
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Iodine passivation facilitates on-surface synthesis of robust regular conjugated two-dimensional organogold networks on Au(111)†
Arash
Badami-Behjat
ab,
Gianluca
Galeotti
ab,
Rico
Gutzler
ab,
Dominik L.
Pastoetter
 c,
Wolfgang M.
Heckl
ab,
Xinliang
Feng
cd and
Markus
Lackinger
*ab
c,
Wolfgang M.
Heckl
ab,
Xinliang
Feng
cd and
Markus
Lackinger
*ab
aDeutsches Museum, Museumsinsel 1, 80538 Munich, Germany. E-mail: markus@lackinger.org
bDepartment of Physics, Technische Universität München, James-Franck-Str. 1, 85748 Garching, Germany
cCenter for Advancing Electronics Dresden & Faculty of Chemistry and Food Chemistry, Technische Universität Dresden, 01069 Dresden, Germany
dMax Planck Institute of Microstructure Physics, Weinberg 2, 06120 Halle, Germany
First published on 11th April 2024
Abstract
Two-dimensional conjugated organogold networks with anthra-tetrathiophene repeat units are synthesized by thermally activated debrominative coupling of 2,5,9,12-tetrabromoanthra[1,2-b:4,3-b′:5,6-b′′:8,7-b′′′]tetrathiophene (TBATT) precursor molecules on Au(111) surfaces under ultra-high vacuum (UHV) conditions. Performing the reaction on iodine-passivated Au(111) surfaces promotes formation of highly regular structures, as revealed by scanning tunneling microscopy (STM). In contrast, coupling on bare Au(111) surfaces results in less regular networks due to the simultaneous expression of competing intermolecular binding motifs in the absence of error correction. The carbon–Au–carbon bonds confer remarkable robustness to the organogold networks, as evidenced by their high thermal stability. In addition, as suggested by density functional theory (DFT) calculations and underscored by scanning tunneling spectroscopy (STS), the organogold networks exhibit a small electronic band gap in the order of 1.0 eV due to their high π-conjugation.
New conceptsDehalogenative couplings as employed for the synthesis of organometallic and covalent nanostructures are the workhorse of on-surface synthesis. These reactions are typically performed on bare metal surfaces whose chemical activity lowers the activation temperature so that the dehalogenation reaction occurs before the reactants thermally desorb. However, metal surfaces can be detrimental to subsequent coupling, because the strong interactions hinder monomer mobility and can also lead to unwanted side reactions. The conceptual idea here is to tune the reactivity of metal surfaces so that they are still sufficiently reactive to promote dehalogenation, but passivated enough to mitigate the detrimental side effects imposed by the strong interactions. This is achieved by chemisorption of an iodine monolayer from the vapor phase. We show that (1) iodine-passivated Au(111) can still be reactive for debromination; (2) the reactivity can be tuned by varying the iodine exposure used for passivation; (3) the two-dimensional organogold networks synthesized on iodine-passivated Au(111) are more regular compared to those synthesized directly on bare Au(111). We unambiguously demonstrate that the presence of iodine induces reversibility of the exceptionally strong organogold bonds and facilitates dynamic error correction, leading to higher structure quality. In addition, less obvious contributions may arise from network formation on (partially) closed iodine layers. |
Introduction
Dehalogenative couplings are arguably the most widely used and important reactions for the on-surface synthesis of extended covalent and organometallic nanostructures on coinage metal surfaces.1–4 Topologically one-dimensional (1D) structures, with graphene nanoribbons being the most prominent example,5,6 are obtained from ditopic precursor molecules. Two-dimensional (2D) networks, on the other hand, can be synthesized from precursors with more than two halogen substituents. But the structural quality obtained in 2D is typically relatively low. Linking monomers in two dimensions is more challenging, for example, because long-range order is often compromised by structural defects such as topological errors or missing repeat units, which are inherently absent in topologically linear structures.In this respect, the organometallic nanostructures that are usually observed on Cu and Ag surfaces,2,7–10 with rare exceptions,11 and less frequently also on Au surfaces12–22 after dehalogenation, hold potential for the on-surface synthesis of regular 2D networks with application-relevant properties. Rather than forming direct carbon–carbon bonds, the dehalogenated monomers are linked by carbon–metal–carbon bonds formed with surface-supplied metal atoms. The organometallic structures are considered metastable because further annealing can result in conversion to thermodynamically more stable carbon–carbon bonded structures by reductive elimination of the organometallic metal atom, unless prevented by steric hindrance or too strong organometallic bonds.12,22–25
Robust and regular nanostructures can be obtained in two ways: (1) organometallic bonds of intermediate strength still allow equilibration into more ordered but still organometallic structures, analogous to supramolecular self-assembly. These are then converted into a similarly regular covalent carbon–carbon bonded structure in a topotactic reaction,8,18,26–30 (2) organometallic self-assembly can directly yield highly regular 2D networks with interesting properties. The organometallic bonds can mediate electronic conjugation, resulting in networks with intriguing properties as electrical conductivity or catalytic activity.31–33 However, this poses a dilemma: in order to achieve high network stability, the organometallic bonds must be sufficiently strong. On the other hand, high bond strength leads to irreversibility, which precludes error correction during growth.
The goal here is to explore coupling on iodine-passivated metal surfaces as a possible complementary new synthesis route to regular and robust 2D nanostructures. The conceptual idea of synthesis on passivated surfaces in general is that the substrate is still chemically active enough to initiate the reaction, while detrimental substrate effects such as limited monomer mobility or side reactions are shielded. The latter is particularly important for the thiophene-containing monomer studied here: while thiophenes are highly relevant for organic electronics,34 their widespread use in on-surface synthesis on metals is limited by their susceptibility to decomposition.19,35 Previous work on metal surfaces has used inorganic 2D materials such as graphene or hexagonal boron nitride as ultimately thin passivation layers.36,37 While it has been shown that either the attenuated reactivity of the underlying metal or co-deposited metal atoms can promote dehalogenative couplings, the quality of the resulting structures has not been significantly improved compared to synthesis directly on metals.
Here, we have chosen passivation of Au(111) by a monolayer of chemisorbed iodine (I–Au(111) in the following) for several reasons: Previously, we have developed a method for post-synthetic decoupling of covalent nanostructures from the metal surfaces on which they were grown by intercalation of an iodine monolayer.38,39 From X-ray standing wave experiments we inferred a relatively large adsorption height in the order of 6 Å with respect to the topmost metal atoms, which indicates efficient decoupling.40 Moreover, in our recent studies of supramolecular self-assembly on I–Au(111), we found that the molecule–surface interactions can be even weaker than on graphite.41,42 Iodine adopts a variety of superstructures on Au(111) where the area density of the chemisorbed iodine atoms ranges from 4.63 nm−2 for the common √3 × √3R ± 30° superstructure up to 5.55 nm−2 for the uniaxially compressed p(5 × √3) superstructure.43,44 Accordingly, even a closed √3 × √3R ± 30° iodine monolayer still has the capacity to additionally incorporate a fair amount of dissociated halogens as the byproduct of the coupling reaction. Finally, the I–Au(111) surface can dynamically and advantageously change its reactivity during heating due to iodine desorption.
In the present work, we demonstrate that iodine-passivated Au(111) surfaces can indeed remain reactive for dehalogenation reactions (cf. ESI† for preparation parameters). Debrominative coupling of the tetra-bromine substituted TBATT precursor leads to highly robust and π-conjugated 2D organogold networks. More importantly, the synthesis on I–Au(111) also offers a crucial advantage over bare Au(111), namely the high regularity of the obtained 2D organogold networks.
Results and discussion
Bare Au(111)
On-surface polymerization of the TBATT precursor (cf.Fig. 1(a) for structure) by debrominative coupling has previously been studied on Ag und Cu surfaces,9,35 where organometallic networks formed with surface-supplied metal atoms. However, the TBATT-derived organometallic networks were mostly irregular with only smaller regular patches due to the kinetic competition between syn- and anti-binding motifs in the irreversible regime. The conversion from organometallic to covalent was averted on Cu(111) by the decomposition of the thiophene moieties at the required temperatures.35 By contrast, decomposition was mitigated on Ag(111), where covalent networks could be obtained via the organometallic intermediate by annealing at 300 °C.9 | ||
| Fig. 1 (a) Chemical structure of TBATT; (b)/(c) STM images of the TBATT self-assembly on bare Au(111) as observed after room temperature deposition; (d) tentative model of the self-assembly derived from the experimental structure (C: gray; Br: red; S: green; H: white); intermolecular halogen bonds are indicated by dashed lines. (e)–(g) STM images of the irregular organogold networks obtained after annealing to 200–220 °C; the image in (g) is overlaid with DFT-calculated organogold dimers and tetramers in which debrominated monomers are linked by carbon–Au–carbon bonds. The scaled overlays confirm the size match and thus the chemical identity of the intermolecular bonds. Covalent carbon–carbon bonds are about 0.26 nm shorter than organometallic carbon–Au–carbon bonds (cf. ESI,† Fig. S7) and would result in an obvious mismatch. (tunneling parameters and scale bars: (b) +1.1 V, 120 pA, 10 nm; (c) +1.1 V, 120 pA, 2 nm; (e) +900 mV, 120 pA, 10 nm; (f) +900 mV, 120 pA, 4 nm; (g) +900 mV, 120 pA, 2 nm, all STM images were acquired at low temperature). | ||
For further reference, we first explore the debrominative coupling of TBATT directly on bare Au(111). Fig. 1(b) and (c) show STM images of the self-assembled structures obtained directly after room temperature deposition of TBATT on Au(111), also confirming the purity of the reactant by the complete absence of impurities. The lattice parameters of the oblique unit cell are a = 1.1 ± 0.1 nm, b = 1.4 ± 0.1 nm and 76° ± 1°, and the arrangement of the intact TBATT molecules is controlled by intermolecular Br⋯S, Br⋯H and Br⋯Br halogen bonds,45,46 as illustrated by the tentative model of the self-assembly in Fig. 1(d).
Annealing at 200–220 °C activates the debromination, resulting in the mostly irregular networks shown in Fig. 1(e)–(g). The measured center-to-center distance of 1.5 ± 0.1 nm between adjacent molecules is incompatible with the DFT-calculated value for direct carbon–carbon bonding of the monomers of 1.203 nm (cf. ESI,† Fig. S7a), but is consistent with corresponding calculations for organometallic carbon–Au–carbon bonds (see overlay in Fig. 1(g)) and the organometallic structures obtained for TBATT on Cu(111) and Ag(111).9 Similarly, organogold networks were observed for tri-bromo-terthieno-benzene on Au(111),19 that is the three-fold symmetric analogue of TBATT. In summary, in view of our calculations and previously reported work,9,19 our experiments consistently indicate the formation of organogold networks after debromination of TBATT on bare Au(111). To quantify the quality of the structure, we performed a statistical analysis of the pore shapes (624 pores were counted in total and only fully closed pores were considered), resulting in a proportion of 28% rhombic pores outlined by four identically bonded molecules with similar azimuthal orientation (see Fig. 2(f) for structure). The remaining 72% of closed pores had deviating shapes and also included pores comprised of either three or more than four molecules, resulting in an energy cost due to bond angle deformations. However, we were not able to obtain better ordered networks directly on bare Au(111).
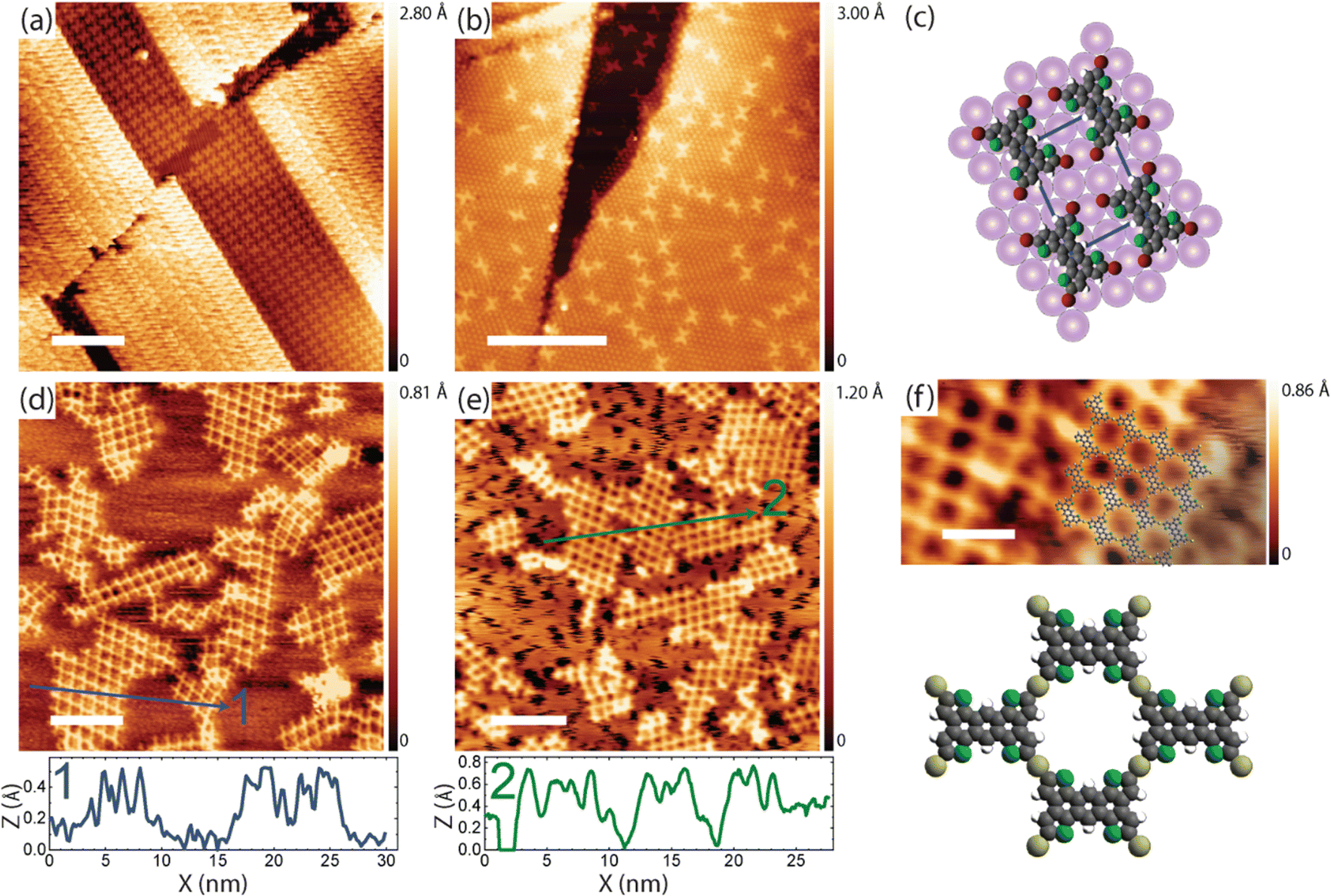 | ||
| Fig. 2 STM images of TBATT structures on iodine-passivated Au(111). (a) self-assembly of intact molecules and (b) dispersed single molecules as similarly observed after room temperature deposition; the brighter appearing areas in (a) correspond to a TBATT bilayer. (c) tentative model of the self-assembled TBATT structure on I–Au(111); pink full circles indicate iodine atoms of the monolayer. (d)–(f) STM images of highly regular organogold networks as obtained after annealing at 200–220 °C; the image in (d) was acquired at room temperature and shows an apparently closed iodine layer. However, the corresponding low-temperature STM image in (e) reveals the fuzzy black areas where iodine was desorbed during the annealing. The apparent height of the networks is illustrated by the line-profiles below the respective STM image. (f) Close-up STM image with scaled overlay of the organogold network; below a model of the organogold structure is shown (Au: yellow). (tunneling parameters and scale bars: (a) +1.8 V, 120 pA, 10 nm; (b) +1.8 V, 120 pA, 10 nm; (d) +1.0 V, 120 pA, 10 nm; (e) +1.0 V, 120 pA, 10 nm; (f) +1.0 V, 120 pA, 3 nm, all STM images were acquired at low temperatures except for (d)). | ||
Iodine-passivated Au(111)
In the next step, TBATT was deposited on I–Au(111) (see ESI,† Materials and methods section for preparation procedures and parameters), where we observed intact molecules in ordered supramolecular structures (Fig. 2(a)) that co-existed with dispersed single TBATT molecules (Fig. 2(b)). Although the principal arrangement of the TBATT molecules in the self-assembly is comparable to that on bare Au(111) (Fig. 1(c)), the rectangular unit cell is substantially larger with a = 1.5 ± 0.1 nm, b = 1.7 ± 0.1 nm. This marked change in lattice parameters compared to bare Au(111) underscores the adsorption of the TBATT molecules onto a closed iodine monolayer. As further illustrated by the tentative model in Fig. 2(c), the intermolecular distances have increased significantly, resulting in a pronounced weakening of the intermolecular interactions. The model is based on a commensurate superstructure (with respect to the iodine monolayer) that agrees with the experimental lattice parameters and reproduces the orientation of both the unit cell and the TBATT molecules with respect to the iodine lattice. It also suggests equivalent adsorption sites for all four bromine substituents of each TBATT molecule. Accordingly, we hypothesize that the self-assembly of fully brominated TBATT on I–Au(111) is governed by registry effects imposed by the highly corrugated iodine monolayer.
superstructure (with respect to the iodine monolayer) that agrees with the experimental lattice parameters and reproduces the orientation of both the unit cell and the TBATT molecules with respect to the iodine lattice. It also suggests equivalent adsorption sites for all four bromine substituents of each TBATT molecule. Accordingly, we hypothesize that the self-assembly of fully brominated TBATT on I–Au(111) is governed by registry effects imposed by the highly corrugated iodine monolayer.
Annealing these samples at 200–220 °C induced a profound structural change from densely packed to the porous grid-like network depicted in Fig. 2(d)–(f) with a centered rectangular unit cell measuring a = 1.8 ± 0.1 nm and b = 2.1 ± 0.1 nm. These experimental dimensions differ substantially from the DFT-calculated values of a = 1.40 nm and b = 1.96 nm for the putative covalent network (cf. ESI,† Fig. S7a). However, there is a good agreement with the DFT-optimized lattice parameters of a = 1.81 nm and b = 2.30 nm of the organogold networks, in which the anthra-tetrathiophene moieties are linked by carbon–Au–carbon bonds (cf. ESI,† Fig. S7b). This structural assignment is also corroborated by the scaled overlay in Fig. 2(f). Moreover, the center-to-center distance between adjacent molecules in the ordered network of 1.4 ± 0.1 nm matches perfectly with the organogold structures obtained on bare Au(111). More importantly, the organogold networks obtained on I–Au(111) are highly regular with 92% of the ideal rhombic pores (1174 closed pores were counted in total).
Interestingly, while the iodine layer appears completely closed when imaged at room temperature (Fig. 2(d)), corresponding STM images taken at low temperatures (Fig. 2(e)) reveal darker appearing, fuzzy patches of missing iodine. We argue that iodine partially desorbs during the thermal treatment used to form the networks. This is supported by STM images of a similarly prepared I–Au(111) surface acquired at low temperatures before the annealing step, which clearly show that the seemingly closed iodine monolayer does not completely cover and passivate the Au(111) surface (cf. ESI,† Fig. S1). Accordingly, a gradually decreasing iodine coverage during heating provides access to the bare Au(111) surface for activation of the debromination. This is also the most plausible pathway by which the Au-atoms are made available for organometallic bond formation. While it is plausible that the iodine monolayer is mostly intact at the onset of debromination, we assume that the progressive iodine desorption will eventually result in iodine vacancy islands that can become large enough to accommodate monomers or even smaller domains of the organogold networks. Indeed, the resulting ruggedness of the iodine passivation layer may also hamper the growth of larger domains of the organogold networks. It also remains presently unclear whether the organogold networks are adsorbed on the remaining patches of the iodine layer or on the bare Au(111). The networks prepared directly on Au(111) (cf.Fig. 1) exhibit apparent STM heights between 0.1 and 0.2 nm. On the iodinated surfaces the apparent height of the networks with respect to the underlying Au(111) can be estimated by line-profiles extending through the iodine vacancy islands (Fig. 2(e)). There we find apparent STM heights either in the order of 0.1 nm, but also occasionally higher values of >0.2 nm. This may indicate a heterogeneous sample state with most organogold domains already adsorbed on bare Au(111) and few remaining on iodine islands. However, we note that apparent STM heights are generally bias- and tip-dependent and thus unreliable. For an accurate assessment of adsorption heights, X-ray standing wave experiments are the gold standard.40
To confirm that a certain permeability of the iodine monolayer is crucial for debromination, we performed a control with a highly iodinated sample prepared with a 20-fold increased iodine exposure (see ESI,† Materials and methods section). After deposition of TBATT and subsequent annealing at 250 °C, which is 30 °C to 50 °C higher than normally used to initiate debromination, a mostly empty I–Au(111) surface was imaged by STM (see ESI,† Fig. S2a and b). Only upon further heating to 350 °C were small remnants of reacted molecules observed at the substrate step-edges, while the iodine was completely desorbed (see ESI,† Fig. S2c and d). This clearly indicates that the initial iodine exposure used to prepare the monolayer must not be too high to obtain reactive I–Au(111) surfaces that facilitate thermally activated debromination. In addition, we also observed that the reactive I–Au(111) surfaces prepared with lower iodine exposure show both areas where the iodine packing is less dense and iodine atom vacancies (see ESI,† Fig. S1 and S3), as similarly observed for electrochemically deposited iodine adlayers.44 Furthermore, we hypothesize that iodine desorption is enhanced in the less densely packed areas, ultimately promoting progressive desorption and the formation of larger vacancy islands.
Robustness of the organogold networks
To explore the robustness of the organogold anthra-tetrathiophene networks, the samples were annealed to successively higher temperatures. After annealing at 300 °C, remnants of the iodine monolayer were still present (cf. ESI,† Fig. S4), which were completely desorbed after annealing at 320 °C. The corresponding STM images in Fig. 3(a) and (b) confirm the persistence of the networks. Yet, the line-profiles in Fig. 3(d) and (e) reveal different pore sizes, clearly indicating the onset of conversion of organometallic carbon–Au–carbon bonds into covalent carbon–carbon bonds as similarly observed on Ag(111).9 However, this conversion is also accompanied by a loss of regularity. When heated to markedly higher temperatures of 400 °C, the degradation becomes apparent in the STM images (Fig. 3(c)).39 Although the stability is not directly tested at high temperatures, subsequent STM imaging allows conclusions to be drawn about the integrity of the networks. Disintegration of the organogold networks at high temperatures and reformation when cooling back to room temperature can be excluded, since individual monomers would desorb at temperatures above 300 °C (vide infra). Despite the onset of conversion to covalent at around 320 °C, the organogold networks exhibit a remarkable thermal stability, which may seem surprising at first glance: Metastable organometallic intermediates upon dehalogenative coupling on metal surfaces are routinely observed on Ag and Cu surfaces,2,7,26 but less commonly on Au surfaces. When observed on Au, the organometallic intermediates are often not very stable: Heating to 200–250 °C already breaks the organogold bonds, resulting in defined14,16,18 or undefined23 covalent products by reductive elimination of the Au-atom. On the contrary, organogold nanostructures with high temperature stability of around 300 °C have been reported previously.20,21 Most closely related are the organogold networks prepared from tri-bromo-terthieno-benzene precursors on Au(111),19 which already allude to the special role of highly conjugated thiophene-rich monomers. Based on our observations and the experimental results published in ref. 19, we formulate the working hypothesis that the high degree of electronic conjugation in the thiophene-containing scaffold as expressed by the relatively small electronic band gap (vide infra) gives rise to remarkably strong organogold bonds. This view is supported by the increase in DFT-calculated binding energies (BE) of organogold aggregates with increasing size (see ESI,† Fig. S8). While the BE of a single carbon–Au bond of −1.8 eV derived for the thiophene dimer with a total of 12 π electrons shows only a moderate bond strength, the BE increases sharply to −2.7 eV already for 52 π electrons in the anthra-tetrathiophene dimer. Doubling the aggregate size to the 2 × 2 tetramer with 104 π electrons results in a modest further increase in BE to −2.8 eV, indicating that a plateau has been reached. In summary, the high BE of the carbon–Au bonds found for larger aggregates can be associated with a high degree of π-conjugation, and is consistent with the experimentally observed high temperature stability of the organogold networks. Moreover, the resulting lack of bond reversibility also explains the limited regularity of the organogold networks obtained on bare Au(111) (Fig. 1(e)–(g)).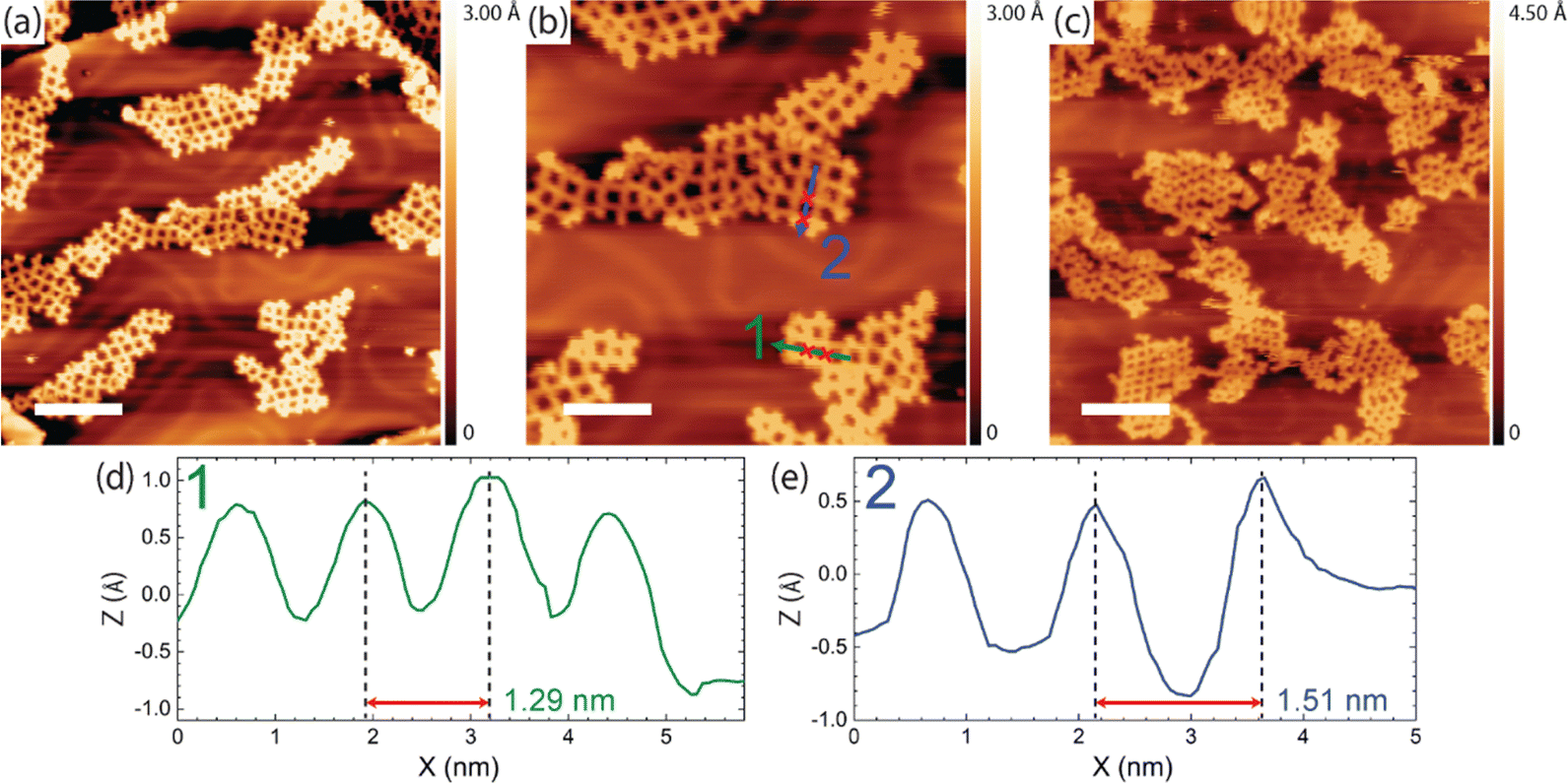 | ||
| Fig. 3 STM images of TBATT-derived organogold networks synthesized on I–Au(111) acquired after additional heating to (a) and (b) 320 °C and (c) 400 °C. (d) and (e) corresponding line-profiles as marked in (b). The iodine is completely desorbed at 320 °C, while the networks persist. The line-profiles reveal two different pore sizes, indicating that annealing at 320 °C partly converts the organogold networks into covalent carbon–carbon bonded networks by reductive elimination of the organometallic Au-atom as similarly observed on Ag(111).9 However, this conversion is accompanied by a loss of regularity. At even higher annealing temperatures of 400 °C, the blurring of the molecular networks in the STM images indicates the onset of decomposition of the molecular building blocks. (tunneling parameters and scale bars: (a) +850 mV, 100 pA, 10 nm; (b) +850 mV, 100 pA, 6 nm; (c) +1.0 V, 120 pA, 10 nm, all STM images were acquired at low temperatures). | ||
Electronic properties of the organogold networks
To further explore the potential of the highly stable and regular organogold networks for electronic applications, we provide a first combined theoretical and experimental assessment of their electronic structure. To this end, DFT calculations were performed on free-standing monolayers with periodic boundary conditions. Valence band and conduction band or shown in Fig. 4(a) (cf. ESI,† Fig. S9 for full band structure). The calculated electronic band structure exhibits a surprisingly small band gap of 0.71 eV (which is presumably underestimated, as is usually the case with DFT) and dispersive valence and conduction bands. These results indicate that the conjugation of the monomers extends through the organogold bonds as previously also reported for carbon–Ag–carbon bonds,31 resulting in an overall conjugated network. The continuous electronic conjugation is already evident in the 2 × 2 anthra-tetrathiophene tetramer from the plots of its frontier molecular orbitals (see ESI,† Fig. S10), which show their delocalization across the organogold bond. To experimentally underscore these theoretical results, scanning tunneling spectroscopy (STS) experiments were performed. The differential conductance dI/dV versus voltage V spectra shown in Fig. 4(c) were acquired above the anthra-tetrathiophene moieties, where all frontier orbitals exhibit intensity (cf. ESI,† Fig. S10), and indicate an electronic band gap in the order of 1.0 eV. However, these STS experiments were conducted at liquid nitrogen temperatures, so thermal broadening cannot be neglected and fine details cannot be interpreted. Nevertheless, the data clearly indicate a comparatively small electronic band gap in accord with the DFT calculations.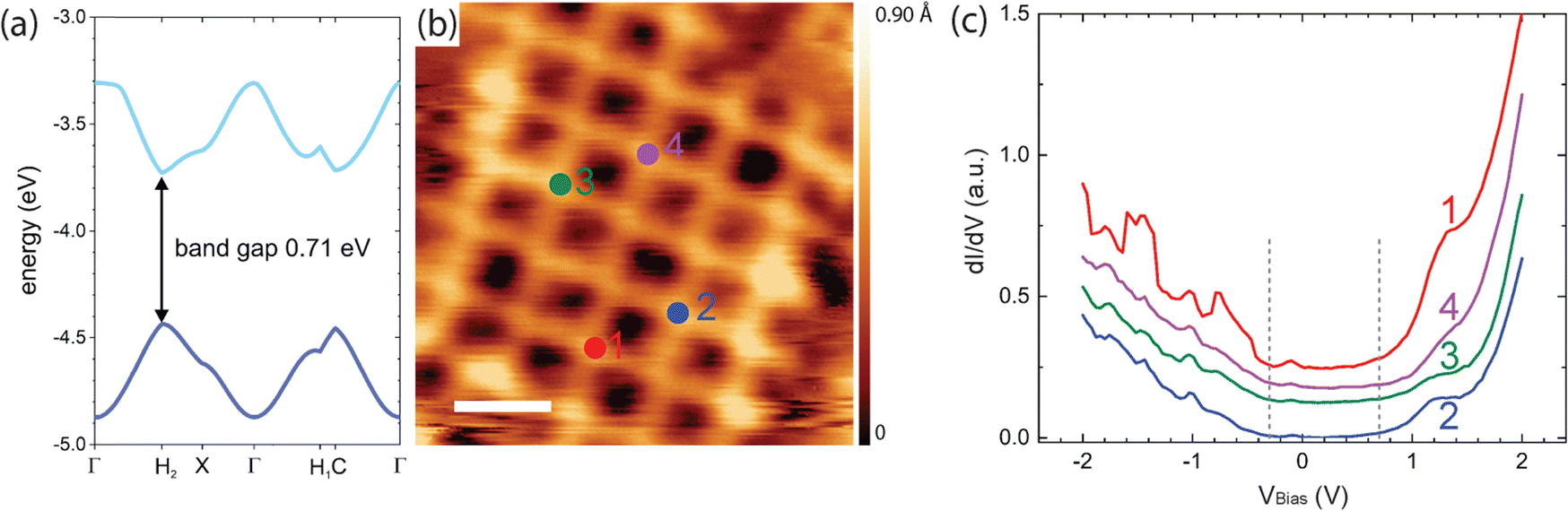 | ||
| Fig. 4 Theoretical and experimental assessment of the electronic structure of the TBATT-derived organogold networks. (a) DFT band structure calculations. Only the valence and conduction band are shown here (cf. ESI,† Fig. S9 for a full band structure). (b) STM image showing the positions where spectra were acquired, i.e. above the anthra-tetrathiophene moieties (tunneling parameters and scale bar: +1.0 V, 120 pA, 2 nm, acquired at low temperature). (c) dI/dV versus voltage curves (lock-in modulation parameters: 329 Hz, 50 mV) acquired at the positions indicated in (b). Although the spectra deviate slightly from each other, the general features are similar: onset of a pronounced feature in the unoccupied states at +0.7 V and onset of a weaker feature in the unoccupied states at −0.3 V as marked by the dashed vertical lines. In addition, all spectra consistently show a weak feature at −0.1 to −0.2 V. | ||
Mechanistic insights
Finally, we address the question of how the iodine monolayer might promote the formation of regular organogold networks. Two effects are conceivable: (1) network formation on top of the iodine layer is advantageous for achieving regularity. Possible mechanisms of action are either an enhanced monomer mobility on I–Au(111) compared to bare Au(111), or registry effects that favor a uniform orientation of the monomers, hence homotacticity. (2) The presence of iodine induces reversibility of the otherwise irreversibly strong carbon–Au–carbon bonds, thereby facilitating dynamic error correction.To further elucidate the possibility of iodine-induced reversibility of the organogold bonds, we performed complementary experiments: TBATT was first deposited on bare Au(111), and then iodine was co-adsorbed. Two different but comparatively small iodine exposures were investigated, small enough that the iodine did not intercalate between Au(111) and TBATT, i.e. the molecules remained directly adsorbed on the metal surface. The sample was then subjected to the usual thermal treatment and heated to temperatures of 200–220 °C. The subsequently acquired STM images in Fig. 5(a) and b unveil formation of organogold networks of intermediate quality as judged by the proportion of 76% ideal rhombic pores (896 closed pores were counted in total) (see ESI,† Fig. S5 for additional data). This is still better than the 28% rhombic pores found for synthesis on bare Au(111) without iodine, but does not reach the 92% achieved on I–Au(111). We interpret this finding as follows: On the one hand, iodine facilitates error correction during growth of organogold networks by inducing reversibility of carbon–Au–carbon bonds. This explains the improved network regularity as compared to synthesis on bare Au(111) in the absence of iodine. On the other hand, co-adsorption of iodine on Au(111) hinders network formation by reducing mobility of the molecules and imposing spatial constraints.47 These are caused by the expansion of the molecular structure during the formation of the porous organogold networks from the more densely packed self-assembly on bare Au(111) when the otherwise free areas are covered with iodine. This explains why the network regularity on Au(111) with co-adsorbed iodine remains lower than that of the networks synthesized on I–Au(111). Conversely, we attribute the high regularity of the organogold networks obtained on I–Au(111) as compared to bare Au(111) with co-adsorbed iodine, at least in part, to initial network formation on a more or less closed iodine layer rather than between co-adsorbed iodine atoms. However, the exact role of the closed iodine layer as a possible venue for formation of extended networks is less clear. First, it is not known how much iodine is already desorbed when debromination proceeds and more extended networks are formed. In addition, it remains presently unclear whether an iodine monolayer facilitates higher or lower mobility for the debrominated species.
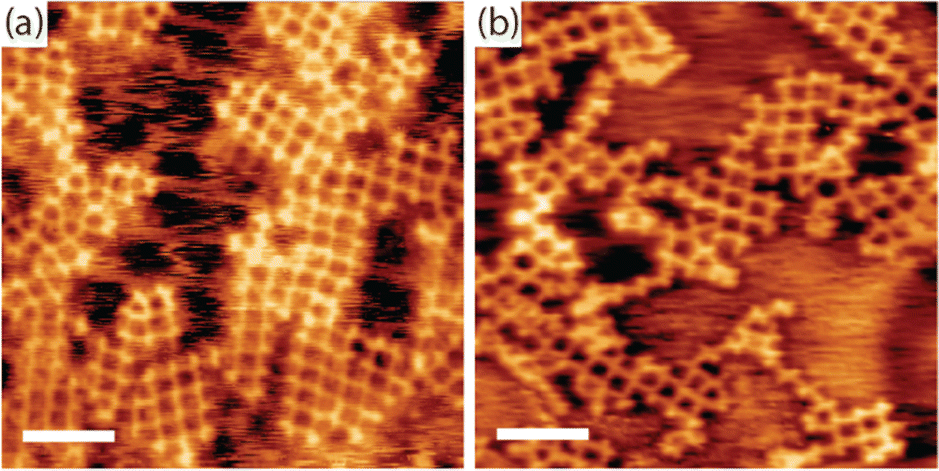 | ||
| Fig. 5 STM images of TBATT-derived organogold networks synthesized by co-deposition of TBATT and iodine on bare Au(111). After deposition of the molecules, the samples were exposed to 1 × 10−7 mbar I2 for (a) 10 min and (b) 1 min and annealed in UHV at 200 °C for 15 min to form the networks. The presence of iodine on the surface mediates the formation of organogold networks that are with 76% rhombic pores more regular than those obtained on bare Au(111) (28% rhombic pores), but still exhibit more defects than those obtained on I–Au(111) (92% rhombic pores) by inducing reversibility in the carbon–Au–carbon bonds. Analogous to Fig. 2, the iodine layer appears more or less closed when imaged at room temperature (b), whereas extended vacancy islands are clearly visible when imaged at low temperature (a). (tunneling parameters and scale bars: (a) +1.2 V, 120 pA, 6 nm, acquired at low temperature; (b) +1.0 V, 120 pA, 6 nm, acquired at room temperature). | ||
But a further control confirmed the iodine-induced reversibility as an experimental fact: Organogold networks synthesized on I–Au(111) as in Fig. 2(d) were annealed at 320 °C in a background pressure of 1 × 10−7 mbar of iodine. Afterwards, only an iodinated but otherwise empty I–Au(111) surface devoid of molecules was observed (cf. ESI,† Fig. S6). By contrast, the networks remained stable during annealing at similar temperatures in ultra-high vacuum, i.e. in the absence of iodine (cf.Fig. 3(a) and (b)). This pronounced difference is attributed to a weakening effect of the iodine on the strength of the carbon–Au–carbon bonds. While without iodine the carbon–Au–carbon bonds withstand the high temperatures according to their inherent strength, with iodine present these bonds can be broken, resulting in decomposition of the organogold networks and the subsequent desorption of the separated monomers.
Conclusion
In summary, we have demonstrated that I–Au(111) surfaces are viable substrates for the on-surface synthesis of 2D organogold networks by debrominative couplings. The organogold networks obtained for the TBATT precursor on I–Au(111) are highly regular with 92% rhombic pores, while those synthesized on bare Au(111) suffer from poor order with only 28% rhombic pores.Although the domains are still comparatively small, the number of repeat units is already sufficient for the anthra-tetrathiophene network to develop an electronic band structure. This is evident from our DFT calculations, where the HOMO–LUMO gap of the isolated 2 × 2 anthra-tetrathiophene tetramer of 0.78 eV has almost reached the electronic band gap of 0.71 eV of the periodic monolayer. Part of the reason is topology: the electronic properties converge faster in 2D than in 1D, essentially because more intermolecular bonds are formed per monomer.48 In this respect, the improved regularity on I–Au(111) is crucial for the emergence of defined electronic properties despite the still limited domain size.
Regarding the coupling mechanism on I–Au(111), we demonstrate that a certain permeability of the iodine passivation layer, as realized by an initially less dense packing in the iodine layer and promoted by a gradual iodine desorption during the thermal treatment, is imperative to facilitate the debromination. This makes the metal surface accessible to activate the reaction, but also to provide the Au-atoms for the formation of organogold networks. Therefore, to facilitate on-surface synthesis on I–Au(111), it is crucial to establish a workable iodine coverage. However, once the appropriate preparation parameters are found, the protocol has proven to be highly reproducible and reliable. An unambiguous effect of the iodine monolayer for the formation of highly regular organogold networks is the iodine-induced reversibility of the strong carbon–Au–carbon bonds to facilitate dynamic error correction. This is confirmed by experiments in which more regular organogold networks were obtained on bare Au(111) when iodine was co-deposited. An additional conceivable effect of the iodine monolayer is that network formation on top of the iodine layer at least initially alleviates the spatial constraints that arise when reactants and iodine are directly co-adsorbed on bare Au(111). The structural quality obtained when the molecules are adsorbed on bare Au(111) and iodine is just co-adsorbed does not reach the high regularity obtained for the synthesis on I–Au(111). So initial adsorption on top of a closed iodine layer must play a role. However, some mechanistic aspects still remain not fully explored. There is a general consensus that higher monomer mobility favors the formation of regular structures by avoiding diffusion-limited aggregation.49,50 But for I–Au(111) it is also unclear whether the mobility of activated, i.e. debrominated, monomers is increased or decreased compared to bare Au(111).
Although the anthra-tetrathiophene repeat units are not directly linked by carbon–carbon bonds, the organometallic carbon–Au–carbon bonds also endow the networks with all the desirable properties, that is electronic conjugation and robustness. We are optimistic that synthesis on I–Au(111) could expand the toolbox of on-surface synthesis and provide a more general approach for cases where direct reaction on bare metal surfaces does not yield the desired products.
Author contributions
M. L. and W. M. H. conceived and designed the study. D. L. P. and X.F. synthesized and purified the compound. A. B.-B. and G. G. carried out the STM experiments. R. G. performed the DFT calculations and analyzed the results. M. L. co-wrote the manuscript, with contributions and approval from all authors.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work has been funded from the Deutsche Forschungsgemeinschaft (DFG) grant 415284307. A. B.-B. acknowledges financial support by the Bayerische Forschungsstiftung (DOK-183-20).References
- L. Grill, M. Dyer, L. Lafferentz, M. Persson, M. V. Peters and S. Hecht, Nano-Architectures by Covalent Assembly of Molecular Building Blocks, Nat. Nanotechnol., 2007, 2, 687–691 CrossRef CAS PubMed.
- M. Lackinger, Surface-Assisted Ullmann Coupling, Chem. Commun., 2017, 53, 7872–7885 RSC.
- S. Clair and D. G. de Oteyza, Controlling a Chemical Coupling Reaction on a Surface: Tools and Strategies for on-Surface Synthesis, Chem. Rev., 2019, 119, 4717–4776 CrossRef CAS PubMed.
- L. Grill and S. Hecht, Covalent on-Surface Polymerization, Nat. Chem., 2020, 12, 115–130 CrossRef CAS PubMed.
- J. M. Cai, P. Ruffieux, R. Jaafar, M. Bieri, T. Braun, S. Blankenburg, M. Muoth, A. P. Seitsonen, M. Saleh, X. L. Feng, K. Müllen and R. Fasel, Atomically Precise Bottom-up Fabrication of Graphene Nanoribbons, Nature, 2010, 466, 470–473 CrossRef CAS PubMed.
- P. Ruffieux, S. Y. Wang, B. Yang, C. Sanchez-Sanchez, J. Liu, T. Dienel, L. Talirz, P. Shinde, C. A. Pignedoli, D. Passerone, T. Dumslaff, X. L. Feng, K. Müllen and R. Fasel, On-Surface Synthesis of Graphene Nanoribbons with Zigzag Edge Topology, Nature, 2016, 531, 489–492 CrossRef CAS PubMed.
- R. Gutzler, H. Walch, G. Eder, S. Kloft, W. M. Heckl and M. Lackinger, Surface Mediated Synthesis of 2d Covalent Organic Frameworks: 1,3,5-Tris(4-Bromophenyl)Benzene on Graphite(001), Cu(111), and Ag(110), Chem. Commun., 2009, 4456–4458 RSC.
- M. Bieri, S. Blankenburg, M. Kivala, C. A. Pignedoli, P. Ruffieux, K. Müllen and R. Fasel, Surface-Supported 2d Heterotriangulene Polymers, Chem. Commun., 2011, 47, 10239–10241 RSC.
- L. Cardenas, R. Gutzler, J. Lipton-Duffin, C. Y. Fu, J. L. Brusso, L. E. Dinca, M. Vondracak, Y. Fagot-Revurat, D. Malterre, F. Rosei and D. F. Perepichka, Synthesis and Electronic Structure of a Two Dimensional Π-Conjugated Polythiophene, Chem. Sci., 2013, 4, 3263–3268 RSC.
- M. Lischka, G. S. Michelitsch, N. Martsinovich, J. Eichhorn, A. Rastgoo-Lahrood, T. Strunskus, R. Breuer, K. Reuter, M. Schmittel and M. Lackinger, Remote Functionalization in Surface-Assisted Dehalogenation by Conformational Mechanics: Organometallic Self-Assembly of 3,3′,5,5′-Tetrabromo-2,2′,4,4′,6,6′-Hexafluorobiphenyl on Ag(111), Nanoscale, 2018, 10, 12035–12044 RSC.
- A. Rastgoo Lahrood, J. Björk, W. M. Heckl and M. Lackinger, 1,3-Diiodobenzene on Cu(111) – an Exceptional Case of on-Surface Ullmann Coupling, Chem. Commun., 2015, 51, 13301–13304 RSC.
- A. Saywell, W. Greń, G. Franc, A. Gourdon, X. Bouju and L. Grill, Manipulating the Conformation of Single Organometallic Chains on Au(111), J. Phys. Chem. C, 2013, 118, 1719–1728 CrossRef.
- H. M. Zhang, J. H. Franke, D. Y. Zhong, Y. Li, A. Timmer, O. D. Arado, H. Mönig, H. Wang, L. F. Chi, Z. H. Wang, K. Müllen and H. Fuchs, Surface Supported Gold-Organic Hybrids: On-Surface Synthesis and Surface Directed Orientation, Small, 2014, 10, 1361–1368 CrossRef CAS PubMed.
- H. M. Zhang, H. P. Lin, K. W. Sun, L. Chen, Y. Zagranyarski, N. Aghdassi, S. Duhm, Q. Li, D. Y. Zhong, Y. Y. Li, K. Müllen, H. Fuchs and L. F. Chi, On-Surface Synthesis of Rylene-Type Graphene Nanoribbons, J. Am. Chem. Soc., 2015, 137, 4022–4025 CrossRef CAS PubMed.
- A. Rastgoo-Lahrood, N. Martsinovich, M. Lischka, J. Eichhorn, P. Szabelski, D. Nieckarz, T. Strunskus, K. Das, M. Schmittel, W. M. Heckl and M. Lackinger, From Au-Thiolate Chains to Thioether Sierpinski Triangles: The Versatile Surface Chemistry of 1,3,5-Tris(4-Mercaptophenyl)Benzene on Au(111), ACS Nano, 2016, 10, 10901–10911 CrossRef CAS PubMed.
- Q. Sun, L. Cai, H. Ma, C. Yuan and W. Xu, Dehalogenative Homocoupling of Terminal Alkynyl Bromides on Au(111): Incorporation of Acetylenic Scaffolding into Surface Nanostructures, ACS Nano, 2016, 10, 7023–7030 CrossRef CAS PubMed.
- J. I. Urgel, H. Hayashi, M. Di Giovannantonio, C. A. Pignedoli, S. Mishra, O. Deniz, M. Yamashita, T. Dienel, P. Ruffieux, H. Yamada and R. Fasel, On-Surface Synthesis of Heptacene Organometallic Complexes, J. Am. Chem. Soc., 2017, 139, 11658–11661 CrossRef CAS PubMed.
- M. Z. Liu, M. X. Liu, L. M. She, Z. Q. Zha, J. L. Pan, S. C. Li, T. Li, Y. Y. He, Z. Y. Cai, J. B. Wang, Y. Zheng, X. H. Qiu and D. Y. Zhong, Graphene-Like Nanoribbons Periodically Embedded with Four- and Eight-Membered Rings, Nat. Commun., 2017, 8, 14924 CrossRef CAS PubMed.
- G. Galeotti, F. De Marchi, T. Taerum, L. V. Besteiro, M. El Garah, J. Lipton-Duffin, M. Ebrahimi, D. F. Perepichka and F. Rosei, Surface-Mediated Assembly, Polymerization and Degradation of Thiophene-Based Monomers, Chem. Sci., 2019, 10, 5167–5175 RSC.
- R. Zuzak, A. Jancarík, A. Gourdon, M. Szymonski and S. Godlewski, On-Surface Synthesis with Atomic Hydrogen, ACS Nano, 2020, 14, 13316–13323 CrossRef CAS PubMed.
- Q. G. Zhong, K. F. Niu, L. Chen, H. M. Zhang, D. Ebeling, J. Björk, K. Müllen, A. Schirmeisen and L. F. Chi, Substrate-Modulated Synthesis of Metal-Organic Hybrids by Tunable Multiple Aryl-Metal Bonds, J. Am. Chem. Soc., 2022, 144, 8214–8222 CrossRef CAS PubMed.
- S. Achilli, F. Tumino, A. Rabia, A. O. Biroli, A. Li Bassi, A. Bossi, N. Manini, G. Onida, G. Fratesi and C. S. Casari, Steric Hindrance in the on-Surface Synthesis of Diethynyl-Linked Anthracene Polymers, Phys. Chem. Chem. Phys., 2022, 24, 13616–13624 RSC.
- M. Lischka, M. Fritton, J. Eichhorn, V. S. Vyas, T. Strunskus, B. V. Lotsch, J. Bjork, W. M. Heckl and M. Lackinger, On-Surface Polymerization of 1,6-Dibromo-3,8-Diiodpyrene-a Comparative Study on Au(111) Versus Ag(111) by Stm, Xps, and Nexafs, J. Phys. Chem. C, 2018, 122, 5967–5977 CrossRef CAS.
- M. Fritton, K. Otte, J. Bjork, P. K. Biswas, W. M. Heckl, M. Schmittel and M. Lackinger, The Influence of Ortho-Methyl Substitution in Organometallic Self-Assembly – a Comparative Study on Cu(111) vs. Ag(111), Chem. Commun., 2018, 54, 9745–9748 RSC.
- K. A. Simonov, A. V. Generalov, A. S. Vinogradov, G. I. Svirskiy, A. A. Cafolla, C. McGuinness, T. Taketsugu, A. Lyalin, N. Martensson and A. B. Preobrajenski, Synthesis of Armchair Graphene Nanoribbons from the 10,10′-Dibromo-9,9′-Bianthracene Molecules on Ag(111): The Role of Organometallic Intermediates, Sci. Rep., 2018, 8, 3506 CrossRef CAS PubMed.
- J. Eichhorn, T. Strunskus, A. Rastgoo-Lahrood, D. Samanta, M. Schmittel and M. Lackinger, On-Surface Ullmann Polymerization Via Intermediate Organometallic Networks on Ag(111), Chem. Commun., 2014, 50, 7680–7682 RSC.
- C. K. Krug, Q. Fan, F. Fillsack, J. Glowatzki, N. Trebel, L. J. Heuplick, T. Koehler and J. M. Gottfried, Organometallic Ring vs. Chain Formation Beyond Kinetic Control: Steering Their Equilibrium in Two-Dimensional Confinement, Chem. Commun., 2018, 54, 9741–9744 RSC.
- C. X. Wang, J. L. Chen, C. H. Shu, K. J. Shi and P. N. Liu, On-Surface Synthesis of 2d Cofs on Cu(111) Via the Formation of Thermodynamically Stable Organometallic Networks as the Template, Phys. Chem. Chem. Phys., 2019, 21, 13222–13229 RSC.
- R. Pawlak, X. S. Liu, S. Ninova, P. D’Astolfo, C. Drechsel, J. C. Liu, R. Häner, S. Decurtins, U. Aschauer, S. X. Liu and E. Meyer, On-Surface Synthesis of Nitrogen-Doped Kagome Graphene, Angew. Chem., Int. Ed., 2021, 60, 8370–8375 CrossRef CAS PubMed.
- X. Y. Li, D. Han, T. C. Qin, J. J. Xiong, J. M. Huang, T. Wang, H. H. Ding, J. Hu, Q. Xu and J. F. Zhu, Selective Synthesis of Kagome Nanoporous Graphene on Ag(111) Via an Organometallic Template, Nanoscale, 2022, 14, 6239–6247 RSC.
- Z. C. Yang, J. Gebhardt, T. A. Schaub, T. Sander, J. Schonamsgruber, H. Soni, A. Gorling, M. Kivala and S. Maier, Two-Dimensional Delocalized States in Organometallic Bis-Acetylide Networks on Ag(111), Nanoscale, 2018, 10, 3769–3776 RSC.
- S. W. Li, R. X. Zhang, L. X. Kang, D. Y. Li, Y. L. Xie, C. X. Wang and P. N. Liu, Steering Metal-Organic Network Structures through Conformations and Configurations on Surfaces, ACS Nano, 2021, 15, 18014–18022 CrossRef CAS PubMed.
- X. L. Hu, N. Q. Su and W. H. Fang, Metallated Graphynes as Efficient Single-Atom Electrocatalysts for Nitric Oxide Reduction to Ammonia, J. Phys. Chem. C, 2023, 127, 11026–11039 CrossRef CAS.
- J. L. Brusso, O. D. Hirst, A. Dadvand, S. Ganesan, F. Cicoira, C. M. Robertson, R. T. Oakley, F. Rosei and D. F. Perepichkat, Two-Dimensional Structural Motif in Thienoacene Semiconductors: Synthesis, Structure, and Properties of Tetrathienoanthracene Isomers, Chem. Mater., 2008, 20, 2484–2494 CrossRef CAS.
- R. Gutzler, L. Cardenas, J. Lipton-Duffin, M. El Garah, L. E. Dinca, C. E. Szakacs, C. Fu, M. Gallagher, M. Vondracek, M. Rybachuk, D. F. Perepichka and F. Rosei, Ullmann-Type Coupling of Brominated Tetrathienoanthracene on Copper and Silver, Nanoscale, 2014, 6, 2660–2668 RSC.
- C. Morchutt, J. Björk, S. Krotzky, R. Gutzler and K. Kern, Covalent Coupling Via Dehalogenation on Ni(111) Supported Boron Nitride and Graphene, Chem. Commun., 2015, 51, 2440–2443 RSC.
- W. Zhao, L. Dong, C. Huang, Z. M. Win and N. A. Lin, Cu- and Pd-Catalyzed Ullmann Reaction on a Hexagonal Boron Nitride Layer, Chem. Commun., 2016, 52, 13225–13228 RSC.
- A. Rastgoo-Lahrood, J. Björk, M. Lischka, J. Eichhorn, S. Kloft, M. Fritton, T. Strunskus, D. Samanta, M. Schmittel, W. M. Heckl and M. Lackinger, Post-Synthetic Decoupling of on-Surface-Synthesized Covalent Nanostructures from Ag(111), Angew. Chem., Int. Ed., 2016, 55, 7650–7654 CrossRef CAS PubMed.
- A. Rastgoo-Lahrood, M. Lischka, J. Eichhorn, D. Samanta, M. Schmittel, W. M. Heckl and M. Lackinger, Reversible Intercalation of Iodine Monolayers between on-Surface Synthesised Covalent Polyphenylene Networks and Au(111), Nanoscale, 2017, 9, 4995–5001 RSC.
- L. Grossmann, D. A. Duncan, S. P. Jarvis, R. G. Jones, S. De, J. Rosen, M. Schmittel, W. M. Heckl, J. Björk and M. Lackinger, Evolution of Adsorption Heights in the on-Surface Synthesis and Decoupling of Covalent Organic Networks on Ag(111) by Normal-Incidence X-Ray Standing Wave, Nanoscale Horiz., 2021, 7, 51–62 RSC.
- A. Badami-Behjat, P. S. Deimel, F. Allegretti, E. Ringel, K. Mahata, M. Schmittel, J. V. Barth, W. M. Heckl and M. Lackinger, Versatile Role of Molecule-Surface Interactions for Monolayer Self-Assembly at Liquid–Solid Interfaces: Substrate-Induced Polymorphism, Thermodynamic Stability, and New Polymorphs, Chem. Mater., 2022, 34, 8876–8884 CrossRef CAS.
- L. Grossmann, E. Ringel, A. Rastgoo-Lahrood, B. T. King, J. Rosen, W. M. Heckl, D. Opris, J. Björk and M. Lackinger, Steering Self-Assembly of Three-Dimensional Iptycenes on Au(111) by Tuning Molecule-Surface Interactions, Angew. Chem., Int. Ed., 2022, 61, e202201044 CrossRef CAS PubMed.
- B. G. Bravo, S. L. Michelhaugh, M. P. Soriaga, I. Villegas, D. W. Suggs and J. L. Stickney, Anodic Underpotential Deposition and Cathodic Stripping of Iodine at Polycrystalline and Single-Crystal Gold – Studies by Leed, Aes, Xps, and Electrochemistry, J. Phys. Chem., 1991, 95, 5245–5249 CrossRef CAS.
- X. P. Gao and M. J. Weaver, Probing Redox-Induced Molecular-Transformations by Atomic-Resolution Scanning Tunneling Microscopy – Iodide Adsorption and Electrooxidation on Au(111) in Aqueous-Solution, J. Am. Chem. Soc., 1992, 114, 8544–8551 CrossRef CAS.
- R. Gutzler, C. Y. Fu, A. Dadvand, Y. Hua, J. M. MacLeod, F. Rosei and D. F. Perepichka, Halogen Bonds in 2d Supramolecular Self-Assembly of Organic Semiconductors, Nanoscale, 2012, 4, 5965–5971 RSC.
- W. Song, N. Martsinovich, W. M. Heckl and M. Lackinger, Thermodynamics of Halogen Bonded Monolayer Self-Assembly at the Liquid–Solid Interface, Chem. Commun., 2014, 50, 13465–13468 RSC.
- S. Schlögl, W. M. Heckl and M. Lackinger, On-Surface Radical Addition of Triply Iodinated Monomers on Au(111)—the Influence of Monomer Size and Thermal Post-Processing, Surf. Sci., 2012, 606, 999–1004 CrossRef.
- R. Gutzler and D. F. Perepichka, Pi-Electron Conjugation in Two Dimensions, J. Am. Chem. Soc., 2013, 135, 16585–16594 CrossRef CAS PubMed.
- M. Bieri, M. T. Nguyen, O. Groning, J. M. Cai, M. Treier, K. Ait-Mansour, P. Ruffieux, C. A. Pignedoli, D. Passerone, M. Kastler, K. Müllen and R. Fasel, Two-Dimensional Polymer Formation on Surfaces: Insight into the Roles of Precursor Mobility and Reactivity, J. Am. Chem. Soc., 2010, 132, 16669–16676 CrossRef CAS PubMed.
- J. Eichhorn, D. Nieckarz, O. Ochs, D. Samanta, M. Schmittel, P. J. Szabelski and M. Lackinger, On-Surface Ullmann Coupling: The Influence of Kinetic Reaction Parameters on the Morphology and Quality of Covalent Networks, ACS Nano, 2014, 8, 7880–7889 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nh00496a |
| This journal is © The Royal Society of Chemistry 2024 |