617–1619 RSC.
- F. Alonso, P. Riente and M. Yus, Tetrahedron Lett., 2009, 50, 3070–3073 CrossRef CAS.
- S. E. Shetgaonkar and F. V. Singh, Synthesis, 2018, 3540–3548 CAS.
- F. V. Singh and P. B. Kole, Synthesis, 2019, 1435–1444 CrossRef CAS.
- S. E. Shetgaonkar and F. V. Singh, Syn. Commun., 2019, 49, 1092–1102 CrossRef CAS.
- P. B. Kole and F. V. Singh, Aust. J. Chem., 2019, 72, 524–532 CrossRef CAS.
- S. E. Shetgaonkar and F. V. Singh, Syn. Commun., 2020, 50, 2511–2521 CrossRef CAS.
- R. Kanimozhi and F. V. Singh, J. Mol. Struct., 2020, 1219, 128633 CrossRef CAS.
- S. Chandrasekar and F. V. Singh, ChemistrySelect, 2020, 5, 7452–7459 CrossRef CAS.
- C. Subashini, L. J. Kennedy and F. V. Singh, J. Mol. Struct., 2021, 1226, 129365 CrossRef.
- A. Goel and V. J. Ram, Tetrahedron, 2009, 65, 7865–7913 CrossRef CAS.
- A. Goel, F. V. Singh, M. Dixit, D. Verma, R. Raghunandan and P. R. Maulik, Chem. – Asian J., 2007, 2, 239–247 CrossRef CAS.
- A. Goel and F. V. Singh, Tetrahedron Lett., 2005, 46, 5585–5587 CrossRef CAS.
- C. P. Sharma, N. M. Gupta, J. Singh, R. A. K. Yadav, D. K. Dubey, K. S. Rawat and A. Goel, J. Org. Chem., 2019, 84, 7674–7684 CrossRef CAS.
- F. V. Singh, S. Chaurasia, M. D. Joshi, A. K. Srivastava and A. Goel, Bioorg. Med. Chem. Lett., 2007, 17, 2425–2429 CrossRef CAS PubMed.
- P. B. Kole and F. V. Singh, J. Mol. Struct., 2022, 1250, 131622 CrossRef CAS.
- M. Krishnan and F. V. Singh, J. Mol. Struct., 2022, 1256, 132544 CrossRef CAS.
- M. Krishnan and F. V. Singh, Synthesis, 2022, 5273–5280 CAS.
- A. G. Pramod, Y. F. Nadaf and C. G. Renuka, J. Mol. Struct., 2019, 1194, 271–283 CrossRef CAS.
- P. Prabukanthan, C. Raveendiran, G. Harichandran and P. Seenuvasakumaran, Results Chem., 2020, 2, 100083 CrossRef CAS.
- N. Mataga, Y. Kaifu and M. Koizumi, Bull. Chem. Soc. Jpn., 1956, 29, 465–470 CrossRef CAS.
- C. Subashini, L. J. Kennedy and F. V. Singh, J. Mol. Struct., 2021, 1226, 129365 CrossRef.
- A. Goel, V. Kumar, S. P. Singh, A. Sharma, S. Prakash, C. Singh and R. S. Anand, J. Mater. Chem., 2012, 22, 14880–14888 RSC.
- P. B. Kole and F. V. Singh, Heterocycl., 2021, 103, 624–659 CrossRef CAS.
- R. Pratap, R. Kumar, P. R. Maulik and V. J. Ram, Tetrahedron Lett., 2006, 47, 2949–2952 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nj03458e |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |