
Ab initio investigation of hot electron transfer in CO2 plasmonic photocatalysis in the presence of hydroxyl adsorbate†
Zelio
Fusco
 *a,
Dirk
Koenig
b,
Sean C.
Smith
b and
Fiona Jean
Beck
a
*a,
Dirk
Koenig
b,
Sean C.
Smith
b and
Fiona Jean
Beck
a
aRenewable Fuel Group, School of Engineering, College of Engineering, Computing and Cybernetics, The Australian National University, Canberra, ACT 2601, Australia. E-mail: zelio.fusco@anu.edu.au
bIntegrated Materials Design Lab, The Australian National University, Canberra, ACT 2601, Australia
First published on 16th April 2024
Abstract
Photoreduction of carbon dioxide (CO2) on plasmonic structures is of great interest in photocatalysis to aid selectivity. While species commonly found in reaction environments and associated intermediates can steer the reaction down different pathways by altering the potential energy landscape of the system, they are often not addressed when designing efficient plasmonic catalysts. Here, we perform an atomistic study of the effect of the hydroxyl group (OH) on CO2 activation and hot electron generation and transfer using first-principles calculations. We show that the presence of OH is essential in breaking the linear symmetry of CO2, which leads to a charge redistribution and a decrease in the O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO angle to 134°, thereby activating CO2. Analysis of the partial density of states (pDOS) demonstrates that the OH group mediates the orbital hybridization between Au and CO2 resulting in more accessible states, thus facilitating charge transfer. By employing time-dependent density functional theory (TDDFT), we quantify the fraction of hot electrons directly generated into hybridized molecular states at resonance, demonstrating a broader energy distribution and an 11% increase in charge-transfer in the presence of OH groups. We further show that the spectral overlap between excitation energy and plasmon resonance plays a critical role in efficiently modulating electron transfer processes. These findings contribute to the mechanistic understanding of plasmon-mediated reactions and demonstrate the importance of co-adsorbed species in tailoring the electron transfer processes, opening new avenues for enhancing selectivity.
CO angle to 134°, thereby activating CO2. Analysis of the partial density of states (pDOS) demonstrates that the OH group mediates the orbital hybridization between Au and CO2 resulting in more accessible states, thus facilitating charge transfer. By employing time-dependent density functional theory (TDDFT), we quantify the fraction of hot electrons directly generated into hybridized molecular states at resonance, demonstrating a broader energy distribution and an 11% increase in charge-transfer in the presence of OH groups. We further show that the spectral overlap between excitation energy and plasmon resonance plays a critical role in efficiently modulating electron transfer processes. These findings contribute to the mechanistic understanding of plasmon-mediated reactions and demonstrate the importance of co-adsorbed species in tailoring the electron transfer processes, opening new avenues for enhancing selectivity.
New conceptsThe development of plasmonic photocatalysts for CO2 reduction is slowed down by a lack of atomistic understanding of the CO2 behaviour on metallic structures under optical bias. Illuminated metallic nanoparticles generate hot electrons which can be transferred to adsorbate molecular species, thereby initiating a chemical reaction. While it is accepted that co-adsorbed species and/or reaction intermediates can influence the reaction pathways, this aspect is often overlooked. We report a first-principles investigation of plasmon-driven CO2 reduction in the presence of a co-adsorbed hydroxyl group (OH) on a gold cluster. By using time-dependent density functional theory, we investigate the dynamics of plasmon excitation and decay and we analyse the energy distribution of hot electrons and quantify their transfer to the molecular adsorbates as a function of the distance, excitation frequency and an increasingly non-monochromatic frequency distribution. We demonstrate the importance of co-adsorbed species in changing the energetics of the system, showing that the OH group mediates the orbital hybridization between gold and CO2 resulting in more accessible states, thus facilitating charge transfer. These findings contribute to the mechanistic understanding of plasmon-mediated reactions and demonstrate the importance of co-adsorbed species in tailoring the electron transfer processes, opening new avenues for enhancing selectivity. |
Introduction
Subwavelength metallic nanoparticles that support plasmonic resonances represent a versatile platform for nanoscale light manipulation. These structures hold great potential in nanophotonic applications that benefit from enhanced near-field light–matter interactions, such as plasmon-enhanced biosensing,1–3 surface-enhanced Raman scattering4,5 and photocatalysis.6–8 Harnessing the hot electrons (HE)9 generated by the non-radiative decay of illuminated metal nanoparticles before they thermalise is an attractive way to initiate and selectively drive chemical reactions,10–13 and has recently attracted significant interest in the heterogeneous catalysis community.14–18 Hot electrons can be rapidly transferred to the orbitals of nearby molecules, thereby driving catalytic chemical reactions. The energy and momentum distribution of the hot carriers depend on multiple factors, including material, particle morphology, crystal orientation, defects and environs,19,20 influencing many aspects of heterogeneous catalysis, including the reaction rate, selectivity, turnover frequency, yield, and reaction order.21 To date, the significant role of HEs provided by photoexcited plasmons has been attributed to many reactions, including water splitting,22 bond cleavage23 or formation,24 and carbon dioxide reduction.25Once generated, HEs can be transferred to nearby molecules directly or indirectly.26,27 In the former process, HEs are directly excited from the metal into the unoccupied molecular orbitals of interacting acceptor adsorbates. The latter process comprises HEs generated within metal nanoparticles, subsequently being scattered to empty states of surface-adsorbed molecules with suitable overlaps in energy and momentum distribution. The direct injection of HEs is a faster and more efficient process compared to the indirect transfer,28 and offers great potential for enabling high selectivities to adsorbed chemical species and their reaction paths.28–30 High selectivities can be achieved by precisely tuning the plasmon resonance and the energy distribution of HEs to align with targeted unoccupied adsorbate states, thereby selectively promoting a specific reaction pathway.6,17,30 The design and engineering of the optoelectronic properties of plasmonic architectures are thus important in particular to achieve selectivity in multiproduct reactions such as CO2 reduction.31
In the context of plasmon-enabled chemical transformations, reaction pathways and activation energies are often being investigated using ground-state density functional theory (DFT) methods.32–35 Although these analyses provide important information on the systems and contribute to the understanding of the action mechanisms, they cannot accurately capture the physics of excited-states and the dynamics of the photogenerated hot carriers. Instead, time-dependent DFT (TDDFT) approaches have been increasingly employed to shed light on plasmon dynamics and hot carrier generation36–38 by providing atomic-scale insights into excitation and transfer processes of electrons. In particular, Rossi et al. have developed a real-time TDDFT methodology to study the dynamics of plasmon-molecule systems,39–43 demonstrating that the generated HEs have an energetic and spatial distribution that depends on the atomic structure, with lower-coordinated atoms exhibiting a higher proportion of HEs.41 These can be directly transferred to adjacent semiconductors40 or molecules,29,42 even when the molecule is not chemisorbed to the plasmonic nanoparticle:42 using Ag–CO as a model system, Fojt et al. demonstrated that HEs can be directly generated on the molecule at distances up to 5 Å.
We extend the approach of Rossi et al. to investigate the energy required to activate carbon dioxide (CO2) on a small Au cluster in the presence of co-adsorbed environmental species and probe the carrier dynamics. It is widely accepted that the reaction environment and intermediate species can steer the reaction pathway44–46 by altering the potential energy landscape of the chemical transformation. However, a mechanistic study of the effect of intermediate adsorbates on HE generation and transfer in CO2 reduction is critical to enhance our understanding of the fundamental processes involved. Here, we focus specifically on the role of adsorbed hydroxyl groups, as they take part in the CO2RR and are crucial for enhanced stability, activity and selectivity.47–49 By using TDDFT calculations, we study the HE transfer process as a function of distance, excitation frequency and an increasingly non-monochromatic frequency distribution, extending the fundamental understanding of HE transfer across nanoparticle–molecule interfaces and their impact on the overall CO2 reduction reaction.
Results and discussion
System description
We consider CO2 as a reactant species because its reduction is of significant industrial and societal importance: artificial synthesis of hydrocarbons and alcohols from CO2, using water to provide hydrogen and sunlight to drive the reaction, has the potential to provide renewable fuels and chemicals at scale.17,50 A considerable challenge in converting CO2 into organic products lies in its activation, due to the notably high reaction barrier linked to the initial electron transfer (−1.9 V vs. standard hydrogen electrode). Adsorption of CO2 on catalytic surfaces forms partially charged CO2δ− species which – due to charge redistribution – results in a change in the OCO bond angle and leads to a bent configuration,51,52 as shown in Fig. 1(a). As a consequence, the lowest unoccupied molecular orbital (LUMO) shifts to lower energies, reducing the energy barrier required for electron transfer and promoting a CO2 reduction reaction.53
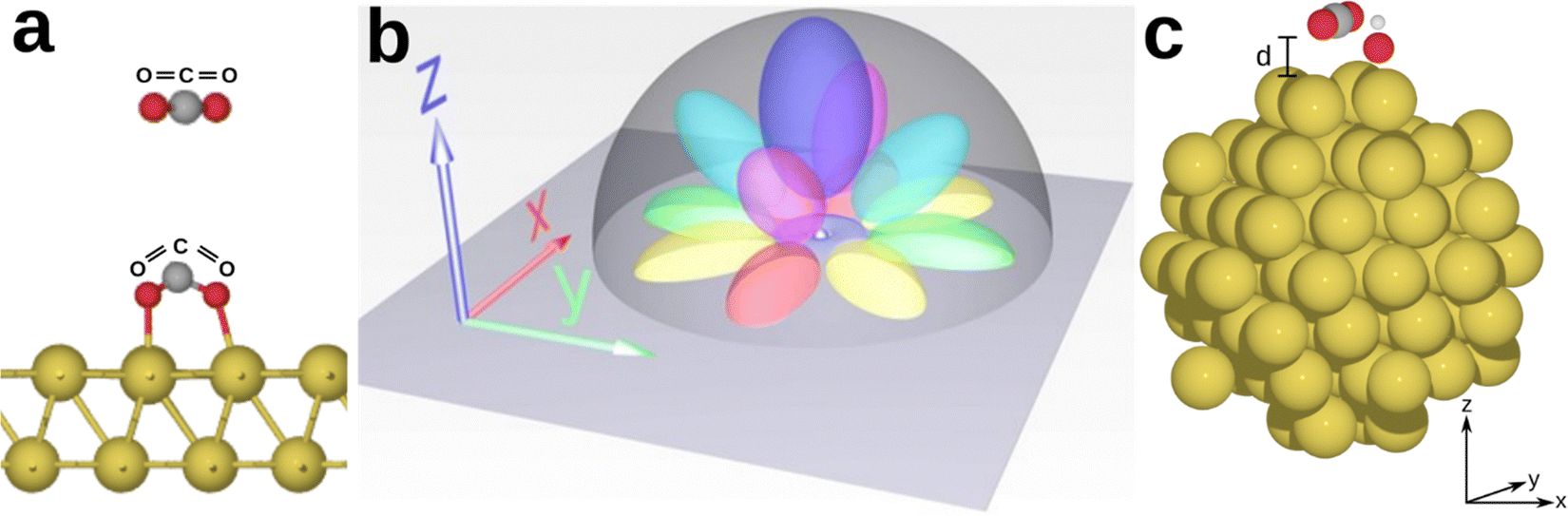 | ||
| Fig. 1 (a) Activation of CO2 on a catalytic surface. The free molecule has a linear configuration with an OCO bond angle of 180°; upon interaction with the surface atoms, the bond angle reduces, thus activating CO2. (b) Spatial scheme of the valence orbitals of a gold atom (bright grey sphere) on the catalytic surface: colour-coding represents the spherical 6s (grey), dxy (yellow), dx2−y2 (red and green), dxz (purple), dyz (cyan) and dz2 (blue). The colours refer to AO lobes aligned to the x-/y-/z-axis (red/green/blue) with superimposed colours indicating bi-axial orientation; xy shown by yellow, xz shown by magenta, and yz shown by cyan. (c) Schematic representation of the system analysed in this work, comprising of a Au87 cluster with an OH group and CO2. | ||
We use gold as a standard plasmonic material because of its unique electronic configuration which allows it to efficiently mediate catalytic reactions.31,54 The electronic configuration of Au is shaped considerably by its atomic orbital arrangement. Gold has a 5d106s1 valence orbital configuration which consists of five filled 5d atomic orbitals that enable hybridization with various reactant molecules, and a partially filled 6s orbital. The five 5d orbitals are strongly lobed and are overlaid by the diffused spherical 6s orbital which does not hybridize with the 5d orbitals. The large nuclear charge of gold leads to a more pronounced nuclear-electron attraction of the valence electron shell: due to relativistic effects, the s-orbitals are found to contract in response to the large nuclear charge, while the d-orbitals are in fact expanded, increasing their involvement in the chemical and physical properties of gold.55,56 These specific atomic orbital lobes allow for various combinations of hybridisation and formation of complexes with molecular species and ligands, making gold a promising material for catalytic applications and HE transfer. It is worth noting that this versatility may pose challenges to spatial selectivity, as gold can couple with numerous molecular species. However, the resulting higher spatial probability of electron transfer due to an increased overlap integral of the atomic and/or molecular orbitals involved promotes catalytic reactions. Selectivity for a specific reaction can be achieved by carefully tuning and aligning the energy distribution of HEs with the energy of the lowest unoccupied molecular orbitals (LUMOs) of the reactant species,28 as discussed at the end of this section.
This is particularly relevant for multibranched chemical transformation like CO2 reduction. Most of the high performing experimental literature on CO2 reduction uses aqueous media that are either near-neutral or slightly alkaline.57–59 While there are recent reports on CO2 reduction under acidic conditions,60,61 neutral and alkaline electrolytes are preferred because they provide a better CO2 activation capacity, improve the suppression of the competing hydrogen evolution reaction and help in further promoting the C−C coupling efficiency.62,63 An example of CO2 reduction to typical products like formiate and ethylene in alkaline media can be represented by the following equations:64,65
| CO2 + H2O + 2e− → HCOO− + OH− |
| 2CO2 + 8H2O + 12e− → C2H4 + 12OH− |
Under these conditions, the competing hydrogen evolution reaction can be drastically suppressed and the generated OH− intermediate can be an active participant in the reaction, speeding up the activation of CO2 and providing a means for further electron transfer.
Electronic ground state properties
We start by investigating the adsorption and activation of a CO2 molecule on a Au87 cluster in presence and absence of an adsorbed hydroxyl group through structural relaxation. The two model systems are Au87 + CO2 and Au87 + OH + CO2, with the CO2 initially placed at a varying distance of 2 Å < d < 5 Å on a bridge-site, as this is the most catalytically active site for CO2 reduction on gold surfaces,66 while the OH group is adsorbed on an on-top site.
Fig. 2(a) shows the OCO bond angle when the systems reached equilibrium (forces <0.05 eV per atom). In the absence of the OH group, CO2 maintains a linear symmetry with a final equilibrium OCO angle which fluctuates around 178°, independent of the distance from the Au cluster (red curve). Contrarily, the presence of an OH group perturbs the charge symmetry of the molecule, leading to structural transformation for d < 3 Å. This results in a bent configuration with a final equilibrium OCO angle of 133°, in line with the previous theoretical reports on activated CO2 on catalysts’ surfaces.51,52 Similar results are also obtained on periodic gold slabs (see ESI,† S1). Simultaneously, as CO2 approaches the Au cluster (d < 3 Å), a redistribution of the charge density occurs, as can be seen by the different LUMO shapes of the system (Fig. 2(b)). A Bader charge analysis67,68 was performed to investigate the charge redistribution which induces a charge dipole on the CO2 molecule, with negative charges on the oxygen atoms (Fig. S2, ESI†). This in turn breaks its linear symmetry and results in an increased likelihood of adsorption on the Au cluster. These results suggest the importance of investigating the effects of co-adsorbed hydroxyl groups – and possibly other environmental species – in the overall activation of CO2, as well as the role of such species in HE transfer for efficiently driving catalytic processes.
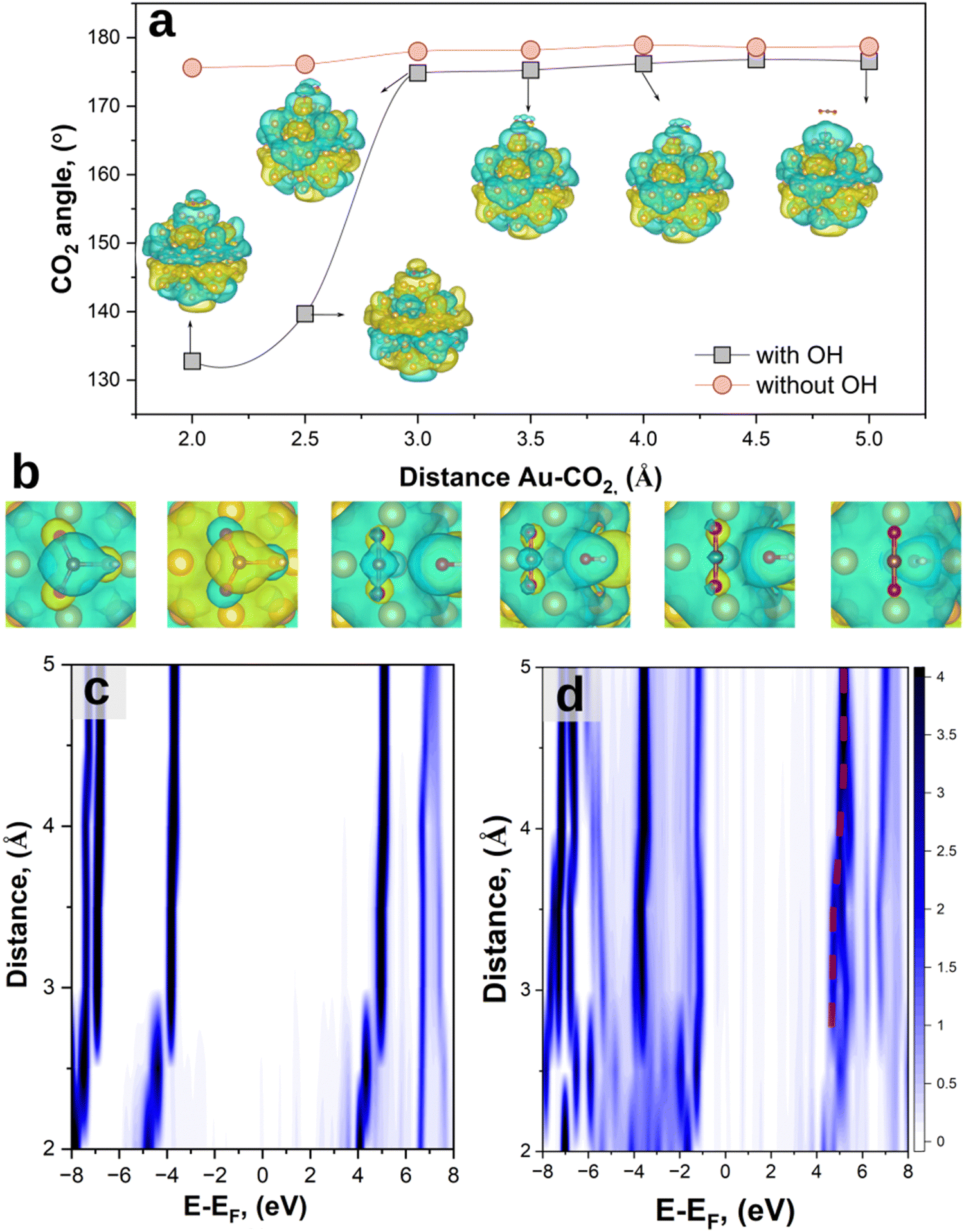 | ||
| Fig. 2 (a) CO2 bond angle as a function of the distance during the adsorption process on Au and Au + OH clusters, represented by red and black curves, respectively. The LUMO wave functions of the systems are also shown at the same isosurface value of 0.05 a.u. (b) Magnification of the top-view of the LUMO at the same distances of (a). At d = 2.5 Å, when transitioning from weak to strong hybridization (from 3 Å to 2.5 Å), we observe a phase flip in the molecular orbital (MO) lobes. This is due to the spin eigenfunction of the MO changing state, i.e. from spin-up to spin-down. When a phase flip happens, it means that the energy difference between both spin states of the MO is very small (≤kBT) so that both electrons can swap their spin eigenfunction (spinor) within the MO. The spin–flip occurrence when going from 3 Å to 2.5 Å and then back to the original spin configuration shows that one spin configuration of the LUMO has a slightly higher ELUMO compared to the ELUMO at d = 2 Å. (c) and (d) Map of the projected density of states as a function of the distance and energy for Au + CO2 and Au + CO2 + OH. | ||
When CO2 approaches the Au and Au + OH clusters, the strength of orbital hybridization increases.42 This can be demonstrated by analysing the projected density of states (pDOS) of the adsorbed molecules as a function of their distance to the Au surface for the different systems (Fig. 2(c) and (d)). At distances d ≳ 4 Å for both Au + CO2 and Au + OH + CO2 systems, the pDOS converges to the molecular density of states (DOS) of the isolated molecules. As d decreases, the pDOS eventually splits into several branches due to hybridisation with Au orbitals.42 The addition of the OH group qualitatively shows a similar trend with a branched pDOS (Fig. 2(b)), but with an increased number of electronic states. In other words, the OH group mediates the orbital hybridization between Au and CO2, allowing for more accessible states.
We anticipate that these states are of significant importance for the further HE transfer process;31 in fact, a HE transfer to the adsorbed molecule requires the presence of acceptor states at suitable energies, i.e. where the pDOS is present,42 thus having more unoccupied levels which can lead to an increase of catalytic efficiency.
Additionally, this adsorption process is also accompanied by a shift towards a lower binding energy of the LUMO, decreasing from ∼4.9 eV (d ≳ 4 Å) to ∼4.6 eV (d ∼ 3.5 Å) in line with the previous literature,14 eventually splitting up into 4.7 eV (LUMO+1) and 4.5 eV (LUMO) for d < 3 Å due to exchange interactions rising with inverse inter-atomic distance (with the overlap integral of associated orbitals forming a bond). This is another indication that the OH group acts as a “bridge” between CO2 and Au in terms of electronic coupling, which can then enable electron transfer from a considerably larger distance.
The total DOS of the different systems is shown in Fig. S3 (ESI†). The introduction of CO2 and OH induces only minor alterations in the total DOS due to the dominant contribution of states originating from the Au cluster. Notably, in the presence of the molecules, the Au cluster exhibits a slightly positive charge, which becomes more pronounced when the OH group is involved. The shift of the HOMO to a higher energy is ∼0.1 eV for Au + CO2 (red curve) and ∼0.16 eV for Au + CO2 + OH (green curve). This indicates that both the adsorbed OH and the CO2 attract negative charges from the Au nanoparticle, which becomes apparent when comparing electronegativities, ionization energies, and electron affinities of C and O vs. Au.69 Consequently, an electron transfer should occur from the Au NP to the molecular species. The CO2 molecule induces a significantly increased shift of the HOMO to higher binding energy as compared to the shift when the OH group is included in the system. This dominant energy shift due to the CO2 molecule suggests that an electron transfer from the Au cluster will eventually reside with the CO2 molecule rather than with the OH group, the latter presenting merely a transient charge path for transferring an electron to the CO2 molecule.
Plasmon dynamics and hot electron (HE) generation
We now study the time evolution of the Au, Au + CO2 and Au + CO2 systems under light illumination; when present, CO2 is placed at a distance of 2 Å from the Au surface. First, the photo absorption spectra of the system are calculated by applying an initial Dirac pulse (δ-kick) in the linear-response regime along the z-direction (and perpendicular to the CO2 molecule, when present). As the Fourier transform of the δ-kick contains all frequencies with equal proportion, this kick equally excites all the optically allowed transitions and allows us to extract the photoabsorption spectrum. Next, we analyse the energy evolution of the systems after excitation with ultra-short optical pulses at the plasmonic resonance energy, which allows us to calculate the energy distribution of hot carriers generated by the decay of the plasmon. This analysis allows us to estimate the plasmon dephasing time after which most of the energy has been transferred to hot carriers.The photoabsorption spectra (Fig. 3(a)) are characterized by a smoothly growing intensity starting at around 1 eV, with a plasmon peak at ∼2.5 eV, followed by a wide modulation up to 4 eV. The latter is in line with previous works on the optical properties of small gold clusters70,71 and can be ascribed to interband transitions, which have a larger probability and a stronger contribution to the photoabsorption spectra than the plasmon excitation.72 As expected, the plasmon peak is relatively weak, in line with the previous theoretical works on small gold clusters73–75 which showed that a clear plasmon emerges only when the cluster size exceeds 2 nm.76 For the combined systems, we observe a slight red-shift (green and red curves in Fig. 3(a)) of the plasmon peak from 2.66 eV for the single Au cluster, to 2.56 eV and 2.52 eV for Au + CO2 Au + CO2 + OH, respectively. This indicates the presence of additional decay channels for the plasmon due to formation of hybridized cluster-molecule states.
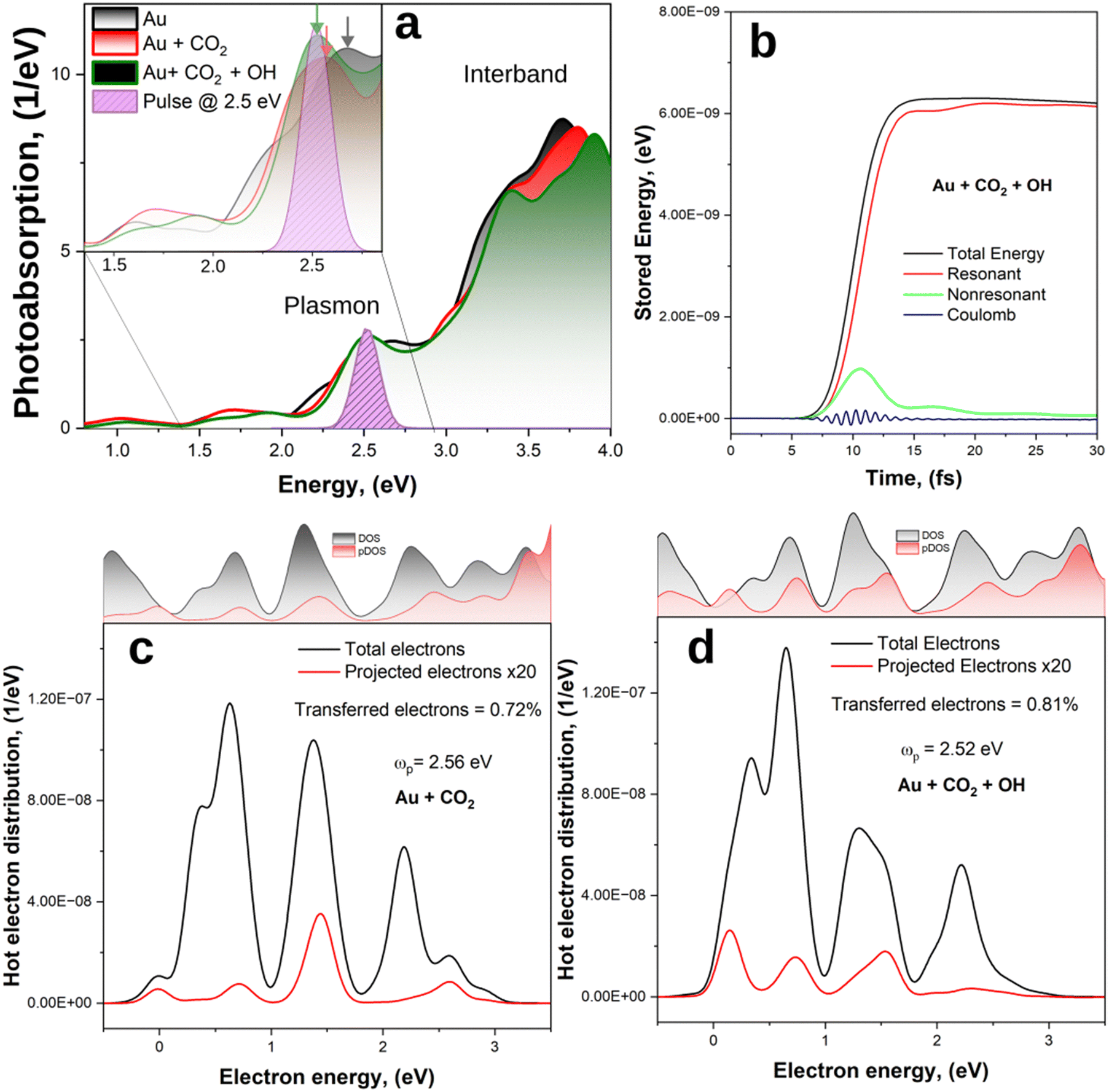 | ||
| Fig. 3 (a) Photoabsorption spectra of Au, Au + CO2 and Au + CO2 + OH. The purple curve represents a typical driving laser pulse in the frequency domain, centered at the LSPR of the Au + CO2 + OH. The colour coded arrows represent the LSPR of the different systems. (b) Time evolution of the stored energy for Au + CO2 + OH, highlighting the different energy contributions. (c) and (d) Generated and transferred HEs energy distribution for the Au + CO2 and Au + CO2 + OH systems, respectively, averaged between 15 and 30 fs. | ||
Next, we excite the systems with a Gaussian pulse – E(t) = E0 cos(ωp(t − t0))exp(−σ2(t − t0)2/2) – with ωp set to be in resonance with the systems (ωp = 2.66 eV for Au, 2.56 eV for Au + CO2 and 2.52 eV for Au + CO2 + OH), centred at 10 fs and with a temporal FWHM of 5 fs (Fig. 3(a), purple peak). In accord with the Heisenberg's uncertainty principle —Δt × ΔE ≥ h/2 – a FWHM of Δt = 5 fs corresponds to an uncertainty in the photon energy range of ΔE = 0.41 eV, presenting a broadband excitation as intended for plasmonic excitation with solar irradiation.
The energy of the light pulse is absorbed by the electrons in the system, promoting them into excited states. The electronic energy is not equally distributed, and can be divided into Coulomb energy, and electron–hole transition energy contributions41,77 as shown for the Au + OH + CO2 system in Fig. 3(b). The latter can be further separated into the energy of transitions resonant with the excitation pulse (i.e. with frequency ω = ωp ± 2σ), constituting hot carriers, and non-resonant transitions, attributed to transitions from d-states in the metal nanoparticle which screen the plasmon. The evolution of the different energy contributions is similar for all the investigated systems (Fig. S4, ESI†), as the plasmon dephasing process is not affected on a significant scale by a single molecular species. Initially, the plasmon is excited via Coulomb interactions78 and non-resonant contributions carry most of the energy during the plasmon excitation. After ∼15 fs, as the plasmon decays, the total energy is redistributed into electron−hole excitations that are resonant with the pulse.
Having established that after ∼15 fs the energy of the plasmon is mostly stored in hot carriers, we proceed to analyse the energy distribution of the photogenerated HEs, as well as the fraction of such HEs directly excited to states localised on the CO2 molecule for the Au + CO2 and Au + CO2 + OH systems. The HE energy distributions are shown in Fig. 3(c) and (d), where we have averaged the energies of the HEs between 15 fs and 30 fs as the resonant transitions have already reached their steady state. Therefore, HEs possess one temperature in accord with Fermi–Dirac statistics relative to the energy of their steady state as given by the convolution of the initial HE population with the total DOS of the system in the relevant energy range. For both the analysed systems, the generated HEs (black lines) can have energies up to the laser pulse frequency (EF + ℏωp) due to the conservation of energy. In addition, their energy distributions are not uniform within this range but show occupation probabilities that match the total DOS of the systems; electrons can only be excited to existing unoccupied states. The total DOS for Au + CO2 and Au + CO2 + OH are similar as they are dominated by the Au contributions. It follows that all systems have similar total HE energy distributions, presenting major peaks at ∼0.7 eV, 1.3 eV and 2.2 eV.
Conversely, the transferred electron energy distributions (red lines), which represent the fraction of HEs directly excited to states localised on the CO2, mirror the pDOS and exhibit differences between the systems. In particular, in the absence of the OH group, the fraction of HEs generated on the CO2 is 0.72%, with most of them having an energy of 1.4 eV. The addition of the OH group increases the density of electronic states (see also Fig. 2(d)), thus opening more channels for electron transfer and effectively improving the charge transfer to 0.81%, which corresponds to an 11% increase with respect to the case without OH.
In the next section, we investigate the effect of the excitation parameters – pulse frequency (ωp) and width (FWHM)– on the direct excitation of HEs to CO2 orbitals. The pulses are visualised in the time domain as shown in Fig. S5 (ESI†) and have varying ωp from 1.6 eV to 2.8 eV, and FWHM from 4 fs (ΔE = 0.52 eV) to 15 fs (ΔE = 0.14 eV). The energy range was chosen to investigate the HE generated from the plasmon excitation as well as from interband excitation, noting that the threshold energy for interband transition (5d to 6sp) for gold is about 2.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) eV.79,80 We have also investigated a pulse at 3.75 eV with a FWHM of 5 fs to match the maximum of the interband transitions within the analysed region (see Fig. 3(a)). The energy distributions of the HEs excited to states localised on the CO2 molecule are reported in Fig. 4(a)–(d) as a function of pulse energy; the distributions for the total amount of HEs in both systems are given in Fig. S6 (ESI†). The amount of generated HEs increases with the increase of pulse width. For FWHM = 4 fs, we obtain a maximum HE density when ωp ∼ 2.8 eV, while for FWHM = 15 fs, the maximum occurs when 2.3 < ωp < 2.7 eV for all systems. In this energy range, the pulse excitation approaches the plasmon resonance of the systems (Fig. 3(a)), thus emphasizing the critical role of spectral overlap in efficiently modulating electron transfer processes.81,82 Furthermore, below ∼2 eV, photoabsorption decreases, thus resulting in a negligible generation of HEs within the molecule.
eV.79,80 We have also investigated a pulse at 3.75 eV with a FWHM of 5 fs to match the maximum of the interband transitions within the analysed region (see Fig. 3(a)). The energy distributions of the HEs excited to states localised on the CO2 molecule are reported in Fig. 4(a)–(d) as a function of pulse energy; the distributions for the total amount of HEs in both systems are given in Fig. S6 (ESI†). The amount of generated HEs increases with the increase of pulse width. For FWHM = 4 fs, we obtain a maximum HE density when ωp ∼ 2.8 eV, while for FWHM = 15 fs, the maximum occurs when 2.3 < ωp < 2.7 eV for all systems. In this energy range, the pulse excitation approaches the plasmon resonance of the systems (Fig. 3(a)), thus emphasizing the critical role of spectral overlap in efficiently modulating electron transfer processes.81,82 Furthermore, below ∼2 eV, photoabsorption decreases, thus resulting in a negligible generation of HEs within the molecule.
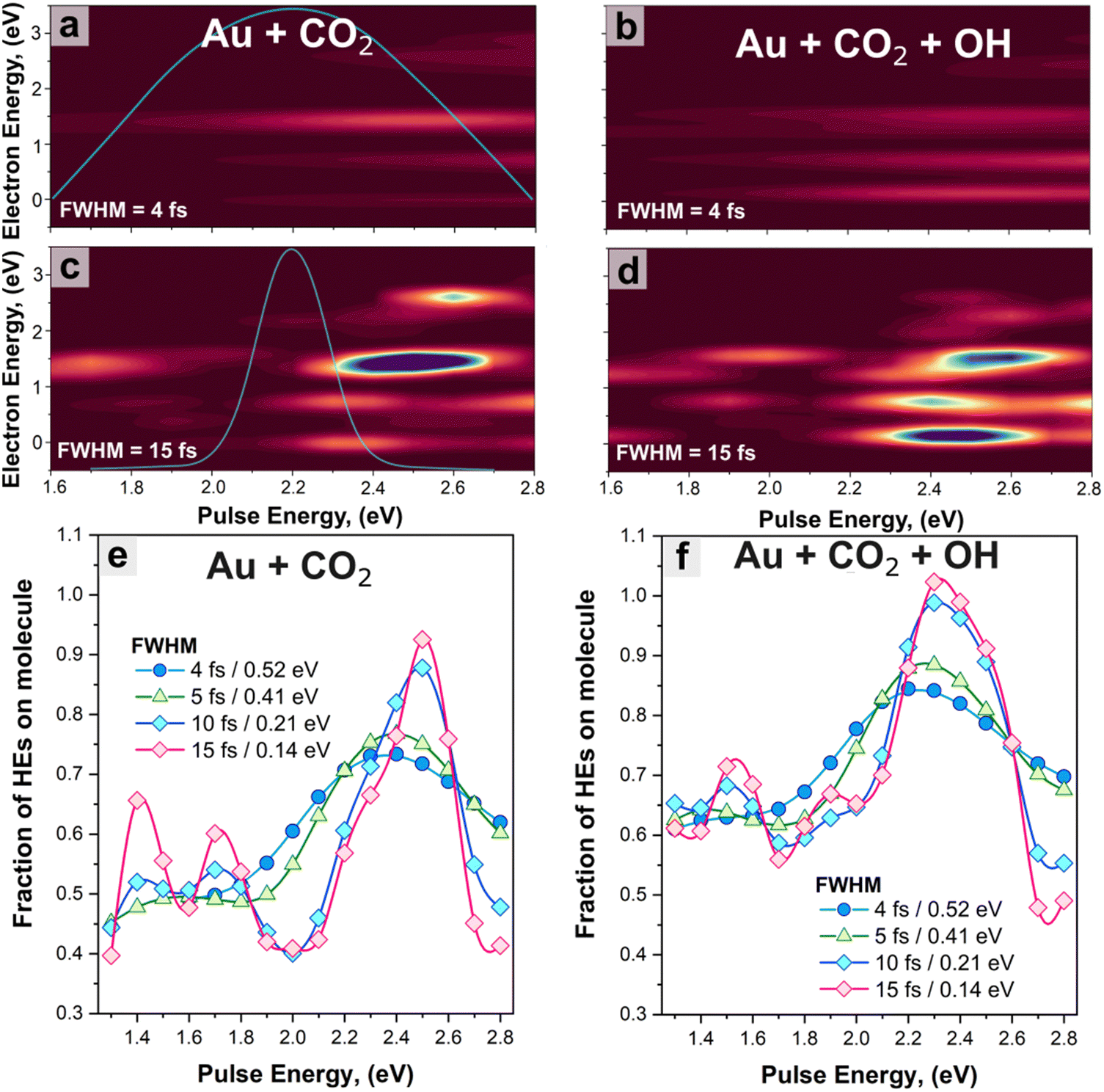 | ||
| Fig. 4 (a) and (b) Amount of HEs generated on CO2 and their energy distribution as a function of the pulse frequency (with a temporal FWHM of 4 fs (0.52 eV)) for Au + CO2 and Au + CO2 + OH, respectively. (c) and (d) Same as a and b, but the excitation has a FWHM of 15 fs (0.14 eV). The light blue lines in (a) and (c) represent a typical laser pulse in the frequency domain. (e) and (f) Fraction of carriers transferred to the molecule as a function of the excitation frequency for multiple FWHM values. | ||
At energies above the plasmon resonance, hot electrons are being generated via interband transitions.72 While the total amount of HEs the systems is higher at the interband transitions (because of the larger photoabsorption72,80), the fraction of those HEs transferred to the CO2 molecule is slightly smaller than at the plasmon resonance (Fig. S7, ESI†). This suggests that using the interband transition to activate and subsequentially reduce CO2 has a little advantage over the plasmon absorption in this specific system. In addition, exciting such transitions would come at the cost of a narrow excitation range and in-phase oscillation, preventing practical applications: using a laser to excite such interband transitions would preclude any use of sustainable energy such as solar irradiation for CO2 reduction. This finding suggests that engineering the photoabsorption, specifically the plasmon resonance energy, should be considered to efficiently leverage photogenerated HEs for tailored reactions.
In the absence of the OH group (Fig. 4(a) and (c)), the energy distribution of HEs exhibits a prominent single peak centred around ∼1.4 eV for pulse energies of 2.4–2.6 eV. Notably, approximately 55% (75%) of the HEs assume this specific energy level when subjected to pulse durations with the FWHM of 4 fs (15 fs) and under resonance conditions. Conversely, the presence of the OH group results in a broader electron energy distribution. This is attributed to the presence of more accessible hybridized states within the energy range of 0–2 eV, which can be populated by HEs. The broadened energy distribution as facilitated by the OH group provides more channels for HEs transfer and promotes the system to a different excited level, potentially leading to a different product distribution.6 This demonstrates that the presence of the OH group in the CO2 photoreduction process alters the energy landscape and transfer behaviour of HEs, thereby possibly affecting the reaction pathway, and highlight the importance of taking into account the influence of environmental and intermediate species for selective HE catalysis.
Furthermore, the excitation width also affects the HE distributions. Narrow band pulses (FWHM = 15 fs; ΔE = 0.14 eV) lead to an overall increase in the generation of HEs on the molecule; conversely, with ‘broadband’ excitation (FWHM = 4 fs; ΔE = 0.52 eV), the pulse energy is redistributed to a wider range of possible transitions in the photoabsorption spectrum, leading to a broader but less intense (less resonant) electron transfer. The transfer probability is given in Fig. 3(f) and 4, calculated as the ratio of the number of HEs generated on the molecule to the total HEs in the systems, and exhibits consistently higher values when the OH group is present, indicating a more efficient transfer. In particular, the addition of the OH group leads to an increase of ∼10 to 15% in the transfer probability, depending on pulse frequencies and FWHMs. Interestingly, when transitioning from the narrowband to broadband excitation (FWHM = 0.14 eV to 0.52 eV), despite a reduced transfer probability, the electron transfer to acceptor hybridized states remains high, with a maximum probability of ca. 0.84% for Au + CO2 + OH at ωp = 2.3 eV. This result suggests that HE transfer can still occur under broadband solar illumination with minor reductions of the yield, which is critical for the practical operation of photocatalysts.
While it is beyond the scope of this work to delve into the quantitative details of Landau damping, it is important to acknowledge the potential contribution of Landau damping from plasmons generated in gold (Au). Plasmons are collective longitudinal electron density waves which align electrons at their wave front to the resonant energy and k-vector of the plasmon wave, thereby providing energy and momentum to electrons below the resonant energy or depleting energy and momentum from electrons with energies above the resonant energy and k-vector.83 Such a resonant HE alignment, in the compound with the adsorbed OH group enabling the HE transfer to CO2 species, may present a resonant charge pump system as briefly outlined below and illustrated schematically in Fig. 5. We note that Au is excellent for plasmon propagation along its cluster surface in an arbitrary direction due to its wide-angle coverage of surface states originating from its hybrid-AO configuration (Fig. 1(b)). With the plasmon wavefront increasing the HE density at the Au atom with an adsorbed OH group, HEs get pushed into OH groups where they get weakly localized, mainly at the outermost tip (towards positively charged H atom). Further advancement of the plasmon wavefront will result in the crest of the HE density wave, providing a dynamic Coulomb force to the HE localized at the end of the OH group, likely facilitating the HE transfer from the OH group to the CO2 molecule, and providing the dynamic energy to overcome the weak localization at the OH group. With the passing of the HE density wave, its back end will dynamically deepen the electron localization potential of the OH group, reaching maximum localization ability at the density minimum of the plasmon. This ability sets the best possible condition to localize another HE in the next oscillation cycle, thereby repeating the HE transfer process.
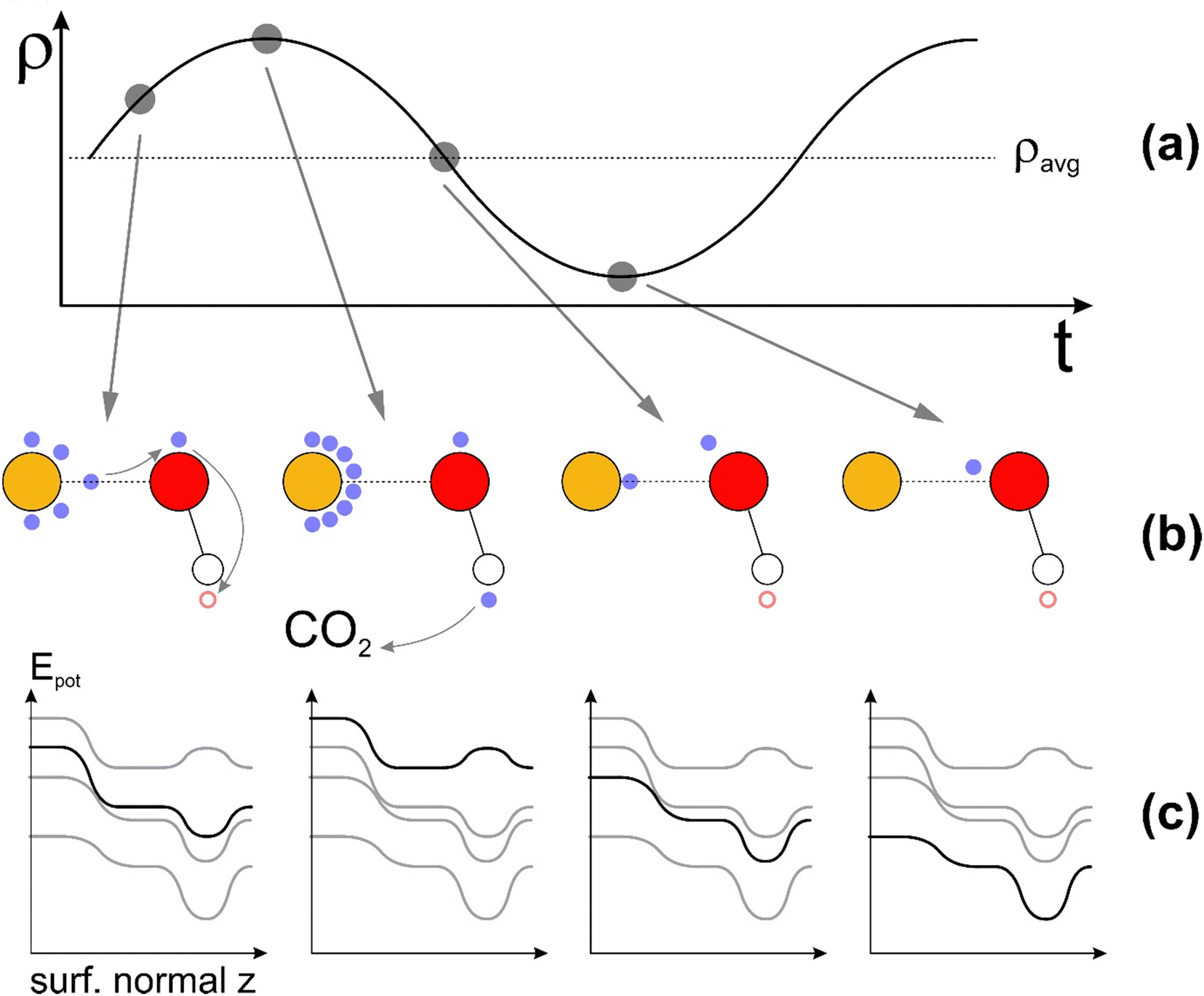 | ||
| Fig. 5 (a) Schematic of the evolution of electron density ρ over time t, featuring a surface plasmon. The grey points refer to specific situations in the charge transfer dynamics brought about by the plasmon. (b) Snapshots of charge distributions at key points of plasmonic oscillation. The gold-yellow sphere represents the Au surface atom, and the red and white spheres represent the O and H atom, respectively, of the adsorbed OH group. The small blue particles represent electrons, and the red empty circle represents the unoccupied valence state of the H atom. (c) Schematic evolution of the potential energy landscape over the Au surface atom with an adsorbed OH group, whereby the black line is the energy landscape referring to the molecule charge snapshot above. | ||
In addition to the resonant HE transfer in the energy spectrum discussed earlier, the charge pump process described above may provide an additional resonance on the time spectrum. While it is beyond the scope of our work to delve into the quantitative details of such a Landau damping-mediated HE pump in the time regime, we point out that the dynamic electric dipole moment of the OH group as the adsorbed species may play a prominent role in further enhancing the HE charge transfer to the CO2 molecule. Furthermore, the Landau damping-mediated resonance will likely have an impact on the selectivity of chemical species to receive the HE, thereby being reduced to a specific hydrocarbon or hydro-oxycarbon compound. With the CO2 molecule and H2 or H2O present as reactants, the testing of analogous adsorbed species to OH groups such as –O–Ag or –O–Li as stable adsorbents with suitable ionization energy and electron affinity of the cation (Ag, Li) provides a clear direction for future research into the ongoing optimization of adsorbent- and plasmon-mediated catalytic reduction of CO2.
This work contributes to the development of a mechanistic and kinetic picture of illuminated nanoparticle systems, which has led to active debate on the role of hot carriers in observed reaction rates.84–87 We note that the quantitative results are specific for the systems considered. However, the fundamental physics and general conclusions elucidated here remain translatable to other systems and co-adsorbed species, as recently shown for electron transfer from Ag and Cu clusters to CO molecules.42 We have studied both CO and CO2 as co-adsorbed species (Fig. S8, ESI†) and performed ground state calculations to investigate the hybridization between the Au cluster and the molecules. However, these species do not appear to hybridize – and thus promoting resonant charge transfer to CO2 molecules – to the same extent as adsorbed OH groups. Importantly, our findings highlight the critical role of OH adsorbates in determining (i) the adsorption and activation of CO2, (ii) metal–molecule hybridization, (iii) importance of matching energy levels to efficiently transfer hot electrons to hybridized states. Consequently, the presence of OH species can promote the CO2 reduction reaction to a different potential energy surface, possibly leading to different product distributions and reaction speeds. It is, therefore, very important to consider adsorbed species during the design of plasmonic catalysts to achieve high efficiency and selectivity for specific reactions, particularly when dealing with plasmonic structures functionalized with ligands.
Conclusions
We investigated the mechanisms of plasmon-driven CO2 reduction in the presence of a co-adsorbed hydroxyl group via real-time time-dependent density functional theory. Our findings show that the OH group induces a redistribution of charges in CO2, leading to favourable absorption and activation on small Au clusters. Critically, adding an OH group significantly increased hybridization between molecular states of CO2 and electronic states of Au as mediated by the OH group, enabling and improving charge transfer. This coupling is distance-dependent, reaching its maximum when CO2 is close to the Au cluster, and this behavior can be predicted by analysing the branching of the partial density of states of the molecules.By applying TDDFT, we are able to study the carrier dynamics of these systems at an atomic level, whereby the quantification of the hot electron (HE) energy distribution is critical. While the total HE distribution is largely unaffected by the presence of the OH group, the additional hybridized states facilitate the transfer of HEs to the CO2 molecule, thereby increasing the resonant direct HE transfer by 11%. Furthermore, our findings confirm the importance of spectral overlap between the excitation energy and the plasmon resonance, and show that direct HE transfer is only slightly decreased (∼1.05% to 0.84%) when the excitation is changed from a narrowband to a broadband. This finding shows that a significant chemical reaction rate still occurs under broadband excitation such as solar light. In addition, Landau dampening of the plasmon wave in the compound with the OH group may present another resonance mechanism on the time scale. This mechanism may further enhance HE transfer to CO2 for specific reductive reactions, whereby the dynamic dipole moment of the OH group coupling the plasmon to a CO2 molecule via HE transfer is a key property.
In summary, this investigation elucidates the role of adsorbed hydroxyl groups in the adsorption and activation of CO2 on plasmonic surfaces. It highlights the importance of considering adsorbed molecules to enhance direct HE transfer, ultimately enabling the design of highly efficient and selective plasmonic photocatalysts with polychromatic excitation.
Conflicts of interest
There are no conflicts to declare.References
- K. M. Mayer and J. H. Hafner, Localized surface plasmon resonance sensors, Chem. Rev., 2011, 111(6), 3828–3857 CrossRef CAS PubMed.
- Z. Fusco, et al., Nanostructured Dielectric Fractals on Resonant Plasmonic Metasurfaces for Selective and Sensitive Optical Sensing of Volatile Compounds, Adv. Mater., 2018, 30(30), 1800931 CrossRef PubMed.
- Z. Fusco, et al., Photonic Fractal Metamaterials: A Metal–Semiconductor Platform with Enhanced Volatile-Compound Sensing Performance, Adv. Mater., 2020, 32(50), 2002471 CrossRef CAS PubMed.
- Z. Fusco, et al., Self-assembly of Au nano-islands with tuneable organized disorder for highly sensitive SERS, J. Mater. Chem. C, 2019, 7(21), 6308–6316 RSC.
- J. Langer, et al., Present and Future of Surface-Enhanced Raman Scattering, ACS Nano, 2020, 14(1), 28–117 CrossRef CAS.
- J. Gargiulo, et al., From Optical to Chemical Hot Spots in Plasmonics, Acc. Chem. Res., 2019, 52(9), 2525–2535 CrossRef CAS PubMed.
- T. Tran-Phu, et al., Multifunctional nanostructures of Au–Bi2O3 fractals for CO2 reduction and optical sensing, J. Mater. Chem. A, 2020, 8(22), 11233–11245 RSC.
- J. S. DuChene, et al., Hot Hole Collection and Photoelectrochemical CO(2) Reduction with Plasmonic Au/p-GaN Photocathodes, Nano Lett., 2018, 18(4), 2545–2550 CrossRef CAS PubMed.
- D. König, et al., Non-equilibrium dynamics, materials and structures for hot carrier solar cells: a detailed review, Semicond. Sci. Technol., 2020, 35(7), 073002 CrossRef.
- L. Zhou, et al., Quantifying hot carrier and thermal contributions in plasmonic photocatalysis, Science, 2018, 362(6410), 69–72 CrossRef CAS PubMed.
- R. Verma, R. Belgamwar and V. Polshettiwar, Plasmonic Photocatalysis for CO2 Conversion to Chemicals and Fuels. ACS, Mater. Lett., 2021, 3(5), 574–598 CAS.
- X. Zhang, et al., Product selectivity in plasmonic photocatalysis for carbon dioxide hydrogenation, Nat. Commun., 2017, 8(1), 14542 CrossRef CAS.
- Y. Zhang, et al., Surface-Plasmon-Driven Hot Electron Photochemistry, Chem. Rev., 2018, 118(6), 2927–2954 CrossRef CAS PubMed.
- S. Yu, et al., Plasmonic Control of Multi-Electron Transfer and C–C Coupling in Visible-Light-Driven CO2 Reduction on Au Nanoparticles, Nano Lett., 2018, 18(4), 2189–2194 CrossRef CAS PubMed.
- G. Tagliabue, et al., Quantifying the role of surface plasmon excitation and hot carrier transport in plasmonic devices, Nat. Commun., 2018, 9(1), 3394 CrossRef PubMed.
- T. Liu, et al., Generation of hot electrons in nanostructures incorporating conventional and unconventional plasmonic materials, Faraday Discuss., 2019, 214, 199–213 RSC.
- S. Ezendam, et al., Hybrid Plasmonic Nanomaterials for Hydrogen Generation and Carbon Dioxide Reduction, ACS Energy Lett., 2022, 7(2), 778–815 CrossRef CAS PubMed.
- L. Yuan, et al., Sustainable chemistry with plasmonic photocatalysts, Nanophotonics, 2023, 12(14), 2745–2762 CrossRef CAS.
- A. Manjavacas, et al., Plasmon-Induced Hot Carriers in Metallic Nanoparticles, ACS Nano, 2014, 8(8), 7630–7638 CrossRef CAS PubMed.
- R. Sundararaman, et al., Theoretical predictions for hot-carrier generation from surface plasmon decay, Nat. Commun., 2014, 5, 5788 CrossRef CAS PubMed.
- E.-R. Newmeyer, J. D. North and D. F. Swearer, Hot carrier photochemistry on metal nanoparticles, J. Appl. Phys., 2022, 132(23), 230901-1–230901-16 CrossRef.
- L. Mascaretti, et al., Plasmon-Enhanced Photoelectrochemical Water Splitting for Efficient Renewable Energy Storage, Adv. Mater., 2019, 31(31), e1805513 CrossRef PubMed.
- S. Mukherjee, et al., Hot-Electron-Induced Dissociation of H2 on Gold Nanoparticles Supported on SiO2, J. Am. Chem. Soc., 2014, 136(1), 64–67 CrossRef CAS PubMed.
- S. Swaminathan, et al., The Pivotal Role of Hot Carriers in Plasmonic Catalysis of C–N Bond Forming Reaction of Amines, Angew. Chem., Int. Ed., 2021, 60(22), 12532–12538 CrossRef CAS PubMed.
- S. Yu and P. K. Jain, Plasmonic photosynthesis of C(1)–C(3) hydrocarbons from carbon dioxide assisted by an ionic liquid, Nat. Commun., 2019, 10(1), 2022 CrossRef PubMed.
- C. Boerigter, et al., Evidence and implications of direct charge excitation as the dominant mechanism in plasmon-mediated photocatalysis, Nat. Commun., 2016, 7(1), 10545 CrossRef CAS PubMed.
- E. Kazuma and Y. Kim, Mechanistic Studies of Plasmon Chemistry on Metal Catalysts, Angew. Chem., Int. Ed., 2019, 58(15), 4800–4808 CrossRef CAS PubMed.
- K. Chen and H. Wang, Plasmon-driven photocatalytic molecular transformations on metallic nanostructure surfaces: mechanistic insights gained from plasmon-enhanced Raman spectroscopy, Mol. Syst. Des. Eng., 2021, 6(4), 250–280 RSC.
- P. V. Kumar, et al., Direct hot-carrier transfer in plasmonic catalysis, Faraday Discuss., 2019, 214, 189–197 RSC.
- Y. Sun and Z. Tang, Photocatalytic hot-carrier chemistry, MRS Bull., 2020, 45(1), 20–25 CrossRef CAS.
- S. Yu, et al., Opportunities and Challenges of Solar-Energy-Driven Carbon Dioxide to Fuel Conversion with Plasmonic Catalysts, ACS Energy Lett., 2017, 2(9), 2058–2070 CrossRef CAS.
- Y. Wu, et al., SERS Study of the Mechanism of Plasmon-Driven Hot Electron Transfer between Gold Nanoparticles and PCBM, J. Phys. Chem. C, 2019, 123(49), 29908–29915 CrossRef CAS.
- T. Le, Y. Shao and B. Wang, Plasmon-Induced CO2 Conversion on Al@Cu2O: A DFT Study, J. Phys. Chem. C, 2021, 125(11), 6108–6115 CrossRef CAS.
- M. G. Sandoval, et al., CO2 adsorption and activation on Ag(111) surfaces in the presence of surface charge density: A static gas phase DFT study, Appl. Surf. Sci., 2023, 610, 155498 CrossRef CAS.
- C. Hu, et al., Near-infrared-featured broadband CO2 reduction with water to hydrocarbons by surface plasmon, Nat. Commun., 2023, 14(1), 221 CrossRef CAS PubMed.
- X.-G. Zhang, et al., Reaction Selectivity for Plasmon-Driven Carbon Dioxide Reduction on Silver Clusters: A Theoretical Prediction, J. Phys. Chem. C, 2019, 123(17), 11101–11108 CrossRef CAS.
- Y. Xiong, et al., Photodriven Catalytic Hydrogenation of CO2 to CH4 with Nearly 100% Selectivity over Ag25 Clusters, Nano Lett., 2021, 21(20), 8693–8700 CrossRef CAS PubMed.
- J. Yan, K. W. Jacobsen and K. S. Thygesen, First-principles study of surface plasmons on Ag(111) and H/Ag(111), Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84(23), 235430 CrossRef.
- T. P. Rossi, et al., Kohn–Sham Decomposition in Real-Time Time-Dependent Density-Functional Theory: An Efficient Tool for Analyzing Plasmonic Excitations, J. Chem. Theory Comput., 2017, 13(10), 4779–4790 CrossRef CAS PubMed.
- P. V. Kumar, et al., Plasmon-Induced Direct Hot-Carrier Transfer at Metal-Acceptor Interfaces, ACS Nano, 2019, 13(3), 3188–3195 CrossRef CAS PubMed.
- T. P. Rossi, P. Erhart and M. Kuisma, Hot-Carrier Generation in Plasmonic Nanoparticles: The Importance of Atomic Structure, ACS Nano, 2020, 14(8), 9963–9971 CrossRef CAS PubMed.
- J. Fojt, et al., Hot-Carrier Transfer across a Nanoparticle–Molecule Junction: The Importance of Orbital Hybridization and Level Alignment, Nano Lett., 2022, 22(21), 8786–8792 CrossRef CAS PubMed.
- M. Kuisma, et al., Localized surface plasmon resonance in silver nanoparticles: Atomistic first-principles time-dependent density-functional theory calculations, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91(11), 115431 CrossRef.
- W. Zhu, et al., Monodisperse Au Nanoparticles for Selective Electrocatalytic Reduction of CO2 to CO, J. Am. Chem. Soc., 2013, 135(45), 16833–16836 CrossRef CAS PubMed.
- D. Gao, et al., Enhancing CO2 Electroreduction with the Metal–Oxide Interface, J. Am. Chem. Soc., 2017, 139(16), 5652–5655 CrossRef CAS PubMed.
- J. H. Lee, et al., Understanding the Role of Functional Groups in Polymeric Binder for Electrochemical Carbon Dioxide Reduction on Gold Nanoparticles, Adv. Funct. Mater., 2018, 28(45), 1804762-1–1804762-6 CrossRef.
- M. Sun, A. Staykov and M. Yamauchi, Understanding the Roles of Hydroxide in CO2 Electroreduction on a Cu Electrode for Achieving Variable Selectivity, ACS Catal., 2022, 12(24), 14856–14863 CrossRef CAS.
- W. Deng, et al., Crucial Role of Surface Hydroxyls on the Activity and Stability in Electrochemical CO2 Reduction, J. Am. Chem. Soc., 2019, 141(7), 2911–2915 CrossRef CAS PubMed.
- S. Singh, et al., Surface plasmon-enhanced photo-driven CO2 hydrogenation by hydroxy-terminated nickel nitride nanosheets, Nat. Commun., 2023, 14(1), 2551 CrossRef CAS PubMed.
- N. N. Vu, S. Kaliaguine and T. O. Do, Plasmonic Photocatalysts for Sunlight-Driven Reduction of CO(2): Details, Developments, and Perspectives, ChemSusChem, 2020, 13(16), 3967–3991 CrossRef CAS PubMed.
- A. Álvarez, et al., CO2 Activation over Catalytic Surfaces, ChemPhysChem, 2017, 18(22), 3135–3141 CrossRef PubMed.
- K. Y. Cohen, et al., Using Light and Electrons to Bend Carbon Dioxide: Developing and Understanding Catalysts for CO2 Conversion to Fuels and Feedstocks, Acc. Chem. Res., 2022, 55(7), 944–954 CrossRef CAS PubMed.
- S. Zhu, et al., A computational study on linear and bent adsorption of CO2 on different surfaces for its photoreduction, Catal. Today, 2019, 335, 278–285 CrossRef CAS.
- D. Cheng, R. Liu and K. Hu, Gold nanoclusters: Photophysical properties and photocatalytic applications, Front. Chem., 2022, 10, 958626 CrossRef CAS PubMed.
- P. Pyykko and J. P. Desclaux, Relativity and the periodic system of elements, Acc. Chem. Res., 1979, 12(8), 276–281 CrossRef CAS.
- R. Meyer, et al., Surface chemistry of catalysis by gold, Gold Bull., 2004, 37(1), 72–124 CrossRef CAS.
- F. P. García de Arquer, et al., CO2 electrolysis to multicarbon products at activities greater than 1 A cm−2, Science, 2020, 367(6478), 661–666 CrossRef PubMed.
- P. De Luna, et al., Catalyst electro-redeposition controls morphology and oxidation state for selective carbon dioxide reduction, Nat. Catal., 2018, 1(2), 103–110 CrossRef CAS.
- C.-T. Dinh, et al., CO2 electroreduction to ethylene via hydroxide-mediated copper catalysis at an abrupt interface, Science, 2018, 360(6390), 783–787 CrossRef CAS PubMed.
- Y. Cao, et al., Surface hydroxide promotes CO2 electrolysis to ethylene in acidic conditions, Nat. Commun., 2023, 14(1), 2387 CrossRef CAS PubMed.
- J. Gu, et al., Modulating electric field distribution by alkali cations for CO2 electroreduction in strongly acidic medium, Nat. Catal., 2022, 5(4), 268–276 CrossRef CAS.
- T. Zhang, et al., Acidic CO2 Electrolysis Addressing the “Alkalinity Issue” and Achieving High CO2 Utilization, Chem. – Eur. J., 2023, 29(46), e202301455 CrossRef CAS PubMed.
- M. Ma, et al., Insights into the carbon balance for CO2 electroreduction on Cu using gas diffusion electrode reactor designs, Energy Environ. Sci., 2020, 13(3), 977–985 RSC.
- M. Ramdin, et al., Electroreduction of CO2/CO to C2 Products: Process Modeling, Downstream Separation, System Integration, and Economic Analysis, Ind. Eng. Chem. Res., 2021, 60(49), 17862–17880 CrossRef CAS PubMed.
- Y. Hori, Electrochemical CO2 Reduction on Metal Electrodes, in Modern Aspects of Electrochemistry, ed. C. G. Vayenas, R. E. White, and M. E. Gamboa-Aldeco, Springer, New York, NY, 2008, pp.89–189 Search PubMed.
- Z. Tao, et al., Bridge Sites of Au Surfaces Are Active for Electrocatalytic CO2 Reduction, J. Am. Chem. Soc., 2022, 144(19), 8641–8648 CrossRef CAS PubMed.
- G. Henkelman, A. Arnaldsson and H. Jónsson, A fast and robust algorithm for Bader decomposition of charge density, Comput. Mater. Sci., 2006, 36(3), 354–360 CrossRef.
- W. Tang, E. Sanville and G. Henkelman, A grid-based Bader analysis algorithm without lattice bias, J. Phys.: Condens. Matter, 2009, 21(8), 084204 CrossRef CAS PubMed.
- E. W. N. Wiberg and A. F. Holleman, Inorganic Chemistry, Academic Press, San Diego, 2001 Search PubMed.
- S. K. Giri and G. C. Schatz, Photodissociation of H2 on Ag and Au Nanoparticles: Effect of Size and Plasmon versus Interband Transitions on Threshold Intensities for Dissociation, J. Phys. Chem. C, 2023, 127(8), 4115–4123 CrossRef CAS.
- R. W. Burgess and V. J. Keast, TDDFT Study of the Optical Absorption Spectra of Bare Gold Clusters, J. Phys. Chem. C, 2014, 118(6), 3194–3201 CrossRef CAS.
- M. Bernardi, et al., Theory and computation of hot carriers generated by surface plasmon polaritons in noble metals, Nat. Commun., 2015, 6, 7044 CrossRef CAS PubMed.
- O. Baseggio, et al., Photoabsorption of Icosahedral Noble Metal Clusters: An Efficient TDDFT Approach to Large-Scale Systems, J. Phys. Chem. C, 2016, 120(23), 12773–12782 CrossRef CAS.
- K. Iida, et al., First-Principles Computational Visualization of Localized Surface Plasmon Resonance in Gold Nanoclusters, J. Phys. Chem. A, 2014, 118(47), 11317–11322 CrossRef CAS PubMed.
- N. Durante, et al., Optical Properties of Au Nanoclusters from TD-DFT Calculations, J. Phys. Chem. C, 2011, 115(14), 6277–6282 CrossRef CAS.
- G. Piccini, et al., Gold Nanowires: A Time-Dependent Density Functional Assessment of Plasmonic Behavior, J. Phys. Chem. C, 2013, 117(33), 17196–17204 CrossRef CAS.
- K. Kluczyk-Korch and T. J. Antosiewicz, Hot carrier generation in a strongly coupled molecule–plasmonic nanoparticle system, Nanophotonics, 2023, 1711–1722 CrossRef CAS.
- J. Ma, Z. Wang and L.-W. Wang, Interplay between plasmon and single-particle excitations in a metal nanocluster, Nat. Commun., 2015, 6(1), 10107 CrossRef CAS PubMed.
- X. Zhang, et al., Transient localized surface plasmon induced by femtosecond interband excitation in gold nanoparticles, Sci. Rep., 2018, 8(1), 10499 CrossRef PubMed.
- P. Lyu, R. Espinoza and S. C. Nguyen, Photocatalysis of Metallic Nanoparticles: Interband vs. Intraband Induced Mechanisms, J. Phys. Chem. C, 2023, 127(32), 15685–15698 CrossRef CAS PubMed.
- Z. Fusco, K. Catchpole and F. J. Beck, Investigation of the mechanisms of plasmon-mediated photocatalysis: synergistic contribution of near-field and charge transfer effects, J. Mater. Chem. C, 2022, 10(19), 7511–7524 RSC.
- Z. Fusco, et al., Cathodoluminescence Spectroscopy of Complex Dendritic Au Architectures for Application in Plasmon-Mediated Photocatalysis and as SERS Substrates, Adv. Mater. Interfaces, 2023, 10(3), 2202236 CrossRef CAS.
- L. Landau, On the vibration of the electronic plasma, J. Exp. Theor. Phys., 1946, 16, 574 Search PubMed.
- P. K. Jain, Comment on “Thermal effects – an alternative mechanism for plasmon-assisted photocatalysis” by Y. Dubi, I. W. Un and Y. Sivan, Chem. Sci., 2020, 11, 5017, Chem. Sci., 2020, 11(33), 9022–9023 RSC.
- Y. Sivan, I. W. Un and Y. Dubi, Assistance of metal nanoparticles in photocatalysis – nothing more than a classical heat source, Faraday Discuss., 2019, 214, 215–233 RSC.
- Y. Dubi and Y. Sivan, “Hot” electrons in metallic nanostructures-non-thermal carriers or heating?, Light: Sci. Appl., 2019, 8, 89 CrossRef PubMed.
- Z. Fusco and F. J. Beck, Advances in fundamentals and application of plasmon-assisted CO2 photoreduction, Nanophotonics, 2024, 13(4), 387–417 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nh00046c |
| This journal is © The Royal Society of Chemistry 2024 |